

SUMMARY
Fibroblasts are major contributors to and regulators of inflammation and dominant producers of interleukin-6 (IL-6) in inflammatory diseases like rheumatoid arthritis. Yet, compared to leukocytes, the regulation of inflammatory pathways in fibroblasts is largely unknown. Here, we report that analyses of genes coordinately upregulated with IL-6 pointed to STAT4 and leukemia inhibitory factor (LIF) as potentially linked. Gene silencing revealed that STAT4 was required for IL-6 transcription. STAT4 was recruited to the IL-6 promoter following fibroblast activation, and LIF receptor (LIFR) and STAT4 formed a molecular complex that together with JAK1 and TYK2 kinases, controlled STAT4 activation. Importantly, a positive feedback loop involving autocrine LIF, LIFR and STAT4 drove sustained IL-6 transcription. Besides IL-6, this autorine loop also drove the production of other key inflammatory factors including IL-8, granulocyte-colony stimulating factor (G-CSF), IL-33, IL-11, IL-1α and IL-1β. These findings define the transcriptional regulation of fibroblast-mediated inflammation as distinct from leukocytes.
Keywords: Interleukin-6, IL-6, IL-33, IL-8, G-CSF, IL-11, IL-1, fibroblasts, stromal cells, inflammation, transcription, STAT4, leukemia inhibitor factor, LIF, leukemia inhibitor factor receptor, LIFR, autocrine, rheumatoid arthritis, autoimmune diseases
eTOC Blurb
Growing evidence implicates fibroblasts as inflammatory cells in sites of peripheral inflammation. Nguyen and colleagues demonstrate that regulation of IL-6 along with a set of other inflammatory cytokines and chemokines is regulated by a positive feedback loop involving LIF, LIF receptor, and STAT4 that selectively operates in fibroblasts.
INTRODUCTION
Fibroblasts function as stromal cells in peripheral tissues and as fibroblastic reticular cells (FRCs) in lymph nodes where they influence the recruitment, retention and survival of leukocytes and the generation and termination of immune responses (Fletcher et al., 2010; Katakai et al., 2004; Link et al., 2007). In chronic inflammation, fibroblasts are increasingly recognized as important contributors and regulators of inflammation through their production of inflammatory cytokines such as IL-6, chemokines such as CCL-2, IL-8 and arachidonate metabolites (Bernardo and Fibbe, 2013; Bottini and Firestein, 2013). Of these factors, IL-6 is a pleiotropic cytokine that plays a major role in the response to injury and infection (Kishimoto, 2005). Elevated expression of IL-6 has been implicated in diverse human diseases including inflammatory and autoimmune disorders such as rheumatoid arthritis, Crohn’s disease, systemic lupus erythematosus, Castleman’s disease, Behcet’s disease and systemic juvenile idiopathic arthritis as well as in coronary artery and neurologic diseases, and neoplasms (Hong et al., 2007; Waldner and Neurath, 2014; Wolf et al., 2014). Genetic studies have implicated IL-6 alleles in the predisposition to several autoimmune disorders (Fishman et al., 1998; Lee et al., 2012; Li et al., 2014; Villuendas et al., 2002). In rheumatoid arthritis (RA) patients, the joint fluids and plasma contain high concentrations of IL-6 and synovial and serum amounts of IL-6 correlate with RA disease activity (Hirano et al., 1988; Houssiau et al., 1988; Madhok et al., 1993). Blockade of IL-6 action by tocilizumab, a humanized anti-IL-6 receptor blocking monoclonal antibody, is an effective treatment for reducing both inflammation and joint destruction in RA and is being tested in a number of other inflammatory disorders (Tanaka et al., 2014). Importantly, fibroblasts are a major source of IL-6 as well as other inflammatory cytokines (Guerne et al., 1989; Okamoto et al., 1997).
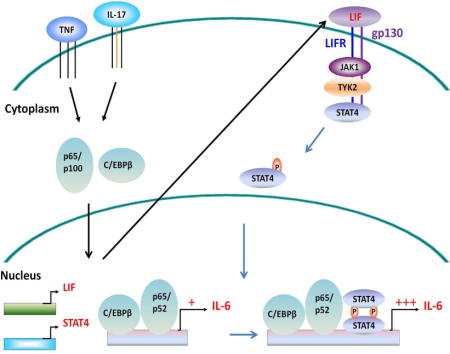
In many pathological states, inflammatory cytokines like TNF, IL-1, IL-17 and IL-6 are part of the overall inflammatory milieu (McInnes and Schett, 2007). All of these cytokines are potent stimulators of fibroblasts and combinations of inflammatory stimuli like TNF and IL-17 can dramatically activate fibroblasts (Zrioual et al., 2009). Much has been learned about the activation and transcriptional regulation of inflammatory factor production in leukocytes, while the regulation of fibroblast production of IL-6 and other inflammatory mediators is poorly understood. To better understand the transcriptional basis for inflammatory factor production by fibroblasts as occurs in chronic inflammatory diseases, we initially focused on IL-6 production following synergistic activation by TNF and IL-17, cytokines typically present in inflammatory disorders (Agarwal et al., 2008; McInnes and Schett, 2007). We designed a screen for differentially expressed genes in primary human fibroblasts to identify the genes and specifically the transcription factors that were induced following fibroblast activation and whose expression patterns correlated with that of IL-6. Analysis of gene expression data showed a marked upregulation of STAT4, a member of the signal transducers and activators of transcription factor family not previously implicated in IL-6 transcription. Importantly, STAT4 silencing reduced IL-6 expression after stimulation and STAT4 was recruited to the IL6 promoter. We found that STAT4 participated in a positive autocrine signaling circuit mediated through leukemia inhibitory factor (LIF) – LIF receptor (LIFR) that is important for sustained IL-6 transcription. LIFR and STAT4 formed a molecular complex that together with JAK1 and TYK2 kinases, controlled STAT4 activation. We found that this LIFR – STAT4 signaling pathway was broadly relevant for the production of a set of other key inflammatory factors including IL-8, G-CSF, IL-33, IL-11, IL-1α and IL-1β in fibroblasts and LIFR is selectively expressed in fibroblasts but not many leukocytes. Taken together, our results implicate the autocrine LIF – LIFR positive feedback loop and STAT4 as essential signaling components in the regulation of key inflammatory mediator production in human fibroblasts. These findings not only underscore how differently IL-6 and other chemokines and cytokines are regulated in mesenchymal compared to hematopoietic cell lineages, but also provide insights for understanding the basis for fibroblast activation and inflammatory factor production in health and diseases.
RESULTS
Human fibroblasts produce IL-6 in response to pro-inflammatory cytokines
To examine directly the ability of fibroblasts to produce IL-6 in the synovium of inflammatory arthritis, we tested both mouse and human samples. We found that fibroblasts were the dominant source for the production of IL-6 compared to leukocytes in the arthritic joints of K/BxN mice, a useful animal model of inflammatory arthritis (Kouskoff et al., 1996) (Supplemental Figure S1D). A substantial portion of IL-6 production in rheumatoid arthritis (RA) also was derived from fibroblasts relative to other leukocytes including B cells, T cells and monocytes (Supplemental Figure S1B). The higher proportional production of IL-6 by fibroblasts in the mouse model likely reflects the fact that mice were in the active phase of arthritis as opposed to human RA synovial samples obtained at the end stage of disease at the time of joint replacement surgery. To confirm this, we placed pieces of freshly isolated synovial tissue in media containing TNF and IL-17, the two cytokines commonly observed in active RA synovial fluids. After 48 hours, we isolated fibroblasts and monocytes, the two cell types that appear to have substantial contribution of IL-6 in the synovium. We observed that under these stimulated inflammatory conditions, fibroblasts produced markedly higher amount of IL6 mRNA while monocytes did not change their IL6 mRNA relative to that from the freshly isolated tissue (Supplemental Figure S1C). This suggests that in inflammatory environments such as those found in the active phase of RA, fibroblasts are a major source of IL-6.
To partially model the inflammatory milieu in RA, we examined the in vitro responses of primary cultured human fibroblasts, including those from synovial, skin and lung tissues after stimulation with TNF or the combination of TNF and IL-17 (Noss et al., 2015; Zrioual et al., 2009). Fibroblasts responded dramatically to the combined stimulation of TNF and IL-17 compared to TNF or IL-17 alone by producing substantial amounts of IL-6 (Figure 1A) that were sustained even after 72 hours (Figure 1B). To examine IL-6 expression over time, we stimulated primary human fibroblast cell lines, OA-4 and RA-32, with TNF + IL-17 and collected supernatant media and RNA samples at various time points. We observed that increases in IL-6 protein (Figure 1C) following stimulation reflected similar increases in IL6 mRNA (Figure 1D). This suggests that a transcriptional and/or post-transcriptional mechanism is involved. Here, we focused on the transcriptional regulation of IL-6 in human fibroblasts.
Figure 1. Expression of IL-6 by human fibroblasts.
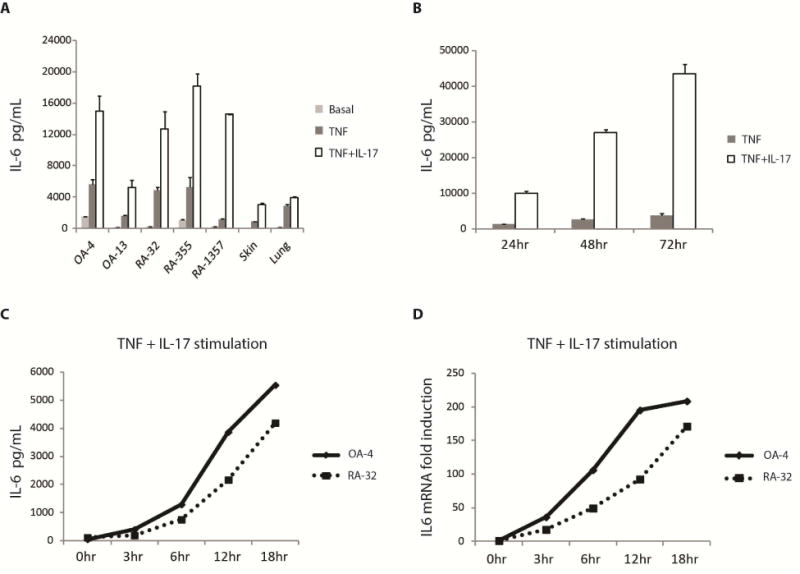
(A) Primary cultured human fibroblasts derived from OA, RA synovium as well as skin and lung fibroblast lines were unstimulated (Basal) or stimulated with TNF (1 ng/mL) or TNF (1 ng/mL) + IL-17 (1 ng/mL) for approximately 20 hours, supernatants were collected and IL-6 release measured by ELISA. (B) Sustained production of IL-6. RA-355 fibroblasts were treated with TNF (1 ng/mL) or TNF (1 ng/mL) + IL-17 (1 ng/mL) for 24, 48 and 72 hours (hr), supernatants were collected and IL-6 secretion measured by ELISA. (C, D) IL-6 protein and mRNA expression over time. OA-4 and RA-32 fibroblasts were stimulated with TNF (1 ng/mL) or TNF (1 ng/mL) + IL-17 (1 ng/mL) for various durations of time as indicated before RNA and supernatants were collected. IL-6 protein was measured by ELISA. The amount of IL6 mRNA was measured by qPCR and normalized to GAPDH. Fold induction was calculated by dividing the normalized IL6 mRNA at a certain time point with that at time 0hr. Error bars represent s.d. of triplicate technical replicates. Data are representative of two independent experiments (A-D). See also Figure S1.
STAT4 is a transcription factor regulating IL-6 expression
To identify transcription factors involved in IL-6 expression, we used four primary human fibroblast cell lines, two derived from RA patients (RA-32, RA-449) and two from OA patients (OA-6, OA-502) and stimulated them with TNF or the combination of TNF and IL-17 for approximately 20 hours before collecting RNA samples for genome-wide expression profiling. As TNF and IL-17 combined stimulation resulted in an enhanced production of IL-6 compared to TNF alone (Figure 1B, 1C), we hypothesized that some transcription factors involved in IL-6 expression would likely have higher expression under the TNF + IL-17 combined stimulation. To identify such transcription factors, using genome-wide expression data, we calculated the fold induction for each gene by dividing its normalized expression after TNF or TNF + IL-17 with its expression in unstimulated samples. Genes whose expression after TNF + IL-17 stimulation were more than 2 fold over TNF alone, across all four fibroblasts lines, were selected for further study (Figure 2A). This group of genes includes four transcription factors (TFs), namely EHF, ELF3, CEBPD and STAT4.
Figure 2. STAT4 and other transcription factors regulating IL-6 expression in human fibroblasts.
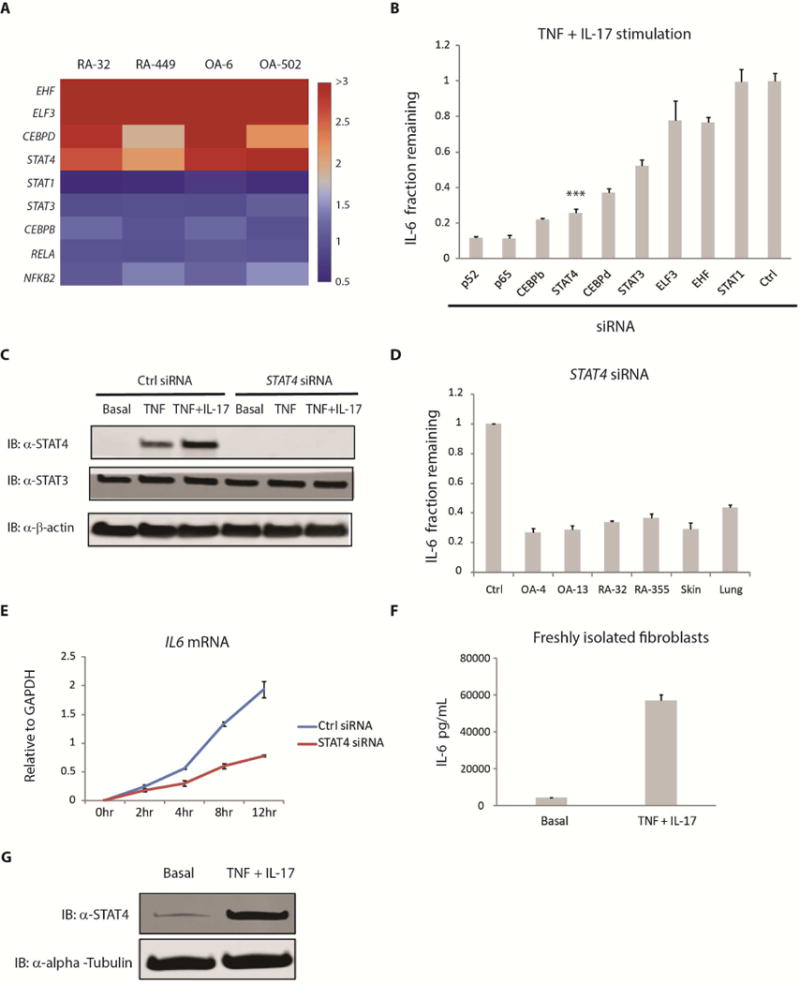
(A) Four different primary human fibroblast lines were stimulated with TNF (1 ng/mL) or the combination of TNF (1 ng/mL) and IL-17 (1 ng/mL) for approximately 20 hours before RNA samples were collected for expression profiling. Using genome-wide expression data, we calculated the fold induction for each gene by dividing its normalized expression after TNF or TNF + IL-17 with that in unstimulated samples. Genes whose expression after TNF + IL-17 stimulation were more than 2 fold over TNF alone, across all four cell lines, were selected. This includes four transcription factors, namely EHF, ELF3, CEBPD and STAT4. The heatmap color used for each gene was based on the fold difference in its expression between TNF + IL-17 versus TNF stimulation alone. (B) RA-1357 fibroblasts expressing siRNA against various transcription factors were stimulated with TNF (0.1 ng/mL) + IL-17 (1 ng/mL) for approximately 20 hours, supernatants were collected and IL-6 was measured by ELISA. Fraction remaining was calculated by dividing the amount of IL-6 release from cells expressing siRNA specific for a transcription factor with that from cells expressing a control (Ctrl) siRNA. Error bars represent s.d. of triplicate technical replicates. Data are representative of 3 independent experiments. P value was calculated using the student t-test (*** = p < 0.001). (C) RA-1357 fibroblasts expressing control (Ctrl) and STAT4 siRNA were stimulated with TNF (0.1 ng/mL) or TNF (0.1 ng/mL) + IL-17 (1 ng/mL) for approximately 20 hours before whole cell lysates were collected. Western blotting was performed using an anti-STAT4 antibody with anti-β-actin as loading controls. Basal represents STAT4 protein expression under unstimulated conditions. (D) Role of STAT4 in IL-6 expression across a series of fibroblast types. Fibroblasts derived from synovium (OA, RA), skin and lung tissues expressing STAT4 siRNA were stimulated with TNF (0.1 ng/mL) + IL-17 (1 ng/mL) for approximately 20 hours, supernatants were collected and IL-6 was measured by ELISA. Fraction remaining was calculated as in (B). A control siRNA was used for each cell line and one representative control siRNA is shown. Error bars represent s.d. of triplicate technical replicates. Data are representative of two independent experiments. (E) Effect of STAT4 silencing on IL6 mRNA expression. RA-1357 fibroblasts expressing control (Ctrl) and STAT4 siRNA were stimulated with TNF (0.1 ng/mL) + IL-17 (1 ng/mL) for different amounts of time before RNA samples were collected. The amount of IL6 mRNA was measured by qPCR and normalized to GAPDH. Error bars represent s.e.m. of biological duplicates. hr, hours. (F) IL-6 and (G) STAT4 protein expression in freshly isolated human synovial fibroblasts rather than cultured cell lines. Synovial fibroblasts freshly disaggregated from human synovium were stimulated with TNF (1 ng/mL) + IL-17 (1 ng/mL) for 20 hours before supernatants (F) and whole cell lysates (G) were collected. IL-6 release was measured by ELISA and STAT4 by Western blotting. Error bars represent s.d. of triplicate technical replicates. Western blotting was performed using an anti-STAT4 antibody with anti-α-alpha-tubulin as loading controls. Efficiency of siRNA silencing was assessed by Western blotting and qPCR. See also Figure S2, S3.
ELF3 and CEBPD TFs have previously been implicated in IL-6 expression in murine dendritic and osteoblastic cells (Kushwah et al., 2011; Ruddy et al., 2004). However, STAT4 and EHF were not known to be directly involved in IL-6 gene expression. To determine if ELF3, EHF, CEBPD and STAT4 played a role in IL-6 regulation in human fibroblasts, we used siRNAs to silence their expression (Figure 2B). We observed that silencing of transcription factors known to regulate IL-6 such as NF-κB/p52, NF-κB/p65, C/EBPβ, C/EBPδ and STAT3 all reduced IL-6 production with p52, p65 and C/EBPβ having the strongest effects. On the other hand, while silencing of ELF3, EHF and STAT1 did not appear to alter IL-6 production, STAT4 silencing resulted in marked reduction of IL-6. Since STAT4 has not previously been implicated in IL-6 regulation, we chose to focus on STAT4 for further study.
Consistent with the microarray data, we observed STAT4 protein upregulation following TNF and TNF + IL-17 stimulation, and combined stimulation resulted in even higher expression of STAT4 (Figure 2C). Of note, STAT4 siRNA was effective in silencing basal and induced expression of STAT4, but it did not affect STAT3 expression (Figure 2C). Importantly, silencing of STAT4 had a strong effect on IL-6 but not on matrix metalloprotease-3 (MMP3) expression (Supplemental Figure S2G). We also observed that although silencing of NF-κB/p65 and C/EBPβ did not affect basal STAT4 protein they prevented STAT4 from being upregulated following stimulation (Supplemental Figure S3A). This suggests that p65 and C/EBPβ may be involved in the induction of STAT4.
To further confirm the role of STAT4 in IL-6 regulation, we transfected the siRNA against STAT4 into additional primary human fibroblast lines derived from synovial, skin and lung tissues. We observed that STAT4 silencing not only affected IL-6 expression in synovial fibroblasts, it also led to a reduction in IL-6 production in primary human lung and skin fibroblasts (Figure 2D). In addition, when analyzing the ability of STAT4 to induce IL-6 expression in a time course experiment, we observed that fibroblasts expressing STAT4 siRNA were less efficient at inducing IL6 mRNA (Figure 2E) and consequently IL-6 protein (Supplemental Figure S3E) than cells expressing a control siRNA. The effect was particularly more pronounced at later time points, which is not the case with other transcripts including CXCL5 and SLC7A2 (Supplemental Figure S3C, D). Furthermore, when examining IL-6 and STAT4 expression in fibroblasts freshly isolated from disaggregated human synovium, we also obtained results similar to those from cultured fibroblast lines, namely both IL-6 and STAT4 were upregulated following TNF + IL-17 combined stimulation (Figure 2F, G). This suggests that STAT4 plays a central role in IL-6 regulation in fibroblasts.
To further study the role of STAT4, we created retrovirus-based overexpression vectors carrying wild type STAT4 and a phospho-mutant STAT4 in which the tyrosine 693 residue was changed to alanine to prevent STAT4 from being phosphorylated, rendering it inactive (Figure 3A). We used a primary human lung fibroblast line to generate cells stably expressing these constructs. Under basal conditions, no phosphorylation of STAT4 was detected. As expected, following TNF + IL-17 stimulation, only wild type STAT4 was phosphorylated but not the mutant (Figure 3B, Supplemental Figure S4A). To examine if STAT4 can be directly recruited to the IL6 promoter, we performed chromatin immunoprecipitation using resins containing an anti-FLAG antibody against the FLAG tag at the C-terminus of the overexpressed STAT4. We observed that following TNF + IL-17 stimulation only wild type STAT4 was recruited to the IL6 promoter but not the phosphorylation mutant (Figure 3C). Moreover, since the IRF1 promoter is a known STAT4 binding target (Galon et al., 1999) and the IRF1 transcript is induced after TNF + IL-17 stimulation (Supplemental Figure S4D), as a positive control, we also quantified the recruitment of STAT4 to this promoter (Figure 3D). Similar to the pattern observed at the IL6 promoter, only wild type STAT4 was able to bind to the IRF1 promoter. In addition, when performing chromatin immunoprecipitation using an anti-STAT4 antibody against endogenous STAT4 from fibroblasts without STAT4 overexpression, we also observed increases in STAT4 recruitment to the IL6 and IRF1 promoters following stimulation (Supplemental Figure S4B). Moreover, of several promoter regions upstream of the IL6 transcription start site (TSS), the most proximal region lying within 500 bp of the TSS has the highest enrichment of STAT4 ChIP-DNA compared to other more distal regions (Supplemental Figure S4C). This suggests that STAT4 is directly involved in the transcriptional regulation of IL-6.
Figure 3. Recruitment of STAT4 to the IL6 promoter.
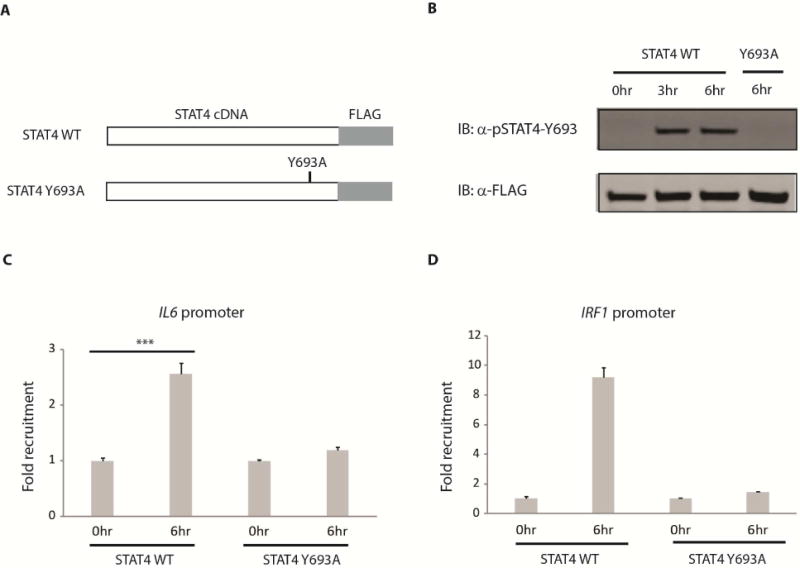
(A) STAT4 overexpression constructs. Wild type (WT) and a phosphorylation defective (Y693A) STAT4 were cloned into a retroviral vector (pQCXIX, Clontech). A FLAG tag was inserted at the C-terminus of STAT4 for affinity purification and Western blotting. Retro viruses carrying these STAT4 constructs were made and used to transduce a human lung fibroblast line that stably expressed the construct after puromycin selection. (B) Lung fibroblast cell lines generated in (A) were stimulated with TNF (1 ng/mL) + IL-17 (1 ng/mL) for 0, 3 and 6 hours (hr) before whole cell lysates were collected. Western blots were performed using anti-pSTAT4-Y693 for phosphorylated STAT4 and anti-FLAG for total STAT4. (C, D) Recruitment of STAT4 to the IL6 (C) and IRF1 (D) promoter. Lung fibroblasts overexpressing wild type (WT) or phospho-defective (Y693A) STAT4 protein were stimulated with TNF (1 ng/mL) + IL-17 (1 ng/mL) for 0 and 6 hours (hr) before cells were fixed with formaldehyde. Chromatin immunoprecipitation (ChIP) was carried out using a FLAG resin. The amount of DNA precipitated was quantified using qPCR. Fold recruitment was calculated by dividing the amount of ChIP DNA at 6hr (normalized with input DNA) with that at 0hr (normalized with input DNA). Error bars represent s.e.m. of triplicate biological replicates. P value was calculated using the student t-test (*** = p < 0.001). See also Figure S4.
LIFR and gp130 play an important role in IL-6 regulation
Activation of STAT transcription factors usually requires an upstream receptor where STATs bind (Schindler, 2002). Traditionally, STAT4 is known to be regulated via the IL-12 and IL-23 cytokine receptors in leukocytes (Bacon et al., 1995; Cho et al., 1996). However, the expression of these receptors across multiple fibroblast lines tested was very low (Figure 4A), implying that STAT4 is likely not regulated by the IL-12 and IL-23 receptor family in fibroblasts. Other receptors including IL6ST (gp130) and IFNAR have been implicated in STAT4 activation (Collison et al., 2012; Tyler et al., 2007). We found that these two receptors as well as the leukemia inhibitory factor receptor (LIFR), an IL-6 family ligand binding and gp130 associated receptor, are substantially expressed in fibroblasts (Figure 4A). Thus, we asked if IFNAR2, gp130 and LIFR play any role in IL-6 expression. We observed that silencing of gp130 or LIFR but not IFNAR2 reduced IL-6 production significantly (Figure 4B), implying that gp130 and LIFR play an important role in IL-6 production in fibroblasts.
Figure 4. Cell surface receptors involved in IL-6 regulation.
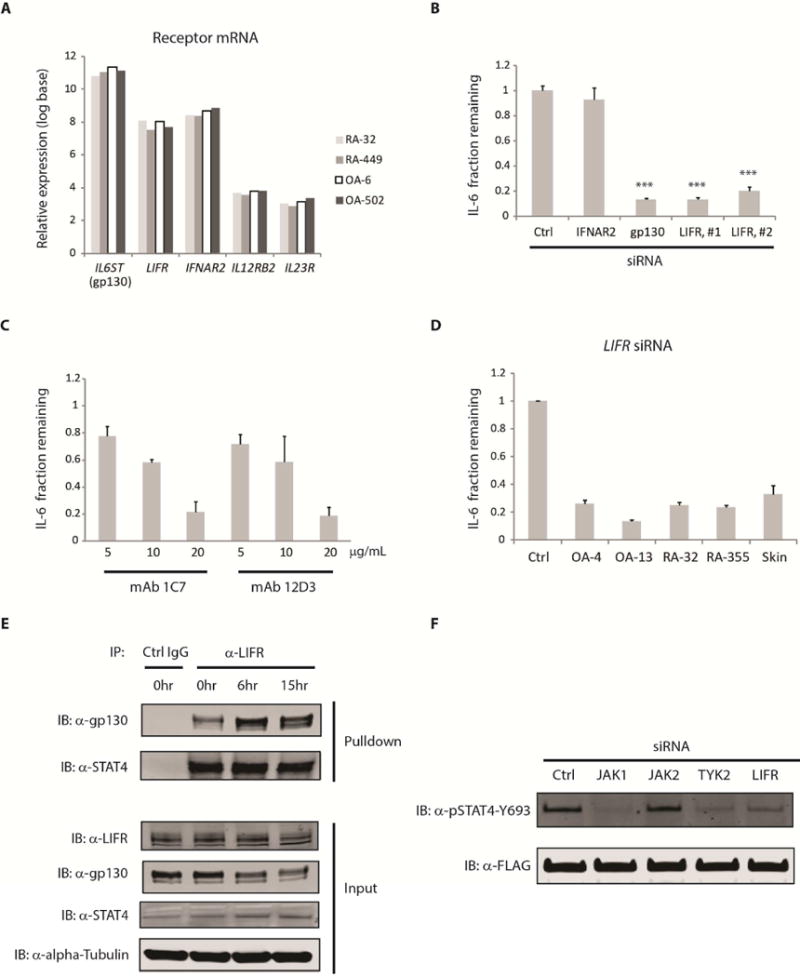
(A) Basal mRNA expression of cell surface receptors implicated in STAT4 activation. Relative expression values from 4 synovial fibroblast lines were derived from microarray data under unstimulated conditions after gene expression have been normalized and converted to log base 2. (B) RA-1357 fibroblasts expressing siRNA against various cell surface receptors were stimulated with TNF (0.1 ng/mL) + IL-17 (1 ng/mL) for approximately 20 hours, supernatants were collected and IL-6 was measured by ELISA. Fraction remaining was calculated by dividing the amount of IL-6 released from cells expressing an siRNA against a receptor with that from cells expressing a control (Ctrl) siRNA. LIFR, #1 and LIFR, #2 represent two different siRNAs against the leukemia inhibitory factor receptor (LIFR). Error bars represent s.d. of triplicate technical replicates. Data are representative of 3 independent experiments. P value was calculated using the student t-test (*** = p < 0.001). (C) Cell surface blocking of LIFR. RA-1357 fibroblasts were stimulated with TNF (0.1 ng/mL) + IL-17 (1 ng/mL) for approximately 20 hours in the presence of mouse monoclonal antibodies (mAb 1C7 and 12D3) targeting different domains of LIFR. Fraction remaining was calculated by dividing the amount of IL-6 released from cells under a blocking condition with that from cells treated with an isotype control antibody at the same concentration. Error bars represent s.d. of triplicate technical replicates. Data are representative of two independent experiments. (D) Role of LIFR across fibroblast types. Synovial and skin fibroblasts expressing a LIFR siRNA were stimulated with TNF (0.1 ng/mL) + IL-17 (1 ng/mL) for approximately 20 hours, supernatants were collected and IL-6 release was measured by ELISA. Fraction remaining was calculated as in (B). A control (Ctrl) siRNA was used for each cell line and one representative control siRNA is shown. Error bars were calculated as in (C). (E) Endogenous interaction of LIFR with gp130 and STAT4. Cell lysates were collected from primary lung fibroblasts stimulated with TNF (1 ng/mL) + IL-17 (1 ng/mL) for 0, 6 and 15 hours (hr). Immunoprecipitation was performed using an anti-LIFR or control antibody (Ctrl IgG). Antibodies against LIFR, STAT4 and gp130 were used to detect the presence of these proteins in the LIFR pulldown fraction. (F) Role of LIFR and JAK kinases in STAT4 phosphorylation. Lung fibroblasts overexpressing wild type STAT4 were transfected with siRNA against JAK1, JAK2, TYK2, LIFR or control (Ctrl) for 48 hours. Cells were then stimulated with TNF (1 ng/mL) + IL-17 (1 ng/mL) for 6 hours before whole cell lysates were collected. Detection of STAT4 phosphorylation was performed using an anti-pSTAT4-Y693 antibody. Total STAT4 was probed using an anti-FLAG antibody. Similar results were observed using synovial fibroblasts overexpressing wild type STAT4 (Data not shown). Efficiency of siRNA silencing was assessed by Western blotting and qPCR. See also Figure S5.
To further confirm the role of LIFR, we used two different anti-LIFR blocking antibodies, namely 1C7 and 12D3. The 1C7 monoclonal antibody (mAb) competes with the ligand binding domain of LIFR whereas the 12D3 mAb blocks the association of LIFR and gp130 (Pitard et al., 1997; Taupin et al., 2001). Both anti-LIFR blocking mAbs were able to reduce IL-6 production in a dose dependent manner (Figure 4C). In addition, we observed that LIFR silencing by siRNA in additional synovium and skin derived fibroblast lines also resulted in marked reduction of IL-6 (Figure 4D).
Since these data suggest that gp130, LIFR and STAT4 play a role in IL-6 regulation, we asked whether or not these proteins interact with one another. Using an antibody against LIFR in immunoprecipitation experiments, we were able to detect the presence of both gp130 and endogenous STAT4 in the LIFR pulldown fraction (Figure 4E). Similarly, using an antibody against gp130, we were also able to detect the presence of both LIFR and STAT4 in the gp130 pulldown fraction (Supplemental Figure S5D). Moreover, we observed that while the total amount of gp130 decreases after stimulation, its amount in the LIFR pulldown fraction actually increases, suggesting that there is additional recruitment of gp130 to the LIFR containing complex following stimulation. On the other hand, both the input amount of LIFR and the pulldown amount of STAT4 do not appear to change, implying that STAT4 binds to LIFR even under basal conditions. Taken together, these data suggests that gp130, LIFR and STAT4 form a molecular complex and STAT4 probably interacts with LIFR directly.
Activation of STAT4 requires phosphorylation of the tyrosine 693 (Y693) residue. To explore the receptor and kinases involved, we examined if STAT4 phosphorylation was altered by siRNA silencing of JAK1, JAK2, TYK2, and LIFR in fibroblasts following TNF + IL-17 stimulation. We found that silencing of JAK1, TYK2 or LIFR but not JAK2 reduced STAT4 phosphorylation, implying that LIFR together with JAK1 and TYK2 are involved in the phosphorylation of STAT4 (Figure 4F). To further confirm the role of LIFR in STAT4 phosphorylation, we used the 1C7 mAb that blocks the interaction between LIF and LIFR. The presence of this mAb prevented STAT4 phosphorylation (Supplemental Figure S5F). To further confirm the role of JAK1 and TYK2 on STAT4 function, we used several JAK inhibitors including filgotinib, tofacitinib, decernotinib and ruxolitinib. We found that only the inhibitors that block both JAK1 and TYK2, namely filgotinib and decernotinib, displayed dose-dependent effects on IL-6 production whereas tofacitinib and ruxolitinib did not since they do not affect TYK2 (Supplemental Figure S5G). Similarly, silencing of JAK1 or TYK2 individually only had partial effects on IL-6 production whereas silencing of both kinases together strongly suppressed IL-6 (Supplemental Figure S5H).
Autocrine LIF is required for IL-6 regulation
LIFR has been shown to bind several IL-6 family ligands including cardiotrophin-1 (CTF-1), ciliary neurotrophic factor (CNTF) and leukemia inhibitory factor (LIF) (Bauer et al., 2007). Of these ligands, only LIF mRNA expression increased in a manner similar to that of IL6 mRNA following TNF or TNF + IL-17 combined stimulation of fibroblasts (Figure 5A). Increases in LIF mRNA after stimulation also could be detected at the protein level (Figure 5B). In addition, we observed that LIF expression increased rapidly as soon as one hour following fibroblast stimulation (Figure 5C, Supplemental Figure S6B). Thus, we hypothesized that LIF may be a key ligand for LIFR. To determine if LIF plays a role in IL-6 production, we used both siRNA and blocking antibody against LIF. Silencing LIF by siRNA or anti-LIF blocking antibody reduced IL-6 production and using both the siRNA and antibody together was even more effective in blocking LIF and subsequent IL-6 expression (Figure 5D). This suggests that LIF produced by fibroblasts following TNF and IL-17 stimulation acts as an autocrine ligand via LIFR in the regulation of IL-6.
Figure 5. Role of LIF ligand in IL-6 production by fibroblasts.
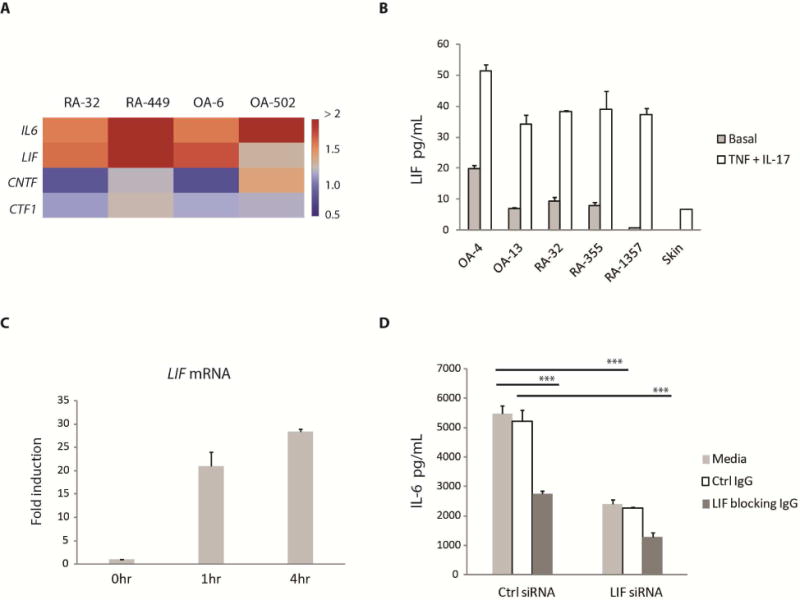
(A) Using genome-wide expression profiling data, we calculated the fold induction for each candidate LIFR ligand by dividing its normalized expression following fibroblast stimulation by TNF or TNF + IL-17 with that in unstimulated samples. The heatmap color used for each gene was based on the fold difference in its expression between TNF + IL-17 versus TNF stimulation. (B) Production of LIF following stimulation. Fibroblasts were stimulated with TNF (0.1 ng/mL) + IL-17 (1 ng/mL) for approximately 20 hours before supernatants were collected. The amount of LIF in the supernatants was measured by ELISA. Basal represents the amount of LIF produced under unstimulated conditions. Error bars represent s.d. of triplicate technical replicates. Data are representative of two independent experiments. (C) Rapid induction of LIF following stimulation. RA-1357 fibroblasts were stimulated with TNF (0.1 ng/mL) + IL-17 (1 ng/mL) for 1 and 4 hours (hr) and RNA samples were collected. The amounts of LIF mRNA were measured by qPCR and normalized to GAPDH. Fold induction was calculated by dividing the normalized LIF mRNA at each time point with that at time 0hr. Error bars were calculated as in (B). (D) Effect of LIF blocking. LIF expression was blocked by using either an siRNA or antibody (IgG) against LIF. Antibody was added approximately 1 hour before cells were stimulated with TNF (0.1 ng/mL) + IL-17 (1 ng/mL) for about 20 hours and IL-6 release in the supernatants was measured by ELISA. Error bars were calculated as in (B). P value was calculated using the student t-test (*** = p < 0.001). Efficiency of LIF silencing was assessed by ELISA. See also Figure S6.
LIFR and STAT4 are involved in IL-6 regulation under various inflammatory conditions
Fibroblasts are versatile cells that can produce IL-6 in the context of various inflammatory conditions. We have shown that LIFR and STAT4 play an important role in IL-6 expression in fibroblasts following TNF + IL-17 stimulation. Next, we asked if silencing of STAT4 or LIFR by siRNA also resulted in reduction of IL-6 release after fibroblast stimulation by IL-1β, TNF, or lipopolysaccharide (LPS) (Figure 6A–C) (all of these stimulations also led to increased LIF production (Supplemental Figure S6C)). These results suggest that autocrine LIF, LIFR and STAT4 form key components in IL-6 transcriptional regulation in fibroblasts under various inflammatory conditions.
Figure 6. Role of LIFR and STAT4 under different stimuli and in the regulation of a range of inflammatory mediators beyond IL-6 relevant to fibroblasts but not hematopoietic cells.
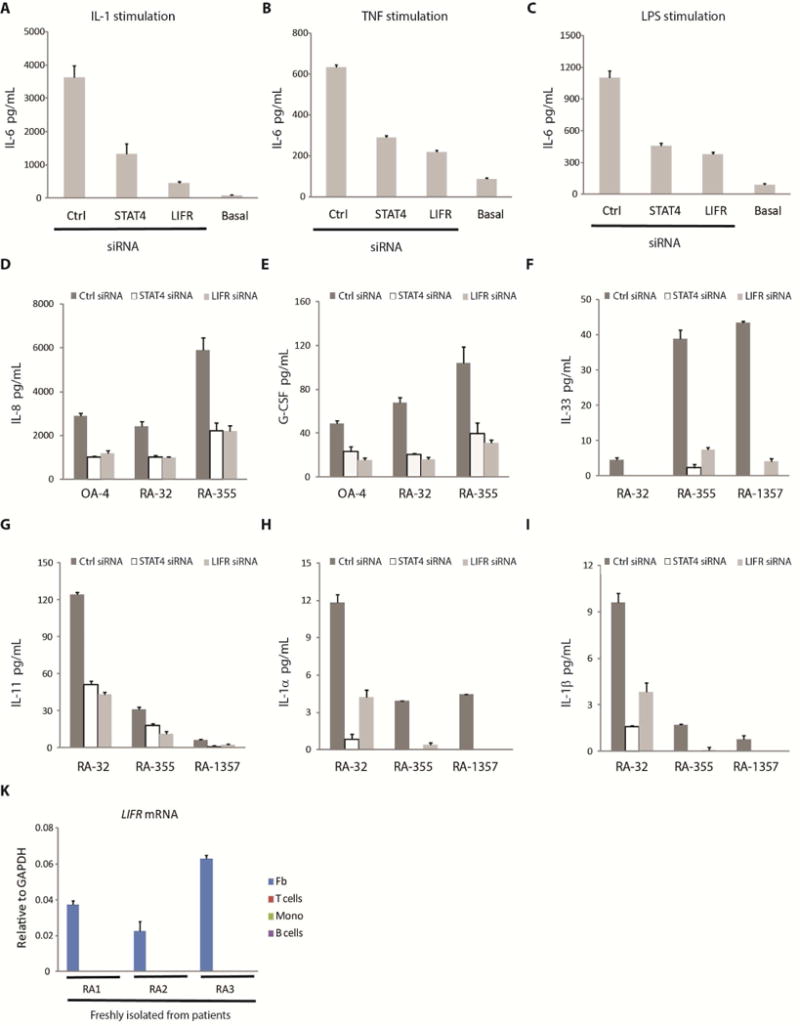
(A, B, C) Involvement of LIFR and STAT4 in IL-6 regulation under various inflammatory conditions. RA-1357 fibroblasts expressing a STAT4, LIFR or a control (Ctrl) siRNA were treated with (A) IL-1β (5 pg/mL); (B) TNF (0.1 ng/mL); or (C) lipopolysaccharide (LPS) (2.5 μg/mL) for approximately 20 hours, supernatants were collected and IL-6 measured by ELISA. Basal represents the amount of IL-6 produced under unstimulated conditions. Error bars represent s.d. of triplicate technical replicates. Data are representative of two independent experiments. (D, E, F, G, H, I) Involvement of LIFR and STAT4 in the regulation of many key inflammatory mediators. Fibroblasts expressing a STAT4, LIFR or a control (Ctrl) siRNA were stimulated with (D, F, G, H, I) TNF (0.1 ng/mL) + IL-17 (1 ng/mL) or (E) TNF (1 ng/mL) + IL-17 (1 ng/mL) for approximately 20 hours, supernatants (D, E) or whole cell lysates (F, G, H, I) were collected and IL-8 and G-CSF protein release (D, E) or IL-33, IL-11, IL-1α and IL-1β protein production (F, G, H, I) were measured by ELISA. Error bars represent s.d. of triplicate technical replicates. Data are representative of two independent experiments. (K) LIFR expression in fibroblasts and leukocytes. Fibroblasts (Fb), B cells, CD4+ T cells (T cells) and monocytes (Mono) were freshly isolated from human rheumatoid arthritis (RA) arthroplasty synovium by flow cytometry sorting (Supplemental Figure S1A) and each number denotes a different donor. The amount of LIFR mRNA was measured by qPCR using GAPDH as the normalization control. Error bars represent s.d. of triplicate technical replicates. Note that LIFR mRNA could not be detected in T cells, B cells and monocytes. See also Figure S6.
LIFR and STAT4 regulate many key inflammatory mediators and LIFR is selectively expressed in fibroblasts
Following TNF + IL-17 stimulation of fibroblasts, besides IL-6, we also observed increases in the production of a series of inflammatory cytokines and chemokines including IL-8, G-CSF, IL-33, IL-11, IL-1α and IL-1β. Importantly, in fibroblasts expressing a STAT4 or LIFR siRNA, the expression of all these inflammatory molecules, like IL-6, were markedly reduced (Figure 6D–I). In addition, LIFR mRNA expression was only detectable in fibroblasts but not leukocytes including B cells, T cells and monocytes freshly isolated from the synovium of RA patients (Figure 6K). Taken together, these results define a critical and selective role for LIFR and STAT4 in the regulation of many key inflammatory mediators produced by fibroblasts under a variety of activation conditions.
DISCUSSION
Fibroblasts are well known for their physiological roles in matrix remodeling and wound healing. Over the past few years, their central roles in regulating and mediating inflammation in a variety of pathological conditions have become clear. In lymph nodes, fibroblasts organize the T cell zones and are called fibroblastic reticular cells (FRCs). These fibroblasts produce chemokines and cytokines that retain and support the survival of T cells and dendritic cells to enable the initiation of potent immune responses (Fletcher et al., 2010). After microbial antigens from peripheral tissues elicit immune responses, the FRCs in lymph nodes produce nitric oxide and surface inhibitory receptors that help dampen and terminate T cell responses (Fletcher et al., 2011). In tumor tissues, fibroblasts referred to as stromal cells, can profoundly inhibit anti-tumor immune responses that are reactivated upon removal of the inhibitory stromal cells (Kraman et al., 2010). In contrast to these inhibitory examples, fibroblasts activated in peripheral tissues like the synovium in RA are key drivers of inflammation as the cytokines and chemokines produced by them have been shown to influence the function of macrophages, lymphocytes and neutrophils (Donlin et al., 2014; Fox et al., 2010; Lally et al., 2005).
Given the key role of IL-6 as a hallmark inflammatory cytokine made by fibroblasts and the relative lack of understanding of IL-6 regulation in mesenchymal cell types, we aimed to explore the transcriptional regulation of IL-6 in human fibroblasts. To model fibroblast activation by cytokines important in RA, we stimulated them with the combination of TNF and IL-17 which results in striking expression of IL-6. We employed global gene expression profiling data from four primary fibroblast lines derived from human synovium, hypothesizing that genes that are involved in IL-6 regulation might have expression patterns related to that of IL6 mRNA. Using this criterion, we identify four transcription factors, namely EHF, ELF3, CEBPD and STAT4. We found that STAT4 silencing strongly affected IL-6 production in fibroblasts derived from multiple tissues including synovium, skin and lung. By chromatin immunoprecipitation, we have shown that only wild type STAT4, but not the phosphorylation defective mutant, was recruited to the IL6 promoter following stimulation, suggesting that STAT4 is directly involved in IL-6 regulation in fibroblasts.
In addition, we found that blocking of LIFR by siRNA or mAb markedly reduced IL-6 production. Further, LIFR silencing affected IL-6 expression in several fibroblast lines derived from skin or synovium. Of the possible LIFR ligands, only LIF is induced rapidly following stimulation of fibroblasts, and its expression exhibits a pattern similar to IL-6 and STAT4. Blocking LIF by either siRNA or blocking antibody was effective in reducing IL-6 production. To establish the physical and functional connection between STAT4, LIFR and LIFR’s co-receptor gp130, we performed immunoprecipitation and revealed that both endogenously expressed STAT4 and gp130 could be detected in the LIFR pull down fraction. This suggests that STAT4, LIFR and gp130 interact with one another. In human vascular smooth muscle and endothelial cells, STAT4 has been shown to be phosphorylated via activation of JAK1 and TYK2 (Dumler et al., 1999; Dumler et al., 1998). Here in fibroblasts, we have shown that silencing of LIFR, JAK1 or TYK2 resulted in a reduction in STAT4 phosphorylation.
We found that silencing of STAT4 or LIFR resulted in reduction of IL-6 following stimulation by inflammatory factors besides TNF and IL-17, including IL-1β, TNF as well as Toll like receptor signaling. Moreover, besides IL-6, fibroblasts produced IL-8, G-CSF, IL-33, IL-11, IL-1α and IL-1β following TNF and IL-17 stimulation; and the production of these cytokines and chemokines is markedly reduced when STAT4 or LIFR expression was silenced by siRNA. Together, these data suggest that LIF, LIFR and STAT4 are important regulators not only of IL-6, but of a module of the major inflammatory mediators in inflammatory reactions involving fibroblasts.
Based on a number of human genetic studies, a common haplotype of the STAT4 gene locus has been shown to be associated with susceptibility to rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), and primary Sjögren’s syndrome leading to suggestions that STAT4 is a pan autoimmune factor (Korman et al., 2008a; Remmers et al., 2007). In the case of RA, several single nucleotide polymorphisms in the large third intron of STAT4 appear to be associated with the risk of developing the disease. Although a variant haplotype of STAT4 and possibly multiple polymorphisms found within it are important in predisposing toward RA and SLE, the questions of how and in which cell types STAT4 acts are not yet known. CD4+ T cells seem to be one likely cellular candidate as STAT4 is known to help promote these cells to differentiate into inflammatory subsets, T helper-1 (Th1) cells via IL-12R and T helper-17 (Th17) cells via IL-23R. Howerver, our work on STAT4 as an important transcription factor for fibroblast-mediated inflammation together with previous studies that implicate the role of fibroblasts in RA (Bottini and Firestein, 2013; Lee et al., 2007; Lefevre et al., 2015) suggest that dysregulation of STAT4 in fibroblasts also may be an important contributor to the risk of disease or disease progression.
LIF is a pleiotropic cytokine of the IL-6 family, and its concentrations in the synovial fluids of RA patients are elevated and correlate with the concentrations of IL-1, IL-8 and IL-6 (Enomoto et al., 2003). In a murine model of arthritis, Lif deficient mice were shown to have a reduction in clinical arthritis severity compared to wild-type disease controls (Upadhyay et al., 2012). Although LIF has been reported to be produced by fibroblasts (Suman et al., 2013), its autocrine role on IL-6 regulation following inflammatory stimulation was not previously known. In addition, other autocrine signaling involving IL-6 – IL-6R and STAT3 circuit has been described for fibroblasts (Ogura et al., 2008). However, IL-6, IL-6R and STAT3 are also commonly expressed by some leukocytes. On the contrary, as LIFR appears to be selectively expressed in fibroblasts and not many leukocytes, this suggests that the autocrine signaling involving LIF, LIFR and STAT4 is rather unique, and may help explain the role of fibroblasts as key drivers in the sustained production of many inflammatory cytokines in RA and other diseases.
Fibroblasts and other mesenchymal cells play a crucial role in regulating and mediating inflammation in lymph nodes, tumors and inflammatory autoimmune diseases. Here, we identify the autocrine LIF, LIFR, STAT4 circuit as a critical driver of IL-6 and a group of other inflammatory mediators that recruit or activate leukocytes and as well as other tissue cells. Our findings underscore how differently IL-6 and other inflammatory factors are regulated in mesenchymal cell lineages. In order to sustain transcription of inflammatory genes, a set of proteins not previously known to work together, form the essential components of a cell-autonomous signaling pathway. Defining stromal cell biology will be important to identify relevant therapeutic targets as well as to understand how drugs may act differently on stromal cells compared to leukocytes.
EXPERIMENTAL PROCEDURES
Cell Lines and Reagents
Human synovial fibroblasts were isolated as previously described (Kiener et al., 2006) from tissues discarded after synovectomy or joint replacement surgery. Each line was derived from a unique donor and used experimentally between passages 5 and 8. Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Gemini), 2 mM L-glutamine, 50 μM 2-mercaptoethanol, antibiotics (penicillin and streptomycin), and essential and nonessential amino acids (Life Technologies). Human skin and lung fibroblasts were purchased from Lonza and cultured using the same media. Freshly isolated synovial fibroblasts, T cells, B cells and monocytes were prepared by digesting pieces of tissues with 4 mg/ml collagenase type 4 (Worthington), 0.8 mg/ml DispaseII (Roche), and 0.2 mg/ml DNase I (Roche) in DMEM at 37°C for 1 hour, followed by sorting cells enriched for CD45−, CD31−, CD235a−, Pdpn+ surface proteins as fibroblasts (Fibro); for CD45+, CD14+ as monocytes (Monos); for CD45+, CD3+, CD4+ as T cells; for CD45+, CD4−, CD19+ as B cells using the FACSAria Fusion (BD). The following antibodies were used: anti-STAT4, anti-pSTAT4-Y693, anti-STAT3, anti-JunB, anti-p52 (Cell Signaling Technology); anti-p65, anti-LIFR, anti-gp130, anti-C/EBPβ (Santa-Cruz); anti-LIFR 1C7 and 12D3 (MBL International); anti-LIF (R&D Systems); anti-β-actin, anti-α-tubulin, anti-FLAG (Sigma); anti-CD45, anti-CD4, anti-CD14, anti-CD19, anti-CD31 (BioLegend); anti-CD235a (Beckman Coulter); anti-Pdpn (eBioscience); anti-CD3 (BD). Other reagents were purchased from the following vendors: IL-6, LIF, IL-8, G-CSF, IL-33, IL-11, IL-1α, and IL-1β ELISA kits, TNF, IL-1β, IL-17 (R&D Systems); LPS (Peprotech); anti-FLAG resins (Sigma). All siRNAs (Silencer Select) were purchased from Life Technologies. All JAK inhibitors were purchased from SelleckChem. qPCR and ChIP primers were purchased from Integrated DNA Technologies and their sequences are listed in Supplemental Table 1. All human sample research was approved by the Brigham and Women’s Hospital Institutional Review Board.
Gene Expression Profiling
Fibroblasts (RA-32, RA-449, OA-6, OA-502) were plated on day 1 at 50,000 cells per well in 24-well plates in 10% FBS containing media. Cells were serum-starved on day 2 by changing to 1% FBS-containing media. Cells were either left unstimulated or stimulated with TNF (1 ng/mL) or TNF (1 ng/mL) + IL-17 (1 ng/mL) for approximately 20 hours on day 3 before samples were collected in TRizol reagent (Life Technologies) and RNA was extracted using the RNeasy Mini Kit (Qiagen). Expression profiling was performed using the Affymetrix HG U133 Plus version 2 arrays. Expression values were normalized using the Robust Multi-array Average (RMA) method. The microarray data used in this publication have been deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GSE93720.
Statistical Analysis
Error bars displayed throughout the manuscript represent standard deviation (s.d.) or standard error of the mean (s.e.m) and were calculated from triplicate technical or triplicate biological replicates as described in figure legends. Data shown are representative of 2 to 3 independent experiments including blots and gels. Statistical significance was determined using unpaired Student’s t-tests; ***p < 0.001. For multiple testing, Bonferroni correction was applied.
Details on Cell Stimulation and Antibody Blocking Assays, siRNA Silencing, Plasmid Constructs, Generation of Cells Expressing STAT4 Protein, Immunoprecipitation, Quantitative Real-Time PCR, Chromatin Immunoprecipitation and Western Blotting can be found in Extended Experimental Procedures.
Supplementary Material
Highlights.
- During inflammation, human fibroblasts upregulate LIF and STAT4
- LIF acts in an autocrine manner via LIF receptor to promote STAT4 activation
- Activated STAT4 together with NF-κB/p65-p52 and C/EBPβ enhances IL-6 transcription
- LIFR/STAT4 circuit also regulates IL-8, G-CSF, IL-33, IL-11, IL-1α and IL-1β
Acknowledgments
This work was supported by grants from the NIH (R01-AR063709). E.H.N. was supported by the Rheumatology Research Foundation New Investigator Award and NIH (K08-AR063696). We thank Dr. Brandon Earp, Dr. Philip Blazar, Dr. John Wright, Dr. Barry Simmons, and Dr. Peter Nigrovic for human and mouse specimen; Dr. Sook Kyung Chang and Dr. I-Cheng Ho for feedback and discussion; and the BWH Human Immunology Center Flow Cytometry Core for cell sorting assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
H. N. N., E. H. N., and M.B.B. conceived and designed the experiments. H. N. N., E. H. N., F. M., C. H., K. S. W., and G. F. M. W. performed the experiments. H. N. N. wrote and M.B.B. and E. H. N. edited the manuscript. M.B.B supervised the study.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Extended Experimental Procedures, six figures and one table.
References
- Agarwal S, Misra R, Aggarwal A. Interleukin 17 levels are increased in juvenile idiopathic arthritis synovial fluid and induce synovial fibroblasts to produce proinflammatory cytokines and matrix metalloproteinases. The Journal of rheumatology. 2008;35:515–519. [PubMed] [Google Scholar]
- Bacon CM, Petricoin EF, 3rd, Ortaldo JR, Rees RC, Larner AC, Johnston JA, O’Shea JJ. Interleukin 12 induces tyrosine phosphorylation and activation of STAT4 in human lymphocytes. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:7307–7311. doi: 10.1073/pnas.92.16.7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer S, Kerr BJ, Patterson PH. The neuropoietic cytokine family in development, plasticity, disease and injury. Nature reviews Neuroscience. 2007;8:221–232. doi: 10.1038/nrn2054. [DOI] [PubMed] [Google Scholar]
- Bernardo ME, Fibbe WE. Mesenchymal stromal cells: sensors and switchers of inflammation. Cell stem cell. 2013;13:392–402. doi: 10.1016/j.stem.2013.09.006. [DOI] [PubMed] [Google Scholar]
- Bottini N, Firestein GS. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nature reviews Rheumatology. 2013;9:24–33. doi: 10.1038/nrrheum.2012.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SS, Bacon CM, Sudarshan C, Rees RC, Finbloom D, Pine R, O’Shea JJ. Activation of STAT4 by IL-12 and IFN-alpha: evidence for the involvement of ligand-induced tyrosine and serine phosphorylation. J Immunol. 1996;157:4781–4789. [PubMed] [Google Scholar]
- Collison LW, Delgoffe GM, Guy CS, Vignali KM, Chaturvedi V, Fairweather D, Satoskar AR, Garcia KC, Hunter CA, Drake CG, et al. The composition and signaling of the IL-35 receptor are unconventional. Nature immunology. 2012;13:290–299. doi: 10.1038/ni.2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donlin LT, Jayatilleke A, Giannopoulou EG, Kalliolias GD, Ivashkiv LB. Modulation of TNF-induced macrophage polarization by synovial fibroblasts. J Immunol. 2014;193:2373–2383. doi: 10.4049/jimmunol.1400486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumler I, Kopmann A, Wagner K, Mayboroda OA, Jerke U, Dietz R, Haller H, Gulba DC. Urokinase induces activation and formation of Stat4 and Stat1-Stat2 complexes in human vascular smooth muscle cells. The Journal of biological chemistry. 1999;274:24059–24065. doi: 10.1074/jbc.274.34.24059. [DOI] [PubMed] [Google Scholar]
- Dumler I, Weis A, Mayboroda OA, Maasch C, Jerke U, Haller H, Gulba DC. The Jak/Stat pathway and urokinase receptor signaling in human aortic vascular smooth muscle cells. The Journal of biological chemistry. 1998;273:315–321. doi: 10.1074/jbc.273.1.315. [DOI] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic acids research. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto H, Saito S, Yabe H, Toyama Y, Tomatu T. The levels of leukemia inhibitory factor in synovial tissues of patients with rheumatoid arthritis: inflammation and other proinflammatory cytokines. Modern rheumatology/the Japan Rheumatism Association. 2003;13:121–128. doi: 10.3109/s10165-002-0210-9. [DOI] [PubMed] [Google Scholar]
- Fishman D, Faulds G, Jeffery R, Mohamed-Ali V, Yudkin JS, Humphries S, Woo P. The effect of novel polymorphisms in the interleukin-6 (IL-6) gene on IL-6 transcription and plasma IL-6 levels, and an association with systemic-onset juvenile chronic arthritis. The Journal of clinical investigation. 1998;102:1369–1376. doi: 10.1172/JCI2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher AL, Lukacs-Kornek V, Reynoso ED, Pinner SE, Bellemare-Pelletier A, Curry MS, Collier AR, Boyd RL, Turley SJ. Lymph node fibroblastic reticular cells directly present peripheral tissue antigen under steady-state and inflammatory conditions. The Journal of experimental medicine. 2010;207:689–697. doi: 10.1084/jem.20092642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher AL, Malhotra D, Turley SJ. Lymph node stroma broaden the peripheral tolerance paradigm. Trends in immunology. 2011;32:12–18. doi: 10.1016/j.it.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox DA, Gizinski A, Morgan R, Lundy SK. Cell-cell interactions in rheumatoid arthritis synovium. Rheumatic diseases clinics of North America. 2010;36:311–323. doi: 10.1016/j.rdc.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galon J, Sudarshan C, Ito S, Finbloom D, O’Shea JJ. IL-12 induces IFN regulating factor-1 (IRF-1) gene expression in human NK and T cells. J Immunol. 1999;162:7256–7262. [PubMed] [Google Scholar]
- Guerne PA, Zuraw BL, Vaughan JH, Carson DA, Lotz M. Synovium as a source of interleukin 6 in vitro. Contribution to local and systemic manifestations of arthritis. The Journal of clinical investigation. 1989;83:585–592. doi: 10.1172/JCI113921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano T, Matsuda T, Turner M, Miyasaka N, Buchan G, Tang B, Sato K, Shimizu M, Maini R, Feldmann M, et al. Excessive production of interleukin 6/B cell stimulatory factor-2 in rheumatoid arthritis. European journal of immunology. 1988;18:1797–1801. doi: 10.1002/eji.1830181122. [DOI] [PubMed] [Google Scholar]
- Hong DS, Angelo LS, Kurzrock R. Interleukin-6 and its receptor in cancer: implications for translational therapeutics. Cancer. 2007;110:1911–1928. doi: 10.1002/cncr.22999. [DOI] [PubMed] [Google Scholar]
- Houssiau FA, Devogelaer JP, Van Damme J, de Deuxchaisnes CN, Van Snick J. Interleukin-6 in synovial fluid and serum of patients with rheumatoid arthritis and other inflammatory arthritides. Arthritis and rheumatism. 1988;31:784–788. doi: 10.1002/art.1780310614. [DOI] [PubMed] [Google Scholar]
- Kaplan MH. STAT4: a critical regulator of inflammation in vivo. Immunologic research. 2005;31:231–242. doi: 10.1385/IR:31:3:231. [DOI] [PubMed] [Google Scholar]
- Katakai T, Hara T, Sugai M, Gonda H, Shimizu A. Lymph node fibroblastic reticular cells construct the stromal reticulum via contact with lymphocytes. The Journal of experimental medicine. 2004;200:783–795. doi: 10.1084/jem.20040254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiener HP, Lee DM, Agarwal SK, Brenner MB. Cadherin-11 induces rheumatoid arthritis fibroblast-like synoviocytes to form lining layers in vitro. The American journal of pathology. 2006;168:1486–1499. doi: 10.2353/ajpath.2006.050999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto T. Interleukin-6: from basic science to medicine–40 years in immunology. Annual review of immunology. 2005;23:1–21. doi: 10.1146/annurev.immunol.23.021704.115806. [DOI] [PubMed] [Google Scholar]
- Korman BD, Alba MI, Le JM, Alevizos I, Smith JA, Nikolov NP, Kastner DL, Remmers EF, Illei GG. Variant form of STAT4 is associated with primary Sjogren’s syndrome. Genes and immunity. 2008a;9:267–270. doi: 10.1038/gene.2008.1. [DOI] [PubMed] [Google Scholar]
- Korman BD, Kastner DL, Gregersen PK, Remmers EF. STAT4: genetics, mechanisms, and implications for autoimmunity. Current allergy and asthma reports. 2008b;8:398–403. doi: 10.1007/s11882-008-0077-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D. Organ-specific disease provoked by systemic autoimmunity. Cell. 1996;87:811–822. doi: 10.1016/s0092-8674(00)81989-3. [DOI] [PubMed] [Google Scholar]
- Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, Gopinathan A, Tuveson DA, Fearon DT. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science. 2010;330:827–830. doi: 10.1126/science.1195300. [DOI] [PubMed] [Google Scholar]
- Kushwah R, Oliver JR, Wu J, Chang Z, Hu J. Elf3 regulates allergic airway inflammation by controlling dendritic cell-driven T cell differentiation. J Immunol. 2011;187:4639–4653. doi: 10.4049/jimmunol.1101967. [DOI] [PubMed] [Google Scholar]
- Lally F, Smith E, Filer A, Stone MA, Shaw JS, Nash GB, Buckley CD, Rainger GE. A novel mechanism of neutrophil recruitment in a coculture model of the rheumatoid synovium. Arthritis and rheumatism. 2005;52:3460–3469. doi: 10.1002/art.21394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DM, Kiener HP, Agarwal SK, Noss EH, Watts GF, Chisaka O, Takeichi M, Brenner MB. Cadherin-11 in synovial lining formation and pathology in arthritis. Science. 2007;315:1006–1010. doi: 10.1126/science.1137306. [DOI] [PubMed] [Google Scholar]
- Lee YH, Bae SC, Choi SJ, Ji JD, Song GG. The association between interleukin-6 polymorphisms and rheumatoid arthritis: a meta-analysis. Inflammation research : official journal of the European Histamine Research Society [et al] 2012;61:665–671. doi: 10.1007/s00011-012-0459-1. [DOI] [PubMed] [Google Scholar]
- Lefevre S, Meier FM, Neumann E, Muller-Ladner U. Role of synovial fibroblasts in rheumatoid arthritis. Current pharmaceutical design. 2015;21:130–141. doi: 10.2174/1381612820666140825122036. [DOI] [PubMed] [Google Scholar]
- Li F, Xu J, Zheng J, Sokolove J, Zhu K, Zhang Y, Sun H, Evangelou E, Pan Z. Association between interleukin-6 gene polymorphisms and rheumatoid arthritis in Chinese Han population: a case-control study and a meta-analysis. Scientific reports. 2014;4:5714. doi: 10.1038/srep05714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link A, Vogt TK, Favre S, Britschgi MR, Acha-Orbea H, Hinz B, Cyster JG, Luther SA. Fibroblastic reticular cells in lymph nodes regulate the homeostasis of naive T cells. Nature immunology. 2007;8:1255–1265. doi: 10.1038/ni1513. [DOI] [PubMed] [Google Scholar]
- Madhok R, Crilly A, Watson J, Capell HA. Serum interleukin 6 levels in rheumatoid arthritis: correlations with clinical and laboratory indices of disease activity. Annals of the rheumatic diseases. 1993;52:232–234. doi: 10.1136/ard.52.3.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nature reviews Immunology. 2007;7:429–442. doi: 10.1038/nri2094. [DOI] [PubMed] [Google Scholar]
- Noss EH, Nguyen HN, Chang SK, Watts GF, Brenner MB. Genetic polymorphism directs IL-6 expression in fibroblasts but not selected other cell types. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:14948–14953. doi: 10.1073/pnas.1520861112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura H, Murakami M, Okuyama Y, Tsuruoka M, Kitabayashi C, Kanamoto M, Nishihara M, Iwakura Y, Hirano T. Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity. 2008;29:628–636. doi: 10.1016/j.immuni.2008.07.018. [DOI] [PubMed] [Google Scholar]
- Okamoto H, Yamamura M, Morita Y, Harada S, Makino H, Ota Z. The synovial expression and serum levels of interleukin-6, interleukin-11, leukemia inhibitory factor, and oncostatin M in rheumatoid arthritis. Arthritis and rheumatism. 1997;40:1096–1105. doi: 10.1002/art.1780400614. [DOI] [PubMed] [Google Scholar]
- Pitard V, Taupin JL, Miossec V, Blanchard F, Cransac M, Jollet I, Vernallis A, Hudson K, Godard A, Jacques Y, et al. Production and characterization of monoclonal antibodies against the leukemia inhibitory factor low affinity receptor, gp190. Journal of immunological methods. 1997;205:177–190. doi: 10.1016/s0022-1759(97)00074-4. [DOI] [PubMed] [Google Scholar]
- Remmers EF, Plenge RM, Lee AT, Graham RR, Hom G, Behrens TW, de Bakker PI, Le JM, Lee HS, Batliwalla F, et al. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. The New England journal of medicine. 2007;357:977–986. doi: 10.1056/NEJMoa073003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruddy MJ, Wong GC, Liu XK, Yamamoto H, Kasayama S, Kirkwood KL, Gaffen SL. Functional cooperation between interleukin-17 and tumor necrosis factor-alpha is mediated by CCAAT/enhancer-binding protein family members. The Journal of biological chemistry. 2004;279:2559–2567. doi: 10.1074/jbc.M308809200. [DOI] [PubMed] [Google Scholar]
- Schindler CW. Series introduction. JAK-STAT signaling in human disease. The Journal of clinical investigation. 2002;109:1133–1137. doi: 10.1172/JCI15644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suman P, Malhotra SS, Gupta SK. LIF-STAT signaling and trophoblast biology. Jak-Stat. 2013;2:e25155. doi: 10.4161/jkst.25155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Narazaki M, Ogata A, Kishimoto T. A new era for the treatment of inflammatory autoimmune diseases by interleukin-6 blockade strategy. Seminars in immunology. 2014;26:88–96. doi: 10.1016/j.smim.2014.01.009. [DOI] [PubMed] [Google Scholar]
- Taupin JL, Legembre P, Bitard J, Daburon S, Pitard V, Blanchard F, Duplomb L, Godard A, Jacques Y, Moreau JF. Identification of agonistic and antagonistic antibodies against gp190, the leukemia inhibitory factor receptor, reveals distinct roles for its two cytokine-binding domains. The Journal of biological chemistry. 2001;276:47975–47981. doi: 10.1074/jbc.M105476200. [DOI] [PubMed] [Google Scholar]
- Tyler DR, Persky ME, Matthews LA, Chan S, Farrar JD. Pre-assembly of STAT4 with the human IFN-alpha/beta receptor-2 subunit is mediated by the STAT4 N-domain. Molecular immunology. 2007;44:1864–1872. doi: 10.1016/j.molimm.2006.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhyay A, Senyschyn D, Santos L, Gu R, Carroll GJ, Jazayeri JA. K/BxN serum transfer arthritis is delayed and less severe in leukaemia inhibitory factor (LIF)-deficient mice. Clinical and experimental immunology. 2012;169:71–78. doi: 10.1111/j.1365-2249.2012.04601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villuendas G, San Millan JL, Sancho J, Escobar-Morreale HF. The -597 G–>A and -174 G–>C polymorphisms in the promoter of the IL-6 gene are associated with hyperandrogenism. The Journal of clinical endocrinology and metabolism. 2002;87:1134–1141. doi: 10.1210/jcem.87.3.8309. [DOI] [PubMed] [Google Scholar]
- Waldner MJ, Neurath MF. Master regulator of intestinal disease: IL-6 in chronic inflammation and cancer development. Seminars in immunology. 2014;26:75–79. doi: 10.1016/j.smim.2013.12.003. [DOI] [PubMed] [Google Scholar]
- Wolf J, Rose-John S, Garbers C. Interleukin-6 and its receptors: a highly regulated and dynamic system. Cytokine. 2014;70:11–20. doi: 10.1016/j.cyto.2014.05.024. [DOI] [PubMed] [Google Scholar]
- Wurster AL, Tanaka T, Grusby MJ. The biology of Stat4 and Stat6. Oncogene. 2000;19:2577–2584. doi: 10.1038/sj.onc.1203485. [DOI] [PubMed] [Google Scholar]
- Zrioual S, Ecochard R, Tournadre A, Lenief V, Cazalis MA, Miossec P. Genome-wide comparison between IL-17A- and IL-17F-induced effects in human rheumatoid arthritis synoviocytes. J Immunol. 2009;182:3112–3120. doi: 10.4049/jimmunol.0801967. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.