

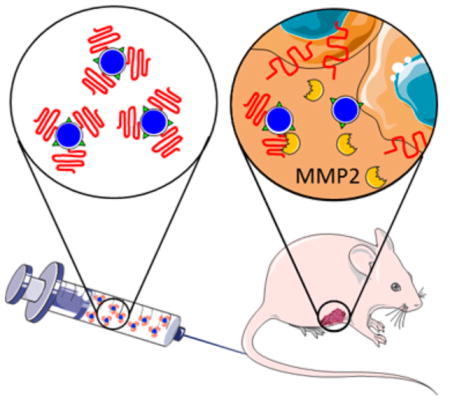
Abstract
Active targeting of nanoparticles through surface functionalization is a common strategy to enhance tumor delivery specificity. However, active targeting strategies tend to work against long polyethylene glycol’s shielding effectiveness and associated favorable pharmacokinetics. To overcome these limitations, we developed a matrix metalloproteinase-2 sensitive surface-converting polyethylene glycol coating. This coating prevents nanoparticle–cell interaction in the bloodstream, but, once exposed to matrix metalloproteinase-2, i.e., when the nanoparticles accumulate within the tumor interstitium, the converting polyethylene glycol coating is cleaved, and targeting ligands become available for binding to tumor cells. In this study, we applied a comprehensive multimodal imaging strategy involving optical, nuclear, and magnetic resonance imaging methods to evaluate this coating approach in a breast tumor mouse model. The data obtained revealed that this surface-converting coating enhances the nanoparticle’s blood half-life and tumor accumulation and ultimately results in improved tumor-cell targeting. Our results show that this enzyme-specific surface-converting coating ensures a high cell-targeting specificity without compromising favorable nanoparticle pharmacokinetics.
Graphical Abstract
INTRODUCTION
Surface functionalization of nanoparticles with targeting ligands such as antibodies, peptides, or nucleic acids has shown significant advantages in preclinical cancer nanotherapy studies.1–4 Arginyl-glycyl-aspartic acid (RGD) and asparagic-glycyl-arginyl acid (NGR) peptides are among the most widely studied targeting ligands5–12 as they have demonstrated strong affinities for, respectively, αvβ3 integrin and CD13, key receptors expressed by breast cancer and tumor endothelial cells involved in cell migration and tumor angiogenesis.13,14 However, in the bloodstream, targeting moieties present on the nanoparticle surface may cause elevated recognition by the mononuclear phagocyte system and thereby compromise nanoparticle pharmacokinetic characteristics and tumor accumulation.15–17 Furthermore, in-depth cellular examination revealed that RGD and NGR decoration mainly induce nanoparticle association with tumor vasculature while marginally reaching the tumor interstitium.10,11
To overcome these limitations, enzyme-sensitive targeting strategies using cleavable peptides18–20 and cleavable polyethylene glycol (PEG) shielding21–26 have been developed. Upon intravenous nanoparticle injection such PEG shielding prevents nanoparticle–cell interactions in the bloodstream, facilitating prolonged blood circulation and increased tumor accumulation through the enhanced permeability and retention effect.2,27,28 Once the nanoparticles accumulate in the tumor microenvironment, the enzymatic cleavage of the peptide linker leads to the surface exposure of the targeting moieties, which enables their association with receptors expressed on the surface of tumor cells.
In the current study, we examined the in vivo efficacy of a matrix metalloproteinase-2 enzyme (MMP2) sensitive surface-converting PEG shielding29 on a recently introduced polymer–lipid nanoparticle platform30,31 (Figure 1A). The MMP2 enzyme was chosen as cleaving agent because this protein is highly expressed and activated during tumor progression in several invasive tumors32,33 and, therefore, a valuable therapeutic and diagnostic target.25,34–39 We developed an extensive multimodal imaging strategy to systematically investigate the effect of our MMP2-cleavable PEG coating by labeling the polymer–lipid nanoparticles with several imaging labels, including radioisotopes, fluorescent dyes, and magnetic resonance imaging (MRI) contrast-generating molecules. This approach allowed us to evaluate nanoparticle pharmacokinetics and distribution, exploiting the strengths of each individual modality. Radionuclide labeling was chosen to systemically evaluate how the different coatings affected nanoparticle pharmacokinetics and biodistribution at tissue level with high sensitivity and accuracy. Nanoparticle tumor distribution was assessed in vivo at high spatial resolution by MRI, and fluorescent labels were used throughout this work to enable the visualization of multiple dyes by fluorescence microscopy and near-infrared fluorescence imaging. Finally, cellular specificity within the tumor as a function of nanoparticle coating was evaluated by flow cytometry analysis of digested tumor tissue.
Figure 1.

Physical characterization of the surface-converting nanoparticles. Design of the surface-converting polymer–lipid platform. (A) Schematic of the nanoparticle structure. The nanoparticle platform is made of a PLGA polymeric core shielded by a phoshopholipid corona. The surface-converting capacity was obtained by introducing MMP2-cleavable mPEG–MMP2p-DSPE (with mPEG being mPEG-2000) in the corona. (B) Dynamic light scattering (DLS) hydrodynamic size and polydispersity measurements of the different nanoparticle platforms (n = 3). (C) Negative staining transmission electron microscopy of: (i) untargeted-NP, (ii) RGD-NP, (iii) PEG-3000/RGD-NP, and (iv) PEG-MMP2p/RGD-NP (scale bar: 100 nm). The discrepancy between the nanoparticle mean hydrodynamic size in solution measured by DLS and the transmission electron microscopy images can be explained by the sample dehydration process required for the latter technique.
RESULTS AND DISCUSSION
Nanoparticle Synthesis and Characteristics
Polymer–lipid nanoparticles were produced using a tailored nano-precipitation method.31,43 The precipitation of poorly water-soluble poly(lactic-co-glycolic acid) (PLGA) and phospholipid capping led to the instantaneous formation of a homogeneous population of small nanoparticles (size: <100 nm and polydispersity index: <0.2) composed of a hydrophobic polymeric core coated by a corona of the selected phospholipid composition (Table 1). After purification and concentration by ultrafiltration, nanoparticle characterization by dynamic light scattering (DLS) and transmission electron microscopy demonstrated that addition of targeting ligands or varying PEG coatings did not significantly alter nanoparticle shape, size, or polydispersity (Figure 1B,C). Nanoparticles were also found to be stable in fetal bovine serum solutions (Figure S1).
Table 1.
Overview of the PEG–DSPE Molar Composition of the Different Formulations Synthesized
| mPEG350–DSPE | RGD–PEG350–DSPE | mPEG3000–DSPE | mPEG2000–MMP2p–DSPE | |
|---|---|---|---|---|
| Untargeted-NP | 100% | 0% | 0% | 0% |
| RGD-NP | 95% | 5% | 0% | 0% |
| PEG-3000/RGD-NP | 85% | 5% | 10% | 0% |
| PEG- | 85% | 5% | 0% | |
| MMP2p/RGD-NP | 10% |
Labeling Strategies for Multimodal Imaging
Next, we developed labeling strategies for nuclear magnetic resonance and fluorescence imaging (Figure 2A). Radiolabeling was employed to determine nanoparticle blood circulation half-life and biodistribution by γ counting and autoradiography. Zirconium-89 (89Zr) was chosen as the radioisotope due to its favorable physical half-life of 78.4 h. Nanoparticle radio-labeling was done in two subsequent steps. First, a hydrophobic custom-made chelator (deferoxamine conjugated to a 34-carbon moiety) was incorporated in nanoparticles (DFO-nanoparticles). After purification, DFO-nanoparticles were complexed with 89Zr, and size-exclusion chromatography was used to confirm labeling success and the absence of free 89Zr (Figure 2B). Gadolinium (Gd) labeling for MRI was achieved by substituting a fraction of 1,2-distearoyl-sn-glycero-3-phosphoethanolamine–N-[methoxy (polyethylene glycol)-350] ammonium salt (mPEG350–DSPE) with a gadolinium phospholipid salt (diethylene triamine penta acetic acid di(stearyl amide), Gd–DTPA–DSA). As shown in Figure 2C, the addition of Gd–DTPA–DSA did not have a significant effect on either the nanoparticle size or polydispersity, while the MRI contrast enhancing properties scaled proportionally (Figure S2). Based on these results, nanoparticles labeled with 25% molar ratio of Gd–DTPA–DSA, with an ionic r1 relaxivity of 24.4 mM−1 s−1 at 1.4 T, were used in subsequent MRI experiments. Fluorescent moieties were introduced by including a fraction of PLGA conjugated with cyanine dyes, such as Cy3.5 (excitation maximum: 591 nm and emission maximum: 604 nm), Cy5 (excitation maximum: 646 nm and emission maximum: 662 nm), and Cy5.5 (excitation maximum: 673 nm and emission maximum: 707 nm). While the near-infrared dye Cy5.5 was primarily used due to its favorable tissue-penetration properties, Cy3.5 and Cy5 were used to label specifically coated nanoparticles for ex vivo flow cytometry analyses. The pair Cy3.5 and Cy5 was chosen as their emission spectra do not overlap at the chosen flow cytometry channels (Figure 2D) and therefore allow a good signal separation and unambiguous nanoparticle identification.
Figure 2.

Multimodal imaging capacity of the nanoparticles. (A) Schematic depiction of the integration of multiple contrast agents within the nanoparticles. Fluorescent cyanine-based molecules were integrated in the polymeric core by using cyanine conjugated PLGA. Magnetic resonance contrast agent gadolinium was introduced by incorporating Gd–DTPA–DSA at the expense of PEG350–DSPE in the phospholipid corona. Nanoparticles were radiolabeled by introducing the hydrophobic chelator C34-DFO during the formulation followed by complexion with 89Zr prior to use. (B) Size-exclusion chromatograms showing fluorescent (green) and radioactive (orange) traces of 89Zr-labeled PEG-3000/RGD-NP. The radioactive trace of free 89Zr (black) is shown as control. (C) Dynamic light scattering hydrodynamic diameter, polydispersity index, r1 measurements, and MR images of the nanoparticles labeled with different amounts of Gd–DTPA–DSA. The T1-weighted MR images were obtained at a field of 7T, and r1 was measured at a field of 1.41T. Size and polydispersity index: mean (n = 3). (D) Fluorescence emission spectra of nanoparticles labeled with Cy3.5 and Cy5 after excitation at 488 (i) and 633 (ii) nm. The excitations and filter pass-bands correspond to the flow cytometry channels used in the study.
Surface-Converting Validation Experiments
As a proof of principle for our surface conversion strategy, nanoparticles decorated with biotin–PEG1000–DSPE were tested in two different (strept)avidin-binding experiments.29 To measure biotin availability of different coatings, nanoparticle solutions were incubated with free avidin for 30 min, and subsequent aggregation was measured by DLS (Figure 3A). Data revealed that nanoparticles exposing unshielded biotin moieties (biotin-NP) that were incubated with avidin exhibited a 10-fold relative size increase, indicative of aggregation. However, this avidin-and biotin-induced aggregation was inhibited for nanoparticles with a coating of 10% of mPEG3000–DSPE (PEG-3000/ biotin-NP) or 10% of cleavable mPEG–MMP2p–DSPE (PEG-MMP2p/biotin-NP). When PEG-MMP2p/biotin-NP were pretreated for 18 h with MMP2,29 they became prone to avidin- and biotin-induced aggregation, demonstrating the MMP2 susceptibility of our coating and validating our surface-converting concept. To corroborate this result and simulate nanoparticle interaction with cellular epitopes, Cy5.5-labeled nanoparticles were incubated in streptavidin-coated plates. Fluorescence-binding measurements (Figure 3B) confirmed that, while both PEG-MMP2p and PEG-3000 coatings prevented nanoparticle specific binding, the PEG-MMP2p shielding was sensitive to MMP2 cleavage. Importantly, further avidin- and biotin-binding assays (Figure S3) demonstrated that the presence of Gd–DTPA–DSA in the phospholipid corona did not impair the targeting, shielding, or surface-converting abilities of the nanoparticles, corroborating the modularity of the different coatings.
Figure 3.

Proof of principle of the surface-converting approach. (A) Avidin-induced aggregation of biotin-functionalized nanoparticles. Nanoparticle relative size was measured by DLS before and after incubation with avidin. (n = 3). (B) Binding of biotin-functionalized, Cy5.5-labeled nanoparticles on a streptavidin-coated plate. (n = 3); *, P < 0.05; ***, P < 0.001.
In Vitro Cell Targeting
Subsequently, the nanoparticles were functionalized with cyclic RGD,29 and their targeting efficiency was tested with two αvβ3 integrin expressing human cell lines: umbilical vein endothelial cells (HUVEC) and green fluorescent protein (GFP) expressing human breast tumor cells (MDA-MB-231-GFP). Fluorescence microscopy images (Figure 4A,B) demonstrated that unshielded RGD-decorated nanoparticles (RGD-NP) possessed a stronger affinity for these αvβ3 integrin expressing cells compared to nondecorated (UntargetedfNP) or decorated but shielded (PEG-3000/RGD-NP) nanoparticles. These results were further confirmed by quantitative T1 MRI measurements of cell pellets incubated with Gd-labeled nanoparticles (Figure S4). Additionally, z-stack confocal microscopy images of GFP expressing MDA-MB-231 revealed red foci patterns within the cells’ cytoplasm consistent with internalization of the nanoparticles and subsequent endosomal and lysosomal accumulation (Figure 4Bv). Our platform’s surface-converting ability was then assessed by flow cytometry. Various Cy5.5-labeled nanoparticle solutions were incubated with HUVEC and MDA-MB-231-GFP cells. After extensive washing, flow cytometry was used to measure the fluorescence of each individual cell. Data presented in Figure 4C,D revealed that cells incubated with PEG-MMP2p/RGD-NP exhibited a reduced fluorescent signal compared to RGD-NP. However, preincubation of PEG-MMP2p/RGD-NP with MMP2 led to a significant increase (P < 0.001) in fluorescent signal for both cell lines, indicative of the coating’s sensitivity to MMP2.
Figure 4.

In vitro characterization of our surface-converting nano-articles. Fluorescence microscopy analysis of αvβ3 integrin-expressing (A) endothelial cells (HUVEC) and (B) breast tumor cells (MDA-MB-231) incubated with Cy5.5-labeled nanoparticles: (i) untreated cells (control), (ii) Untargeted-NP, (iii) PEG-3000/RGD-NP, and (iv) RGD-NP. Staining: blue, DAPI and red, Cy5.5. (v) Same field of view as (Biv) imaged with the green channel (GFP). Scale bar: 50 μm. Flow cytometry analysis of HUVEC (C) and MDA-MB-231 (D) cells incubated with Cy5.5-labeled nanoparticles. n = 4; ***, P < 0.001.
Pharmacokinetics and Biodistribution
To assess the effect of the different coatings on pharmacokinetics and tumor accumulation, nanoparticles were labeled with 89Zr and intravenously injected in female mice bearing orthotropic breast tumors (MDA-MB-231-GFP). For these experiments, radiolabeling was chosen to take advantage of the high sensitivity and accuracy of radioisotope detection measurements. Pharmacokinetic experiments (Figure 5A) revealed that the two nanoparticles with a long PEG-shielding (PEG-3000/ RGD-NP and PEG-MMP2p/RGD-NP) exhibited longer weighted blood half-lives than RGD-NP (t1/2 = 2.5 and 3.5 h, compared to 1.4 h, respectively). Those improved circulation times directly translated into higher tumor accumulation of both PEG-3000/RGD-NP and PEG-MMP2p/RGD-NP (Figure 5B–D) as compared to RGD-NP (P < 0.05). Furthermore, comparison between the biodistribution profiles of PEG-3000/ RGD-NP and PEG-MMP2p/RGD-NP revealed no significant differences between the two long PEG-shielded platforms. These results indicate that in circulation, similar to traditional long PEG, the MMP2-sensitive PEG corona of the PEG-MMP2p/RGD-NP is preventing nanoparticles’ mononuclear phagocyte system recognition, leading to effective passive tumor accumulation. Nanoparticle tumor accumulation was further confirmed in vivo by near-infrared imaging and MRI using a dual-labeled Cy5.5–gadolinium platform (Figure S5). Furthermore, T1-mapping revealed that nanoparticles primarily accumulated within the rim of the tumors.
Figure 5.

Pharmacokinetics and biodistribution of the surface-converting nanoparticles. Nanoparticles were injected intravenously in nude mice bearing orthotopic MDA-MB-231 GFP breast cancer tumors. (A) Blood time–activity curve for the different 89Zr-labeled nanoparticles (n = 3 per nanoparticle type; ID: injected dose). (B) Ex vivo autoradiography of selected tissues 24 h after the administration of 89Zr-labeled nanoparticles. Organs are Liv: liver, Kid: kidneys, Lun: lung, Spl: spleen, Tum: tumor, Mus: muscle, Bra: brain, Hea: heart. (C) Radioactivity distribution in selected tissues of 89Zr-labeled nanoparticles expressed as percentage of injected dose per gram of tissue (%ID/g). (n = 3 per nanoparticle type). (D) Radioactivity concentration for tumor-to-muscle ratio. n = 3 per nanoparticle type; vs RGD-NP; *, P < 0.05.
In Vivo Cell-Targeting Specificity
Finally, the cell targeting specificity of the differently coated nanoparticles was tested in vivo. To reduce the effect of the high variability in tumor permeability that exists between animals,42,44 PEG-MMP2p/RGD-NP were compared to control platforms within the same animal (Figure 6A). Therefore, mice were injected with Cy3.5-labeled PEG-MMP2p/RGD-NP and Cy5-labeled RGD-NP (n = 5) or Cy3.5-labeled PEG-MMP2p/RGD-NP and Cy5-labeled PEG-3000/RGD-NP (n = 5). 24 h after injection, tumors were excised, processed, and analyzed by f;low cytometry to measure the uptake of the different nanoparticles in three specific cell types: tumor cells, tumor-associated leukocytes, and endothelial cells (Figure S6). Flow cytometry results (Figure 6B,C) revealed a significant 10-fold higher ratio of tumor cells positive for PEG-MMP2p/RGD-NP than for unshielded RGD-NP, whereas leukocytes and endothelial cells showed a slight nonsignificant preference for RGD-NP. A similar pattern was observed when comparing PEG-MMP2p/ RGD-NP to PEG-3000/RGD-NP (Figure 6D,E). Because biodistribution data showed similar whole-tumor accumulation for both PEG-MMP2p/RGD-NP and PEG-3000/RGD-NP, these results seem to indicate that once in the tumor microenvironment the two platforms behave differently at the cellular level, with PEG-MMP2p/RGD-NP exhibiting higher efficiency targeting tumor cells.
Figure 6.

Tumor cell targeting by surface-converting nanoparticles. (A) Schematic of the experiment: mice bearing orthotopic breast cancer tumors were co-injected with a mixture of two nanoparticle formulations, either Cy3.5-labeled PEG-MMP2p/RGD-NP and Cy5-labeled RGD-NP or Cy3.5-labeled PEG-MMP2p/RGD-NP and Cy5-labeled PEG-3000/RGD-NP. 24 h after injection, the animals were sacrificed, and the fluorescence in the different cell types (tumor cells, leukocytes, and endothelial cells) was measured by flow cytometry. (B) Percentages of PEG-MMP2p/RGD-NP-positive (red) and RGD-NP-positive (blue) cells in each animal for tumor cells (Tum), leukocytes (Leu), and endothelial cells (End), respectively. n = 5; *, P < 0.05. (C) Ratio between percentages of PEG-MMP2p/RGD-NP-positive cells and RGD-NP-positive cells for each cell type (n = 5). (D) Percentages of PEG-MMP2p/RGD-NP-positive (green) and PEG-3000/RGD-NP-positive (blue) cells in each animal for tumor cells (Tum), leukocytes (Leu), and endothelial cells (End), respectively (n = 5). (E) Ratio between percentages of PEG-MMP2p/RGD-NP-positive cells and PEG-3000/RGD-NP-positive cells for each cell type (n = 5).
Taken together, the in vivo pharmacokinetic, biodistribution, and cell-specificity data for the MMP2-sensitive coated nanoparticles are compatible with the hypothesized properties and targeting mechanism. First, upon intravenous injection, PEG-MMP2p/RGD-NP exhibited enhanced blood half-life and tumor accumulation similar to MMP2-insensitive PEG-3000/ RGD-NP. Then, once in the tumor microenvironment, the nanoparticles are subjected to MMP2-driven PEG cleavage leading to the surface exposure of RGD and subsequent active targeting of αvβ3 expressing tumor cells.
CONCLUSIONS
We developed a comprehensive multimodal imaging strategy to monitor the in vitro and in vivo behavior of a PLGA–lipid hybrid nanoparticle that is shielded with an MMP2-cleavable PEG coating at the whole-body, tissue, and cellular levels. The inclusion of various contrast agents and the use of a multimodal imaging approach was key to demonstrate that this new enzyme specific surface-converting coating strategy ensures a high nanoparticle cell-targeting specificity without compromising favorable nanoparticle pharmacokinetics. We believe that this highly modular surface-converting coating strategy can easily be tailored by modifying the cleavable amino acid sequence, the targeting ligands, or both to target other types of cancer and inflammatory conditions such as rheumatoid arthritis, bowel diseases, or atherosclerosis.
EXPERIMENTAL PROCEDURES
Materials
1,2-Distearoyl-sn-glycero-3-phosphoethanol-amine–N-[methoxy (polyethylene glycol)-3000] ammonium salt (mPEG3000–DSPE), 1,2-distearoyl-sn-glycero-3-phos-phoethanolamine-N-[methoxy (polyethylene glycol)-350] ammonium salt (mPEG350–DSPE), and DTPA–dis-(stearyleamide) (gadolinium salt) (Gd–DTPA–DSA) were from Avanti Polar Lipids Inc. Soybean lecithin was purchased from MP Biomedicals, LLC. 1,2-Distearoyl-sn-glycero-3-phos-phoethanolamine–N-[methoxy (polyethylene glycol)-1000] biotin (biotin–PEG1000–DSPE) was purchased from Creative PEGWorks. MMP2-cleavable lipid (mPEG2000-MMP2p-DSPE) and c[RGDfK]-PEG18-DSPE were synthesized by SyMO-Chem (Eindhoven, The Netherlands, www.symo-chem.nl) as previously reported.29 4-Amino-phenyl-mercuric acetate (APMA) and Poly(D,L-lactide-co-glycolide) (PLGA) with a 50:50 monomer ratio and a 30 000–60 000 Da molecular weight were acquired from Sigma-Aldrich. PLGA conjugation with Cy5.5, Cy5, or Cy3.5 was obtained as previously reported.40 Dulbecco’s modified Eagle medium (DMEM) was from Gibco and Invitrogen. Human umbilical vein endothelial cells and bullet kit endothelial basal medium (EBM-2) were purchased from Lonza (Allendale, NJ). Human MMP2 (hMMP2) was purchased from R&D Systems. MDA-MB-231 expressing green fluorescent protein (GFP) was a gift from Dr. Aaronson (Icahn School of Medicine, New York, NY).
Nanoparticle Synthesis
The phospholipid–PEGs were dissolved in 10% ethanol, Gd–DTPA–DSA and soybean lecithin (40% weight of phospholipid–PEGs) were dissolved in absolute ethanol, and all lipids were mixed together in a solution of deionized (DI) water with 10% ethanol. PLGA was dissolved in acetonitrile to reach a concentration of 2 mg/mL. The polymer–lipid nanoparticles were synthesized by dripping the PLGA solution at a flow rate of 0.2 mL/min into a hot (50 °C) phospholipid water/ethanol solution under stirring to reach a molar ratio for PLGA/phospholipids of 1.76:10. Once the dripping finished, the samples were sonicated using an ultrasonic bath for 5 min on ice and left overnight under a hood at room temperature while stirring to let the organic solvents evaporate. The nanoparticle solution was then centrifuged at 400g for 30 min to remove large aggregates. Nanoparticles were then washed three times with deionized water and concentrated using centrifugal filtration (molecular weight cutoff: 100 000 Da) followed by sterile filtration with a 0.45 μm filter under aseptic conditions. Nanoparticle phospholipid compositions are shown in Table 1. Soybean lecithin was included in all formulations at 40% weight of the total lipid content. For MRI experiments, Gd–DTPA–DSA was included at the expense of a fraction of mPEG350–DSPE. For avidin- and streptavidin-based experiments, biotin-labeled nanoparticles were produced by including 5% of biotin–PEG1000–DSPE at the expense of RGD–PEG350–DSPE.
Synthesis of C34-DFO
2-Hexadecyl-octanoic acid (TCI America, 30 mg, 59 μmol), (benzotriazol-1-yloxy) Tris-(dimethylamino)phosphonium hexafluorophosphate (Sigma-Aldrich, 29 mg, 66 μmol), and N,N-diisopropylethylamine (Sigma-Aldrich, 10 μL) were dissolved in anhydrous dichloro-methane (2 mL) and stirred at 40 °C for 10 min under nitrogen atmosphere. A solution of deferoxamine mesylate (Sigma-Aldrich, DFO, 30 mg, 46 μmol) and N,N-diisopropylethyl-amine (10 μL) in anhydrous dimethyl sulfoxide (Sigma-Aldrich, 0.7 mL) was then added and the resulting mixture stirred for 4 h at 40 °C under nitrogen. The cloudy suspension was allowed to cool down to room temperature, and dichloromethane was removed under reduced pressure. Hydrochloric acid (0.1 M, 1 mL) was added, and the mixture was stirred for 10 min at room temperature. The solid was filtered with 0.1 M HCl (3 × 1 mL), dimethyl sulfoxide (3 × 1 mL), water (3 × 1 mL), and, finally, dichloromethane (3 × 1 mL) and dried to yield a white solid (31 mg, 64%). MS–ES+: 1074 (M+Na)+. MS–ES−: 1050 (M–H)−, 1086 (M+Cl)−. 1H NMR (CDCl3/CD3OD, δ, ppm): 0.55 (t, 6H), 0.92 (br, 60H), 1.06 (br, 6H), 1.27 (m, 6H), 1.32 (m, 6H), 1.76 (m, 1H), 1.80 (s, 3H), 2.15 (t, 4H), 2.46 (t, 4H), 2.87 (m, 6H), 3.29 (m, 6H).
Radiolabeling
DFO-bearing nanoparticles (containing 1 mol % C34-DFO) were labeled as previously reported.41 Briefly, a nanoparticle solution was reacted with 89Zr-oxalate (0.2 mCi) in phosphate-buffered saline (PBS) (pH: 7.1–7.4) for 2 h at 37 °C. The mixture was allowed to cool, and the nanoparticles were purified by centrifugal filtration (molecular weight cutoff: 100 000 Da) and washing with deionized water. The retentate was diluted with water to the desired concentration. The radiochemical yield was 20–25%, and the radiochemical purity >95%, as determined by size-exclusion chromatography.
Dynamic Light Scattering
Nanoparticle solutions were diluted in deionized water, and the size and corresponding polydispersity index measurements were done in triplicates with 10 runs per sample using a DLS device (Brookhaven Instruments Corporation).
Nanoparticle Stability in Serum
A total of 50 μL of nanoparticles solution were added to 1 mL of 0%, 20%, 40%, 80%, and 100% fetal bovine serum (FBS, F4135 Sigma-Aldrich). DLS was used to determine the size of the nanoparticles in the different solutions. The samples were measured in triplicates with 10 runs per sample, and the data were normalized to 0% FBS.
Determination of the Nanoparticle Gadolinium Content
The content of gadolinium in the different nanoparticles was analyzed using inductively coupled plasma optical emission spectroscopy (Spectro Arcos). Each sample was diluted in 1% HNO3 and measured three times and the mean value interpolated in a calibration curve obtained with standard solutions.
Relaxometry
The longitudinal (r1) relaxivities of nanoparticles containing different amounts of Gd–DTPA–DSA (0%, 6.25%, 12.5%, or 25%) were determined using a 60 MHz Bruker Minispec (Bruker Medical GmbH) operating at 40 °C. Relaxivities r1 were calculated from the slope of the graph of 1/ T1 plotted against the gadolinium concentration measured with inductively coupled plasma optical emission spectroscopy.
Transmission Electron Microscopy
Nanoparticle sample solutions were transferred to an ammonium acetate buffer solution and then negatively stained using an ammonium acetate buffer containing 2% sodium phosphotungstate. Drops of samples were then placed onto 100 mesh Formvar-coated nickel grids (Electron Microscopy Sciences) and allowed to air-dry. Grids were imaged using a Hitachi 7650 microscope operated at 80 kV and connected to a digital camera (Scientific Instruments and Applications) controlled by Maxim CCD software.
MMP2 Activation
Human MMP2 (hMMP2) was activated by incubation in 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid buffered saline (HBS) + 5 mM CaCl2 pH 7.5 for 1 h at 37 °C in the presence of 1 mM 4-aminophenylmercuric acetate following our previously established method.29
Avidin-Induced Aggregation Experiment
Biotin-labeled nanoparticle solutions corresponding to 0.1065 μmol phospholipids were mixed with 525 ng of activated hMMP2 and HBS buffer up to 10 μL and incubated overnight at 37 °C.29 Nanoparticles incubated without hMMP2 were used as the negative control. The nanoparticles were then mixed with 40 pmol of avidin and HBS buffer up to 100 μL and incubated for 30 min at room temperature. The size was measured in 1 mL of water by DLS and compared to the size at t = 0 (diametert=30 min/diametert=0 min).
Binding to Streptavidin-Coated Plate
Cy5.5-labeled nanoparticles that were either preincubated with hMMP2 as described earlier or untreated were added to a streptavidin-coated 96 well plate for 1 h at room temperature. After incubation, the plate was washed three times with water, and the fluorescence from the wells was measured using a near-infrared imaging system (IVIS system, PerkinElmer) with excitation and emission wavelengths set at 640 and 700 nm, respectively. The Living Image (PerkinElmer) software was used for the analysis.
Cell Culture
MDA-MB-231-GFP cell lines were grown in DMEM supplemented with 10% fetal bovine serum and 1% penicillin and streptomycin. HUVEC were grown in EBM-2 medium supplemented with bullet kit. All cells were grown at 5% CO2 and 37 °C.
In Vitro Flow Cytometry
Cy5.5-labeled nanoparticles that were either preincubated with hMMP2 in the same ratio as described in 2.11 or untreated were incubated in the MDA-MB-231 and HUVEC cells for 2 h at 37 °C. The cells were washed three times with PBS, detached, centrifuged, and resuspended in 0.5 mL of PBS. Subsequently, Cy5.5 fluorescence in individual cells was analyzed by flow cytometery (LSRII system, BD Biosciences).
Magnetic Resonance Imaging of Cell Pellets
MDA-MB-231 and HUVEC, cells were seeded in T75 flasks. After 2 days, the cells were incubated for 5 h at 37 °C with the different nanoparticle solutions. Cells were then washed three times with PBS and detached using trypsin. The cells were washed once again and fixed with 200 μL of PFA (4%) overnight at room temperature. The PFA was then exchanged with 200 μL of PBS, and the cells were added to PCR tubes. Cell pellets were formed by gravity. To prepare an agarose phantom, a 2% agarose gel was made and PCR tubes with cell pellets were inserted into the agarose gel. Magnetic resonance imaging (MRI) was performed on a 7 T magnetic resonance scanner (Bruker) using a mouse coil (30 mm). T1 was measured using a multislice spin–echo sequence, with TR ranging between 100 and 5000 ms in 16 steps. All acquired images had a matrix size of 256 × 256, FOV of 3 × 3 cm2, 0.50 mm slice thickness, and four averages. Circular regions of interest (ROIs) were drawn encompassing the cell pellets on the T1-weighted images, and ROI mean values as a function of TR were fitted using Mathematica (Wolfram Research) to the signal equation S ≈ (1 − Exp[−TR/T1]) to obtain T1 map.
Orthotropic Breast Tumor Model
A total of 3 000 000 MDA-MB-231 expressing GFP cells were injected in the mammary fat pad of female NCR/NU mice (NCI Frederick) under isoflurane anesthesia. The animals had access to standard chow and water ad libitum. Measurable tumors were detected 10 days after injection, and tumor size was determined by measuring the length, width, and depth. Tumor volume was calculated as (max × min × min)/2, with max being the maximum diameter measured and min the minimum diameter measured. Animals used in the experiments were selected to present a tumor volume of ≥28 mm3 with no sign of necrotic core or general distress. All in vivo experiments and animal handling were approved by the Icahn School of Medicine at Mount Sinai Institutional Animal Care and Use Committee.
In Vivo Magnetic Resonance Imaging
Mice bearing MDA-MB-231-GFP orthotropic tumors were injected intravenously with 56 nmol of gadolinium per g of body weight of Cy5.5/gadolinium-labeled nanoparticles (n = 4). Magnetic resonance imaging was performed right before and 24 h after the injection of gadolinium labeled nanoparticles on a 7 T magnetic resonance scanner (Bruker) using a mouse coil (30 mm). The animals were anesthetized with a 3% isoflurane/O2 gas mixture and maintained with a 1.5% isoflurane/O2 gas mixture delivered through a nose cone. A respiratory sensor connected to a monitoring and gating system was placed on the abdomen to monitor the depth and frequency of respiration. T1 was measured using a multislice spin–echo sequence (13 slices), with TR ranging between 100 and 5000 ms in 16 steps. T2 was measured using a multislice multispin–echo sequence (13 slices) with a TR of 2000 and a TE ranging between 8 and 128 ms in 18 steps. All acquired images had a matrix size of 256 × 256, a FOV of 3 × 3 cm2, 0.50 mm slice thickness, and a 0.55 interslice distance. Regions of interest delimiting the contours of the tumor were manually drawn on each slide using the T2-weighted images. ROI mean values as a function of TR were fitted in MATLAB to the signal equation S ≈ (1 − Exp[−TR/T1]) to obtain T1 maps.
In Vivo Near-Infrared Imaging
Near-infrared imaging was performed 24 h after injection of Cy5.5-labeled nanoparticles. Mice were anesthetized with a 3% isoflurane/O2 gas mixture and maintained with a 1.5% isoflurane/O2 gas delivered through a nose cone. A near-infrared imaging system (IVIS system, PerkinElmer) with excitation and emission wavelengths set at 640 and 700 nm, respectively, was used. The Living Image (PerkinElmer) software was used for the analysis.
Pharmacokinetics and Biodistribution Studies
Mice bearing MDA-MB-231-GFP orthotropic tumors were injected with 10 ± 2 μCi of 89Zr labeled nanoparticle solutions (n = 3). Blood was sampled at predetermined time points (1 min, 30 min, 2 h, 4 h, 8 h, and 24 h) and radioactivity measured on a Wizard2 Automatic Gamma Counter (PerkinElmer, Waltham, MA). Radioactivity concentration was calculated as the mean percentage of injected dose per gram of tissue (%ID/g) ± SD. The data were plotted on a time activity curved and fitted using a two-phase decay nonlinear regression42 calculated by Prism GraphPad (GraphPad Software Inc.). From this regression, half-life(fast) [t1/2 (fast)], half-life(slow) [t1/2 (slow)], and percent fast (xf) parameters were obtained for each nanoparticle. A weighted blood radioactivity half-life (t1/2) was finally calculated using the following equation:
For biodistribution studies, animals were sacrificed 24 h after injection. The mice were perfused with 30 mL of PBS. The radioactive content in specific tissues (liver, kidneys, lung, spleen, tumor, muscle, brain, and heart) was measured using a 2470 Wizard2 automatic γ counter (PerkinElmer, Waltham, MA) and radioactivity concentration was calculated as the mean percentage injected dose per gram of tissue (%ID/g) ± SD. Following radioactivity counting, liver, kidneys, lung, spleen, tumor, muscle, brain, and heart tissues were placed in a film cassette against a phosphorimaging plate (BASMS-2325, Fujifilm, Valhalla, NY) for 24 h at −20 °C. The plates were read at a pixel resolution of 25 μm using a Typhoon 7000IP plate reader (GE Healthcare, Pittsburgh, PA).
In Vivo Cell Specificity
Mice bearing MDA-MB-231-GFP orthotropic tumors were injected intravenously with a solution containing a mixture of equivalent amounts (based on gadolinium content) of either Cy3.5/1% Gd-labeled PEG-MMP2p/RGD-NP and Cy5/1% Gd-labeled PEG-3000/RGD-NP (n = 5) or Cy3.5/1% Gd-labeled PEG-MMP2p/RGD-NP and Cy5/1% Gd-labeled RGD-NP (n = 5). Animals were sacrificed 24 h after injection and perfused with 30 mL of PBS. Tumors were collected, cut in small pieces, and then digested with 4 U/ml colagenase (Sigma-Aldrich) in RPMI-1640 media at 37 °C for 45 min. The suspensions were filtered through a 70 μm filter and treated with red blood cells lysis bufier. Cell suspensions obtained were incubated with mouse Fc Block (BD Pharmingen) and then incubated with Viability Dye eFluor 780 (Affymetrix) and a mixture of monoclonal antibodies for 30 min at 4 °C. Antibodies used were: anti-mouse CD45 eFluor 450 clone 30-F11 (Affymetrix) and anti-mouse CD31 PE/Cy7, clone 390 (Biolegend). Fluorescence was detected using a flow cytometer (BD Biosciences LSR II), and the data were analyzed using the FlowJO software (Tree Star). Viability Dye eFluor 780 fluorescence signal was first used to exclude dead cells. Tumor cells were identified as GFP+ and CD45−; leukocytes were identified as GFP− and CD45+, and endothelial cells were identified as GFP−, CD45−, and CD31+ (Figure S6); next, the percentage (relative to cell type population) of cells positive for each nanoparticle signal were quantified for tumor cells, leukocytes, and endothelial cells. Data presented in Figure 6C,E are the ratios of percentage of PEG-MMP2p/RGD-NP-positive cells to the percentage of RGD-NP-positive cells and the percentage of PEG-MMP2p/RGD-NP-positive cells to the percentage of PEG-3000/RGD-NP-positive cells.
Statistical Analysis
Unless specified, data are expressed as mean ± standard deviation (SD). The Student’s t-test was used to analyze statistical significance between two distinct sample groups assuming equal variance. For multiple groups (three or more), data were analyzed using one-way variance analysis, followed by Boneferroni multiple comparison test. For the in vivo cell specificity study, a two-tailed paired t-test was used to test the statistical difierences between the percentages of nanoparticle-positive cells. All statistical analyses were performed with Prism GraphPad (GraphPad Software Inc.), and P values <0.05 were considered significant.
Supplementary Material
Acknowledgments
Fluorescence microscopy was performed at the Microscopy Core at Icahn School of Medicine at Mount Sinai. Flow cytometry was performed at the Flow Cytometry Core at Icahn School of Medicine at Mount Sinai. Near-infrared imaging, autoradiography, γ counting, and magnetic imaging resonance were all done at the TMII Imaging Core at Icahn School of Medicine at Mount Sinai. Radiolabeling of the nanoparticles was supported by the MSKCC Radiochemistry Core Facility (in part through NIH Cancer Center Support grant no. P30 CA008748-48). We also greatly thank P. Hald and A. Mamakhel (Aarhus University) for their help with inductively coupled plasma optical emission spectroscopy measurements. This research was supported by the National Heart, Lung, and Blood Institute (NHLBI) and the National Institutes of Health (NIH) as a Program of Excellence in Nanotechnology (PEN) Award, contract nos. HHSN268201000045C, R01 HL118440 (W.J.M.M.), R01 HL125703 (W.J.M.M.), R01 CA155432 (W.J.M.M.), R01 EB009638 (Z.A.F.), a Harold S. Geneen Charitable Trust Award (Z.A.F.); NWO Vidi (W.J.M.M.), as well as The Lundbeck Foundation Nanomedicine Centre for Individualized Management of Tissue Damage and Regeneration (L.H and J.K). Some of the artwork present in the Abstract graphic and Figure 6 are adapted from Servier Medical Art (used under a Creative Commons Attribution 3.0 Unported License).
ABBREVIATIONS
- APMA
4-amino-phenyl-mercuric acetate
- biotin–PEG1000–DSPE
1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethylene glycol)-1000] biotin
- DFO
deferox-amine mesylate
- DLS
dynamic light scattering
- FBS
fetal bovine serum
- Gd
gadolinium
- Gd–DTPA–DSA
diethylene triamine penta acetic acid di(stearyl amide) (gadolinium salt)
- GFP
green fluorescent protein
- HBS
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid buffered saline
- hMMP2
human MMP2
- HUVEC
human umbilical vein endothelial cells
- MMP2
matrix metalloproteinase-2 enzyme
- mPEG3000–DSPE
1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethylene glycol)-3000] ammonium salt
- mPEG350–DSPE
1,2-distearoyl-sn-glycero-3-phosphoethanol-amine-N-[methoxy (polyethylene glycol)-350] ammonium salt
- MRI
magnetic resonance imaging
- NGR
asparagic-glycyl-arginyl acid
- PDI
polydispersity index
- PEG
polyethylene glycol
- PLGA
poly(lactic-co-glycolic acid)
- PBS
phosphate-buffered saline
- RGD
arginyl-glycyl-aspartic acid
- 89Zr
zirconium-89
Footnotes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bioconj-chem.7b00086.
Figures showing nanoparticles’ stability, T1-weighted MR images, binding of biotin-functionalized Cy5.5/Gd-labeled nanoparticles, in vitro targeting of the nanoparticles, biodistribution of the Cy5.5/Gd-labeled nanoparticles, and gating procedures used in the flow cytometry analysis. (PDF)
References
- 1.Schmid D, Fay F, Small DM, Jaworski J, Riley JS, Tegazzini D, Fenning C, Jones DS, Johnston PG, Longley DB, et al. Efficient drug delivery and induction of apoptosis in colorectal tumors using a death receptor 5-targeted nanomedicine. Mol Ther. 2014;22:2083–2092. doi: 10.1038/mt.2014.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: The impact of passive and active targeting in the era of modern cancer biology. Adv Drug Delivery Rev. 2014;66:2–25. doi: 10.1016/j.addr.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kamaly N, Xiao Z, Valencia PM, Radovic-Moreno AF, Farokhzad OC. Targeted polymeric therapeutic nanoparticles: Design, development and clinical translation. Chem Soc Rev. 2012;41:2971–3010. doi: 10.1039/c2cs15344k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hrkach J, Von Hoff D, Mukkaram Ali M, Andrianova E, Auer J, Campbell T, De Witt D, Figa M, Figueiredo M, Horhota A, et al. Preclinical development and clinical translation of a PSMA-targeted docetaxel nanoparticle with a differentiated pharm \acological profile. Sci Transl Med. 2012;4:128ra39. doi: 10.1126/scitranslmed.3003651. [DOI] [PubMed] [Google Scholar]
- 5.Ruoslahti E, Pierschbacher MD. New perspectives in cell adhesion: RGD and integrins. Science. 1987;238:491–497. doi: 10.1126/science.2821619. [DOI] [PubMed] [Google Scholar]
- 6.Arap W, Pasqualini R, Ruoslahti E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science. 1998;279:377–380. doi: 10.1126/science.279.5349.377. [DOI] [PubMed] [Google Scholar]
- 7.Chen W, Jarzyna PA, van Tilborg GA, Nguyen VA, Cormode DP, Klink A, Griffioen AW, Randolph GJ, Fisher EA, Mulder WJ, et al. RGD peptide functionalized and reconstituted high-density lipoprotein nanoparticles as a versatile and multimodal tumor targeting molecular imaging probe. FASEB J. 2010;24:1689–1699. doi: 10.1096/fj.09-139865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hak S, Cebulla J, Huuse EM, Davies CdL, Mulder WJ, Larsson HB, Haraldseth O. Periodicity in tumor vasculature targeting kinetics of ligand-functionalized nanoparticles studied by dynamic contrast enhanced magnetic resonance imaging and intravital microscopy. Angiogenesis. 2014;17:93–107. doi: 10.1007/s10456-013-9380-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murphy EA, Majeti BK, Barnes LA, Makale M, Weis SM, Lutu-Fuga K, Wrasidlo W, Cheresh DA. Nanoparticle-mediated drug delivery to tumor vasculature suppresses metastasis. Proc Natl Acad Sci U S A. 2008;105:9343–9348. doi: 10.1073/pnas.0803728105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith BR, Cheng Z, De A, Koh AL, Sinclair R, Gambhir SS. Real-time intravital imaging of RGD-quantum dot binding to luminal endothelium in mouse tumor neovasculature. Nano Lett. 2008;8:2599–2606. doi: 10.1021/nl080141f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kunjachan S, Pola R, Gremse F, Theek B, Ehling J, Moeckel D, Hermanns-Sachweh B, Pechar M, Ulbrich K, Hennink WE, et al. Passive versus active tumor targeting using RGD- and NGR-modified polymeric nanomedicines. Nano Lett. 2014;14:972–981. doi: 10.1021/nl404391r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luo LM, Huang Y, Zhao BX, Zhao X, Duan Y, Du R, Yu KF, Song P, Zhao Y, Zhang X, et al. Anti-tumor and anti-angiogenic effect of metronomic cyclic NGR-modified liposomes containing paclitaxel. Biomaterials. 2013;34:1102–1114. doi: 10.1016/j.biomaterials.2012.10.029. [DOI] [PubMed] [Google Scholar]
- 13.Zhao F, Li L, Guan L, Yang H, Wu C, Liu Y. Roles for GP IIb/IIIa and alphavbeta3 integrins in MDA-MB-231 cell invasion and shear flow-induced cancer cell mechanotransduction. Cancer Lett. 2014;344:62–73. doi: 10.1016/j.canlet.2013.10.019. [DOI] [PubMed] [Google Scholar]
- 14.Liu Z, Wang F, Chen X. Integrin targeted delivery of radiotherapeutics. Theranostics. 2011;1:201–210. doi: 10.7150/thno/v01p0201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gu F, Zhang L, Teply BA, Mann N, Wang A, Radovic-Moreno AF, Langer R, Farokhzad OC. Precise engineering of targeted nanoparticles by using self-assembled biointegrated block copolymers. Proc Natl Acad Sci U S A. 2008;105:2586–2591. doi: 10.1073/pnas.0711714105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reuter KG, Perry JL, Kim D, Luft JC, Liu R, DeSimone JM. Targeted PRINT hydrogels: The role of nanoparticle size and ligand density on cell association, biodistribution, and tumor accumulation. Nano Lett. 2015;15:6371–6378. doi: 10.1021/acs.nanolett.5b01362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McNeeley KM, Karathanasis E, Annapragada AV, Bellamkonda RV. Masking and triggered unmasking of targeting ligands on nanocarriers to improve drug delivery to brain tumors. Biomaterials. 2009;30:3986–3995. doi: 10.1016/j.biomaterials.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 18.Veiman KL, Kunnapuu K, Lehto T, Kiisholts K, Parn K, Langel U, Kurrikoff K. PEG shielded MMP sensitive CPPs for efficient and tumor specific gene delivery in vivo. J Controlled Release. 2015;209:238–247. doi: 10.1016/j.jconrel.2015.04.038. [DOI] [PubMed] [Google Scholar]
- 19.Olson ES, Jiang T, Aguilera TA, Nguyen QT, Ellies LG, Scadeng M, Tsien RY. Activatable cell penetrating peptides linked to nanoparticles as dual probes for in vivo fluorescence and MR imaging of proteases. Proc Natl Acad Sci U S A. 2010;107:4311–4316. doi: 10.1073/pnas.0910283107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang B, Zhang Y, Liao Z, Jiang T, Zhao J, Tuo Y, She X, Shen S, Chen J, Zhang Q, et al. UPA-sensitive ACPP-conjugated nanoparticles for multi-targeting therapy of brain glioma. Biomaterials. 2015;36:98–109. doi: 10.1016/j.biomaterials.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 21.Zhu L, Kate P, Torchilin VP. Matrix metalloprotease 2-responsive multifunctional liposomal nanocarrier for enhanced tumor targeting. ACS Nano. 2012;6:3491–3498. doi: 10.1021/nn300524f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu L, Perche F, Wang T, Torchilin VP. Matrix metalloproteinase 2-sensitive multifunctional polymeric micelles for tumor-specific co-delivery of siRNA and hydrophobic drugs. Biomaterials. 2014;35:4213–4222. doi: 10.1016/j.biomaterials.2014.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu L, Wang T, Perche F, Taigind A, Torchilin VP. Enhanced anticancer activity of nanopreparation containing an MMP2-sensitive PEG-drug conjugate and cell-penetrating moiety. Proc Natl Acad Sci U S A. 2013;110:17047–17052. doi: 10.1073/pnas.1304987110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang S, Shao K, Kuang Y, Liu Y, Li J, An S, Guo Y, Ma H, He X, Jiang C. Tumor targeting and microenvironment-responsive nanoparticles for gene delivery. Biomaterials. 2013;34:5294–5302. doi: 10.1016/j.biomaterials.2013.03.043. [DOI] [PubMed] [Google Scholar]
- 25.Yu H, Chen J, Liu S, Lu Q, He J, Zhou Z, Hu Y. Enzyme sensitive, surface engineered nanoparticles for enhanced delivery of camptothecin. J Controlled Release. 2015;216:111–120. doi: 10.1016/j.jconrel.2015.08.021. [DOI] [PubMed] [Google Scholar]
- 26.Mok H, Bae KH, Ahn CH, Park TG. PEGylated and MMP-2 specifically dePEGylated quantum dots: Comparative evaluation of cellular uptake. Langmuir. 2009;25:1645–1650. doi: 10.1021/la803542v. [DOI] [PubMed] [Google Scholar]
- 27.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46:6387–6392. [PubMed] [Google Scholar]
- 28.Maeda H. Macromolecular therapeutics in cancer treatment: The EPR effect and beyond. J Controlled Release. 2012;164:138–144. doi: 10.1016/j.jconrel.2012.04.038. [DOI] [PubMed] [Google Scholar]
- 29.Gianella A, Mieszawska AJ, Hoeben FJ, Janssen HM, Jarzyna PA, Cormode DP, Costa KD, Rao S, Farokhzad OC, Langer R, et al. Synthesis and in vitro evaluation of a multifunctional and surface-switchable nanoemulsion platform. Chem Commun (Cambridge, U K) 2013;49:9392–9394. doi: 10.1039/c3cc43618g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mieszawska AJ, Kim Y, Gianella A, van Rooy I, Priem B, Labarre MP, Ozcan C, Cormode DP, Petrov A, Langer R, et al. Synthesis of polymer-lipid nanoparticles for image-guided delivery of dual modality therapy. Bioconjugate Chem. 2013;24:1429–1434. doi: 10.1021/bc400166j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang L, Chan JM, Gu FX, Rhee JW, Wang AZ, Radovic-Moreno AF, Alexis F, Langer R, Farokhzad OC. Self-assembled lipid–polymer hybrid nanoparticles: A robust drug delivery platform. ACS Nano. 2008;2:1696–1702. doi: 10.1021/nn800275r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakopoulou L, Tsirmpa I, Alexandrou P, Louvrou A, Ampela C, Markaki S, Davaris PS. MMP-2 protein in invasive breast cancer and the impact of MMP-2/TIMP-2 phenotype on overall survival. Breast Cancer Res Treat. 2003;77:145–155. doi: 10.1023/a:1021371028777. [DOI] [PubMed] [Google Scholar]
- 33.Stetler-Stevenson WG. Type IV collagenases in tumor invasion and metastasis. Cancer Metastasis Rev. 1990;9:289–303. doi: 10.1007/BF00049520. [DOI] [PubMed] [Google Scholar]
- 34.Jastrzebska B, Lebel R, Therriault H, McIntyre JO, Escher E, Guerin B, Paquette B, Neugebauer WA, Lepage M. New enzyme-activated solubility-switchable contrast agent for magnetic resonance imaging: From synthesis to in vivo imaging. J Med Chem. 2009;52:1576–1581. doi: 10.1021/jm801411h. [DOI] [PubMed] [Google Scholar]
- 35.Gallo J, Kamaly N, Lavdas I, Stevens E, Nguyen QD, Wylezinska-Arridge M, Aboagye EO, Long NJ. CXCR4-targeted and MMP-responsive iron oxide nanoparticles for enhanced magnetic resonance imaging. Angew Chem, Int Ed. 2014;53:9550–9554. doi: 10.1002/anie.201405442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yue X, Wang Z, Zhu L, Wang Y, Qian C, Ma Y, Kiesewetter DO, Niu G, Chen X. Novel 19F activatable probe for the detection of matrix metalloprotease-2 activity by MRI/MRS. Mol Pharmaceutics. 2014;11:4208–4217. doi: 10.1021/mp500443x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Groves MD, Puduvalli VK, Conrad CA, Gilbert MR, Yung WK, Jaeckle K, Liu V, Hess KR, Aldape KD, Levin VA. Phase II trial of Temozolomide plus marimastat for recurrent anaplastic gliomas: A relationship among efficacy, joint toxicity and anticonvulsant status. J Neuro-Oncol. 2006;80:83–90. doi: 10.1007/s11060-006-9160-y. [DOI] [PubMed] [Google Scholar]
- 38.Gabelloni P, Da Pozzo E, Bendinelli S, Costa B, Nuti E, Casalini F, Orlandini E, Da Settimo F, Rossello A, Martini C. Inhibition of metalloproteinases derived from tumours: New insights in the treatment of human glioblastoma. Neuroscience. 2010;168:514–522. doi: 10.1016/j.neuroscience.2010.03.064. [DOI] [PubMed] [Google Scholar]
- 39.Hua N, Baik F, Pham T, Phinikaridou A, Giordano N, Friedman B, Whitney M, Nguyen QT, Tsien RY, Hamilton JA. Identification of high-risk plaques by MRI and fluorescence imaging in a rabbit model of atherothrombosis. PLoS One. 2015;10:e0139833. doi: 10.1371/journal.pone.0139833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim Y, Lobatto ME, Kawahara T, Lee Chung B, Mieszawska AJ, Sanchez-Gaytan BL, Fay F, Senders ML, Calcagno C, Becraft J, et al. Probing nanoparticle translocation across the permeable endothelium in experimental atherosclerosis. Proc Natl Acad Sci U S A. 2014;111:1078–1083. doi: 10.1073/pnas.1322725111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perez-Medina C, Abdel-Atti D, Zhang Y, Longo VA, Irwin CP, Binderup T, Ruiz-Cabello J, Fayad ZA, Lewis JS, Mulder WJ, et al. A modular labeling strategy for in vivo PET and near-infrared fluorescence imaging of nanoparticle tumor targeting. J Nucl Med. 2014;55:1706–1711. doi: 10.2967/jnumed.114.141861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perez-Medina C, Abdel-Atti D, Tang J, Zhao Y, Fayad ZA, Lewis JS, Mulder WJ, Reiner T. Nanoreporter PET predicts the efficacy of anti-cancer nanotherapy. Nat Commun. 2016;7:11838. doi: 10.1038/ncomms11838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chan JM, Zhang L, Yuet KP, Liao G, Rhee JW, Langer R, Farokhzad OC. PLGA-lecithin-PEG core-shell nanoparticles for controlled drug delivery. Biomaterials. 2009;30:1627–1634. doi: 10.1016/j.biomaterials.2008.12.013. [DOI] [PubMed] [Google Scholar]
- 44.Miller MA, Gadde S, Pfirschke C, Engblom C, Sprachman MM, Kohler RH, Yang KS, Laughney AM, Wojtkiewicz G, Kamaly N, et al. Predicting therapeutic nanomedicine efficacy using a companion magnetic resonance imaging nanoparticle. Sci Transl Med. 2015;7:314ra183. doi: 10.1126/scitranslmed.aac6522. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.