

Abstract
Emerging viral infections are difficult to control as heterogeneous members periodically cycle in and out of humans and zoonotic hosts, complicating the development of specific antiviral therapies and vaccines. Coronaviruses (CoVs) have a proclivity to spread rapidly into new host species causing severe disease. SARS-CoV and MERS-CoV successively emerged causing severe epidemic respiratory disease in immunologically naïve human populations throughout the globe. Broad-spectrum therapies capable of inhibiting CoV infections would address an immediate unmet medical need and could be invaluable in the treatment of emerging and endemic CoV infections. Here we show that a nucleotide prodrug GS-5734, currently in clinical development for treatment of Ebola virus disease, can inhibit SARS-CoV and MERS-CoV replication in multiple in vitro systems including primary human airway epithelial cell cultures with submicromolar IC50 values. GS-5734 was also effective against bat-CoVs, prepandemic bat-CoVs and circulating contemporary human CoV in primary human lung cells, thus demonstrating broad-spectrum anti-CoV activity. In a mouse model of SARS-CoV pathogenesis, prophylactic and early therapeutic administration of GS-5734 significantly reduced lung viral load and improved clinical signs of disease as well as respiratory functions. These data provide substantive evidence that GS-5734 may prove effective against endemic MERS-CoV in the Middle East, circulating human CoV, and possibly most importantly, emerging CoV of the future.
Introduction
The genetically diverse coronavirus (CoV) family, currently comprised of four genogroups: 1 (alpha), 2 (beta), 3 (gamma) and 4 (delta), infects birds and a variety of mammals. Thus far, only CoV groups 1 and 2 are known to infect humans. Although CoV replication machinery exhibits substantial proof reading activity, replication of viral genomic RNA is inherently error prone, driving the existence of genetically related yet diverse quasispecies(1). Most CoV strains are narrow in their host range but zoonotic CoV have a proclivity to jump into new host species(2). Severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV) are recent examples of newly emerging CoV that caused severe disease in immunologically naïve human populations. SARS-CoV emerged in Guangdong China in 2002 and with the aid of commercial air travel, spread rapidly throughout the globe causing over 8000 cases with 10% mortality(2). In 2012, it was discovered that MERS-CoV evolved to infect humans from bats by way of an intermediate camel host causing over 1700 cases in twenty-seven countries with almost 40% mortality and like SARS-CoV air travel has fueled global spread to twenty seven countries(2). MERS-CoV is endemic in the Middle East and serologic studies in the Kingdom of Saudi Arabia and Kenya suggest fairly frequent infections in humans (>45,000 persons) (3, 4). The SARS-CoV epidemic ended over a decade ago but several SARS-like CoVs have been isolated from bats that efficiently use the human angiotensin converting enzyme 2 (ACE2) receptor, replicate to high titer in primary human airway cells and are resistant to existing therapeutic antibodies and vaccines(5, 6). With increasing overlap of human and wild animal ecologies, the potential for novel CoV emergence into humans is great(2). Broad-spectrum CoV therapies capable of inhibiting known human CoV would address an immediate unmet medical need and could be an invaluable treatment in the event of novel CoV emergence in the future.
Currently, there are no approved specific antiviral therapies for CoV in humans. Attempts made to treat both SARS- and MERS-CoV patients with approved antivirals (i.e. ribavirin, lopinivir-ritonavir) and immune modulators (i.e. corticosteroids, interferons, etc.) has not been effective in randomized controlled trials(7). Clinical development of effective CoV-specific direct acting antivirals (DAAs) has been elusive even though there are several conserved druggable CoV enzyme targets including 3CL protease, PL protease, and NSP12 replicase (7). In 2016, Warren et. al reported the in vivo antiviral efficacy of a small molecule monophosphoramidate prodrug of an adenosine analogue, GS-5734, against Ebola virus in non-human primates(8). Since the mechanism of action (MOA) of GS-5734 for Ebola virus is inhibition of the viral RNA-dependent RNA polymerase (RdRp) and prior work had suggested weak activity of the nucleoside component of GS-5734 against SARS-CoV (9), we sought to assess the antiviral potency and breadth of activity of GS-5734 against a diverse panel of human and zoonotic CoV.
Results
GS-5734 prevents SARS- and MERS-CoV replication in human airway epithelial cells
GS-5734 is a prodrug that requires metabolism by the host cell to the pharmacologically active triphosphate (TP) to inhibit virus replication (9). To determine if GS-5734 could inhibit replication of highly pathogenic human CoV, we first evaluated antiviral activity and cytotoxicity in the continuous human lung epithelial cell line Calu-3 2B4 (2B4)(10). GS-5734 inhibited MERS-CoV replication in 2B4 cells with an average half-maximum effective concentration (IC50) value of 0.025 μM (Fig. 1A and S1). Importantly, we did not observe any measureable cytotoxicity at concentrations up to 10 μM (Fig. 1B and S1) thus demonstrating the 50% cytotoxic concentration (i.e. CC50) for GS-5734 is in excess of 10μM (CC50/IC50 = therapeutic index > 400) in 2B4 cells. With incubation of 1 μM GS-5734, the average intracellular concentration of the pharmacologically active TP of GS-5734 in Calu-3-2B4 was 2.79 μM during 48hr treatment (Fig. 1C). Taken together, these results suggest substantial inhibition of CoV replication will be achieved at low micromolar concentrations of the TP in lung.
Figure 1. MERS-CoV antiviral efficacy, toxicity and metabolism of GS-5734 in 2B4 cells.

(A) Mean percent inhibition of MERS-CoV replication by GS-5734. 2B4 cells were infected in triplicate with MERS-CoV nLuc at an MOI of 0.08 in the presence of varying concentrations of GS-5734 for 48hr after which replication was measured through quantitation of MERS-CoV expressed nano luciferase (nLUC). (B) Cytoxicity in 2B4 cells treated similarly to those in A. Viability was measured via CellTiter-Glo. Data for A and B is representative of three independent experiments. (C) Measurement of intracellular nucleotide triphosphate (NTP) in 2B4 cells. In three independent experiments, triplicate wells of cells were treated with 1 μM GS-5734 and harvested over time to measure NTP via LC/MS.
Primary human airway epithelial (HAE) cell cultures are among the most biologically relevant in vitro models of the lung, recapitulating the cellular complexity and physiology of the human conducting airway(11). We assessed the antiviral activity of GS-5734 against SARS- and MERS-CoV in HAE cultures. A dose-dependent reduction in replication approaching 1 log10 was observed at 0.1μM and exceeded 2 log10 at 1μM GS-5734 as compared to untreated controls (Fig. 2A and B) with average IC50 values of 0.069 μM (SARS-CoV) and 0.074 μM (MERS-CoV). In parallel, we assessed the abundance of intracellular genomic (ORF1a) and subgenomic viral RNA (ORFN) via quantitative reverse transcriptase polymerase chain reaction (qRT-PCR). A dose-dependent reduction in both ORF1a and ORFN was observed for both SARS- and MERS-CoV (Fig. 2C and D), consistent with titer reduction. The numbers of MERS-CoV infected HAE cells also diminished with increasing dose of GS-5734 as observed by microscopy (Fig. 2E). To assess cytotoxicity of GS-5734 in HAE, we first we measured the transcript levels of multiple pro- and anti-apoptotic factors within the signaling cascades of two different death receptors, TNF and FAS (Fig. S2A and B). Unlike the positive control drug, staurosporine, which uniformly upregulated transcription of all apoptosis factors measured, we did not observe a dose-dependent effect with GS-5734 treatment at concentrations that inhibit CoV replication. We then measured cytotoxicity via CellTiter-Glo assay in HAE treated with 10 μM or 0.1 μM GS-5734, or DMSO for 48hr. As expected, GS-5734 treatment was similar to that of DMSO (Fig. S2C, CC50 in HAE >10 μM, therapeutic index >100). To assess cytotoxicity of GS-5734 in an additional primary human lung cell type, we exposed normal human bronchiolar epithelial (NHBE) cell cultures to dilutions of GS-5734 or two known cytotoxic compounds, puromycin or staurosporin (Fig. S3). In NHBE, the average CC50 for GS-5734 was determined to be 45 μM, which is 1800-fold above the observed for IC50 value for MERS-CoV in 2B4 cells (0.025 μM) and 600-fold above the observe IC50 value for MERS-CoV in HAE cells (0.074 μM) (Fig. S3A and B). Together, we demonstrate antiviral efficacy in HAE against both SARS- and MERS-CoV at concentrations that are at least 100-fold lower than those with observable cytotoxicity.
Figure 2. GS-5734 prevents SARS- and MERS-CoV replication in human airway epithelial cells.

(A) Antiviral efficacy of GS-5734 against MERS-CoV in primary human airway epithelial (HAE) cell cultures. HAE cells were infected with MERS-CoV RFP at an MOI of 0.5 in duplicate in the presence of GS-5734 for 48hr after which apical washes were collected for virus titration. Representative data from two separate experiments with three different cell donors is displayed. (B) Antiviral efficacy of GS-5734 against SARS-CoV in HAE cells. Cultures were infected with SARS-CoV GFP and treated and analyzed as described in A. (C) Quantitative RT-PCR for MERS-CoV ORF1 and ORFN mRNA. Total RNA was isolated from the cultures in panel A for RT-PCR analysis. (D) Quantitative RT-PCR for SARS-CoV ORF1 and ORFN in cells from C as described in B. (E) HAE cells were infected with MERS-CoV RFP and treated with GS-5734 as in A. Nuclei were stained with Hoechst 33258 prior to fluorescent imaging.
GS-5734 is effective against a diverse array of human and zoonotic CoV in HAE
CoV host specificity and entry into host cells is guided by the viral spike glycoprotein (S) whose extensive genetic variation is a reflection of host species diversity and variation in virus receptor usage (Fig. 3A). Conversely, the CoV RdRp nsp12 is highly conserved between CoV especially within genogroups (Fig. 3A), making it a potentially broadly applicable drug target.
Figure 3. GS-5734 is effective against a diverse array of human and zoonotic CoV in HAE.

(A) Neighbor joining trees created with representatives from all four CoV genogroups showing the genetic similarity of CoV nsp12 (RNA dependent RNA polymerase) and CoV Spike glycoprotein, which mediates host tropism and entry into cells. Text color of virus strain label corresponds to virus host species on left. (B) Top panel, antiviral efficacy of GS-5734 in HAE cells against group 1 human circulating CoV (hCoV-NL63) and bat CoV from group 2b (HKU3, SHC014, WIV1) and 2c (HKU5). HAE cells were infected at an MOI of 0.5 in the presence of GS-5734 in duplicate. After 48hr, virus produced was titrated via plaque assay. Each data point represents the titer per culture. Bottom panel, quantitative RT-PCR for CoV ORF1 and ORFN mRNA in total RNA from the cultures in top panel.
Since we observed an antiviral effect against two members of genogroup 2 (SARS- and MERS-CoV), we sought to assess the breadth of antiviral activity against a genetically diverse array of human and zoonotic bat CoV in HAE cells. Treatment of HAE cultures with GS-5734 infected with HCoV-NL63, a circulating group 1 human CoV that typically causes bronchitis(12), resulted in a pronounced 3 log10 reduction in virus production at 0.1μM and undetectable virus at higher concentrations (Fig. 3B). Likewise, GS-5734 treatment inhibited replication of very diverse SARS-CoV-like group 2b (HKU3, WIV1 and SHC014) and MERS-CoV-like group 2c (HKU5) bat coronaviruses. Among these, WIV1 and SHC014 pose particular concern as “prepandemic strains”, which can infect HAE cultures without adaptation and are thus, poised for emergence in humans(5, 6). With 1μM GS-5734, infectious virus production of bat CoV was reduced by 1.5–2 log10 and levels of viral genomes and subgenomic transcripts were reduced 1–2 log10 (Fig. 3B). Together, these data suggest that GS-5734 can inhibit a broad range of diverse CoV including circulating human, zoonotic bat CoV and prepandemic zoonotic CoV.
Prophylactic treatment with GS-5734 reduces SARS-CoV disease
GS-5734 has relatively poor plasma stability in mice (i.e. half-life < 5min) due to expression of a secreted carboxylesterase 1c (Ces1c) absent in humans(13). Plasma stability of GS-5734 was markedly increased (half-life ~25min, Fig. S4) in mice genetically deleted for Ces1c (Ces1c−/−). We confirmed that SARS-CoV pathogenesis as measured by weight loss and lung viral titers was similar in wild-type (WT) C57BL/6J and Ces1c−/− mice through the infection of age and sex matched mice from both strains (Fig. S4). We then assessed the pharmacokinetic (PK) profile in Ces1c−/− mice dosed subcutaneously (s.c.) with 50 mg/kg once daily (QD) or 25 mg/kg twice daily (BID). Plasma concentrations of prodrug diminished rapidly, accompanied by transient exposure to the alanine metabolite (Ala-Met) and more persistent exposure to the nucleoside analog (Fig. 4A). The plasma PK profile in esterase deficient mice was similar to that reported previously in monkeys (8) but tissue accumulation of metabolites was ~10-fold less efficient in mice, suggesting that high doses and corresponding plasma exposures are necessary to obtain lung TP levels similar to those predicted in human. The metabolite profile in the lung showed the TP to be the dominant intracellular metabolite, establishing the less technically challenging assessment of total lung metabolite levels as a close approximation of TP (Fig. 4B). Although both 50mg/kg QD and 25mg/kg BID resulted in target maximal lung levels, tissue activation was not only less efficient in mouse but also, the TP had a substantially shorter half-life in mouse lung (~3hr) relative to that observed in human lung cells in vitro or non-human primate lung in vivo (half-life ~20hr; Fig. S5). Therefore, only the BID-dosing regimen was able to maintain lung levels between 12 and 24hr consistent with those anticipated in humans and sufficient to maintain CoV inhibition over the dosing interval (Fig 4C).
Figure 4. Pharmacokinetics of GS-5734 in Ces1c−/− mice.
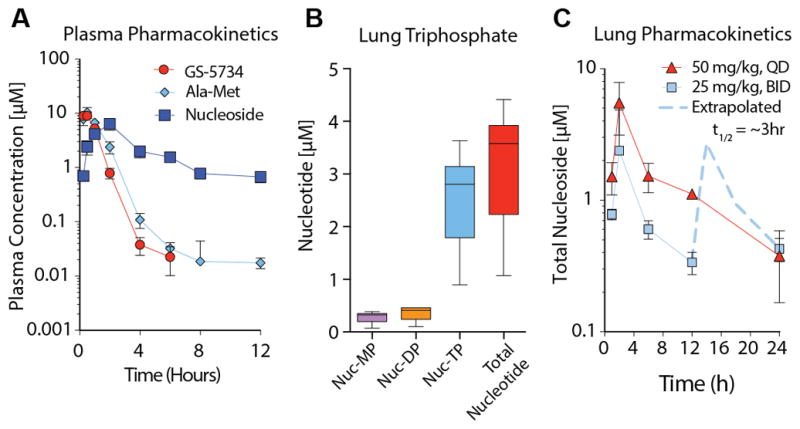
(A) Pharmacokinetics in Ces1c−/− mouse plasma following subcutaneous administration of 25mg/kg GS-5734. Longitudinal plasma samples were taken to measure prodrug GS-5734, intermediate metabolites Ala-Met, and nucleoside by LC/MS. (B) Lung triphosphate in Ces1c−/− mouse lung 4 hr following subcutaneous administration of 50mg/kg GS-5734. Nucleotide monophosphate (Nuc-MP), diphosphate (Nuc-DP), triphosphate (Nuc-TP) and total nucleotide (sum of Nuc-MP, DP and TP) is displayed. (C) Pharmacokinetics of total nucleotide in Ces1c−/− mouse lung following subcutaneous administration of GS-5734 at 25mg/kg twice daily (BID) or 50mg/kg once daily (QD).
Mouse models of SARS-CoV disease faithfully recapitulate many aspects of SARS-CoV pathogenesis in humans including anorexia, high titers of virus replication in the lung, the development of acute respiratory distress syndrome (ARDS) as well as an age related exacerbation of disease(14). As shown in Figure 5A, prophylactic administration once per day at 50 mg/kg or twice daily at 25 mg/kg ameliorated SARS-CoV induced weight loss seen with vehicle treatment. Virus titers in the lung were significantly reduced (P < 0.05) on both 2 and 5 dpi in GS-5734-treated mice as compared to vehicle control animals (Fig. 5B). In addition, levels of viral antigen staining in lung sections of GS-5734-treated animals were significantly lower (P < 0.05) as compared to vehicle-treated animals (Fig 5C and D). Treatment with GS-5734 also reduced SARS-CoV-induced lung pathology including denuding bronchiolitis, perivascular accumulation of inflammatory infiltrates (i.e. “cuffing”), and intra-alveolar edema associated with diffuse alveolar damage as compared to vehicle-treated animals (Fig 5E and S6).
Figure 5. Prophylactic treatment with GS-5734 reduces SARS-CoV disease.

(A) Percent starting weight of Ces1c−/− mice infected with 104 pfu SARS-CoV MA15 treated beginning -1dpi with either vehicle (n = 42), 25mg/kg twice daily (BID, n = 25) or 50mg/kg once daily (QD, n = 28) GS-5734. (B) SARS-CoV lung titer in panel A mice at 2dpi (left) (Vehicle N = 11, 50mg/kg N = 11, 25mg/kg N = 5) or 5dpi (right) (Vehicle N = 13, 50mg/kg N = 13, 25mg/kg N = 4). (C) Quantitation of SARS-CoV antigen in lung sections in panel A mice at 2dpi (left) (Vehicle N = 15, 50mg/kg N = 12, 25mg/kg N = 7) or 5dpi (right) (Vehicle N = 10, 50mg/kg N = 12, 25mg/kg N = 4). (D) Photomicrographs of SARS-CoV antigen staining (brown) and nuclei (blue) in lung sections from 2 and 5 dpi. (E) Photomicrographs of hematoxylin and eosin stained mouse lung sections from 2dpi highlighting the conducting airway lumen (aw). (F) Whole-body plethysmography (WBP) to measure pulmonary function in panel A mice. Penh is a surrogate measure of bronchoconstriction. Expiration time is the time taken to release one breath. End of expiratory pause is the time between breaths. Symbols and error bars for panels A, B, D, and I represent the mean and standard deviation. The boxes encompass the 25–75th percentile while the whiskers represent the range in C, E and F. Asterisk indicates statistical significance (p<0.05) by two-way ANOVA with Tukey’s multiple comparison test for D, F, and I and Kruskal-Wallis in E.
Prophylactic treatment with GS-5734 prevented defects in pulmonary function seen in SARS-CoV infected vehicle-treated animals as measured by whole body plethysmography (WBP), an extremely sensitive means of quantitating pulmonary function (15). Penh, a surrogate measure of airway resistance or accumulation of debris in the airway, was significantly (P < 0.05) elevated in vehicle treated animals as compared to those treated with GS-5734 (Fig. 5F). Metrics of labored breathing such as increased exhalation time and extended pause in between the end of one breath and the beginning of the next were increased in vehicle-treated animals as compared to those administered GS-5734 (Fig. 5F).
Therapeutic post-exposure administration of GS-5734 mitigates disease
Since prophylactic administration of GS-5734 reduced virus lung titers, improved lung function and ameliorated symptoms of SARS-CoV disease, it was of interest to determine if therapeutic treatment would also be effective. First, we compared the antiviral efficacy of 25 mg/kg GS-5734 BID beginning at −1 dpi (i.e. prophylactic) or +1 dpi (i.e. therapeutic). Therapeutic GS-5734 substantially reduced the SARS-CoV induced weight loss in infected animals (Fig. 6A and B) and significantly suppressed virus lung titers (P = 0.0059) thus demonstrating that therapeutic administration of GS-5734 can reduce disease and suppress replication during an ongoing infection (Fig. 6C). Importantly, therapeutic treatment significantly (P = 0.003) improved pulmonary function (i.e. reduced Penh scores) as compared to vehicle-treated controls (Fig. 6D). We then explored the therapeutic potential of GS-5734 given 2dpi, which is after virus replication and lung airway epithelial damage have peaked (Fig. S7). Disease severity and survival did not differ with treatment but we observed a significant reduction (P < 0.05) in SARS-CoV lung titers of GS-5734 treated animals at 6dpi. These data suggest that reductions in viral load after peak lung titers were achieved were insufficient to improve outcomes after the immunopathological phase of disease had been initiated. Thus in the mouse, if given prior to the peak of SARS-CoV replication and peak damage to the airway epithelium, GS-5734 can improve pulmonary function, reduce viral loads and diminish disease.
Figure 6. Therapeutic post-exposure administration of GS-5734 mitigates disease.

(A) Percent starting weight of 27–28 week old female Ces1c−/− infected with 103 pfu SARS-CoV MA15 treated twice daily with vehicle or 25mg/kg GS-5734 beginning on either -1dpi (vehicle n = 5, GS-5734 n = 10) or +1dpi (vehicle n = 4, GS-5734 n = 11). GS-5734-treated animal weights were statistically different (p<0.05) from vehicle-treated 3 and 4 dpi for prophylactic and 4dpi for therapeutic groups by two-way ANOVA with Tukey’s multiple comparison test. (B) Percent starting weights of mice in panel A at 4 dpi. (C) SARS-CoV lung titer in mice infected and treated as described in panel A. Asterisks indicate statistical significance (p<0.05) by Mann-Whitney test for panels B and C. (D) Whole-body plethysmography (WBP) was used to measure pulmonary function in mice infected and treated as described in panel A. Penh is a surrogate measure of bronchoconstriction or airway obstruction. Asterisks indicate statistical significance by two-way ANOVA with Sidek’s multiple comparison test.
Discussion
Emerging viral infections represent a critical global health concern as specific antiviral therapies and vaccines are usually lacking. To maximize the potential public health benefit of therapeutics against emerging viruses, they should be efficacious against past (i.e. SARS-CoV), current (i.e. MERS-CoV), and future emerging viral threats. Knowledge of the spectrum of therapeutic efficacy is essential for making informed clinical decisions, especially in the early stages of an outbreak. Since zoonotic CoV emergence is driven by an amalgamation of human, wild-animal and viral factors, it is difficult to gauge zoonotic CoV emergence potential based on viral genome sequences alone(2). In this manuscript, we provide an example of a successful public-private partnership that combines metagenomics, synthetic biology, primary human cell culture models, drug metabolism, pharmacokinetics, and in vivo models of viral pathogenesis to demonstrate broad-spectrum activity of a drug candidate against a virus family prone to emergence (Fig. S8). Although we demonstrated broad-spectrum efficacy against human and zoonotic CoV from multiple CoV genogroups, we have not yet assessed antiviral efficacy for all CoV genogroups, which is a limitation of our current study. Nevertheless, our panel of reconstructed human and zoonotic bat CoV was essential to determine if GS-5734 would be efficacious against highly divergent emerged (SARS-CoV), emerging (MERS-CoV) and circulating zoonotic strains with pandemic potential (i.e. WIV1, SHC014) (5, 6, 16, 17). In the future, the rapid development of vaccines, therapies and diagnostics for emerging viruses will be dependent on the reconstruction and the in vitro and in vivo adaptation of these viruses in the laboratory.
Here we report the broad-spectrum antiviral efficacy of a small molecule against multiple genetically distinct CoV in vitro and in vivo. Current vaccine and human monoclonal antibody approaches have proven to be effective but typically have limited breadth of protection due to antigenic diversity in the CoV S glycoprotein(5, 6). Conversely, RdRp-targeting therapies like GS-5734 are more likely to be broadly active against past, current and future CoV due to the inherent genetic conservation of the CoV replicase. As evidenced by the failure of the nucleoside prodrug, balapirivir, to translate in vitro efficacy into in vivo efficacy in mice and humans, antiviral drug candidates should be thoroughly evaluated in the most biologically relevant models of pathogenesis to maximize clinical translatability(18). Cell-type specific differences in the active transport and/or metabolism of nucleoside analogs may affect the antiviral profile(19). Thus, we aimed to expand upon previous in vitro studies of SARS- and MERS-CoV antiviral efficacy that were limited to monkey kidney cancer cell lines (8, 9). Similar in cellular complexity and physiology to the human conducting airway, the HAE cell culture contains mucus-secreting cells, basolateral cells and some of the main target cells of SARS-CoV (ciliated epithelial cells) and MERS-CoV (non-ciliated epithelial cells) in vivo(11, 20). With our HAE cell antiviral efficacy data, we provide strong evidence that GS-5734 will be taken up and metabolized in cells targeted by multiple human and zoonotic CoV in the human lung.
Preclinical in vivo antiviral efficacy studies provide insight into the pharmacokinetic-pharmacodynamic (PK/PD) relationship of a drug from which effective dosing regimens can be extrapolated for human clinical trial. To maximize the utility of preclinical PK/PD studies, the use of animal models that accurately recapitulate human disease is essential. Multiple aspects of the human disease are captured by the mouse adapted SARS-CoV (SARS-CoV MA15) model used herein, including high titer virus replication limited to the lung, the development of ARDS, age related exacerbation of disease and death. In contrast to humans infected with SARS-CoV where viral titers peak 7–10 days after the onset of symptoms, virus titers in the lungs of SARS-CoV MA15 infected C57BL/6 mice rapidly increase 4–5 logs and peak at 2dpi, concurrent with maximal damage to the conducting airway epithelium and alveoli(14). After 2dpi, virus titers wane and the remainder of the disease course is driven by immunopathology. Thus, the 7–10 days prior to peak replication in humans is compressed into the first 48hr of our mouse model. Similar to humans, disease severity in SARS-CoV infected mice is directly correlated with lung viral load, which can be modulated through increasing dose of input virus (14, 21). With both prophylactic and therapeutic (+1dpi) dosing of GS-5734, we demonstrate a reduction in replication below a disease-causing threshold. Therapeutic treatment beginning at 2dpi reduced lung viral loads yet did not improve disease outcomes, suggesting that antivirals initiated after virus replication and immunopathology have reached their tipping point were not clinically beneficial. This result is not surprising given the precedent set by the influenza antiviral, oseltamivir, where treatment efficacy diminishes with time after the onset of symptoms(22). Like SARS-CoV, MERS-CoV titers in the respiratory tract peak in the second week after the onset of symptoms(23). Thus, the window in which to administer antiviral treatment after the onset of symptoms but prior to achieving peak virus titers should be prolonged in humans as compared to experimentally infected mice. Unfortunately, the differences in SARS-CoV pathogenesis among mice and humans noted above limit our ability to determine the time at which treatment no longer will provide a clinical benefit in humans. Nevertheless, our studies provide data which strongly support the testing of GS-5734 in non-human primates and suggest that therapeutic treatment of MERS-CoV-infected humans with GS-5734 will help diminish virus replication and disease if administered early enough during the course of infection.
Currently, there are no approved antiviral treatments for SARS- or MERS-CoV that specifically target the virus. Multiple therapeutic approaches against SARS- and MERS-CoV are currently in development including immune modulation, vaccination, direct-acting antivirals (DAAs) and host-targeted antivirals(7). Known antivirals like ribavirin and lopinavir-ritonavir, as well as immune modulators like interferon and corticosteroids have been used to treat both SARS- and MERS-CoV patients but none were proven effective in randomized controlled trials(7). Cell culture studies in multiple cell lines have demonstrated antiviral effects of several FDA approved drugs (ritonavir, lopinavir, nelfinavir, mycophenolic acid, and ribavirin) but contradictory results and experimental incongruities make interpretation difficult(24–29). Small molecules targeting SARS- and MERS-CoV have been assessed in cancer cell lines in vitro but their antiviral efficacy against other human or zoonotic CoV remains unknown(16, 30). Very few small molecules have been assessed in CoV animal models of viral pathogenesis and some have even been shown to exacerbate disease (e.g. ribavirin, mycophenolic acid)(31, 32). Although the Ces1c−/− mice used herein foster increased drug stability, they are not suitable for MERS-CoV efficacy studies because the murine ortholog of the MERS-CoV receptor, dipeptidyl peptidase 4 (DPP4), does not facilitate MERS-CoV infection (33). Thus, our in vivo studies were limited to SARS-CoV and future studies assessing MERS-CoV efficacy in double transgenic humanized DPP4/Ces1c−/− mice are planned. Human safety testing for GS-5734 is ongoing and the drug has already been used to treat a small number of Ebola virus infected patients under the “compassionate use” clause(34). Overall, our work provides evidence that GS-5734 may protect CoV-infected patients from progression to severe disease, could prophylactically protect health care workers in areas with existing endemic MERS-CoV, and its broad-spectrum activity may prove valuable when a novel CoV emerges in the future.
Materials and Methods
Study Design
The primary goal of this study was to determine if the small molecule nucleoside analog, GS-5734, exhibited broad-spectrum antiviral activity against the CoV family. Using multiple in vitro models including human primary cells we measured the antiviral effect of GS-5734 on multiple CoV encompassing much of the inherent family-wide genetic diversity. Data presented are representative of those from three human donors. Cytotoxicity was assessed in the 2B4 cell line as well as two human primary lung cell types. Experiments were performed in triplicate unless otherwise stated. Drug effects were measured relative to vehicle controls. The secondary goal of this study was to assess antiviral efficacy in vivo within mouse models of severe CoV disease. The in vivo efficacy studies were intended to gain the data required to justify further testing in non-human primates and collectively inform future human clinical trials. Mice were age and sex matched and randomly assigned into groups prior to infection and prior to treatment. The prophylactic and therapeutic in vivo studies presented in the main text were repeated at least once. Pathology and SARS-CoV antigen scoring was performed in a blinded manner. Exclusion criteria for in vivo studies were as follows: If a given mouse unexpectedly did not lose weight after infection and their virus lung titers were more than 2 Log10 lower the mean of the group, this indicated that infection was inefficient and all data related to that mouse were censored.
Viruses
SARS-CoV expressing green fluorescent protein (SARS-GFP, GFP replaces ORF 7) and MERS-CoV expressing red fluorescent protein (MERS-RFP, RFP replaces ORF3) were created from molecular cDNA clones according as described(11, 20). To create SARS- and MERS-CoV expressing nanoluciferase (nLUC), the genes for GFP and RFP were replaced with nLUC and isolated as referenced above. Recombinant human coronavirus NL63 (NL63) and recombinant bat coronavirus for strains HKU3, HKU5, WIV1 and SHC014 were created as described(5, 6, 12, 16, 17).
GS-5734
GS-5734 was synthesized at Gilead Sciences, Inc., and chemical identity and purity was determined by NMR, HRMS, and HPLC analysis(9). GS-5734 was solubilized in 100% DMSO for in vitro studies and in vehicle containing 12% sulfobutylether-β-cyclodextin in water (with HCl/NaOH) at pH 5.0 for in vivo studies. GS-5734 was made available to The University of North Carolina at Chapel Hill under a material transfer agreement with Gilead Sciences.
In vitro efficacy and cytotoxicity in 2B4 cells
The human lung epithelial cell line, Calu-3 2B4 (2B4), was maintained in DMEM (Gibco), 20% fetal bovine serum (Hyclone) and 1X Antibiotic-Antimycotic (A.A., Gibco) (10). 24hr after plating 5×104 cells/well, fresh medium was added. In triplicate, cells were infected for 1hr with MERS-nLUC diluted in growth medium (MOI 0.08) after which virus was removed, cultures were rinsed once and fresh medium containing dilutions of GS-5734 or vehicle was added. DMSO (0.05%) was constant in all conditions. At 48hrs post infection (hpi), virus replication was measured by nLUC assay (Promega) and cytotoxicity was measured via CellTiter-Glo (Promega) assay and then read on a Spectramax plate reader (Molecular Devices). The IC50 value was defined in Graphpad Prism 7.0 (Graphpad) as the concentration at which there was a 50% decrease in viral replication using UV-treated MERS-nLUC (100% inhibition) and vehicle only (0% inhibition) as controls. 50% cytotoxic concentration (CC50) was determined through comparison of data with that from cell-free (100% cytotoxic) and vehicle only (0% cytotoxic) samples.
In vitro efficacy and toxicity in human airway epithelial (HAE) cells
Human airway epithelial (HAE) cell cultures were obtained from the Tissue Procurement and Cell Culture Core Laboratory in the Marsico Lung Institute/CF Research Center at UNC. Prior to infection, HAE were washed with PBS and moved into ALI media containing a dose response of GS-5734 ranging from 10μM to 0.00260μM (Final DMSO < 0.05%)(11). HAE were infected at an MOI of 0.5 for 3 hours at 37°C after which virus was removed, cultures were washed with PBS and then incubated at 37°C for 48hr. Fluorescent images of MERS-RFP were taken at 48 hpi after nuclear staining with Hoechst 33258. Virus replication/titration was performed as previously described(11). Similar data was obtained using cells from three different patient donors. Cytotoxicity was measured via CellTiter-Glo (Promega) in duplicate HAE cell cultures treated with 10 or 0.1μM GS-5734, or DMSO at 0.05%.
In vivo pharmacokinetic analysis in plasma following GS-5734 administration in Ces1c−/− mice and marmosets
Mice were subcutaneously (s.c.) administered 25mg/kg GS-5734 after which plasma was isolated from triplicate mice at 0.25, 0.5, 1, 2, 4, 6, 8, and 12hr post administration. Three male marmosets were administered a single dose of GS-5734 intravenously (i.v.) at 10mg/kg after which plasma was isolated at 0.083, 0.25, 0.5, 1, 2, 4, 8 and 24hr post administration. For both mouse and marmoset, 25 μL of plasma was treated and analyzed as described in Supplemental Materials and Methods “Stability of GS-5734 in WT or Ces1c−/− mouse plasma.” Plasma concentrations of GS-5734, alanine metabolite (Ala-Met) and nucleoside monophosphate (“Nuc-MP”) were determined using 8 to 10-point calibration curves spanning at least 3 orders of magnitude with quality control samples to ensure accuracy and precision, prepared in normal mouse plasma. Analytes were separated by a 75 × 2.0 mm, 4.0 μm Synergi Hydro-RP 30A column (Phenomenex) using a multi-stage linear gradient from 0.2% to 99.0% acetonitrile in mobile phase A at a flow rate of 260 μL/min.
Quantitation of GS-5734 metabolites in the lung following GS-5734 administration in Ces1c−/− mice and marmosets
Mice were dosed with 25 mg/kg or 50 mg/kg GS-5734 as described above. Lungs from triplicate mice were isolated at 1, 2, 6, 12 and 24 hr post administration and snap frozen. Four male marmosets were dosed with GS-5734 as described above and lungs were isolated at 2 and 24hr post administration and snap frozen. On dry ice, frozen lung samples were pulverized and weighed. Dry ice cold extraction buffer containing 0.1% potassium hydroxide and 67 mM ethylenediamine tetraacetic acid (EDTA) in 70% methanol, containing 0.5μM chloro-adenosine triphosphate as internal standard was added and homogenized. After clarifying centrifugation at 20,000 × g for 20 minutes, supernatants were dried in a centrifuge evaporator. Dried samples were then reconstituted with 60 μL of mobile phase A, containing 3 mM ammonium formate (pH 5) with 10 mM dimethylhexylamine (DMH) in water, centrifuged at 20,000 × g for 20 minutes with final supernatants transferred to HPLC injection vials. An aliquot of 10 μL was subsequently injected onto an API 5000 LC/MS/MS system for analysis performed using a similar method as described for intracellular metabolism studies.
Prophylactic and therapeutic efficacy of GS-5734 against SARS-CoV in Ces1c−/− mice
25–28 week old male and female mice genetically deleted for carboxylesterase 1C (Ces1c−/−) (Jackson Laboratories stock 014096) were anaesthetized with ketamine/xylazine and infected with 104pfu/50μl (prophylactic studies) or 103pfu/50μl (therapeutic studies) SARS-CoV MA15. Animals were weighed daily to monitor virus-associated weight loss and to determine the appropriate dose volume of GS-5734 or vehicle. GS-5734 or vehicle was administered s.c. BID 12hr apart. On 2 and 5 dpi (prophylactic) or 2 and 4 or 6 dpi (therapeutic), animals were sacrificed by isofluorane overdose and the large left lobe was frozen at −80°C for viral titration via plaque assay as described(14). The inferior right lobe was placed in 10% buffered formalin and stored at 4°C until histological analysis. Aberrations in lung function were determined by whole body plethysmography (WBP, Data Sciences International) as described(15).
Biocontainment and Biosafety
Reported studies were initiated after the University of North Carolina Institutional Biosafety Committee approved the experimental protocols under the Baric Laboratory Safety Plan: 20167715. SARS-CoV is a select agent. All work for these studies was performed with approved standard operating procedures for SARS-CoV, MERS-CoV and other related CoVs in facilities conforming to requirements recommended in Biosafety in Microbiological and Biomedical Laboratories, the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and the NIH.
Animal Care
Efficacy studies were performed in animal biosafety level 3 (ABSL3) facilities at UNC Chapel Hill. All work was conducted under protocols approved by the Institutional Animal Care and Use Committee at UNC Chapel Hill (Protocol # 13-288.0, continued on # 16-284) according to guidelines set by the Association for the Assessment and Accreditation of Laboratory Animal Care and the United States Department of Agriculture.
Statistics
All statistical calculations were performed in Graphad Prism 7. Specific tests to determine statistical significance are noted in each figure legend.
Supplementary Material
Figure S1: In vitro toxicity and efficacy of GS-5734 in Calu-3 2B4 cells.
Figure S2: In vitro toxicity of GS-5734 in primary human airway epithelial (HAE) cell cultures.
Figure S3: In vitro toxicity of GS-5734 in primary normal human bronchiolar epithelial (NHBE) cell cultures.
Figure S4: SARS-CoV in vivo pathogenesis is similar in WT and Ces1c−/− mice.
Figure S5: In vitro metabolism in NHBE and in vivo pharmacokinetic analysis in non-human primates.
Figure S6: GS-5734 diminishes SARS-CoV induced lung pathology.
Figure S7: Therapeutic administration of GS-5734 beginning 2dpi does not provide a therapeutic benefit.
Figure S8: A comprehensive platform approach to evaluate therapeutics against emerging viral infections.
Table S1. Coronavirus genomic and subgenomic real-time primer sets.
Table S2. Real-time primer/probe sets for indicators of cellular apoptosis/toxicity.
Table S3. Primary data.
Acknowledgments
Funding: We would like to acknowledge the following funding sources, Antiviral Drug Discovery and Development Center (5U19AI109680), grants from the National Institutes of Health (AI108197, AI109761) and Cystic Fibrosis and Pulmonary Research and Treatment Center (BOUCHE15RO and NIH P30DK065988). Additionally, compound formulation, pharmacokinetic and metabolism studies were performed and paid for by Gilead Sciences.
Footnotes
Author Contributions: A.C.S., J.Y.F., T.P.S., T.C., R.J. and R.B. designed in vitro efficacy studies. A.C.S., T.P.S. and S.R.L. executed and analyzed in vitro efficacy studies. T.P.S., J.Y.F., R.J., T.C., R.S.B. and A.S.R., designed in vivo efficacy studies. T.P.S. executed and analyzed in vivo efficacy studies. R.B. scored pathology and virus lung antigen staining. R.L.G., and K.P. performed quantitative RT-PCR. V.D.M. and L.E.G. performed whole body plethysmography for some in vivo studies. J.B.C. and M.R.D. designed and performed pilot studies initially demonstrating efficacy against coronavirus. C.P., R.B., Y.P., D.B., and A.S.R., designed, executed and analyzed metabolism, pharmacokinetics, stability or toxicity studies. M.O.C., D.S., R.L.M., J.E.S. and I.T. were responsible for synthesis, scale-up and formulation of small molecules. T.P.S., A.C.S., J.Y.F., T.C., R.S.B., M.R.D., D.B., R.J., and A.S.R. wrote the manuscript. We would also like to thank Ande West and D. Trevor Scobey for excellent technical expertise.
Competing interests: The authors affiliated with Gilead Sciences are employees of the company and may own company stock. M.O.C., J.Y.F, R.J., R.L.M., A.S.R., and D.S. are listed as inventors on International Application No. PCT/US2016/052092 filed by Gilead Sciences, Inc., directed to methods of treating coronaviridae virus infections. Travel of M.R.D. to Gilead Sciences, Inc. to discuss this project was paid for by Gilead Sciences.
References and Notes
- 1.Eckerle LD, Becker MM, Halpin RA, Li K, Venter E, Lu X, Scherbakova S, Graham RL, Baric RS, Stockwell TB, Spiro DJ, Denison MR. Infidelity of SARS-CoV Nsp14-exonuclease mutant virus replication is revealed by complete genome sequencing. PLoS Pathog. 2010;6:e1000896. doi: 10.1371/journal.ppat.1000896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Wit E, van Doremalen N, Falzarano D, Munster VJ. SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol. 2016;14:523–534. doi: 10.1038/nrmicro.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liljander A, Meyer B, Jores J, Muller MA, Lattwein E, Njeru I, Bett B, Drosten C, Corman VM. MERS-CoV Antibodies in Humans, Africa, 2013–2014. Emerg Infect Dis. 2016;22:1086–1089. doi: 10.3201/eid2206.160064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muller MA, Meyer B, Corman VM, Al-Masri M, Turkestani A, Ritz D, Sieberg A, Aldabbagh S, Bosch BJ, Lattwein E, Alhakeem RF, Assiri AM, Albarrak AM, Al-Shangiti AM, Al-Tawfiq JA, Wikramaratna P, Alrabeeah AA, Drosten C, Memish ZA. Presence of Middle East respiratory syndrome coronavirus antibodies in Saudi Arabia: a nationwide, cross-sectional, serological study. Lancet Infect Dis. 2015;15:559–564. doi: 10.1016/S1473-3099(15)70090-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Menachery VD, Yount BL, Jr, Debbink K, Agnihothram S, Gralinski LE, Plante JA, Graham RL, Scobey T, Ge XY, Donaldson EF, Randell SH, Lanzavecchia A, Marasco WA, Shi ZL, Baric RS. A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nat Med. 2015;21:1508–1513. doi: 10.1038/nm.3985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Menachery VD, Yount BL, Jr, Sims AC, Debbink K, Agnihothram SS, Gralinski LE, Graham RL, Scobey T, Plante JA, Royal SR, Swanstrom J, Sheahan TP, Pickles RJ, Corti D, Randell SH, Lanzavecchia A, Marasco WA, Baric RS. SARS-like WIV1-CoV poised for human emergence. Proc Natl Acad Sci U S A. 2016 doi: 10.1073/pnas.1517719113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zumla A, Chan JF, Azhar EI, Hui DS, Yuen KY. Coronaviruses - drug discovery and therapeutic options. Nat Rev Drug Discov. 2016;15:327–347. doi: 10.1038/nrd.2015.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Warren TK, Jordan R, Lo MK, Ray AS, Mackman RL, Soloveva V, Siegel D, Perron M, Bannister R, Hui HC, Larson N, Strickley R, Wells J, Stuthman KS, Van Tongeren SA, Garza NL, Donnelly G, Shurtleff AC, Retterer CJ, Gharaibeh D, Zamani R, Kenny T, Eaton BP, Grimes E, Welch LS, Gomba L, Wilhelmsen CL, Nichols DK, Nuss JE, Nagle ER, Kugelman JR, Palacios G, Doerffler E, Neville S, Carra E, Clarke MO, Zhang L, Lew W, Ross B, Wang Q, Chun K, Wolfe L, Babusis D, Park Y, Stray KM, Trancheva I, Feng JY, Barauskas O, Xu Y, Wong P, Braun MR, Flint M, McMullan LK, Chen SS, Fearns R, Swaminathan S, Mayers DL, Spiropoulou CF, Lee WA, Nichol ST, Cihlar T, Bavari S. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature. 2016;531:381–385. doi: 10.1038/nature17180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cho A, Saunders OL, Butler T, Zhang L, Xu J, Vela JE, Feng JY, Ray AS, Kim CU. Synthesis and antiviral activity of a series of 1′-substituted 4-aza-7,9-dideazaadenosine C-nucleosides. Bioorg Med Chem Lett. 2012;22:2705–2707. doi: 10.1016/j.bmcl.2012.02.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sims AC, Tilton SC, Menachery VD, Gralinski LE, Schafer A, Matzke MM, Webb-Robertson BJ, Chang J, Luna ML, Long CE, Shukla AK, Bankhead AR, 3rd, Burkett SE, Zornetzer G, Tseng CT, Metz TO, Pickles R, McWeeney S, Smith RD, Katze MG, Waters KM, Baric RS. Release of severe acute respiratory syndrome coronavirus nuclear import block enhances host transcription in human lung cells. J Virol. 2013;87:3885–3902. doi: 10.1128/JVI.02520-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sims AC, Baric RS, Yount B, Burkett SE, Collins PL, Pickles RJ. Severe acute respiratory syndrome coronavirus infection of human ciliated airway epithelia: role of ciliated cells in viral spread in the conducting airways of the lungs. J Virol. 2005;79:15511–15524. doi: 10.1128/JVI.79.24.15511-15524.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Donaldson EF, Yount B, Sims AC, Burkett S, Pickles RJ, Baric RS. Systematic assembly of a full-length infectious clone of human coronavirus NL63. J Virol. 2008;82:11948–11957. doi: 10.1128/JVI.01804-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li B, Sedlacek M, Manoharan I, Boopathy R, Duysen EG, Masson P, Lockridge O. Butyrylcholinesterase, paraoxonase, and albumin esterase, but not carboxylesterase, are present in human plasma. Biochem Pharmacol. 2005;70:1673–1684. doi: 10.1016/j.bcp.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 14.Gralinski LE, Bankhead A, 3rd, Jeng S, Menachery VD, Proll S, Belisle SE, Matzke M, Webb-Robertson BJ, Luna ML, Shukla AK, Ferris MT, Bolles M, Chang J, Aicher L, Waters KM, Smith RD, Metz TO, Law GL, Katze MG, McWeeney S, Baric RS. Mechanisms of severe acute respiratory syndrome coronavirus-induced acute lung injury. MBio. 2013;4 doi: 10.1128/mBio.00271-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Menachery VD, Gralinski LE, Baric RS, Ferris MT. New Metrics for Evaluating Viral Respiratory Pathogenesis. PLoS One. 2015;10:e0131451. doi: 10.1371/journal.pone.0131451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agnihothram S, Yount BL, Jr, Donaldson EF, Huynh J, Menachery VD, Gralinski LE, Graham RL, Becker MM, Tomar S, Scobey TD, Osswald HL, Whitmore A, Gopal R, Ghosh AK, Mesecar A, Zambon M, Heise M, Denison MR, Baric RS. A mouse model for Betacoronavirus subgroup 2c using a bat coronavirus strain HKU5 variant. MBio. 2014;5:e00047–00014. doi: 10.1128/mBio.00047-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Becker MM, Graham RL, Donaldson EF, Rockx B, Sims AC, Sheahan T, Pickles RJ, Corti D, Johnston RE, Baric RS, Denison MR. Synthetic recombinant bat SARS-like coronavirus is infectious in cultured cells and in mice. Proc Natl Acad Sci U S A. 2008;105:19944–19949. doi: 10.1073/pnas.0808116105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nguyen NM, Tran CN, Phung LK, Duong KT, Huynh Hle A, Farrar J, Nguyen QT, Tran HT, Nguyen CV, Merson L, Hoang LT, Hibberd ML, Aw PP, Wilm A, Nagarajan N, Nguyen DT, Pham MP, Nguyen TT, Javanbakht H, Klumpp K, Hammond J, Petric R, Wolbers M, Nguyen CT, Simmons CP. A randomized, double-blind placebo controlled trial of balapiravir, a polymerase inhibitor, in adult dengue patients. J Infect Dis. 2013;207:1442–1450. doi: 10.1093/infdis/jis470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ibarra KD, Pfeiffer JK. Reduced ribavirin antiviral efficacy via nucleoside transporter-mediated drug resistance. J Virol. 2009;83:4538–4547. doi: 10.1128/JVI.02280-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scobey T, Yount BL, Sims AC, Donaldson EF, Agnihothram SS, Menachery VD, Graham RL, Swanstrom J, Bove PF, Kim JD, Grego S, Randell SH, Baric RS. Reverse genetics with a full-length infectious cDNA of the Middle East respiratory syndrome coronavirus. Proc Natl Acad Sci U S A. 2013;110:16157–16162. doi: 10.1073/pnas.1311542110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peiris JS, Chu CM, Cheng VC, Chan KS, Hung IF, Poon LL, Law KI, Tang BS, Hon TY, Chan CS, Chan KH, Ng JS, Zheng BJ, Ng WL, Lai RW, Guan Y, Yuen KY Group HUSS. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet. 2003;361:1767–1772. doi: 10.1016/S0140-6736(03)13412-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu H, Feng Z, Uyeki TM, Liao Q, Zhou L, Feng L, Ye M, Xiang N, Huai Y, Yuan Y, Jiang H, Zheng Y, Gargiullo P, Peng Z, Feng Y, Zheng J, Xu C, Zhang Y, Shu Y, Gao Z, Yang W, Wang Y. Risk factors for severe illness with 2009 pandemic influenza A (H1N1) virus infection in China. Clin Infect Dis. 2011;52:457–465. doi: 10.1093/cid/ciq144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oh MD, Park WB, Choe PG, Choi SJ, Kim JI, Chae J, Park SS, Kim EC, Oh HS, Kim EJ, Nam EY, Na SH, Kim DK, Lee SM, Song KH, Bang JH, Kim ES, Kim HB, Park SW, Kim NJ. Viral Load Kinetics of MERS Coronavirus Infection. N Engl J Med. 2016;375:1303–1305. doi: 10.1056/NEJMc1511695. [DOI] [PubMed] [Google Scholar]
- 24.Chan JF, Lau SK, To KK, Cheng VC, Woo PC, Yuen KY. Middle East respiratory syndrome coronavirus: another zoonotic betacoronavirus causing SARS-like disease. Clin Microbiol Rev. 2015;28:465–522. doi: 10.1128/CMR.00102-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Falzarano D, de Wit E, Martellaro C, Callison J, Munster VJ, Feldmann H. Inhibition of novel beta coronavirus replication by a combination of interferon-alpha2b and ribavirin. Sci Rep. 2013;3:1686. doi: 10.1038/srep01686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hart BJ, Dyall J, Postnikova E, Zhou H, Kindrachuk J, Johnson RF, Olinger GG, Jr, Frieman MB, Holbrook MR, Jahrling PB, Hensley L. Interferon-beta and mycophenolic acid are potent inhibitors of Middle East respiratory syndrome coronavirus in cell-based assays. J Gen Virol. 2014;95:571–577. doi: 10.1099/vir.0.061911-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morgenstern B, Michaelis M, Baer PC, Doerr HW, Cinatl J., Jr Ribavirin and interferon-beta synergistically inhibit SARS-associated coronavirus replication in animal and human cell lines. Biochem Biophys Res Commun. 2005;326:905–908. doi: 10.1016/j.bbrc.2004.11.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamamoto N, Yang R, Yoshinaka Y, Amari S, Nakano T, Cinatl J, Rabenau H, Doerr HW, Hunsmann G, Otaka A, Tamamura H, Fujii N, Yamamoto N. HIV protease inhibitor nelfinavir inhibits replication of SARS-associated coronavirus. Biochem Biophys Res Commun. 2004;318:719–725. doi: 10.1016/j.bbrc.2004.04.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chan JF, Chan KH, Kao RY, To KK, Zheng BJ, Li CP, Li PT, Dai J, Mok FK, Chen H, Hayden FG, Yuen KY. Broad-spectrum antivirals for the emerging Middle East respiratory syndrome coronavirus. J Infect. 2013;67:606–616. doi: 10.1016/j.jinf.2013.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ratia K, Pegan S, Takayama J, Sleeman K, Coughlin M, Baliji S, Chaudhuri R, Fu W, Prabhakar BS, Johnson ME, Baker SC, Ghosh AK, Mesecar AD. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc Natl Acad Sci U S A. 2008;105:16119–16124. doi: 10.1073/pnas.0805240105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barnard DL, Day CW, Bailey K, Heiner M, Montgomery R, Lauridsen L, Winslow S, Hoopes J, Li JK, Lee J, Carson DA, Cottam HB, Sidwell RW. Enhancement of the infectivity of SARS-CoV in BALB/c mice by IMP dehydrogenase inhibitors, including ribavirin. Antiviral Res. 2006;71:53–63. doi: 10.1016/j.antiviral.2006.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chan JF, Yao Y, Yeung ML, Deng W, Bao L, Jia L, Li F, Xiao C, Gao H, Yu P, Cai JP, Chu H, Zhou J, Chen H, Qin C, Yuen KY. Treatment With Lopinavir/Ritonavir or Interferon-beta1b Improves Outcome of MERS-CoV Infection in a Nonhuman Primate Model of Common Marmoset. J Infect Dis. 2015;212:1904–1913. doi: 10.1093/infdis/jiv392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cockrell AS, Yount BL, Scobey T, Jensen K, Douglas M, Beall A, Tang XC, Marasco WA, Heise MT, Baric RS. A mouse model for MERS coronavirus-induced acute respiratory distress syndrome. Nat Microbiol. 2016;2:16226. doi: 10.1038/nmicrobiol.2016.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jacobs M, Rodger A, Bell DJ, Bhagani S, Cropley I, Filipe A, Gifford RJ, Hopkins S, Hughes J, Jabeen F, Johannessen I, Karageorgopoulos D, Lackenby A, Lester R, Liu RS, MacConnachie A, Mahungu T, Martin D, Marshall N, Mepham S, Orton R, Palmarini M, Patel M, Perry C, Peters SE, Porter D, Ritchie D, Ritchie ND, Seaton RA, Sreenu VB, Templeton K, Warren S, Wilkie GS, Zambon M, Gopal R, Thomson EC. Late Ebola virus relapse causing meningoencephalitis: a case report. Lancet. 2016 doi: 10.1016/S0140-6736(16)30386-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: In vitro toxicity and efficacy of GS-5734 in Calu-3 2B4 cells.
Figure S2: In vitro toxicity of GS-5734 in primary human airway epithelial (HAE) cell cultures.
Figure S3: In vitro toxicity of GS-5734 in primary normal human bronchiolar epithelial (NHBE) cell cultures.
Figure S4: SARS-CoV in vivo pathogenesis is similar in WT and Ces1c−/− mice.
Figure S5: In vitro metabolism in NHBE and in vivo pharmacokinetic analysis in non-human primates.
Figure S6: GS-5734 diminishes SARS-CoV induced lung pathology.
Figure S7: Therapeutic administration of GS-5734 beginning 2dpi does not provide a therapeutic benefit.
Figure S8: A comprehensive platform approach to evaluate therapeutics against emerging viral infections.
Table S1. Coronavirus genomic and subgenomic real-time primer sets.
Table S2. Real-time primer/probe sets for indicators of cellular apoptosis/toxicity.
Table S3. Primary data.