

Abstract
Selective A3 adenosine receptor agonists have been shown to induce apoptosis in a variety of cell types. In this study we examined the effects of adenosine receptor agonists selective for A1, A2A, or A3 receptors on the induction of apoptosis in primary cultures of rat astrocytes and in C6 glial cells. Treatment of the cells with the A3 receptor agonist Cl-IB-MECA (10 μM) induced apoptosis in both cell types. The effects of Cl-IB-MECA were partially antagonized by the A3 receptor-selective antagonist MRS 1191. In contrast, the A1 and A2A receptor agonists, CPA and CGS 21680, respectively, did not have significant effects on apoptosis in these cells. Cl-IB-MECA reduced the expression of endogenous Bcl-2, whereas it did not affect the expression of Bax. Overexpression of Bcl-2 in C6 cells abrogated the induction of apoptosis induced by the A3 agonist. Cl-IB-MECA also induced an increase in caspase 3 activity and caspase inhibitors decreased the apoptosis induced by the A3 agonist. These findings suggest that intense activation of the A3 receptor is pro-apoptotic in glial cells via bcl2 and caspase-3 dependent pathways.
Index Entries: Astrocytes, agonists, adenosine receptor, apoptosis, Bcl-2
Introduction
Adenosine, which acts as a neurotransmitter in the brain, activates four specific G-protein-coupled receptors: the A1, A2A, A2B, and A3 receptors (Fredholm et al., 1994). The A1 receptor has been shown to mediate neuroprotection (MacGregor et al., 1993; Schubert et al., 1997; Von Lubitz et al., 1999), via blockade of Ca2+ influx, which results in inhibition of glutamate release and reduction of its excitatory effects. Similarly, the selective A2A receptor antagonists (Ongini et al., 1997) have been demonstrated to markedly reduce cell death associated with brain ischemia in the rat. In contrast, activation of the A3 receptor can act as either pro- or anti-apoptotic signal (Ceruti et al., 1996; Yao et al., 1997), depending on the degree of receptor activation and the specific pathophysiological conditions.
Selective A3 adenosine receptor agonists have been shown to induce apoptosis in a variety of cell cultures. Selective adenosine receptor agonists and antagonists have marked effects on the outcome of models of cerebral ischemia (Von Lubitz et al., 1994; Ongini et al., 1997). Acute administration of the selective A1 agonists, ADAC and CPA (Von Lubitz et al., 1999), or chronic administration of the selective A3 agonist, IB-MECA (Von Lubitz et al., 1994), improved hippocampal cell survival following global ischemia in gerbils, and reduced lethality and behavioral and learning impairment. Astroglial adenosine receptors are potentially involved in these effects.
Glial cells express functional adenosine receptors of all four subtypes. Activation of A1 receptors was found to inhibit adenylyl cyclase in type-2 rat astrocytes (Peakman and Hill, 1996), and activation of A2A receptors stimulated adenylate cyclase in astrocytes and inhibited the induction of nitric oxide induced by Lipopoly Saccharide (LPS)/interferon-γ (IFN-γ) (Brodie et al., 1998a). A3 selective agonists, IB-MECA and Cl-IB-MECA, were found to induce morphological changes in primary rat astroglial cultures and to induce cell spreading and reorganization of the actin cytoskeleton of ADF human astrocytoma cells (Ceruti et al., 1996; Abbracchio et al., 1997).
In this study we examined the effects of various adenosine agonists on the induction of apoptosis in primary astrocytes and in C6 glial cells and the role of Bcl-2 and Bax in these effects. Our results indicate that the A3 agonist Cl-IB-MECA at micromolar concentrations induces apoptosis and exert differential effects on the expression of Bcl2 and Bax. In addition, we found that overexpression of Bcl-2 and inhibition of caspase 3 activity suppressed the apoptotic effects of Cl-IB-MECA in glial cells.
Materials and Methods
Materials
CPA, CPX, CSC, MRS 1191 (Jacobson et al., 1997), and CGS 21680 were obtained from Sigma-RBI (St. Louis, MO). Cl-IB-MECA was synthesized as reported (Kim et al., 1994). Anti-Bcl2 and anti-Bax antibodies were obtained from Transduction Laboratories (Lexington, KY). The vector pCMV5, which contains the full-length mouse bcl2, was a gift from Dr. Reuven Stein (Tel-Aviv University, Israel).
Glial Cell Cultures
Primary glial cultures were obtained from cerebral cortices of 2-d-old newborn rats as previously described (Brodie et al., 1997). Following the removal of the meninges, the hemispheres were mechanically dissociated with fire-polished Pasteur pipets. The growth medium consisted of Dulbecco’s modified Eagle’s medium (DMEM) containing 10% heat-inactivated fetal calf serum (FCS), 2 mM glutamine, penicillin (50 U/mL), and streptomycin (0.05 mg/mL). Cells were plated into plastic petri dishes (Nunc) and incubated at 37°C in a humidified 5% CO2/95% air atmosphere. After 12–14 d, confluent cultures were shaken for 4 h at 250 rpm and the medium was discarded. This procedure was repeated three times. The adherent astrocytes were removed by trypsinization and replated with a density of 106 cells/mL. The secondary cultures consisted of highly purified astrocytes as determined by GFAP staining and of less than 1% microglia as determined by MAC-1 immunostaining.
C6 cells of late passages (50–60), which exhibit astrocytic phenotypes, were used in this study. Cells (1 × 105 cells/mL) were seeded on tissue-culture dishes (10 cm) and were grown in medium consisting of DMEM containing 10% heat-inactivated FCS, 2 mM glutamine, penicillin (50 U/mL), and streptomycin (0.05 mg/mL). Cells were split once a week by short exposure to 0.25% trypsin.
Measurement of Apoptosis
Cell apoptosis was measured using Hoechst staining and by enzyme-linked immunosorbent assay (ELISA) using anti-histone1 antibodies. Cells (1 × 106/mL) were plated in 6-well plates and treated with the indicated treatments for 24 h. For Hoechst staining, cells were fixed with methanol and incubated for 10 min with 1 μg/mL Hoechst 33258. Cells were then viewed and counted under UV illumination for the visualization of the Hoechst stained nuclei. For anti-histone1 ELISA, fragmented DNA was extracted from the cells and was incubated in 96-well plates coated with anti-histone antibodies for 2 h. Plates were then washed and incubated with anti-DNA antibodies conjugated to peroxidase for an additional 2 h. Substrate solution was added and absorbance was measured at 405 nm.
Caspase-3 Activity
Caspase-3 activity was measured using the ApoAlert CPP32 Colorimetric Assay Kit according to the manufacturer’s instructions (CLONTECH Laboratories Inc.).
Cell Transfection
C6 Cells (1 × 105 cells/mL) were seeded on tissue culture dishes (10 cm) and were grown in medium consisting of DMEM containing 10% heat-inactivated FCS, 2 mM glutamine, penicillin (50 U/mL), and streptomycin (0.05 mg/mL). The cells were transfected either with the empty vector or with the Bcl-2 expression vectors using LipofectAMINE (Gibco-BRL Life Technologies, Gaithersburg, MD) according to the manufacturer’s instructions. Experiments were routinely carried out on two clones of the transfected cells, but all the results were confirmed on one pool and two different individual clones.
Preparation of Cell Homogenates
Cells were washed and resuspended in serum-free medium. The plates were placed on ice, scraped with a rubber policeman, and centrifuged at 1,400 rpm for 10 min. The supernatants were aspirated and the cell pellets were resuspended in 100 μL of lysis buffer (25 mM Tris-HCl, pH 7.4, 50 mM NaCl, 0.5% Na deoxycholate; 2% NP-40; 0.2% SDS; 1 mM PMSF; 50 μg/mL aprotinin; 50 μM leupeptin; 0.5 mM Na3VO4) on ice for 15 min. The cell lysates were centrifuged for 15 min at 14,000 rpm in an Eppendorf microcentrifuge, supernatants were removed, and 2X sample buffer was added.
Immunoblot Analysis
Lysates (20 μg protein) were resolved by SDS-PAGE (12%) and were transferred to nitrocellulose membranes. The membranes were blocked with 5% dry milk in phosphate-buffered saline and subsequently stained with the primary antibody (mouse Bcl-2). Specific reactive bands were detected using a goat anti-mouse IgG conjugated to horseradish peroxidase (BioRad, Hercules, CA), and the immunoreactive bands were visualized using an ECL Western blotting detection kit (Amersham, Arlington Heights, IL).
Immunoprecipitation
Immunoprecipitation of Bcl-2 was performed as previously described (Brodie et al., 1998b). Briefly, C6 cells overexpressing Bcl-2 were treated with Cl-IB-MEAC (25 μM) for 12 and 24 h. The samples were preabsorbed with 25 μLProtein A/G-Sepharose (50%) for 10 min and immunoprecipitation was performed using 4 μg/mL anti-Bcl-2 antibody and 30 μL of A/G Sepharose at 4°C. Following washes, the pellets were resuspended in 25 μL SDS sample buffer and boiled for 5 min. The entire supernatants were subjected to Western blotting. Membranes were probed with an anti-Bcl-2 antibody.
Results
Induction of Apoptosis by A3 Adenosine Receptor Agonist
Primary cultures of astrocytes and C6 glial cells were treated with adenosine agonists selective for A1, A2A, or A3 receptors for 24–48 h, and the induction of apoptosis was determined as indicated by anti-histone ELISA and by Hoechst staining. Treatment of both cell types with the A3 receptor agonist Cl-IB-MECA (10–50 μM) stimulated apoptosis in a concentration-dependent manner albeit to a different degree (Fig. 1A, B). Thus, Cl-IB-MECA at a concentration of 10 μM induced 46% apoptosis in C6 glial cells, whereas it induced only 17% apoptosis in primary astrocytes. Maximal apoptotic effects were observed with 50 μM Cl-IB-MECA. The apoptotic effects were partially blocked by the A3-selective antagonist MRS 1191 (10 μM), a dihydropyridine derivative known to be weak at rat A3 receptors but moderately selective. The A1 and A2A receptor agonists, CPA (10 μM) and CGS 21680 (10 μM), respectively, did not have significant effects on the apoptosis of either primary astrocytes or C6 cells (data not shown).
Fig. 1.
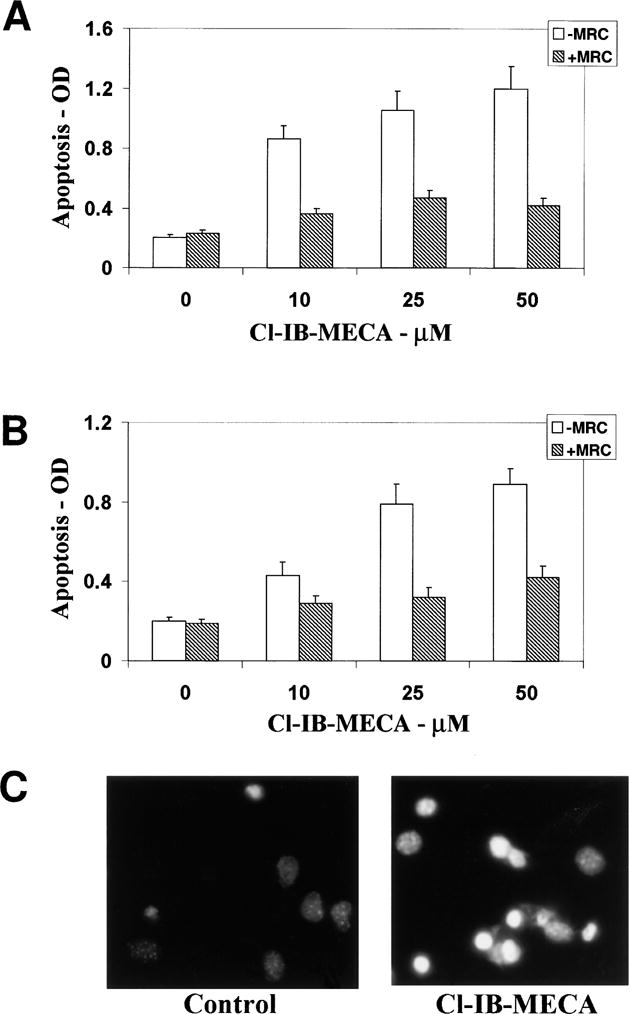
Effects of the A3 receptor agonist Cl-IB-MECA on apoptosis in C6 glial cells. C6 cells (A) and astrocytes (B) were treated with different concentrations of Cl-IB-MECA for 48 h in the presence and absence of the A3 antagonist MRS 1191 (10 μM). Apoptosis was measured by ELISA using anti-histone antibodies. Fragmented DNA was extracted from the cells and was incubated in 96-well plates coated with anti-histone antibodies for 2 h followed by incubation with anti-DNA antibodies conjugated to peroxidase for an additional 2 h. Absorbance was measured at 405 nm. Apoptosis of primary astrocytes was also measured using Hoechst staining (C). Astrocytes were treated with 50 μM Cl-IB-MECA for 48 h and were stained with Hoechst as described in Methods. Results represent the means ± SD of three separate experiments.
Differential Effects of the A3 Adenosine Receptor Agonist on Bcl-2 and Bax Expression
To study the mechanisms involved in the apoptotic effects of Cl-IB-MECA, we examined the effects of the A3 agonist on the expression of the apoptotic-related proteins, Bcl-2, and Bax. Primary astrocytes or C6 cells were treated with 25 μM Cl-IB-MECA for 12 and 24 h and the expression of Bcl-2 and Bax was measured using immunoprecipitation and Western blot analysis, respectively. As presented in Fig. 2, untreated astrocytes and C6 cells expressed basal levels of Bcl-2 and Bax. Treatment of astrocytes with Cl-IB-MECA induced a marked decease in Bcl-2 expression after 12 h of treatment, and a further decrease was observed after 24 h. Similar results were observed in C6 cells, except that the basal level of Bcl2 was higher in these cells as compared to that of astrocytes. In contrast, treatment of astrocytes and C6 cells with Cl-IB-MECA for 12 and 24 h did not induce significant changes in the expression of Bax in either cell type.
Fig. 2.
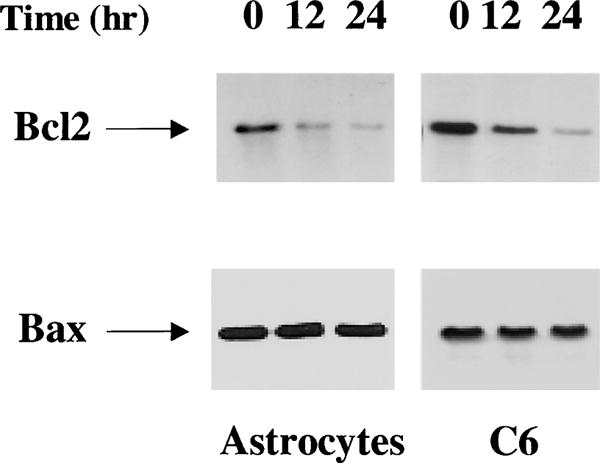
Effect of the A3 receptor agonist Cl-IB-MECA on the expression of Bcl-2 and Bax. Cells were treated with Cl-IB-MECA (25 μM) for 12 and 24 h. The cells were then harvested and immunoprecipitation of Bcl-2 was performed as described in the Methods or cells were extracted for Western-blot analysis. Following SDS-PAGE, membranes were stained with anti-Bcl-2 antibody or with anti-Bax antibody. The results represent one of three separate experiments, which gave similar results.
Overexpression of Bcl-2 Reduced Cell Apoptosis Induced by Cl-IB-MECA
To further study the role of Bcl-2 in the apoptotic effects of Cl-IB-MECA, we overexpressed Bcl-2 in C6 cells and examined the effects of Cl-IB-MECA. C6 cells were stably transfected with an empty vector and with Bcl-2. Figure 3A illustrates a representative Western blot of C6 cells overexpressing Bcl-2 and the vector control. The levels of the overexpressed Bcl-2 in the transfected cells were 10–15-fold higher than the endogenous Bcl-2, as determined using an anti-Bcl-2 specific antibody (Fig. 3A).
Fig. 3.
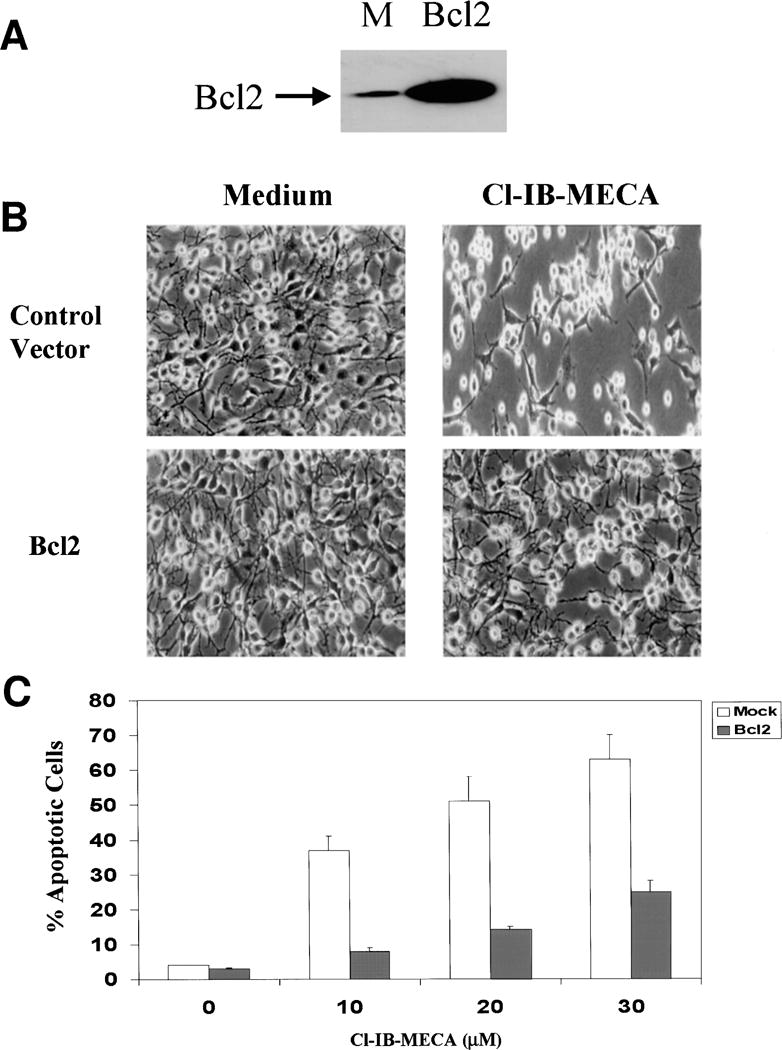
Effect of Bcl-2 overexpression on the apoptosis induced by the A3 receptor agonist Cl-IB-MECA. Stable transfectants of Bcl-2 or control vector were harvested and subjected to SDS-PAGE and Western blot analysis. Membranes were probed with anti-Bcl-2 antibody (A). Cells overexpressing Bcl2 or empty vector were treated with Cl-IB-MECA for 48 h and the morphology of the cells was examined using a phase-contrast light microscope (B). Cell apoptosis was determined using Hoechst staining and the apoptotic cells were viewed and counted under UV illumination (C). The results represent means ± SD from three separate experiments.
C6 cells overexpressing the anti-apoptotic protein Bcl-2 exhibited similar morphology to cells overexpressing empty vector. Treatment of empty vector-transfected cells with the A3 agonist resulted in lower cell number and in the appearance of rounded and detached cells, which are characteristic of apoptotic cells. In contrast, treatment of cells overexpressing Bcl-2 with the A3 agonist exhibited morphology similar to untreated cells (Fig. 3B). Similar results were obtained when apoptosis was measured using Hoechst Staining. Thus, the A3 agonist induced apoptosis in only 22% of the Bcl-2-overexpressing cells following 48 h of treatment (Fig. 3C), whereas it induced apoptosis in 66% of the empty vector-transfected cells.
Caspase 3 is Involved in the Apoptotic Effect of Cl-IB-MECA
We also examined the role of caspase 3 in the apoptotic effect of Cl-IB-MECA. We found that treatment of C6 cells and astrocytes with Cl-IB-MECAinduced a marked increase in the activity of caspase 3. This effect was observed after 8 h of treatment. The increased activity of caspase 3 in the Cl-IB-MECA treated cells was partially blocked by the A3 antagonist (data not shown). To further explore the role of caspase 3 in the apoptosis induced by Cl-IB-MECA, we examined the effect of the caspase inhibitors DEVD.FMK and Z-VAD.FMK on cell apoptosis and found that pre-treatment of the cells with these inhibitors significantly decreased the apoptotic response induced by Cl-IB-MECA (data not shown).
Discussion
In this study we examined the effect of A3 receptor activation on the apoptosis of glial cells and the role of Bcl-2 in this effect. Our results demonstrate that treatment of glial cells with μM concentrations of the selective A3 receptor agonist, Cl-IB-MECA, induced apoptosis in C6 glial cells. The apoptotic effect was observed only with activation of the A3 receptor, whereas activation of the A1 or A2A receptors did not induce significant apoptotic effects.
Activation of A3 receptors may either promote apoptosis (micromolar agonist concentrations) or protect against apoptosis (nanomolar agonist concentrations) in a variety of cell lines (Yao et al., 1997). Indeed, at concentrations higher than 10 μM, the A3 selective agonist Cl-IB-MECA induced cell death in rat cerebellar granule neurons in primary culture in a concentration-dependent fashion, whereas agonists of A1, A2A, and A2B receptors (CPA, CGS21680, and NECA) had no effect on neuronal cell apoptosis (Abbracchio et al., 1997). In addition, subcytotoxic concentration of Cl-IB-MECA (1 μM) significantly augmented the receptor-mediated neurotoxicity of 50 μM glutamate. In ADF astrocytoma cells, activation of the A3 receptor by 200 nM Cl-IB-MECAresulted in formation of stress fibers and elongation of astrocytic processes (Abbracchio et al., 1997). This was accompanied by changes in the intracellular distribution of the antiapoptotic protein Bcl-XL, i.e., from the perinuclear area to cell protrusions. Similarly, Cl-IB-MECAinduced apoptosis of rat cardiomyocytes, and this effect was antagonized by β-adrenergic stimulation (Shneyvays et al., 2000).
Apoptosis is controlled by pro-apoptotic and anti-apoptotic proteins. The cellular oncogene Bcl-2, which encodes an outer mitochondrial membrane protein of Mr26,000 has been shown to delay or block apoptosis induced by numerous, often unrelated physiological and pathological stimuli. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death (Korsmeyer, 1999). We found that Cl-IB-MECA decreased the expression of Bcl-2 in astrocytes and C6 cells, whereas it did not affect the expression of Bax in these cells. These results suggest that activation of A3 receptor evokes signals that are involved in the regulation of Bcl-2. There have been a number of studies showing changes in endogenous levels of Bcl-2 in response to apoptotic stimuli. For example, both p53 and transforming growth factor-β1 (TGF-β1) downregulated endogenous Bcl-2 expression (Miyashita et al., 1994; Selvakumaran et al., 1994). In addition, the expression of HIV-1 tat protein induced downregulation of Bcl-2 at both the transcriptional and translational levels (Sastry et al., 1996). The signals involved in the inhibitory effect of A3 receptor on Bcl-2 expression are currently being explored.
Overexpression of Bcl-2 protected C6 cells from Cl-IB-MECA-induced apoptosis, suggesting that the inhibitory effect of Cl-IB-MECA on endogenous Bcl-2 expression is involved in the apoptotic effect of this drug on C6 cells. The incomplete protection by the exogenous Bcl-2 can be due to the induction of pro-apoptotic proteins or the activation of caspases, which cleave Bcl2 by Cl-IB-MECA. Indeed, we found that Cl-IB-MECA induced activation of caspase 3 in C6 cells and that inhibition of caspase 3 abolished the apoptosis induced by Cl-IB-MECA.
The biological significance of adenosine A3 receptors in neuronal systems has been discussed. Activation of A3 receptors elicits depression of locomotor activity in mice (Jacobson et al., 1993), and plays a significant role in the generation of postischemic brain damage in gerbils (Von Lubitz et al., 1994) and in a seizure model (Von Lubitz et al., 1995). A3 agonists show dual regulation of astrocyte cell viability (Ceruti et al., 1996), in which changes in structural components of the cytoskeleton and intracellular distribution of Bcl-XL were found (Abbracchio et al., 1997). It was also suggested that A3 receptors function in brain in conjunction with other adenosine receptor subtypes (Dunwiddie et al., 1997).
In summary, the A3 adenosine agonist Cl-IB-MECA exerts apoptotic effects in gial cells, in which the anti-apoptotic protein Bcl-2 and caspase 3 are apparently involved. The effects of Cl-IB-MECA were partially antagonized by the A3 receptor-selective antagonist MRS 1191, suggesting that the apoptotic effects of this adenosine derivative in C6 cells were largely receptor-mediated. Thus, while multiple subtypes of adenosine receptors are present on glial cells, only the A3 receptor has a critical role in apoptosis, suggesting this receptor as a prospective therapeutic target in pathological conditions of the brain.
Fig. 4.
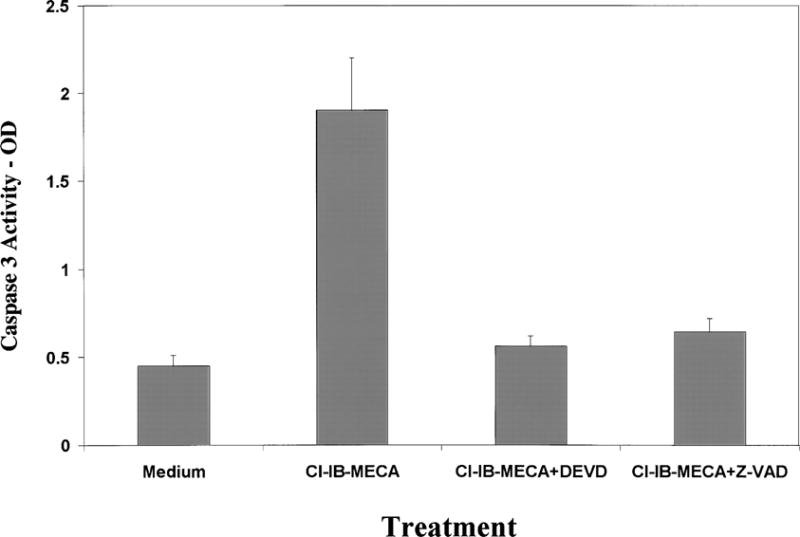
Caspase 3 activity. Primary astrocytes or C6 cells treated with 25 μM for 12 h and the activity of caspase 3 (CPP32) was measured using spectrophotometric detection of the cleaved product of the labeled substrate DEVD-p-nitroanilide. The results represent the means ± SD from three separate experiments.
References
- Abbracchio MP, Rainaldi G, Giammarioli AM, Ceruti S, Brambilla R, Cattabeni F, et al. The A3 adenosine receptor mediates cell spreading, reorganization of actin cytoskeleton, and distribution of Bcl-XL: studies in human astroglioma cells. Biochem Biophys Res Commun. 1997;241:297–304. doi: 10.1006/bbrc.1997.7705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodie C, Blumberg PM, Jacobson KA. Activation of the A2A adenosine receptor inhibits nitric oxide production in glial cells. FEBS Lett. 1998a;429:139–142. doi: 10.1016/s0014-5793(98)00556-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodie C, Bogi K, Acs P, Lorenzo PS, Baskin L, Blumberg PM. Protein kinase C delta (PKCdelta) inhibits the expression of glutamine synthetase in glial cells via the PKCdelta regulatory domain and its tyrosine phosphorylation. J Biol Chem. 1998b;273:30,713–30,718. doi: 10.1074/jbc.273.46.30713. [DOI] [PubMed] [Google Scholar]
- Brodie C, Weizman N, Katzoff A, Lustig S, Kobiler D. Astrocyte activation by Sindbis virus: expression of GFAP, cytokines, and adhesion molecules. Glia. 1997;19:275–285. [PubMed] [Google Scholar]
- Ceruti S, Barbieri D, Franceschi C, Giammarioli AM, Rainaldi G, Malorni W, et al. Effects of adenosine A3 receptor agonists on astrocytes: induction of cell protection at low and cell death at high concentration. Drug Dev Res. 1996;37:177. [Google Scholar]
- Dunwiddie TV, Diao L, Kim HO, Jiang JL, Jacobson KA. Activation of hippocampal adenosine A3 receptors produces a desensitization of A1 receptor-mediated responses in rat hippocampus. J Neurosci. 1997;17:607–614. doi: 10.1523/JNEUROSCI.17-02-00607.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB, Abbracchio MP, Burnstock G, Daly JW, Harden TK, Jacobson KA, et al. Nomenclature and classification of purinoceptors. Pharmacol Rev. 1994;46:143–156. [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Nikodijevic O, Shi D, Gallo-Rodriguez C, Olah ME, Stiles GL, Daly JW. A role for central A3-adenosine receptors. Mediation of behavioral depressant effects. FEBS Lett. 1993;336:57–60. doi: 10.1016/0014-5793(93)81608-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Park KS, Jiang JL, Kim YC, Olah ME, Stiles GL, Ji XD. Pharmacological characterization of novel A3 adenosine receptor-selective antagonists. Neuropharmacology. 1997;36:1157–1165. doi: 10.1016/s0028-3908(97)00104-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HO, Ji XD, Siddiqi SM, Olah ME, Stiles GL, Jacobson KA. 2-Substitution of N6-benzyladenosine-5′-uronamides enhances selectivity for A3 adenosine receptors. J Med Chem. 1994;37:3614–3621. doi: 10.1021/jm00047a018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsmeyer SJ. BCL-2 gene family and the regulation of programmed cell death. Cancer Res. 1999;59:1693s–1700s. [PubMed] [Google Scholar]
- MacGregor DG, Miller WJ, Stone TW. Mediation of the neuroprotective action of R-phenylisopropyl-adenosine through a centrally located adenosine A1 receptor. Br J Pharmacol. 1993;110:470–476. doi: 10.1111/j.1476-5381.1993.tb13834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita T, Harigai M, Hanada M, Reed JC. Identification of a p53-dependent negative response element in the bcl-2 gene. Cancer Res. 1994;54:3131–3135. [PubMed] [Google Scholar]
- Ongini E, Adami M, Ferri C, Bertorelli R. Adenosine A2Areceptors and neuroprotection. Ann NY Acad Sci. 1997;825:30–48. doi: 10.1111/j.1749-6632.1997.tb48412.x. [DOI] [PubMed] [Google Scholar]
- Peakman MC, Hill SJ. Adenosine A1 receptor-mediated inhibition of cyclic AMP accumulation in type-2 but not type-1 rat astrocytes. Eur J Pharmacol. 1996;306:281–289. doi: 10.1016/0014-2999(96)00202-6. [DOI] [PubMed] [Google Scholar]
- Sastry KJ, Marin MC, Nehete PN, McConnell K, Naggar AK, McDonnell TJ. Expression of human immunodeficiency virus type I tat results in down-regulation of bcl-2 and induction of apoptosis in hematopoietic cells. Oncogene. 1996;13:487–493. [PubMed] [Google Scholar]
- Schubert P, Ogata T, Marchini C, Ferroni S, Rudolphi K. Protective mechanisms of adenosine in neurons and glial cells. Ann NY Acad Sci. 1997;825:1–10. doi: 10.1111/j.1749-6632.1997.tb48409.x. [DOI] [PubMed] [Google Scholar]
- Selvakumaran M, Lin HK, Miyashita T, Wang HG, Krajewski S, Reed JC, et al. Immediate early up-regulation of bax expression by p53 but not TGF beta 1: a paradigm for distinct apoptotic pathways. Oncogene. 1994;9:1791–1798. [PubMed] [Google Scholar]
- Shneyvays V, Jacobson KA, Li AH, Nawrath H, Zinman T, Isaac A, Shainberg A. Induction of apoptosis in rat cardiocytes by A3 adenosine receptor activation and its suppression by isoproterenol. Exp Cell Res. 2000;257:111–126. doi: 10.1006/excr.2000.4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Lubitz DK, Carter MF, Deutsch SI, Lin RC, Mastropaolo J, Meshulam Y, Jacobson KA. The effects of adenosine A3 receptor stimulation on seizures in mice. Eur J Pharmacol. 1995;275:23–29. doi: 10.1016/0014-2999(94)00734-o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Lubitz DK, Lin RC, Bischofberger N, Beenhakker M, Boyd M, Lipartowska R, Jacobson KA. Protection against ischemic damage by adenosine amine congener, a potent and selective adenosine A1 receptor agonist. Eur J Pharmacol. 1999;369:313–317. doi: 10.1016/s0014-2999(99)00073-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Lubitz DK, Lin RC, Popik P, Carter MF, Jacobson KA. Adenosine A3 receptor stimulation and cerebral ischemia. Eur J Pharmacol. 1994;263:59–67. doi: 10.1016/0014-2999(94)90523-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Sei Y, Abbracchio MP, Jiang JL, Kim YC, Jacobson KA. Adenosine A3 receptor agonists protect HL-60 and U-937 cells from apoptosis induced by A3 antagonists. Biochem Biophys Res Commun. 1997;232:317–322. doi: 10.1006/bbrc.1997.6290. [DOI] [PMC free article] [PubMed] [Google Scholar]