

Abstract
Although the octapeptide hormone angiotensin II (Ang II) regulates cardiovascular and renal homeostasis through the Ang II type 1 receptor (AT1R), overstimulation of AT1R causes various human diseases, such as hypertension and cardiac hypertrophy. Therefore, AT1R blockers (ARBs) have been widely used as therapeutic drugs for these diseases. Recent basic research and clinical studies have resulted in the discovery of interesting phenomena associated with AT1R function. For example, ligand-independent activation of AT1R by mechanical stress and agonistic autoantibodies, as well as via receptor mutations, has been shown to decrease the inverse agonistic efficacy of ARBs, though the molecular mechanisms of such phenomena had remained elusive until recently. Furthermore, although AT1R is believed to exist as a monomer, recent studies have demonstrated that AT1R can homodimerize and heterodimerize with other G-protein coupled receptors (GPCR), altering the receptor signaling properties. Therefore, formation of both AT1R homodimers and AT1R-GPCR heterodimers may be involved in the pathogenesis of human disease states, such as atherosclerosis and preeclampsia. Finally, biased AT1R ligands that can preferentially activate the β-arrestin-mediated signaling pathway have been discovered. Such β-arrestin-biased AT1R ligands may be better therapeutic drugs for cardiovascular diseases. New findings on AT1R described herein could provide a conceptual framework for application of ARBs in the treatment of diseases, as well as for novel drug development. Since AT1R is an extensively studied member of the GPCR superfamily encoded in the human genome, this review is relevant for understanding the functions of other members of this superfamily.
Keywords: angiotensin II type 1 receptor, G-protein coupled receptor, inverse agonism, receptor dimerization, biased agonism, conformational change
Chemical compounds described in this article: Losartan (PubChem CID: 3961), EXP3174 (PubChem CID: 108185), Candesartan (PubChem CID: 2541), Valsartan (PubChem CID: 60846), Irbesartan (PubChem CID: 3749), Olmesartan (PubChem CID: 158781), Azilsartan (PubChem CID: 9825285), Telmisartan (PubChem CID: 65999), Eprosartan (PubChem CID: 5281037)
Graphical abstract
1. Introduction
G protein-coupled receptors (GPCRs) are characterized by a seven-transmembrane α-helical architecture. GPCRs constitute one of the largest gene superfamilies, encoding more than 800 GPCR genes in the human genome [1]. Activation of GPCRs promotes intracellular signaling cascades and regulates numerous physiological and pathological processes. Therefore, GPCRs are known to be major targets for treating human diseases. In fact, approximately 26% of clinically available drugs target GPCRs [2].
Physiological levels of the octapeptide hormone angiotensin II (Ang II) regulate blood pressure and body fluid homeostasis, and also maintain cardiovascular and renal homeostasis by activation of the Ang II type 1 receptor (AT1R), which belongs to the GPCR superfamily. Human disease states, such as hypertension, coronary artery disease, cardiac hypertrophy, heart failure, arrhythmia, stroke, diabetic nephropathy, and ischemic heart and renal diseases are associated with overstimulation of AT1R [3–6]. These disease conditions can be treated with AT1R blocking drugs, known as ARBs. Recently, a number of interesting phenomena have been discovered regarding AT1R function. For example, studies have demonstrated that mechanical stress and AT1R-directed agonistic autoantibodies can activate AT1R without Ang II binding (Fig. 1) [7–11]. Such ligand-independent activation of AT1R may occur clinically, as in hypertension, cardiac overload conditions or in preeclampsia [12, 13]. Inverse agonists such as candesartan can inhibit ligand-independent activation of AT1R and may exhibit enhanced therapeutic effects for these disease states [12, 13].
Figure 1.
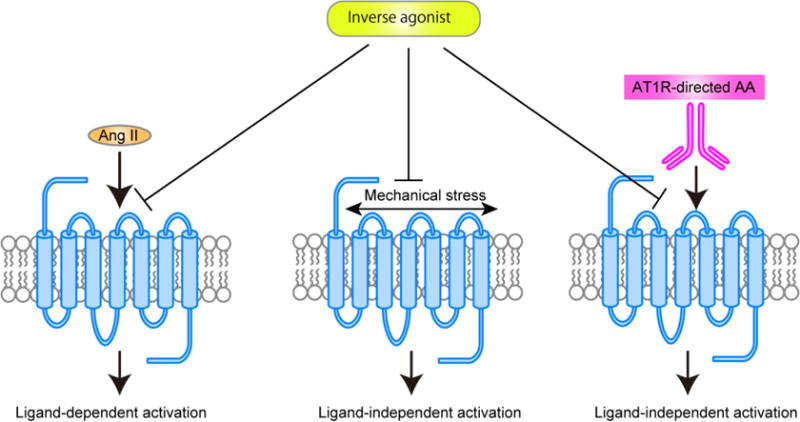
Ligand-dependent and ligand-independent activation of AT1R. Ang II binds to AT1R and causes ligand-dependent AT1R activation. On the other hand, mechanical stress and AT1R-directed autoantibodies (AT1R-directed AA) cause ligand-independent AT1R activation. Inverse agonists block not only ligand-dependent activation, but also ligand-independent AT1R activation.
Experiments on constitutively active AT1R mutants that mimic the active AT1R conformation have shown that the ability of ARBs to decrease the inositol phosphate (IP) accumulation of the constitutively active mutant receptor is reduced [14, 15]. We recently identified the molecular mechanism associated with the decrease in inverse agonism [16]. Furthermore, studies revealed that AT1R can form both homodimers and AT1R-GPCR heterodimers, which can alter ligand binding and receptor function [17–25]. Both AT1R homodimers and AT1R-GPCR heterodimers play important roles in the pathogenesis of human disease states, such as atherosclerosis and preeclampsia [18, 24, 26]. Finally, GPCR ligands that can preferentially activate one signaling pathway, referred to as biased agonists, have recently been discovered [27], including biased AT1R agonists that preferentially activate the β-arrestin signaling pathway [28, 29]. Such β-arrestin-biased AT1R agonists may exhibit enhanced therapeutic potential for cardiovascular diseases [30, 31]. In this review, we describe the current state of AT1R research, with a specific focus on inverse agonism, receptor dimerization, and biased agonism.
2. Structural classification of ARBs
ARBs are non-peptide small molecular weight compounds with high specificity for AT1R. Eight ARBs (Fig. 2) are clinically used as antihypertensive drugs, all of which function as competitive inhibitors of Ang II binding to AT1R [32–37]. Furthermore, ARBs harbor variable inverse agonist activity for AT1R, which may be due to differences in their molecular structures. Five of the eight ARBs (losartan, candesartan, olmesartan, valsartan, irbesartan) share a common biphenyl-tetrazole scaffold and are referred to as biphenyl-tetrazole ARBs. Azilsartan is derived from candesartan by substitution of the oxadiazole for tetrazole, and thus contains a biphenyl-oxadiazole scaffold. Telmisartan and eprosartan exhibit structural differences compared to the biphenyl-tetrazole ARBs. In particular, telmisartan contains a carboxyl group instead of a tetrazole at the 2’ position of the biphenyl moiety, in addition to bulky bis-benzimidazole rings. Eprosartan contains both carboxyphenyl and thiophenepropanoic acid groups.
Figure 2.

Chemical structures of losartan, olmesartan, valsartan, irbesartan, candesartan, azilsartan, telmisartan and eprosartan. Five of the eight ARBs share a common biphenyl tetrazole scaffold structure. Azilsartan contains a biphenyl-oxadiazole scaffold. Telmisartan and eprosartan do not belong to the class of biphenyl-tetrazole ARBs. The biphenyl-tetrazole is outlined by a red dashed box. The bis-benzimidazole rings of telmisartan are outlined by a light green dashed box. The carboxyphenyl and thiophenepropanoic acid groups of eprosartan are outlined by yellow and light blue dashed boxes, respectively.
We recently analyzed the structure-function relationships of losartan, EXP3174 (an active metabolite of losartan), valsartan and irbesartan for the basal and constitutively active states of AT1R, and identified the molecular mechanism of decreased maximal inverse agonist activity of the ARBs for the active state of AT1R compared to the ground state [16]. Details of the mechanism are described in Sections 3 through 5.
3. Crystal structure of AT1R provides precise ligand-receptor interactions
As the crystal structure of AT1R is not available, ligand-receptor interactions were previously analyzed using models of AT1R based on the crystal structures of other GPCRs, such as bovine rhodopsin, β2-adrenergic receptor and CXCR4 [32, 38–42]. An understanding of the structural basis for AT1R function is therefore limited. However, the crystal structures of human AT1R bound to the biphenyl-tetrazole ARBs ZD7155 and olmesartan were recently solved (Fig. 3) [43, 44], providing insight into understanding the structural basis for AT1R function, and demonstrating considerable differences from the previous models of AT1R based on the crystal structures of other GPCRs (Fig. 4). This landmark achievement paved the way for determination of the precise docking models of various ARBs based on the solved crystal structure [16, 43]. Energy-based docking simulations of the ARBs using the crystal structure of human AT1R revealed that the interactions of the biphenyl-tetrazole ARBs with AT1R were completely different from previously proposed ARB-AT1R interactions [16, 43, 44]. For example, differences of losartan binding mode between in a previously proposed AT1R model based on bovine rhodopsin structure and crystal structure of human AT1R are shown in Fig. 4.
Figure 3.
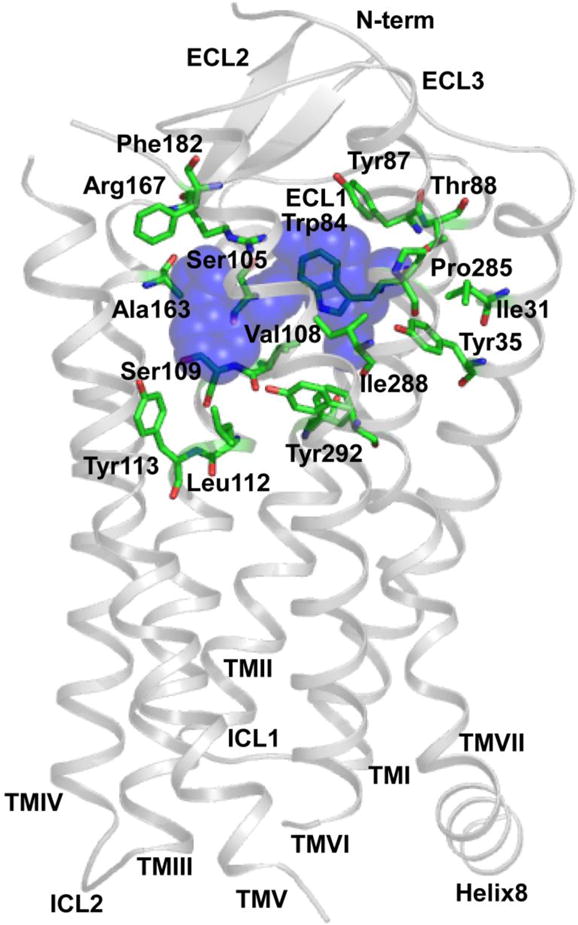
Crystal structure of human AT1R bound to ZD7155. ZD7155 is shown as blue spheres. The following residues interact with ZD7155: Ile31TM1, Tyr35TM1, Trp84TM2, Tyr87TM2, Thr88TM2, Ser105TM3, Val108TM3, Ser109TM3, Leu112TM3, Tyr113TM3, Ala163TM4, Arg167TM4, Phe182ECL2, Pro285TM7, Ile288TM7 and Tyr292TM7. Modified with permission from Molecular Pharmacology [16].
Figure 4.
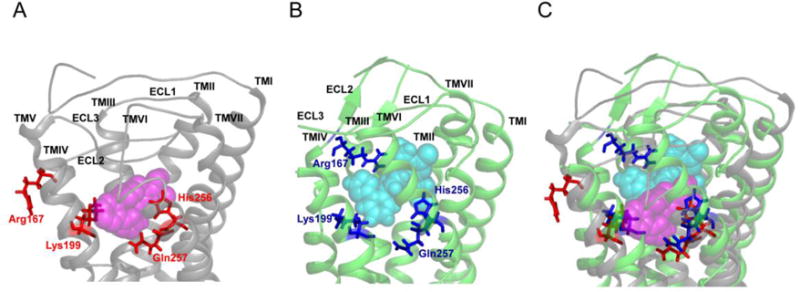
Differences in losartan binding to an AT1R model and the crystal structure of human AT1R. (A) Docking model of losartan bound to an AT1R model based on the crystal structure of bovine rhodopsin (AT1R model). Modified with permission from the Journal of Biological Chemistry [32]. (B) Docking model of losartan bound to the crystal structure of human AT1R. Modified with permission from Molecular Pharmacology [16]. (C) Superposition of losartan bound to both the AT1R model (gray ribbon structure) and the crystal structure of human AT1R (green ribbon structure); losartan is shown as magenta spheres in the former, and as cyan spheres in the latter, while the residues Arg167, Lys199, His256 and Gln257 are denoted as red sticks in the former, and as blue sticks in latter.
The docking results showed that all biphenyl-tetrazole ARBs were bound in similar orientations in the ligand-binding pocket (Figs. 5A–E). Although the previous models of AT1R based on the crystal structures of other GPCRs proposed that the tetrazole group, a common acidic moiety present in the biphenyl-tetrazole ARBs, interacts with Lys199 in the transmembrane (TM) 5 domain, or doubly interacts with Lys199TM5 and His256TM6, or triply interacts with Lys199TM5, His256TM6 and Gln257TM6 [32, 38–40], the ARB/AT1R crystal structure suggests that these previously proposed interactions are incorrect (Fig. 4) [32, 43, 44].
Figure 5.

AT1R interaction with losartan (A), EXP3174 (B), candesartan (C), valsartan (D) and irbesartan (E) in the AT1R crystal structure. The ARBs are shown as sticks with blue carbons. Residues with the side-chains within 10 Å from each ARB are shown. The hydrogen bonds are shown as black dashed lines. Modified with permission from Cell [43].
Docking results using the crystal structure of human AT1R indicate that the tetrazole moiety of the biphenyl-tetrazole ARBs actually forms salt bridges/hydrogen bonds with Arg167 located in the second extracellular loop (ECL2) (Figs. 5A–E). Although EXP3174 and candesartan bind in orientations similar to that of losartan, the carboxyl groups of EXP3174 and candesartan could form additional salt bridges with Arg167ECL2 (Figs. 5B and C) [16, 43]. Moreover, the tetrazole group of candesartan is predicted to form a second salt bridge with Lys199TM5 (Fig. 5C) [43]. These additional salt bridges may be responsible for the differences between the surmountable behavior of losartan and the insurmountable behavior of EXP3174 and candesartan (discussed in Section 4).
The imidazole rings of losartan and EXP3174, as well as equivalent substituents in irbesartan and candesartan, form π-π contacts with Trp84 in TM2 (Figs. 5A–C, E) and interact with the following residues that constitute the common floor of the binding pocket: Val108TM3, Ser109TM3, Tyr113TM3, Ala163TM4, Gln257TM6, Tyr292TM7 and Asn295TM7 (Fig. 6). In contrast, the N-acylated valine residue of valsartan, that substitutes the imidazole ring, does not interact with Trp84 (Fig. 5D), but does interact with the above residues (Fig. 6). The short alkyl tails of the biphenyl-tetrazole ARBs interact with Tyr35TM1, and their biphenyl rings interact with Val108TM3 and Ser109TM3, as well as Trp253TM6 and Gln257TM6. In addition, the biphenyl-tetrazole ARBs may interact hydrophobically with Tyr113TM3, Phe182ECL2, Tyr184ECL2 and His256TM6. The residues Phe77TM2, Tyr87TM2, Ser105TM3, Leu112TM3 and Ile288TM7 also contribute to ARB-AT1R interactions and the structure of the ligand-binding pocket [43].
Figure 6.

The common floor of the ARB binding pocket. The ARBs are shown as sticks with blue (losartan), red (EXP3174), magenta (candesartan), cyan (valsartan) and yellow (irbesartan) carbons. The residues that comprise the common floor of the binding pocket are shown as green spheres.
4. Mechanism of insurmountable AT1R antagonism
The ARBs are classified as either surmountable or insurmountable according to their inhibitory patterns (based on the Ang II concentration-response curve) and dissociation rates from the receptor [45]. The surmountable ARBs result in a parallel rightward shift of the Ang II concentration-response curve without affecting the maximal response, and exhibit a fast dissociation rate from the receptor. On the other hand, the insurmountable ARBs result in a partial to complete waning of the response accompanied by a parallel rightward shift of the Ang II concentration-response curve and slow dissociation rates from the receptor. Losartan and eprosartan are classified as surmountable ARBs [46], whereas EXP3174, valsartan, irbesartan, candesartan, olmesartan, telmisartan and azilsartan are classified as insurmountable ARBs [32–34, 37].
Herein, the molecular mechanism of insurmountable AT1R antagonism is discussed based on the results of previous site-directed mutagenesis studies for losartan, EXP3174 and candesartan [32, 47], as well as docking simulations using the crystal structure of human AT1R [43]. Docking simulations indicated that the tetrazole moieties of all three ARBs form salt bridges/hydrogen bonds with Arg167ECL2 (Fig. 5A–C). Furthermore, EXP3174 and candesartan, but not losartan, form additional salt bridges with Arg167ECL2. During EXP3174 binding, one oxygen atom of the carboxyl group forms a salt bridge with Arg167ECL2 (Fig. 5B), whereas two oxygen atoms of the carboxyl group form two salt bridges with Arg167ECL2 during candesartan binding (Fig. 5C). Differences in the distances and angles between the carboxyl group and Arg167ECL2 are partly due to differences in the structures of EXP3174 and candesartan. These results suggest that the insurmountable ARBs form at least two salt bridges with Arg167ECL2, with the strength of insurmountable antagonism correlating with the number of salt bridges formed. In addition, the docking simulations also suggested that the flexible side chain of Lys199TM5 provides some conformational heterogeneity in AT1R. The amino group of the side chain of this residue may reach the carboxyl group of candesartan by forming a salt bridge (Fig. 5C), or it may interact with the biphenyl scaffold in losartan and EXP3174 via water-mediated interactions, which may explain the stronger insurmountable antagonism of candesartan compared to EXP3174, as well as the increased dissociation rate of candesartan from the receptor and decreased binding affinity of losartan, EXP3174 and candesartan upon mutation of Lys199 [47, 48].
As described in Section 3, the following residues constitute the common floor of the binding pocket among the different ARBs: Val108TM3, Ser109TM3, Tyr113TM3, Ala163TM4, Gln257TM6, Tyr292TM7 and Asn295TM7 (Fig. 6) [16]. Therefore, mutation of these residues may alter the conformation of the binding pocket floor, which may explain the decreased binding affinity of losartan, EXP3174 and candesartan upon mutation of these residues [16, 32, 49–52]. In addition, mutation of Gln257TM6 has been shown to transform the binding mode of candesartan and EXP3174 from insurmountable antagonism to surmountable antagonism [32]. Conformational alteration of the binding pocket floor upon mutation of Gln257TM6 may cause attenuation of the interaction of EXP3174 and candesartan with Arg167ECL2 and Lys199TM5. Although it has not been examined whether mutations of Val108TM3, Ser109TM3, Tyr113TM3, Ala163TM4, Tyr292TM7 or Asn295TM7 transform the binding property of the ARBs from insurmountable antagonism to surmountable antagonism, these residues, along with Gln257TM6, may play an important role in the mechanism of insurmountable antagonism. Taken together, we propose that the above residues support the structure of the binding pocket, enabling EXP3174 and candesartan to strongly interact with Arg167ECL2, which could explain their insurmountable behavior. Additional interactions of candesartan with Arg167 ECL2 and Lys199TM5 make it a stronger insurmountable antagonist than EXP3174.
5. Basis of inverse agonism of biphenyl-tetrazole ARBs for AT1R in the ground state
As wild-type AT1R (WT-AT1R) displays only modest constitutive activity, previous studies could not examine the molecular mechanism of inverse agonism of the biphenyl-tetrazole ARBs for AT1R in the ground state [38, 39]. However, we found that constitutive IP accumulation was increased in a linear fashion along with the incubation period, reaching levels adequate to measure the constitutive activity of WT-AT1R, and enabling us to examine the inverse agonist efficacy using WT-AT1R. Thus, we identified the molecular mechanism of inverse agonism of the biphenyl-tetrazole ARBs for the ground state of AT1R [16].
A hydrogen bond (H-bond) between Asn111TM3 and Asn295TM7 (Asn111-Asn295 H-bond) was previously suggested to stabilize AT1R in the inactive state, which is confirmed by the crystal structure of human AT1R bound to ZD7155 [43]. A recent molecular dynamics simulation proposed that AT1R activation disrupts this H-bond, leading to Asn295TM7 interaction with the conserved Asp74TM2 to form the Asn46-Asp74-Asn295 H-bond network [42]. This H-bond network in the active state involves additional residues, namely Trp253TM6 from the “toggle-switch” motif [53, 54], and Phe77TM2, Val108TM3, Ile288TM7, Tyr292TM7 and Asn298TM7 from the NPxxY motif [42, 43]. Thus, the network of interacting residues around Asn111TM3 and Asn295TM7 likely conveys conformational changes in the ligand-binding pocket to the cytoplasmic domain coupling to the G proteins, which plays an essential role in the AT1R activation process. This network may also be responsible for the inter-helical interactions required for the binding and functional properties of the biphenyl-tetrazole ARBs, as well as consequent inactivation of AT1R. However, since biphenyl-tetrazole ARBs exhibit slightly different structures, e.g., the carboxyl group of EXP3174, valsartan and candesartan, the cyclopentane group of irbesartan and the N-acylated valine in valsartan (Fig. 2), differences in ligand-receptor interactions may explain the differences in their binding affinities and inverse agonist activities. Therefore, the inverse agonism potential of the biphenyl-tetrazole ARBs toward AT1R must be based on differences in energy gained through their binding with residues. However, we propose that all biphenyl-tetrazole ARBs tightly interact with the set of residues, namely Ser109TM3, Phe182ECL2, Gln257TM6, Tyr292TM7 and Asn295TM7, that stabilize the Asn111-Asn295 H-bond and the network of the above-mentioned residues around Asn111TM3 and Asn295TM7 in the inactive conformation, thereby leading to stabilization of AT1R in an inactive state, and resulting in robust inverse agonism in the ground state (Fig. 7A). All residues involved in the inverse agonism of the biphenyl-tetrazole ARBs toward AT1R, with the exception of Phe182ECL2, are conserved in many other GPCRs, implying that this may be a general mechanism for inverse agonism in GPCRs. On the other hand, the role of Phe182ECL2 in the inverse agonism of the biphenyl-tetrazole ARBs may be unique to AT1R, as supported by previous functional studies [55, 56], as well as the crystal structure of AT1R [43].
Figure 7.
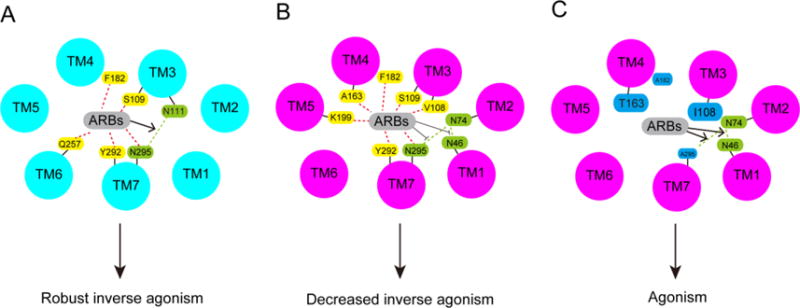
ARB-receptor interactions in the ground and active states of AT1R, and possible mechanism by which mutation of the residues Val108, Ala163, Asn295 and Phe182 switch the efficacy of biphenyl-tetrazole ARBs from inverse agonism to agonism in the active state of AT1R. (A) The ARBs stabilize the Asn111-Asn295 H-bond in the ground state of AT1R and cause robust inverse agonism. (B) The ARBs destabilize the Asn46-Asp74-Asn295 H-bond network in the active state of AT1R and cause inverse agonism. (C) Bulky substitution of Val108 and Ala163, and removal of the side chains of Asn295 and Phe182, may alter ARB-AT1R interactions, which may hydrate the hydrophobic core and stabilize the Asn46-Asp74-Asn295 H-bond network, causing agonism. The ground and active states of AT1R are colored light blue and light purple, respectively. The residues that interact with the ARBs are colored yellow, the residues Asn46, Asp74 and Asn295 (Ala295 substitution) are colored green, and mutated residues are colored cyan. The H-bonds between Asn46 and Asp74 and between Asn46 and Asn295 (Ala295 substitution) are indicated by green dotted lines.
6. Mechanism of decreased maximal inverse agonist activity of the biphenyl-tetrazole ARBs upon transition of AT1R toward the activated state
Similar to reports of various GPCRs, modeling the active state of AT1R is problematic because long time scales are untenable in such molecular dynamics simulations [57]. In addition, superposition of multiple active and inactive crystal structures of the adenosine A2a receptor, bovine rhodopsin and the β2 adrenergic receptor have been reported to show only modest changes in the ligand-binding pocket residues in each receptor, with the two states being remarkably similar in the ligand-binding pocket [58]. Therefore, although the crystal structure of the active state of AT1R is not currently available, results obtained for GPCRs suggest that the active structure of AT1R should show only modest conformational changes in the ligand-binding pocket compared to the inactive structure of AT1R. Therefore, we identified the molecular mechanism by which the AT1R transition toward the activated state decreased maximal inverse agonist activity of the ARBs by superimposing the experimental data (effect of mutations on binding affinity and inverse agonist activity) for each ARB in the ground and active states of AT1R using the inactive crystal structure of AT1R [16].
Previous AT1R structure-function studies have suggested both rotational and translational motion of TM2, TM3, TM5, TM6 and TM7 in the constitutively active mutant N111G-AT1R, which mimics the active state of AT1R [59–62]. In the active state, formation of the Asn46-Asp74-Asn295 H-bond network is proposed, which involves additional interacting residues around Asn111TM3 and Asn295TM7 (see Section 5). The active state of AT1R was also proposed to hydrate the hydrophobic core and facilitate the interaction of the “toggle switch” residue Trp253TM6 with Ala291TM7 and Leu112TM3 [42]. The biphenyl-tetrazole ARBs may destabilize the Asn46-Asp74-Asn295 H-bond network, as well as the residues around AsnmTM3 and Asn295TM7, and reduce hydration of the TM core due to their hydrophobic characteristics. In our recent structure-function study, we obtained evidence that the active-state transition of AT1R indeed decreased maximal inverse agonist activity of four biphenyl-tetrazole ARBs to different extents via changes in specific ligand-receptor interactions. In the ground state, four ARBs interacted with a set of residues, namely Ser109TM3, Phe182ECL2, Gln257TM6, Tyr292TM7, and Asn295TM7. In N111G-AT1R, the ARB interactions were shifted to a different set of residues (Val108TM3, Ser109TM3, Ala163TM4, Phe182ECL2, Lys199TM5, Tyr292TM7, and Asn295TM7), resulting in decreased inverse agonism (Fig. 6). Further, we observed that differences in the chemical structures of the ARBs indeed switched the interacting residues in the activated state. The effects of different mutations on the inverse agonism of the four ARBs in the active state of N111G-AT1R were quite different from those observed in the basal sate of WT-AT1R, which may also account for efficacy differences (discussed in detail later). Thus, we propose that rearrangement of the ARB-AT1R interactions in the active-state of AT1R insufficiently destabilize the Asn46-Asp74-Asn295 H-bond network, as well as the interacting residues around Asn111TM3 and Asn295TM7, and may insufficiently reduce hydration of the TM core, resulting in decreased inverse agonism of the biphenyl-tetrazole ARBs (Fig. 7B).
7. Efficacy switching residues in the active state of AT1R
We observed that mutation of the residues Val108TM3, Ala163TM4, Asn295TM7 and Phe182ECL2 in N111G-AT1R (active state of AT1R), but not in WT-AT1R (ground state of AT1R), switched the biphenyl-tetrazole ARBs from inverse agonist to agonist in the active state of AT1R [16]. Substitution of Val108 for an Ile in the N111G-AT1R switched valsartan and EXP3174 from inverse agonist to agonist, and substitutions of Ala163 for a Thr, Phe182 for an Ala and Asn295 for an Ala in the N111G-AT1R switched losartan and irbesartan from inverse agonist to agonist. Furthermore, simultaneous substitutions of Ala163 for a Thr and Asn295 for an Ala in the N111G-AT1R additively increase agonist activity of losartan and irbesartan. Although the exact mechanism for this phenomenon is unclear, one possible mechanism is described as follows. Bulky substitution of the Val108TM3 and Ala163TM4 residues may cause steric hindrance for the biphenyl-tetrazole ARBs, which may hydrate the hydrophobic core and stabilize the Asn46-Asp74-Asn295 H-bond network (Fig. 7C). On the other hand, removal of the side chains of Asn295TM7 and Phe182ECL2 may weaken interactions with the biphenyl-tetrazole ARBs, which may also hydrate the hydrophobic core and stabilize the Asn46-Asp74-Asn295 H-bond network (Fig. 7C). Elucidating the precise mechanism of such transformation of the pharmacological behavior of the ligands requires additional biophysical experiments, such as visualization of bound water molecules in both the active and inactive states. However, the current resolution of the crystal structure of human AT1R is not sufficient for this type of analysis. Site-saturation mutagenesis of Val108TM3, Ala163TM4, Asn295TM7 and Phe182ECL2, followed by assessment of binding affinity and receptor function may be an indirect method that would elucidate a potential mechanism for this phenomenon. Ultimately, both types of analyses should provide insights into the regulatory mechanism of GPCR function.
8. Homo- and heterodimerization of AT1R
GPCRs were previously believed to exist as monomers. However, recent studies have demonstrated that GPCRs can form homodimers as well as heterodimers with other GPCRs, which can alter receptor signaling properties, and result in the progression of human diseases [63]. AT1R is not an exception. In fact, levels of the AT1R homodimer are reported to be increased in monocytes of hypertensive patients as well as hypercholesterolemic Apo E deficient mice [18]. Factor XIIIA transglutaminase induces covalently crosslinked homodimers of AT1R, which enhance Ang II signaling efficacy. Inhibition of factor XIIIA transglutaminase activity, on the other hand, decreases the level of AT1R homodimers in monocytes and reduces atherosclerotic lesions in Apo E deficient mice. Furthermore, AT1R can form complexes (i.e., heterodimers) with the bradykinin B2 receptor (B2R), namely AT1R-B2R heterodimers [17, 26]. Increased potency and efficacy of AT1R-B2R heterodimers toward Ang II was shown to cause hypersensitivity to Ang II in patients with preeclampsia [17, 26]. Although independent groups confirmed formation of AT1R-B2R heterodimers by fluorescent resonance energy transfer (FRET) and bioluminescence resonance energy transfer (BRET) [64, 65], other groups failed to detect AT1R-B2R heterodimer formation using BRET [66, 67]. The most plausible explanation for the failure to detect the formation of AT1R-B2R heterodimers is the lack of essential chaperones in the cellular system, which are required for receptor-heterodimer folding [68–70], thus resulting in apoptosis of the cells by AT1R-B2R heterodimer-mediated overstimulation of Gq/11 signaling [71]. AT1R is also known to heterodimerize with other GPCRs. Heterodimerization of AT1R with the Ang II type 2 receptor, the apelin receptor, and the MAS receptor antagonize AT1R signaling [25, 72, 73]. Heterodimerization of AT1R with the β1- and β2-adrenergic receptors (β1AR and β2AR, respectively) causes cross-inhibition of opposing receptor signaling by either valsartan or βAR blockers [74]. Furthermore, AT1R may also heterodimerize with dopamine D1, D3, D4 and D5 receptors and alter receptor signaling [75–78].
Previous studies proposed that ligand binding to a GPCR protomer controls activation of both GPCRs in the dimer, thus fine-tuning signaling by both molecules [79–83]. However, a recent study indicated that the two protomers collaborate to form a unique conformational state of the heterodimer [20]. This study examined structural and functional properties of a heterodimer formed by Gq protein-coupled AT1R and Gi protein-coupled α2c adrenergic receptor (α2cAR). The Ang II bound AT1R protomer stabilized the α2cAR-AT1R heterodimer in the Gq activating conformation (Fig. 8A), while the norepinephrine (NE) bound α2cAR protomer stabilized the α2cAR-AT1R heterodimer in the Gi activating conformation (Fig. 8B). Interestingly, simultaneous binding of NE and Ang II to each protomer stabilized the α2cAR-AT1R heterodimer in a new Gs activating conformation that was different from the single agonist bound conformation (Fig. 8C). This study suggests that dual occupancy of the heterodimer by two selective agonists stabilizes GPCR heterodimers in a unique conformation, which may play a different role in both physiological and pathophysiological conditions compared to the GPCR monomer.
Figure 8.
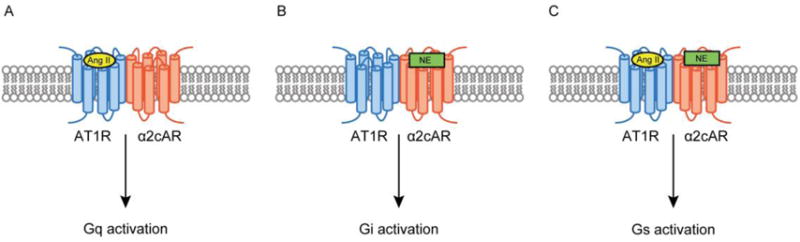
Dual agonist occupancy of AT1R-α2cAR heterodimers activates different types of G-proteins from a single agonist occupancy of each protomer. (A) Binding of Ang II to the AT1R protomer (light blue) causes Gq activation. (B) Binding of NE to α2cAR (orange) causes Gi activation. (C) Simultaneous binding of Ang II and NE to AT1R-α2cAR heterodimers causes Gs activation.
9. Biased agonism of AT1R
Activation of GPCRs is known to promote not only G protein-dependent signaling pathways, but also G protein-independent signaling pathways, such as β-arrestin signaling. Generally, natural GPCR agonists are known to activate all signaling pathways. However, recent studies have demonstrated that GPCR ligand analogs can preferentially activate one pathway, and are therefore referred to as biased agonists [84–86]. Some AT1R ligands have been identified as biased agonists. Although the Ang II analog [Sar1, Ile4, Ile8]Ang II (SII-Ang II) is known as an antagonist, recent studies demonstrated that this ligand can activate the β-arrestin-dependent signaling pathway but not the Gq-dependent signaling pathway, and was thus identified as a β-arrestin-biased AT1R agonist (Fig. 9) [28]. However, as SII-Ang II exhibits very low affinity for AT1R, the high affinity β-arrestin-biased AT1R agonists TRV120023 and TRV027 were developed based on the structure of SII-Ang II [29]. These β-arrestin-biased AT1R agonists have been reported to cause beneficial cardiovascular effects in experimental settings. For example, SII-Ang II activates β-arrestin 2 and promotes anti-apoptotic effects in rat vascular smooth muscle cells endogenously expressing AT1R [87]. TRV120023 diminishes myocyte apoptosis caused by mechanical stress and ischemia/reperfusion injury in mice [88], and increases cardiac performance in a transgenic mouse model of familial dilated cardiomyopathy [89]. TRV027 causes cardiac unloading action while preserving renal function in a canine model of acute heart failure [30]. However, as studies comparing the preventive effects of β-arrestin-biased AT1R agonists with those of the ARBs for cardiovascular diseases have not yet been performed, it is not clear whether β-arrestin-biased AT1R agonists demonstrate more beneficial effects in this setting. Therefore, a comparative study of the effects of both β-arrestin-biased AT1R agonists and the ARBs in cardiovascular disease is needed.
Figure 9.

β-arrestin-biased agonists. Ang II activates both the Gq and β-arrestin-mediated signaling pathways. β-arrestin-biased agonists preferentially activate the β-arrestin-mediated signaling pathway and may exhibit enhanced therapeutic effects for cardiovascular diseases.
10. Conclusion
The biphenyl-tetrazole ARBs are important drugs used for the treatment of hypertension and cardiovascular diseases. Although the biphenyl-tetrazole ARBs demonstrate robust inverse agonist activity for the ground state of AT1R, transitioning toward the activated state decreases the inverse agonist activity of the biphenyl-tetrazole ARBs. Since AT1R can be activated independent of ligands in disease states by mechanical stress and auto-antibodies, as well as by key receptor mutations, biphenyl-tetrazole ARBs may show different degrees of reduced therapeutic effects in clinical settings such as hypertension, preeclampsia and renal transplantation. Therefore, novel ARBs need to be developed with enhanced inverse agonist activity, relative to the current biphenyl-tetrazole ARBs, for the active state of AT1R. Moreover, AT1R can form both homodimers and AT1R-GPCR heterodimers, which results in altered receptor signaling associated with human diseases such as hypertension, atherosclerosis and preeclampsia. Finally, as AT1R-GPCR heterodimers adopt different conformational states compared to the AT1R monomer, the binding mode of ARBs may be changed, and may alter their pharmacological properties. Therefore, the effect of ARBs on signaling of AT1R-GPCR heterodimers needs to be examined. Further, β-arrestin-biased AT1R agonists have demonstrated beneficial cardiovascular effects in experimental conditions. However, whether such β-arrestin-biased AT1R agonists exhibit enhanced therapeutic effects for cardiovascular diseases, relative to ARBs, has not yet been elucidated, so that comparative studies are needed. Taken together, this review provides significant information for the development of a new class of drugs targeting AT1R.
Acknowledgments
This work was supported by a Grant-in-Aid for Research Activity Start-up [18890141] to T. T. from the Ministry of Education, Culture, Sports, Science and Technology in Japan. This work was supported in part by National Institutes of Health Grants [R56HL132351, HL132351] to S. K. and TUBITAK Grants [115C023 and 115S624] to H. U.
Abbreviations
- Ang II
angiotensin II
- SII-Ang II
[Sar1, Ile4, Ile8]Ang II
- AT1R
Angiotensin II type 1 receptor
- ARB
AT1R blocker
- AT1R-directed autoantibody
AT1R-directed AA
- GPCR
G protein-coupled receptor
- H-bond
hydrogen bond
- IP
inositol phosphate
- WT-AT1R
wild-type AT1R
- ECL2
second extracellular loop
- TM
transmembrane
- NE
norepinephrine
- α2cAR
α2c adrenergic receptor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
K.N. was financially supported by the amount of contributions of Merck & Co., Inc., Shionogi & Co., Ltd. Novartis Pharma K.K.
References
- 1.Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63(6):1256–72. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- 2.Garland SL. Are GPCRs still a source of new targets? J Biomol Screen. 2013;18(9):947–66. doi: 10.1177/1087057113498418. [DOI] [PubMed] [Google Scholar]
- 3.Khan BV. The effect of amlodipine besylate, losartan potassium, olmesartan medoxomil, and other antihypertensives on central aortic blood pressure and biomarkers of vascular function. Ther Adv Cardiovasc Dis. 2011;5(5):241–73. doi: 10.1177/1753944711420464. [DOI] [PubMed] [Google Scholar]
- 4.Vijayaraghavan K, Deedwania P. Renin-angiotensin-aldosterone blockade for cardiovascular disease prevention. Cardiol Clin. 2011;29(1):137–56. doi: 10.1016/j.ccl.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 5.Vejakama P, Thakkinstian A, Lertrattananon D, Ingsathit A, Ngarmukos C, Attia J. Reno-protective effects of renin-angiotensin system blockade in type 2 diabetic patients: a systematic review and network meta-analysis. Diabetologia. 2012;55(3):566–78. doi: 10.1007/s00125-011-2398-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee M, Saver JL, Hong KS, Hao Q, Chow J, Ovbiagele B. Renin-Angiotensin system modulators modestly reduce vascular risk in persons with prior stroke. Stroke. 2012;43(1):113–9. doi: 10.1161/STROKEAHA.111.632596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mederos M, Schnitzler Y, Storch U, Gudermann T. AT1 receptors as mechanosensors. Curr Opin Pharmacol. 2011;11(2):112–6. doi: 10.1016/j.coph.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 8.Wallukat G, Schimke I. Agonistic autoantibodies directed against G-protein-coupled receptors and their relationship to cardiovascular diseases. Semin Immunopathol. 2014;36(3):351–63. doi: 10.1007/s00281-014-0425-9. [DOI] [PubMed] [Google Scholar]
- 9.Unal H, Jagannathan R, Karnik SS. Mechanism of GPCR-directed autoantibodies in diseases. Adv Exp Med Biol. 2012;749:187–99. doi: 10.1007/978-1-4614-3381-1_13. [DOI] [PubMed] [Google Scholar]
- 10.Liu F, Wang Y, Wang X, Zheng Y, Jin Z, Zhi J. Role of agonistic autoantibodies against type-1 angiotensin II receptor in the pathogenesis of retinopathy in preeclampsia. Sci Rep. 2016;6:29036. doi: 10.1038/srep29036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Storch U, Mederos M, Schnitzler Y, Gudermann T. G protein-mediated stretch reception. Am J Physiol Heart Circ Physiol. 2012;302(6):H1241–9. doi: 10.1152/ajpheart.00818.2011. [DOI] [PubMed] [Google Scholar]
- 12.Zou Y, Akazawa H, Qin Y, Sano M, Takano H, Minamino T, Makita N, Iwanaga K, Zhu W, Kudoh S, Toko H, Tamura K, Kihara M, Nagai T, Fukamizu A, Umemura S, Iiri T, Fujita T, Komuro I. Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat Cell Biol. 2004;6(6):499–506. doi: 10.1038/ncb1137. [DOI] [PubMed] [Google Scholar]
- 13.Wei F, Jia XJ, Yu SQ, Gu Y, Wang L, Guo XM, Wang M, Zhu F, Cheng X, Wei YM, Zhou ZH, Fu M, Liao YH, Group SAS. Candesartan versus imidapril in hypertension: a randomised study to assess effects of anti-AT1 receptor autoantibodies. Heart. 2011;97(6):479–84. doi: 10.1136/hrt.2009.192104. [DOI] [PubMed] [Google Scholar]
- 14.Noda K, Feng YH, Liu XP, Saad Y, Husain A, Karnik SS. The active state of the AT1 angiotensin receptor is generated by angiotensin II induction. Biochemistry. 1996;35(51):16435–42. doi: 10.1021/bi961593m. [DOI] [PubMed] [Google Scholar]
- 15.Le MT, Vanderheyden PM, Szaszak M, Hunyady L, Kersemans V, Vauquelin G. Peptide and nonpeptide antagonist interaction with constitutively active human AT1 receptors. Biochem Pharmacol. 2003;65(8):1329–38. doi: 10.1016/s0006-2952(03)00072-8. [DOI] [PubMed] [Google Scholar]
- 16.Takezako T, Unal H, Karnik SS, Node K. Structure-Function Basis of Attenuated Inverse Agonism of Angiotensin II Type 1 Receptor Blockers for Active-State Angiotensin II Type 1 Receptor. Mol Pharmacol. 2015;88(3):488–501. doi: 10.1124/mol.115.099176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.AbdAlla S, Lother H, Quitterer U. AT1-receptor heterodimers show enhanced G-protein activation and altered receptor sequestration. Nature. 2000;407(6800):94–8. doi: 10.1038/35024095. [DOI] [PubMed] [Google Scholar]
- 18.AbdAlla S, Lother H, Langer A, el Faramawy Y, Quitterer U. Factor XIIIA transglutaminase crosslinks AT1 receptor dimers of monocytes at the onset of atherosclerosis. Cell. 2004;119(3):343–54. doi: 10.1016/j.cell.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 19.Goupil E, Fillion D, Clement S, Luo X, Devost D, Sleno R, Petrin D, Saragovi HU, Thorin E, Laporte SA, Hebert TE. Angiotensin II type I and prostaglandin F2alpha receptors cooperatively modulate signaling in vascular smooth muscle cells. J Biol Chem. 2015;290(5):3137–48. doi: 10.1074/jbc.M114.631119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bellot M, Galandrin S, Boularan C, Matthies HJ, Despas F, Denis C, Javitch J, Mazeres S, Sanni SJ, Pons V, Seguelas MH, Hansen JL, Pathak A, Galli A, Senard JM, Gales C. Dual agonist occupancy of AT1-R-alpha2C-AR heterodimers results in atypical Gs-PKA signaling. Nat Chem Biol. 2015;11(4):271–9. doi: 10.1038/nchembio.1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rozenfeld R, Gupta A, Gagnidze K, Lim MP, Gomes I, Lee-Ramos D, Nieto N, Devi LA. AT1R-CB(1)R heteromerization reveals a new mechanism for the pathogenic properties of angiotensin II. EMBO J. 2011;30(12):2350–63. doi: 10.1038/emboj.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wnorowski A, Jozwiak K. Homo- and hetero-oligomerization of beta2-adrenergic receptor in receptor trafficking, signaling pathways and receptor pharmacology. Cell Signal. 2014;26(10):2259–65. doi: 10.1016/j.cellsig.2014.06.016. [DOI] [PubMed] [Google Scholar]
- 23.Martinez-Pinilla E, Rodriguez-Perez AI, Navarro G, Aguinaga D, Moreno E, Lanciego JL, Labandeira-Garcia JL, Franco R. Dopamine D2 and angiotensin II type 1 receptors form functional heteromers in rat striatum. Biochem Pharmacol. 2015;96(2):131–42. doi: 10.1016/j.bcp.2015.05.006. [DOI] [PubMed] [Google Scholar]
- 24.Gonzalez-Hernandez Mde L, Godinez-Hernandez D, Bobadilla-Lugo RA, Lopez-Sanchez P. Angiotensin-II type 1 receptor (AT1R) and alpha-1D adrenoceptor form a heterodimer during pregnancy-induced hypertension. Auton Autacoid Pharmacol. 2010;30(3):167–72. doi: 10.1111/j.1474-8673.2009.00446.x. [DOI] [PubMed] [Google Scholar]
- 25.Siddiquee K, Hampton J, McAnally D, May L, Smith L. The apelin receptor inhibits the angiotensin II type 1 receptor via allosteric trans-inhibition. Br J Pharmacol. 2013;168(5):1104–17. doi: 10.1111/j.1476-5381.2012.02192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.AbdAlla S, Lother H, el Massiery A, Quitterer U. Increased AT(1) receptor heterodimers in preeclampsia mediate enhanced angiotensin II responsiveness. Nat Med. 2001;7(9):1003–9. doi: 10.1038/nm0901-1003. [DOI] [PubMed] [Google Scholar]
- 27.Luttrell LM. Minireview: More than just a hammer: ligand “bias” and pharmaceutical discovery. Mol Endocrinol. 2014;28(3):281–94. doi: 10.1210/me.2013-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei H, Ahn S, Shenoy SK, Karnik SS, Hunyady L, Luttrell LM, Lefkowitz RJ. Independent beta-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc Natl Acad Sci USA. 2003;100(19):10782–7. doi: 10.1073/pnas.1834556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Violin JD, DeWire SM, Yamashita D, Rominger DH, Nguyen L, Schiller K, Whalen EJ, Gowen M, Lark MW. Selectively engaging beta-arrestins at the angiotensin II type 1 receptor reduces blood pressure and increases cardiac performance. J Pharmacol Exp Ther. 2010;335(3):572–9. doi: 10.1124/jpet.110.173005. [DOI] [PubMed] [Google Scholar]
- 30.Boerrigter G, Lark MW, Whalen EJ, Soergel DG, Violin JD, Burnett JC., Jr Cardiorenal actions of TRV120027, a novel ss-arrestin-biased ligand at the angiotensin II type I receptor, in healthy and heart failure canines: a novel therapeutic strategy for acute heart failure. Circ Heart Fail. 2011;4(6):770–8. doi: 10.1161/CIRCHEARTFAILURE.111.962571. [DOI] [PubMed] [Google Scholar]
- 31.Boerrigter G, Soergel DG, Violin JD, Lark MW, Burnett JC., Jr TRV120027, a novel beta-arrestin biased ligand at the angiotensin II type I receptor, unloads the heart and maintains renal function when added to furosemide in experimental heart failure. Circ Heart Fail. 2012;5(5):627–34. doi: 10.1161/CIRCHEARTFAILURE.112.969220. [DOI] [PubMed] [Google Scholar]
- 32.Takezako T, Gogonea C, Saad Y, Noda K, Karnik SS. “Network leaning” as a mechanism of insurmountable antagonism of the angiotensin II type 1 receptor by non-peptide antagonists. J Biol Chem. 2004;279(15):15248–57. doi: 10.1074/jbc.M312728200. [DOI] [PubMed] [Google Scholar]
- 33.Vanderheyden PM, Fierens FL, De Backer JP, Fraeyman N, Vauquelin G. Distinction between surmountable and insurmountable selective AT1 receptor antagonists by use of CHO-K1 cells expressing human angiotensin II AT1 receptors. Br J Pharmacol. 1999;126(4):1057–65. doi: 10.1038/sj.bjp.0702398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Le MT, Pugsley MK, Vauquelin G, Van Liefde I. Molecular characterisation of the interactions between olmesartan and telmisartan and the human angiotensin II AT1 receptor. Br J Pharmacol. 2007;151(7):952–62. doi: 10.1038/sj.bjp.0707323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Verheijen I, Fierens FL, Debacker JP, Vauquelin G, Vanderheyden PM. Interaction between the partially insurmountable antagonist valsartan and human recombinant angiotensin II type 1 receptors. Fundam Clin Pharmacol. 2000;14(6):577–85. doi: 10.1111/j.1472-8206.2000.tb00443.x. [DOI] [PubMed] [Google Scholar]
- 36.Edwards RM, Aiyar N, Ohlstein EH, Weidley EF, Griffin E, Ezekiel M, Keenan RM, Ruffolo RR, Weinstock J. Pharmacological characterization of the nonpeptide angiotensin II receptor antagonist, SK&F 108566. J Pharmacol Exp Ther. 1992;260(1):175–81. [PubMed] [Google Scholar]
- 37.Ojima M, Igata H, Tanaka M, Sakamoto H, Kuroita T, Kohara Y, Kubo K, Fuse H, Imura Y, Kusumoto K, Nagaya H. In vitro antagonistic properties of a new angiotensin type 1 receptor blocker, azilsartan, in receptor binding and function studies. J Pharmacol Exp Ther. 2011;336(3):801–8. doi: 10.1124/jpet.110.176636. [DOI] [PubMed] [Google Scholar]
- 38.Miura S, Kiya Y, Kanazawa T, Imaizumi S, Fujino M, Matsuo Y, Karnik SS, Saku K. Differential bonding interactions of inverse agonists of angiotensin II type 1 receptor in stabilizing the inactive state. Mol Endocrinol. 2008;22(1):139–46. doi: 10.1210/me.2007-0312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miura S, Fujino M, Hanzawa H, Kiya Y, Imaizumi S, Matsuo Y, Tomita S, Uehara Y, Kamik SS, Yanagisawa H, Koike H, Komuro I, Saku K. Molecular mechanism underlying inverse agonist of angiotensin II type 1 receptor. J Biol Chem. 2006;281(28):19288–95. doi: 10.1074/jbc.M602144200. [DOI] [PubMed] [Google Scholar]
- 40.Miura S, Nakao N, Hanzawa H, Matsuo Y, Saku K, Karnik SS. Reassessment of the unique mode of binding between angiotensin II type 1 receptor and their blockers. PLoS One. 2013;8(11):e79914. doi: 10.1371/journal.pone.0079914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fillion D, Cabana J, Guillemette G, Leduc R, Lavigne P, Escher E. Structure of the human angiotensin II type 1 (AT1) receptor bound to angiotensin II from multiple chemoselective photoprobe contacts reveals a unique peptide binding mode. J Biol Chem. 2013;288(12):8187–97. doi: 10.1074/jbc.M112.442053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cabana J, Holleran B, Beaulieu ME, Leduc R, Escher E, Guillemette G, Lavigne P. Critical hydrogen bond formation for activation of the angiotensin II type 1 receptor. J Biol Chem. 2013;288(4):2593–604. doi: 10.1074/jbc.M112.395939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang H, Unal H, Gati C, Han GW, Liu W, Zatsepin NA, James D, Wang D, Nelson G, Weierstall U, Sawaya MR, Xu Q, Messerschmidt M, Williams GJ, Boutet S, Yefanov OM, White TA, Wang C, Ishchenko A, Tirupula KC, Desnoyer R, Coe J, Conrad CE, Fromme P, Stevens RC, Katritch V, Karnik SS, Cherezov V. Structure of the Angiotensin receptor revealed by serial femtosecond crystallography. Cell. 2015;161(4):833–44. doi: 10.1016/j.cell.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang H, Unal H, Desnoyer R, Han GW, Patel N, Katritch V, Karnik SS, Cherezov V, Stevens RC. Structural Basis for Ligand Recognition and Functional Selectivity at Angiotensin Receptor. J Biol Chem. 2015;290(49):29127–39. doi: 10.1074/jbc.M115.689000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van Liefde I, Vauquelin G. Sartan-AT1 receptor interactions: in vitro evidence for insurmountable antagonism and inverse agonism. Mol Cell Endocrinol. 2009;302(2):237–43. doi: 10.1016/j.mce.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 46.Panek RL, Lu GH, Overhiser RW, Major TC, Hodges JC, Taylor DG. Functional studies but not receptor binding can distinguish surmountable from insurmountable AT1 antagonism. Journal of Pharmacology and Experimental Therapeutics. 1995;273(2):753–761. [PubMed] [Google Scholar]
- 47.Fierens FL, Vanderheyden PM, Gaborik Z, Minh TL, Backer JP, Hunyady L, Ijzerman A, Vauquelin G. Lys(199) mutation of the human angiotensin type 1 receptor differentially affects the binding of surmountable and insurmountable non-peptide antagonists. J Renin Angiotensin Aldosterone Syst. 2000;1(3):283–8. doi: 10.3317/jraas.2000.044. [DOI] [PubMed] [Google Scholar]
- 48.Vauquelin G, Fierens FL, Gaborik Z, Le Minh T, De Backer JP, Hunyady L, Vanderheyden PM. Role of basic amino acids of the human angiotensin type 1 receptor in the binding of the non-peptide antagonist candesartan. J Renin Angiotensin Aldosterone Syst. 2001;2(1_suppl):S32–S36. doi: 10.1177/14703203010020010501. [DOI] [PubMed] [Google Scholar]
- 49.Ji H, Leung M, Zhang Y, Catt KJ, Sandberg K. Differential structural requirements for specific binding of nonpeptide and peptide antagonists to the AT1 angiotensin receptor. Identification of amino acid residues that determine binding of the antihypertensive drug losartan. J Biol Chem. 1994;269(24):16533–6. [PubMed] [Google Scholar]
- 50.Ji H, Zheng W, Zhang Y, Catt KJ, Sandberg K. Genetic transfer of a nonpeptide antagonist binding site to a previously unresponsive angiotensin receptor. Proc Natl Acad Sci USA. 1995;92(20):9240–4. doi: 10.1073/pnas.92.20.9240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gosselin MJ, Leclerc PC, Auger-Messier M, Guillemette G, Escher E, Leduc R. Molecular cloning of a ferret angiotensin II AT(1) receptor reveals the importance of position 163 for Losartan binding. Biochim Biophys Acta. 2000;1497(1):94–102. doi: 10.1016/s0167-4889(00)00046-x. [DOI] [PubMed] [Google Scholar]
- 52.Noda K, Saad Y, Kinoshita A, Boyle TP, Graham RM, Husain A, Karnik SS. Tetrazole and carboxylate groups of angiotensin receptor antagonists bind to the same subsite by different mechanisms. J Biol Chem. 1995;270(5):2284–9. doi: 10.1074/jbc.270.5.2284. [DOI] [PubMed] [Google Scholar]
- 53.Ahuja S, Smith SO. Multiple switches in G protein-coupled receptor activation. Trends Pharmacol Sci. 2009;30(9):494–502. doi: 10.1016/j.tips.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 54.Holst B, Nygaard R, Valentin-Hansen L, Bach A, Engelstoft MS, Petersen PS, Frimurer TM, Schwartz TW. A conserved aromatic lock for the tryptophan rotameric switch in TM-VI of seven-transmembrane receptors. J Biol Chem. 2010;285(6):3973–85. doi: 10.1074/jbc.M109.064725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Unal H, Jagannathan R, Bhat MB, Karnik SS. Ligand-specific conformation of extracellular loop-2 in the angiotensin II type 1 receptor. J Biol Chem. 2010;285(21):16341–50. doi: 10.1074/jbc.M109.094870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Unal H, Jagannathan R, Bhatnagar A, Tirupula K, Desnoyer R, Kamik SS. Long range effect of mutations on specific conformational changes in the extracellular loop 2 of angiotensin II type 1 receptor. J Biol Chem. 2013;288(1):540–51. doi: 10.1074/jbc.M112.392514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Manglik A, Kobilka B. The role of protein dynamics in GPCR function: insights from the beta2AR and rhodopsin. Curr Opin Cell Biol. 2014;27:136–43. doi: 10.1016/j.ceb.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Katritch V, Cherezov V, Stevens RC. Structure-function of the G protein-coupled receptor superfamily. Annu Rev Pharmacol Toxicol. 2013;53:531–56. doi: 10.1146/annurev-pharmtox-032112-135923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boucard AA, Roy M, Beaulieu ME, Lavigne P, Escher E, Guillemette G, Leduc R. Constitutive activation of the angiotensin II type 1 receptor alters the spatial proximity of transmembrane 7 to the ligand-binding pocket. J Biol Chem. 2003;278(38):36628–36. doi: 10.1074/jbc.M305952200. [DOI] [PubMed] [Google Scholar]
- 60.Martin SS, Boucard AA, Clement M, Escher E, Leduc R, Guillemette G. Analysis of the third transmembrane domain of the human type 1 angiotensin II receptor by cysteine scanning mutagenesis. J Biol Chem. 2004;279(49):51415–23. doi: 10.1074/jbc.M407965200. [DOI] [PubMed] [Google Scholar]
- 61.Martin SS, Holleran BJ, Escher E, Guillemette G, Leduc R. Activation of the angiotensin II type 1 receptor leads to movement of the sixth transmembrane domain: analysis by the substituted cysteine accessibility method. Mol Pharmacol. 2007;72(1):182–90. doi: 10.1124/mol.106.033670. [DOI] [PubMed] [Google Scholar]
- 62.Domazet I, Martin SS, Holleran BJ, Morin ME, Lacasse P, Lavigne P, Escher E, Leduc R, Guillemette G. The fifth transmembrane domain of angiotensin II Type 1 receptor participates in the formation of the ligand-binding pocket and undergoes a counterclockwise rotation upon receptor activation. J Biol Chem. 2009;284(46):31953–61. doi: 10.1074/jbc.M109.051839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Karnik SS, Unal H, Kemp JR, Tirupula KC, Eguchi S, Vanderheyden PM, Thomas WG. International Union of Basic and Clinical Pharmacology. XCIX. Angiotensin Receptors: Interpreters of Pathophysiological Angiotensinergic Stimuli [corrected] Pharmacol Rev. 2015;67(4):754–819. doi: 10.1124/pr.114.010454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Quitterer U, Pohl A, Langer A, Koller S, Abdalla S. A cleavable signal peptide enhances cell surface delivery and heterodimerization of Cerulean-tagged angiotensin II AT1 and bradykinin B2 receptor. Biochem Biophys Res Commun. 2011;409(3):544–9. doi: 10.1016/j.bbrc.2011.05.041. [DOI] [PubMed] [Google Scholar]
- 65.Wilson PC, Lee MH, Appleton KM, El-Shewy HM, Morinelli TA, Peterson YK, Luttrell LM, Jaffa AA. The arrestin-selective angiotensin AT1 receptor agonist [Sar1,Ile4,Ile8]-AngII negatively regulates bradykinin B2 receptor signaling via AT1-B2 receptor heterodimers. J Biol Chem. 2013;288(26):18872–84. doi: 10.1074/jbc.M113.472381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hansen JL, Hansen JT, Speerschneider T, Lyngso C, Erikstrup N, Burstein ES, Weiner DM, Walther T, Makita N, Iiri T, Merten N, Kostenis E, Sheikh SP. Lack of evidence for AT1R/B2R heterodimerization in COS-7, HEK293, and NIH3T3 cells: how common is the AT1R/B2R heterodimer? J Biol Chem. 2009;284(3):1831–9. doi: 10.1074/jbc.M804607200. [DOI] [PubMed] [Google Scholar]
- 67.See HB, Seeber RM, Kocan M, Eidne KA, Pfleger KD. Application of G protein-coupled receptor-heteromer identification technology to monitor beta-arrestin recruitment to G protein-coupled receptor heteromers. Assay Drug Dev Technol. 2011;9(1):21–30. doi: 10.1089/adt.2010.0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Abd Alla J, Reeck K, Langer A, Streichert T, Quitterer U. Calreticulin enhances B2 bradykinin receptor maturation and heterodimerization. Biochem Biophys Res Commun. 2009;387(1):186–90. doi: 10.1016/j.bbrc.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 69.Abd Alla J, Pohl A, Reeck K, Streichert T, Quitterer U. Establishment of an in vivo model facilitates B2 receptor protein maturation and heterodimerization. Integr Biol (Camb) 2010;2(4):209–17. doi: 10.1039/b922592g. [DOI] [PubMed] [Google Scholar]
- 70.Bulenger S, Marullo S, Bouvier M. Emerging role of homo- and heterodimerization in G-protein-coupled receptor biosynthesis and maturation. Trends Pharmacol Sci. 2005;26(3):131–7. doi: 10.1016/j.tips.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 71.Quitterer U, AbdAlla S. Vasopressor meets vasodepressor: The AT1-B2 receptor heterodimer. Biochem Pharmacol. 2014;88(3):284–90. doi: 10.1016/j.bcp.2014.01.019. [DOI] [PubMed] [Google Scholar]
- 72.AbdAlla S, Lother H, Abdel-tawab AM, Quitterer U. The angiotensin II AT2 receptor is an AT1 receptor antagonist. J Biol Chem. 2001;276(43):39721–6. doi: 10.1074/jbc.M105253200. [DOI] [PubMed] [Google Scholar]
- 73.Kostenis E, Milligan G, Christopoulos A, Sanchez-Ferrer CF, Heringer-Walther S, Sexton PM, Gembardt F, Kellett E, Martini L, Vanderheyden P, Schultheiss HP, Walther T. G-protein-coupled receptor Mas is a physiological antagonist of the angiotensin II type 1 receptor. Circulation. 2005;111(14):1806–13. doi: 10.1161/01.CIR.0000160867.23556.7D. [DOI] [PubMed] [Google Scholar]
- 74.Barki-Harrington L, Luttrell LM, Rockman HA. Dual inhibition of beta-adrenergic and angiotensin II receptors by a single antagonist: a functional role for receptor-receptor interaction in vivo. Circulation. 2003;108(13):1611–8. doi: 10.1161/01.CIR.0000092166.30360.78. [DOI] [PubMed] [Google Scholar]
- 75.Zeng C, Luo Y, Asico LD, Hopfer U, Eisner GM, Felder RA, Jose PA. Perturbation of D1 dopamine and AT1 receptor interaction in spontaneously hypertensive rats. Hypertension. 2003;42(4):787–92. doi: 10.1161/01.HYP.0000085334.34963.4E. [DOI] [PubMed] [Google Scholar]
- 76.Zeng C, Asico LD, Wang X, Hopfer U, Eisner GM, Felder RA, Jose PA. Angiotensin II regulation of AT1 and D3 dopamine receptors in renal proximal tubule cells of SHR. Hypertension. 2003;41(3 Pt 2):724–9. doi: 10.1161/01.HYP.0000047880.78462.0E. [DOI] [PubMed] [Google Scholar]
- 77.Zeng C, Yang Z, Wang Z, Jones J, Wang X, Altea J, Mangrum AJ, Hopfer U, Sibley DR, Eisner GM, Felder RA, Jose PA. Interaction of angiotensin II type 1 and D5 dopamine receptors in renal proximal tubule cells. Hypertension. 2005;45(4):804–10. doi: 10.1161/01.HYP.0000155212.33212.99. [DOI] [PubMed] [Google Scholar]
- 78.Chen K, Deng K, Wang X, Wang Z, Zheng S, Ren H, He D, Han Y, Asico LD, Jose PA, Zeng C. Activation of D4 dopamine receptor decreases angiotensin II type 1 receptor expression in rat renal proximal tubule cells. Hypertension. 2015;65(1):153–60. doi: 10.1161/HYPERTENSIONAHA.114.04038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Goudet C, Kniazeff J, Hlavackova V, Malhaire F, Maurel D, Acher F, Blahos J, Prezeau L, Pin JP. Asymmetric functioning of dimeric metabotropic glutamate receptors disclosed by positive allosteric modulators. J Biol Chem. 2005;280(26):24380–5. doi: 10.1074/jbc.M502642200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hlavackova V, Goudet C, Kniazeff J, Zikova A, Maurel D, Vol C, Trojanova J, Prezeau L, Pin JP, Blahos J. Evidence for a single heptahelical domain being turned on upon activation of a dimeric GPCR. EMBO J. 2005;24(3):499–509. doi: 10.1038/sj.emboj.7600557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Han Y, Moreira IS, Urizar E, Weinstein H, Javitch JA. Allosteric communication between protomers of dopamine class A GPCR dimers modulates activation. Nat Chem Biol. 2009;5(9):688–95. doi: 10.1038/nchembio.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kaupmann K, Malitschek B, Schuler V, Heid J, Froestl W, Beck P, Mosbacher J, Bischoff S, Kulik A, Shigemoto R, Karschin A, Bettler B. GABA(B)-receptor subtypes assemble into functional heteromeric complexes. Nature. 1998;396(6712):683–7. doi: 10.1038/25360. [DOI] [PubMed] [Google Scholar]
- 83.Maurice P, Kamal M, Jockers R. Asymmetry of GPCR oligomers supports their functional relevance. Trends Pharmacol Sci. 2011;32(9):514–20. doi: 10.1016/j.tips.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 84.Azzi M, Charest PG, Angers S, Rousseau G, Kohout T, Bouvier M, Pineyro G. Beta-arrestin-mediated activation of MAPK by inverse agonists reveals distinct active conformations for G protein-coupled receptors. Proc Natl Acad Sci USA. 2003;100(20):11406–11. doi: 10.1073/pnas.1936664100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Drake MT, Violin JD, Whalen EJ, Wisler JW, Shenoy SK, Lefkowitz RJ. beta-arrestin-biased agonism at the beta2-adrenergic receptor. J Biol Chem. 2008;283(9):5669–76. doi: 10.1074/jbc.M708118200. [DOI] [PubMed] [Google Scholar]
- 86.Shukla AK, Violin JD, Whalen EJ, Gesty-Palmer D, Shenoy SK, Lefkowitz RJ. Distinct conformational changes in beta-arrestin report biased agonism at seven-transmembrane receptors. Proc Natl Acad Sci USA. 2008;105(29):9988–93. doi: 10.1073/pnas.0804246105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ahn S, Kim J, Hara MR, Ren XR, Lefkowitz RJ. {beta}-Arrestin-2 Mediates Anti-apoptotic Signaling through Regulation of BAD Phosphorylation. J Biol Chem. 2009;284(13):8855–65. doi: 10.1074/jbc.M808463200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kim KS, Abraham D, Williams B, Violin JD, Mao L, Rockman HA. beta-Arrestin-biased AT1R stimulation promotes cell survival during acute cardiac injury. Am J Physiol Heart Circ Physiol. 2012;303(8):H1001–10. doi: 10.1152/ajpheart.00475.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tarigopula M, Davis RT, 3rd, Mungai PT, Ryba DM, Wieczorek DF, Cowan CL, Violin JD, Wolska BM, Solaro RJ. Cardiac myosin light chain phosphorylation and inotropic effects of a biased ligand, TRV120023, in a dilated cardiomyopathy model. Cardiovasc Res. 2015;107(2):226–34. doi: 10.1093/cvr/cvv162. [DOI] [PMC free article] [PubMed] [Google Scholar]