

Cubic frame–like superstructures are constructed via the assembly of nanocuboids on the edges of preformed nanocubes.
Abstract
It is fascinating but challenging for nanoscientists to organize nanoparticles (NPs) into ordered architectures just as it is for chemists to manipulate atoms and molecules to form functional molecules and supramolecules. We explore a strategy to assemble anisotropic NPs into open frame-like superstructures via oriented attachment (OA), experimentally realizing a nanoscale analog to the bonding behavior in M8L12-type supramolecular cubes. We highlight the role of NP shape in the OA-involved assembly for constructing predictable superstructures. In addition, the frame-like superstructures can retain their basic structure when undergoing postcrystallization of the building blocks as well as annealing for conversion toward functional electrocatalytic materials. Our work enables fundamental insights into directional “bonding” among NPs and adds to the growing body of knowledge for bottom-up assembly of anisotropic NPs into sophisticated functional materials.
INTRODUCTION
A grand challenge in the realms of chemistry and materials science is to construct assemblies with precise control over the placement of each component for tailoring a given property. As chemists can manipulate atomic or molecular bonds to synthesize complex and functional molecules or supramolecules, bottom-up assembly and organization of colloids allow nanoscientists to build sophisticated architectures to address materials needs (1). Apart from the functionalization goal (2, 3), nanoparticles (NPs) also demonstrate the ability to mimic atomic habits when they assemble into ordered structures, such as superlattices (4–10) and crystals (11–13). It is appealing to draw the analogy between assembly from NPs and atoms/molecules to better understand the interactions between NPs and thus achieve desirable assemblies with guidelines from well-developed atomic and molecular chemistry. A notable assembly technique to address this analogy is the DNA-driven self-assembly of NPs (8–13). In general, this approach uses spherical NPs (for example, Au) as templates for the immobilization and orientation of surface-bound DNA strands, followed by fabricating programmable constructions, where building blocks of NPs act as the “atoms” and DNA hybridization as the “bonds.” Although these spherical NP-based DNA bonds allow regular network structures and well-ordered superlattices to be assembled by fairly isotropic interactions that mimic metallic or ionic bonding, some effort has also been devoted to developing methods to break the spherical symmetry and achieve greater directional interactions between the building blocks that more closely mimic covalent bonding (14, 15). One conceptually straightforward, albeit experimentally challenging, way to achieve this is to replace the spherical cores with anisotropic ones (16, 17). The organized assembly of anisotropic particles is typically governed by specific interactions or directing factors associated with the particle building blocks. For example, self-assembly of nanorods into sparse triclinic lattices can be realized by electrostatic interactions in nematic liquid crystal hosts (18). The oriented attachment (OA) of colloidal NPs can also be regarded as an assembly process but is different from the aforementioned NP self-assemblies in terms of the strength of the driving forces, being interatomic bonding of opposing crystal facets in OA (19) and van der Waals interactions or entropic factors in others (20–24). Some remarkable examples have shown that one-dimensional (1D) and 2D assemblies can be fabricated by attaching different facets of PbSe NPs along linear, square, or honeycomb pathways (25, 26). Other alternative strategies have also been developed for engineering assembly of anisotropic NPs, such as chiral self-sorting (27), spray-drying (28), and capillarity (29).
In principle, the addition of the shape and interaction anisotropy to NPs could not only mimic the atomic directional bonds but also extend the possible assemblies to motifs as complex as those molecules with more open and anisotropic structures formed from molecular directional bonding, for example, supramolecular coordination complexes (SCCs) (16). The SCCs encompass discrete systems in which selected metal centers or metal clusters are bridged by organic ligands through metal-ligand coordination bonds oriented with particular angularity to generate a finite supramolecular complex (30, 31). In contrast to the atomic building blocks of simple molecules, metal clusters and organic linkers become primary structural components to participate in the supramolecular assembly system. With predictable symmetry of the known molecular geometries in hand, chemists are able to rationally select these molecular building blocks to design a broad range of 2D systems [triangles (32), squares (33), and higher polygons (34)] and 3D systems [tetrahedra (35), cubes (36), and dodecahedra (37)] with required symmetries. For instance, the geometries of supramolecular polyhedra and their general formula of MnL2n can be predicted when metal ions (M) with square planar coordination sphere and rigid bent ligands (L) are mapped onto the vertices and edges, respectively, of the polyhedra (38). This symmetry consideration provides an inspiring strategy to govern the formation of discrete open constructs assembled from highly directional building blocks of molecules (30, 31) and even biomacromolecules (39, 40). However, it remains poorly developed from NP building blocks due to the daunting synthetic challenge. A compelling demonstration of NP assembly that closely mimics the oriented coordination bonds among molecules in SCCs would be even more difficult to produce.
Here, we explore and demonstrate the extension of the symmetry consideration to fabricate open and discrete architectures from anisotropic NPs, in which single crystal–like, 3D frame-like superstructures can be constructed via the assembly of nanocuboids (NCBs) on the edges of a cube. Toward this goal, we choose Prussian blue analogs (PBAs) as the target materials in view of the limited success in exploiting these PBA-type materials for colloidal assembly and their transformation versatility toward functional materials with retainable structures (41–43). Besides, the Co-Fe combination is one of the most important PBA-type materials with switchable magnetic and electronic properties (44–49), which enables promising application in catalysis to be realized (50). Comprehensive investigations reveal that phase transition, epitaxial growth, confined assembly, and OA are involved in the formation process for the superstructures reported here. Specifically, we show that the typical NCB-assembled frame-like superstructure (NAFS) is a 20-component cubic ensemble, with a high degree of similarity to a proposed supramolecular M8L12-type cube (31). We then expound how the natural symmetry effect derived from NP shape can be considered and applied rationally as the design principle for predictable OA-involved assembly. In addition, the synthetic strategy in this work allows us to realize the postcrystallization of the building blocks into single-crystalline Co-Fe PBA nanoframes (NFs) at a proper temperature. After the thermal treatment in air, the Co-Fe PBA NAFSs and NFs are further transformed into Co-Fe mixed oxides with preserved structures, which manifest as efficient electrocatalysts for the oxygen evolution reaction (OER).
RESULTS
Characterization of the discrete frame-like superstructures
X-ray powder diffraction (XRD) pattern (fig. S1A) is consistent with a face-centered cubic (fcc) structure (a = 9.82 Å). Energy-dispersive x-ray spectroscopy (EDX) spectrum reveals the atomic ratio of K/Co/Fe = 0.67:1:0.71 (fig. S1B), hereafter denoted as KCoFe-1. The morphology of the KCoFe-1 NAFSs is characterized by field-emission scanning electron microscopy (FESEM). As shown in Fig. 1A (also see fig. S2), these KCoFe-1 particles are highly uniform with a cubic geometry, a 3D frame-like architecture, and an average size of ca. 350 nm. Closer inspection of a representative NAFS (inset of Fig. 1A and fig. S3) reveals an ensemble braced by small NCBs with different side lengths (70 to 150 nm) serving as building blocks. These unique NAFSs display distinctive architectural features as follows: (i) A typical superstructure comprises 20 components (NCBs) that distributed onto 12 edges, (ii) three adjacent (two or four in a few cases) NCBs attach to one another in an orderly way to build up an edge, and (iii) the NCBs on the corners provide three facets for attachment, whereas those in the middle of edges offer two. On the basis of the above observation, we could illustrate this structure with schematic models, as provided in Fig. 1 (B to D). The models display, from three directional orientations, a cubic frame superstructure constructed by 12 orthogonal edges, on each of which the NCBs have a 1D assembly. Transmission electron microscopy (TEM) images (Fig. 1, E to G) of the superstructures in different directions on the substrate are all similar to the corresponding schematic models (Fig. 1, B to D), further confirming that well-knit NCBs constitute the 3D frame-like architectures with a highly open structure. Each of these NAFSs appears as a single crystal, as evidenced by selected area electron diffraction (SAED) patterns from common zone axes of <100>, <110>, and <111> of a cubic system in Fig. 1 (H to J). Small arcs of the spots seen in the SAED patterns imply that only slight crystallographic rotation exists among the NCBs. We can infer that these building blocks hold highly similar crystallographic orientation, although they may not align well on every edge of the frames. An integrated analysis of the TEM image (Fig. 1E) and related SAED pattern (Fig. 1H) allows us to conclude that the NCBs are dominated by six {100} facets because the <100> zone axis is positioned perpendicularly across the side faces of these NCBs, in conformity with an fcc crystal habit. Meanwhile, the corresponding high-resolution TEM (HRTEM) and fast Fourier transform (FFT) images imply that each NCB should be a single crystal (Fig. 1, K to M). We also observe that adjacent NCBs on edges could attach and coalesce at their interparticle interfaces (marked by arrows), as depicted in Fig. 1N. The lattice fringes around the attachment region (square 1 for the NCB-1/NCB-2 couple, square 2 for the upper/nether edge couple, and square 3 for the NCB-3/NCB-4 couple) share a common crystallographic orientation in a continuous manner. Moreover, the interplanar spacing (4.9 Å for {200} planes) of the upper edge is identical to that of the nether edge, as evidenced in Fig. 1N and further confirmed by the FFT patterns in Fig. 1O. These observations further prove the single crystal–like feature of the 3D NAFSs, in line with the SAED result (Fig. 1H) and our related inference above. Therefore, each KCoFe-1 frame-like structure could be regarded as a supercrystal formed from the ordered assembly of small NCB single crystals via a face-to-face ({100} facets) OA arrangement.
Fig. 1. Discrete frame-like superstructures of Co-Fe PBA assembled from the attachment of NCBs.

(A) FESEM images of the KCoFe-1 NAFSs. Inset: A magnified FESEM image displays a single NAFS. Schematic models of a NAFS from three orientations (B to D), and their corresponding TEM images (E to G) and SAED patterns (H to J) from different zones. The spots located on each ring in (H) to (J) (semirings are marked by blue dots) represent those families of lattice planes with the same interplanar spacing (from the inner to outer ring): {200}, {022}, {400}, {024}, and {600} in (H); {200}, {220}, {222}, {400}, and {600} in (I); and {220}, {440}, and {660} in (J). (K to M) HRTEM images of the white square shown in (E) to (G), respectively. Interplanar spacing of 4.9 and 3.5 Å represents {200} and {220} planes, respectively. Insets: Corresponding FFT images. (N) HRTEM image of a part of two separate edges within a NAFS (marked by a red rectangle) in (E). The dashed line delineates the boundary profile of the upper edge (high contrast on the left) from the nether one (lower contrast on the right). The left arrow reveals the attachment interface between NCB-1 and NCB-2 on the upper edge, whereas the right one reveals that of NCB-3 and NCB-4 on the nether edge. The squares display continuous lattice fringes across the region around the attachment interface of the adjacent NCBs. (O) FFT images from the three squares shown in (N).
Formation process and mechanism of the NAFSs
To obtain a direct observation of the evolution for the unique KCoFe-1 NAFSs, we monitor reaction products at different stages. We observe that truncated nanocubes (NCs) around 250 nm in size form in 5 s (Fig. 2A). Further characterization reveals that these uniform NCs are Co-Fe PBA single crystals with the fcc structure (a = 10.10 Å) and are dominated by {100} facets (atomic ratio of K/Co/Fe = 0.07:1:0.67, hereafter denoted as KCoFe-2) (fig. S4). As the reaction time reaches 2 hours, small NPs preferentially form on the edges and corners of the NCs. However, these NPs cannot be easily defined as cuboid in shape or well detected as the KCoFe-1 phase until the reaction time reaches 6 hours, as revealed by related FESEM images (Fig. 2A) and XRD patterns (fig. S5). Most of the NCBs are still discretely distributed on the NCs as satellites at this stage. They then would arrange in an orderly way one by one on each edge of the NCs after reaction for 9 hours (Fig. 2A and fig. S6). From the FESEM images of the intermediate at 18 hours, we find that the attachment of the NCBs results in a cubic frame–like structure by taking advantage of the NC core template. Simultaneously, these cores gradually dissolve, leaving cavities and holes in the central regions of these core@satellite superstructures. As the reaction time goes to 36 hours, the complete removal of the KCoFe-2 core eventually leads to the formation of 3D NAFSs with a pure KCoFe-1 phase (Fig. 2A and fig. S5). On the basis of the foregoing observations, the following points are crucial for us to disclose the formation mechanism of the NAFSs and thus appealing for further investigation.
Fig. 2. Formation process and mechanism of the KCoFe-1 NAFSs.
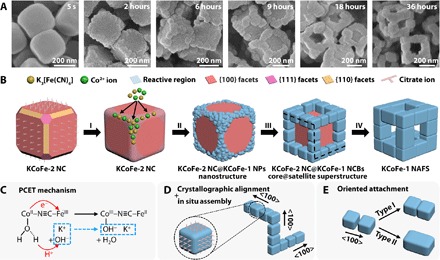
(A) FESEM images of the samples obtained at 5 s and 2, 6, 9, 18, and 36 hours of the reaction process, showing the morphological change from NCs to NAFSs. (B) Schematic illustration of the formation mechanism of a NAFS, in which four stages were involved: I, site-selected deposition; II, the formation of the new KCoFe-1 phase; III, epitaxial growth of the NCBs; IV, core@satellite superstructure transform to NAFS. (C) Mechanism of the hydroxyl-potassium ion insertion triggered PCET. (D) Ordered arrangement of NCBs on three orthogonal edges of the core@satellite superstructure in (B) (marked by black dashed lines). (E) Two types of outcome from the OA of adjacent NCBs: type I, neck attaching; type II, total integration.
First, why does the KCoFe-1 form only on selected regions of the KCoFe-2 NCs? Control experiments reveal that trisodium citrate should play a significant role here by controlling the reaction kinetics (table S1). It could promote the formation of cobalt-citrate complexes in aqueous solution, which adjusts the release of cobalt ions from the complexes and thus the precipitation rate of the Co-Fe PBA. In addition, citrate ions might selectively cap on the {100} facets of the Co-Fe PBA as the shape-directing agent, facilitating the formation of {100}-dominated NCs. This role might be similar to that of bromide ions for the formation of palladium NCs (51). Thus, a proper amount of trisodium citrate is of importance for obtaining the desired template, meaning an amount that could lead to the formation of the cube-like template with passivated side faces and exposed truncated edges and corners that are highly reactive. Note that the size of the NCs hardly changes after they form (in 5 s) when only about one-third amount of K3[Fe(CN)6] is added into the reaction system. Therefore, a site-selected deposition of the remaining reactants on those reactive regions of the NCs can be expected (stage I in Fig. 2B). As a consequence, we can observe that the KCoFe-1 NPs form restrictedly in these regions and grow into a frame rather than a shell structure.
Second, what is the origin for the generation of the new phase (KCoFe-1) in the presence of a preformed phase (KCoFe-2) in a one-pot synthesis? We obtain some useful clues by comparing the structure of the initially formed KCoFe-2 phase and the newly generated KCoFe-1 phase. The K proportion in the KCoFe-1 phase is conspicuously higher than that in the KCoFe-2 phase, that is, 0.67 versus 0.07 (atomic ratio to Co), as revealed by the EDX mentioned before. This difference is also evident in the elemental mapping of a typical core@satellite superstructure (fig. S7). Also, the shift of the peaks in XRD patterns indicates a cell parameter contraction of the KCoFe-1 phase (9.82 Å) relative to the KCoFe-2 phase (10.10 Å) (fig. S5). Such a contraction is observed in Co-Fe PBAs when CoII–N≡C–FeIII is transformed to CoIII–N≡C–FeII (46). Fourier transform infrared (FTIR) spectra further prove this transformation (fig. S8). Compared with the KCoFe-2 phase, we observe a significant decrease of the ν (CN) peak for CoII–N≡C–FeIII and the emergence of a new strong ν (CN) peak for CoIII–N≡C–FeII in the KCoFe-1 phase (table S2). Note that we use Co(II)- and Fe(III)-based compounds as the reactants, which means that the intramolecular electron transfer from CoII to FeIII sites occurs after the generation of CoII–N≡C–FeIII molecules. Hydroxyl ions are also detected in the KCoFe-1 phase by the FTIR measurement. These observations allow us to propose a formation mechanism for the KCoFe-1 phase (stage II in Fig. 2B): Hydroxyl-potassium ion insertion triggered proton-coupled electron transfer (PCET) (52), as shown in Fig. 2C. As the local concentration of K+ ions in the solution increases with the addition of the remaining K3[Fe(CN)6], K+ ions would migrate into the nanoporous framework of the Co-Fe PBA deposition on the edges and corners of the NCs, driven by the concentration gradient. Synchronously, the hydroxyl ions provided from hydrolysis of trisodium citrate would also transport into the framework as the charge-balance counterions of the K+ ions. PBA frameworks are well known for containing water molecules in terms of zeolitic water and ligand water (coordinate to the Co sites in our work). Hydroxyl ions in the pore channels could attack the H atoms of the weakly acidic ligand water molecules bound to the CoII species, which would reinforce the H2O to CoII σ donation and thus lower the CoIII/CoII redox potential (46). Moreover, the insertion of K+ ions might also decrease the CoIII/CoII redox potential while increasing the FeIII/FeII potential by interacting with the cyanide species (47). These two factors could trigger the electron transfer from CoII to FeIII sites through the cyanide bridges. The oxidation of CoII sites would enhance the acidity of their ligand water and cause the deportonation reaction, forming zeolitic water molecules with the neighboring hydroxyl ions. The Co-H2O sites act at the same time both as electron donor toward the FeIII sites (electron acceptors) and as proton donor toward the hydroxyl ions (proton acceptors). This type of electron transfer in PBAs which is mediated by chemical modification (specifically, ion insertion) has been reported before (46, 53), and is distinct from other means using external stimuli, such as photoradiation (44), temperature (48), and pressure (49).
Third, how do the NCBs assemble into single crystal–like superstructures? It is well reported that core-shell nanostructures of PBAs can be synthesized via epitaxial growth (54, 55). Here, the relatively small lattice mismatch between KCoFe-1 and KCoFe-2 (2.64, 2.63, and 2.71% for the {100}, {110}, and {111} planes, respectively) encourages us to hypothesize an epitaxial growth process for the KCoFe-1 phase. As verified by TEM and SAED results, the morphological and crystallographic alignments (fig. S9A) of the KCoFe-2 NC and the KCoFe-1 NCBs support our hypothesis above. The cube-based shape of the NCBs indicates that the growth of different crystal planes is also manipulated by the citrate ions, leading to the formation of {100}-dominated NCBs similar to the KCoFe-2 NCs. Here, the epitaxial growth enables the immobilization of these NCBs on the NCs and thus markedly constrains the movement and rotation freedom of the NCBs (stage III in Fig. 2B). Because of the confined growth on the NC template, anchored NCBs can achieve in situ assembly spontaneously on each edge of the NC once they form (Fig. 2D). As a result, all NCBs are directed with the <100> zone axis along the edges and ready for subsequent attachment, although some spatial misalignment may exist (fig. S3). As the reaction proceeds, a classical coarsening mechanism would favor the growth of each single NCB via the addition of ions or molecules to particle surfaces from the reaction solution. When the NCBs grow to be close enough to each other, the confined assembly would regulate these prealigned NCB satellites into a cubic frame–like structure by OA. A further coarsening could extend the fusing interface of the as-attached NCBs to make a robust architecture. However, this process is limited by the low temperature (35°C), resulting in a partial coalescence of the NCB building blocks (“type I” in Fig. 2E and revealed in Fig. 1N). Note that the core NCs form quickly at the very beginning of the reaction and thus have many defects that make them weak and “soft,” and cause them to gradually dissolve during the process (55), which transforms the core@satellite superstructures to the NAFSs (stage IV in Fig. 2B). Apparently, the frame-like structures would be loose and easily collapse into free-floating NCBs if there is no attachment involved. Moreover, we observe that an OA event leads to the fusion of the nearly full interface of two small NCBs (fig. S9B). An integral larger NCB is expected if these two small NCBs could undergo further attachment and coarsening. This observation indicates that some of the NCBs in the NAFSs could be grown from relatively small NCBs found in the intermediates (for example, obtained at 6 hours) by the OA and subsequent coarsening process (“type II” in Fig. 2E). In a general process for OA, the NPs need to continuously diffuse into sufficient proximity by means of Brownian motion and then rotate to find an optimum configuration before enduring attachment takes place (19). Our assembly strategy bypasses these conventional steps. More specifically, the “optimum configuration” in the present system is achieved instead in a template-assisted manner. The NC templates serve as the substrates for the epitaxial growth of the NCBs, thus offering intrinsic crystallographic alignment of the NCBs when they are initially formed. Subsequently, the additive and crystal structure–dependent coarsening process renders a cube-based shape for the NCBs as they grow further. Besides, the growth of the NCBs would be restricted because they have been anchored on the oriented edges of the NCs. The anisotropy of the NCs prevents the random growth of the NCBs and guarantees the growth of these NCBs along the initial crystallographic orientation (<100> direction), followed by attachment via their prealigned {100} facets. Therefore, the pathway of this OA assembly is quite different from those using free-floating NPs in solution (19, 25, 26). We also expect a gradual detachment of the capping molecules on the bonding regions when the attachment of NCBs starts (25).
Supramolecular chemistry–inspired design principle for OA-involved NP assembly
An early survey has provided us a hint from the growth kinetics point of view that the OA process has “molecule-like” features, in which the nanocrystal growth via OA behaves as the collision and reaction between molecules (56). Here, we notice that the topological structure of our assembled superstructures is similar to that of the proposed M8L12 supramolecular cubes. This motivates us to learn from the supramolecular chemistry (from a spatial layout perspective) to attempt to obtain better insight into the role of the anisotropy of NPs and their OA in governing the motifs of the final architectures. A typical example of a 3D molecule assembly of M8L12 supramolecular cube is given by Roche et al. (57), as illustrated in Fig. 3A. When [Ru([9]ane-S3)Cl2(DMSO)]2+ is combined with 4,4′-bipyridine (bpy), the chlorides and dimethyl sulfoxide (DMSO) ligands are displaced. The capping nature of the [9]ane-S3 ligand endows availability of the remaining three binding sites in the octahedral Ru coordination environment for occupation by incoming ligands. Thus, this Ru-based compound can be considered as a “tritopic” building block, where the bond angles hold at 90° with respect to one another. Similarly, the 4,4′-bpy linker can be regarded as a linear “ditopic” building block because its two lone pairs of electrons are 180° apart and ready for donating. The vector of the M-L bond is simply from the ligand to the metal, collinear with both the coordination vector of the metal-based center and the linker. Consequently, thermodynamics of the system delivers the favored 3D supramolecular cubic cage from 8 tritopic 90° corner units and 12 ditopic linear edge linkers, following the directional coordination bonding approach. Here, the NP assembly seems to comply with conceptually similar rules (Fig. 3B). Specifically, each NCB has a particular shape dominated by six {100} facets whose normal vectors stand at 90° or 180° with respect to one another. This configuration enables each NCB to serve as the molecule equivalent of the metal-based complex or the linear linker for nanoscale assembly, providing either three (tritopic) or two (ditopic) facets for attachment. As for the bonding manner, the directionality of OA originated from the high selectivity of facet bonding is quite similar to that in the coordination bonds. Therefore, the possible attachment angle of the adjoining NCBs can only be 90° or 180°. In the present system, the OA of these NCB building blocks leads to the M8L12-type NAFS, within which 8 tritopic NCBs locate on the vertexes, whereas 12 ditopic NCBs stretch as the edges of the cubic frame.
Fig. 3. Schematic illustration of the analogy of 3D assembly from molecules and NPs.
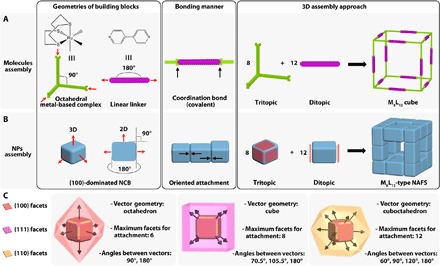
(A) Assembly of a typical M8L12 supramolecular cube from the directional coordination bonding of specific molecules. Red arrows represent the coordination vectors. (B) Assembly of an M8L12-type NAFS from the OA of anisotropic NPs. Red arrows represent the normal vectors to the {100} facets. Red polygons represent the facets of NCB for attachment. (C) Features of diverse types of facets of a typical truncated NC for the OA-involved assembly. Arrows represent normal vectors to the corresponding facets. Colored polyhedra outside the NC represent the normal vector geometries of the relevant facets.
Inspired by the concept of “symmetry-interaction self-assembly” in supramolecular chemistry (30, 31), we propose here that the symmetry from the morphological anisotropy of the NPs might serve as the cornerstone in the design principle for the nanoscale assemblies via OA. This is because the inherent preferences for particular geometries and highly directional binding motifs (dipole-induced electrostatic interaction) are “encoded” in certain NPs depending on the crystal system (58, 59); these moieties help control the way in which the building blocks assemble into well-defined discrete superstructures. Although the use of the symmetry interaction has not been exploited well by nanoscientists, it is nonetheless a potential tool (60) and, in some cases, is actually used without notice. In the process of OA-involved assembly, the geometry of the normal vectors to the attachment facets is critical to defining the symmetry of the NP building blocks. We take the typical truncated NCs as the building blocks to illustrate the concept for simplicity (Fig. 3C). A truncated cubic NP can be enclosed by 6 {100}, 8 {111}, and 12 {110} facets. The normal vector angle of every two facets within one family of crystal planes is rigidly fixed, that is, 90° or 180° for {100} facets; 70.5°, 109.5°, or 180° for {111} facets; and 60°, 90°, 120°, or 180° for {110} facets. Thus, the normal vector geometries are octahedron, cube, and cuboctahedron for {100}, {111}, and {110} facets, respectively. In general, only one type of facet will be involved in the OA in a certain system, and the OA takes place along the direction parallel to the normal vectors of the attachment facets. The number of facets on a certain NC that participated in the attachment can theoretically be in the range of 1 to 6 for {100} facets, 1 to 8 for {111} facets, and 1 to 12 for {110} facets. On the basis of these habits encoded in the NCs, we generalize some guidelines from the relationship among five elements—the type of attachment facets, attachment angle, normal vector geometry, number of attachment facets of a certain NP, and possible attachment motif (symmetry)—for the cubic system (table S3). For a certain NC, the attachment angle for the incoming NCs is predictable if the type of attachment facet is given and vice versa. Meanwhile, the short-range attachment geometries of NCs can be diverse and predicted as well because they are located at different positions of the ensembles where a particular number of the facets for attachment are required. For example, the NCBs in the present study serve as both tritopic at the vertexes and ditopic on the edges, manifesting the versatility of anisotropic NPs as the building blocks for assembly. One can expect that both linear and square assemblies could be realized by the attachment of the building blocks through the {100} facets (26). Furthermore, this relationship allows us to see that only using {100} facets for OA can guarantee the building blocks as both 90° tritopic and linear ditopic in one system, which is a key precondition for facilitating the assembly of cubic superstructures in this work (Fig. 3B). That is, the cubic geometry of these superstructures cannot be reached if the NCBs attach to one another through other types of facets, even in the present template-assisted system. For instance, we model the OA of {110} facets of NCs and find that a frame-like superstructure can possibly be acquired but with a cuboctahedral geometry (fig. S10). Ideally, divergent nanoscale assemblies analogous to the metal-organic frameworks could be constructed by octahedral hexatopic and linear ditopic NP building blocks. This relationship also suggests that a particular entity assembled by this OA-involved strategy should logically manifest as a geometrical ensemble of all included motifs of the short-range attachment. A thorough investigation of the PbSe NC assembly via OA confirms this assertion (25).
Related structures and catalysis of the frame-like architectures
On further investigation, we find that the average sizes of both the NAFSs and respective building blocks (NCBs) can be tuned by controlling the reaction conditions (fig. S11). Also, that this strategy can be performed at a higher temperature of 80°C (versus 35°C for NAFSs) to synthesize another type of frame-like/cage-like structure (61). As shown in Fig. 4A (also see fig. S12), the product is composed of uniform NFs with an average size of ca. 350 nm. A magnified FESEM image in Fig. 4B reveals the detailed structure of the NFs. We did not find any footprint about assembled NCBs within the NFs like those in NAFSs. Instead, we saw intact edges with a smooth surface and an average thickness of ≈115 nm. XRD and EDX results reveal that these NFs are also Co-Fe PBAs but with a different atomic ratio as K/Co/Fe = 0.97:1:0.84, hereafter denoted as KCoFe-3 (fig. S13, A and B). As the K proportion (relative to Co element) increases in the order of KCoFe-2 (0.07), KCoFe-1 (0.67), and KCoFe-3 (0.97), the color of the corresponding products changes from red to blue, and then violet due to the violet spectral lines of the potassium ions (62). The FTIR spectrum of KCoFe-3 NFs is closer to that of the KCoFe-1 NAFSs compared to that of the KCoFe-2 NCs (fig. S13C and table S2). Time-dependent experiments (fig. S14) verify that the NFs are formed by a very similar mechanism to that of the NAFSs (Fig. 2B). In addition, the higher temperature offers more energy that drives the postcrystallization of the as-attached NCBs by a higher degree of supplementary crystal growth and interparticle ripening in the coarsening process, which leads to the complete coalescence of all these building blocks into the final NF architecture. TEM images (Fig. 4, C and D) further confirm the open structure of the NFs and their integrity throughout the edges, in line with the FESEM images. As expected, a typical NF (Fig. 4D) is displayed as a high-quality single crystal, as demonstrated in the SAED pattern in Fig. 4E. Moreover, the HRTEM image in Fig. 4F reveals clear lattice fringes, implying the high crystallinity of the NF. The interplanar spacing is ca. 5.0 Å, which can be assigned to {200} planes, consistent with the SAED analysis (Fig. 4E).
Fig. 4. Morphology and structural characterization of single-crystalline KCoFe-3 NFs.

(A and B) FESEM images. Inset of (B) is a schematic model of the NF. (C and D) TEM images. (E) SAED pattern. (F) HRTEM image and the corresponding FFT image (inset).
OER is an important half reaction in electrochemical water splitting. In practice, however, the high overpotential caused by sluggish OER markedly diminishes the overall energy efficiency of the water splitting. Therefore, catalysts are always required to lower the activation energy and enhance conversion rate. Because of their high activity, low cost, and earth-abundant storage, transition metal oxides have been considered as promising catalysts for OER and well developed in the past few years (43, 63, 64). Here, we convert the KCoFe-1 NAFSs and KCoFe-3 NFs to Co-Fe oxides by simple annealing in air at 350°C for 2 hours. XRD analysis shows that the oxide products are composites composed mainly of Co3O4, CoFe2O4, and K2CoO3 for both of the derived samples (fig. S15). We observed that the as-prepared Co-Fe oxides retain the uniform 3D cubic frame–like structures of their precursors well but with rougher surfaces (Fig. 5, A and D, and fig. S16). TEM images further indicate that these frame-like structures are porous and composed of very small NPs (Fig. 5, B, C, E, and F). Notably, the interface of the particles can still be seen in the structures derived from KCoFe-1 NAFSs, demonstrating that these ensembles could also be regarded as superstructures constructed by small Co-Fe oxide NCBs (Fig. 5, A to C). We then evaluate the feasibility of using both the NAFSs and NFs of Co-Fe oxides as electrocatalysts for OER. We conduct the measurements in a standard three-electrode system in 1.0 M KOH electrolyte. Figure 6A shows the linear sweep voltammetry (LSV) curves for OER. To reach a current density of 10 mA cm−2, which is a common criterion to evaluate the OER activity, the Co-Fe oxide NAFSs need a potential of 1.57 V corresponding to an overpotential of 0.34 V, whereas the NFs require an overpotential of 0.35 V. These values are comparable with those advanced electrocatalysts based on transition metal oxides (table S4), which indicates that the Co-Fe oxide NAFSs and NFs have high electrocatalytic activity toward OER. From the Tafel plots (Fig. 6B), the NAFSs and NFs exhibit a Tafel slope of 57 and 61 mV dec−1, respectively, suggesting the enhanced kinetics for the NAFSs. Note that the “gaps” observed at the particle interface of the Co-Fe oxide NAFSs might provide a larger electrochemical active surface area (ECSA) than that of the Co-Fe oxide NFs (fig. S17), suggesting that the improved electrocatalytic activity of the former should come from a larger ECSA. In addition to the activity, the stability of the catalysts is also crucial for their practical applications. As seen from the chronopotentiometry (CP) curves in Fig. 6C, the NFs could maintain the current density of 10 mA cm−2 for 5.7 hours with only ~7% increase required for the potential, whereas the NAFSs could even last for 8.6 hours with the same potential increase.
Fig. 5. Morphology and structural characterization of Co-Fe PBA–derived composites.

(A to C) FESEM (A) and TEM (B and C) images of Co-Fe mixed oxides derived from KCoFe-1 NAFSs. Arrows show the interparticle interfaces within the NAFSs. (D to F) FESEM (D) and TEM (E and F) images of Co-Fe mixed oxides derived from KCoFe-3 NFs. The details of the edges of the frame-like structures in the left column of (B), (C), (E), and (F) (red dashed squares) are displayed in the right column.
Fig. 6. Electrocatalytic oxygen evolution on Co-Fe mixed oxides.
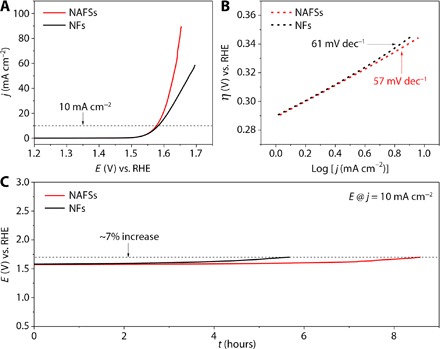
(A) LSV curves of NAFS and NF catalysts. (B) Tafel slopes of NAFS and NF catalysts. RHE, reversible hydrogen electrode. (C) CP response of NAFS and NF catalysts at the current density of 10 mA cm−2.
DISCUSSION
In summary, we have successfully developed a synthetic strategy to create discrete 3D assemblies with highly open structure from NPs. The combination of the template-mediated epitaxial growth, confined assembly, and the OA mechanism in this strategy opens an avenue that promises access to the unique frame- or cage-like superstructures. As a demonstration, we successfully synthesized Co-Fe PBA NAFSs, which extends both the diversity of the NP building blocks and the ensemble motifs. We have also elucidated how to seize the symmetry features that are intrinsically encoded in the shape of the building blocks as the design principle. The present work provides fundamental insights into directional bonding among NPs, which mimics directional molecular assembly, and also adds to the growing body of knowledge for assembling superstructures with anisotropic building blocks. Furthermore, we have demonstrated that high-quality single-crystal Co-Fe PBA NFs can be obtained by this strategy. To demonstrate the practical application, we converted both NAFSs and NFs of Co-Fe PBA into corresponding Co-Fe mixed oxides with little alteration in structure. These NAFSs and NFs of Co-Fe oxides exhibit excellent electrocatalytic activity for the OER with an overpotential of about 0.34 V to reach the current density of 10 mA cm−2.
MATERIALS AND METHODS
Synthesis of Co-Fe PBA NAFSs and NFs with an average size of 350 nm
In a typical synthesis of Co-Fe PBA NAFSs, 0.15 g of cobalt(II) acetate tetrahydrate and 0.147 g of trisodium citrate dihydrate were dissolved in 40 ml of deionized (DI) water to form solution A. Potassium hexacyanoferrate(III) (0.132 g) was dissolved in 60 ml of DI water to form solution B. Then, solution B was added into solution A under magnetic stirring in 15 s. After continuous stirring for 1 min, the obtained mixed solution was aged at 35°C for 36 hours. The precipitate was collected by centrifugation, washed with DI water and ethanol, and dried at 70°C overnight. Co-Fe PBA NFs were obtained by a similar procedure except that they were aged at 80°C for 6 hours.
Synthesis of Co-Fe PBA NAFSs with an average size of 600 nm
In a typical synthesis, 0.15 g of cobalt(II) acetate tetrahydrate and 0.221 g of trisodium citrate dihydrate were dissolved in 40 ml of DI water to form solution A. Potassium hexacyanoferrate(III) (0.132 g) was dissolved in 60 ml of DI water to form solution B. Then, solution B was added into solution A under magnetic stirring in 15 s. After continuous stirring for 1 min, the obtained mixed solution was aged at 35°C for 36 hours. The precipitate was collected by centrifugation, washed with DI water and ethanol, and dried at 70°C overnight.
Synthesis of Co-Fe PBA NAFSs with an average size of 200 nm
In a typical synthesis, 0.15 g of cobalt(II) acetate tetrahydrate and 0.147 g of trisodium citrate dihydrate were dissolved in 20 ml of DI water to form solution A. Potassium hexacyanoferrate(III) (0.100 g) was dissolved in 30 ml of DI water to form solution B. Then, solution B was added into solution A under magnetic stirring in 15 s. After continuous stirring for 1 min, the obtained mixed solution was aged at 35°C for 18 hours. The precipitate was collected by centrifugation, washed with DI water and ethanol, and dried at 70°C overnight.
Synthesis of Co-Fe mixed oxides
In a typical synthesis, both the as-prepared Co-Fe PBA NAFSs and NFs were annealed in a muffle furnace at 350°C for 2 hours with a heating rate of 2°C min−1 in air.
Materials characterization
XRD patterns were collected on a Bruker D2 Phaser x-ray diffractometer with Ni-filtered Cu Kα radiation (λ = 1.5406 Å) at a voltage of 30 kV and a current of 10 mA. The morphology and structure of the products were characterized using FESEM (JEOL, JSM-6700F) and TEM (JEOL, JEM-1400/JEM-2010) equipped with EDX. FTIR spectra were collected by PerkinElmer FTIR spectrometers.
Electrochemical measurements
We first conducted a RHE calibration. We used a Pt sheet, a Pt wire, and a Ag/AgCl electrode as the working electrode, counter electrode, and reference electrode, respectively. For all the electrochemical measurements, an aqueous solution of 1.0 M KOH was used as the electrolyte. Before calibration, the solution was bubbled with hydrogen for 0.5 hour to obtain a hydrogen-saturated electrolyte. Cyclic voltammetry was recorded at a scan rate of 2.0 mV s−1 (fig. S18). The average of the two potentials, where the current crossed zero, was taken to be the thermodynamic potential for the hydrogen electrode reactions. All the potentials shown in our tests were calibrated and transformed to RHE: E(RHE) = E(Ag/AgCl) + 0.977 V. For the OER measurement, 2.0 mg of catalysts (for example, Co-Fe mixed oxide NAFSs) was dispersed in 700 μl of DI water, 268 μl of ethanol, and 32 μl of 5 weight % Nafion solution to form a homogeneous ink. For fabrication of the working electrodes, 10 μl of catalyst inks was loaded onto a 5-mm-diameter glassy carbon rotating disk electrode (loading amount was about 0.1 mg cm−2). Then, the electrode was dried at room temperature. The electrochemical studies were carried out in a standard three-electrode system using a Pt wire and a Ag/AgCl electrode as the counter and reference electrode, respectively (controlled by a Pine Instruments electrochemistry workstation). N2 was used to purge the solution to achieve a N2-saturated condition, while the working electrode was continuously rotated at 1600 rpm to get rid of the oxygen bubbles. LSV was carried out at 5.0 mV s−1 for the polarization curves and 0.1 mV s−1 for Tafel plots. All polarization curves were corrected with iR compensation.
Acknowledgments
Funding: X.W.L. acknowledges the funding support from the National Research Foundation (NRF) of Singapore via the NRF investigatorship (NRF-NRFI2016-04). Author contributions: J.N. and X.W.L. conceived the idea and cowrote the manuscript. J.N. designed and performed the experiments and analyzed the data. B.Y.G. helped in the analysis of the TEM data. B.Y.G. and L.Y. assisted in materials characterizations. All authors discussed the results and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/8/e1700732/DC1
fig. S1. Characterization of the KCoFe-1 NAFSs.
fig. S2. Overview FESEM image of the KCoFe-1 NAFSs.
fig. S3. FESEM images of two NAFSs.
fig. S4. Characterization of the KCoFe-2 NCs.
fig. S5. XRD patterns of different samples.
fig. S6. FESEM image of the product obtained at 9 hours of the reaction process.
fig. S7. TEM and EDX mapping images of a typical core@satellite superstructure.
fig. S8. FTIR spectra of different samples.
fig. S9. TEM and HRTEM images of a typical core@satellite superstructure.
fig. S10. Model of the cuboctahedral frame-like superstructure.
fig. S11. Characterization of the NAFSs with different sizes.
fig. S12. Overview FESEM image of the KCoFe-3 NFs.
fig. S13. Characterization of the KCoFe-3 NFs.
fig. S14. FESEM images of the samples obtained from different stages of the growth of NFs.
fig. S15. XRD patterns of the samples derived from KCoFe-1 NAFSs and KCoFe-3 NFs.
fig. S16. Overview FESEM images of the Co-Fe oxide composites.
fig. S17. Estimation of the ECSA of the catalysts.
fig. S18. Reference electrode calibration in 1.0 M KOH solution.
table S1. Observations from the FESEM images for different samples.
table S2. FTIR peak positions of the samples of KCoFe-1, KCoFe-2, and KCoFe-3 and their assignment.
table S3. Some typical examples of the relationship of the type of attachment facets, attachment angle, normal vector geometry, the number of attachment facet of a certain NP, and possible attachment motif (symmetry) in a cubic system.
table S4. Comparison of Co-Fe mixed oxide NAFSs and NFs prepared in this work with some recently reported transition metal oxide–based catalysts.
REFERENCES AND NOTES
- 1.Jones M. R., Seeman N. C., Mirkin C. A., Programmable materials and the nature of the DNA bond. Science 347, 1260901 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Tang Z., Kotov N. A., Giersig M., Spontaneous organization of single CdTe nanoparticles into luminescent nanowires. Science 297, 237–240 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Nie Z., Petukhova A., Kumacheva E., Properties and emerging applications of self-assembled structures made from inorganic nanoparticles. Nat. Nanotechnol. 5, 15–25 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Zhang Y., Lu F., Yager K. G., van der Lelie D., Gang O., A general strategy for the DNA-mediated self-assembly of functional nanoparticles into heterogeneous systems. Nat. Nanotechnol. 8, 865–872 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Shevchenko E. V., Talapin D. V., Kotov N. A., O’Brien S., Murray C. B., Structural diversity in binary nanoparticle superlattices. Nature 439, 55–59 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Talapin D. V., Schevchenko E. V., Bodnarchuk M. I., Ye X., Chen J., Murray C. B., Quasicrystalline order in self-assembled binary nanoparticle superlattices. Nature 461, 964–967 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Cargnello M., Johnston-Peck A. C., Diroll B. T., Wong E., Datta B., Damodhar D., Doan-Nguyen V. V. T., Herzing A. A., Kagan C. R., Murray C. B., Substitutional doping in nanocrystal superlattices. Nature 524, 450–453 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Macfarlane R. J., Lee B., Jones M. R., Harris N., Schatz G. C., Mirkin C. A., Nanoparticle superlattice engineering with DNA. Science 334, 204–208 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Macfarlane R. J., Jones M. R., Lee B., Auyeung E., Mirkin C. A., Topotactic interconversion of nanoparticle superlattices. Science 341, 1222–1225 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Zhang C., Macfarlane R. J., Young K. L., Choi C. H. J., Hao L., Auyeung E., Liu G., Zhou X., Mirkin C. A., A general approach to DNA-programmable atom equivalents. Nat. Mater. 12, 741–746 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Nykypanchuk D., Maye M. M., van der Lelie D., Gang O., DNA-guided crystallization of colloidal nanoparticles. Nature 451, 549–552 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Park S. Y., Lytton-Jean A. K., Lee B., Weigand S., Schatz G. C., Mirkin C. A., DNA-programmable nanoparticle crystallization. Nature 451, 553–556 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Auyeung E., Li T. I. N. G., Senesi A. J., Schmucker A. L., Pals B. C., Olvera de la Cruz M., Mirkin C. A., DNA-mediated nanoparticle crystallization into Wulff polyhedra. Nature 505, 73–77 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Wang Y., Wang Y., Breed D. R., Manoharan V. N., Feng L., Hollingsworth A. D., Weck M., Pine D. J., Colloids with valence and specific directional bonding. Nature 491, 51–55 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Liu W., Tagawa M., Xin H. L., Wang T., Emamy H., Li H., Yager K. G., Starr F. W., Tkachenko A. V., Gang O., Diamond family of nanoparticle superlattices. Science 351, 582–586 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glotzer S. C., Solomon M. J., Anisotropy of building blocks and their assembly into complex structures. Nat. Mater. 6, 557–562 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Damasceno P. F., Engel M., Glotzer S. C., Predictive self-assembly of polyhedra into complex structures. Science 337, 453–457 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Mundoor H., Senyuk B., Smalyukh I. I., Triclinic nematic colloidal crystals from competing elastic and electrostatic interactions. Science 352, 69–73 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Niederberger M., Cölfen H., Oriented attachment and mesocrystals: Non-classical crystallization mechanisms based on nanoparticle assembly. Phys. Chem. Chem. Phys. 8, 3271–3287 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Wang T., Zhuang J., Lynch J., Chen O., Wang Z., Wang X., LaMontagne D., Wu H., Wang Z., Cao Y. C., Self-assembled colloidal superparticles from nanorods. Science 338, 358–363 (2012). [DOI] [PubMed] [Google Scholar]
- 21.Chiu C.-Y., Chen C.-K., Chang C.-W., Jeng U.-S., Tan C.-S., Yang C.-W., Chen L.-J., Yen T.-J., Huang M. H., Surfactant-directed fabrication of supercrystals from the assembly of polyhedral Au–Pd core–shell nanocrystals and their electrical and optical properties. J. Am. Chem. Soc. 137, 2265–2275 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Henzie J., Grünwald M., Widmer-Cooper A., Geissler P. L., Yang P., Self-assembly of uniform polyhedral silver nanocrystals into densest packings and exotic superlattices. Nat. Mater. 11, 131–137 (2012). [DOI] [PubMed] [Google Scholar]
- 23.O’brien M. N., Jones M. R., Lee B., Mirkin C. A., Anisotropic nanoparticle complementarity in DNA-mediated co-crystallization. Nat. Mater. 14, 833–839 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Jones M. R., Macfarlane R. J., Lee B., Zhang J., Young K. L., Senesi A. J., Mirkin C. A., DNA-nanoparticle superlattices formed from anisotropic building blocks. Nat. Mater. 9, 913–917 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Boneschanscher M. P., Evers W. H., Geuchies J. J., Altantzis T., Goris B., Rabouw F. T., van Rossum S. A. P., van der Zant H. S. J., Siebbeles L. D. A., Van Tendeloo G., Swart I., Hilhorst J., Petukhov A. V., Bals S., Vanmaekelbergh D., Long-range orientation and atomic attachment of nanocrystals in 2D honeycomb superlattices. Science 344, 1377–1380 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Evers W. H., Goris B., Bals S., Casavola M., de Graaf J., van Roij R., Dijkstra M., Vanmaekelbergh D., Low-dimensional semiconductor superlattices formed by geometric control over nanocrystal attachment. Nano Lett. 13, 2317–2323 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Feng W., Kim J.-Y., Wang X., Calcaterra H. A., Qu Z., Meshi L., Kotov N. A., Assembly of mesoscale helices with near-unity enantiomeric excess and light-matter interactions for chiral semiconductors. Sci. Adv. 3, e1601159 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carné-Sánchez A., Imaz I., Cano-Sarabia M., Maspoch D., A spray-drying strategy for synthesis of nanoscale metal–organic frameworks and their assembly into hollow superstructures. Nat. Chem. 5, 203–211 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Flauraud V., Mastrangeli M., Bernasconi G. D., Butet J., Alexander D. T. L., Shahrabi E., Martin O. J. F., Brugger J., Nanoscale topographical control of capillary assembly of nanoparticles. Nat. Nanotechnol. 12, 73–80 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Chakrabarty R., Mukherjee P. S., Stang P. J., Supramolecular coordination: Self-assembly of finite two- and three-dimensional ensembles. Chem. Rev. 111, 6810–6918 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cook T. R., Zheng Y.-R., Stang P. J., Metal–organic frameworks and self-assembled supramolecular coordination complexes: Comparing and contrasting the design, synthesis, and functionality of metal–organic materials. Chem. Rev. 113, 734–777 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zangrando E., Casanova M., Alessio E., Trinuclear metallacycles: Metallatriangles and much more. Chem. Rev. 108, 4979–5013 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Würthner F., You C.-C., Saha-Moller C. R., Metallosupramolecular squares: From structure to function. Chem. Soc. Rev. 33, 133–146 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Stang P. J., Persky N. E., Manna J., Molecular architecture via coordination: Self-assembly of nanoscale platinum containing molecular hexagons. J. Am. Chem. Soc. 119, 4777–4778 (1997). [Google Scholar]
- 35.Ward M. D., Polynuclear coordination cages. Chem. Commun. 0, 4487–4499 (2009). [DOI] [PubMed] [Google Scholar]
- 36.Lang J.-P., Xu Q.-F., Chen Z.-N., Abrahams B. F., Assembly of a supramolecular cube, [(Cp*WS3Cu3)8Cl8(CN)12Li4] from a preformed incomplete cubane-like compound [PPh4][Cp*WS3(CuCN)3]. J. Am. Chem. Soc. 125, 12682–12683 (2003). [DOI] [PubMed] [Google Scholar]
- 37.Levin M. D., Stang P. J., Insights into the mechanism of coordination-directed self-assembly. J. Am. Chem. Soc. 122, 7428–7429 (2000). [Google Scholar]
- 38.Sun Q.-F., Iwasa J., Ogawa D., Ishido Y., Sato S., Ozeki T., Sei Y., Yamaguchi K., Fujita M., Self-assembled M24L48 polyhedra and their sharp structural switch upon subtle ligand variation. Science 328, 1144–1147 (2010). [DOI] [PubMed] [Google Scholar]
- 39.He Y., Ye T., Su M., Zhang C., Ribbe A. E., Jiang W., Mao C., Hierarchical self-assembly of DNA into symmetric supramolecular polyhedra. Nature 452, 198–201 (2008). [DOI] [PubMed] [Google Scholar]
- 40.Lai Y.-T., Reading E., Hura G. L., Tsai K.-L., Laganowsky A., Asturias F. J., Tainer J. A., Robinson C. V., Yeates T. O., Structure of a designed protein cage that self-assembles into a highly porous cube. Nat. Chem. 6, 1065–1071 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang L., Wu H. B., Lou X. W., Metal–organic-frameworks-derived general formation of hollow structures with high complexity. J. Am. Chem. Soc. 135, 10664–10672 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Yu X.-Y., Yu L., Wu H. B., Lou X. W., Formation of nickel sulfide nanoframes from metal–organic frameworks with enhanced pseudocapacitive and electrocatalytic properties. Angew. Chem. Int. Ed. 54, 5331–5335 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Han L., Yu X.-Y., Lou X. W., Formation of Prussian-blue-analog nanocages via a direct etching method and their conversion into Ni–Co-mixed oxide for enhanced oxygen evolution. Adv. Mater. 28, 4601–4605 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Sato O., Iyoda T., Fujishima A., Hashimoto K., Photoinduced magnetization of a cobalt-iron cyanide. Science 272, 704–705 (1996). [DOI] [PubMed] [Google Scholar]
- 45.Hoshino N., Iijima F., Newton G. N., Yoshida N., Shiga T., Nojiri H., Nakao A., Kumai R., Murakami Y., Oshio H., Three-way switching in a cyanide-bridged [CoFe] chain. Nat. Chem. 4, 921–926 (2012). [DOI] [PubMed] [Google Scholar]
- 46.Higel P., Villain F., Verdaguer M., Rivière E., Bleuzen A., Solid-state magnetic switching triggered by proton-coupled electron-transfer assisted by long-distance proton-alkali cation transport. J. Am. Chem. Soc. 136, 6231–6234 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Cafun J.-D., Champion G., Arrio M.-A., Cartier dit Moulin C., Bleuzen A., Photomagnetic CoFe Prussian blue analogues: Role of the cyanide ions as active electron transfer bridges modulated by cyanide–alkali metal ion interactions. J. Am. Chem. Soc. 132, 11552–11559 (2010). [DOI] [PubMed] [Google Scholar]
- 48.Bleuzen A., Escax V., Ferrier A., Villain F., Verdaguer M., Münsch P., Itié J.-P., Thermally induced electron transfer in a CsCoFe Prussian blue derivative: The specific role of the alkali-metal ion. Angew. Chem. Int. Ed. 43, 3728–3731 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Cafun J.-D., Lejeune J., Baudelet F., Dumas P., Itié J.-P., Bleuzen A., Room-temperature photoinduced electron transfer in a Prussian blue analogue under hydrostatic pressure. Angew. Chem. Int. Ed. 51, 9146–9148 (2012). [DOI] [PubMed] [Google Scholar]
- 50.Pintado S., Goberna-Ferrón S., Escudero-Adán E. C., Galán-Mascarós J. R., Fast and persistent electrocatalytic water oxidation by Co–Fe Prussian blue coordination polymers. J. Am. Chem. Soc. 135, 13270–13273 (2013). [DOI] [PubMed] [Google Scholar]
- 51.Xiong Y., Cai H., Wiley B. J., Wang J., Kim M. J., Xia Y., Synthesis and mechanistic study of palladium nanobars and nanorods. J. Am. Chem. Soc. 129, 3665–3675 (2007). [DOI] [PubMed] [Google Scholar]
- 52.Weinberg D. R., Gagliardi C. J., Hull J. F., Murphy C. F., Kent C. A., Westlake B. C., Paul A., Ess D. H., McCafferty D. G., Meyer T. J., Proton-coupled electron transfer. Chem. Rev. 112, 4016–4093 (2012). [DOI] [PubMed] [Google Scholar]
- 53.Ohkoshi S.-i., Arai K.-i., Sato Y., Hashimoto K., Humidity-induced magnetization and magnetic pole inversion in a cyano-bridged metal assembly. Nat. Mater. 3, 857–861 (2004). [DOI] [PubMed] [Google Scholar]
- 54.Catala L., Brinzei D., Prado Y., Gloter A., Stéphan O., Rogez G., Mallah T., Core–multishell magnetic coordination nanoparticles: Toward multifunctionality on the nanoscale. Angew. Chem. Int. Ed. 48, 183–187 (2009). [DOI] [PubMed] [Google Scholar]
- 55.Hu M., Belik A. A., Imura M., Yamauchi Y., Tailored design of multiple nanoarchitectures in metal-cyanide hybrid coordination polymers. J. Am. Chem. Soc. 135, 384–391 (2013). [DOI] [PubMed] [Google Scholar]
- 56.Zhang J., Huang F., Lin Z., Progress of nanocrystalline growth kinetics based on oriented attachment. Nanoscale 2, 18–34 (2010). [DOI] [PubMed] [Google Scholar]
- 57.Roche S., Haslam C., Heath S. L., Thomas J. A., Self-assembly of a supramolecular cube. Chem. Commun. 0, 1681–1682 (1998). [Google Scholar]
- 58.Li D., Nielsen M. H., Lee J. R. I., Frandsen C., Banfield J. F., De Yoreo J. J., Direction-specific interactions control crystal growth by oriented attachment. Science 336, 1014–1018 (2012). [DOI] [PubMed] [Google Scholar]
- 59.Lv W., He W., Wang X., Niu Y., Cao H., Dickerson J. H., Wang Z., Understanding the oriented-attachment growth of nanocrystals from an energy point of view: A review. Nanoscale 6, 2531–2547 (2014). [DOI] [PubMed] [Google Scholar]
- 60.Agarwal U., Escobedo F. A., Mesophase behaviour of polyhedral particles. Nat. Mater. 10, 230–235 (2011). [DOI] [PubMed] [Google Scholar]
- 61.González E., Arbiol J., Puntes V. F., Carving at the nanoscale: Sequential galvanic exchange and Kirkendall growth at room temperature. Science 334, 1377–1380 (2011). [DOI] [PubMed] [Google Scholar]
- 62.Samain L., Grandjean F., Long G. J., Martinetto P., Bordet P., Strivay D., Relationship between the synthesis of Prussian blue pigments, their color, physical properties, and their behavior in paint layers. J. Phys. Chem. C 117, 9693–9712 (2013). [Google Scholar]
- 63.Liang Y., Li Y., Wang H., Zhou J., Wang J., Regier T., Dai H., Co3O4 nanocrystals on graphene as a synergistic catalyst for oxygen reduction reaction. Nat. Mater. 10, 780–786 (2011). [DOI] [PubMed] [Google Scholar]
- 64.Trotochaud L., Ranney J. K., Williams K. N., Boettcher S. W., Solution-cast metal oxide thin film electrocatalysts for oxygen evolution. J. Am. Chem. Soc. 134, 17253–17261 (2012). [DOI] [PubMed] [Google Scholar]
- 65.Lee H.-W., Wang R. Y., Pasta M., Woo Lee S., Liu N., Cui Y., Manganese hexacyanomanganate open framework as a high-capacity positive electrode material for sodium-ion batteries. Nat. Commun. 5, 5280 (2014). [DOI] [PubMed] [Google Scholar]
- 66.Song F., Hu X., Ultrathin cobalt–manganese layered double hydroxide is an efficient oxygen evolution catalyst. J. Am. Chem. Soc. 136, 16481–16484 (2014). [DOI] [PubMed] [Google Scholar]
- 67.Martínez-García R., Knobel M., Balmaseda J., Yee-Madeira H., Reguera E., Mixed valence states in cobalt iron cyanide. J. Phys. Chem. Solids 68, 290–298 (2007). [Google Scholar]
- 68.Wang D., Chen X., Evans D. G., Yang W., Well-dispersed Co3O4/Co2MnO4 nanocomposites as a synergistic bifunctional catalyst for oxygen reduction and oxygen evolution reactions. Nanoscale 5, 5312–5315 (2013). [DOI] [PubMed] [Google Scholar]
- 69.Gao M.-R., Xu Y.-F., Jiang J., Zheng Y.-R., Yu S.-H., Water oxidation electrocatalyzed by an efficient Mn3O4/CoSe2 nanocomposite. J. Am. Chem. Soc. 134, 2930–2933 (2012). [DOI] [PubMed] [Google Scholar]
- 70.Grewe T., Deng X., Weidenthaler C., Schüth F., Tüysüz H., Design of ordered mesoporous composite materials and their electrocatalytic activities for water oxidation. Chem. Mater. 25, 4926–4935 (2013). [Google Scholar]
- 71.Chen S., Qiao S.-Z., Hierarchically porous nitrogen-doped graphene–NiCo2O4 hybrid paper as an advanced electrocatalytic water-splitting material. ACS Nano 7, 10190–10196 (2013). [DOI] [PubMed] [Google Scholar]
- 72.Liu X., Chang Z., Luo L., Xu T., Lei X., Liu J., Sun X., Hierarchical ZnxCo3–xO4 nanoarrays with high activity for electrocatalytic oxygen evolution. Chem. Mater. 26, 1889–1895 (2014). [Google Scholar]
- 73.Xiao C., Lu X., Zhao C., Unusual synergistic effects upon incorporation of Fe and/or Ni into mesoporous Co3O4 for enhanced oxygen evolution. Chem. Commun. 50, 10122–10125 (2014). [DOI] [PubMed] [Google Scholar]
- 74.Li Y., Hasin P., Wu Y., NixCo3−xO4 nanowire arrays for electrocatalytic oxygen evolution. Adv. Mater. 22, 1926–1929 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/8/e1700732/DC1
fig. S1. Characterization of the KCoFe-1 NAFSs.
fig. S2. Overview FESEM image of the KCoFe-1 NAFSs.
fig. S3. FESEM images of two NAFSs.
fig. S4. Characterization of the KCoFe-2 NCs.
fig. S5. XRD patterns of different samples.
fig. S6. FESEM image of the product obtained at 9 hours of the reaction process.
fig. S7. TEM and EDX mapping images of a typical core@satellite superstructure.
fig. S8. FTIR spectra of different samples.
fig. S9. TEM and HRTEM images of a typical core@satellite superstructure.
fig. S10. Model of the cuboctahedral frame-like superstructure.
fig. S11. Characterization of the NAFSs with different sizes.
fig. S12. Overview FESEM image of the KCoFe-3 NFs.
fig. S13. Characterization of the KCoFe-3 NFs.
fig. S14. FESEM images of the samples obtained from different stages of the growth of NFs.
fig. S15. XRD patterns of the samples derived from KCoFe-1 NAFSs and KCoFe-3 NFs.
fig. S16. Overview FESEM images of the Co-Fe oxide composites.
fig. S17. Estimation of the ECSA of the catalysts.
fig. S18. Reference electrode calibration in 1.0 M KOH solution.
table S1. Observations from the FESEM images for different samples.
table S2. FTIR peak positions of the samples of KCoFe-1, KCoFe-2, and KCoFe-3 and their assignment.
table S3. Some typical examples of the relationship of the type of attachment facets, attachment angle, normal vector geometry, the number of attachment facet of a certain NP, and possible attachment motif (symmetry) in a cubic system.
table S4. Comparison of Co-Fe mixed oxide NAFSs and NFs prepared in this work with some recently reported transition metal oxide–based catalysts.