

Abstract
Purpose
The PI3K/Akt/mTOR pathway is dysregulated in metastatic renal cell carcinoma (mRCC). Buparlisib is a pan-PI3K inhibitor with activity in advanced solid tumors. The primary aim of this study was to determine the maximum tolerated dose (MTD) and dose limiting toxicities (DLTs) of buparlisib and bevacizumab in mRCC. Secondary objectives included efficacy, biomarker discovery and additional toxicity.
Methods
This was a standard 3+3 dose-escalation study of buparlisib (60–100 mg/day) and bevacizumab (10 mg/kg every 2 weeks). After the MTD was defined, 15 patients were accrued to the expansion cohort.
Results
Thirty-two patients were accrued (3 treated at 60 mg/day, 21 at 80 mg/day, 6 at 100 mg/day, and 2 never received therapy). The majority had clear-cell histology (87%) and 50% had ≥2 prior lines of therapy. The MTD of buparlisib was 80 mg/day and bevacizumab 10 mg/kg every 2 weeks. Twenty-eight patients discontinued therapy: n=17 progression, n=7 toxicity, and n=4 other reasons. DLTs included rash/pruritis, elevated lipase/amylase, anorexia and psychiatric disorders (suicidal ideation, depression, and cognitive disturbances). Of the 30 patients who received at least one dose, 13% had a partial response (95% CI 4%, 31%). Two patients harboring activating PI3KA mutations achieved 42% and 16% maximal tumor shrinkage.
Conclusions
Buparlisib (80 mg/day) with bevacizumab was a tolerable regimen with preliminary activity in VEGF-refractory mRCC. The benefit of this combination may be of interest for future mRCC trials, possibly in a selected population.
Keywords: Bevacizumab, Buparlisib, Phase I, PI3K inhibitor, Renal cell carcinoma
Introduction
Angiogenesis is an essential driver of tumorogenesis and progression in metastatic renal cell carcinoma (mRCC) (1). Vascular endothelial growth factor (VEGF) and its receptors are critical in the process of angiogenesis and numerous studies have demonstrated that the VEGF signaling pathway drives RCC pathogenesis (2,3). The importance of the VEGF signally pathway in RCC is made evident by the success of VEGF targeting agents – axitinib, bevacizumab, pazopanib, sorafeninb and sunitinib – which have improved the overall survival for patients with this disease (4–8). However, mRCC remains mostly an incurable disease and the majority of patients develop resistance to VEGF targeted therapy (9).
In addition to the commonly altered Von Hippel-Lindau (VHL)/Hypoxia Inducible Factor (HIF) pathway, which activates its downstream target VEGF, the phosphatidylinositol-3 kinase (PI3K) pathway is recurrently altered in both clear cell and non-clear cell RCC and is thought to play a central role in cancer progression (10,11). Although the exact mechanism of HIF pathway activation is not well characterized, it is postulated that PI3K activation results in an increase in HIF-1α gene expression, leading to resistance to VEGF targeted therapy (12). Thus, the PI3K pathway represents a potential therapeutic target in RCC, supporting the rationale for combinatorial PI3K and VEGF inhibition (10).
Buparlisib (BKM-120) is an orally bioavailable pan-inhibitor of PI3K. Buparlisib specifically inhibits class I PI3K in an ATP-competitive manner, reducing the production of the secondary messenger phosphatidylinositol-3,4,5-trisphosphate. Buparlisib reduces the phosphorylation of direct downstream effector AKT (Protein kinase B) (13). In addition to inhibiting all isoforms of wild type PI3K, this compound has been demonstrated to inhibit PI3Kα harboring somatic mutations. Few data is available regarding its ability to inhibit the other mutated isoforms. (14). In the first-in-human phase I clinical trial, buparlisib demonstrated a tolerable safety profile and antitumor activity in several solid tumors independent of PI3K and phosphatase and tensin homolog (PTEN) mutational status and showed a high correlation between buparlisib exposure and inhibition of PI3K signaling (15).
Bevacizumab, a monoclonal VEGF binding antibody, has already been approved by the United States (US) Food and Drug Administration for use in seven different tumors types, including RCC (16). Given the potential to overcome intrinsic and acquired resistance to VEGF targeted therapy, combination strategies of targeted therapies are being explored to improve long-term outcomes for patients with metastatic disease (17). To date, these combinations have demonstrated modest clinical benefits and the majority of studies have shown significant toxicity (18). As a monoclonal antibody, bevacizumab’s side effect profile differs from the kinase inhibitors. Given less expected overlapping toxicities, bevacizumab has been tested in combination with several compounds such as temsirolimus or erlortinib with demonstrated feasibility and tolerability (18–20).
Herein, we report the results of an open-label phase I clinical study assessing the safety, tolerability, and antitumor activity of buparlisib given concurrently with bevacizumab in a cohort of previously treated mRCC patients.
Material and Methods
Patient population
Patients with mRCC with a clear cell component or papillary or chromophobe histologies were eligible. Participants must have received at least one prior anti-VEGF systemic therapy for mRCC (excluding bevacizumab and a PI3K inhibitor). Prior everolimus or temsirolimus was allowed. Other eligibility criteria included: age ≥18 years, Eastern Cooperative Oncology Group (ECOG) performance status ≤ 2, absolute neutrophil count ≥ 1.5 × 109/L, platelets ≥ 100 × 109/L, hemoglobin ≥ 9 g/dL, serum creatinine ≤ 1.5 upper limit of normal (ULN), fasting glucose ≤ 120 mg/dL or HgA1c ≤ 6.7%, urine protein/creatinine ratio < 1.5 or ≤ 1+ protein on urinalysis, and amylase, lipase, aspartate aminotransferase (or ≤ 3 × ULN if liver metastases were present), alanine aminotransferase (or ≤ 3 × ULN if liver metastases were present), bilirubin (or ≤ 1.5 × ULN if liver metastases were present), calcium (corrected for serum albumin), potassium and magnesium within the institutional normal range. Patients with untreated brain metastases, active liver disease, active pancreatic disease including poorly controlled diabetes, cardiac disease, and poorly controlled hypertension (defined as a systolic blood pressure > 140 or diastolic blood pressure > 90) were excluded. Patients with Common Terminology Criteria for Adverse Events (CTCAE) version 4.0 anxiety or active psychiatric illness were excluded unless disease has been stable without modifications to medical therapy within six weeks of therapy initiation.
Study design and treatment
This was a multi-center, open-label, investigator-sponsored, single-arm, phase I study with a standard 3+3 dose-escalation design followed by dose-expansion at the maximum tolerated dose (MTD) of buparlisib and bevacizumab (NCT01283048) (Figure 1). The study was conducted at three institutions in the US: Dana-Farber Cancer Institute (DFCI, Boston, MA), Beth Israel Deaconess Medical Center (Boston, MA), and Karmanos Cancer Institute (Detroit, MI). Patients received oral buparlisib once daily in a continuous schedule of 28-day cycles. Bevacizumab was administered intravenously every two weeks. Buparlisib was administered at escalating doses (60 mg, 80 mg, and 100 mg once daily). Bevacizumab was administered at a fixed dose of 10 mg/kg. Once the MTD was achieved, an expansion cohort including 15 patients was conducted at the MTD for additional safety and efficacy evaluation.
Figure 1.

Study design. DLTs=Dose limiting toxicities.
Routine clinical and laboratory assessments were conducted at baseline, and weekly for the first cycle and then days 1 and 15 of each subsequent cycle. Patients were evaluated for adverse events (AE) on the basis of CTCAE version 4.0. Radiologic assessments were performed every eight weeks. Hematologic dose-limiting toxicities (DLTs) included persistent grade ≥3 neutropenia or thrombocytopenia, any febrile neutropenia, and grade 4 thrombocytopenia. Non-hematologic DLTs included grade ≥1 neurocognitive disorder, persistent grade 2 or any grade ≥3 mood alteration, persistent grade ≥2 phototoxicity or skin toxicity, persistent elevations in creatinine or grade > 3 creatinine elevations, persistent elevations in bilirubin or any grade > 3 bilirubin elevations, persistent grade 3 or any grade 4 AST or ALT elevations, persistent grade 2 or any grade ≥3 hyperglycemia, grade ≥2 pancreatitis or persistent grade 3 or any grade 4 amylase and/or lipase elevations. Any other grade ≥3 toxicity was also considered a DLT, with the following exceptions: anemia, lymphopenia, and elevations in alkaline phosphatase. Patients who experienced grades 3–4 AE had dose adjustments as specified in the clinical study. Dose reductions for bevacizumab or buparlisib below the starting dose of 60 mg were not allowed. Patient remained on study until intolerable toxicity, disease progression, or withdrawal.
Primary endpoints
The primary objective was to determine the MTD and DLTs of the combination of buparlisib and bevacizumab in patients with mRCC. The MTD was defined as the highest dose at which no more than 1 of 6 new participants in a dosing cohort experienced a DLT possibly related to the study drug during the first cycle of treatment. Secondary objectives included: safety and tolerability, objective response rate (ORR), time-to-treatment failure (TTF), overall survival (OS). All participants who received at least one dose of study treatment were evaluable for toxicity. Disease response was defined by Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 (21). TTF was defined as the time from enrollment to treatment discontinuation for any reason. OS was defined as the time from enrollment to death from any cause.
Mental safety assessment
Due to the central nervous system (CNS) penetration of the drug and the incidence of mood disorders, two different evaluation test were performed to analyze effects of buparlisib on depression and anxiety: The Patient Health Questionnaire (PHQ-9), a diagnostic tool to assess mental health disorders, and the Generalized Anxiety Disorder 7-item (GAD-7), a scale for anxiety, were performed at baseline, cycle 1 and cycle 2 to assess mental health during the study period. PHQ-9 scores from 0 to 27 and the cutoffs of 5, 10, 15 and 20 represent the thresholds towards severe depression with higher scores representing worse symptoms (22). GAD-7 scores from 0 to 21 and the cutoffs of 5, 10 and 15 represent the thresholds towards severe anxiety with higher scores representing worse symptoms (23).
Biomarker assessments
To examine the effects of buparlisib on metabolism, fasting serum lipids and fasting glucose levels were measured on day 1 and 15 of every cycle until therapy discontinuation. To identify genetic mechanisms of sensitivity to the buparlisib-based combination, nine DFCI treated patients underwent genomic profiling of tumor tissue as part of a separate local Institutional Review Board approved protocol. Mutation profiling was performed using massively parallel sequencing technology (OncoPanel) as previously described (24,25). OncoPanel assay surveys exonic DNA sequences of 275 cancer genes and detects copy number variations and structural variants in tumor DNA.
Statistical analysis
Patient and clinical characteristics at baseline were summarized as numbers and percentages for categorical variables and median with interquartile ranges for continuous variables. All reported toxicities by toxicity type and maximum grade were summarized and sorted by number of patients experiencing the toxicity. Toxicities were summarized regardless of attributions as well as with regard to study treatments. ORR was summarized as number and percentage with 95% confidence interval (CI). The 95% CI was calculated using exact bionomial method. Kaplan Meier estimate was used to summarize median TTF and OS. Association of glucose and total cholesterol changes from baseline to cycle 1 and response was summarized using Wilcoxon’s rank sum test.
Results
Patient and disease characteristics
Between September 2011 and December 2013, the study enrolled 32 patients. Two patients who consented were not able to receive treatment. The majority of patients had clear cell histology (87%) and 15 patients (50%) had received two or more prior lines of systemic therapy (Table 1). With regard to International Metastatic RCC Database Consortium (IMDC) risk groups, the majority of patients had intermediate-risk disease.
Table 1.
Baseline patient and disease characteristics.
| N=30 | % or Median (q1, q3) | |
|---|---|---|
| Age at baseline | 30 | 61 (52, 68) |
| Male gender | 27 | 90% |
| Histology | ||
| Clear-Cell | 26 | 87% |
| Papillary | 2 | 7% |
| Unknown | 2 | 7% |
| Baseline sites of metastases | ||
| Lung | 17 | 57% |
| Lymph nodes | 18 | 60% |
| Bone | 10 | 33% |
| Liver | 9 | 30% |
| Brain | 5 | 17% |
| Baseline number of metastatic sites | ||
| 1 | 6 | 20% |
| >1 | 24 | 80% |
| Prior lines of systemic therapy | ||
| 1 | 15 | 50% |
| 2 | 8 | 27% |
| 3 | 3 | 10% |
| ≥4 | 4 | 13% |
| Time from original diagnosis to treatment | ||
| < 1 year | 25 | 83% |
| ≥1 year | 5 | 17% |
| ECOG performance status | ||
| 0 | 13 | 43% |
| 1 | 16 | 53% |
| Unknown | 1 | 3% |
| Hemoglobin (g/dL) | 30 | 11.9 (10.1, 13.7) |
| Corrected calcium (mg/dL) | 30 | 9.6 (9.4, 10.3) |
| IMDC risk group | ||
| Favorable | 2 | 7% |
| Intermediate | 17 | 57% |
| Poor | 10 | 33% |
| Unknown | 1 | 3% |
ECOG= Eastern Cooperative Oncology Group, IMDC=International Metastatic Renal Cell Carcinoma Database Consortium
DLTs and determination of MDT
The first six patients treated in cohorts 1 or 2 did not develop DLTs at the dose of buparlisib 60 mg (n=3) or 80 mg (n=3). At the dose of buparlisib 100 mg, one patient in cohort 3 and another one in cohort 4 developed DLTs (Table 2). Subsequently, the dose was de-escalated and another three patients were enrolled in cohort 5 at buparlisib 80 mg per day, which was determined to be the MTD when given in combination with bevacizumab 10 mg/kg, meeting the primary endpoint of the study. Fifteen additional mRCC patients were enrolled at this dose level and among them, two patients developed DLTs (Table 2). In summary, three patients were treated at the initial dose level of buparlisib 60 mg, 21 patients at 80 mg and 6 patients at 100 mg.
Table 2.
Dose limiting toxicities as graded by Common Terminology Criteria for Adverse Events version 4.0.
| Dose Level | N | DLTs | PatientswithDLTs | CaseNumber | Type | Grade | AttributionBuparlisib | Attribution Bevacizumab |
|---|---|---|---|---|---|---|---|---|
| Dose Escalation | ||||||||
| 60 mg daily | 3 | No | 0 | |||||
| 80 mg daily | 6 | Yes | 1 | 15 | Cognitive disturbance | 2 | Possible | Possible |
| 100 mg daily | 6 | Yes | 2 | 11 | Depression | 3 | Definite | No Relation |
| Suicidal ideation | 3 | Possible | No Relation | |||||
| Anxiety | 3 | Possible | No Relation | |||||
| 12 | Anorexia | 3 | Possible | No Relation | ||||
| Weight loss | 3 | Possible | No Relation | |||||
| Dose Expansion | ||||||||
| 80 mg daily | 15 | Yes | 2 | 26 | Lipase increase | 3 | Possible | No Relation |
| Amylase increase | 2 | Possible | No Relation | |||||
| Depression | 2 | Possible | No Relation | |||||
| 31 | Pruritus | 3 | Possible | No Relation | ||||
| Maculopapular rash | 3 | Possible | No Relation | |||||
DLTs=dose limiting toxicities.
Safety and tolerability
Of the 30 patients who received at least one dose of treatment, the median number of cycles administered was four (range 1–9). Overall, 28 patients discontinued therapy: n=17 due to disease progression, n=7 due to toxicity, and 4 due to other reasons. One death occurred within 30 days of treatment discontinuation, which was attributed to progression disease.
The most frequently reported AE (any grade) for all patients included AST elevation (n=16), fatigue (n=16), ALT elevation (n=13), hyperglycemia (n=13), diarrhea (n=12) and nausea (n=11) (Table S1). The most frequently reported grade 3–4 AE included hypertension (n=4), ALT elevation (n=4), AST elevation (n=4) and lipase elevation (n=4) (Table S1). The most common AE attributed to buparlisib were AST elevation (n=14), fatigue (n=12) and hyperglycemia (n=11). The most common reported AE attributed to bevacizumab were hypertension (n=9), fatigue (n=8) and proteinuria (n=6) (Table 3). At the MTD, the most common toxicities included hyperglycemia (n=6), ALT elevation (n=5) and AST increase (n=5).
Table 3.
Any grade treatment associated toxicities attributed to buparlisib or bevacizumab.
| Toxicity | Grade 1 | Grade 2 | Grade 3 | Grade 4 | Total |
|---|---|---|---|---|---|
| Attributed to Bevacizumab | |||||
| Hypertension | 0 | 5 | 4 | 0 | 9 |
| Fatigue | 8 | 0 | 0 | 0 | 8 |
| Proteinuria | 1 | 5 | 0 | 0 | 6 |
| Epistaxis | 4 | 1 | 0 | 0 | 5 |
| Anorexia | 3 | 0 | 0 | 0 | 3 |
| Attributed to Buparlisib | |||||
| AST increase | 7 | 3 | 2 | 2 | 14 |
| Fatigue | 7 | 4 | 1 | 0 | 12 |
| Hyperglycemia | 5 | 5 | 1 | 0 | 11 |
| ALT increase | 5 | 1 | 3 | 1 | 10 |
| Diarrhea | 6 | 1 | 1 | 0 | 8 |
| Nausea | 7 | 0 | 1 | 0 | 8 |
| Anorexia | 6 | 1 | 0 | 0 | 7 |
| Anxiety | 5 | 0 | 1 | 0 | 6 |
| Depression | 4 | 1 | 1 | 0 | 6 |
| Lipase increase | 1 | 2 | 3 | 0 | 6 |
| Oral mucositis | 5 | 1 | 0 | 0 | 6 |
| Amylase increase | 4 | 1 | 1 | 0 | 6 |
| Hypertriglyceridemia | 2 | 2 | 1 | 0 | 5 |
| Weight loss | 3 | 2 | 0 | 0 | 5 |
| Rash – acneiform | 4 | 0 | 0 | 0 | 4 |
| Cognitive disturbance | 0 | 3 | 0 | 0 | 3 |
| Dry skin | 2 | 1 | 0 | 0 | 3 |
| Thrombocytopenia | 3 | 0 | 0 | 0 | 3 |
| Rash – maculopapular | 1 | 1 | 1 | 0 | 3 |
| Vomiting | 3 | 0 | 0 | 0 | 3 |
AST=aspartate aminotransferase; ALT= alanine aminotransferase.
Median levels of anxiety, measured by GAD-7, improved from a score of 1 at baseline to 0 in both cycles 1 and 2. In addition, median PHQ score improved from a score of 2 at baseline and cycle 1, to 1 at cycle 2.
Clinical activity
Of the 30 treated patients, four (13%; 95% CI 4%, 31%) achieved a partial response and 15 (50%; 95% CI 31%, 69%) achieved stable disease (Figure 2). Six patients (20%) had progressive disease as best response and five patients were unevaluable (17%). Of the 21 patients treated at the MTD, two (10%; 95% CI 1%, 30%) achieved a partial response and 10 (48%; 95% CI 26%, 70%) achieved stable disease as the best response, with 14 patients experiencing some degree of tumor shrinkage. For the overall cohort, the median TTF was 4 months (95% CI 2, 9) and median OS was not reached. Of the 21 patients who were treated at MTD, median TTF was 3 months (95% CI 2, 8) and median OS was 13 months (95% CI 4, not reached). The four patients with partial response had either intermediate (n=2) or poor IMDC risk criteria (n=2), and achieved a median duration of response of 9.2 months (range 5,5–15). A total of 10 patients were able to receive subsequent therapy, of whom six patients received more than two subsequent lines of treatment following discontinuation of buparlisib and bevacizumab.
Figure 2.
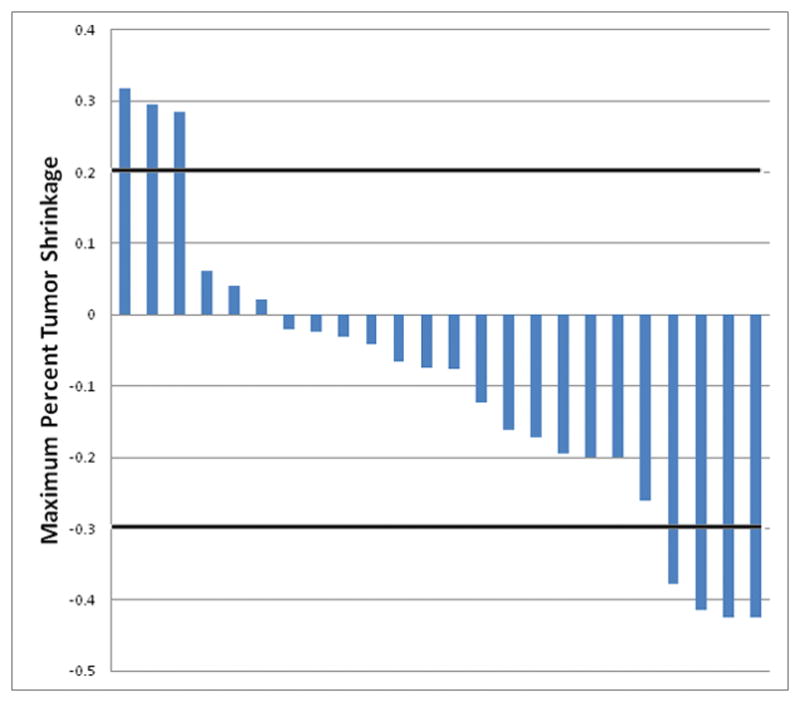
Waterfall plot of percent maximal tumor shrinkage of measurable target lesions by RECIST 1.1.
Biomarker assessment
Of the nine patients evaluated, two had activating PI3KCA mutations. Both hotspot mutations were c.3140A>G (H1047R). In term of outcomes, one achieved a partial response with 42% tumor shrinkage (TTF 13 months) and the other achieved stable disease with 16% tumor shrinkage (TTF 9 months). Both patients had clear cell histology and poor IMDC risk criteria.
Biomarker samples were available for metabolic analysis in 27 of 30 patients. A correlation between percentage change in glucose with ORR was observed. Patients who developed elevations in fasting glucose levels from baseline (n=4, median change +30% [7%, 48%)] were more likely to have an objective response compared with patients without an increase from baseline (n=23, median change −4% [9%, −16%]) (p= 0.04). There was no significant correlation between changes in lipids (total cholesterol, triglycerides or low-density lipoprotein) and tumor response.
Discussion
The results from this phase 1b study provide evidence that buparlisib at the MTD of 80 mg daily combined with bevacizumab 10 mg/kg every two weeks is safe and tolerable in patients with mRCC. We demonstrate that therapy with combination buparlisib and bevacizumab was feasible and toxicity was manageable.
The most common serious adverse events were hepatic enzyme abnormalities, fatigue and hypertension. While hepatitis was mainly attributed to buparlisib it is uncertain to what degree fatigue and hypertension may be attributed to buparlisib, bevacizumab or both. The rates of grade 3–4 fatigue and hypertension demonstrated in phase III studies of bevacizumab with interferon were 12% and 3%, which is similar to our study of the bevacizumab in combination with buparlisib (4).
With regard to all grade toxicities, hyperglycemia and diarrhea, attributed to buparlisib, were common but well tolerated. Psychiatric adverse events, which have been reported with PI3K inhibitors in approximately 30% of patients in early phase clinical trials, consisted of anxiety and depression. In our study the rate of psychiatric adverse events was 43% (n=13), though 11 out of 13 patients had grade 1 anxiety or depression. The mood questionnaires did not show major changes with time, although mood disorders produced two DLTs in the escalation cohort. Additional DLTs seen in the study were anorexia and pruritus which were manageable and previously described with PI3K inhibitors (26).
To overcome resistance to VEGF-targeted therapies and improve clinical outcomes, a wide variety of combinatorial treatment strategies have been investigated in patients with mRCC. Efforts to combine agents including tyrosine kinase inhibitors and mTOR inhibitors have been generally limited due to serious and overlapping toxicity. Combinations of sunitinib with temsirolimus and bevacizumab showed intolerable toxicity: DLTs were observed at low starting doses of both agents with all grade hypertension ~ 92% (19,27). In our study, we demonstrate that the combination of buparlisip and bevacizumab was safe and tolerable. Indeed, bevacizumab has been shown to be a feasible compound to combine with other agents given that as a monoclonal antibody it has less overlapping toxicities. It has been used safety with high dose IL-2, mTOR inhibitors or novel compounds such as TRC105 (anti-endoglin IgG1 monoclonal antibody) (15,28–30). In future combinatorial studies, bevacizumab may be an agent of choice for VEGF inhibition. Currently, bevacizumab is also being explored in combination with immune checkpoint inhibitors in mRCC; a phase 3 clinical trial of MPDL3280A (anti-programmed death ligand 1 [PD-L1] antibody) in combination with bevacizumab versus sunitinib is already ongoing in patients with untreated advanced RCC (NCT02420821).
PI3K inhibitors may be promising drugs to overcome VEGF resistance in mRCC. The three main classes of PI3K inhibitors, according to the substrate they target, are dual pan-Class I PI3K/mTOR inhibitors, pan-Class I PI3K inhibitors lacking significant mTOR activity and isoform-selective PI3K inhibitors (31). PI3K inhibitors are also being studied with different combinations including chemotherapy (e.g. lung cancer: combined with carboplatin-pemetrexed), hormonal therapy (e.g. combined with fulvestrant or letrozol in breast cancer), targeted therapy (e.g. combined cetuximab in head and neck tumors) or radiotherapy (e.g. thymoma) (32). Specifically buparlisib is being tested across several tumor types such as breast cancer, lung cancer and thymomas (32). In solid tumors including mRCC, preliminary data demonstrated that buparlisib combined with the MEK1/2 inhibitor trametinib showed early signs of antitumor activity but toxicity was significant (33). Though combined PI3K/mTOR inhibitors have demonstrated a more profound effect on cellular proliferation in preclinical RCC models, their clinical progress has been limited by toxicity in early phase studies (34,35).
In the current study, the combination of buparlisib and bevacizumab demonstrated antitumor activity in this cohort of heavily pretreated mRCC patients. Although comparisons to other clinical trials can be flawed, the objective responses were somewhat lower compared to the combination of bevacizumab and mTOR inhibitors. Both everolimus and temsirolimus with bevacizumab have shown objective responses in approximately 23% of pretreated mRCC patients compared to only 13% in our study (36,37), although our population is more heavily pre-treated.
Precision medicine platforms can identify molecular drivers of disease for the application of personalized approaches for cancer therapeutics (25). Recent efforts have been devoted to characterizing the genomic landscape of mRCC with the goal of discovering potentially clinically relevant actionable genes (38). Despite identification of the recurrent mutations beyond VHL, such as PBRM1, SETD2 or BAP1, we currently have no evidence that clinical practice should be modified based on the presence of these mutations. Integrated molecular analysis (e.g. performing whole exome sequencing) may be the required step for appropriate biomarker selection (39). Indeed, Janku et al. showed that heavily pretreated patients with advanced solid tumors harbor activating PIK3CA mutations may be sensitive to therapeutic targeting with PI3K/AKT/mTOR pathway inhibitors (40). Overall, based on data from The Cancer Genome Atlas and other sources, PIK3CA mutations in both localized and metastatic disease seem to be uncommon (less than 5%) in RCC (41,42). In our study, two out of nine patients with genomic data (22%), harbored PIK3CA mutations and achieved clinical benefit from treatment. Additionally, some patients demonstrated mutations known to be harbored in RCC including VHL and BAP1 but not associations with response were found. Unfortunately, given the limited number of patients with genomic data, no other alterations were identified which may and serve as predictors of response to therapy. Experimental data suggests that additional biomarkers, yet to be defined, are required beyond PI3K status to predict response to these compounds (43).
The PI3K/Akt/mTOR pathway has a critical role in insulin signaling and glucose homeostasis (44). Hyperglycemia is a class effect observed with PI3K/Akt/mTOR inhibition. This metabolic alteration is secondary to a fasting state characterized by reduced utilization of glucose and predilection for fatty acid metabolism (45). We measured metabolic adverse events – hyperlipidemia and hyperglycemia –and correlated levels with response. Glycemic changes appeared to correlate with response and may be considered a predictive clinical biomarker of response. This observation parallels the association of early onset of hyperglycemia and clinical benefit described with everolimus (46).
In recent years, RCC treatments have focused largely on inhibition of VEGF pathways. The PI3K pathway, which is recurrently altered in RCC, may be an escape mechanism for resistance to anti-VEGF therapies. The safety profile and antitumor activity of this combination leads us to believe that a subset of patients harboring PI3K mutations may derive benefit from buparlisib and that increased fasting blood sugars may be an early predictor of activity. The results of this study provide important pharmacologic and toxicity data to explore this combination further potentially in a preselected patient population.
Supplementary Material
Translational Relevance.
An improved understanding of the pathogenesis of renal cell carcinoma (RCC) has identified the vascular-endothelial growth factor (VEGF) pathway as a key target in this disease. Though VEGF-targeted therapies have improved survival for patients with metastatic RCC, nearly all patients develop resistance. Consequently, novel and combinatorial treatment strategies, which provide durable responses in patients refractory to current therapies, are warranted. The PI3K/Akt/mTOR pathway is dysregulated in patients with metastatic RCC and targeting this pathway, in addition to the VEGF pathway, is a potential therapeutic strategy in the management of RCC. Elucidation of the impact of combinatorial PI3K and VEGF inhibition on outcomes in patients with RCC is therefore highly relevant to optimizing the current treatment armamentarium for patients with metastatic RCC.
Acknowledgments
Funding: This study was funded in part by Novartis. Additionally, this research was funded in part by the Dana-Farber/Harvard Cancer Center Kidney SPORE (DM and TKC), and the Trust Family, Michael Brigham, and Loker Pinard Funds for Kidney Cancer Research at Dana-Farber Cancer Institute for TKC.
Grant: P50 CA101942-01.
References
- 1.Seizinger BR, Rouleau GA, Ozelius LJ, et al. Von Hippel-Lindau disease maps to the region of chromosome 3 associated with renal cell carcinoma. Nature. 1988 Mar 17;332(6161):268–9. doi: 10.1038/332268a0. [DOI] [PubMed] [Google Scholar]
- 2.Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011 Sep 16;146(6):873–87. doi: 10.1016/j.cell.2011.08.039. [DOI] [PubMed] [Google Scholar]
- 3.Massari F, Ciccarese C, Santoni M, et al. Metabolic alterations in renal cell carcinoma. Cancer Treat Rev. 2015 Jul 9; doi: 10.1016/j.ctrv.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 4.Escudier B, Pluzanska A, Koralewski P, et al. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: a randomised, double-blind phase III trial. Lancet Lond Engl. 2007 Dec 22;370(9605):2103–11. doi: 10.1016/S0140-6736(07)61904-7. [DOI] [PubMed] [Google Scholar]
- 5.Escudier B, Eisen T, Stadler WM, et al. Sorafenib for treatment of renal cell carcinoma: Final efficacy and safety results of the phase III treatment approaches in renal cancer global evaluation trial. J Clin Oncol Off J Am Soc Clin Oncol. 2009 Jul 10;27(20):3312–8. doi: 10.1200/JCO.2008.19.5511. [DOI] [PubMed] [Google Scholar]
- 6.Motzer RJ, Hutson TE, McCann L, Deen K, Choueiri TK. Overall survival in renal-cell carcinoma with pazopanib versus sunitinib. N Engl J Med. 2014 May 1;370(18):1769–70. doi: 10.1056/NEJMc1400731. [DOI] [PubMed] [Google Scholar]
- 7.Motzer RJ, Hutson TE, Cella D, et al. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N Engl J Med. 2013 Aug 22;369(8):722–31. doi: 10.1056/NEJMoa1303989. [DOI] [PubMed] [Google Scholar]
- 8.Motzer RJ, Escudier B, Tomczak P, et al. Axitinib versus sorafenib as second-line treatment for advanced renal cell carcinoma: overall survival analysis and updated results from a randomised phase 3 trial. Lancet Oncol. 2013 May;14(6):552–62. doi: 10.1016/S1470-2045(13)70093-7. [DOI] [PubMed] [Google Scholar]
- 9.Ellis LM, Hicklin DJ. Resistance to Targeted Therapies: Refining Anticancer Therapy in the Era of Molecular Oncology. Clin Cancer Res. 2009 Dec 15;15(24):7471–8. doi: 10.1158/1078-0432.CCR-09-1070. [DOI] [PubMed] [Google Scholar]
- 10.Creighton CJ, Morgan M, Gunaratne PH, et al. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013 Jun 23;499(7456):43–9. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.cBioPortal for Cancer Genomics. 2015 Available from: http://www.cbioportal.org/cross_cancer.
- 12.Koshikawa N, Hayashi J-I, Nakagawara A, Takenaga K. Reactive Oxygen Species-generating Mitochondrial DNA Mutation Up-regulates Hypoxia-inducible Factor-1 Gene Transcription via Phosphatidylinositol 3-Kinase-Akt/Protein Kinase C/Histone Deacetylase Pathway. J Biol Chem. 2009 Nov 27;284(48):33185–94. doi: 10.1074/jbc.M109.054221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fransecky L, Mochmann LH, Baldus CD. Outlook on PI3K/AKT/mTOR inhibition in acute leukemia. Mol Cell Ther [Internet] 2015 Dec;3(1) doi: 10.1186/s40591-015-0040-8. [cited 2015 Jul 17] Available from: http://www.molcelltherapies.com/content/3/1/2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fritsch C, Huang A, Chatenay-Rivauday C, et al. Characterization of the Novel and Specific PI3K Inhibitor NVP-BYL719 and Development of the Patient Stratification Strategy for Clinical Trials. Mol Cancer Ther. 2014 May 1;13(5):1117–29. doi: 10.1158/1535-7163.MCT-13-0865. [DOI] [PubMed] [Google Scholar]
- 15.Bendell JC, Gordon MS, Hurwitz HI, et al. Safety, pharmacokinetics, pharmacodynamics, and antitumor activity of dalantercept, an activin receptor-like kinase-1 ligand trap, in patients with advanced cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2014 Jan 15;20(2):480–9. doi: 10.1158/1078-0432.CCR-13-1840. [DOI] [PubMed] [Google Scholar]
- 16.FDA Approval for Bevacizumab [Internet] [cited 2015 Jul 16]. Available from: http://www.cancer.gov/about-cancer/treatment/drugs/fda-bevacizumab.
- 17.Ravaud A, Gross-Goupil M, Bellmunt J. Combination therapy in metastatic renal cell cancer. Semin Oncol. 2013 Aug;40(4):472–81. doi: 10.1053/j.seminoncol.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 18.Amit L, Ben-Aharon I, Vidal L, Leibovici L, Stemmer S. The impact of Bevacizumab (Avastin) on survival in metastatic solid tumors--a meta-analysis and systematic review. PloS One. 2013;8(1):e51780. doi: 10.1371/journal.pone.0051780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson BE, Kabbinavar F, Fehrenbacher L, et al. ATLAS: randomized, double-blind, placebo-controlled, phase IIIB trial comparing bevacizumab therapy with or without erlotinib, after completion of chemotherapy, with bevacizumab for first-line treatment of advanced non-small-cell lung cancer. J Clin Oncol Off J Am Soc Clin Oncol. 2013 Nov 1;31(31):3926–34. doi: 10.1200/JCO.2012.47.3983. [DOI] [PubMed] [Google Scholar]
- 20.Hobday TJ, Qin R, Reidy-Lagunes D, et al. Multicenter Phase II Trial of Temsirolimus and Bevacizumab in Pancreatic Neuroendocrine Tumors. J Clin Oncol Off J Am Soc Clin Oncol. 2015 May 10;33(14):1551–6. doi: 10.1200/JCO.2014.56.2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer Oxf Engl 1990. 2009 Jan;45(2):228–47. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 22.Kroenke K, Spitzer RL, Williams JBW. The PHQ-9: Validity of a brief depression severity measure. J Gen Intern Med. 2001 Sep;16(9):606–13. doi: 10.1046/j.1525-1497.2001.016009606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kroenke K, Spitzer RL, Williams JBW, Monahan PO, Löwe B. Anxiety disorders in primary care: prevalence, impairment, comorbidity, and detection. Ann Intern Med. 2007 Mar 6;146(5):317–25. doi: 10.7326/0003-4819-146-5-200703060-00004. [DOI] [PubMed] [Google Scholar]
- 24.MacConaill LE, Campbell CD, Kehoe SM, et al. Profiling critical cancer gene mutations in clinical tumor samples. PloS One. 2009;4(11):e7887. doi: 10.1371/journal.pone.0007887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wagle N, Berger MF, Davis MJ, et al. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov. 2012 Jan;2(1):82–93. doi: 10.1158/2159-8290.CD-11-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bendell JC, Rodon J, Burris HA, et al. Phase I, Dose-Escalation Study of BKM120, an Oral Pan-Class I PI3K Inhibitor, in Patients With Advanced Solid Tumors. J Clin Oncol. 2012 Jan 20;30(3):282–90. doi: 10.1200/JCO.2011.36.1360. [DOI] [PubMed] [Google Scholar]
- 27.Feldman DR, Baum MS, Ginsberg MS, et al. Phase I trial of bevacizumab plus escalated doses of sunitinib in patients with metastatic renal cell carcinoma. J Clin Oncol Off J Am Soc Clin Oncol. 2009 Mar 20;27(9):1432–9. doi: 10.1200/JCO.2008.19.0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dandamudi UB, Ghebremichael M, Sosman JA, et al. A Phase II Study of Bevacizumab and High-dose Interleukin-2 in Patients With Metastatic Renal Cell Carcinoma: A Cytokine Working Group (CWG) Study. J Immunother. 2013;36(9):490–5. doi: 10.1097/CJI.0000000000000003. [DOI] [PubMed] [Google Scholar]
- 29.Harshman LC, Barbeau S, McMillian A, Srinivas S. A Phase II Study of Bevacizumab and Everolimus as Treatment for Refractory Metastatic Renal Cell Carcinoma. Clin Genitourin Cancer. 2013 Jun;11(2):100–6. doi: 10.1016/j.clgc.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 30.Rini BI, Bellmunt J, Clancy J, et al. Randomized Phase III Trial of Temsirolimus and Bevacizumab Versus Interferon Alfa and Bevacizumab in Metastatic Renal Cell Carcinoma: INTORACT Trial. J Clin Oncol. 2014 Mar 10;32(8):752–9. doi: 10.1200/JCO.2013.50.5305. [DOI] [PubMed] [Google Scholar]
- 31.Bauer TM, Patel MR, Infante JR. Targeting PI3 kinase in cancer. Pharmacol Ther. 2015 Feb;146:53–60. doi: 10.1016/j.pharmthera.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 32.ClinicalTrials.gov. A service of the U.S. National Institutes of Health [Internet] Clinicaltrials.gov. [cited 2015 Aug 4]. Available from: https://clinicaltrials.gov/ct2/results?term=buparlisib&Search=Search.
- 33.Bedard PL, Tabernero J, Janku F, et al. A Phase Ib Dose-Escalation Study of the Oral Pan-PI3K Inhibitor Buparlisib (BKM120) in Combination with the Oral MEK1/2 Inhibitor Trametinib (GSK1120212) in Patients with Selected Advanced Solid Tumors. Clin Cancer Res. 2015 Feb 15;21(4):730–8. doi: 10.1158/1078-0432.CCR-14-1814. [DOI] [PubMed] [Google Scholar]
- 34.Serova M, de Gramont A, Tijeras-Raballand A, et al. Benchmarking effects of mTOR, PI3K, and dual PI3K/mTOR inhibitors in hepatocellular and renal cell carcinoma models developing resistance to sunitinib and sorafenib. Cancer Chemother Pharmacol. 2013 May;71(5):1297–307. doi: 10.1007/s00280-013-2129-6. [DOI] [PubMed] [Google Scholar]
- 35.Cho DC, Cohen MB, Panka DJ, et al. The efficacy of the novel dual PI3-kinase/mTOR inhibitor NVP-BEZ235 compared with rapamycin in renal cell carcinoma. Clin Cancer Res Off J Am Assoc Cancer Res. 2010 Jul 15;16(14):3628–38. doi: 10.1158/1078-0432.CCR-09-3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Merchan JR, Qin R, Pitot H, et al. Safety and activity of temsirolimus and bevacizumab in patients with advanced renal cell carcinoma previously treated with tyrosine kinase inhibitors: a phase 2 consortium study. Cancer Chemother Pharmacol. 2015 Mar;75(3):485–93. doi: 10.1007/s00280-014-2668-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hainsworth JD, Spigel DR, Burris HA, Waterhouse D, Clark BL, Whorf R. Phase II trial of bevacizumab and everolimus in patients with advanced renal cell carcinoma. J Clin Oncol Off J Am Soc Clin Oncol. 2010 May 1;28(13):2131–6. doi: 10.1200/JCO.2009.26.3152. [DOI] [PubMed] [Google Scholar]
- 38.Chin L, Hahn WC, Getz G, Meyerson M. Making sense of cancer genomic data. Genes Dev. 2011 Mar 15;25(6):534–55. doi: 10.1101/gad.2017311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones SJ, Laskin J, Li YY, et al. Evolution of an adenocarcinoma in response to selection by targeted kinase inhibitors. Genome Biol. 2010;11(8):R82. doi: 10.1186/gb-2010-11-8-r82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Janku F, Wheler JJ, Naing A, et al. PIK3CA mutation H1047R is associated with response to PI3K/AKT/mTOR signaling pathway inhibitors in early-phase clinical trials. Cancer Res. 2013 Jan 1;73(1):276–84. doi: 10.1158/0008-5472.CAN-12-1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Creighton CJ, Morgan M, Gunaratne PH, et al. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013 Jun 23;499(7456):43–9. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pal SK, Ali SM, Chalmers Z, et al. Comprehensive genomic profiling of 443 cases of renal cell carcinoma to reveal frequent clinically relevant genomic alterations. San Francisco: J Clin Oncol. 2015;33(suppl 7) (abstr 433); [cited 2016 Mar 11]. Available from: http://meetinglibrary.asco.org/content/142083-159. [Google Scholar]
- 43.Arques O, Chicote I, Puig I, et al. Tankyrase inhibition blocks Wnt/β-catenin pathway and reverts resistance to PI3K and AKT inhibitors in the treatment of colorectal cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2015 Jul 29; doi: 10.1158/1078-0432.CCR-14-3081. [DOI] [PubMed] [Google Scholar]
- 44.Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene. 2005 Nov 14;24(50):7455–64. doi: 10.1038/sj.onc.1209085. [DOI] [PubMed] [Google Scholar]
- 45.Busaidy NL, Farooki A, Dowlati A, et al. Management of Metabolic Effects Associated With Anticancer Agents Targeting the PI3K-Akt-mTOR Pathway. J Clin Oncol. 2012 Aug 10;30(23):2919–28. doi: 10.1200/JCO.2011.39.7356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stone R, Schlumbrecht M, Johnston T, et al. Sweet Response: Hyperglycemia and Hypertriglyceridemia as Biomarkers of Clinical Benefit for Everolimus in Patients with Recurrent Endometrial Cancer. Gynecol Oncol. 2012 May;125(2):S189. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.