

Abstract
Neuronal nitric oxide synthase (nNOS) inhibition is a promising strategy to treat neurodegenerative disorders, but development of nNOS inhibitors is often hindered by poor pharmacokinetics. We previously developed a class of membrane-permeable 2-aminoquinoline inhibitors and later rearranged the scaffold to decrease off-target binding. However, the resulting compounds had decreased permeability, low human nNOS activity, and low selectivity versus human eNOS. In this study, 5-substituted phenyl ether-linked aminoquinolines and derivatives were synthesized and assayed against purified NOS isoforms. 5-Cyano compounds are especially potent and selective rat and human nNOS inhibitors. Activity and selectivity are mediated by the binding of the cyano group to a new auxiliary pocket in nNOS. Potency was enhanced by methylation of the quinoline and by introduction of simple chiral moieties, resulting in a combination of hydrophobic and auxiliary pocket effects that yielded high (∼500-fold) n/e selectivity. Importantly, Caco-2 assay also revealed improved membrane permeability over previous compounds.
Graphical abstract
Introduction
Neurodegenerative disorders (Alzheimer's and Parkinson's diseases, amyotrophic lateral sclerosis, Huntington's disease, and others) are characterized by the gradual loss of neuronal function and structure. The resulting symptoms cause great suffering not only to patients, but also to their caretakers, the economy, and to global health in general. Effective treatments for neurodegenerative diseases are limited, and the development of novel therapeutics to treat neurodegeneration is a highly desirable unmet medical need.
Neuronal nitric oxide synthase (nNOS) is an enzymatic target under investigation for the treatment of neurodegenerative disorders (as well as other conditions characterized by neuronal damage, such as stroke, ischemic events, cerebral palsy, and neuropathic pain).1 Three NOS isoenzymes produce nitric oxide (NO), a free-radical second-messenger molecule, in the human body: endothelial NOS (eNOS) produces the NO employed in blood pressure regulation and smooth muscle tone, inducible NOS (iNOS) plays a role in immune activation, and in the CNS, the NO produced by nNOS is required for normal neuronal signaling.2
Under neuroinflammatory or neurodegenerative phenotypes, however, nNOS can become overactive or overexpressed, and NO levels surge several orders of magnitude, where NO can cause damage or combine to form other damaging species like peroxynitrite.3 These species can cause protein nitration and aggregation,4 depletion of cellular energy and glutathione reserves,5,6 damage to various cellular structures, and the eventual apoptosis or necrosis of neurons, leading progressively to the symptoms characteristic of neurodegeneration. Studies have shown that hyperactive nNOS and dysfunctional nitrergic signaling are affiliated with or directly implicated in the pathology of many neurodegenerative disorders7,8, 9, 10 making nNOS a desirable target for therapeutic intervention.9, 11, 12
nNOS functions by converting l-arginine to l-citrulline and NO via an electron relay proceeding through five cofactors. nNOS is only functional as a homodimer with each monomer containing an oxygenase domain and a reductase domain that are joined by a linker domain where calmodulin, in response to elevated calcium levels, binds and activates the enzyme. Once activated, electron flow proceeds from the reductase domain-bound reduced nicotinamide adenine dinucleotide phosphate (NADPH), to flavin adenine dinucleotide (FAD), to flavin mononucleotide (FMN), and then from the FMN subdomain of one monomer to the other monomer's oxygenase domain,13 through (6R)-5,6,7,8-tetrahydrobiopterin (H4B), and finally to the heme active center, where the bound l-arginine is oxidized in the presence of molecular oxygen.14
Most nNOS inhibitors are competitive with the substrate and resemble l-arginine in their physicochemical properties. Unfortunately, these polar, high-pKa, ionizable molecules often suffer from low bioavailability and low CNS permeation, which severely limits their therapeutic use. An additional challenge is that an inhibitor must be selective for nNOS over eNOS and iNOS, as inhibition of eNOS could cause cardiovascular liabilities,15 whereas iNOS inhibition could disrupt immune system activation. This is a daunting task, however, as all three NOS isoforms share very similar sequences and structure.16
Previously, we reported several classes of nNOS inhibitors based on a 2-aminoquinoline scaffold.17, 18 Our first generation of inhibitors, such as 1 and 2 (Figure 1), were potent, selective, and possessed excellent cellular and in vivo pharmacokinetics.17 Unfortunately, 2 was selective for rat nNOS (rnNOS) over human nNOS (hnNOS), displayed low selectivity for human nNOS over human eNOS (heNOS), caused toxic side effects in rats, and was extremely promiscuous in CNS counterscreens. The second-generation,18 rearranged phenyl ether 4 (optimized from lead 3), preserved the potency and selectivity of 1 and 2 while drastically decreasing the off-target binding, but this compound had significantly decreased Caco-2 permeability, low human nNOS activity, and similarly low selectivity for hnNOS over heNOS.
Figure 1.

Previous use of 2-aminoquinolines as nNOS inhibitors.
We chose to continue investigating this cleaner-binding phenyl ether scaffold in an attempt to improve n/e selectivity, hnNOS inhibitory potency, and possibly cellular permeability. First, the 5-position of the phenyl ring (Figure 2) was substituted with a variety of groups, leading to analogues 5-9. Previously, the 1,3,5-trisubstituted phenyl or pyridyl moieties of 2-aminopyridine inhibitors19, 20, 21 were able to access nNOS-specific residues such as Asp597 (Asp602 in hnNOS), or other nNOS-specific regions, and lead to high n/e selectivity. It was proposed that analogous substituents on the phenyl ether scaffold could reach potentially similar nNOS-specific regions that could improve hnNOS potency, such as the hnNOS-specific residue His342.
Figure 2.

Design strategy utilized and compounds synthesized in this study. All molecules have a CLogP between 2.5-4 (lower for cyano compounds and higher for deoxy compounds), and tPSA (total polar surface area) of 50-83 Å2 (higher for cyano compounds and lower for deoxy compounds).
Second, it was previously reported that for 2-aminopyridines, installation of a methyl group at the 4-position of the pyridine could drastically improve potency, and in some cases, selectivity.22 A fragment screen then showed that 2-amino-4-methylquinoline bound nearly 7-fold tighter (Ki = 94 nM) to rat nNOS than the unmethylated 2-aminoquinoline (630 nM). X-ray crystallography indicated that the 4-methyl compound acted as a competitive l-arginine antagonist. To this end, the methylated analogues of compounds 8 and 9 (11 and 13, respectively) and of the original phenyl ether leads 3 and 4 (10 and 12, respectively) were prepared.
We also investigated removal of the oxygen from the ether linkage entirely (as seen in many previous aminopyridines),19, 21 as the methylene adjacent to the phenyl ether could serve as a potential site of metabolism, and removal of the oxygen would lower the tPSA of the molecule (it is 50.41 Å2 for 14). Therefore, analogues 14 and 16, and desmethyl analogue 15 (deoxygenated analogues of active compounds 10, 11, and 8, respectively) were synthesized.
Finally, more specific efforts to optimize the most potent and selective scaffold (the 5-cyanophenyl ether, as in 8 and 11) were made, all centered on improving n/e selectivity and overall hydrophobicity. The short methylamine tail of 11 was replaced with the ethylamine of 18 and the chiral alpha-methyl ethylamine of 19, to direct an extra alkyl group in the area of the nNOS-specific hydrophobic residue, Met336/Met341 (rnNOS/hnNOS).23 Van der Waals contact between inhibitors and this residue has been implicated in improved n/e selectivity, as this residue is replaced by a smaller valine in eNOS isoforms.18 Similarly, compound 17 combines a 4-substitution pattern (as in 4) with the 5-cyano group.
All compounds were assayed against rnNOS, and select compounds were also assayed against hnNOS. To shift our structure-activity relationship (SAR) work toward increasingly more human systems, human eNOS (heNOS) was employed instead of previously used bovine eNOS. Murine iNOS (miNOS) was used, and n/i selectivity is reported as the ratio of miNOS/rnNOS. Finally, as most of these molecules have excellent chemical properties (Figure 1), potent and selective compounds were assayed in a Caco-2 assay to approximate their cellular permeability.
Chemistry
To prepare the initial set of 5-substituted quinoline analogues (5-9), a series of 3,5-disubstituted phenols was first synthesized (Scheme 1). Commercially available aldehydes 20-22 were reductively aminated and Boc-protected18 to afford phenols 23-25, respectively. Brominated compound 24 was converted, via palladium-catalyzed cyanation, into cyanophenol 26.24 Sonogashira coupling between 24 and ethynyltrimethylsilane yielded 27, which was desilylated (to give 28) and reduced to give ethylphenol 29. The requisite phenols were then treated with quinolinemethyl bromide 30 (prepared by previously reported procedures18 (Scheme 2) under basic conditions to afford the phenyl ether cores (31-35). Compounds 31-35 were first deacetylated using K2CO3 in hot methanol, and the Boc groups were then cleaved using HCl (31, 32, and 34) or TFA (33 and 35) to yield pure final compounds 5-9 as water-soluble hydrochloride or trifluoroacetate salts.
Scheme 1.

aReagents and conditions: (a) i. MeNH2 in THF, cat. AcOH, CHCI3/MeOH, Na2SO4, r.t., ii. NaBH4, MeOH, 0 °C - r.t., iii. Boc2O, THF, r.t.; (b) K4Fe(CN)6 × 3 H2O, t-BuXPhos, t-BuXPhos G3, KOAc, H2O/dioxane, 100 °C; (c) [(PPh3)2PdCI2], Cul, Et3N, 70 °C; (d) TBAF in THF, THF, 0 °C; (e) H2, Pd/C, MeOH, r.t.
Scheme 2.

aReagents and conditions: (a) i. Phenols 23-26, 29, NaH, DMF, 0 °C, ii. 30 (in DMF), 0 °C; (b) i. K2CO3, MeOH, reflux, ii. HCI/MeOH, ether, r.t., or TFA/DCM (for 35), after isolation.
Because of the reactivity and acidity of 4-methylquinolines, the analogous bromide (43) could not be prepared by our previous synthetic route (which utilized both free-radical bromination of a 7-methylquinoline and basic conditions that are incompatible with the 4-methyl group). A new synthetic route had to be devised,25 and we envisioned that installing a readily derivatizable handle at position 7 and then functionalizing the 2-position would be the most viable strategy. To this end, 7-bromoquinoline 37 was prepared by the Doebner-Miller condensation of 7-bromoaniline (36) with methyl vinyl ketone26 (Scheme 3). Treatment with m-CPBA afforded N-oxide 38, which readily underwent deoxygenative amination upon treatment with Ts2O and t-BuNH2;27, 28 heating with TFA removed the t-butyl group to yield the free aminoquinoline (39) in good yields (even on a multigram scale) following neutralization and column chromatography. The obtained aminoquinoline was then protected as acetamidoquinoline 40 as previously described. To further functionalize 40, the bromide was converted into aldehyde 41 using Ueda et al.'s palladium-catalyzed hydrocarbonylation,29 which employs N-formylsaccharin as the CO donor and Et3SiH as the reductant. Reduction of the aldehyde afforded 42, which could be brominated (Appel conditions18 or chlorinated with SOCl2) to yield bromide 43 or chloride 44, respectively. Bromide 43 was treated with phenols 26 and 29, as well as with 45 and 46 (prepared by literature procedures;18 not in Scheme 1), to yield assembled cores 47-50, which were deprotected as described above to yield 4-methylated analogues 10-13.
Scheme 3.

aReagents and conditions: (a) 2-buten-1-one, FeCI3· 6 H2O, AcOH, reflux; (b) m-CBPA, CH2CI2, r.t.; (c) i. t-BuNH2, Ts2O, PhCF3/CH2Cl2, 0 °C, ii. TFA, 75 °C; (d) N-acetylimidazole, DMAP, THF, reflux; (e) N-formylsaccharin, Pd(OAc)2, dppb, Et3SiH, Na2CO3, DMF, 75 °C; (f) NaBH4, MeOH, 50 °C-r.t.; (g) PPh3, CBr4, THF, 0 °C-r.t. (for 43), SOCI2, 0 °C (for 44); (h) i. Phenols 26, 29, 45, 46, NaH, DMF, 0 °C, ii. 43 (in DMF), 0 °C; (i) i. K2CO3, MeOH, reflux, ii. HCI/MeOH, ether, r.t., or TFA/DCM (for 48), after isolation.
To prepare the deoxygenated analogues 14-16, bromide 40 was employed (to prepare 4-methyl analogues) and compound 51 (to prepare compounds with no substituent at the 4-position), via aminoquinoline 52,30 was converted into desmethyl 7-bromoquinoline 53 (Scheme 4A). Next, suitable Sonogashira coupling partners were prepared. To prepare 14, 3-iodobenzyl bromide (54, Scheme 4B) was converted to carbamate 55, and coupling with ethynyltrimethylsilane afforded 56 in excellent yields, which was then desilylated to yield 57. Synthesis of cyanated analogues 15 and 16 began with bromination of commercially available cyanotoluene 58 (Scheme 4C); bromide 59 was subsequently aminated and protected to yield 60. As described above, Sonogashira coupling and desilylation of 61 yielded 62. Phenylacetylene 62 is sensitive and polymerizes at room temperature, so it must be kept cold until use.
Scheme 4.

aReagents and conditions: (a) AcNH2, K2CO3, reflux (230 °C); (b) N-acetylimidazole, DMAP, THF, reflux; (c) i. MeNH2 in THF, CH2CI2, r.t., ii. Boc2O, CH2CI2, r.t.; (d) [(PPh3)2PdCI2], Cul, Et3N, 70 °C; (e) TBAF in THF, THF, 0 °C; (f) NBS, (PhCO2)2, CCI4, reflux.
With the quinolines and phenylacetylenes in hand, copper-free Sonogashira conditions31 were used to join the halves (Scheme 5). The quinolinyl-acetylenes 63-65 were readily identifiable by TLC because of their bright blue fluorescence. After purification, the triple bonds were hydrogenated to alkanes 66-68. For 63, this was readily accomplished with palladium on carbon (to yield 66), but these conditions also reduced the nitriles of 64 and 65. For these compounds, a Pd/C-ethylenediamine complex32, 33 was used to reduce the alkyne, which showed excellent chemoselectivity, yielding 67 and 68 despite requiring extended reaction times or higher pressures. Finally, 66-68 were deprotected to afford 14-16.
Scheme 5.

aReagents and conditions: (a) XPhos, Pd(MeCN)2CI2, Cs2CO3, MeCN, 80 °C; (b) H2, Pd/C, MeOH, r.t. (for 66), H2, Pd/C(en), THF, atmospheric pressure or 50 PSI (for 67 and 68); (c) i. K2CO3, MeOH, reflux, ii. HCI/MeOH, ether, r.t., after isolation.
Preparing the phenols required for analogues 17-19 proved more challenging, as phenolic (or other) precursors with these particular 1,3,5-substitution patterns are not commercially available or readily synthesized. Nonetheless, the meta-borylation/oxidation strategy has been employed by Smith, Maleczka, and others34, 35 to prepare similar 3,5-disubstituted phenols. In our case, to prepare a borylation substrate, commercially available toluene 69 (Scheme 6) was brominated (to give 70) and converted into protected amine 71. Cyanation afforded 72. To prepare the phenol, iridium-catalyzed borylation with Pin2B2 afforded an intermediate boronic ester (not isolated), which was then treated with Oxone in aqueous acetone to yield phenol 73 in moderate yield. The phenoxide of 73 was then treated with chloride 44. Because of 44's lower reactivity, the etherification reaction was performed at 50 °C (instead of 0 °C as for 43), and deprotection of 74 afforded analogue 17.
Scheme 6.

aReagents and conditions: (a) NBS, (PhCO2)2, CCI4, reflux; (b) i. MeNH2 in THF, CH2CI2, r.t.; ii. Boc2O, CH2CI2, r.t.; (c) K4Fe(CN)6 × 3 H2O, t-BuXPhos, t-BuXPhos G3, KOAc, H2O/dioxane, 100 °C; (d) i. [lr(OCH3)(COD]2, dtbpy, Pin2B2, hexanes, 50 °C; ii. Oxone, H2O/acetone, 0 °C; (e) i. phenol, NaH, DMF, 0 °C; ii. 44 (in DMF), 0 °C-50 °C; (f) i. K2CO3, MeOH, reflux, ii. HCI/MeOH, ether, r.t., after isolation.
Similarly, phenethylamine analogues 18 and (R,S)-19 were also prepared via meta-borylation (Scheme 7). The substrates (unsubstituted 79 and alpha-methylated (R,S)-80) were prepared by Boc-protection and methylation of 75 (to yield 77) or reductive amination and Boc-protection of 76 (to yield (R,S)-78). Cyanation (as described above) afforded benzonitriles 79 and (R,S)-80, and the borylation-oxidation strategy produced, respectively, 81 and (R,S)-82. These phenols were treated with 44, and deprotection of 83 and (R,S)-84 afforded final compounds 18 and (R,S)-19, respectively.
Scheme 7.

aReagents and conditions: (a) i. Boc2O, THF, r.t., ii. NaH, DMF, then Mel, 0 °C-60 °C; (b) i. MeNH2 in THF, Na(OAc)3BH, AcOH, CH2CI2, ii. Boc2O, THF, r.t.; (c) K4Fe(CN)6 × 3 H2O, t-BuXPhos G3, t-BuXPhos, 1:1 KOAc in H2O: dioxane, 100 °C; (d) i. [lr(OCH3)(COD)]2, dtbpy, Pin2B2, hexanes, r.t. or 50 °C; ii. Oxone, H2O/acetone, 0 °C-r.t. or r.t. (e) i. NaH, DMF, 0 °C; ii. 44 (in DMF), 0 °C-50 °C; (f) i. K2CO3, MeOH, reflux, ii, HCI/MeOH, ether, r.t.
As derivatives of 78 proved difficult to resolve into its enantiomers, an asymmetric synthesis of the two enantiomers of 19 (Scheme 8) was developed, based on the Ellman auxiliary.36, 37 Commercially available (R)- or (S)-t-butylsulfinamides (R- and S-85) were condensed with 76. Low-temperature reduction of the imine with NaBH4 afforded the expected (R,R)-diastereomer (R,R)-86 (when (R)-85 was used), and (S,S)-diastereomer (S,S)-86 (when S-85 was used) in satisfactory (>7:1) diastereomeric ratios. After purification, cleavage of the sulfinamide auxiliaries and Boc-protection of the free amines yielded (R)- and (S)-87, which were both methylated to yield (R)- and (S)-78. The remainder of the synthesis proceeded as with the racemic material: cyanation afforded (R)- and (S)-80, which were borylated and treated with oxone to yield the phenols (R)- and (S)-82. Enantomeric purity of these phenols (as assessed by chiral HPLC) was very high (>99%). Subsequent phenyl ether formation and deprotection of (R)- and (S)-84 afforded (R)- and (S)-19, which were confirmed as single enantiomers via derivatization (at the secondary amine) with (S)-camphanic chloride.
Scheme 8.

aReagents and conditions: (a) i. Ti(OEt)4, THF, reflux, ii. NaBH4, THF, -48 °C; (b) i. HCI/MeOH, ether, r.t., ii. Boc2O, Et3N, MeOH/THF, r.t.; (c) NaH, THF, °C - r.t., then Mel, 0°C - r.t.; (d) K4Fe(CN)6 × 3 H2O, t-BuXPhos G3, t-BuXPhos, 1:1 KOAc in H2O: dioxane, 100 °C; (e) i. [lr(OCH3)(COD)]2, dtbpy, Pin2B2, hexanes, 50 °C, ii. Oxone, H2O/acetone, 0 °C-r.t.; (e) i. NaH, DMF, 0 °C; ii. 44 (in DMF), 0 °C-50 °C; (f) i. K2CO3, MeOH, reflux, ii, HCI/MeOH, ether, r.t.
Results and Discussion
Compounds 5-19 were assayed against purified rat nNOS (rnNOS). Select compounds were then assayed against murine iNOS (miNOS) and also against hnNOS and heNOS. With recent advances,38 it is now possible to obtain and crystallize both hnNOS and heNOS, so these enzymes were used as part of a shift to obtain more human SAR data. RnNOS and miNOS were still used, as historically, they are the easiest to express and purify, and for clinical purposes, it is desirable to prove efficacy and selectivity in lower animals. As miNOS is often difficult to crystallize, the majority of the structural discussion will concentrate on nNOS and eNOS. Inhibition data are summarized in Table 1, where iNOS (n/i) selectivity is reported as the ratio of the Ki values obtained for miNOS/rnNOS, whereas eNOS (n/e) selectivity is for heNOS/hnNOS. Values for compounds 1-4 are included for comparison.
Table 1. Inhibition of NOS enzymes by synthesized compounds.
| Compound | Ki (μM)a | Selectivity | ||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| rnNOS | hnNOS | miNOS | heNOS | rn/mi | hn/he | |
| 1 | 0.049 | 0.318 | 44.0 | 9.49 | 899 | 30 |
| 2 | 0.066 | 0.440 | 28.4 | 11.8 | 431 | 27 |
| 3 | 0.142 | 0.911 | 33.2 | 17.3 | 237 | 19 |
| 4 | 0.058 | 0.295 | 27.7 | 7.41 | 478 | 25 |
| 5 | 0.079 | NT | 16.8 | NT | 213 | ND |
| 6 | 0.071 | NT | 15.2 | NT | 214 | ND |
| 7 | 0.082 | 0.136 | 32.7 | NT | 399 | ND |
| 8 | 0.035 | 0.036 | 12.5 | 12.3 | 357 | 342 |
| 9 | 0.054 | 0.125 | 40.7 | 43.0 | 754 | 344 |
| 10 | 0.039 | 0.033 | 5.75 | 6.66 | 147 | 202 |
| 11 | 0.033 | 0.031 | 6.7 | 5.63 | 203 | 181 |
| 12 | 0.028 | 0.052 | 7.44 | 4.19 | 266 | 81 |
| 13 | 0.037 | 0.078 | 13.0 | 21.0 | 351 | 269 |
| 14 | 0.033 | 0.051 | 4.54 | 6.09 | 138 | 119 |
| 15 | 0.063 | NT | 19.9 | NT | 316 | ND |
| 16 | 0.036 | 0.056 | 3.42 | 5.04 | 95 | 90 |
| 17 | 0.032 | 0.054 | 3.35 | 6.08 | 105 | 113 |
| 18 | 0.042 | 0.061 | 25.7 | 18.9 | 612 | 310 |
| (RS)-19 | 0.050 | 0.074 | 10.8 | 15.0 | 216 | 203 |
| (R)-19 | 0.061 | 0.065 | 7.34 | 16.1 | 120 | 248 |
| (S)-19 | 0.050 | 0.046 | 25.6 | 22.2 | 512 | 483 |
The compounds were assayed for in vitro inhibition against four purified NOS isoforms: rat nNOS (rnNOS), human nNOS (hnNOS), murine iNOS (miNOS), and human eNOS (heNOS) using known literature methods (see Experimental Section for details), and Ki values are calculated directly from IC50 values. IC50 values are the average of at least two replicates from 6-9 data points; all experimental standard error values (for the LogIC50) are less than ±0.012, and all correlation coefficients are good (R2>0.85). Specific LogIC50 and SE data can be found in the Supporting Information, SI Table 2. Selectivity values are ratios of respective Ki values. NT = not tested; ND = not determined.
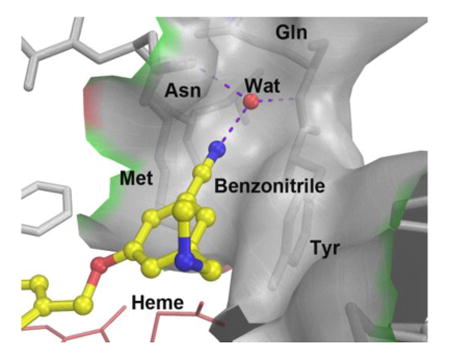
Compared to unsubstituted compound 3, the 5-substituted phenyl ethers (5-9) all have considerably greater potencies against rnNOS. The standout among this series is the 5-cyano compound 8, which is approximately 4 times more potent than 3 and 1.6 times more potent than previous phenyl ether lead 4. The X-ray crystal structure of 8 bound to rnNOS (Figure 3A) was examined to determine what causes this dramatic effect. As observed for similar aminoquinoline-containing compounds17, 18 the 2-aminoquinoline moiety of 8 acts as a competitive arginine mimic by hydrogen bonding with the conserved glutamate residue (Glu592). As also previously observed, the phenyl ring extends beyond the heme-binding pocket, where the methylamine tail H-bonds the water bridging a heme propionate and the cofactor H4B. In most nNOS-phenyl ether-linked structures (such as with 3), the phenyl ring region is flexible, as evidenced by poor electron density in this region in many crystal structures.18 The electron density of 8, by contrast, is intact throughout the inhibitor, indicating greatly reduced flexibility. This stabilization comes from the cyano group of 8, which fits into a narrow pocket formed by the side chains of Tyr706, Met570, and Asn569. The backbone carbonyl of Asn569, the amide of Gln707, and the side-chain of Asn569 also surround 8's cyano group. While other nNOS inhibitors previously studied contain a 1,3,5-trisubstituted cyanophenyl motif,19,20 those portions of the inhibitors occupy entirely different spaces within the enzyme; this newly-revealed “auxiliary pocket” has never been previously reported to interact with any nNOS inhibitor. It is possible that the increased stabilization caused by the nitrile (and as a result, improved potency) is partially electrostatic in nature. One outcome of the nitrile fitting into this pocket is that the electron-poor benzonitrile ring is now anchored in close proximity (<4 Å) to the electron-rich Tyr706, enhancing pi-stacking interactions with this residue.
Figure 3.
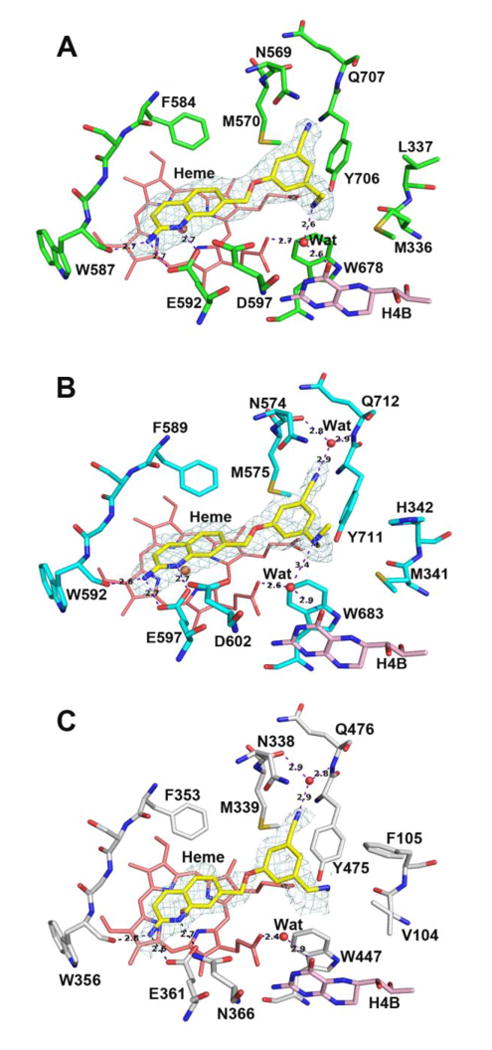
Active site structures of 8 bound to rat nNOS (A), human nNOS (B), and human eNOS (C). In this and all the following figures for crystal structures the omit Fo – Fc density map for the inhibitor is shown at the 2.5 σ contour level. Major hydrogen bonds are shown as dashed lines with distances in Å. Figures were prepared with PyMol (www.pymol.org).
This “nitrile effect” also extends to human nNOS. In the hnNOS-8 structure (Figure 3B), Leu337 of rat nNOS is replaced by His342,38 but the small pocket is still present (bounded by Tyr711, Met575, and Asn574), which similarly accommodates the nitrile of 8. The binding modes of 8 between the two isozymes are virtually identical, and as expected, 8 is equipotent against rat and human nNOS. This represents a remarkable improvement in hnNOS potency over leads 1-4, all of which are highly selective (5-7-fold) for rnNOS over hnNOS. With the improved data quality, a water molecule now is visible inside the auxiliary pocket in the hnNOS-8 structure, forming a bridging H-bond between the nitrile and both the carbonyl of Asn574 and amide of Gln712 (Asn569 and Gln707 in rnNOS). There is no doubt that this water molecule is structural, as it has previously been observed in nNOS crystal structures with l-arginine, as well as many other inhibitor molecules.39, 40 The solvent structure is highly sensitive to crystal quality, however, and this is likely why the water is not present in the rnNOS-8 structure. This suggests that the effects of the nitrile may be multifaceted by providing enhanced electrostatic and H-bond acceptor stabilization.
In addition to the improvements in potency, the nitrile-auxiliary pocket interaction also enhances n/e selectivity. Compound 8 is over 300-fold selective for hnNOS over heNOS – high selectivity in a human-based system. We recently obtained some of our first heNOS-inhibitor crystal structures, belonging to a new P21 space group (see Experimental Section). The chief differences between hnNOS and heNOS are that the Asp602, His342, and Met341 residues of the former isoform are replaced with Asn366, Phe105, and Val104 in the latter.16 As previously reported, the Asp/Asn difference does not appear to play any role in n/e selectivity for these phenyl ether-linked compounds,18 whereas the Met/Val difference can cause dramatic changes in Ki values. In the heNOS-8 structure (Figure 3C), the smaller Val104 residue causes the methylamine tail to assume an alternate conformation, where it is accommodated next to Phe105, breaking the H-bond present between the amine and H4B-site water molecule in hnNOS. Although the small auxiliary pocket (into which the nitrile fits) is conserved between hnNOS and heNOS, the stabilization from the nitrile appears to be less pronounced in heNOS, as evidenced by the incomplete electron density of the benzonitrile ring. The bulky Phe105 residue in heNOS pushes Tyr475 down toward O1D of the heme propionate (away from the inhibitor), whereas in hnNOS, Tyr711 H-bonds O2D of the same propionate, and can thus interact more tightly with the benzonitrile ring. Tyrosine residues in nNOS isoforms are also reported to be more flexible than those in eNOS isoforms,41 suggesting a greater possibility for induced fit in nNOS.
While the nitrile-containing molecule confers the highest potency, there is a general trend of 5-substituted compounds having greater potency than 3; the 5-ethylated analogue (9) shows the second-highest potency after 8. Although the density near the ethyl group indicates some flexibility, the rnNOS-9 structure (Figure 4A) suggests that the 5-ethyl group does not bind in the small auxiliary pocket, likely due to its different shape from the linear nitrile and lack of a H-bonding partner. Instead, the phenyl ring swings over toward the H4B site, and the ethyl group reaches the nNOS-specific hydrophobic pocket, where it likely makes favorable van der Waals contact with Met336, Leu337, and Tyr706, although the phenyl-Tyr706 pi-stack is broken, and the amine-H4B site interaction is much weaker (compared to that in the nNOS-8 structure) with less defined density. It is likely that the other 5-substituents of 5, 6, and 7 (with lower potency than 8) might also fit into this region of the enzyme as 9 does, which reflects the requirement for a linear and polar substituent for auxiliary pocket access.
Figure 4.
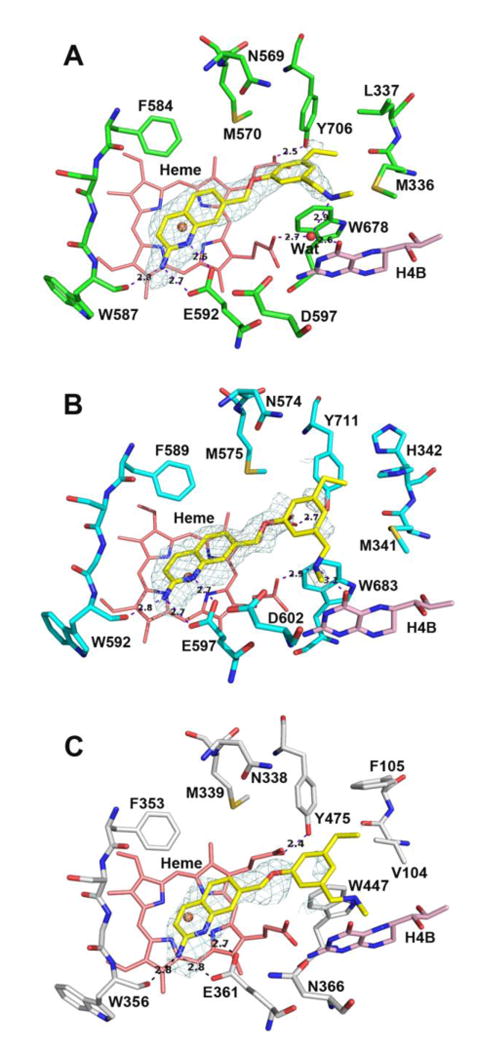
Active site structures of 9 bound to rat nNOS (A), human nNOS (B), and human eNOS (C). Note the alternate rotamer assumed by His342 as a result of a possible clash with the ethyl group in hnNOS (B).
Compared to 8, compound 9 is also a poorer human nNOS inhibitor. In the hnNOS-9 structure (Figure 4B), the nitrile pocket is again unoccupied, but there is a possible clash between the hydrophobic ethyl group and the side chain of His342. Indeed, an alternate rotamer of His342 can be modeled in this structure, where the bulky, polar imidazole ring faces away from the ethyl group. The bulk of the histidine forces the amine of 9 to displace the H4B-site water molecule. This interaction is lacking in the heNOS-9 structure (Figure 4C); the combination of the missing H4B-amine interaction, along with different contact between the ethyl group and Val104 (vs. Met341 of hnNOS), may be responsible for the high n/e selectivity for 9. Interestingly, 9 (and to a lesser degree, the structurally similar compound 7) is highly selective for rnNOS over miNOS. Murine iNOS has a polar asparagine residue (Asn115, instead of Leu337 in rat nNOS) next to the conserved substrate access channel tyrosine (Tyr485 in miNOS or Tyr706 in rnNOS),42 and it is likely that large, hydrophobic groups (such as methoxyl or ethyl) would disfavor binding near Asn115.
In our second series of compounds (10-13), a methyl group was installed at position 4 of the 2-aminoquinoline. Previous studies with 2-aminopyridines indicated that 4-methylation improved nNOS inhibitory potency (and in some cases, n/e selectivity). Computer modeling predicted (and X-ray crystallography confirmed) that the 4-methyl groups fit into a sterically small, hydrophobic region (termed the “S-pocket”)22, 43 located along the “back wall” of the heme-binding site. Originally, we believed that the 2-aminoquinoline group may have been too bulky for this positive modification to be successfully translated to this scaffold, but this hypothesis appears disproven, both from fragment screening and crystallographic results (vide supra) and from the observation that the 4-methyl group in these phenyl ether analogues has positive effects on potency for both rat nNOS and human nNOS when compared to their unmethylated congeners (cf. 3 vs. 10 and 4 vs. 12). As expected, the rnNOS-10 crystal structure (Figure 5) shows that, as with the aminopyridines, the methyl group fits into the S-pocket, bounded by Phe584, Val567, and the backbone of Ser585. The bulk of the aminoquinoline is still well accommodated in the binding mode: 4-methylation forces the 2-aminoquinoline into a more parallel orientation above the heme, instead of the tilted conformation assumed by unmethylated compounds to avoid steric clashes with Phe584 and Val567.17
Figure 5.
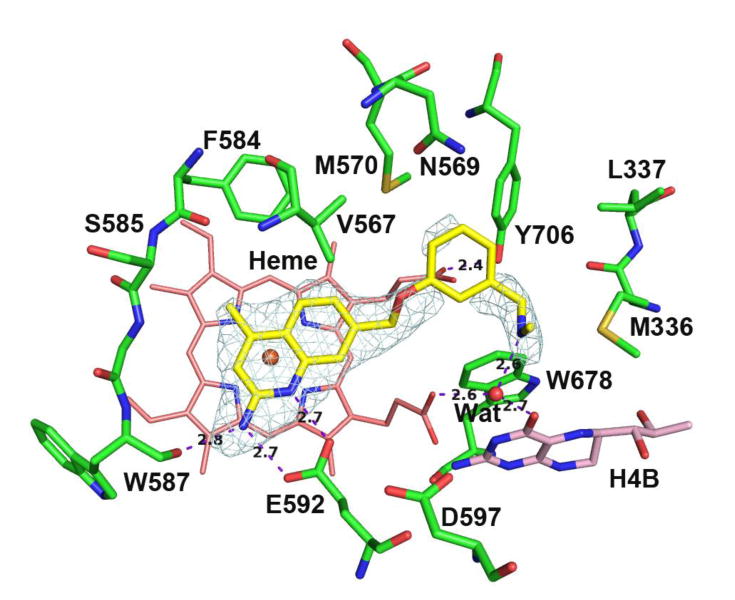
Active site structure of 10 bound to rat nNOS. The phenyl ring shows weaker density.
The enhancing effects that the 4-methyl group have on the 5-substituted compounds are much less pronounced (cf. 8 and 11, where there is little effect, vs. 3 and 10), which indicates that the combination of the 4-methyl and 5-substituent is not additive. Nonetheless, compound 11, with its combination of S-pocket and nitrile-auxiliary pocket interactions, is a very potent dual rnNOS/hnNOS inhibitor. As with 8, the binding mode is identical in the rnNOS-11 (Figure 6A) and hnNOS-11 (Figure 6B) crystal structures, with clear electron density throughout and the bridging structural water molecule present in both the rnNOS and hnNOS structures.
Figure 6.
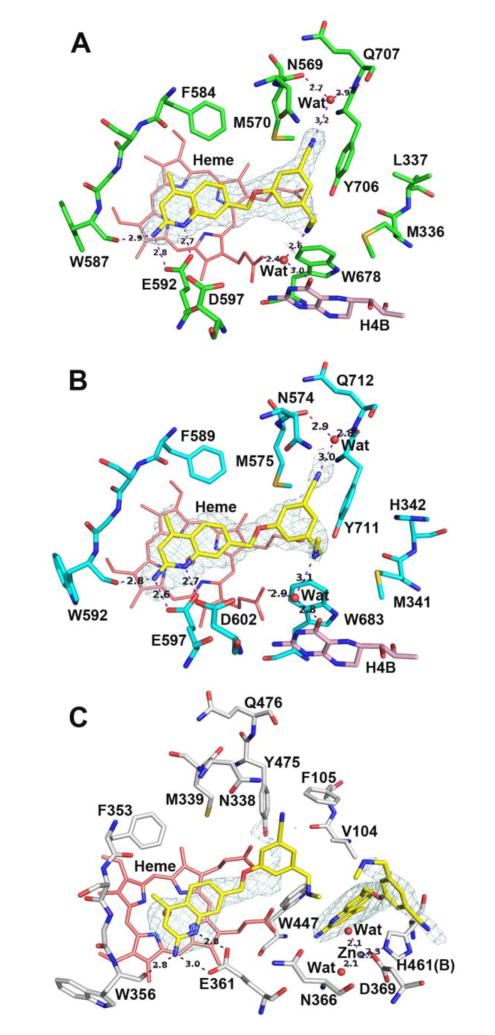
Active site structures of 11 bound to rat nNOS (A), human nNOS (B), and human eNOS (C). There are two molecules of 11 bound in heNOS (C), the first one with a disordered benzonitrile portion while the second one displacing H4B and creating a new Zn site.
The effects of methylation on nNOS/eNOS selectivity are more complicated. It was previously reported that 4-methylation of 2-aminopyridine inhibitors can considerably improve n/e selectivity,22 but this is not consistently observed with 2-aminoquinolines. Compounds 10 and 12 have higher selectivity because of the greatly improved hnNOS activities relative to 3 and 4 (respectively), but for 5-substituted compounds 11 and 13, the n/e selectivity decreases upon methylation compared to unmethylated compounds 8 and 9, respectively. In all cases, the heNOS Ki value for the methylated analogue is lower than the desmethyl one. This (along with the universal decrease in n/i selectivity) could reflect a nonspecific increase in inhibitor binding, as the S-pocket is conserved among all isoforms. The X-ray crystal structure of heNOS-11 (Figure 6C), however, is surprising – two molecules of 11 are present in the binding site. One binds in the “usual” mode (l-arginine mimicry with nitrile-auxiliary pocket interactions), while the other displaces the H4B cofactor and pi-stacks with Trp477. Arg365 (not shown in the figure), which normally H-bonds with H4B, swings away as H4B is displaced and a Zn2+ ion is coordinated by Asp369, His461 (from the other monomer), and two water molecules. The H4B displacement occurs even when micromolar amounts of H4B are added to the crystal soaking solution, suggesting that 11 outcompetes H4B binding in the pterin pocket. Supporting this observation is the fact that the Ki value for 11 for heNOS does not change significantly when the concentration of H4B in the assay is increased from 10 μM to 50 μM (5.6 μM for the former vs. 4.8 μM for the latter). The 4-methyl group on the quinoline of 11 likely enhances this stacking with Trp477, which explains why H4B displacement is not observed with 8 or other desmethyl compounds. Although this is the first report of an aminoquinoline-based inhibitor binding a NOS enzyme in this way, dual inhibitor binding, H4B displacement, and extra Zn2+-coordination were previously observed in rat nNOS with double-headed aminopyridine inhibitors,44 but has not been observed in bovine eNOS owing to its more rigid dimer interface,45 and thus was proposed to be exploitable for selective inhibitor design. Indeed, the bovine eNOS-11 structure (SI Figure S1) only shows one inhibitor bound to the substrate site (even when Zn2+ is added to the crystal soaking solution), with a binding mode similar to the heNOS-8 structure (where the methylamine can face away from the H4B site). It is possible that some global structural difference between bovine eNOS and human eNOS simply causes a decrease in H4B affinity (or it may only occur under certain conditions), and an extra bound inhibitor could cause the decrease in Ki value obtained upon methylation (cf. 8 and 11). However, the poor electron density of 11 in the substrate site in the heNOS-11 structure (Figure 6C), especially in the amine and benzonitrile portions of the substrate-site molecule that are crucial for binding, suggests that this is an unstable complex. This could occur because binding of the second 11 in the H4B site may weaken the binding of the first 11 in the substrate site (i.e., negative cooperativity), and as such, result in a much higher heNOS Ki value than that observed for hnNOS.
To examine these different binding modes (and the requirements for the nitrile to reach the auxiliary pocket) further, deoxygenated derivatives 14-16 were prepared. These compounds are also desirable because of their high cLogP value and lack of the polar oxygen, which increases tPSA and could be a potential site of metabolism in vivo. Interestingly, 14 behaves similarly to compound 10 – the Ki values for rnNOS, miNOS, or heNOS for 14 are very close to those for 10, suggesting that the oxygen is not a crucial requirement for selectivity for this compound, although the hnNOS activity has decreased slightly. The rnNOS-14 structure (Figure 7A), however, indicates that removal of the oxygen changes the binding mode. While the 4-methylaminoquinoline is fixed in the same position as it is in 10, the larger ethylene linker of 14, compared to the oxaethylene linker of 10, prefers an “upward” position away from Met570 and Val567. However, the phenyl ether linker snugly abuts these two residues, and the phenyl ring swings over toward the H4B site where the methylamine tail displaces the H4B-site water instead of H-bonding it. As the rnNOS Ki value of 14 is close to that of 10, this is obviously a favorable conformation.
Figure 7.
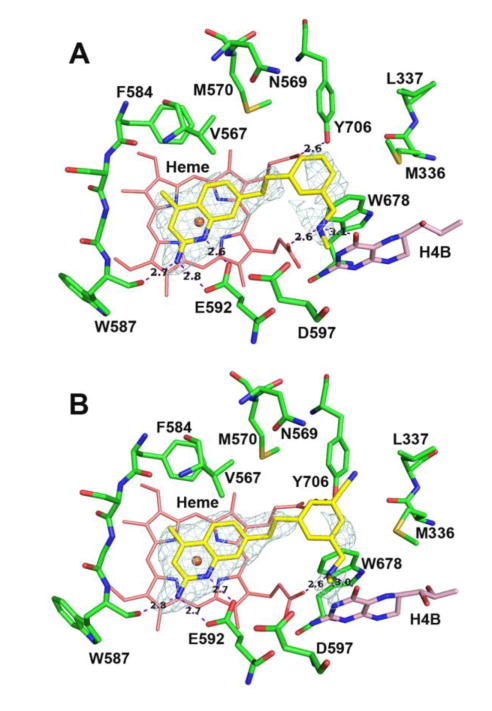
Active site structures of 14 (A) and 16 (B) bound to rat nNOS. Note that unlike 11 (Figure 6A) the benzonitrile of 16 is not bound in the auxiliary pocket because of the repulsion between the bulkier ethylene linker and Val567.
As in the phenyl ether scaffold, methylation of the quinoline in the alkyl scaffold (15 to 16) also improves potency. However, the rnNOS-16 (Figure 7B) structure indicates that 16 is less stable when bound than is 11. The nitrile is not bound in the auxiliary pocket, and the electron density indicates greatly increased flexibility. This suggests that the geometric and steric requirements to place a substituent into the auxiliary pocket are quite strict. The methylene bridge again assumes the “upward” position observed in the rnNOS-14 structure to avoid steric clashes with Val567, breaking the Tyr706-aryl interaction and pointing the nitrile toward Leu337 instead.
Despite the hypothesis that a 4-substituent, such as fluorine, could improve potency, compound 17 does not have improved rnNOS and hnNOS activity compared to 11, and the selectivity values are lower. Previously, it was also shown that fluorine pointing roughly toward the region of the nNOS-specific hydrophobic pocket was deleterious to n/i selectivity.18 This same decrease is also observed here; the fluorine-Asn115 interaction may be favorable in miNOS, as indicated by a lower Ki of 3.34 μM for 17 (compared to 6.7 μM for 11). Compound 18 represents an interesting example where cyanation and methylation can be used to “rescue” a substitution pattern with low activity. Previously, an uncyanated, unmethylated version of this compound18 was reported to have a rat nNOS Ki value of 332 nM – the methyl and cyano substituents together decrease its Ki value 8-fold to 42 nM. The rnNOS-18 structure (Figure 8A) shows that while the nitrile-auxiliary pocket interaction is retained, the longer phenethylamino group displaces the H4B-site water, whereas the hnNOS-18 structure (Figure 8B) indicates that the amine H-bonds the existing H4B site water.
Figure 8.
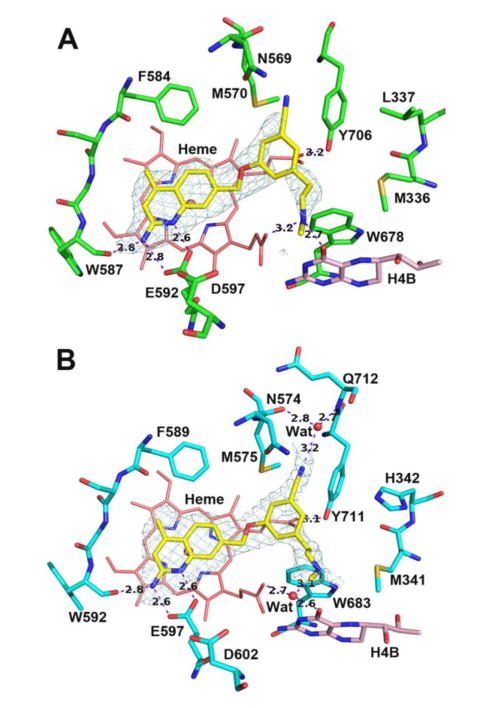
Active site structures of 18 bound to rat nNOS (A) and human nNOS (B). The structural water is missing in rnNOS likely due to the diffraction data quality.
Compound 19, assayed initially as its racemate (R,S)-19, is also very close in rnNOS and hnNOS potency to those of 18. Interestingly, when rnNOS and hnNOS crystals were soaked with (R,S)-19, the electron density of the resulting crystal structures (hnNOS and rnNOS, SI Figures S2A and S2B) was consistent with both enantiomers being bound, suggesting that the two are similar in potency, which was confirmed by assaying them separately. The Ki value for (R)-19 is 61 nM against rnNOS and 65 nM against hnNOS, whereas those for (S)-19 are 50 nM and 46 nM, respectively. Compounds 18 and 19 (and isomers) were designed to place longer tail groups in the vicinity of Met336/Met341 (rnNOS/hnNOS). In the hnNOS-18 structure, the hnNOS-(R)-19, and hnNOS-(S)-19 structures (Figures 9A and 9B), and the rnNOS-(S)-19 and rnNOS-(R)-19 structures (SI Figures S3A and S3B, respectively) large, flexible portions of the alkylamine tails are all < 5Å away from, and could contact, Met336 or Met341, with the rest of the molecules anchored by aminoquinoline-glutamate, nitrile-auxiliary pocket, and H4B-site-amine interactions. No obvious differences in binding, other than the positioning of the alpha-methyl group, are observable between (R)-19, and (S)-19.
Figure 9.
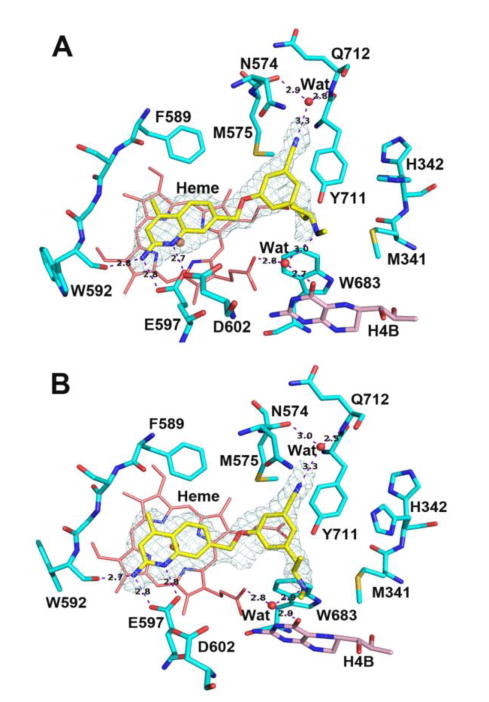
Active site structures of (R)-19 (A) and (S)-19 (B) bound to human nNOS. The only difference between the two enantiomers is the α-methyl position. Alternate rotamers for His342 were observed.
As these methionine residues are not present in eNOS enzymes, extra van der Waals contact with these methionine residues should improve n/e selectivity, as the analogous eNOS valines have a smaller surface area and thus make less contact with inhibitors. As predicted, the hnNOS/heNOS selectivities for these bulkier compounds (18 and the enantiomers of 19) exceed that of 11. Despite the similar potencies, there is a clear difference in selectivity between (R)- and (S)-19; the n/e selectivity for (S)-19 is nearly twice that of (R)-19. Approaching 500-fold, (S)-19 has the highest n/e selectivity ever observed for a 2-aminoquinoline-based inhibitor.
We examined the heNOS crystal structures with the two enantiomers bound for insights into this disparity. In the (S)-19-heNOS structure (Figure 10A), only one molecule of (S)-19 is bound, with the density in the H4B site largely consistent with H4B being bound, although there is some ambiguity about the occupancy of the new Zn2+-site in several chains (not shown in the figure). As observed for 8, Tyr475 in heNOS is pushed by the bulky Phe105 from O2D toward O1D of the heme propionate and appears to form a weaker electrostatic/van der Waals interaction with the benzonitrile ring than the analogous Tyr711 in hnNOS. The amine tail of (S)-19 is disordered in this structure compared to the relatively intact density of the tail in the hnNOS-(S)-19 structure, suggesting destabilization of the amine binding, possibly the result of reduced interactions between Val104 and the alpha-methyl group, compared to those stronger interactions with nNOS's methionine. By contrast, the (R)-19-heNOS structure (Figure 10B) resembles that of 11, with two molecules of inhibitor present in the binding site. The heNOS Ki value for (R)-19 (16.1 μM) is not substantially increased from (S)-19 (22.1 μM), suggesting that the heNOS-dual-inhibitor complex, like that of 11, may be relatively weak or unstable. As much of the binding of these phenyl ether-linked compounds is dependent on tail amine-H4B-site interactions, any complex that lacks these interactions, as a result of displacement of H4B and its attendant water molecule, may destabilize inhibitor binding. A similar dual-inhibitor complex is observed for heNOS-18 (SI Figure S4), suggesting that the ability to displace H4B is ligand-dependent, but may be common among certain classes of these benzonitrile compounds. Although far from the first record of stereochemical influence on the potency or selectivity of nNOS inhibitors,46, 47 the case of 19 reveals that even very simple chiral groups, such as the tail of 19, can impart excellent n/e selectivity for 2-aminoquinolines.
Figure 10.
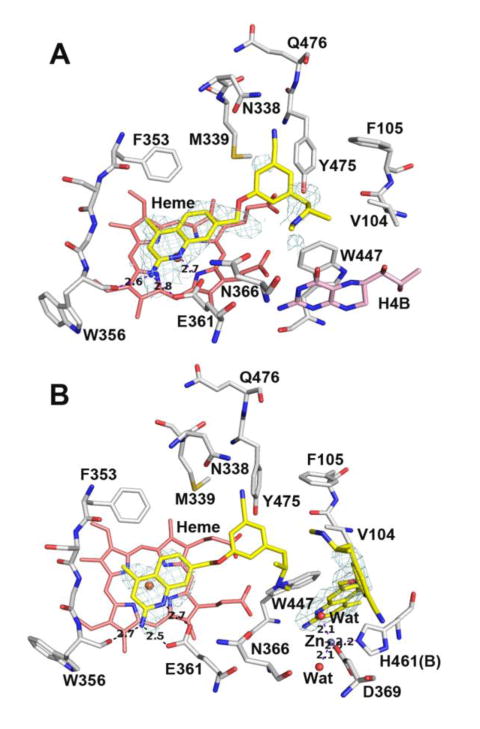
Active site structures of (S)-19 (A) and (R)-19 (B) bound to human eNOS. There are two molecule of (R)-19 bound in human eNOS. The first one in the active site shows a disordered tail portion, while the second one displaces H4B and creates a new Zn site.
Finally, two potent dual rnNOS/hnNOS inhibitors with different structural motifs and high n/e selectivity (8 and 10) were assayed for membrane permeability in a Caco-2 assay. In this assay, the permeability of a compound through a monolayer of cells resembling the intestinal epithelium is measured. Caco-2 assays have been used to approximate the potential for both oral bioavailability and blood-brain barrier permeation,48, 49 although the latter is generally less accurate. Although compounds 1 and 2 were unfavorably promiscuous binders, they were highly permeable in this assay. Compound 4, despite its significantly cleaner off-target profile, was much less permeable and had higher efflux, indicating that the structural rearrangement had detrimental effects on cellular pharmacokinetics. Compared to 4, both 8 and 10 have modestly increased permeability, lower efflux, and higher recovery values (Table 2; values for compounds 2 and 4 are given for comparison), although their permeability is still considerably lower than 2. Interestingly, the cyano compound 8 is more permeable than 10, despite having a higher tPSA (83.4 Å2, increased because of the nitrile, vs. 59.6 Å2) and lower cLogP (2.5 vs. 3.2), which suggests that these parameters may not be entirely predictive of cellular permeability. Regardless, 4-methylation and 5-cyanation might be favored modifications for improving the bioavailability of future compounds.
Table 2.
Caco-2 permeability summary for select compounds.
| Compound | Apparent Permeability (Papp, 10−6 cm s−1)b | Efflux ratiof | Recovery | ||
|---|---|---|---|---|---|
|
|
|
||||
| Mean A-->B | Mean B-->A | A-->B | B-->A | ||
| 2 | 16.9 | 41.9 | 2.5 | 63% | 103% |
| 4 | 2.3 | 12.6 | 5.5 | 37% | 79% |
| 8 | 9.2 | 32.1 | 3.5 | 63% | 89% |
| 10 | 7.1 | 34.4 | 4.8 | 70% | 100% |
| Warfarinc | 26.0 | 17.7 | 0.68 | - | - |
| Ranitidined | 0.33 | 2.4 | 7.3 | - | - |
| Talinolole | 0.12 | 8.2 | 68.3 | - | - |
All assays were performed over 2 h at a concentration of 10 μM. See Experimental Section for details.
Apparent permeability value.
High permeability control;
low permeability control;
high efflux control.
Efflux ratio is defined as Papp (B-->A)/Papp (A-->B); efflux ratio values >3 indicate that a compound may be a substrate for P-gp or other active transport systems.
Conclusions
To summarize, we undertook further modification of our “cleaner-binding” phenyl ether-linked aminoquinoline scaffold with hopes that we could improve human nNOS inhibition, hnNOS/heNOS selectivity, and possibly the cellular permeability of this promising class of compounds. We discovered that 5-substitution of the phenyl ring results in greatly improved hnNOS activity and hnNOS/heNOS selectivity, especially when the substituent is cyano. X-ray crystallographic studies revealed that the 5-cyano group fits into a small, previously unreported, auxiliary pocket located next to the heme-binding sites of both rnNOS and hnNOS, resulting in good potency against both isoforms. The nitrile H-bonds to a structural water within this pocket and anchors an electron-deficient aryl ring next to an electron-rich tyrosine, an interaction that is weaker in heNOS. Additionally, the methylamine tails of 8 and 11 favor a water-mediated H-bond to H4B and a heme propionate in nNOS, an interaction that is missing in heNOS. These combined interactions resulted in the first 2-aminoquinolines with high selectivity for human nNOS over human eNOS. Although this new auxiliary pocket is a promising site for further optimization, the structural requirements to fit a substituent into this region may be fairly strict; replacement of the nitrile with an ethyl group resulted in the 5-substituent being bound elsewhere, and replacement of the phenyl ether oxygen of 11 with a slightly larger methylene (15) resulted in increased flexibility because of the increased steric bulk, as observed by crystallography. Additionally, we found that methylation of the 4-position of the aminoquinoline could greatly improve rnNOS and hnNOS potency further, although it decreased hnNOS/heNOS selectivity in some cases. 4-Methylation also caused some inhibitors to displace H4B in heNOS in addition to mimicking l-arginine, although this appears ligand-dependent. In spite of the decrease, further modification of the cyanoaryl-containing tail (elongation to 18, or introduction of a stereocenter, such as 19) could also be used to disfavor binding to heNOS, and one compound, (S)-19, had about 500-fold selectivity over both eNOS and iNOS, exerted through a mixture of differential hydrophobic pocket and tyrosine-benzonitrile interactions, although the role of H4B antagonism in heNOS cannot entirely be excluded. Additionally, both cyanation and 4-methylation showed some ability to improve the Caco-2 permeability of the phenyl ether scaffold. These results, taken together, highlight the promise of this new auxiliary pocket in nNOS as a novel “hot spot” for further structure-based design of nNOS inhibitors.
Experimental Section
General Procedures
Anhydrous solvents (THF, CH2Cl2, MeOH, Et3N, MeCN, and DMF) were distilled prior to use. All other solvents, reactants, and reagents were purchased from commercial vendors and were used without further purification. Methanolic HCl (3 M, for ammonium hydrochloride salt formation and Boc-deprotection) was freshly prepared by the reaction of acetyl chloride and anhydrous MeOH at 0 °C. Melting points were determined in capillary tubes using a Buchi melting point B-540 apparatus and are uncorrected. 1H-NMR spectra were recorded at 500 MHz, using a Bruker Avance III 500 (direct cryoprobe), and 13C-NMR spectra were obtained at 126 MHz using the same instrument. Low-resolution ESIMS were obtained on a Bruker AmaZon SL Ion Trap mass spectrometer system. High-resolution mass spectral data were obtained at the Integrated Molecular Structure Education and Research Center (IMSERC, Northwestern University) on an Agilent 6210A TOF mass spectrometer in positive ion mode using electrospray ionization with an Agilent G1312A HPLC pump and an Agilent G1367B autoinjector. Data were processed using MassHunter software version B.04.00. Flash column chromatography was performed using an Agilent 971-FP automated flash purification system with a Varian column station and SiliCycle cartridges (12-80 g, both normal and High Performance). Analytical HPLC was performed using an Agilent Infinity 1260 HPLC system and injection volumes of 5-10 μL. A Phenomenex Luna 5 μm C-8(2) 100 Å column, 50 × 4.60 mm, was used for all HPLC experiments, using a 10-min gradient of 95% H2O/5% acetonitrile + 0.05% TFA to 95% acetonitrile/5% H2O + 0.05% TFA, at 1.5 mL/min. Chiral analytical HPLC was performed using a Chiralpak AD-H 5 μm column, 250 × 4.60 mm, using 25-min isocratic elution at 5% isopropanol in hexanes, at 1 mL/min. The purity of all final target compounds was found to be ≥95% by HPLC. Analytical thin-layer chromatography was performed on Silicycle extra-hard 250 μm TLC plates. Compounds were visualized with short-wavelength UV light, and with ninhydrin, FeCl3, CAM, and KMnO4 stains, where appropriate. Compounds 30,17,18 45,18 46,18 and 5150 were prepared by known literature procedures, and their spectral data are consistent with those data reported for them. The optical rotation of chiral compounds was measured using a Rudolph Research Analytical Autopol IV automatic polarimeter, using a 50 mm cell and the sodium D-line (589 nm). The preparation of quinoline precursors and assembly of final compounds is described below, while the preparation of phenols and precursors 23-25, 26-29, 55-57, 59-62, 70-73 and 77-82 is discussed in the Supporting Information.
General Procedure for Synthesis and Deprotection of Phenyl Ether-Linked Aminoquinolines
The procedure is similar to that recently reported:18 sodium hydride (60% suspension in mineral oil, 1 equiv) was diluted with anhydrous DMF (1-2 mL) and cooled to 0 °C under argon. A solution of the required phenol (1 equiv) in anhydrous DMF (1-2 mL) was added slowly to the suspension and stirred at 0 °C for 10-30 min (typically ∼25 min), following which bromides 30 or 43 (1 equiv) or chloride 44 (0.90- equiv) was added as a solution in anhydrous DMF. If 30 or 43 were employed, the reaction mixture was stirred at 0 °C for 40 min-1 h (typically ∼50 min), and if 44 was employed, the reaction mixture was warmed to r.t. and then heated to 50 °C for 45 min-1 h. In both cases, the reaction mixture was then quenched at r.t. by addition of a 1:1 sat. aq. NaCl/H2O mixture or a sat. aq. NaHCO3 solution (∼15 mL). The mixture was extracted with EtOAc (usually 3 × 20 mL was sufficient for these lipophilic carbamates), and the organic phase was washed with 5% aq. NaCl (3-4 × 30-80 mL) and sat aq. NaCl (30-50 mL). The organic layer was dried over anhydrous sodium sulfate, concentrated, and purified by flash column chromatography (12 g SiO2 cartridge), using gradients as described for individual compounds below. The resulting intermediate acetamides were not characterized or purified further, but were diluted with anhydrous MeOH (5-10 mL), and anhydrous K2CO3 (∼2 equiv) was added. The mixture was heated at reflux for 2-2.5 h, cooled, and concentrated. The resulting residue was partitioned between EtOAc and 1:1 H2O/sat. aq. NaCl, and the aqueous layer was extracted with EtOAc (2-3 × 5-20 mL). The organic layers were washed with sat. aq. NaCl and dried over anhydrous sodium sulfate. Purification is detailed under subheadings for individual compounds. The free aminoquinoline was diluted in dry ether (or 10:1-4:1 ether/MeOH) or dichloromethane (for 31, 35, and 48) and filtered to remove any particulate matter. To the filtered solution, methanolic HCl (3 M, 1-2 mL) was added (except for 31, 35, and 48, where 150-300 μL trifluoroacetic acid was added instead), and the mixture was stirred at room temperature either overnight or for 20-30 min (for 31, 35, and 48). The salts were isolated by filtration or precipitation, and the final purification was performed as described below for individual compounds.
7-[(3-Chloro-5-((methylamino)methyl)phenoxy)methyl]quinolin-2-amine Dihydrochloride (5)
This compound was prepared from 30 (0.100 g, 0.358 mmol) and phenol 23 (0.097 g, 0.358 mmol). Workup and purification by flash column chromatography, eluting with a gradient of 2% EtOAc in CH2Cl2 to 35% EtOAc in CH2Cl2, afforded intermediate acetamide 31 as a colorless foam (0.138 g, 82%), which was immediately deprotected using K2CO3 (0.081 g, 0.587 mmol), as described in the General Procedure. After workup, the resulting gum was purified by flash column chromatography, eluting with EtOAc to yield the free aminoquinoline as a semisolid residue that was diluted in 10:1 ether: MeOH (∼12 mL) and treated with methanolic HCl (2 mL). After being stirred overnight, filtration afforded 5 (0.081 g, 69% from 31) as a white flocculent solid after precipitation from hot MeOH (1 mL) with ether (5 mL) and washing with ether: mp 298-299 °C. 1H-NMR (500 MHz; DMSO-d6): δ 14.36 (br s, 1 H), 9.31 (br s, 2 H), 9.20 (br s, 1 H) 8.37 (d, J = 9.3 Hz, 1 H), 8.30 (br s, 1 H), 7.96 (d, J = 8.2 Hz, 1 H), 7.76 (s, 1 H), 7.53 (dd, J = 8.2, 1.2 Hz, 1 H), 7.30 (t, J = 1.7 Hz, 1 H), 7.25 (t, J = 1.4 Hz, 1 H), 7.22 (t, J = 2.0 Hz, 1 H), 7.10 (d, J = 9.3 Hz, 1 H), 5.38 (s, 2 H), 4.09 (s, 2 H), 2.55-2.50 (m, 3 H, partially obscured by solvent peak). 13C-NMR (126 MHz; DMSO-d6): δ 159.2, 154.8, 143.1, 141.7, 135.6, 134.3, 129.4, 124.2, 122.7, 120.8, 116.3, 115.7, 115.4, 114.2, 69.3, 50.7, 32.3; one of the quinoline carbons is not visible due to baseline broadening; ESIMS m/z (rel. intensity) 328/330 (MH+, 100/33); HRMS calcd for C18H19ClN3O+: 328.1211; found, 328.1215.
7-[(3-Bromo-5-((methylamino)methyl)phenoxy)methyl]quinolin-2-amine Dihydrochloride (6)
This compound was prepared from 30 (0.085 g, 0.305 mmol) and phenol 24 (0.096 g, 0.305 mmol). Workup and purification by flash column chromatography, eluting with a gradient of 2% EtOAc in CH2Cl2 to 35% EtOAc in CH2Cl2, afforded intermediate acetamide 32 as an off-white foam (0.110 g, 70%), which was immediately deprotected using K2CO3 (0.059 g, 0.427 mmol) as described in the General Procedure. After workup, the free aminoquinoline was obtained as a gummy residue that was diluted in 4:1 ether: MeOH and treated with methanolic HCl (2 mL). After being stirred overnight, filtration afforded 6 (0.081 g, 84% from 32) as a white flocculent solid after precipitation from hot MeOH (2 mL) with ether (10 mL) and washing with ether: mp 286-287.5 °C. 1H-NMR (500 MHz; DMSO-d6): δ 14.32 (br s, 1 H), 9.26 (br s, 2 H), 9.20 (br s, 1 H), 8.37 (d, J = 9.3 Hz, 1 H), 8.21 (br s, 1 H), 7.96 (d, J = 8.2 Hz, 1 H), 7.76 (s, 1 H), 7.53 (dd, J = 8.2, 1.0 Hz, 1 H), 7.37 (t, J = 1.3 Hz, 1 H), 7.35 (t, J = 2.0 Hz, 1 H), 7.34 (t J = 1.3 Hz, 1 H), 7.10 (d, J = 9.3 Hz, 1 H), 5.38 (s, 2 H), 4.09 (t, J = 4.7 Hz, 2 H), 2.52-2.50 (m, 3 H, partially obscured by solvent peak); 13C-NMR (126 MHz; DMSO-d6): δ 158.88, 154.51, 142.59, 141.30, 135.56, 129.03, 125.19, 123.77, 122.22, 120.49, 117.81, 116.34, 115.51, 113.83, 68.97, 50.27, 32.00; one of the quinoline carbons is not visible due to baseline broadening; ESIMS m/z (rel. intensity) 372/374 (MH+, 100/100); HRMS calcd for C18H19BrN3O+: 372.0706; found, 372.0709.
7-[(3-Methoxy-5-((methylamino)methyl)phenoxy)methyl]quinolin-2-amine Ditrifluoroacetate (7)
This compound was prepared from 30 (0.054 g, 0.193 mmol) and phenol 25 (0.052 g, 0.193 mmol). Workup and purification by flash column chromatography, eluting with a gradient of 3% EtOAc in CH2Cl2 to 43% EtOAc in CH2Cl2, afforded intermediate acetamide 33 as a pale-yellow foam (0.064 g, 71%), which was immediately deprotected using K2CO3 (0.038 g, 0.273 mmol) as described in the General Procedure. After workup, the free aminoquinoline was diluted with CH2Cl2, filtered, and concentrated. The residue was diluted with anhydrous CH2Cl2 (3 mL), and trifluoroacetic acid (200 μL) was added. The mixture was stirred for 25 min and concentrated, and ether (10 mL) was added to the residue. The mixture was sonicated until a solid formed, which was collected to yield 7 (0.061 g, 81% from 33) as a white solid after precipitation from hot MeOH (1 mL) with ether (10 mL) and washing with ether: mp 130 °C (softens), 156-158 °C (melts). 1H-NMR (500 MHz; DMSO-d6): δ 14.16 (br s, 1 H), 8.79 (s, 2 H), 8.23 (s, 1 H), 7.87-7.82 (m, 1 H), 7.64 (s, 1 H), 7.43 (s, 1 H), 6.98-6.97 (m, 1 H), 6.76 (t, J = 1.5 Hz, 1 H), 6.69 (s, 1 H), 6.68 (t, J = 2.1 Hz, 1 H), 5.29 (s, 2 H), 4.05 (s, 2 H), 3.76 (s, 3 H), 2.55 (s, 3 H); the aminoquinoline protons are not visible, but are broadened into the baseline and cause an overall broadening of the signals from 7-8 ppm; 13C-NMR (126 MHz; DMSO-d6): δ 160.6, 159.4, (158.5 + 158.3 + 158.0 + 157.8, 1 C), 134.1, 128.6, 121.0, (118.5 + 116.1, 1 C), 113.5, 108.7, 107.9, 101.3, 68.9, 55.4, 51.3, 32.1; several of the aminoquinoline and trifloroacetate carbon signals are not visible due to baseline broadening; ESIMS m/z (rel. intensity) 324 (MH+,100); HRMS calcd for C19H22N3O2+: 324.1707; found, 324.1711.
3-[(2-Aminoquinolin-7-yl)methoxy]-5-((methylamino)methyl)benzonitrile Dihydrochloride (8)
This compound was prepared from 30 (0.085 g, 0.305 mmol) and phenol 26 (0.080 g, 0.305 mmol). Workup and purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 40% EtOAc in CH2Cl2, afforded intermediate acetamide 34 as a yellow foam (0.127 g, 90%), which was immediately deprotected using K2CO3 (0.076 g, 0.551 mmol) as described in the General Procedure. After workup, the free aminoquinoline was purified by flash column chromatography (eluting with a gradient of EtOAc to 2% MeOH in EtOAc), and the resultant white solid was diluted in 4:1 ether: MeOH and treated with methanolic HCl (1.5 mL). After being stirred overnight, filtration afforded 8 (0.073 g, 68% from 34) as a white flocculent solid after precipitation from hot MeOH (2 mL) with ether (10 mL) and washing with ether: mp 304-305 °C (dec). 1H-NMR (500 MHz; DMSO-d6): δ 14.24 (br s, 1 H), 9.30 (br s, 2 H), 9.20 (br s, 1 H), 8.37 (d, J = 9.2 Hz, 1 H), 8.21 (br s, 1 H), 7.96 (d, J = 8.2 Hz, 1 H), 7.76 (s, 1 H), 7.67 (t, J = 1.8 Hz, 1 H), 7.64 (t, J = 1.8 Hz, 1 H), 7.61 (s, 1 H), 7.54 (d, J = 7.9 Hz, 1 H), 7.09 (d, J = 9.3 Hz, 1 H), 5.42 (s, 2 H), 4.15 (s, 2 H), 2.53-2.50 (m, 3 H); 13C-NMR (126 MHz; DMSO-d6): δ 158.2, 154.5, 142.7, 141.0, 135.3, 129.1, 126.3, 125.1, 123.9, 122.4, 120.6, 118.19, 118.00, 115.6, 113.9, 112.4, 69.2, 50.1, 32.0; ESIMS m/z (rel. intensity) 319 (MH+, 100); HRMS calcd for C19H19N4O+: 319.1553; found, 319.1559.
7-[(3-Ethyl-5-((methylamino)methyl)phenoxy)methyl]quinolin-2-amine Ditrifluoroacetate (9)
This compound was prepared from 30 (0.042 g, 0.152 mmol) and phenol 29 (0.041 g, 0.152 mmol). Workup and purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 30% EtOAc in CH2Cl2, afforded intermediate acetamide 35 as a translucent, colorless gum (0.052 g, 74%), which was immediately deprotected using K2CO3 (0.031 g, 0.224 mmol) as described in the General Procedure. After workup, the free aminoquinoline was diluted with anhydrous CH2Cl2 (3 mL), and trifluoroacetic acid (150 μL) was added. The mixture was stirred for 15 min and concentrated, and ether (10 mL) was added to the residue. The mixture was stirred until a solid formed, which was collected to yield 9 (0.027 g, 67% from 35) as a white solid after being washed with ether: mp 154-156 °C. 1H-NMR (500 MHz; DMSO-d6): δ 14.23 (br s, 1 H), 8.81-8.70 (m, 4 H), 8.36 (d, J = 9.3 Hz, 1 H), 7.95 (d, J = 8.2 Hz, 1 H), 7.70 (s, 1 H), 7.53 (d, J = 8.1 Hz, 1 H), 7.06 (d, J = 9.3 Hz, 1 H), 7.00 (d, J = 1.2 Hz, 1 H), 6.98 (s, 1 H), 6.94 (s, 1 H), 5.31 (s, 2 H), 4.07 (s, 2 H), 2.61 (q, J = 7.6 Hz, 2 H), 2.56 (s, 3 H), 1.19 (t, J = 7.6 Hz, 3 H); 13C-NMR (126 MHz; DMSO-d6): δ (158.8 + 158.5 + 158.30 + 158.0, 1 C), 158.27, 154.7, 146.1, 142.6, 142.0, 133.3, 129.0, 123.7, 121.8, 120.5, (118.3 + 115.9), 114.8, 113.8, 113.5, 68.5, 51.3, 32.2, 28.1, 15.3; the trifluoroacetate carbon signals are only partially visible; two of the aminoquinoline carbons are not visible due to baseline broadening; ESIMS m/z (rel. intensity) 322 (MH+, 100); HRMS calcd for C20H24N3O+: 322.1914; found, 322.1916.
4-Methyl-7-[(3-((methylamino)methyl)phenoxy)methyl]quinolin-2-amine Dihydrochloride (10)
This compound was prepared from 43 (0.065 g, 0.221 mmol) and phenol 46 (0.052 g, 0.221 mmol). Workup and purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 30% EtOAc in CH2Cl2, afforded intermediate acetamide 50 as a white solid (0.076 g, 77%), which was immediately deprotected using K2CO3 (0.047 g, 0.338 mmol) as described in the General Procedure. After workup, the free aminoquinoline was triturated with hexanes and filtered, and the resultant white solid was diluted in 10:1 ether: MeOH (8 mL) and treated with methanolic HCl (1.5 mL). After being stirred overnight, filtration afforded 10 (0.051 g, 80% from 50) as a white flocculent solid, after precipitation from hot MeOH (0.5 mL) with ether (5 mL) and being washed with ether: mp 249-251 °C. 1H-NMR (500 MHz; DMSO-d6): δ 14.12 (br s, 1 H), 9.19 (br s, 2 H), 9.00 (br s, 1 H), 8.10 (br s, 1 H), 8.03 (d, J = 8.4 Hz, 1 H), 7.78 (s, 1 H), 7.57 (dd, J = 8.4, 1.3 Hz, 1 H), 7.39 (t, J = 7.9 Hz, 1 H), 7.32 (t, J = 1.8 Hz, 1 H), 7.12-7.09 (m, 2 H), 6.94 (d, J = 0.8 Hz, 1 H), 5.37 (s, 2 H), 4.09 (t, J = 4.8 Hz, 2 H), 2.65 (d, J = 0.9 Hz, 3 H), 2.54-2.50 (m, 3 H, partially obscured by solvent peak); 13C-NMR (126 MHz; DMSO-d6): δ 158.1, 153.8, 141.8, 133.6, 130.0, 125.8, 123.6, 122.4, 120.6, 116.6, 115.5, 115.1, 112.6, 68.4, 51.0, 32.0, 19.0; two of the aminoquinoline carbons are not visible due to baseline broadening; ESIMS m/z (rel. intensity) 308 (MH+, 100); HRMS calcd for C19H22N3O+: 308.1757; found, 308.1757.
3-[(2-Amino-4-methylquinolin-7-yl)methoxy]-5-((methylamino)methyl)benzonitrile Dihydrochloride (11)
This compound was prepared from 43 (0.065 g, 0.221 mmol) and phenol 26 (0.058 g, 0.221 mmol). Workup and purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 38% EtOAc in CH2Cl2, afforded intermediate acetamide 47 as a yellow foam (0.043 g, 41%), which was immediately deprotected using K2CO3 (0.025 g, 0.181 mmol) as described in the General Procedure. After workup, the free aminoquinoline was purified by flash column chromatography (eluting with a gradient of EtOAc to 5% MeOH in EtOAc), and the resultant residue was diluted in 10:1 ether: MeOH (10 mL) and treated with methanolic HCl (1.5 mL). After being stirred overnight, filtration afforded 8 (0.023 g, 68% from 47) as a pale yellow flocculent solid after precipitation twice from hot MeOH (0.4 mL) with ether (10 mL) and being washed with ether: mp 185-188 °C. 1H-NMR (500 MHz; DMSO-d6): δ 14.14 (br s, 1 H), 9.38 (br s, 2 H), 8.99 (br s, 1 H), 8.10 (br s, 1 H), 8.03 (d, J = 8.4 Hz, 1 H), 7.76 (s, 1 H), 7.69 (d, J = 1.4 Hz, 1 H), 7.64-7.63 (m, 1 H), 7.62 (s, 1 H), 7.56 (dd, J = 8.4, 1.2 Hz, 1 H), 6.94 (s, 1 H), 5.43 (s, 2 H), 4.15 (s, 2 H), 2.63 (d, J = 0.7 Hz, 3 H), 2.55-2.50 (m, 3 H, partially obscured by solvent peak); 13C-NMR (126 MHz; DMSO-d6): δ 158.2, 153.8, 152.2, 140.8, 135.8, 135.3, 126.3, 125.9, 123.8, 122.5, 120.8, 118.2, 118.0, 115.8, 112.7, 112.4, 69.0, 50.1, 32.0, 19.0; ESIMS m/z (rel. intensity) 333 (MH+, 100); HRMS calcd for C20H21N4O+: 333.1710; found, 333.1716.
7-[(4-Chloro-3-((methylamino)methyl)phenoxy)methyl]-4-methylquinolin-2-amine Dihydrochloride (12)
This compound was prepared from 43 (0.065 g, 0.220 mmol) and phenol 45 (0.060 g, 0.220 mmol). Workup and purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 37% EtOAc in CH2Cl2, afforded intermediate acetamide 49 as a white solid (0.084 g, 79%), which was immediately deprotected using K2CO3 (0.048 g, 0.347 mmol) as described in the General Procedure. After workup, the free aminoquinoline was diluted in ether (10 mL, with a few drops of MeOH added) and treated with methanolic HCl (1 mL). After being stirred overnight, filtration afforded 12 (0.058 g, 81% from 49) as a white flocculent solid after precipitation from hot MeOH (1 mL) with ether (10 mL) and being washed with ether: mp 280-282 °C. 1H-NMR (500 MHz; DMSO- d6): δ 1H-NMR (500 MHz; DMSO-d6): δ 14.13 (s, 1H), 8.84-8.70 (s, 4 H), 8.02 (d, J = 8.4 Hz, 1 H), 7.70 (s, 1 H), 7.55 (d, J = 8.3 Hz, 1 H), 7.00 (s, 1 H), 6.98 (s, 1 H), 6.94 (s, 1 H), 6.91 (s, 1 H), 5.38 (s, 2 H), 4.21 (s, 2 H), 2.64-2.58 (m, 5 H), 2.55 (s, 3 H), 1.19 (t, J = 7.5 Hz, 3 H); 13C-NMR (126 MHz; DMSO-d6): δ 156.9, 153.8, 152.3, 141.3, 135.7, 130.9, 130.5, 125.8, 124.9, 123.7, 120.7, 118.3, 117.0, 115.6, 112.6, 68.9, 48.3, 32.4, 19.0; ESIMS m/z (rel. intensity) 342/344 (MH+, 100/30); HRMS calcd for C19H21ClN3O+: 342.1368; found, 342.1371.
7-((3-Ethyl-5-((methylamino)methyl)phenoxy)methyl)-4-methylquinolin-2-amine Ditrifluoroacetate (13)
This compound was prepared from 43 (0.065 g, 0.22 mmol) and phenol 29 (0.058 g, 0.22 mmol). Workup and purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 37% EtOAc in CH2Cl2, afforded intermediate acetamide 48 as a white solid (0.076 g, 72%), which was immediately deprotected using K2CO3 (0.044 g, 0.318 mmol) as described in the General Procedure. After workup, the free aminoquinoline was diluted with anhydrous CH2Cl2 (6 mL), and trifluoroacetic acid (300 μL) was added. The mixture was stirred for 30 min and concentrated, and ether (10 mL) was added to the residue. The mixture was sonicated until a solid formed and filtered to yield 13 (0.079 g, 89% from 48) as a white solid after being washed with ether: mp 199.5-201 °C. 1H-NMR (500 MHz; DMSO-d6): 14.13 (br s, 1 H), 8.02 (d, J = 8.4 Hz, 1 H), 7.70 (s, 1 H), 7.55 (d, J = 8.3 Hz, 1 H), 7.00 (s, 1 H), 6.98 (s, 1 H), 6.94 (s, 1 H), 6.91 (s, 1 H), 5.31 (s, 2 H), 4.06 (S, 2 H), 2.64-2.58 (m, 5 H), 2.55 (s, 3 H), 1.19 (t, J = 7.5 Hz, 3 H). 13C-NMR (126 MHz; DMSO-d6): δ (158.9 + 158.7 + 158.43 + 158.19, 1 C), 158.25, 154.1, 152.2, 146.0, 141.7, 136.2, 133.3, 125.7, 123.6, 121.9, 120.7, (118.3 + 115.9, 1 C), 115.6, 114.8, 113.5, 112.7, 68.4, 51.3, 32.1, 28.1, 18.9, 15.3; the trifluoroacetate carbon signals are only partially visible; ESIMS m/z (rel. intensity) 336 (MH+, 100); HRMS calcd for C21H25N3O+: 336.2070; found, 336.2075.
4-Methyl-7-[3-((methylamino)methyl)phenethyl]quinolin-2-amine dihydrochloride (14)
Compound 66 (0.058 g, 0.129 mmol) was deprotected using K2CO3 (0.036 g, 0.259 mmol) as described in the General Procedure. After workup, the free aminoquinoline was purified by flash column chromatography, eluting with a gradient of EtOAc to 7% MeOH in EtOAc, to yield a colorless gum, which was diluted in 20:1 ether: MeOH (10 mL) and treated with methanolic HCl (0.7 mL). After being stirred overnight, filtration afforded 14 (0.026 g, 54% from 66) as a white solid, after precipitation from hot MeOH (0.5 mL) with ether (3 mL) and being washed with ether: mp 135-137 °C; 1H-NMR (500 MHz; DMSO-d6): δ 14.05 (br s, 1 H), 9.13-9.10 (br m, 3 H), 9.00 (br s, 1 H), 8.10 (br s, 1 H), 7.90 (d, J = 8.4 Hz, 1 H), 7.50 (s, 1 H), 7.47 (s, 1 H), 7.41 (dd, J = 8.4, 1.1 Hz, 1 H), 7.35-7.31 (m, 2 H), 7.28-7.26 (m, 1 H), 6.87 (d, J = 0.4 Hz, 1 H), 4.08 (s, 2 H), 3.10 (t, J = 7.8 Hz, 2 H), 2.98 (t, J = 7.8 Hz, 2 H), 2.60 (s, 3 H), 2.55-2.50 (m, 3 H, obscured by solvent peak); 13C-NMR (126 MHz; DMSO-d6): δ 153.6, 152.3, 146.4, 141.3, 135.6, 132.0, 130.0, 129.0, 128.7, 127.5, 125.8, 125.4, 119.5, 116.6, 111.8, 51.2, 36.5, 36.2, 31.9, 18.9; ESIMS m/z (rel. intensity) 306 (MH+, 100); HRMS calcd for C20H24N3+: 306.1965; found, 306.1969.
3-[2-(2-Aminoquinolin-7-yl)ethyl]-5-((methylamino)methyl)benzonitrile Dihydrochloride (15)
Compound 67 (0.026 g, 0.057 mmol) was deprotected using K2CO3 (0.016 g, 0.114 mmol) as described in the General Procedure. After workup, the free aminoquinoline was passed through a short SiO2 plug (eluting with EtOAc) to yield a colorless gum that was diluted in ether (10 mL) and treated with methanolic HCl (0.5 mL). After being stirred for 18 h, filtration afforded 15. The filtrate was concentrated, and the residue was re-suspended in ether and treated with methanolic HCl (0.5 mL); after 24 h, additional 15 was obtained. The total obtained solid was precipitated from hot MeOH (0.5 mL) with ether (3 mL) and washed with ether to afford 15 as a cream-colored solid (0.014 g, 62% from 67): mp 260-261.5 °C. 1H-NMR (500 MHz; DMSO-d6): δ 14.01 (br s, 1 H), 9.14-9.10 (br m, 3 H), 8.32 (d, J = 9.3 Hz, 1 H), 8.10 (br s, 1 H), 7.85-7.81 (m, 4 H), 7.49 (s, 1 H), 7.38 (d, J = 8.3 Hz, 1 H), 7.01 (d, J = 9.3 Hz, 1 H), 4.14 (s, 2 H), 3.12 (dd, J = 9.2, 6.2 Hz, 2 H), 3.03 (dd, J = 9.3, 6.3 Hz, 2 H), 2.55-2.50 (m, 3 H, obscured by solvent peak); 13C-NMR (126 MHz; DMSO-d6): δ 154.30. 146.24, 143.00, 142.84, 135.89, 135.22, 133.57, 132.46, 131.33, 128.75, 125.86, 119.33, 118.47, 116.36, 112.92, 111.38, 50.18, 36.13, 35.61, 31.93; ESIMS m/z (rel. intensity) 317 (MH+, 100); HRMS calcd for C20H21N4+: 317.1761; found, 317.1764.
3-[2-(2-Amino-4-methylquinolin-7-yl)ethyl]-5-((methylamino)methyl)benzonitrile Dihydrochloride (16)
Compound 68 (0.045 g, 0.095 mmol) was deprotected using K2CO3 (0.026 g, 0.190 mmol) as described in the General Procedure. After workup, the free aminoquinoline was purified by flash column chromatography, eluting with a gradient of EtOAc to 3% MeOH in EtOAc, to yield a colorless gum that was diluted in 5:1 ether:MeOH (10 mL) and treated with methanolic HCl (1 mL). After being stirred overnight, filtration afforded 16 (0.019 g, 50% from 68) as a white solid after precipitation from hot MeOH (0.75 mL) with ether (13 mL) and being washed with 30% MeOH in ether, with ether, and dried: mp 213-215 °C. 1H-NMR (500 MHz; DMSO-d6): δ 13.99 (s, 1 H), 9.29 (s, 2 H), 8.91 (br s, 1 H), 7.91 (d, J = 8.4 Hz, 1 H), 7.85-7.82 (m, 2 H), 7.83-7.81 (m, 1 H), 7.49 (d, J = 1.2 Hz, 1 H), 7.41 (dd, J = 8.4, 1.5 Hz, 1 H), 6.87 (d, J = 1.0 Hz, 1 H), 4.14 (s, 2 H), 3.13 (dd, J = 9.1, 6.4 Hz, 2 H), 3.03 (dd, J = 9.2, 6.5 Hz, 2 H), 2.61 (d, J = 0.8 Hz, 3 H), 2.55-2.50 (m, 3 H, obscured by solvent peak). One of the aminoquinoline –NH protons is not visible due to baseline broadening; 13C-NMR (126 MHz; DMSO-d6): δ 153.7, 152.3, 146.0, 143.0, 135.7, 135.2, 133.6, 132.4, 131.3, 125.7, 125.5, 119.6, 118.5, 116.6, 111.8, 111.4, 50.2, 35.9, 35.6, 31.9, 18.9; ESIMS m/z (rel. intensity) 331 (MH+, 100); HRMS calcd for C21H23N4+: 331.1917; found, 331.1922.
5-[(2-Amino-4-methylquinolin-7-yl)methoxy]-2-fluoro-3-((methylamino)methyl)benzonitrile Dihydrochloride (17)
This compound was prepared from 44 (0.050 g, 0.202 mmol) and phenol 73 (0.054 g, 0.192 mmol) at 50 °C. Workup and purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 40% EtOAc in CH2Cl2, afforded intermediate acetamide 74 as a colorless glass (0.072 g, 73%), which was immediately deprotected using K2CO3 (0.040 g, 0.291 mmol) as described in the General Procedure. After workup, the free aminoquinoline was purified by flash column chromatography, eluting with a gradient of EtOAc to 3% MeOH in EtOAc, and the obtained residue was diluted in 5:1 ether: MeOH (10 mL) and treated with methanolic HCl (1 mL). After being stirred overnight, filtration afforded 17 (0.047 g, 77% from 74) as a white flocculent solid after precipitation from hot MeOH (0.75 mL) with ether (10 mL) and being washed with 30% MeOH in ether and then with ether: mp 279.5-281 °C (dec). 1H-NMR (500 MHz; DMSO-d6): δ 14.15 (br s, 1 H), 9.44 (br s, 2 H), 9.00 (br s, 1 H), 8.10 (br s, 1 H), 8.03 (d, J = 8.4 Hz, 1 H), 7.85-7.84 (m, 1 H), 7.76-7.74 (m, 2 H), 7.56 (dd, J = 8.4, 1.4 Hz, 1 H), 6.94 (d, J = 0.9 Hz, 1 H), 5.40 (s, 2 H), 4.21 (s, 2 H), 2.63 (d, J = 0.9 Hz, 3 H), 2.59 (s, 3 H); 13C-NMR (126 MHz; DMSO-d6): δ (156.7 + 154.7, 1 C), (153.92 + 153.90, 1 C), 153.80, 152.2, 140.7, 135.7, 125.9, (124.92 + 124.89, 1C), 123.8, (121.92 + 121.80, 1 C), 120.8, 119.2, 115.9, 113.6, 112.7, (100.83 + 100.69, 1 C) 69.5, (43.84 + 43.82, 1 C), 32.2, 19.0; ESIMS m/z (rel. intensity) 351 (MH+, 100); HRMS calcd for C20H20FN4O+: 351.1616; found, 351.1623.
3-[(2-Amino-4-methylquinolin-7-yl)methoxy]-5-(2-(methylamino)ethyl)benzonitrile Dihydrochloride (18)
This compound was prepared from 44 (0.024 g, 0.0949 mmol) and phenol 81 (0.025 g, 0.0904 mmol). The reaction mixture was stirred at r.t. for four days. Workup and purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 35% EtOAc in CH2Cl2, afforded intermediate acetamide 83 as a white semisolid (0.025 g, 57%), which was immediately deprotected using K2CO3 (0.014 g, 0.102 mmol) as described in the General Procedure. After workup, the free aminoquinoline was purified by flash column chromatography, eluting with a gradient of EtOAc to 7% MeOH in EtOAc, and the obtained clear gum was diluted in 10:1 ether: MeOH (10 mL) and treated with methanolic HCl (0.5 mL). After being stirred overnight, filtration afforded 18 (0.010 g, 49% from 83) as a white flocculent solid after precipitation from hot MeOH (1 mL) with ether (10 mL) and being washed with ether: mp 213-215 °C. 1H-NMR (500 MHz; DMSO-d6): δ 14.07 (br s, 1 H), 9.10-8.79 (br m, 3 H), 8.10 (br m, 1 H), 8.03 (d, J = 8.4 Hz, 1 H), 7.75 (s, 1 H), 7.56 (dd, J = 8.4, 1.1 Hz, 1 H), 7.47 (dd, J = 2.3, 1.3 Hz, 1 H), 7.41-7.49 (m, 1 H), 7.37 (t, J = 1.8 Hz, 1 H), 6.93 (s, 1 H), 5.41 (s, 2 H), 3.22-3.17 (m, 2 H), 2.98 (t, J = 7.7 Hz, 2 H), 2.63 (d, J = 0.8 Hz, 3 H), 2.56 (t, J = 5.3 Hz, 3 H). 13C-NMR (126 MHz; DMSO-d6): δ 158.3, 153.8, 141.1, 140.6, 125.8, 125.4, 123.7, 121.4, 120.8, 118.5, 116.0, 115.7, 112.7, 112.4, 68.8, 48.3, 32.5, 30.8, 19.0; ESIMS m/z (rel. intensity) 347 (MH+, 100); HRMS calcd for C21H23N4O+: 347.1866; found, 347.1872.
(RS)-3-[(2-amino-4-methylquinolin-7-yl)methoxy]-5-(2-(methylamino)propyl)benzonitrile Dihydrochloride (R,S-19)
This compound was prepared from 44 (0.054 g, 0.217 mmol) and phenol 82 (0.060 g, 0.206 mmol) at 50 °C. Workup and purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 40% EtOAc in CH2Cl2, afforded intermediate acetamide 84 as a yellow foam (0.063 g, 61%), which was immediately deprotected using K2CO3 (0.035 g, 0.251 mmol) as described in the General Procedure. After workup, the free aminoquinoline was purified by flash column chromatography, eluting with a gradient of EtOAc to 2% MeOH in EtOAc, and the obtained residue was diluted in 4:1 ether: MeOH (10 mL) and treated with methanolic HCl (0.5 mL). After being stirred overnight, filtration afforded (R,S)-19 (0.041 g, 76% from 84) as a white flocculent solid after precipitation from hot MeOH (1 mL) with ether (10 mL) and being washed with ether: mp 190-200 °C (softens); 246-250 °C (melts). 1H-NMR (500 MHz; DMSO-d6): δ 14.10 (s, 1 H), 9.00-8.90 (m, 3 H), 8.10 (br s, 1 H), 8.03 (d, J = 8.4 Hz, 1 H), 7.75 (d, J = 0.8 Hz, 1 H), 7.56 (dd, J = 8.4, 1.4 Hz, 1 H), 7.47 (dd, J = 2.4, 1.3 Hz, 1 H), 7.41 (s, 1 H), 7.38 (dd, J = 2.1, 1.6 Hz, 1 H), 6.93 (d, J = 0.8 Hz, 1 H), 5.40 (s, 2 H), 3.48-3.44 (m, 1 H), 3.17 (dd, J = 13.6, 4.5 Hz, 1 H), 2.73 (dd, J = 13.4, 9.4 Hz, 1 H), 2.63 (d, J = 0.9 Hz, 3 H), 2.57 (t, J = 5.4 Hz, 3 H), 1.10 (d, J = 6.5 Hz, 3 H); 13C-NMR (126 MHz; DMSO-d6): δ 158.3, 153.8, 141.1, 140.1, 125.88, 125.84, 123.8, 121.8, 120.7, 118.5, 116.2, 115.7, 112.7, 112.4, 68.8, 54.7, 37.6, 29.7, 19.0, 15.1; two of the aminoquinoline carbons are not visible due to baseline broadening; ESIMS m/z (rel. intensity) 361 (MH+, 100); HRMS calcd for C22H25N4O+: 361.2023; found, 361.2028.
(R)-3-[(2-amino-4-methylquinolin-7-yl)methoxy]-5-(2-(methylamino)propyl)benzonitrile Dihydrochloride ((R)-19)
This compound was prepared from 44 (0.057 g, 0.228 mmol) and phenol (R)-82 (0.070 g, 0.241 mmol) at 50 °C. Workup and purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 40% EtOAc in CH2Cl2, afforded intermediate acetamide (R)-84 as a white foam (0.079 g, 69%), which was immediately deprotected using K2CO3 (0.043 g, 0.312 mmol) as described in the General Procedure. After workup, the free aminoquinoline was purified by flash column chromatography, eluting with a gradient of EtOAc to 2% MeOH in EtOAc, and the obtained residue was diluted in 4:1 ether: MeOH (10 mL) and treated with methanolic HCl (0.75 mL). The mixture was stirred for 20 h at r.t., and concentrated. The residue was triturated with 5% MeOH in ether (10 mL) and collected by filtration. The precipitate was recrystallized from MeOH: ether (5:1, 15 mL) to yield (R)-19 as a white solid ((0.049 g, 72% from (R)-84)) after washing with ether: mp 146-148 °C (softens), 170-173 °C (melts). 1H-NMR (500 MHz; DMSO-d6): δ 14.03 (s, 1 H), 8.90-8.80 (m, 3 H), 8.10 (br s, 1 H), 8.03 (d, J = 8.4 Hz, 2 H), 7.75 (s, 1 H), 7.56 (dd, J = 8.4, 0.7 Hz, 1 H), 7.48 (dd, J = 2.3, 1.3 Hz, 1 H), 7.41 (s, 1 H), 7.38 (dd, J = 2.0, 1.6 Hz, 1 H), 6.93 (s, 1 H), 5.40 (s, 2 H), 3.48-3.44 (m, 1 H), 3.17 (dd, J = 13.4, 4.5 Hz, 1 H), 2.73 (dd, J = 13.4, 9.4 Hz, 1 H), 2.63 (d, J = 0.8 Hz, 3 H), 2.57 (t, J = 5.2 Hz, 3 H), 1.10 (d, J = 6.5 Hz, 3 H); 13C-NMR (126 MHz; DMSO-d6): δ 158.3, 153.8, 141.0, 140.1, 125.88, 125.81, 123.7, 121.8, 120.8, 118.5, 116.2, 115.8, 112.7, 112.4, 68.8, 54.7, 37.6, 29.7, 19.0, 15.1; two of the aminoquinoline carbons are not visible due to baseline broadening; ESIMS m/z (rel. intensity) 361 (MH+, 100); HRMS calcd for C22H25N4O+: 361.2023; found, 361.2030. The compound displays little optical rotation at a concentration of 10 mg/mL, but was confirmed as a single enantiomer via chiral derivatization.
(S)-3-[(2-amino-4-methylquinolin-7-yl)methoxy]-5-(2-(methylamino)propyl)benzonitrile Dihydrochloride ((S)-19)
This compound was prepared from 44 (0.047 g, 0.190 mmol) and phenol (S)-82 (0.058 g, 0.200 mmol) at 50 °C. Workup and purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 40% EtOAc in CH2Cl2, afforded intermediate acetamide (S)-84 as a white foam (0.059 g, 62%), which was immediately deprotected using K2CO3 (0.033 g, 0.239 mmol) as described in the General Procedure. After workup, the free aminoquinoline was purified by flash column chromatography, eluting with a gradient of EtOAc to 2% MeOH in EtOAc, and the obtained residue was diluted in 6:1 ether: MeOH (7 mL) and treated with methanolic HCl (0.75 mL). The mixture was stirred for 20 h at r.t., ether (6 mL) was added, and the mixture was filtered. The precipitate was recrystallized from MeOH: ether (5:1, 10 mL) to yield (S)-19 as a white solid ((0.042 g, 83% from (S)-84)) after washing with ether: mp 146-148 °C (softens), 170-173 °C (melts); 1H-NMR (500 MHz; DMSO-d6): δ 14.00 (br s, 1 H), 8.90-8.80 (m, 3 H), 8.10 (br s, 1 H), 8.03 (d, J = 8.4 Hz, 2 H), 7.75 (s, 1 H), 7.56 (dd, J = 8.4, 0.6 Hz, 1 H), 7.48 (dd, J = 2.4, 1.3 Hz, 1 H), 7.41 (s, 1 H), 7.38 (t, J = 1.8 Hz, 1 H), 6.93 (s, 1 H), 5.41 (s, 2 H), 3.48-3.44 (m, 1 H), 3.17 (dd, J = 13.4, 4.6 Hz, 1 H), 2.73 (dd, J = 13.4, 9.4 Hz, 1 H), 2.64 (d, J = 0.8 Hz, 3 H), 2.58 (t, J = 5.3 Hz, 3 H), 1.10 (d, J = 6.5 Hz, 3 H); 13C-NMR (126 MHz; DMSO-d6): δ 158.8, 154.3, 141.5, 140.6, 126.37, 126.32, 124.3, 122.3, 121.3, 119.0, 116.6, 116.3, 113.2, 112.9, 69.3, 55.2, 38.0, 30.2, 19.4, 15.6; two of the aminoquinoline carbons are not visible due to baseline broadening; ESIMS m/z (rel. intensity) 361 (MH+, 100); ESIMS m/z (rel. intensity) 361 (MH+, 100); HRMS calcd for C22H25N4O+: 361.2023; found, 361.2033. The compound displays little optical rotation at a concentration of 10 mg/mL, but was confirmed as a single enantiomer via chiral derivatization.
7-Bromo-4-methylquinoline (37)26
3-Bromoaniline (36, 13.8 g, 80 mmol) and FeCl3. 6 H2O (22.8 g, 84 mmol) were diluted with glacial acetic acid (200 mL). The mixture was heated to 60 °C to ensure dissolution of the solids (ca. 15 min), and then methyl vinyl ketone (8 mL, 88 mmol) was added slowly. The mixture was heated at reflux (∼140 °C) for 3 h, during which time a yellow precipitate formed. The mixture was then cooled to r.t., and the precipitate was filtered from the solution and washed with EtOAc; the filtrate was colorless (this filtrate was discarded). The filter cake was then diluted with EtOAc (400 mL), and 1 M NaOH was added until the pH of the resulting suspension was >9. The resulting emulsion was filtered through a coarse fritted funnel to remove iron salts, and the organic and aqueous layers were separated. The aqueous layer was extracted with EtOAc (4 × 400 mL). The combined organic layers (∼1.5 L) were washed with H2O (2 × 400 mL) and sat. aq. NaCl (400 mL), dried over anhydrous sodium sulfate, and concentrated to 1/10th of the original volume. The solution was filtered through a pad of Celite and re-concentrated to afford 37 as a yellow-green solid (7.71 g, 44%). The analytical data for this compound are consistent with those previously reported.26
7-Bromo-4-methylquinoline N-Oxide (38)
Compound 37 (7.71 g, 34.7 mmol) was diluted in anhydrous CH2Cl2, and m-chloroperoxybenzoic acid (8.39 g of 70% m-CPBA, 48.6 mmol) was added. The mixture was stirred at r.t. for 19 h, and 1 M NaOH (∼100 mL) was added. The layers were separated, and the aqueous layer was extracted exhaustively with CH2Cl2 (3 × 100 mL), following which the combined organic layers were washed with sat. aq. NaHCO3 (100 mL) and sat. aq. NaCl (100 mL) and dried over anhydrous sodium sulfate. Concentration afforded a residue that was washed with 5% CH2Cl2 in hexanes to afford the desired compound (7.58 g, 92%) as a yellow crystalline solid after washing with hexanes and drying: mp 179-181 °C. 1H NMR (500 MHz; DMSO-d6): δ 9.01 (d, J = 2.0 Hz, 1 H), 8.48 (d, J = 6.0 Hz, 1 H), 7.86 (d, J = 9.0 Hz, 1 H), 7.79 (dd, J = 9.0, 2.0 Hz, 1 H), 7.18 (d, J = 6.0 Hz, 1 H), 2.69 (s, 3 H); 13C-NMR (126 MHz; DMSO-d6): δ 140.8, 135.4, 133.5, 131.6, 128.4, 127.8, 123.7, 122.7, 121.7, 17.5; ESIMS m/z (rel. intensity) 260/262 (MNa+, 13/13).
7-Bromo-4-methylquinolin-2-amine (39)
N-Oxide 38 (3.81 g, 16 mmol) was diluted in 2:1 trifluorotoluene: CH2Cl2 (120 mL) and cooled to 0 °C. t-Butylamine (8.41 mL, 80 mmol) was added, followed by p-toluenesulfonic anhydride (10.4 g, 32 mmol) in small portions. After 10 min, additional t-BuNH2 (1 mL) and Ts2O (1.0 g) were added, and after a total of 20 min, TLC indicated the presence of a less polar product (intermediate t-butylaminoquinoline). The suspension was concentrated until the CH2Cl2 was removed, trifluoroacetic acid (34 mL) was added, and the mixture was heated until the internal temp reached 80 °C, where it was held for 5 h. The mixture was then cooled and stirred 17 h at r.t. The dark red mixture was concentrated and neutralized with 1 N NaOH until the pH was ∼12. The suspension was extracted with EtOAc (3 × 150 mL), and the organic layers were washed with H2O and sat. aq. NaCl (100 mL each). The organic layers were dried over anhydrous sodium sulfate and concentrated to yield a residue that was adsorbed onto silica gel and purified by flash column chromatography, eluting with a gradient of 50% EtOAc in CH2Cl2 to EtOAc, to yield 39 as a tan crystalline solid (2.26 g, 60%) after washing with hexanes: mp 180-182 °C. 1H-NMR (500 MHz; DMSO-d6): δ 7.68 (d, J = 8.7 Hz, 1 H), 7.57 (d, J = 2.0 Hz, 1 H), 7.27 (dd, J = 8.7, 2.1 Hz, 1 H), 6.62 (d, J = 1.0 Hz, 1 H), 6.53 (s, 2 H), 2.46 (d, J = 1.0 Hz, 3 H); 13C-NMR (126 MHz; CDCl3): δ 157.6, 148.5, 145.9, 128.5, 125.6, 125.1, 123.5, 122.6, 112.3, 18.7; ESIMS m/z (rel. intensity) 237/239 (MNa+, 100/94).
N-(7-Bromo-4-methylquinolin-2-yl)acetamide (40)
Compound 39 (2.20 g, 9.28 mmol) was diluted in anhydrous THF (50 mL), and N-acetylimidazole (1.53 g, 13.9 mmol) and a catalytic amount of DMAP (∼20 mg) were added. The mixture was heated at reflux for 17 h. The mixture was cooled and concentrated, and the residue was dissolved in EtOAc (200 mL) and washed with H2O (100 mL). The aqueous layer was extracted with EtOAc (2 × 200 mL), and the organic layers were washed with H2O (200 mL) and sat. aq. NaCl (200 mL). The combined organic layers were dried over anhydrous sodium sulfate and concentrated, and the residue was suspended in EtOAc (5 mL) and precipitated by addition of hexanes (150 mL). The precipitate was collected and washed with hexanes to yield 40 as an off-white iridescent solid (2.32 g, 90%): mp 246-248.5 °C. 1H-NMR (500 MHz; CDCl3): δ 8.75 (br s, 1 H), 8.33 (s, 1 H), 8.01 (d, J = 1.9 Hz, 1 H), 7.81 (d, J = 8.8 Hz, 1 H), 7.59 (dd, J = 8.8, 1.5 Hz, 1 H), 2.73 (s, 3 H), 2.31 (s, 3 H). 13C-NMR (126 MHz; CDCl3): δ 169.9, 151.7, 147.9, 146.4, 129.4, 128.4, 125.4, 124.8, 124.0, 114.9, 24.8, 19.2; ESIMS m/z (rel. intensity) 301/303 (MNa+, 20/19).
N-(7-Formyl-4-methylquinolin-2-yl)acetamide (41)
Compound 40 (1.08 g, 3.6 mmol), Na2CO3 (0.576 g, 5.38 mmol), dppb (0.070 g, 0.164 mmol), Pd(OAc)2 (0.024 g, 0.107 mmol), and N-formylsaccharin (1.15 g, 5.44 mmol) were placed in a sealable, heavy-walled glass tube, which was evacuated and backfilled with argon several times. To the tube was added anhydrous, degassed DMF (20 mL) containing Et3SiH (0.744 mL, 4.66 mmol). The tube was sealed, and the reaction mixture was allowed to stir at r.t. for 10 min before being heated to 70 °C for 22 h. The reaction mixture was cooled and quenched with 1:1 H2O: sat. aq. NaCl (50 mL) and extracted with EtOAc (3 × 70 mL). The organic layer was washed with 5% aq. NaCl (3 × 100 mL) and sat. aq. NaCl (100 mL), dried over anhydrous sodium sulfate, and concentrated to yield a residue that was adsorbed onto silica gel and purified by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 40% EtOAc in CH2Cl2 to yield 41 as a white flocculent solid (0.514 g, 63%); mp 209-211 °C (softens), 218-221 °C (melts). 1H-NMR (500 MHz; CDCl3): δ 10.20 (d, J = 0.5 Hz, 1 H), 8.40 (s, 1 H), 8.25 (d, J = 1.3 Hz, 1 H), 8.05 (d, J = 8.6 Hz, 2 H), 8.00 (br s, 1 H), 7.95 (dd, J = 8.6, 1.6 Hz, 1 H), 2.76 (d, J = 1.0 Hz, 3 H), 2.29 (s, 3 H). 13C-NMR (126 MHz; CDCl3): δ 192.2, 169.3, 151.7, 147.7, 146.2, 137.3, 133.3, 130.1, 125.3, 122.1, 116.8, 25.2, 19.5; ESIMS m/z (rel. intensity) 229 (MH+, 5).
N-(7-(Hydroxymethyl)-4-methylquinolin-2-yl)acetamide (42)
Aldehyde 41 (0.514 g, 2.25 mmol) was diluted in MeOH (30 mL) and heated gently to 50 °C to effect solution. When the mixture was just dissolved, NaBH4 (0.111 g, 2.92 mmol) was added in one portion, and the mixture was stirred for 20 min and concentrated. The residue was partitioned between EtOAc and 1:1 sat. aq. NaHCO3:H2O (30 mL each). The aqueous layer was extracted with EtOAc (3 × 30 mL), and the organic layers were washed with sat. aq. NaCl and dried over anhydrous sodium sulfate. Concentration afforded a residue that was partially dissolved in EtOAc (5 mL) and precipitated with hexanes (100 mL). Filtration afforded 42 as a white crystalline powder (0.493 g, 95%) that was used without further purification. 1H-NMR (500 MHz; CDCl3): δ 9.75 (br s, 1 H), 8.22 (s, 1 H), 7.89 (d, J = 8.5 Hz, 1 H), 7.78 (s, 1 H), 7.49 (dd, J = 8.5, 1.2 Hz, 1 H), 4.83 (s, 2 H), 3.50 (br s, 1 H) 2.69 (s, 3 H), 2.28 (s, 3 H).
N-(7-(Bromomethyl)-4-methylquinolin-2-yl)acetamide (43)
Alcohol 42 (0.147 g, 0.64 mmol) was diluted in anhydrous THF (5 mL) and cooled to 0 °C. CBr4 (0.255 g, 0.768 mmol) and PPh3 (0.201 g, 0.768 mmol) were added. The mixture was stirred at 0 °C for 15 min, warmed to r.t., and stirred for 3.5 h. The mixture was then concentrated and diluted in CH2Cl2 and filtered to remove insoluble materials. The filtrate was concentrated and purified by flash column chromatography, eluting with a gradient of 3% EtOAc in CH2Cl2 to 30% EtOAc in CH2Cl2 to yield 43 as a white solid (0.135 g, 72%). This crude product (containing some by-products arising from over-bromination of the acetamide) was suitable for formation of phenyl ethers and was used without further purification or characterization; 1H-NMR (500 MHz; CDCl3): δ 8.38 (s, 1 H), 7.98 (d, J = 8.6 Hz, 1 H), 7.85 (d, J = 1.4 Hz, 1 H), 7.60 (dd, J = 8.6, 1.4 Hz, 1 H), 4.63 (s, 2 H), 2.77 (d, J = 0.4 Hz, 3 H), 2.36 (s, 3 H).
N-(7-(Chloromethyl)-4-methylquinolin-2-yl)acetamide (44)
Alcohol 42 (0.500 g, 2.17 mmol) was cooled to 0 °C under argon. Chilled SOCl2 (2.1 mL) was added, and the mixture was stirred at 0 °C for 1 h. The mixture was then concentrated, and the residue was azeotroped with toluene (2 × 5 mL) to remove traces of SOCl2. The residue was suspended in H2O, and sat. aq. NaHCO3 was added until a white solid precipitated (at pH ∼8). The suspension was extracted with EtOAc (3 × 50 mL). Sat. aq. NaHCO3 (50 mL) was added to the organic layer, and the mixture was sonicated vigorously until the EtOAc became cloudy, and the mixture was allowed to sit overnight for 17 h to ensure hydrolysis of any by-products arising from over-chlorination at the acetyl group. The layers were then separated, and the organic layer was washed with sat. aq. NaCl (100 mL), dried over anhydrous sodium sulfate, and concentrated. The resulting residue was washed with 1% EtOAc in hexanes to yield 51 as a white iridescent solid (0.508 g, 94%); mp 211-212.5 °C. 1H-NMR (500 MHz; CDCl3): δ 8.28 (s, 1 H), 8.15 (br s, 1 H), 7.94 (d, J = 8.6 Hz, 1 H), 7.79 (d, J = 1.4 Hz, 1 H), 7.51 (dd, J = 8.6, 1.8 Hz, 1 H), 4.75 (s, 2 H), 2.72 (d, J = 0.9 Hz, 3 H), 2.27 (s, 3 H); 13C-NMR (126 MHz; CDCl3): δ 169.4, 151.1, 148.1, 139.5, 127.0, 126.1, 125.5, 124.9, 114.9, 46.0, 25.2, 19.4; one of the quinoline carbons is not visible due to baseline broadening; ESIMS m/z (rel. intensity) 271/273 (MNa+, 41/12).
7-Bromoquinolin-2-amine (52)
Compound 51 (0.700 g, 2.89 mmol), anhydrous K2CO3 (1.99 g, 14.4 mmol) and dry acetamide (13.7 g, 231 mmol) were heated under argon to reflux (∼230 °C) for 18 h. The mixture was cooled, diluted with H2O (60 mL), and the resulting suspension was extracted with EtOAc (3 × 70 mL). The organic layers were washed with H2O and sat. aq. NaCl (70 mL each), dried over anhydrous sodium sulfate, and concentrated. The resulting solid was dissolved in hot EtOAc (5 mL) and precipitated by the addition of hexanes (100 mL). The yellow solid was collected and dried to yield crude compound 52 (0.481 g, 75%), which was used without any further purification; analytical data for this compound are consistent with those previously reported.28
N-(7-Bromoquinolin-2-yl)acetamide (53)
Compound 52 (0.481 g, 2.15 mmol) was diluted in anhydrous THF (25 mL). N-Acetylimidazole (0.284 g, 2.58 mmol) was added, and the mixture was heated to reflux under argon for 20 h, after which additional N-acetylimidazole (0.284 g, 2.58 mmol) and a catalytic amount of DMAP (∼20 mg) were added. After a total of 23 h at reflux, the mixture was cooled and concentrated, and the residue was partitioned between H2O and EtOAc (30 mL each). The aqueous layer was then extracted with EtOAc (3 × 30 mL), and the organic layers were washed with H2O and sat. aq. NaCl (100 mL each). The organic phase was dried over anhydrous sodium sulfate and concentrated, and the residue was purified by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 25% EtOAc in CH2Cl2 to yield 53 as a flocculent, off-white solid (0.262 g, 46%) after washing with hexanes; mp 220-222 °C. 1H-NMR (500 MHz; CDCl3): δ 8.42 (br d, J = 8.1 Hz, 1 H), 8.20 (br s, 1 H), 8.14 (d, J = 9.1 Hz, 1 H), 7.99 (s, 1 H), 7.64 (d, J = 8.6 Hz, 1 H), 7.54 (d, J = 8.7 Hz, 1 H), 2.29 (s, 3 H); 13C-NMR (126 MHz; CDCl3): δ 169.3, 151.6, 147.0, 138.9, 129.6, 128.96, 128.94, 125.0, 124.5, 114.5, 25.1; 287/289 (MNa+, 21/20).
General Procedure for Sonogashira Coupling Between 7-Bromoquinolines and Phenylacetylenes31
A sealable vial or tube was charged with the requisite 7-bromoquinoline (1 equiv) and phenylacetylene (1.1-1.5 equiv), XPhos (6 mol%), Pd(MeCN)2Cl2 (3 mol%), and Cs2CO3 (1.5 equiv). The vial was evacuated and backfilled with argon several times, and then anhydrous MeCN (2 mL/100 mg bromoquinoline) was added, and the mixture was heated to 75-80 °C overnight (typically 16-18 h). The mixture was then cooled, diluted with water, and extracted with EtOAc (typically 3 × 30 mL is sufficient), and the organic layers were washed with 5% sat. aq. NaCl (3 × 50 mL) and sat. aq. NaCl (50 mL). The organic phases were dried over anhydrous sodium sulfate and concentrated to yield residues that were purified using flash column chromatography (SiO2) with gradients as described for individual compounds below.
tert-Butyl (3-((2-Acetamido-4-methylquinolin-7-yl)ethinyl)benzyl)-(methyl)carbamate (63)
This compound was prepared from 40 (0.059 g, 0.179 mmol) and 57 (0.048 g, 0.197 mmol) at 75 °C. Workup and purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 30% EtOAc in CH2Cl2 gave the desired product as a yellow powder (0.058 g, 73%). The identity of the product was confirmed by NMR spectroscopy, and the product was carried on to the next step without further purification or characterization.
tert-Butyl (3-((2-Acetamidoquinolin-7-yl)ethinyl)-5-cyanobenzyl)-(methyl)carbamate (64)
This compound was prepared from 53 (0.071 g, 0.268 mmol) and 62 (0.083 g, 0.307 mmol) at 80 °C. Workup and purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 30% EtOAc in CH2Cl2,gave the desired product as a tan powder (0.031 g, 28%). The product was carried on to the next step without further purification or characterization.
tert-Butyl (3-((2-Acetamido-4-methylquinolin-7-yl)ethinyl)-5-cyanobenzyl)-(methyl)carbamate (65)
This compound was prepared from 40 (0.120 g, 0.430 mmol) and 62 (0.180 g, 0.665 mmol) at 80 °C. Workup and purification by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 30% EtOAc in CH2Cl2 gave the desired product as a brownish-yellow semisolid (0.191 g, 95%). The product was carried on to the next step without further purification or characterization.
tert-Butyl (3-(2-(2-Acetamido-4-methylquinolin-7-yl)ethyl)benzyl) (methyl)carbamate (66)
Compound 63 (0.385 g, 0.868 mmol) was diluted with MeOH (40 mL) and heated gently to affect solution. A catalytic amount of 10% Pd/C (∼20 mg) was added, and the mixture was hydrogenated (using a balloon) for 20 h at r.t. The Pd/C was then removed via syringe filter, and the filtrate was concentrated. The residue was diluted in minimal CH2Cl2 (2 mL) and precipitated by the addition of hexanes (50 mL). The suspension was filtered to yield 66 as a white solid (0.362 g, 93%) after washing with hexanes and drying in vacuo. This compound was deprotected immediately without further characterization or purification.
tert-Butyl (3-(2-(2-Acetamidoquinolin-7-yl)ethyl)-5-cyanobenzyl) (methyl)carbamate (67)
Compound 64 (0.035 g, 0.077 mmol) was diluted in THF (5 mL), and a catalytic amount of Pd/C(en) was added. The mixture was hydrogenated (using a balloon) for 66 h, with another equivalent of catalyst being added after 18 h. The Pd/C(en) was removed via filtration, and the filtrate was concentrated. The residue was purified by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 45% EtOAc in CH2Cl2, to yield 67 as a yellow gum (0.026 g, 76%), which was deprotected immediately without further characterization or purification.
tert-Butyl (3-(2-(2-Acetamido-4-methylquinolin-7-yl)ethyl)-5-cyanobenzyl) (methyl)carbamate (68)
Compound 65 (0.100 g, 0.213 mmol) was diluted in THF (12 mL), and a catalytic amount of Pd/C(en) was added. The mixture was hydrogenated at 50 PSI on a Parr shaker for 48 h. The Pd/C(en) was removed via filtration, and the filtrate was concentrated. The residue was purified by flash column chromatography, eluting with a gradient of 5% EtOAc in CH2Cl2 to 40% EtOAc in CH2Cl2, to yield 68 as a yellow gum (0.045 g, 45%), which was immediately deprotected without further characterization or purification.
Purified NOS Enzyme Assays
Rat and human nNOS, murine macrophage iNOS, and human eNOS were recombinant enzymes (expressed in E. coli and purified as reported previously).38, 51, 52, 53 To test for NOS inhibition, the hemoglobin capture assay was used to measure nitric oxide production. The assay was performed at 37 °C in HEPES buffer (100 mM with 10% glycerol, pH 7.4) in the presence of 10 μM l-arginine (used because a) it is close to the Km values all three isoforms, where detection of competitive inhibitors is most sensitive, and b) significant NOS uncoupling does not occur at this concentration). Also included were 100 μM NADPH, 0.83 mM CaCl2, approximately 320 units/mL of calmodulin, 10 μM H4B, and human oxyhemoglobin (3 μM). For iNOS, the CaCl2 and calmodulin were omitted and replaced with HEPES buffer (neither are required for activation of iNOS). The assay was performed in 96-well plates using a Synergy 4 BioTek hybrid reader. The dispensing of NOS enzyme and hemoglobin were automated, and after 30 sec (maximum delay), NO production was read by monitoring the absorbance at 401 nm (resulting from conversion of oxyhemoglobin to methemoglobin). Kinetic readouts were performed for 5 min. Each compound was assayed at least in duplicate, and six to nine concentrations (500 μM-50 nM or 100 μM-10 nM for eNOS and iNOS; 50 μM to 5 nM for rat and human nNOS) were used to construct dose-response curves. IC50 values were calculated by nonlinear regression (variable slope, four parameters) using GraphPad Prism software (standard error is reported for the LogIC50), and Ki values were obtained from IC50 values using the Cheng-Prusoff54 equation [Ki = IC50/(1+[S]/Km)] with the following Km values: 1.3 μM (rat nNOS), 1.6 μM (human nNOS), 8.2 μM (murine macrophage iNOS), and 3.9 μM (human eNOS).55
Inhibitor Complex Crystal Preparation
The sitting drop vapor diffusion methods were used to grow crystals at 4°C for the heme domains of rat nNOS (8 mg/mL containing 20 mM histidine), the human nNOS K301R/R354A/G357D mutant (10 mg/mL), and human eNOS (7 mg/mL). The well solutions are: for rat nNOS, 20-24% PEG3350, 0.1 M MES, pH 5.8, 140-200 mM ammonium acetate, 10% ethylene glycol, 5 mM GSH, and 30 μM SDS; for human nNOS, 8% PEG3350, 35 mM citric acid, 65 mM Bis-Tris-Propane, pH 7.2, 10% glycerol, and 5 mM TCEP; for human eNOS, 12-15% PEG3350, 0.1 M Bis-Tris, pH 6.5, 200-250 mM magnesium acetate, 100 mM GdCl3, 10% glycerol, and 5 mM TCEP. Fresh crystals were first passed stepwise through cryoprotectant solutions and then soaked with 5-10 mM inhibitor for 3-4 h at 4 °C before being flash cooled with liquid nitrogen and stored until data collection. The presence of an acetate ion near the heme active site in bovine eNOS had caused interference in the binding mode of some phenyl ether-linked aminoquinoline compounds.18 The high concentration of magnesium acetate in the heNOS growth conditions may also introduce an acetate near the active site that can influence the binding mode of inhibitors. To avoid having this acetate in the structure the magnesium acetate in the cryoprotectant solution was replaced with either MgSO4 or MgCl2.
X-ray Diffraction Data Collection, Data Processing, and Structural Refinement
The cryogenic (100 K) X-ray diffraction data were collected remotely at the Stanford Synchrotron Radiation Lightsource (SSRL) or Advanced Light Source (ALS) through the data collection control software Blu-Ice56 and a crystal-mounting robot. When a Q315r CCD detector was used, 100-125° of data were typically collected with 0.5° per frame. If a Pilatus pixel array detector was used, 140-160° of fine-sliced data were collected with 0.2° per frame. Raw CCD data frames were indexed, integrated, and scaled using iMOSFLM57, but the pixel array data were processed with XDS58 and scaled with Aimless.59 The binding of inhibitors was detected by initial difference Fourier maps calculated with REFMAC.60 The inhibitor molecules were then modeled in Coot61 and refined using REFMAC or PHENIX.62 Replacement of magnesium acetate in the cryoprotectant with MgSO4 or MgCl2 for heNOS crystals caused a shrinkage of cell dimensions (MgSO4) or a distortion of the symmetry (MgCl2) compared to heNOS crystal structures reported previously.38 Therefore, a molecular replacement calculation with PHASER-MR63was needed to solve the structure. The crystal packing of the MgCl2 soaked heNOS crystals was changed slightly, resulting in a symmetry change from orthorhombic P212121 to monoclinic P21, with a β angle only 0.6-0.7° off compared to the original 90°. In the P21 space group, there are two heNOS dimers in the asymmetric unit. Disordering in portions of inhibitors bound in the NOS active sites was often observed, sometimes resulting in poor density quality. However, partial structural features were usually still visible if the contour level of the sigmaA weighted 2m|Fo| – D|Fc| map was dropped to 0.5 σ, which afforded the building of reasonable models into the disordered regions. Water molecules were added in PHENIX and checked by Coot. The TLS64 protocol was implemented in the final stage of refinements with each subunit as one TLS group. The omit Fo – Fc density maps were calculated by removing inhibitor coordinates from the input PDB file before running one more round of TLS refinement in PHENIX (simulated annealing protocol with a 2000 K initial temperature). The resulting map coefficients DELFWT and PHDELWT were used to generate maps. The refined structures were validated in Coot before deposition in the wwPDB protein data bank.
Caco-2 Permeability Assay
Caco-2 monolayer assays were performed by Cyprotex US, LLC (Watertown, MA), using standard procedures, as previously reported for compound 4.18
Supplementary Material
Acknowledgments
The authors thank the National Institutes of Health (R01GM049725, to R.B.S., GM057353 to T.L.P., and F32GM109667 to M.A.C.), for generous support of this work. M.A.C. wishes to thank Mr. Saman Shafaie and Dr. S. Habibi Goudarzi for assistance with HRMS experiments, Mr. Ben McDonald for his help with chiral HPLC, and Dr. Gashaw Goshu for his help with polarimetry measurements. This work made use of IMSERC at Northwestern University, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF NNCI-1542205), the State of Illinois, and the International Institute for Nanotechnology (IIN). H.L. thanks Carla Plaza for her assistance in protein purifications that provided samples for both enzyme activity assays and crystallization. We also wish to thank the SSRL and ALS beamline staff for their support during remote X-ray diffraction data collection.
Abbreviations
- NO
nitric oxide
- nNOS
neuronal nitric oxide synthase
- iNOS
inducible nitric oxide synthase
- eNOS
endothelial nitric oxide synthase
- rnNOS
rat neuronal nitric oxide synthase
- hnNOS
human neuronal nitric oxide synthase
- heNOS
human endothelial nitric oxide synthase
- miNOS
murine inducible nitric oxide synthase
- FMN
flavin mononucleotide
- H4B
(6R)-5,6,7,8-tetrahydrobiopterin
- tPSA
total polar surface area
- Papp
apparent permeability
- Bis-Tris
bis(2-hydroxyethyl)amino-tris(hydroxymethyl)-methane
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- GSH
glutathione, reduced
- TCEP
tris(2-carboxyethyl)phosphine
- dppb
1,4-bis(diphenylphosphino)butane
Footnotes
PDB ID Codes. PDB codes for X-ray crystal structures described in this study have been deposited in the Protein Data Bank. Authors will release the atomic coordinates and experimental data upon article publication under the following accession codes: rnNOS-8: 5UNR; rnNOS-9: 5UNS; rnNOS-10: 5UNT; rnNOS-11: 5UNU; rnNOS-14: 5UNV; rnNOS-16: 5UNW; rnNOS-18: 5UNX; rnNOS-(R,S)-19: 5UNY; rnNOS-(R)-19: 5UNZ; rnNOS-(S)-19: 5UO0; hnNOS-8: 5UO1; hnNOS-9: 5UO2; hnNOS-11: 5UO3; hnNOS-18: 5UO4; hnNOS-(R,S)-19: 5UO5; hnNOS-(R)-19: 5UO6; hnNOS-(S)-19: 5UO7; heNOS-8: 5UO8; heNOS-9: 5UO9; heNOS-11: 5UOA; heNOS-(R)-19: 5UOB; heNOS-(S)-19: 5UOC; beNOS-11: 5UOD.
Supporting Information. Crystallographic data collection and refinement statistics for rat and human nNOS, human eNOS, and bovine eNOS; bovine eNOS-11, rnNOS and hnNOS-(R,S)-19, rnNOS (R)- and (S)-19, and heNOS-18 Table S1) crystal structures (Figures S1-S4), NOS inhibition LogIC50 values and standard errors for new compounds (Table S2) and synthesis and analytical data for compounds 23-25, 26-29, 55-57, 59-62, 70-73 and 77-82.
References Cited
- 1.Maccallini C, Amoroso R. Targeting neuronal nitric oxide synthase as a valuable strategy for the therapy of neurological disorders. Neural Regen Res. 2016;11:1731–1734. doi: 10.4103/1673-5374.194707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Förstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2012;33:829–837. doi: 10.1093/eurheartj/ehr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Torreilles F, Salman-Tabcheh S, Guerin M, Torreilles J. Neurodegenerative disorders: the role of peroxynitrite. Brain Res Brain Res Rev. 1999;30:153–163. doi: 10.1016/s0165-0173(99)00014-4. [DOI] [PubMed] [Google Scholar]
- 4.Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, Ma Y, Masliah E, Nomura Y, Lipton SA. S-Nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. 2006;441:513–517. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- 5.Clementi E, Brown GC, Feelisch M, Moncada S. Persistent inhibition of cell respiration by nitric oxide: Crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc Natl Acad Sci U S A. 1998;95:7631–7636. doi: 10.1073/pnas.95.13.7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Canals S, Casarejos MJ, de Bernardo S, Rodríguez-Martín E, Mena MA. Nitric oxide triggers the toxicity due to glutathione depletion in midbrain cultures through 12-lipoxygenase. J Biol Chem. 2003;278:21542–21549. doi: 10.1074/jbc.M213174200. [DOI] [PubMed] [Google Scholar]
- 7.Zhang L, Dawson VL, Dawson TM. Role of nitric oxide in Parkinson's disease. Pharmacol Ther. 2006;109:33–41. doi: 10.1016/j.pharmthera.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 8.Dreschel DA, Estevez AG, Barbeito L, Beckman JS. Nitric oxide-mediated oxidative damage and the progressive demise of motor neurons in ALS. Neurotoxic Res. 2012;22:251–264. doi: 10.1007/s12640-012-9322-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang Z, Huang PL, Panahian N, Dalkara T, Fishman MC, Moskowitz MA. Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science. 1994;265:1883–1885. doi: 10.1126/science.7522345. [DOI] [PubMed] [Google Scholar]
- 10.Dorheim MA, Tracey WR, Pollock JS, Grammas P. Nitric oxide synthase activity is elevated in brain microvessels in Alzheimer's disease. Biochem Biophys Res Commun. 1994;205:659–665. doi: 10.1006/bbrc.1994.2716. [DOI] [PubMed] [Google Scholar]
- 11.Hantraye P, Brouillet E, Ferrante R, Palfi S, Dolan R, Matthews RT, Beal MF. Inhibition of neuronal nitric oxide synthase prevents MPTP-induced Parkinsonism in baboons. Nat Med. 1996;2:1017–1021. doi: 10.1038/nm0996-1017. [DOI] [PubMed] [Google Scholar]
- 12.Ikeda K, Iwasaki Y, Kinoshita M. Neuronal nitric oxide synthase inhibitor, 7-nitroindazole, delays motor dysfunction and spinal motoneuron degeneration in the wobbler mouse. J Neurol Sci. 1998;160:9–15. doi: 10.1016/s0022-510x(98)00224-x. [DOI] [PubMed] [Google Scholar]
- 13.Siddhanta U, Presta A, Fan B, Wolan D, Rousseau DL, Stuehr DJ. Domain swapping in inducible NO synthase: electron transfer cccurs between flavin and heme groups located on adjacent subunits in the dimer. J Biol Chem. 1998;273:18950–18958. doi: 10.1074/jbc.273.30.18950. [DOI] [PubMed] [Google Scholar]
- 14.Rosen GM, Tsai P, Pou S. Mechanism of free-radical generation by nitric oxide synthase. Chem Rev. 2002;102:1191–1199. doi: 10.1021/cr010187s. [DOI] [PubMed] [Google Scholar]
- 15.Kobayashi Y, Ikeda K, Shinozuka K, Nara Y, Yamori Y, Hattori K. L-nitroarginine increases blood pressure in the rat. Clin Exp Pharmacol Physiol. 1991;18:397–399. doi: 10.1111/j.1440-1681.1991.tb01470.x. [DOI] [PubMed] [Google Scholar]
- 16.Mukherjee P, Cinelli MA, Kang S, Silverman RB. Development of nitric oxide synthase (NOS) inhibitors for neurodegenerative diseases and neuropathic pain. Chem Soc Rev. 2014;43:6814–6838. doi: 10.1039/c3cs60467e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cinelli MA, Li H, Chreifi G, Martásek P, Roman LJ, Poulos TL, Silverman RB. Simplified 2-aminoquinoline-based scaffold for potent and selective neuronal nitric oxide synthase inhibition. J Med Chem. 2014;57:1513–1530. doi: 10.1021/jm401838x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cinelli MA, Li H, Pensa AV, Kang S, Martásek P, Roman LJ, Poulos TL, Silverman RB. Phenyl ether- and aniline-containing 2-aminoquinolines as potent and selective inhibitors of neuronal nitric oxide synthase. J Med Chem. 2015;58:8694–8712. doi: 10.1021/acs.jmedchem.5b01330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang H, Li H, Martasek P, Roman LJ, Poulos TJ, Silverman RB. Structure-guided design of selective inhibitors of neuronal nitric oxide synthase. J Med Chem. 2013;56:3024–3032. doi: 10.1021/jm4000984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kang S, Li H, Tang W, Martásek P, Roman LJ, Poulos TL, Silverman RB. 2-Aminopyridines with a truncated side chain to improve human neuronal nitric oxide synthase inhibitory potency and selectivity. J Med Chem. 2015;58:5548–5560. doi: 10.1021/acs.jmedchem.5b00573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang HY, Qin Y, Li H, Roman LJ, Martásek P, Poulos TL, Silverman RB. Potent and selective human neuronal nitric oxide synthase inhibition by optimization of the 2-aminopyridine-based scaffold with a pyridine linker. J Med Chem. 2016;59:4913–4925. doi: 10.1021/acs.jmedchem.6b00273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ji H, Li H, Martasek P, Roman LJ, Poulos TL, Silverman RB. Discovery of highly potent and selective inhibitors of neuronal nitric oxide synthase by fragment hopping. J Med Chem. 2009;52:779–797. doi: 10.1021/jm801220a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xue F, Li H, Fang J, Roman LJ, Martasek P, Poulos TL, Silverman RB. Peripheral but crucial: A hydrophobic pocket (Tyr706, Leu337, and Met336) for potent and selective inhibition of neuronal nitric oxide synthase. Biooorg Med Chem Lett. 2010;20:6258–6261. doi: 10.1016/j.bmcl.2010.08.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Senecal TD, Shu W, Buchwald SL. A general, practical palladium-catalyzed cyanation of (hetero)aryl chlorides and bromides. Angew Chem Int Ed. 2013;52:10035–10039. doi: 10.1002/anie.201304188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Silverman RB, Cinelli MA, Pensa AV. 2-Aminoquinoline compounds for potent and selective neuronal nitric oxide synthase inhibition. US Pat Appl Publ. 2016 doi: 10.1021/jm401838x. 20160368877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kawashima K, Honda T, Tajima H, Okamoto K, Yamamoto M. Preparation of novel cyclic compounds having a quinolinylalkylthio group. US Pat Appl Publ. 2008 20080021064. [Google Scholar]
- 27.Yin J, Xiang B, Huffman MA, Raab CE, Davies IW. A general and efficient 2-amination of pyridines and quinolines. J Org Chem. 2007;72:4554–4557. doi: 10.1021/jo070189y. [DOI] [PubMed] [Google Scholar]
- 28.Cheng Y, Judd TC, Bartberger MD, Brown J, Chen K, Fremeau RT, Hickman D, Hitchcock SA, Jordan B, Li V, Lopez P, Louie SW, Luo Y, Michelsen K, Nixey T, Powers TS, Rattan C, Sickmier EA, St Jean DJ, Wahl RC, Wen PH, Wood S. From fragment screening to in vivo efficacy: optimization of a series of 2-aminoquinolines as potent inhibitors of beta-site amyloid precursor protein cleaving enzyme 1 (BACE1) J Med Chem. 2011;54:5836–5857. doi: 10.1021/jm200544q. [DOI] [PubMed] [Google Scholar]
- 29.Ueda T, Konishi H, Manabe K. Palladium-catalyzed reductive carbonylation of aryl halides with N-formylsaccharin as a CO source. Angew Chem Int Ed. 2013;52:8611–8615. doi: 10.1002/anie.201303926. [DOI] [PubMed] [Google Scholar]
- 30.Inglis S, Jones R, Fritz D, Stojkoski C, Booker G, Pyke S. Synthesis of 5-, 6- and 7-substituted-2-aminoquinolines as SH3 domain ligands. Org Biomol Chem. 2005;3:2543–2557. doi: 10.1039/b504498g. [DOI] [PubMed] [Google Scholar]
- 31.Hirner JJ, Zacuto MJ. 7-Chloroquinoline: a versatile intermediate for the synthesis of 7-substituted quinolines. Tetrahedron Lett. 2009;50:4989–4993. [Google Scholar]
- 32.Sajiki H, Hattori K, Hirota K. The formation of a novel Pd/C-ethylenediamine catalyst: chemoselective hydrogenation without deprotection of the O-benzyl and N-CBz groups. J Org Chem. 1998;63:7990–7992. [Google Scholar]
- 33.Maegawa T, Fujita Y, Sakurai A, Akashi A, Sato M, Oono J, Sajiki H. Pd/C(en) catalyzed chemoselective hydrogenation in the presence of aryl nitriles. Chem Pharm Bull. 2007;55:837–839. doi: 10.1248/cpb.55.837. [DOI] [PubMed] [Google Scholar]
- 34.Maleczka RE, Shi F, Holmes D, Smith MR., III C-H activation/borylation/oxidation: a one-pot unified route to meta-substituted phenols bearing ortho-/para-directing groups. J Am Chem Soc. 2003;125:7792–7793. doi: 10.1021/ja0349857. [DOI] [PubMed] [Google Scholar]
- 35.Norberg AM, Smith MR, Maleczka RE. Practical one-pot C-H activation/borylation/oxidation: preparation of 3-bromo-5-methyphenol on a multigram scale. Synthesis. 2011;6:857–859. doi: 10.1055/s-0030-1258443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Borg G, Cogan DA, Ellman JA. One-pot asymmetric synthesis of tert-butanesulfinyl-protected amines from ketones by the in situ reduction of tert-butanesulfinyl ketimines. Tetrahedron Lett. 1999;40:6709–6712. [Google Scholar]
- 37.Llabres S, Garcia-Rates S, Cristobal-Lecina E, Riera A, Borrell JI, Camarasa J, Pubill D, Luque FJ, Escubedo E. Molecular basis of the selective binding of MDMA enantiomers to the alpha-4-beta-2 nicotinic receptor subtype: Synthesis, pharmacological evaluation and mechanistic studies. Eur J Med Chem. 2014;81:35–46. doi: 10.1016/j.ejmech.2014.04.044. [DOI] [PubMed] [Google Scholar]
- 38.Li H, Jamal J, Plaza C, Pineda SH, Chreifi G, Jing Q, Cinelli MA, Silverman RB, Poulos TL. Crystal structures of human constitutive nitric oxide synthases. Acta Crystallogr Sect D: Biol Crystallogr. 2014;D70:2667–2674. doi: 10.1107/S1399004714017064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H, Martasek P, Shimizu H, Masters BSS, Poulos TL, Raman CS. Unpublished results, PDB accession no. 1OM4 [Google Scholar]
- 40.Flinspach ML, Li H, Jamal J, Yang W, Huang H, Ha JM, Gomez-Vidal JA, Litzinger EA, Silverman RB, Poulos TL. Structural basis for dipeptide amide isoform-selective inhibition of neuronal nitric oxide synthase. Nat Struct Mol Biol. 2004;11:54–59. doi: 10.1038/nsmb704. see supporting information for structure. [DOI] [PubMed] [Google Scholar]
- 41.Li H, Jamal J, Delker S, Plaza C, Ji H, Jing Q, Huang H, Kang S, Silverman RB, Poulos TL. The mobility of a conserved tyrosine residue controls isoform-dependent enzyme-inhibitor interactions in nitric oxide synthases. Biochemistry. 2014;53:5272–5279. doi: 10.1021/bi500561h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fedorov R, Vasan R, Ghosh DK, Schlichting I. Structures of nitric oxide synthase isoforms complexed with the inhibitor AR-R17477 suggest a rational basis for specificity and inhibitor design. Proc Nat Acad Sci U S A. 2004;101:5892–5897. doi: 10.1073/pnas.0306588101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ji H, Delker SL, Li H, Martásek P, Roman LJ, Poulos TL, Silverman RB. Exploration of the active site of neuronal nitric oxide synthase by the design and synthesis of pyrrolidinomethyl 2-aminopyridine derivatives. J Med Chem. 2010;53:7804–7824. doi: 10.1021/jm100947x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xue F, Fang J, Delker SL, Li H, Martásek P, Roman LJ, Poulos TL, Silverman RB. Symmetric double-headed aminopyridines, a novel strategy for potent and membrane-permeable inhibitors of neuronal nitric oxide synthase. J Med Chem. 2011;54:2039–2048. doi: 10.1021/jm101071n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Delker SL, Xue F, Li H, Jamal J, Silverman RB, Poulos TL. Role of zinc in isoform-selective inhibitor binding to neuronal nitric oxide synthase. Biochemistry. 2010;49:10803–10810. doi: 10.1021/bi1013479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Delker SL, Ji H, Li H, Jamal J, Fang J, Xue F, Silverman RB, Poulos TL. Unexpected binding modes of nitric oxide synthase inhibitors effective in the prevention of cerebral palsy. J Am Chem Soc. 2010;132:5437–5442. doi: 10.1021/ja910228a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jing Q, Li H, Chreifi G, Roman LJ, Martásek P, Poulos TL, Silverman RB. Chiral linkers to improve selectivity of double-headed neuronal nitric oxide synthase inhibitors. Bioorg Med Chem Lett. 2013;23:5674–5679. doi: 10.1016/j.bmcl.2013.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lohmann C, Huwel S, Galla HJ. Predicting blood-brain barrier permeability of drugs: evaluation of different in vitro assays. J Drug Targeting. 2002;10:263–276. doi: 10.1080/10611860290031903. [DOI] [PubMed] [Google Scholar]
- 49.Stewart BH, Chan OH, Lu RH, Reyner EL, Shmid HL, Hamilton HW, Steinbaugh BA, Taylor MD. Comparison of intestinal permeabilities determined in multiple in vitro and in situ models: relationship to absorption in humans. Pharm Res. 1995;12:693–699. doi: 10.1023/a:1016207525186. [DOI] [PubMed] [Google Scholar]
- 50.Hazeldine ST, Polin L, Kushner J, White K, Bouregeois NM, Crantz B, Palomino E, Corbett TH, Horwitz JP. II. Synthesis and biological evaluation of some bioisosteres and congeners of the antitumor agent, 2-{4-[(7-chloro-2-quinoxalinyl)oxy]phenoxy}propionic acid (XK469) J Med Chem. 2002;45:3130–3137. doi: 10.1021/jm0200097. [DOI] [PubMed] [Google Scholar]
- 51.Roman LJ, Sheta EA, Martásek P, Gross SS, Liu Q, Masters BSS. High-level expression of functional rat neuronal nitric oxide synthase in Escherichia coli. Proc Natl Acad Sci USA. 1995;92:8428–8432. doi: 10.1073/pnas.92.18.8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hevel JM, White KA, Marletta MA. Purification of the inducible murine macrophage nitric oxide synthase: identification as a flavoprotein. J Biol Chem. 1991;266:22789–22791. [PubMed] [Google Scholar]
- 53.Gerber NC, Ortiz de Montellano PR. Neuronal nitric oxide synthase: expression in Escherichia coli, irreversible inhibition by phenyldiazene, and active site topology. J Biol Chem. 1995;270:17791–17796. doi: 10.1074/jbc.270.30.17791. [DOI] [PubMed] [Google Scholar]
- 54.Cheng YC, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of the inhibitor which causes 50 per cent inhibition (IC50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 55.Leber A, Hemmens B, Klosch B, Goessler W, Raber G, Mayer B, Schmidt K. Characterization of recombinant human endothelial nitric-oxide synthase purified from the yeast pichia pastoris. J Biol Chem. 1999;274:37658–37665. doi: 10.1074/jbc.274.53.37658. [DOI] [PubMed] [Google Scholar]
- 56.McPhillips TM, McPhillips SE, Chiu HJ, Cohen AE, Deacon AM, Ellis PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP, Soltis SM, Kuhn P. Blu-Ice and the Distributed Control System: software for data acquisition and instrument control at macromolecular crystallography beamlines. J Synchrotron Radiat. 2002;9:401–406. doi: 10.1107/s0909049502015170. [DOI] [PubMed] [Google Scholar]
- 57.Battye TGG, Kontogiannis L, Johnson O, Powell HR, Leslie AGW. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D. 2001;67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kabsch WXDS. Acta Crystallogr Sect D: Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Evans PR. Scaling and assessment of data quality. Acta Crystallogr Sect D: Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 60.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 61.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 62.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr Sect D: Biol Crystallogr. 2010;D66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. 2001;D57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.