

Abstract
The acetylation status of lysine residues on histone proteins has long been attributed to a balance struck between the catalytic activity of histone acetyl transferases and histone deacetylases (HDAC). HDACs were identified as the sole removers of acetyl post-translational modifications (PTM) of histone lysine residues. Studies into the biological role of HDACs have also elucidated their role as removers of acetyl PTMs from lysine residues of nonhistone proteins. These findings, coupled with high-resolution mass spectrometry studies that revealed the presence of acyl-group PTMs on lysine residues of nonhistone proteins, brought forth the possibility of HDACs acting as removers of both acyl- and acetyl-based PTMs. We posited that HDACs fulfill this dual role and sought to investigate their specificity. Utilizing a fluorescence-based assay and biologically relevant acyl-substrates, the selectivities of zinc-dependent HDACs toward these acyl-based PTMs were identified. These findings were further validated using cellular models and molecular biology techniques. As a proof of principal, an HDAC3 selective inhibitor was designed using HDAC3’s substrate preference. This resulting inhibitor demonstrates nanomolar activity and >30 fold selectivity toward HDAC3 compared to the other class I HDACs. This inhibitor is capable of increasing p65 acetylation, attenuating NF-κB activation, and thereby preventing downstream nitric oxide signaling. Additionally, this selective HDAC3 inhibition allows for control of HMGB-1 secretion from activated macrophages without altering the acetylation status of histones or tubulin.
Graphical abstract
Increasing evidence suggests that lysine post-translational modifications (PTMs) play multiple and extensive roles in cell signaling, akin to the well-studied phosphorylation, methylation, or ubiquitinylation PTMs.1 Initial proteomic studies using high-resolution mass spectrometry have identified at least 3600 lysine acetylation sites on over 1750 proteins.2 In addition to lysine acetylation, a wider array of lysine acylations has gradually become recognized as important PTMs that control key cellular processes.3 These modifications include lysine formylation, acetylation, propionylation, butyrylation, crotonylation, glutarylation, malonyl/succinylation, and myristoyl/palmitoylation.4–13 A common feature of these lysine acylations is that most of them originate from coenzyme A (CoA) metabolites. The even numbered acyl groups such as acetyl and butyryl are likely derived from β-oxidation pathways, and the more complex succinyl modification stems from succinyl-CoA, most commonly used in regulation of cellular energy homeostasis. This crosstalk between metabolism and PTM status suggests a role for these lysine modifications to regulate enzymes in metabolic pathways.14 Further, the identification of the diverse acyl-based PTMs has sparked studies focusing on the conditions under which they are attached and removed, leading to the demonstration that acylation of lysine residues is a nonspecific process performed either through promiscuous histone acetyl transferases or simply by noncatalytic chemical ligation.15 Unlike the promiscuous and even equilibrium-based ligation of acyl groups to lysine residues, the removal of these groups seems to be more carefully controlled.15 One enzyme family capable of removing glutaryl, malonyl/succinyl, and myristoyl/palmitoyl groups is the sirtuins.9,10,13 As class III members of the histone deacetylase (HDAC) family of enzymes, sirtuins are NAD+-dependent deacylases. Contrarily, class I, II, and IV HDACs are metal-containing deacetylases. It has been suggested that class I, II, and IV HDACs possess the ability to also deacylate lysines rather than an isolated ability to deacetylate them. With this knowledge, we asked if these relatively novel acyl groups were substrates for any zinc-dependent HDAC and, if so, questioned whether there was any level of specificity these isozymes displayed toward certain acyl substrates.
RESULTS AND DISCUSSION
HDAC Isozyme Deacylase Kinetic Profiling
Our study began by developing 12 different aminomethylcoumarin-based fluorogenic substrates that would mimic biologically relevant acyl-group PTMs (Supporting Information Figure 1a). These substrates were developed based on either known acids/acyl-CoA bound esters that have been found in the bloodstream in high concentrations and are likely to be ligated to the ε-N terminus of lysine residues or known PTMs shown to exist via mass spectrometry ex vivo. The nonbiologically relevant trifluoroacetyl (TFA) substrate was utilized as a positive control for class IIa HDACs and HDAC8, as it is the best-known substrate to be efficiently removed by these HDAC isozymes.16 Utilizing these substrates and recombinant human HDACs, all zinc-dependent HDAC isozymes were tested en bloc for their ability to deacylate each substrate, with particular interest for substrate cleaved over time with constant enzyme and substrate concentrations (Supporting Information Figure 1b).
As previously reported, HDACs 1, 2, 3, and 6 demonstrated the most robust deacetylase activity compared to all other HDAC isozymes.17 Also in line with external findings, class IIa HDACs and HDAC8 only displayed the ability to deacylate the TFA-based substrate.18 No appreciable deacylase activity was seen for HDACs 10 and 11, which falls in line with a similarly performed study.17 In addition to this, we saw no appreciable activity of any isozyme toward our heptanoyl-, octanoyl-, glutaryl-, or adipoyl-based substrates (Supporting Information Figure 1b). As such, the results of this experiment directed our focus toward more rigorous interrogation of the deacylase capacity of HDACs 1, 2, 3, and 6.
HDACs 3 and 6 demonstrated appreciable deformylase activity with HDAC6 demonstrating higher catalytic activity as a deformylase than as a deacetylase with the concentrations of enzyme and substrate used. HDAC3 possessed by far the most diverse ability to deacylate a variety of substrates, including the TFA-based substrate, with a particular preference for deacylating the butyryl-, crotonyl-, and valeryl-based substrates compared to HDACs 1 and 2. Last, HDACs 1–3 were able to depropionylate with high catalytic efficiency (Figure 1a and Supporting Information Figure 1b). While there have been previous reports of HDAC3 possessing the ability to deacylate the TFA-based substrate,17 we sought to determine if this finding was due to an impurity of one or more class IIa HDACs in our HDAC3 solution. Briefly, HDAC3 was coincubated with TFA substrate and vorinostat or diphenyl acetic hydroxamic acid (dPAHA). It has been previously shown that vorinostat possesses no appreciable inhibitory activity for class IIa HDACs,17 while dPAHA only possesses the ability to inhibit class IIa HDACs.19 As expected, and in line with previous publications,20 vorinostat, but not dPAHA, was capable of altering HDAC3’s ability to deacylate the TFA substrate (Supporting Information Figure 2). Therefore, we are confident in associating this deacylase ability with HDAC3.
Figure 1.
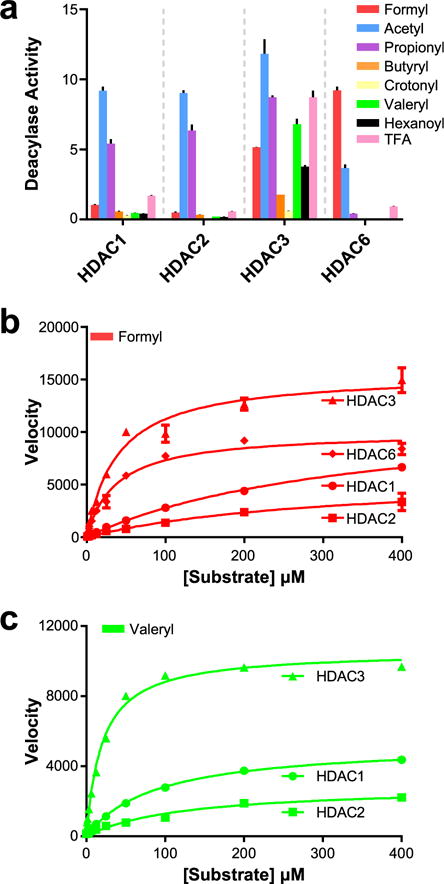
Acyl-substrate profiling. (a) Results of en bloc acyl-substrate profiling screen against HDACs 1, 2, 3, and 6. y-axis units in μmol substrate cleaved·μmol enzyme−1·min−1. (b) Vmax study of formyl-substrate vs HDACs 1, 2, 3, and 6. y-axis units in pmol substrate cleaved·s−1·mg enzyme−1. (c) Vmax study of valeryl-substrate vs HDACs 1, 2, and 3. y-axis units in pmol substrate cleaved·s−1·mg enzyme−1. n = 3; error bars are SEM.
To further investigate the key findings from our initial screen, we performed Vmax kinetic analyses on HDACs 1, 2, 3, and 6 versus substrates that were deacylated by one or more of these isozymes. We determined values of Km, Vmax, kcat, and kcat/Km, the latter being the most well accepted measurement of catalytic efficiency (Table 1). Interestingly, the Km values of our formyl-based substrate vs HDACs 3 and 6 is nearly a log lower than the corresponding values for HDACs 1 and 2. The Vmax value for HDAC1, despite being higher than the value for HDAC6, is not likely to be achieved in vivo, as the concentration required to achieve this would be in the millimolar range (Figure 1b and Table 1). HDAC6 is the most catalytically efficient deformylase; however, it is still a more efficient deacetylase. Surprisingly, HDACs 1–3 displayed remarkable catalytic efficiency as depropionylases. Despite this, there appears to be very little difference in selectivity or efficiency between the three isozymes. The last intriguing finding from this kinetic study was the deacylation ability of HDAC3 toward butyryl-, crotonyl-, and valeryl-based substrates. It has been previously reported that HDAC3 was capable of deacylating crotonyl-substrates.21 Unlike the depropionylase ability of HDACs 1–3, the deacylase activity toward these substrates was very specific to HDAC3. This seems to stem from HDAC3’s ability to both bind these substrates more efficiently (lower Km values) and efficiently cleave these substrates (higher kcat/Km values) from the ε-N of lysine residues relative to HDACs 1 or 2 (Figure 1c and Table 1). Most interestingly, the crotonyl-substrate binds to HDAC3 with very high affinity; however, there is little substrate turnover (kcat) compared to the canonical acetyl substrate (Table 1).
Table 1.
Kinetic profile of HDACs 1, 2, 3, and 6a
| HDAC1 | Km (μM) | Vmax (pmol·s−1·mg protein−1) | kcat (min−1) | kcat/Km (M−1·s−1) |
|---|---|---|---|---|
| formyl | 342 | 12300 | 8.24 | 402 |
| acetyl | 36.6 | 24800 | 16.6 | 7580 |
| propionyl | 13.1 | 9850 | 6.62 | 8410 |
| butyryl | 49.1 | 1910 | 1.28 | 435 |
| crotonyl | 2.40 | 802 | 0.380 | 2670 |
| valeryl | 102 | 5330 | 3.56 | 582 |
|
| ||||
| HDAC1 | Km (μM) | Vmax (pmol·s−1·mg protein−1) | kcat (min−1) | kcat/Km (M−1·s−1) |
|
| ||||
| formyl | 313 | 5980 | 4.01 | 214 |
| acetyl | 36.6 | 4.00 × 103 | 2.69 | 1230 |
| propionyl | 8.53 | 4.90 × 103 | 3.29 | 6440 |
| butyryl | 21.9 | 804 | 0.540 | 412 |
| crotonyl | 3.20 | 1010 | 0.485 | 2520 |
| valeryl | 122 | 2880 | 1.93 | 264 |
|
| ||||
| HDAC1 | Km (μM) | Vmax (pmol·s−1·mg protein−1) | kcat (min−1) | kcat/Km (M−1·s−1) |
|
| ||||
| formyl | 39.1 | 15600 | 9.30 | 3970 |
| acetyl | 17.3 | 26900 | 16.0 | 15400 |
| propionyl | 8.06 | 13900 | 8.31 | 17200 |
| butyryl | 1.03 | 2560 | 1.53 | 24700 |
| crotonyl | 0.114 | 758 | 0.362 | 53700 |
| valeryl | 9.99 | 11100 | 6.63 | 11100 |
|
| ||||
| HDAC1 | Km (μM) | Vmax (pmol·s−1·mg protein−1) | kcat (min−1) | kcat/Km (M−1·s−1) |
|
| ||||
| formyl | 37.2 | 1.00 × 104 | 19.1 | 8580 |
| acetyl | 1.93 | 2130 | 4.04 | 34800 |
| propionyl | 161 | 9.00 × 102 | 1.72 | 178 |
| butyryl | N.D. | N.D. | N.D. | N.D. |
| crotonyl | N.D. | N.D. | N.D. | N.D. |
| valeryl | N.D. | N.D. | N.D. | N.D. |
Results are mean values of n = 3 experiments. SEM < 10% of mean in all cases.
Interrogation of HDACs 3 and 6 As Deformylases
Utilizing Hek293 cell lysates and various HDAC inhibitors, we sought to determine if both this newly discovered deformylase activity translated into a more robust cellular-based model and if it was affected by traditional small molecule inhibitors. Vorinostat, a class I and HDAC6 inhibitor;17 Tubastatin A (tubA), an HDAC6 specific inhibitor;22 and PD-106, an HDACs 1–3 inhibitor23 were used to interrogate the individual and combined inhibitory effects of these small molecules against HDAC isozymes’ cellular deformylase activity (Figure 2a). The pan-inhibition of vorinostat at 1 μM demonstrated the ability to inhibit both deacetylation and deformylation, in line with our kinetics study. The selective HDAC6 inhibitor Tubastatin A at 0.5 μM, well above its IC50 of <50 nM, did not affect overall deacetylation activity but did lower deformylation activity (Figure 2b). This is due to the ability of HDACs 1–3 to fulfill the deacetylase role in the cell even with an inhibited HDAC6.24 The HDACs 1–3 inhibitor PD-106 at 1 μM also showed a deformylase inhibition profile akin to Tubastatin A, once again showing that selective inhibition of HDAC3 or HDAC6 is not enough to affect global deacetylation but can affect deformylation. Last, when Tubastatin A and PD-106 were combined, additional lowering in deformylation activity was seen while leaving deacetylation activity nearly unaffected (Figure 2b). Despite inhibiting HDACs 1, 2, 3, and 6, the combined treatment only affected deformylation. This, unlike vorinostat’s effect on deacetylation, can be explained through the stoichiometry used to achieve this result. At 1 μM, PD-106 is below its IC50 values for HDACs 1 and 2 with sufficient incubation time to account for its slow-on/slow-off kinetics. As such, it is not likely to be inhibiting these enzymes strongly enough to induce a physiologic effect that would be seen with less discriminant class I HDAC inhibition at higher doses.
Figure 2.
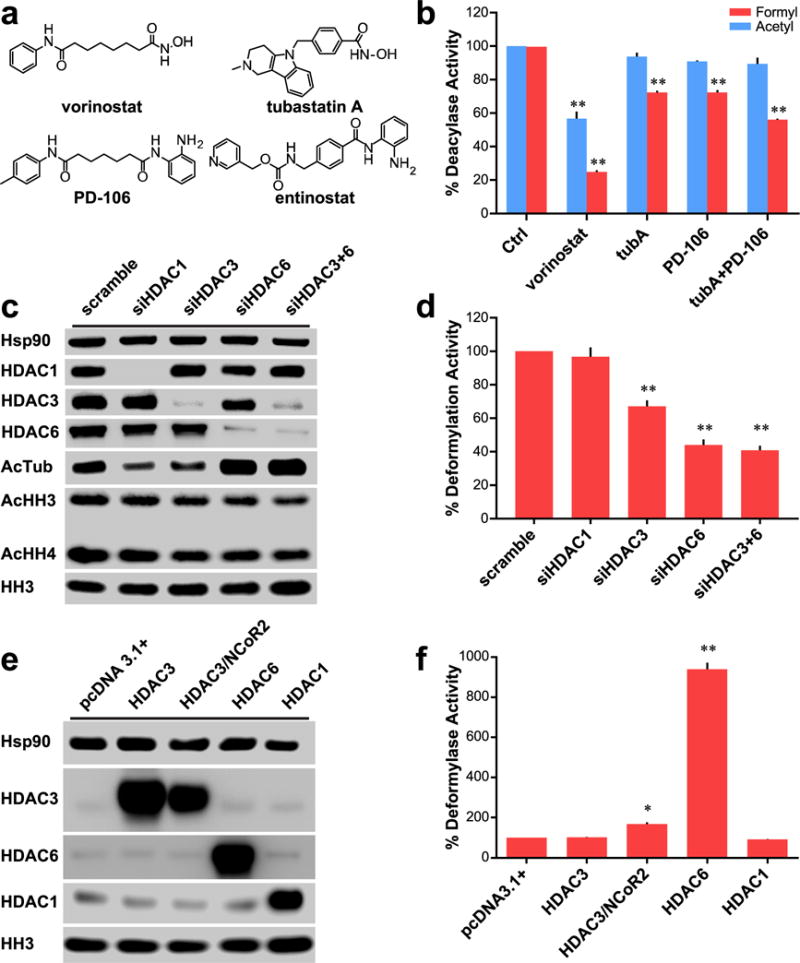
Role of HDACs 3 and 6 in cellular deformylation. (a) Chemical structures of small molecule inhibitors of HDACs 1, 2, 3, and 6. (b) Comparison of effect on deformylase and deacetylase activities of Hek293 lysates with various inhibitors for 24 h; data normalized to control deacylase activity. Vorinostat and PD-106 used at 1 μM; tubA used at 0.5 μM. (c) Western blot analysis of Hek293 cells with various siRNA transfections demonstrating selective and specific knockdown of targeted HDACs. (d) Deformylase activity of siRNA treated Hek293 lysates; data normalized to scramble deacetylase activity. (e) Western blot analysis of Hek293 cells transfected with various vectors to overexpress targeted HDACs. (f) Deformylase activity of transfected Hek293 cells; data normalized to pcDNA3.1+ vector transfected cells. Parts b, d, and f are n = 3; error bars are SEM. Parts c and e are representative of n ≥ 2 experiments. *p-value < 0.01. **p-value < 0.0001. All statistical analyses were Dunnett’s multiple comparisons of means to the means of their respective controls.
With the results of small molecule inhibition matching the data from our initial screening, we next interrogated the roles of HDACs 3 and 6 as deformylases utilizing siRNA knockdown and plasmid transfection for overexpression in Hek293 cells (Figure 2c–f). Utilizing HDAC1 as a negative control, we demonstrated that its knockdown bore no impact on deformylation activity. Knockdown of HDAC3, however, led to an approximate 35% decrease in deformylation activity of the cell lysate. Knockdown of HDAC6 led to a more robust 55% decrease in deformylation, and the concomitant knockdown of both isozymes led to a similar effect (Figure 2d). These data support the previous findings that HDACs 3 and 6 are the majority, or possibly even sole, controllers of deformylation in the cell with their suppression leading to extensive loss of activity. Following up on these studies, we performed overexpression of HDACs 1, 3, and 6 (Figure 2e). Using HDAC1 as a negative control, its overexpression bore no effect on deformylase activity similar to its knockdown. Initial overexpression of HDAC3 without its required corepressor NCoR2 led to no appreciable increase in deformylase activity. However, simultaneous overexpression of both HDAC3 and its corepressor led to an approximate doubling in activity. Overexpression of HDAC6 in similar fashion displayed a much more robust increase in deformylation activity, leading to a near 10-fold increase in activity. These increases were not seen in the control pcDNA3.1 plasmid (Figure 2f). Taken together, these data suggest that HDACs 3 and 6 are both controllers of deformylation; however, HDAC6 is a more active deformylase in vitro.
Interrogation of HDAC3’s Role As a Selective Devalerylase
We next chose to probe HDAC3’s seemingly selective ability to remove short chain fatty acids of four to five carbons in length. As carbon chain length for our synthesized substrates exceeded three, deacylation activity decreased for all isozymes but HDAC3 (Supporting Information Figure 1b and Table 1). In particular, we were interested in the potential devalerylase activity of this isozyme. Traditionally, short chain fatty acids created in vivo contain an even number of carbons. Valeryl groups, confirmed to exist in human serum as its fatty acid form in the high micromolar range,25 are the result of bacterial-based metabolism of sloughed intestinal cells from gut dwelling flora.26 Combining these data with our findings that HDAC3 may serve as a devalerylase, we discovered a potentially clinically relevant finding and chose to interrogate this observation further.
We proceeded to study the effect of HDAC isozyme inhibition with small molecule inhibitors on HDAC3’s potential devalerylase activity using Hek293 lysates. Similar to the prior experiment, the pan-HDAC inhibition of vorinostat led to a lowering in both deacetylation and devalerylation (Figure 3a). The HDAC6 selective inhibitor Tubastatin A displays no effect on deacetylation or devalerylation, in line with our previous experiments that suggest HDAC3 is the only HDAC isozyme capable of devalerylation (Figure 1a). The utilization of the HDACs 1–3 inhibitor PD-106 demonstrates selective inhibition of devalerylation. The addition of Tubastatin A leads to seemingly no difference in overall effect. Taken together, these data give further validity to our initial discovery that HDAC3 possesses devalerylase activity. To further test this finding, we again utilized our Hek293 lysates with individual HDAC isozymes knocked down (Figure 2c). With the selective knockdown of HDAC3, but not HDACs 1 or 6, we see an approximately 40% decrease in global deformylation activity (Figure 3b). Using the Hek293 lysates with overexpressed HDAC1, HDAC3, HDAC3/NCoR2, and HDAC6 (Figure 2e), we indeed see that increased cellular concentrations of HDAC3 and its corepressor NCoR2 led to substantial increases in the devalerylation activity of our Hek293 lysates while overexpression of other HDACs had no appreciable effect (Figure 3c).
Figure 3.
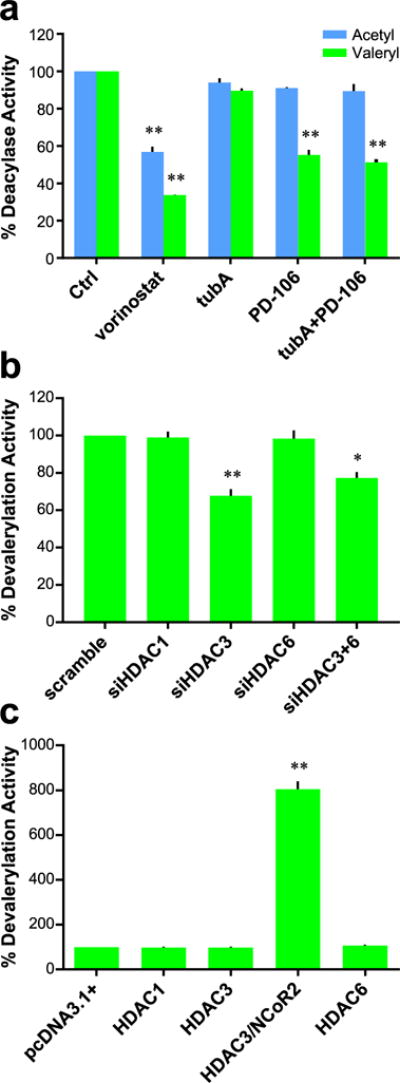
Role of HDAC3 in cellular devalerylation. (a) Comparison of effect on devalerylase and deacetylase activities of Hek293 lysates with various inhibitors; data normalized to control deacylase activity. Vorinostat and PD-106 used at 1 μM; tubA used at 0.5 μM. (b) Devalerylase activity of siRNA treated Hek293 lysates; data normalized to scramble deacetylase activity. (c) Devalerylase activity of transfected Hek293 cells; data normalized to pcDNA3.1+ vector transfected cells. Data collected after 24 h of treatment. n = 3; error bars are SEM. *p-value < 0.01. **p-value < 0.0001. All statistical analyses were Dunnett’s multiple comparisons of means to the means of their respective controls.
Interrogation of HDAC Inhibitors on Global Protein Formylation and Valerylation
A potential pitfall for all previous experiments was that they relied upon artificially synthesized fluorogenic substrates rather than naturally occurring substrates. To assuage this possible confounding factor, we utilized antibodies specific toward acetyl-, formyl-, and valeryl-lysine PTMs and measured the effect of small molecule inhibition and siRNA knockdown of individual HDAC isozymes on these levels. In agreement with previous results, the pan-HDAC inhibitor vorinostat induced hyperacetylation of tubulin and histones H3 and H4. Also in line with published research, we see that entinostat27 and PD-106 induced hyperacetylation of histones without affecting tubulin acetylation levels. The HDAC6 specific inhibitor Tubastatin A, conversely, only affected acetylated tubulin levels (Figure 4a).
Figure 4.
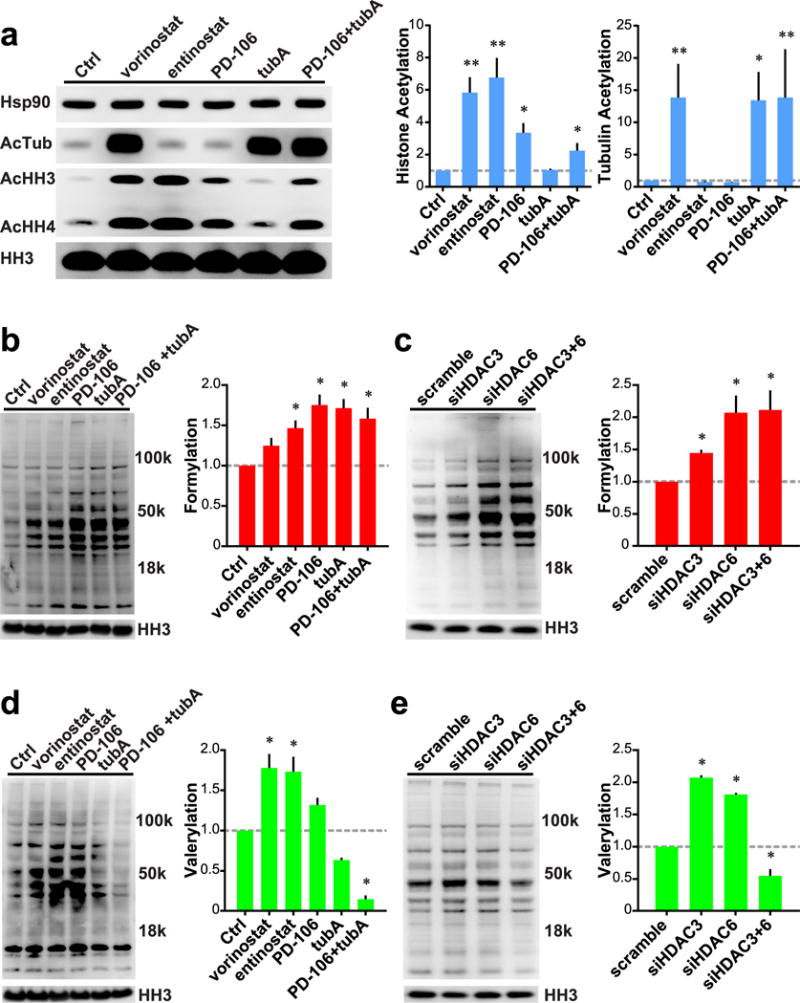
Roles of HDACs 3 and 6 on global cellular formylation and valerylation. (a) Left: Western blot analysis of Hek293 cells treated with small molecule HDAC inhibitors. Right: Quantification of histone and tubulin acetylation normalized to HH3 levels. (b) Left: Global formylation levels of Hek293 cells treated with HDAC inhibitors. Right: Quantification of global formylation normalized to HH3 levels. (c) Left: Global formylation levels of Hek293 cells treated with siRNA. Right: Quantification of global formylation normalized to HH3 levels. (d) Left: Global valerylation levels of Hek293 cells treated with HDAC inhibitors. Right: Quantification of global valerylation normalized to HH3 levels. (e) Left: Global valerylation levels of Hek293 cells treated with siRNA. Right: Quantification of global valerylation normalized to HH3 levels. All inhibitors used at 1 μM, except tubA at 0.5 μM. Data recorded after 24 h of treatment; representative Western blots of n ≥ 3 experiments. Error bars are SEM. *p-value < 0.01. **p-value < 0.0001. All statistical analyses were Dunnett’s multiple comparisons of means to the means of their respective controls.
Translating from our previous findings, we see that the pan-HDAC inhibitor vorinostat, caused global protein hyperformylation due to its concomitant inhibition of HDACs 3 and 6; however, its increase was not statistically significant with a p-value of 0.072. Entinostat and PD-106 are also capable of this effect, although their mechanism is likely to be through HDAC3 inhibition, without inhibition of HDAC6 at the concentrations used. Last, the HDAC6-specific inhibitor Tubastatin A also induced global protein hyperformylation through its inhibition of HDAC6 (Figure 4b). The combination of PD-106 and Tubastatin A led to a nonsignificant decrease in global protein formylation compared to either agent alone.
Following these experiments, we utilized siRNA knockdowns of HDACs 3 and 6 to determine the effect of each isozyme more specifically. Knockdown of HDAC3 alone led to a significant rise in global protein formylation levels. Compared to HDAC3, knockdown of HDAC6 led to an even greater, arguably significant, increase in global formylation with a p-value of 0.049. The concomitant knockdown of HDACs 3 and 6 together had no significant effect compared to HDAC6 alone, further suggesting HDAC6’s dominant role as a deformylase (Figure 4c).
Moving our attention toward global protein valerylation, we repeated these experiments using antibodies specific for valerylated lysine. Inhibition of HDAC3 with vorinostat, entinostat, or PD-106 led to global protein hypervalerylation; however, the increase for PD-106 was nonsignificant with a p-value of 0.120. We also see that selective inhibition of HDAC6 with Tubastatin A led to a nonsignificant hypovalerylation (p-value = 0.073). Further, combined HDAC3 and HDAC6 inhibition with PD-106+Tubastatin A led to an even more pronounced, significant hypovalerylation effect (Figure 4d). One explanation for this discrepancy was off-target effects of either Tubastatin A or PD-106. As such, we used siRNA to study this, hypothesizing that specific knockdown of either isozyme would more readily determine each isozyme’s effect on valerylation levels. Knockdown of HDAC3 led to the expected result of hypervalerylation; however, knockdown of HDAC6 alone led to a hypervalerylation as well. Even more interesting still is the hypovalerylation that occurred when both HDACs 3 and 6 are knocked down concomitantly (Figure 4e). One possible explanation for this discrepancy could lie with the translatability of our valeryl-based substrate in vitro and in cellular lysate models versus the natural substrate. Another explanation is that knockdown of HDAC6 may lead to a dissolution of a key complex required for maintenance of lysine acylation levels.28 Last, another possibility may be a non-selective antibody. To assuage this possibility, we performed dot blot analyses with each antibody against formylated-, acetylated-, or valerylated-bovine serum albumin. The results demonstrate, in line with the manufacturer’s ELISA specifications, that each antibody is highly selective for its appropriate acyl group (Supporting Information Figure 3).
Substrate Driven Development of HDAC3 Specific Inhibitor
We realized that the crotonyl-substrate possessed very unique properties with HDAC3, but not HDACs 1, 2, or 6. Its nanomolar Km value led to HDAC3 binding very tightly to the substrate; however, very little substrate turnover (kcat) was seen. Further, the crotonyl substrate’s binding affinity was over 20-fold more selective for HDAC3 compared to HDACs 1 or 2 (Table 1). Taken together, we hypothesized that the crotonyl substrate could act as a competitive inhibitor against the canonical acetyl substrate. Further, we wondered if this specificity in Km translated to specificity in inhibition. Our data show that indeed the crotonyl substrate is a notably selective HDAC3 inhibitor (Figure 5a). Encouraged by this result, and recognizing the potential of developing a novel, selective chemical tool to study the HDAC3 isozyme selectively,29,30 we performed a rudimentary SAR analysis based around the trans-geometry of the crotonyl acyl group; the length of the HDAC3 selective valeryl acyl group; and the HDACs 1, 2, and 3 selective benzamide metal-chelation moiety (Supporting Information Table 1).
Figure 5.
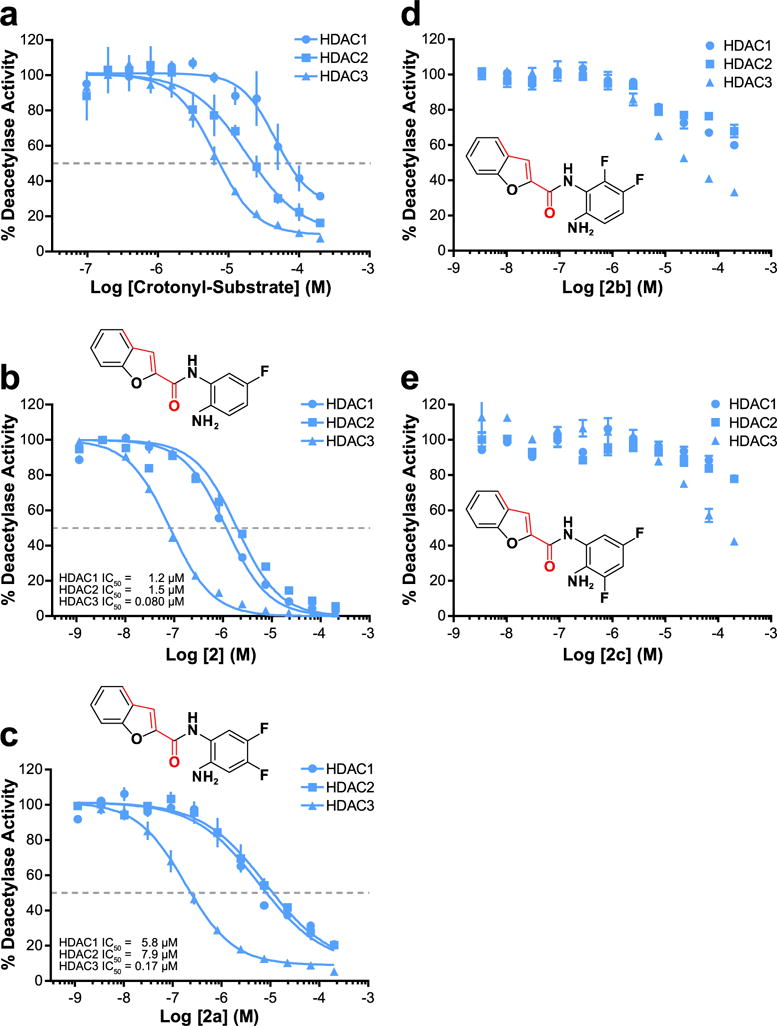
Substrate-specificity driven development of HDAC3 selective inhibitor. (a) IC50 of HDACs 1–3. The crotonyl subtrate serves as a fairly potent and selective HDAC3 inhibitor. IC50 values for HDACs 1–3 respectively: 46.5 μM, 18.1 μM, 6.42 μM. (b) IC50 of HDACs 1–3 vs 2. (c) IC50 of HDACs 1–3 vs 2a. (d) IC50 of HDACs 1–3 vs 2b. (e) IC50 of HDACs 1–3 vs 2c. All data normalized to vehicle (DMSO) control. Graphs a–c fit via GraphPad Prism log(inhibitor) vs normalized response–variable slope parameters. n ≥ 3; error bars are SEM.
We began simplistically with a benzamide structure with an attachment to mimic the crotonyl and valeryl modifications simultaneously (compound 1). We gained potency by utilizing a trans-cyclopentadiene ring which further forced proper conformation. Between all heterocycles generated, the oxygen-containing furan ring of 1a seemed most promising from its potency and mild selectivity toward HDAC3. Building off of 1a, the addition of a fused benzene ring led to compound 1f, which demonstrated nanomolar HDAC1 and HDAC3 IC50 values with its benzofuran group. While 1f was our best lead from the first round of SAR, it lacked the selectivity we believed possible for HDAC3, only demonstrating a 2-fold preference for HDAC3 over HDAC1. Looking back at our initial acyl-substrate screen, we also noted HDAC3’s ability to deacylate the TFA substrate, which HDACs 1 and 2 lacked. Additionally, a recent publication also reported on fluorination-based HDAC3 selectivity.31 As such, we utilized both of these findings by incorporation of a fluorine to 1f, which resulted in compound 2. This inhibitor possessed 15-fold selectivity for HDAC3 compared to HDAC1 (Figure 5b) while also increasing its inhibitory potency further toward HDAC3. We added a second fluorine to 2 to generate 2a, which resulted in a slightly weaker HDAC3 inhibitor but established even greater selectivity of the molecule for HDAC3 (Figure 5c). Curious to test the effect of fluorination in other positions around the benzene ring, we also developed and interrogated 2b and 2c. These inhibitors, possessing different fluorine positioning than 2a, showed very little efficacy in vitro (Figure 5d and e). Concluding our substrate driven SAR, compound 2a possessed an IC50 for HDAC3 of 170 nM and was over 30-fold more selective for HDAC3 compared to other class I HDACs. Further testing of 2a revealed it possessed interestingly selective slow-binding toward HDAC3 but not HDACs 1 or 2 (Supporting Information Figure 4).
Effects of 2a on NF-κB Acetylation, NO Signaling, and HMGB-1 Secretion
A previous study showed that cytosolic localization of HDAC3 depends on its interaction with IκBα within an intact NF-κB complex, thereby suggesting that it plays a potential role in the NF-κB signaling.32 Using 2a, 2b, 2c, and other known HDAC inhibitors with different HDAC isozyme selectivities, we examined these HDAC inhibitors’ relative effects on acetylation of NF-κB p65, p53, histones H3 and H4, and tubulin. The selective HDAC3 inhibitor 2a induced NF-κB p65 Lys122/Lys123 and p53 Lys382 acetylation without the induction of histone H3/H4 or tubulin acetylation (Figure 6a). This result is consistent with previous studies that demonstrated that deacetylation of Lys122/Lys123 of NF-κB p65 is controlled by HDAC3, and that inhibition of HDACs 1 or 2 has little effect on their acetylation status.33 However, our finding that selective HDAC3 inhibition with a small molecule is capable of inducing hyperacetylation of Lys382 on p53 is novel. Ryu et al. recently demonstrated that HDAC6 is also capable of controlling the acetylation status of Lys382 on p53.34 Unfortunately, they did not have an HDAC inhibitor capable of targeting HDAC3 without HDAC6 to delineate out the impact of HDAC3 inhibition vs HDAC6 inhibition on this residue’s acetylation status. Further, they demonstrated the use of tubA at 2 μM for 24 h was able to induce hyperacetylation of Lys382 in their tested HCT116 and HT29 cells. Our findings do not demonstrate this increase (Figure 6a) but could be a result of a cell-line specific effect or our use of a lower dose comparatively. Further, and against the findings by Ryu et al., the class I selective inhibitor, romidepsin, was shown to induce hyperacetylation of Lys373 and Lys382 at reasonable concentrations in the A549 cell line.35
Figure 6.
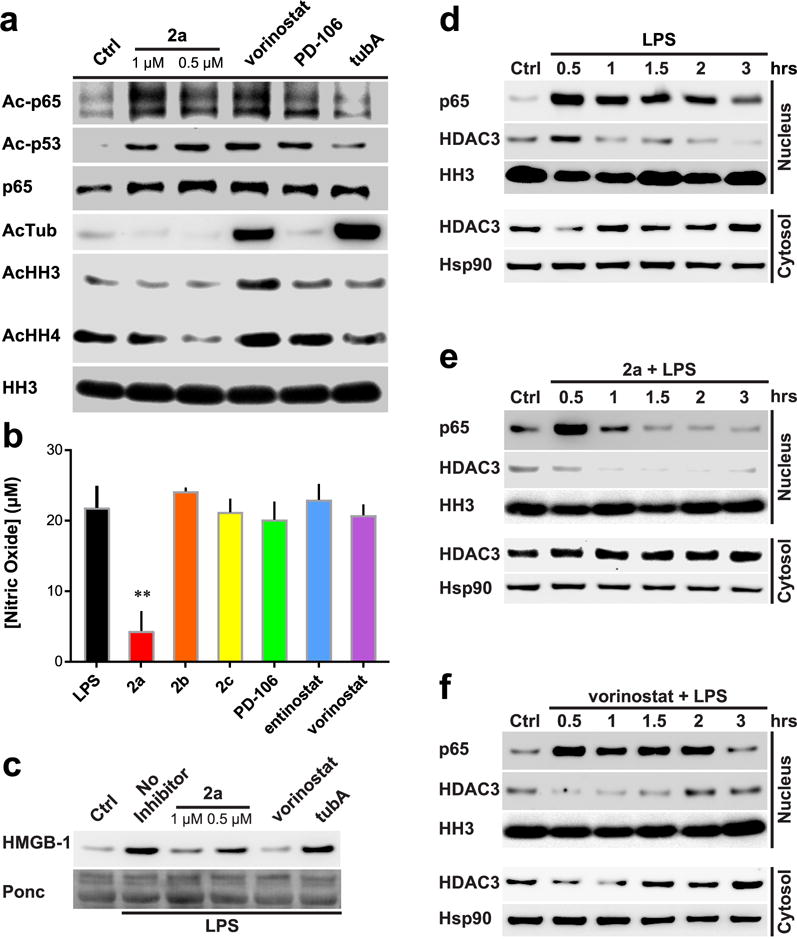
Effects of HDAC inhibition on NF-κB p65 acetylation and inflammatory responses. (a) Western blot analysis of RAW264.7 cells treated with various HDAC inhibitors. Vorinostat and PD-106 used at 1 μM; tubA at 0.5 μM. (b) NO concentration secreted from RAW264.7 treated cells; normalized to control treated viable cell concentration. Molarity of 1 μM used for all inhibitors. n ≥ 3. (c) Western blot analysis of RAW264.7 cells. HMGB-1 secretion monitoring with ponceau stain as loading control. Cells were treated for 6 h. Molarity at 1 μM vorinostat and 0.5 μM tubA used. (d–f) Western blot analysis of LPS-treated RAW264.7 cells. Nuclear and cytosolic fractions split. 2a and vorinostat used to assess regulation of HDACs on Ac-p65 subcellular localization. Representative Westerns of n ≥ 2 experiments; error bars are SEM. **p-value < 0.0001.
We furthered our investigation on the significance of HDAC3 inhibition and its effects on NF-κB activation, nitric oxide (NO) production, and HMGB-1 secretion in RAW264.7 macrophages challenged with bacterial lipopolysaccharide (LPS). Compound 2a decreased NO production after LPS induction in RAW264.7, while all other inhibitors, including 2b and 2c, failed to affect NO production (Figure 6b). It should be noted that PD-106, vorinostat, and entinostat, being HDAC3 inhibitors, did lower NO release. However, when cell viability was taken into account, as these inhibitors proved to be more lethal than 2a–2c, it was found this decrease was completely offset when we normalized the data to cell count/viability. LPS also induces HMGB-1 secretion, a late mediator of lethality in sepsis, in RAW264.7 cells.36,37 Previous studies have shown that the HMGB-1 protein contains multiple acetylation and formylation modifications, which control its cellular localization and secretion.5,38 2a, but not 2b or 2c, also blocked HMGB-1 secretion from the activated macrophage cells (Figure 6c and Supporting Information Figure 5). These data suggest that HDAC3 activity is required for proper inflammatory activation and response.
As HDAC3 activity is linked to IκBα and NF-κB activation, we further investigated the effect of 2a on NF-κB p65 and HDAC3 after LPS induction. As previously reported, LPS induced the nuclear localization of p65 and HDAC3.32 Cells treated with 2a had significantly shorter p65 nuclear retention times as well as diminished nuclear localization of HDAC3 (Figure 6d). Interestingly, in the 2a-treated cells, the nuclear localization of HDAC3 decreased slowly with time after LPS treatment (Figure 6e). This effect was not as pronounced in the vorinostat treated cells (Figure 6f). This suggests that HDAC3 activity is likely involved in multiple regulatory functions, such as its own cellular localization, in addition to p65 acetylation status and gene transcription regulation. Further, selective inhibition of HDAC3, but not pan-HDAC inhibition as seen with vorinostat, may be key to controlling HDAC3 and p65 subcellular localization.
DISCUSSION
In this study, we investigated HDAC isozyme-specific deacylase activity and demonstrated a biological relevancy of these findings. We have shown that HDACs 3 and 6 possess deformylation activity, confirmed previous findings that HDAC3 has decrotonylase activity, presented preliminary evidence that HDACs 1–3 can remove propionyl groups, and demonstrated that HDAC3 has specific activity toward butyryl and valeryl substrates (Figure 1a). We developed a selective HDAC3 inhibitor 2a based on the results from our initial deacylase profiling. This inhibitor induced NF-κB p65 Lys122/Lys123 and p53 Lys382 acetylation without affecting histone or tubulin acetylation. Additionally, 2a shortened NF-κB p65 nuclear localization duration and attenuated NO production and HMGB-1 secretion in activated macrophages (Figure 6).
The presented data suggest that lysine deformylation is likely to be coregulated by HDACs 3 and 6, with HDAC6 driving more deformylation activity in vitro than HDAC3 (Figure 1b and Figure 2). This process is likely to occur in the cytosol as HDAC6 shows high subcellular localization fidelity to this compartment.39 This result explains why a previous study utilizing mass spectrometry indicates that HDAC activity does not affect nuclear and histone formylation and is refractory to HDAC inhibitor treatments.40 Additionally, upon treatment with HDAC inhibitors, total protein formylation levels increased in a manner dependent on the selectivity of the inhibitor (Figure 4b).
The presented data also suggest HDAC3 has the ability to remove butyryl-, crotonyl-, and valeryl-based PTMs (Figure 1a and Supporting Information Figure 1b). Only HDAC inhibitors targeting HDAC3 were capable of inducing an increase in protein valerylation, which is consistent with our in vitro activity profiling (Figures 1a and 3a). Interestingly, the highly selective HDAC6 inhibitor Tubastatin A blocked induction of valerylated protein by other HDAC inhibitors. Global levels of valerylated cellular proteins also decreased when both HDACs 3 and 6 were concomitantly silenced, similar to the HDAC inhibitor results. Silencing of either HDAC3 or HDAC6 resulted in an increase in valerylated protein levels, suggesting that the elimination of HDAC6 likely affects HDAC3 devalerylase activity (Figure 4d). This may be explained through the known complexes in which HDACs 3 and 6 both have been shown to reside.28,41 Another possible explanation involves acetyl groups and valeryl groups competing for the same lysine residues. If HDAC6 is inhibited and acetyl groups are either more ubiquitous or are more favorably ligated to free lysine residues than valeryl groups, we would expect the lysine residues to be stabilized with more acetyl groups rather than valeryl. This is due to the cross coverage that a multitude of HDACs perform in the removal of acetyl groups, whereas only HDAC3 seems to be able to remove valeryl modifications. This would in turn lead to an overall decrease in valerylated lysine residues compared to acetylated. With respect to the knockdown data demonstrating hypervalerylation, this may be in part due to a compensatory mechanism of the cell that behaves differently than reversible small molecule inhibition.
Traditionally, developing selective HDAC3 inhibitors has led to very limited success, the development of entinostat, PD-106, and T32642 being the most notable. This is largely in part due to the high level of homology showcased between HDACs 1–3 (Supporting Information Figure 6). In this study, we have shown that by maintaining the trans-pentene geometry of the crotonyl-based PTMs and introducing fluorines around the ortho-aminoanilide ring (Figure 5 and Supporting Information Figure 4), a highly potent and selective HDAC3 inhibitor can be achieved. This inhibitor defines a chemotype that promotes selective HDAC3 inhibition. Interestingly, 2a also showed selective slow-on/slow-off kinetics for HDAC3 but not for HDACs 1 or 2 (Supporting Information Figure 4). This is in contrast to previous findings that described benzamide-containing HDAC inhibitors to possess time-dependent binding kinetics vs class I HDACs.23 It is interesting to note that a previous study presented the rationale behind why fluorination in certain positions around the benzamide ring may lead to HDAC3 selectivity, postulating that the intramolecular constraints of a pocket dwelling leucine was different in HDAC3 compared to HDAC2.31 This difference allowed the authors to develop an inhibitor that was selective for HDAC3. However, their explanation does not adequately cover the striking similarities between this conserved leucine when comparing HDAC3 with HDAC1 (Supporting Information Figure 7). While it is undoubted that fluorination plays a key part in both their inhibitors’ design as well as ours, there is still much to be studied. Further, with the discovery of potent HDAC inhibitors possessing internal metal chelation groups, the potential to develop even more potent and selective inhibitors could become more realized.43,44
The effects on NF-κB, NO production, and HMGB-1 secretion in activated macrophages by inhibitor 2a indicate that HDAC3 activity plays a key role in proinflammatory signaling. HDAC3 itself has been implicated in the regulation of inflammatory gene transcription in macrophages.45 However, based on our results, HDAC3 activity also plays other roles, such as regulating its own cellular localization and the duration of NF-κB p65 nuclear localization (Figure 6d). Our findings are consistent with previous observations,46 which suggest that HDAC3 controls NF-κB p65 Lys122/Lys123 acetylation status, its cellular localization, and rate of recovery to its latent state complex in the cytosol. Additionally, NO production is closely linked to NF-κB activation in macrophages;47–49 thus it is not surprising that NO production is attenuated by 2a. Our findings also demonstrate the ability of HDAC3 to control the acetylation of Lys382 of p53. There are several reports of the effects of inhibitors demonstrating different effects on this lysine residue. Further study controlling for cell type, concentrations of inhibitors, and length of treatment is undoubtedly needed.
HMGB-1 contains multiple PTMs, including acetylation and formylation of lysine.38 Upon stimulation, acetylated HMGB-1 translocates from the nucleus to the cytosol primed for secretion38 and sequential activation of inflammasomes is required for HMGB-1 secretion.50–52 Selective HDAC3 inhibition prevents HMGB-1 secretion similar to the pan-HDAC inhibition of vorinostat (Figure 6c). A similar result was observed for IL-1B secretion in macrophages, which also requires a two-step process for its secretion.53 Since acetylated HMGB-1 accumulates in the cytosol and HDAC3 activity is required for its secretions, this suggests HDAC3 plays a role in inflammasome activation during the second phase of secretion. Since HDAC3 has both deacetylase and select deacylase activity, it is likely that the presented anti-inflammatory effects are due to an increase in lysine acetylation or certain acyl-PTMs of specific lysine substrates.
METHODS
All final compounds were purified via Teledyne Isco Combiflash technology and confirmed to be >95% pure via LC/MS and 1H and 13C NMR. All HDAC isozymes were purchased from BPS biosciences with >90% purity with the exceptions of HDACs 4 and 7. Chemical reagents were purchased from Bachem, Sigma-Aldrich, or Fisher Scientific. Cells were purchased through ATCC and cultured according to ATCC guidelines. Detailed methods can be found in the Supporting Information.
Supplementary Material
Acknowledgments
Funding This work was supported by the National Institutes of Health/National Cancer Institute (CA163452 to C.J.C.), the National Institute of General Medical Sciences (P20GM103542) from the National Institutes of Health, South Carolina Clinical and Translational Research Institute UL1TR001450, and in part by pilot research funding from an American Cancer Society Institutional Research Grant awarded to the Hollings Cancer Center, Medical University of South Carolina.
The authors would like to thank C. Beeson and R. Schnellmann for their comments and discussions. We also thank the Medical University of South Carolina Mass Spectrometry and NMR core facilities.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschem-bio.7b00321.
Supporting Figures 1–7, Supporting Table 1, Supporting Methods, and Supporting Chemistry and Characterization(PDF)
ORCID
Jesse J. McClure: 0000-0002-5689-4731
Cheng Zhang: 0000-0002-8760-8152
Notes
The authors declare no competing financial interest.
References
- 1.Zencheck WD, Xiao H, Weiss LM. Lysine post-translational modifications and the cytoskeleton. Essays Biochem. 2012;52:135–145. doi: 10.1042/bse0520135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 3.Lin Y-Y, Kiihl S, Suhail Y, Liu SY, Chou Y-H, Kuang Z, Lu J-Y, Khor CN, Lin CL, Bader JS, Irizarry R, Boeke JD. Functional dissection of lysine deacetylases reveals that HDAC1 and p300 regulate AMPK. Nature. 2012;482:251–255. doi: 10.1038/nature10804. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 4.Jiang T, Zhou X, Taghizadeh K, Dong M, Dedon PC. N-formylation of lysine in histone proteins as a secondary modification arising from oxidative DNA damage. Proc Natl Acad Sci U S A. 2007;104:60–65. doi: 10.1073/pnas.0606775103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wisniewski JR, Zougman A, Mann M. N ε-formylation of lysine is a widespread post-translational modification of nuclear proteins occurring at residues involved in regulation of chromatin function. Nucleic Acids Res. 2008;36:570–577. doi: 10.1093/nar/gkm1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Y, Sprung R, Tang Y, Ball H, Sangras B, Kim SC, Falck JR, Peng J, Gu W, Zhao Y. Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol Cell Proteomics. 2007;6:812–819. doi: 10.1074/mcp.M700021-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang K, Chen Y, Zhang Z, Zhao Y. Identification and verification of lysine propionylation and butyrylation in yeast core histones using PTMap software. J Proteome Res. 2009;8:900–906. doi: 10.1021/pr8005155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, Lu Z, Ye Z, Zhu Q, Wysocka J, Ye Y, Khochbin S, Ren B, Zhao Y. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146:1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tan M, Peng C, Anderson KA, Chhoy P, Xie Z, Dai L, Park J, Chen Y, Huang H, Zhang Y, Ro J, Wagner GR, Green MF, Madsen AS, Schmiesing J, Peterson BS, Xu G, Ilkayeva OR, Muehlbauer MJ, Braulke T, Muhlhausen C, Backos DS, Olsen CA, McGuire PJ, Pletcher SD, Lombard DB, Hirschey MD, Zhao Y. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 2014;19:605–617. doi: 10.1016/j.cmet.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, Kim J, Woo J, Kim JH, Choi BH, He B, Chen W, Zhang S, Cerione RA, Auwerx J, Hao Q, Lin H. Sirt5 Is an NAD-Dependent Protein Lysine Demalonylase and Desuccinylase. Science. 2011;334:806–809. doi: 10.1126/science.1207861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peng C, Lu Z, Xie Z, Cheng Z, Chen Y, Tan M, Luo H, Zhang Y, He W, Yang K, Zwaans BM, Tishkoff D, Ho L, Lombard D, He TC, Dai J, Verdin E, Ye Y, Zhao Y. The first identification of lysine malonylation substrates and its regulatory enzyme. Mol Cell Proteomics. 2011;10:M111012658. doi: 10.1074/mcp.M111.012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xie Z, Dai J, Dai L, Tan M, Cheng Z, Wu Y, Boeke JD, Zhao Y. Lysine succinylation and lysine malonylation in histones. Mol Cell Proteomics. 2012;11:100–107. doi: 10.1074/mcp.M111.015875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang H, Khan S, Wang Y, Charron G, He B, Sebastian C, Du J, Kim R, Ge E, Mostoslavsky R, Hang HC, Hao Q, Lin H. SIRT6 regulates TNF-alpha secretion through hydrolysis of long-chain fatty acyl lysine. Nature. 2013;496:110–113. doi: 10.1038/nature12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin H, Su X, He B. Protein lysine acylation and cysteine succination by intermediates of energy metabolism. ACS Chem Biol. 2012;7:947–960. doi: 10.1021/cb3001793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang YY, Ascano JM, Hang HC. Bioorthogonal chemical reporters for monitoring protein acetylation. J Am Chem Soc. 2010;132:3640–3641. doi: 10.1021/ja908871t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bürli RW, Luckhurst CA, Aziz O, Matthews KL, Yates D, Lyons KA, Beconi M, McAllister G, Breccia P, Stott AJ, Penrose SD, Wall M, Lamers M, Leonard P, Müller I, Richardson CM, Jarvis R, Stones L, Hughes S, Wishart G, Haughan AF, O’Connell C, Mead T, McNeil H, Vann J, Mangette J, Maillard M, Beaumont V, Munoz-Sanjuan I, Dominguez C. Design, synthesis, and biological evaluation of potent and selective Class IIa Histone Deacetylase (HDAC) inhibitors as a potential therapy for Huntington’s Disease. J Med Chem. 2013;56:9934–9954. doi: 10.1021/jm4011884. [DOI] [PubMed] [Google Scholar]
- 17.Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T, Mazitschek R. Chemical phylogenetics of histone deacetylases. Nat Chem Biol. 2010;6:238–243. doi: 10.1038/nchembio.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lahm A, Paolini C, Pallaoro M, Nardi MC, Jones P, Neddermann P, Sambucini S, Bottomley MJ, Lo Surdo P, Carfí A, Koch U, De Francesco R, Steinkühler C, Gallinari P. Unraveling the hidden catalytic activity of vertebrate Class IIa histone deacetylases. Proc Natl Acad Sci U S A. 2007;104:17335–17340. doi: 10.1073/pnas.0706487104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tessier P, Smil DV, Wahhab A, Leit S, Rahil J, Li Z, Deziel R, Besterman JM. Diphenylmethylene hydroxamic acids as selective Class IIa histone deacetylase inhibitors. Bioorg Med Chem Lett. 2009;19:5684–5688. doi: 10.1016/j.bmcl.2009.08.010. [DOI] [PubMed] [Google Scholar]
- 20.Inks ES, Josey BJ, Jesinkey SR, Chou CJ. A novel class of small molecule inhibitors of HDAC6. ACS Chem Biol. 2012;7:331–339. doi: 10.1021/cb200134p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Madsen AS, Olsen CA. Profiling of substrates for zinc-dependent lysine deacylase enzymes: HDAC3 exhibits decrotonylase activity in vitro. Angew Chem, Int Ed. 2012;51:9083–9087. doi: 10.1002/anie.201203754. [DOI] [PubMed] [Google Scholar]
- 22.Butler KV, Kalin J, Brochier C, Vistoli G, Langley B, Kozikowski AP. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, Tubastatin A. J Am Chem Soc. 2010;132:10842–10846. doi: 10.1021/ja102758v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chou CJ, Herman D, Gottesfeld JM. Pimelic diphenylamide 106 is a slow, tight-binding inhibitor of class I histone deacetylases. J Biol Chem. 2008;283:35402–35409. doi: 10.1074/jbc.M807045200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci U S A. 2003;100:4389–4394. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.El-Ansary AK, Ben Bacha AG, Al-Ayahdi LY. Plasma fatty acids as diagnostic markers in autistic patients from Saudi Arabia. Lipids Health Dis. 2011;10:62–62. doi: 10.1186/1476-511X-10-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cardona ME, Stern S, Tjellstrom B, Norin E, Midtvedt T, Collinder E. Correlation between faecal iso-butyric and iso-valeric acids in different species. Microb Ecol Health Dis. 2005;17:177–182. [Google Scholar]
- 27.Saito A, Yamashita T, Mariko Y, Nosaka Y, Tsuchiya K, Ando T, Suzuki T, Tsuruo T, Nakanishi O. A synthetic inhibitor of histone deacetylase, MS-275, with marked in vivo antitumor activity against human tumors. Proc Natl Acad Sci U S A. 1999;96:4592–4597. doi: 10.1073/pnas.96.8.4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jayne S, Zwartjes CG, van Schaik FM, Timmers HT. Involvement of the SMRT/NCoR-HDAC3 complex in transcriptional repression by the CNOT2 subunit of the human Ccr4-Not complex. Biochem J. 2006;398:461–467. doi: 10.1042/BJ20060406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Richon VM, Emiliani S, Verdin E, Webb Y, Breslow R, Rifkind RA, Marks PA. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc Natl Acad Sci U S A. 1998;95:3003–3007. doi: 10.1073/pnas.95.6.3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discovery. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 31.Wagner FF, Lundh M, Kaya T, McCarren P, Zhang YL, Chattopadhyay S, Gale JP, Galbo T, Fisher SL, Meier BC, Vetere A, Richardson S, Morgan NG, Christensen DP, Gilbert TJ, Hooker JM, Leroy M, Walpita D, Mandrup-Poulsen T, Wagner BK, Holson EB. An isochemogenic set of inhibitors to define the therapeutic potential of Histone Deacetylases in β-cell protection. ACS Chem Biol. 2016;11:363–374. doi: 10.1021/acschembio.5b00640. [DOI] [PubMed] [Google Scholar]
- 32.Gao Z, He Q, Peng B, Chiao PJ, Ye J. Regulation of nuclear translocation of HDAC3 by IkappaBalpha is required for tumor necrosis factor inhibition of peroxisome proliferator-activated receptor gamma function. J Biol Chem. 2006;281:4540–4547. doi: 10.1074/jbc.M507784200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kiernan R, Brès V, Ng RWM, Coudart MP, El Messaoudi S, Sardet C, Jin DY, Emiliani S, Benkirane M. Post-activation turn-off of NF-κB-dependent transcription is regulated by acetylation of p65. J Biol Chem. 2003;278:2758–2766. doi: 10.1074/jbc.M209572200. [DOI] [PubMed] [Google Scholar]
- 34.Ryu HW, Shin DH, Lee DH, Choi J, Han G, Lee KY, Kwon SH. HDAC6 deacetylates p53 at lysines 381/382 and differentially coordinates p53-induced apoptosis. Cancer Lett. 2017;391:162–171. doi: 10.1016/j.canlet.2017.01.033. [DOI] [PubMed] [Google Scholar]
- 35.Zhao Y, Lu S, Wu L, Chai G, Wang H, Chen Y, Sun J, Yu Y, Zhou W, Zheng Q, Wu M, Otterson GA, Zhu WG. Acetylation of p53 at lysine 373/382 by the Histone Deacetylase inhibitor depsipeptide induces expression of p21Waf1/Cip1. Mol Cell Biol. 2006;26:2782–2790. doi: 10.1128/MCB.26.7.2782-2790.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang D, Shi Y, Jang L, Wang K, Xiao W, Xiao X. Heat shock response inhibits release of high mobility group box 1 protein induced by endotoxin in murine macrophages. Shock. 2005;23:434–440. doi: 10.1097/01.shk.0000159556.95285.df. [DOI] [PubMed] [Google Scholar]
- 37.Wang H, Yang H, Czura CJ, Sama AE, Tracey KJ. HMGB1 as a late mediator of lethal systemic inflammation. Am J Respir Crit Care Med. 2001;164:1768–1773. doi: 10.1164/ajrccm.164.10.2106117. [DOI] [PubMed] [Google Scholar]
- 38.Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, Rubartelli A, Agresti A, Bianchi ME. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003;22:5551–5560. doi: 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 40.Edrissi B, Taghizadeh K, Dedon PC. Quantitative analysis of histone modifications: formaldehyde is a source of pathological n(6)-formyllysine that is refractory to histone deacetylases. PLoS Genet. 2013;9:e1003328. doi: 10.1371/journal.pgen.1003328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Palijan A, Fernandes I, Bastien Y, Tang L, Verway M, Kourelis M, Tavera-Mendoza LE, Li Z, Bourdeau V, Mader S, Yang XJ, White JH. Function of Histone Deacetylase 6 as a cofactor of nuclear receptor coregulator LCoR. J Biol Chem. 2009;284:30264–30274. doi: 10.1074/jbc.M109.045526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suzuki T, Kasuya Y, Itoh Y, Ota Y, Zhan P, Asamitsu K, Nakagawa H, Okamoto T, Miyata N. Identification of highly selective and potent Histone Deacetylase 3 inhibitors using click chemistry-based combinatorial fragment assembly. PLoS One. 2013;8:e68669. doi: 10.1371/journal.pone.0068669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Y, Stowe RL, Pinello CE, Tian G, Madoux F, Li D, Zhao LY, Li JL, Wang Y, Wang Y, Ma H, Hodder P, Roush WR, Liao D. Identification of HDAC Inhibitors with Benzoylhydrazide scaffold that selectively inhibit Class I HDACs. Chem Biol. 2015;22:273–284. doi: 10.1016/j.chembiol.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McClure JJ, Zhang C, Inks ES, Peterson YK, Li J, Chou CJ. Development of allosteric hydrazide-containing Class I Histone Deacetylase inhibitors for use in Acute Myeloid Leukemia. J Med Chem. 2016;59:9942–9959. doi: 10.1021/acs.jmedchem.6b01385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen X, Barozzi I, Termanini A, Prosperini E, Recchiuti A, Dalli J, Mietton F, Matteoli G, Hiebert S, Natoli G. Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc Natl Acad Sci U S A. 2012;109:E2865–2874. doi: 10.1073/pnas.1121131109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kiernan R, Bres V, Ng RW, Coudart MP, El Messaoudi S, Sardet C, Jin DY, Emiliani S, Benkirane M. Post-activation turn-off of NF-kappa B-dependent transcription is regulated by acetylation of p65. J Biol Chem. 2003;278:2758–2766. doi: 10.1074/jbc.M209572200. [DOI] [PubMed] [Google Scholar]
- 47.Hatano E, Bennett BL, Manning AM, Qian T, Lemasters JJ, Brenner DA. NF-kappaB stimulates inducible nitric oxide synthase to protect mouse hepatocytes from TNF-alpha- and Fas-mediated apoptosis. Gastroenterology. 2001;120:1251–1262. doi: 10.1053/gast.2001.23239. [DOI] [PubMed] [Google Scholar]
- 48.Griscavage JM, Wilk S, Ignarro LJ. Inhibitors of the proteasome pathway interfere with induction of nitric oxide synthase in macrophages by blocking activation of transcription factor NF-kappa B. Proc Natl Acad Sci U S A. 1996;93:3308–3312. doi: 10.1073/pnas.93.8.3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jones E, Adcock IM, Ahmed BY, Punchard NA. Modulation of LPS stimulated NF-kappaB mediated Nitric Oxide production by PKCepsilon and JAK2 in RAW macrophages. J Inflammation. 2007;4:23. doi: 10.1186/1476-9255-4-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lu B, Wang H, Andersson U, Tracey KJ. Regulation of HMGB1 release by inflammasomes. Protein Cell. 2013;4:163–167. doi: 10.1007/s13238-012-2118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundback P, Valdes-Ferrer SI, Olofsson PS, Kalb T, Roth J, Zou Y, Erlandsson-Harris H, Yang H, Ting JP, Wang H, Andersson U, Antoine DJ, Chavan SS, Hotamisligil GS, Tracey KJ. Novel role of PKR in inflammasome activation and HMGB1 release. Nature. 2012;488:670–674. doi: 10.1038/nature11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Willingham SB, Allen IC, Bergstralh DT, Brickey WJ, Huang MT, Taxman DJ, Duncan JA, Ting JP. NLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways. J Immunol. 2009;183:2008–2015. doi: 10.4049/jimmunol.0900138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stammler D, Eigenbrod T, Menz S, Frick JS, Sweet MJ, Shakespear MR, Jantsch J, Siegert I, Wolfle S, Langer JD, Oehme I, Schaefer L, Fischer A, Knievel J, Heeg K, Dalpke AH, Bode KA. Inhibition of Histone Deacetylases Permits Lipopolysaccharide-Mediated Secretion of Bioactive IL-1beta via a Caspase-1-Independent Mechanism. J Immunol. 2015;195:5421–5431. doi: 10.4049/jimmunol.1501195. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.