

SUMMARY
G protein-coupled receptors (GPCRs) mediate diverse signaling in part through interaction with arrestins, whose binding promotes receptor internalization and signaling through G protein-independent pathways. High-affinity arrestin binding requires receptor phosphorylation, often at the receptor’s C-terminal tail. Here we report an X-ray free electron laser (XFEL) crystal structure of the rhodopsin–arrestin complex, in which the phosphorylated C-terminus of rhodopsin forms an extended intermolecular β-sheet with the N-terminal β-strands of arrestin. Phosphorylation was detected at rhodopsin C-terminal tail residues T336 and S338. These two phospho-residues, together with E341, form an extensive network of electrostatic interactions with three positively charged pockets in arrestin in a mode that resembles binding of the phosphorylated vasopressin-2 receptor tail to β-arrestin-1. Based on these observations, we derived and validated a set of phosphorylation codes that serve as a common mechanism for phosphorylation-dependent recruitment of arrestins by GPCRs.
ETOC blurb
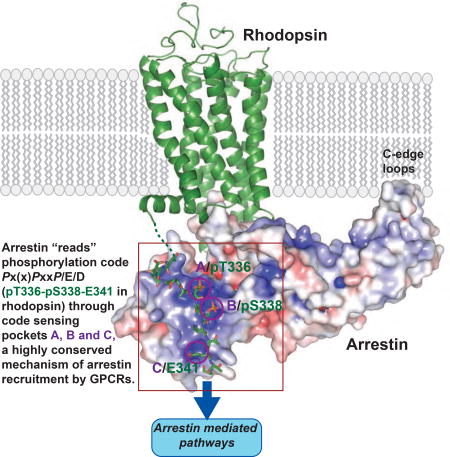
A crystal structure of a fully-engaged rhodopsin-arrestin complex identifies phosphorylation codes as a common mechanism of arrestin recruitment by GPCRs
Introduction
G protein-coupled receptors (GPCRs) are a family of membrane receptors that mediate transmembrane signaling through G proteins and arrestins (Pierce et al., 2002; Shukla et al., 2011). In response to stimuli, GPCRs activate G proteins to regulate the generation of second messengers, which then modulate downstream signaling effectors. To turn off the response, GPCR kinases (GRKs) are recruited to phosphorylate the GPCRs and prepare them for arrestin binding that blocks G proteinmediated signaling and directs the receptors to internalization (Pitcher et al., 1998). Upon binding to GPCRs, arrestins also serve as signaling scaffolds to initiate alternative pathways independent of G proteins. Thus, arrestin binding to receptors is a pivotal event that orchestrates the GPCR signaling network (Gurevich and Gurevich, 2013).
Arrestin recruitment by a GPCR is thought to be initiated by the interaction between inactive arrestin and phosphorylated receptor C-tail, which displaces arrestin’s C-tail and converts the inactive arrestin into a pre-activated state for high affinity GPCR binding (Gurevich and Gurevich, 2006b). Our understanding of how arrestins are recruited by activated GPCRs has been enhanced by recent structural studies (Kang et al., 2015; Kim et al., 2013; Shukla et al., 2013; Shukla et al., 2014). Electron microscopy analysis of β2 adrenergic receptor (β2AR) in complex with β-arrestin-1 revealed a dynamic equilibrium of two states: one is the fully-engaged state with arrestin bound to the central cavity of the receptor 7TM helix bundle; and the other is a partially-engaged state with arrestin bound only to the receptor C-terminal tail (Shukla et al., 2014). The crystal structure of phosphorylated vasopressin-2 receptor (V2R) tail bound to β-arrestin-1 provided a basis for arrestin activation induced by the phosphorylated receptor tail and the conformation of the activated arrestin, which closely resembles the activated state of visual arrestin (Kim et al., 2013; Shukla et al., 2013). A recent XFEL crystal structure of rhodopsin bound to activated visual arrestin revealed a near-atomic resolution interaction of arrestin with the receptor’s 7TM helix bundle (Kang et al., 2015). However, the C-terminal tail of rhodopsin was unresolved in that structure. Thus, how arrestin fully engages with a phosphorylated receptor remains an unanswered fundamental question in GPCR signaling. Here we report a structure of rhodopsin-arrestin complex with additional structural features including the phosphorylated rhodopsin C-terminal tail bound to the N-terminal domain of arrestin. The phosphorhodopsin-arrestin interface displays an extended intermolecular β-sheet together with an extensive network of electrostatic interactions formed between positive residues at the N-terminal domain of arrestin and the phosphorylated C-tail of rhodopsin. These structural features were extensively validated by biochemical and biophysical experiments and computer modeling to provide a basis for understanding phosphorylation-mediated arrestin recruitment by GPCRs.
Results and Discussion
Structure determination and the overall structure
The original XFEL crystal structure of rhodopsin bound to arrestin was solved by serial femtosecond crystallography (Kang et al., 2015; Zhou et al., 2016). Using the recently released 0.6.2 version of CrystFEL (White et al., 2016), we reprocessed diffraction images of 22,462 micro-crystals collected at the CXI beamline of the Linac Coherent Light Source at SLAC National Laboratory, which yielded 22% more reflections than those used for the original structure. The reprocessed data were of higher quality with better Pearson correlation coefficient (CC*) and overall signal-to-noise ratio (Tables S1–2). The resolution of the new dataset were improved from 3.8 Å to 3.6 Å along the a* and b* axes and from 3.5 Å to 3.0 Å along the c* axis. The crystals were pseudo-merohedrally twinned in P212121 space group with a twin fraction of 0.5 based on L-test analysis (Kang et al., 2015). The structure was refined with twin law (k, h, -l) to Rwork and Rfree of 23.4% and 27.2%, respectively, with excellent geometry (Table S3). The overall structure and the newly resolved elements are well supported by electron density maps (Figures S1).
The crystal structure contains four T4 lysozyme (T4L)-rhodopsin-arrestin fusion complexes as presented in our previous model (Kang et al., 2015), denoted as A, B, C and D, respectively (Figure 1). The four rhodopsin-arrestin complexes in each asymmetric unit adopt nearly identical structures with root-mean-square deviations (RMSDs) of Cα atoms among four complexes are between 0.5~0.7 Å, in agreement with the previous structure in the modelled regions (Kang et al., 2015). Importantly, we observed electron density for the membrane-touching loop of arrestin (C-edge loop) from residue 340 through 342 (Figure S1C). Molecular dynamics simulation indicated that C-edge loops of arrestin function as membrane anchors, whose engagement with membrane is required for GPCR binding by arrestins (Lally et al., 2017). We also observed electron density for poly N-acetyl-D-glucosamine at the N15 glycosylation site of each rhodopsin molecule (Figure S1D), and most of the rhodopsin C-terminal tail (residues 330–343) including two phosphate groups at T336 and S338 (Figures 1, 2 and S1E–F). These structural elements were missing in our previous model (Kang et al., 2015), indicating that the reprocessed data have improved the structure and revealed additional structural features that are important to rhodopsin-arrestin interaction.
Figure 1.

Crystal structure of rhodopsin-arrestin complex with rhodopsin C-terminal tail interacting with arrestin N-terminal domain. See also Figure S1.
(A–B) Two views of T4L-rhodopsin-arrestin fusion complex with rhodopsin in green and arrestin in brown (T4L not shown). (C–D) Two views of rhodopsin C-terminal tails of overlayed four rhodopsin-arrestin complexes in an asymmetric unit. Complex A is shown in green, B in magenta, C in cyan, and D in yellow.
Figure 2.

Interface between rhodopsin C-terminal tail and arrestin N-terminal domain. See also Figures S1–2 and Table S3.
(A) The interface between rhodopsin C-tail and arrestin N-terminal domain with the rhodopsin C-tail covered with a 2mFo-DFc density map contoured at 1 σ. Rhodopsin is in green, and arrestin in brown. (B) Interface residues between rhodopsin C-tail and arrestin N-terminal domain, with rhodopsin residues colored green and arrestin residues brown. (C) A schematic diagram of the interactions between rhodopsin C-tail residues (green) and arrestin residues (brown). Solid lines and arrows indicate hydrogen bonds and salt bridges, respectively. (D) Charge-distribution surface of the arrestin N-terminal domain in the rhodopsin-arrestin complex with an electrostatic scale from −3 to +3 eV corresponding to red to blue colors. Labeled are the rhodopsin C-tail residues involved in the charge interaction network.
The phosphorylated rhodopsin C-terminal tail-arrestin interface
The most important new feature of the structure is the rhodopsin C-terminal tail that serves as an interface for arrestin recruitment (Figure 2). While all four complexes in the structure are similar, we focus on molecule A because it has the best electron density for phosphorylated T336 and S338, which are critical for arrestin binding (Figures 1–2). Of the resolved rhodopsin C-tail, the C-terminal portion from K339 to T342 adopts a β-strand that forms an extended intermolecular β-sheet with β-strand I, from V12 to K16, of the N-terminal domain of arrestin (Figure 2). Residues N-terminal to the C-tail β-strand of rhodopsin, D330 through S338, form an extended stretch that interacts with arrestin and connects the β-strand to helix 8 through a flexible linker region (K325 through G329) that is disordered in the crystal structure. Of this region, residues D330 through A333 make a turn that is positioned alongside the turn between the β-strands I and II of arrestin, with its D330 and E332 electrostatically associated with R19, K167 and K168 of arrestin (Figure 2C). The conformation and position of residues D330-S335 are slightly different among the four complexes in the crystal structure, indicating the dynamic nature of this region. In contrast, the β-strand (K339-T342) of the rhodopsin C-tail adopts the same structure in all four complexes (Figures 1C–D), indicating its stability and its role as one of the anchor points for rhodopsin-arrestin interaction.
While the rhodopsin C-tail contains six serine or threonine residues, mass spectrometry analysis detected phosphate groups at S334, T336 and S338 in our protein samples (Figure S2). In our crystal structure, however, we observed the phosphate group at S338 in all four rhodopsin molecules and at T336 only in molecules A and B. The phosphate group at S334 was not observed in the structure, possibly due to a high disorder in this part of the C-terminal tail (Figure 2).
The interaction between rhodopsin and arrestin is primarily maintained by the formation of the intermolecular anti-parallel β-sheet between the rhodopsin C-tail β-strand and β-strand I of arrestin (Figures 1–2), and further stabilized by an extensive electrostatic interaction network between the phosphorylated T336 and S338 and the negatively charged residue E341 of rhodopsin, and three positively charged pockets on the arrestin surface denoted as A, B and C (Figure 2D). The three positively charged pockets are formed by three groups of basic residues: K16, R19 and R172 (pocket A), K16, R30 and K301 (pocket B), and K15 and K111 (pocket C), which accommodate phosphate groups at T336 and S338, and the negatively charged E341, respectively, of the rhodopsin C-tail (Figures 2B–D).
In addition, there is another electrostatic interaction interface between negatively charged rhodopsin residues D330, E332 and potentially phosphorylated S334, and a positively charged patch on arrestin formed by R19, K167 and K168 at the tips of the two β-sheets of the N-terminal domain of arrestin (Figures 2B and 2D). It is interesting to note that the total number of positive charges on this arrestin N-terminal surface is nine (Figure 2C), which is equivalent to eight or ten total negative charges of the phosphate groups and acidic amino acid residues at the interface (depending on the phosphorylation of S334, detected by mass spectrometry but not observed in our crystal structure), indicating an overall electrostatic balance at this rhodopsin-arrestin interface (Figure 2D).
Structure validation
To validate the structure of the C-terminal tail of rhodopsin in the complex, we performed Double Electron–Electron Resonance (DEER) experiments, which are widely used to determine the distances between spin-labeled residues in protein complexes (Altenbach et al., 2008; Kang et al., 2015). We determined the distances from rhodopsin C-tail residues 335, 337 and 342 to arrestin residues 106 or 107, which are located at arrestin helix I. Figure 3 shows the distances from residues 335 and 337 of rhodopsin to residue 107 of arrestin (Figures 3A–B), and the distances between residue 342 of rhodopsin, and residues 106 and 107 of arrestin (Figures 3C–D). These DEER distances are in agreement with those of the structural model, indicating the correct positioning of the C-terminal tail of rhodopsin in the crystal structure.
Figure 3.

Validation of the interface between rhodopsin C-tail and arrestin N-terminal domain by DEER spectroscopy.
(A) Interface between the rhodopsin C-tail and arrestin showing the intermolecular distances between pairs of R1 nitroxide spin-labeled side chains at arrestin position 107 and rhodopsin position 335 or 337. (B) Experimental distance distributions between the R1 nitroxide pairs shown in (A). (C) Interface between rhodopsin C-tail and arrestin showing the intermolecular distances between pairs of R1 nitroxide spin-labeled side chains at rhodopsin position 342 and arrestin position 106 or 107. (D) Experimental distance distributions between the R1 nitroxide pairs shown in (C).
We further validated the rhodopsin C-tail–arrestin interface by site-specific disulfide cross-linking through engineering cysteine pairs across the interface (Kang et al., 2015). A total of 13 cysteine mutants of arrestin together with seven cysteine mutants of rhodopsin were expressed. A total of 63 co-expression combinations were tested and monitored by SDS-PAGE followed by Western blotting, and the results are summarized in Figure S3A. Examples show that rhodopsin E332 strongly cross-linked with R19, K167 and K168 of arrestin (Figure 4A), as E332 faces toward these positively charged residues, while D331 of rhodopsin weakly cross-linked, because it faces away from the arrestin surface, with longer distances and unfavorable geometry to form crosslinking with these arrestin residues (Figure 4A). Rhodopsin residues A333, S334 and A335 cross-linked with only R19, and T336 crosslinked with both K16 and R19 of arrestin, indicating that all these residues are in close proximity to R19, while T336 is closer to K16 than A333, S334 and A335 (Figures 4B–C). Rhodopsin E341 cross-linked with K111 on helix I, but did not cross-link with residues on β-strand I of arrestin, probably due to unfavorable geometry (Figure 4C). Collectively these cross-linking data support the positioning of the rhodopsin C-tail in the crystal structure.
Figure 4.

Validation of the interface between rhodopsin C-tail and arrestin by disulfide cross-linking. Crystal structures showing interface residues (left), and Western blots showing disulfide cross-linking data (right). The black asterisk indicates arrestin, and the arrowhead the rhodopsin-arrestin crosslinking product. See also Figure S3.
(A) The disulfide cross-linking between D330 of rhodopsin and K167 of arrestin, D331 of rhodopsin and R19 of arrestin, and E332 of rhodopsin and R19, K167 and K168 of arrestin. (B) The disulfide cross-linking between A333, S334 and A335 of rhodopsin and R19 of arrestin. (C) The disulfide cross-linking between T336 of rhodopsin and K16 and R19 of arrestin, and E341 of rhodopsin and K111 of arrestin.
The interface revealed by the crystal structure was also confirmed by hydrogen-deuterium exchange mass spectrometry (HDX), which probes the dynamics of protein-protein interaction in solution (West et al., 2013). Compared to the free pre-activated arrestin, the binding of rhodopsin protects arrestin against hydrogen-deuterium exchange in several regions (Figure S3B). Notably, the arrestin regions protected from hydrogen-deuterium exchange include the N-terminal β-strands (residues 11 to 52, and 115 to 120) and helix I (residues 104 to 110) (Figure S3B). The protection of these structural elements in the arrestin N-domain could not be explained by our previous structure that lacks the rhodopsin C-tail (Kang et al., 2015), but it is consistent with the structure we report here, in which these arrestin regions are stabilized by binding to the C-terminal tail of rhodopsin (Figure S3C).
We further analyzed the conformational stability between rhodopsin C-terminal tail and arrestin N-terminal β-sheet by performing two independent, three microsecond-long all-atom molecular dynamics simulations. Throughout both simulations the charged interaction network connecting the phosphorylated T336 and S338, and the negatively charged E341 of rhodopsin with the positively charged arrestin residues comprising pockets A, B and C were well maintained with average interaction frequencies upwards of 80% for most crystallographic contacts (Figures 5A–B and S4A). Overall, the RMSDs of pT336, pS338 and E341 generally remained within 3.0 Å of their crystallographic positions, consistent with maintenance of the interwoven ionic network, while residues D330 and S334 located N-terminally to pT336 exhibit a number of metastable states whose RMSDs reached values upwards of 19 Å (Figure 5C). This differential stability of the C-tail was further characterized by the root mean square fluctuation (RMSF) of the C-tail alpha carbons around their average simulation positions. By defining an RMSF cut-off of 1.7 Å, both simulations suggest a key interaction range comprising C-tail residues A335 to E341 (Figures 5D–E). Subsequent comparison of average simulation B-factors calculated from RMSF to the XFEL room temperature structure B-factors exhibits a remarkable agreement of stability within the key interactions mediated by the C-tail residues A335 to E341 (Figure S4B).
Figure 5.

Molecular dynamics simulations suggest a key interface between rhodopsin C-tail and arrestin. See also Figure S5.
(A–B) Interaction frequencies of pT336, pS338 and E341 to key residues within arrestin’s A, B, and C phosphate binding pockets for simulation 1 (A) and simulation 2 (B). (C) Displacement of selected residues within the C-tail region away from their crystallographic position measured by RMSDs of their heavy atoms. RMSD traces were smoothed over a 10 ns window. Residues comprising the phosphorylation code region remain stable within 3 Å of their initial positions (pT336: 2.72 ± 0.5 Å, pS338: 2.01 ± 0.78 Å, E341: 2.98 ± 0.66 Å), while residues out of the code region (D330 and S334) exhibit a number of metastable states reaching values upwards of 19 Å away. (D) Root mean square fluctuation (RMSF) of C-tail alpha carbons around their average position between 1 and 3 µs suggests a key interaction region comprising residues 335 to 341 in agreement with previous RMSD observations. (E) Average RMSF values for each residue converted into B-factor values (B-factor=[8*π2]/3*(RMSF)2) and mapped onto the initial simulation model for C-tail simulations.
A common mechanism for phosphorylation-mediated recruitment of arrestins by GPCRs
Phosphorylation-dependent recruitment of arrestins is a common feature for GPCRs (Lefkowitz and Shenoy, 2005; Shukla et al., 2011). The structure of the phosphorylated rhodopsin-arrestin complex, together with the previous structure of β-arrestin-1 in complex with phosphorylated V2R C-tail peptide (Shukla et al., 2013), provides mechanistic insights into phosphorylation-dependent arrestin recruitment by GPCRs. Superposition of the V2R-β-arrestin-1 structure (PDB code: 4JQI) with our rhodopsin-arrestin complex structure reveals a noticeable similarity between the β-strand of V2R (residues 360–365) and the C-terminal β-strand (residues 338–343) of rhodopsin (Figures 6A–B). The phosphorylated residues S357 and T360 of the V2R C-tail are nearly superposable with the phosphorylated T336 and S338 of rhodopsin C-tail (Figure 6B). In addition, the ionic interaction between the phosphorylated S363 of V2R and K107 of β-arrestin-1 is comparable to the interaction of rhodopsin E341 with K111 of visual arrestin (Figures 6B and S5A). The V2R residues N-terminal to the phosphorylated S357, however, are located differently compared to those proceeding T336 in rhodopsin. The V2R residues N-terminal to phosphorylated S357 are positioned close to a location corresponding to the middle loop and the finger loop of β-arrestin-1 (Figures 6B and S5A). As revealed by electron microscopic studies of β2AR-V2R C-tail chimeric receptor in complex with β-arrestin-1, arrestin can bind either to the central cytoplasmic cavity of the receptor TM bundle in a fully-engaged state or to the receptor’s C-terminal tail in a partially tail-engaged state (Shukla et al., 2014). The position of those V2R residues would clash with the corresponding residues in the finger loop of visual arrestin in the fully-engaged state, thus the structure of β-arrestin-1 in complex with the phosphorylated V2R C-tail likely represents a tail-engaged state, whereas the structure of the phosphorylated rhodopsin–arrestin complex represents the fully-engaged state.
Figure 6.

Phosphorylation codes derived from the interfaces of the rhodopsin C-tail with visual arrestin and the V2R C-tail peptide with β-arrestin-1. See also Figures S5–7 and Table S4.
(A) A sequence alignment of rhodopsin C-tail with the V2R peptide based on the phospho-residues that bind to the positively charged pockets of arrestin. (B) An overlay of the rhodopsin C-tail (green) with the V2R C-tail (4JQI, magenta) binding at arrestin N-terminal surfaces. Only visual arrestin surface is shown. Blue indicates positive charges, and red negative charges at arrestin surfaces. (C) A model depiction of the conserved arrestin N-terminal surface “reading” the phosphorylation code of rhodopsin or V2R. (D) Mutations of phosphorylation code residues on rhodopsin C-tail (left panel), and the binding (normalized luminescence) of the mutant rhodopsin to visual arrestin determined by Tango assay (right panel). (E) Mutations that abolish the phosphorylation codes on the V2R C-tail of the β2AR-V2R C-tail chimera (left panel), and the binding (normalized luminescence) of the mutant chimeric receptors to β-arrestin-1 determined by Tango assay (right panel) in the presence or absence of 1 µM salmeterol xinafoate (SX). In (D) and (E), phosphorylation codes are highlighted in red boxes. The relative activity values obtained from three experiments are represented as mean ± s.d. (n=3, *P<0.05, **P<0.01, and ***P<0.001).
Despite the two seemingly different states represented by the structures of β-arrestin-1 in complex with the phosphorylated V2R C-tail peptide and the phosphorylated rhodopsin–visual arrestin complex, the mechanism of sensing receptor phosphorylation by the two arrestins is very similar. The positions of phosphorylated T336 and S338 and E341 of rhodopsin overlap well with phosphorylated S357, T360, and S363 of V2R, which interact with three corresponding positively charged pockets in arrestins denoted as A, B and C (Figures 6B and S5A). The positively charged residues of the pockets (K16, R19 and R172 of pocket A, K16, R30 and K301 of pocket B, and K15 and K111 of pocket C) that interact with those phospho-residues or negatively charged residue are highly conserved between those two arrestins (Figure S5B–C), which is also consistent with the previous notes that positively charged residues implicated in phosphate binding are highly conserved in arrestins from C. elegans to mammals (Gurevich and Gurevich, 2006a).
The three conserved positively charged pockets in arrestin N-domain would suggest that three phosphorylation sites in GPCRs (two phosphorylation sites plus E341 in rhodopsin) are needed to mediate high affinity interaction with arrestin. However, there are often more than three phosphorylation sites in a receptor C-terminal tail (six for human rhodopsin and eight for V2R), presenting a possibility of multiple combinations of three phospho-residues. The three positive pockets at the N-terminal domain of an activated arrestin (Figures 6B and S5C) would require specific phosphorylation patterns, i.e. the phosphate groups (including a glutamic acid in rhodopsin) on the C-tail of a GPCR should be separated by certain distances or by a specific number of residues (Figure 6B). In our rhodopsin–arrestin complex, the space between the first and the second phosphate groups on the C-tail (Space 1 in Figure 6C) is one residue, while the space between the second and the third phosphate groups (the third one is glutamic acid in this case) (Space 2 in Figure 6C) is two residues. The V2R C-tail–β-arrestin-1 complex, however, displays two-residue separations between both the first to the second (Space 1) and the second to the third phosphate groups (Space 2, Figures 6B–C). The phosphate groups bound to pockets A and B of the arrestin are located at a flexible loop region. Thus the space between those two phosphate groups can accommodate either one or two residues (Figures 6B–C). The space between pockets B and C, however, can only accommodate two residues because they are a part of a five-residue β-strand that forms the intermolecular β-sheet with arrestin (Figures 6B–C). Based on these space constraints, a phosphorylation code required for high-affinity arrestin binding is predicted to be PxPxxP/E/D (denoted as short code) or PxxPxxP/E/D (denoted as long code), in which P represents a phospho-serine or phospho-threonine, and x any amino acid residue, except proline in the second xx occurrence. The phosphorylation codes proposed here are consistent with the requirement for three receptor-attached phosphates for high-affinity arrestin binding in vivo (Mendez et al., 2000) and in vitro (Vishnivetskiy et al., 2007). Sequence alignment reveals that there are only two phosphorylation codes in both rhodopsin and V2R C-tails (Figure 6C).
To confirm our hypothesis that a full phosphorylation code is critical for a GPCR to bind to an arrestin, we generated multiple mutations T336A/S338A/T340A and T336A/S338A/T343A in rhodopsin C-terminal tail to test their effect on arrestin recruitment using a cell-based Tango assay (Barnea et al., 2008; Kang et al., 2015). Those mutations significantly reduced recruitment efficiency of visual arrestin by rhodopsin (Figure 6D). Indeed, similar rhodopsin mutants with single or multiple serine/threonine residues substituted with alanine have been reported to prolong reproducible single-photon response, i.e. rhodopsin deactivation and recovery (Azevedo et al., 2015; Mendez et al., 2000). We further tested the effects of the phosphorylation code of V2R C-tail on arrestin binding by mutations of S357A/T360A/S363A (3A) or S357A/T360A/S362A/S363A/S364A (6A) that respectively disrupt either the first phosphorylation code SCTTAS or both codes SCTTAS and SCTTASS on the V2R C-tail of a β2AR-V2R chimeric receptor. Tango assay showed significant reduction of binding efficiency of the mutated chimeric receptors with β-arrestin-1 (Figure 6E). Full dose-response curve on key mutations suggested that the phosphorylation code mutations do not affect the shape of the dose-response curve (EC50), but affect the maximal activity (Emax) observed in Tango assay (Figure S7). Since the EC50 is mainly determined by the ligand binding capacity of the ligand binding pocket in the TM bundle of β2AR, the phosphorylation code mutations at the C-terminal tail is expected to only affect the Emax and the basal activity of arrestin recruitment (Figure S7). Taken together, these data support the critical role of phosphorylation codes in arrestin recruitment.
The residues that form the positive pockets at the N-terminal domain are overall conserved between β-arrestin-1 and visual arrestin with three exceptions (Figure S5B–C). The first exception is R19 in visual arrestin (mouse numbering), which is proline (P14) in β-arrestin-1 (Figure S5B). R19 in visual arrestin increases the overall positive charge density in this area, which may explain the presence of several additional negative residues in rhodopsin C-tail (D330, D331, E332 and pS334 in rhodopsin, which correspond to S350, L351, G352 and D354 in V2R) to balance the extra positive charge from R19 of the visual arrestin (Figure 6A). Consistently, R19 has been proposed to make visual arrestin the most dependent on receptor-attached phosphates for binding, as compared to other arrestin subtypes (Sutton et al., 2005). The second exception is V12 of visual arrestin, which corresponds to an arginine in both β-arrestins. This likely leads to a higher positive charge density at pocket C of β-arrestins (Figure S5), giving receptors with a phosphate group at the third position of their phosphorylation codes a preference for recruiting β-arrestins. Similarly, the third exception occurs at the position of S107 of mouse visual arrestin, which corresponds to an arginine in both β-arrestins, located on helix I and immediately next to pocket C (Figure S5B). The pocket C of visual arrestin is formed by only two positive residues (K15 and K111) and thus has less positive charge density, which can be balanced by a glutamic or an aspartic acid residue. Despite the overall structural similarity between visual arrestin and β-arrestins, these subtle differences in the surface charge distribution may contribute to the specificity of visual arrestin recruitment by rhodopsin and β-arrestin recruitment by other GPCRs such as V2R (Oakley et al., 2000).
Phosphorylation-mediated recruitment of β-arrestin-1 by β2AR
We analyzed the C-terminal sequences of several well-studied GPCRs for potential phosphorylation codes as listed in Table S4. All these receptors have at least one complete phosphorylation code except for β2AR, whose C-terminal tail does not contain any complete phosphorylation code but a series of overlapping partial codes (two matches out of three phosphate positions in the complete codes) (Figure 7A). It has been well documented that the C-terminal tail of β2AR has low binding affinity to arrestins, and construction of a chimeric receptor with V2R C-tail increases its arrestin-binding ability (Oakley et al., 2000; Shukla et al., 2014; Wisler et al., 2007). The low arrestin binding affinity by β2AR is consistent with our observation that this receptor has no full phosphorylation code on its C-tail.
Figure 7.

Mutations turning partial phosphorylation codes at the β2AR C-tail to full codes increase the binding affinity of the receptor to β-arrestin-1. See also Figure S7.
(A) Amino acid sequence of β2AR C-tail. Partial phosphorylation codes are highlighted with red lines (same as in panel (B) and (C)), and serine, threonine or glutamic acid residues that form the partial codes are colored in red. Two clusters of phosphorylation sites are noted. (B) Mutations that turn partial phosphorylation codes to full codes (in red box) or replace the partial code serine or threonine residues with alanine at the β2AR C-tail (left panel), and the binding affinity (normalized luminescence) of the mutant receptor to β-arrestin-1 determined by Tango assay in the presence or absence of 1 µM SX (right panel). (C) Mutations that turn partial phosphorylation codes to full codes (in red box) on the β2AR C-tail (left panel), and the binding affinity (normalized luminescence) of the mutant receptor to β-arrestin-1 determined by Tango assay in the presence or absence of 1 µM SX (right panel). The relative activity values obtained from three experiments are represented as mean ± s.d. (n=3, *P<0.05, **P<0.01, and ***P<0.001).
The partial codes of β2AR occur within two clusters (clusters 1 and 2) of densely distributed serine, threonine as well as glutamic acid or aspartic acid residues (Figure 7A). Most of those serine or threonine residues have been demonstrated to be phosphorylated by GRKs (Seibold et al., 2000), and residues S355, S356 and S364 of cluster 1 have been demonstrated to be important for GRK-mediated arrestin recruitment and desensitization (Gimenez et al., 2012; Krasel et al., 2008; Vaughan et al., 2006). In agreement with those observations, mutations that disrupt the partial codes in cluster 1 (S355A, S356A, T360A and E362A in Figure 7B) significantly reduced β-arrestin binding by the receptor. In contrast, the N359S mutation that turns partial code SNGNTGE in cluster 1 of the C-tail into a full code SNGSTGE enhanced arrestin recruitment by the mutated receptor in the presence or absence of a β2AR agonist, salmeterol xinafoate (SX). Similarly, mutations I399S and N405S that turn the partial codes TVPSDNI and NCSTNDS in cluster 2 into full codes TVPSDNS and SCSTNDS, respectively, increased the arrestin interaction with the mutant receptors (Figure 7C). Together, these data suggest that the partial codes in β2AR are important for its interaction with β-arrestin and mutations that turn the partial codes into full codes increase the binding efficiency of the mutated receptor to β-arrestin (Zindel et al., 2015).
Phosphorylation codes are found in all major GPCR subfamilies
We next sought to determine if phosphorylation codes were conserved across the major GPCR subfamilies as a unifying mechanism of arrestin recruitment. To scan protein sequences for potential phosphorylation codes we developed both a web GUI and Python-based command line tool termed PhosCoFinder (http://tools.vai.org/phoscofinder/). By analyzing the GPCR database with PhosCoFinder, we found that phosphorylation codes exist in all major GPCR subfamilies (Figures S6A–B). The percentage of the GPCR members that have either short, long or both phosphorylation codes is 52.3% for class A, 66.6% for class B, 59.1% for class C, and 45.5% for class F (Figure S6A). The majority of remaining receptors have partial codes, while only a small group of receptors have neither a complete nor a partial code in their C-terminal tails. In addition, we searched the GPCR proteome as annotated in UniProt. Among 825 annotated GPCRs, 436 were found to contain full or partial codes in their C-terminal tails (http://tools.vai.org/phoscofinder/downloads.php). Of the 389 receptors without codes in their tails, 308 (~79%) were found to belong to the olfactory receptor family, which has been shown to recruit arrestin through phosphorylation sites within their ICL3 just N-terminal to TM6 (Mashukova et al., 2006). Analysis of the remaining 81 receptors found that 48 contained full or partial codes within their ICL3 and comprised many members from the serotonin, dopamine, muscarinic, and α-adrenergic families, which have been shown to recruit β-arrestins in a ICL3-dependent manner (Smith and Rajagopal, 2016; Yang et al., 2017). In total, our phosphorylation code analysis accounts for 96% of the GPCR proteome.
Oakley et al. analyzed agonist-mediated arrestin binding to multiple GPCRs and classified those receptors as “class A” (α1- and β2-adrenergic, μ-opioid, endothelin ETA and dopamine D1A receptors) and “class B” (angiotensin II type 1A receptor, neurotensin receptor 1, vasopressin-2 receptor, thyrotropin-releasing hormone receptor, and substance P receptor) to indicate their distinct arrestin-binding properties (Oakley et al., 2000). In particular, “class A” GPCRs bind β-arrestin-2 more tightly than β-arrestin-1, and do not bind visual arrestin. In contrast, “class B” GPCRs bind both β-arrestins as well as visual arrestin (Lohse and Hoffmann, 2014). Our search for phosphorylation codes revealed that the classification of GPCRs based on their arrestin-binding properties strongly correlates with the occurrence of complete phosphorylation codes in their C-terminal tails (Figure S6C). All “class A” GPCRs possess none or at most one complete phosphorylation code, whereas all “class B” receptors possess at least two complete phosphorylation codes. Consistent with the experimental classification, analysis of the β3-adrenergic receptor that does not directly interact with β-arrestins failed to identify any full or partial phosphorylation code (Cao et al., 2000).
We further expanded our search for complete phosphorylation codes to non-GPCR membrane proteins that contain C-terminal cytoplasmic tails, and interestingly found numerous proteins that were experimentally shown to bind β-arrestins, including TRPV1 (Por et al., 2012; Por et al., 2013), insulin-like growth factor 1 receptor (IGF1R) (Girnita et al., 2005), transforming growth factor beta receptor type 3 (TGFβR3) (Chen et al., 2003), VE-cadherin (Hebda et al., 2013), LRP4 (Xiao et al., 2007), T-cell surface glycoprotein CD3 zeta chain (CD247) (Fernandez-Arenas et al., 2014), killer cell immunoglobulin-like receptor 2DL1 and 2DL4 (Yu et al., 2008), and the membrane protein associated cytoplasmic voltage-gated potassium channel subunit beta-1 (Xiao et al., 2007) (http://tools.vai.org/phoscofinder/downloads.php). The presence of the Px(x)PxxP motifs in these non-GPCR membrane proteins suggests a common mechanism of arrestin recruitment. The role of these motifs in arrestin binding has to be tested experimentally.
It has been proposed that a phosphorylation pattern in GPCR tails could serve as a “barcode” to mediate distinct signaling through interaction with different effectors (Liggett, 2011; Nobles et al., 2011; Ostermaier et al., 2014b; Reiter et al., 2012). For example, phosphorylation of the C-terminal partial code of β2AR would mediate its interaction with SNX27 and retromer, which is required for recycling the receptor to the plasma membrane (Yang et al., 2015). Whereas any phosphorylation pattern may relate to distinct GPCR functions, our structural and biochemical analyses indicate that only a full phosphorylation code with a pattern of Px(x)PxxP can facilitate high-affinity arrestin binding, which is likely required for specific direction of arrestin-dependent signaling for many GPCRs. Importantly, in case of GPCRs with multiple phosphorylation codes (the majority of the receptors, Figure S6), phosphorylation of a particular code would determine the “pose” of the arrestin in the complex, which is likely to dictate functional consequences of arrestin binding (Nobles et al., 2011; Yang et al., 2015).
Detailed analysis of phosphorylated rhodopsin-arrestin interface also provides important insight into dynamic assembly of the rhodopsin-arrestin complex. Including the C-terminal tail, the rhodopsinarrestin complex buries 2,420 Å2 of surface area in the interface (complex A), compared with 1,300 Å2 in the previous structure (Kang et al., 2015), illustrating the crucial role of the C-terminal tail in arrestin recruitment. Consistent with the C-tail exchange model (Kim et al., 2012; Ostermaier et al., 2014a), the binding of the phosphorylated rhodopsin C-tail displaces the arrestin C-terminal tail, releasing the intramolecular N-C lock, which leads to a domain twist that activates arrestin, allowing it to engage with the TM bundle of rhodopsin. The core rhodopsin-arrestin interface is, however, quite discontinuous with several small patches that comprise arrestin finger loop, middle loop and the back loop, and therefore is relatively weak (Kang et al., 2015). The discontinuous patchy interfaces between rhodopsin and arrestin may explain the dynamic and unstable nature of many GPCR-arrestin complexes. The dynamic nature of GPCR-arrestin complex may be required for the complexity of arrestin functions in GPCR signaling, including the perplexing capability of arrestin to mediate desensitization of some receptors but also to promote G protein signaling at other GPCRs (Thomsen et al., 2016; Wehbi et al., 2013).
Arrestin recruitment by GPCRs requires specific phosphorylation patterns on GPCR C-terminal tails, which has been one of the most puzzling problems due to the highly diverse sequences of the GPCR tails. The XFEL crystal structure presented here reveals a complete interface between phosphorylated rhodopsin and arrestin. The structure not only provides an integrated view for the assembly of the rhodopsin–arrestin complex but also uncovers a common mechanism of arrestin recruitment, in which a set of phosphorylation codes in the C-terminal tail of a GPCR is used for high-affinity arrestin binding by the receptor. The elucidation of the phosphorylation codes provides a guiding template to shed light on phosphorylation-dependent recruitment of arrestins by highly diverse GPCRs and possibly other membrane proteins.
STAR*METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Please contact the Lead Contact, H. Eric Xu (Eric.Xu@vai.org), with any requests regarding reagents used in this study.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture
HEK 293 cells were cultured in CDM4HEK293 medium with 4mM Glutamax-l and 20mM HEPES. HTL and AD293 cells were cultured in DMEM (Invitrogen) medium with 10% FBS (Invitrogen) in 37°C incubator with 5% CO2.
METHOD DETAILS
Protein Preparation, crystallization, and XFEL data collection
Human rhodopsin and mouse visual arrestin-1 were used in this study. The protein preparation, crystallization and XFEL data collection have been described in our previously published paper (Kang et al., 2015). The fusion protein expressed in HEK293 system was demonstrated by LC-MS/MS endogenously phosphorylated in the mammalian expression system (see the method section and Figure S2).
XFEL data reprocessing and structure solving
The XFEL data collected at LCLS in November 2014 were first processed using Cheetah (Barty et al., 2014) to identify images with crystal hits, as described in our previous paper (XFEL data were deposited in Coherent X-ray Imaging Data Bank http://dx.doi.org/10.11577/1241101, and are accessible via web service at http://www.cxidb.org with CXIDB ID 32) (Kang et al., 2015; Zhou et al., 2016). A total of 22,462 images with more than 40 diffraction peaks were reprocessed using the program “indexamajig” from the CrystFEL package (White et al., 2016) version 0.6.2 with indexing methods of MOSFLM (Battye et al., 2011), DirAx (Duisenberg, 1992), and XDS (Kabsch, 2010). 22,462 diffraction patterns were successfully indexed, from which 17,730 patterns were merged by the CrystFEL program “partialator”, using one cycle of scaling without partiality modelling, a minimum of 5 measurements per unique reflection, a saturation cutoff of 13,500 detector units, and merging reflections up to 1.5 nm−1 above the resolution limit conservatively estimated by indexamajig for each pattern. The dataset was anisotropically truncated using CrystFEL program “get_hkl” to 3.6 Å along both a* and b* axes, and to 3.0 Å along c* axis so that the Pearson correlation coefficient CC* is greater than 0.6 in all resolution shells. The truncated dataset contains 76,360 reflections, which represent 22% more reflections than the data used for the determination of the previously published rhodopsin-arrestin complex structure (Kang et al., 2015). The crystals were pseudo-merohedrally twinned in P212121 with a twin law of k, h, -l and a twin fraction of 0.5 based on L-test analysis described in our previous paper (Kang et al., 2015). The structural model (4JWZ), described in our previous publication (Kang et al., 2015), was used as the starting point for further refinement against the reprocessed data, and building of the C-terminal tail and the poly N-acetyl-D-glucosamine of the rhodopsin molecules, which were missing in our previous model. Building of the C-terminal tail of rhodopsin was based on simulated-annealing composite omit maps calculated with the Phenix program package (Adams et al., 2010). While the model was more completely built, the electron density map was improved so that more structural details became visible, including the phosphate groups at the rhodopsin C-terminal tail residues T336 and S338. The structural model was extensively refined using Phenix programs including deformable elastic network (DEN) refinement and rosetta refinement. The final model was refined with the twin law (k, h, -l) to an R factor of 23.3 % and free R factor of 27.2 % using automatic NCS restraints and 13 TLS groups, with 100% of the residues in favorable or allowed Ramachandran regions, an all-atom-clash score of 1.7 and the MolProbity score of 0.9 (Table S3).
Liquid-chromatography-tandem mass spectrometry (LC-MS/MS)
The protein sample (total 10 µg loaded in each well) and a protein ladder (Spectra Multicolor Broad Range Protein Ladder) were subjected in parallel to SDS-PAGE at 150 V (gel run full length). The gel was Coomassie stained for 1 hour at room temperature with shaking, followed by destaining in water overnight. The gel band corresponding to the protein of interest was cut, treated with 10 mM DTT followed by 50 mM iodoacetamide, and subjected to trypsin digestion overnight at 37°C. The peptide pool was acidified, desalted through a Zip-Tip C18 tip column and dried down. Sample was reconstructed in 100 µl of 0.1% formic acid and 13 µl were loaded into the system for liquid-chromatography-tandem mass spectrometry analysis (LC-MS/MS). The sample was analyzed by an Orbitrap Fusion™ Tribrid™ Mass Spectrometer (Thermo Fisher Scientific) coupled to an EASY-nLC 1000 system. Peptides were concentrated and desalted on an reverse phase (RP) pre-column (0.075 × 20 mm Acclaim PepMap 100 nano Viper, Thermo Fisher Scientific) and on-line eluted on an analytical RP column (0.075 × 250 mm Acclaim PepMap RLSC nano Viper, Thermo Fisher Scientific), operating at 300nl/min using the following gradient: 5% B for 5 min, 5–40% B in 60 min, 40–80% B in 10 sec, 80% B for 5 min, 80-5% B in 10 sec, and 5% B for 20 min (solvent A: 0.1% formic acid (v/v); solvent B: 0.1% formic acid (v/v) and 80% CH3CN (v/v) (Fisher Scientific)). The Orbitrap Fusion was operated in a data-dependent MS/MS mode using the 10 most intense precursors detected in a survey scan from 380 to 1,400 m/z performed at 120K resolution. Tandem MS was performed by HCD fragmentation with normalized collision energy (NCE) of 30.0%. Protein identification was carried out using Mascot and Sequest algorithms, allowing Oxidation (Met) and Phosphorylation (Ser, Thr, Tyr) as variable modifications. Other settings included Carbamidomethylation of Cys as fixed modification, three missed cleavages, and mass tolerance of 10 and 20 ppm for precursor and fragment ions, respectively. MS/MS raw files were searched against a homemade database containing the protein of interest, human keratins and porcine trypsin.
Hydrogen-Deuterium Exchange Mass Spectrometry (HDX)
HDX was carried out as described (Goswami et al., 2013; Kang et al., 2015). All stock solutions and dilutions were made using the 7TM HDX buffer (50 mM HEPES (pH 7.5), 150 mM NaCl, 2% (v/v) glycerol, 0.01% (m/v) CHS and 0.05% (m/v) DMNG in either H2O or in D2O for on-exchange). All HDX protein stock solutions were prepared at 15 µM in the 7TM HDX H2O buffer. On-exchange was carried out in triplicate for predetermined times (10, 30, 60, 900 and 3600 sec) at 4 °C by mixing 5 µL of stock protein solution with 20 µL of D2O on-exchange buffer. Exchange was quenched by adding 25 µL of quench solution (100 mM NaH2PO4, 0.02% DMNG, and 15 mM TCEP at pH 2.4) to the reaction. Digestion was performed in line with chromatography using an in-house packed pepsin column. Peptides were captured and desalted on a C8 trap. Peptides were then separated across a 5 µ 10×1 mm Betasil C8 column (Thermo Fisher Scientific) with a linear gradient of 12–40% acetonitrile in 0.3% formic acid over a short 5 min gradient to limit back exchange with the solvent. Mass spectra were acquired in the range of m/z 300–2000 at a resolution of 60,000 for 8 min in positive ion mode on a Q Exactive mass spectrometer (Thermo Fisher Scientific) equipped with an ESI source operated at a capillary temperature of 225 °C and spray voltage of 3.5 kV. The intensity weighted average m/z value (centroid) of each peptide’s isotopic envelope was calculated with the Workbench program (Pascal et al., 2012) and converted to % deuterium values. Back-exchange correction was based on an estimated 70% deuterium recovery and accounting for the known 80% deuterium content of the on-exchange buffer. The Workbench software used P-values lower than 0.05 for two consecutive time points to determine significance. Sequence coverage experiments for these proteins were carried out in the 7TM HDX buffer and LC system described above, but with a longer 60 min gradient. 75 pmol of protein were loaded onto the column. For sequencing, tandem mass spectra were obtained using data-dependent acquisition with 30 s dynamic exclusion, where the top five most abundant ions in each scan were selected and subjected to CID fragmentation. Each scan was the average of 3 microscans under normal scan mode in both MS and MS/MS. Peptides were identified by searching spectra against an in-house database using the mascot search engine as described previously (West et al., 2013).
DEER Spectroscopy of the rhodopsin-arrestin fusion complex
Preparations of the rhodopsin-(A335C)-arrestin-(S107C), rhodopsin-(V337C)-arrestin-(S107C), rhodopsin-(T342C)-arrestin-(E106C), and rhodopsin-(T342C)-arrestin-(S107C) fusion complexes were performed using a mammalian expression system as described previously (Kang et al., 2015). For DEER measurements, the phosphorhodopsin-arrestin fusion complexes were reacted with the sulfhydryl spin label S-(1-oxy-2,2,5,5,-tetramethylpyrroline-3-methyl)-methanethiosulfonate to generate R1 nitroxide side chains (Altenbach et al., 2008). Deuterated glycerol (20%) was added to the samples as a cryoprotectant. The spin labeled double mutants were loaded into quartz capillaries (1.5 mm ID and 1.8 mm OD) and flash frozen using a dry ice / ethanol bath. After freezing, they were loaded into an ER 5107D2 Q-band flexline resonator, and Q-band measurements were performed at 80 K on a Bruker Elexsys 580 spectrometer (University of Toronto) with a Super Q-FTu Bridge. A 32-ns π-pump pulse was applied to the low field peak of the nitroxide field swept spectrum, and the observer π/2 (16 ns) and π (32 ns) pulses were positioned 50 MHz (17.8 G) upfield, which corresponds to the nitroxide center line. Distance distributions were obtained from the raw DEER data using the LabVIEW (National Instruments) program “LongDistances” (developed by Christian Altenbach) that can be downloaded from http://www.biochemistry.ucla.edu/biochem/Faculty/Hubbell/. The modeled distances between nitroxide spin labels are based on the crystal structure of the phosphorhodopsin-arrestin complex. The R1 nitroxide side chains were modeled into the structure using common R1 rotamers (Fleissner et al., 2009). The X1-X3 dihedrals of 106R1, 335R1, and 342R1 were {m,m,m}, 107R1 was {m,m,p}, and 337R1 was {t,p,p}.
Disulfide cross-linking
Full-length arrestin with C-terminal Flag tag and full-length rhodopsin with C-terminal haemagglutinin (HA) tag were cloned into pcDNA6. Cysteine mutations were introduced into arrestin and rhodopsin in the DNA vectors. AD293 cells were applied to 50,000 cells per well in a 24-well plate, and were grown for one day before transfection with 100 ng rhodopsin constructs (pcDNA6-rho-3HA) plus 100 ng arrestin plasmid (pcDNA6-Arr-3Flag) using Lipofectamine 2000 (DNA/Lipofectamine 2000 ratio of 1:2) in each well. Transfected cells were grown for two days, and then treated with hydrogen peroxide (H2O2) that was freshly diluted in the cell culture medium to a final concentration of 1 mM. After 5 min incubation at room temperature with H2O2, the medium was removed and 100 µl of CelLytic M (Sigma C2978) were added to each well and the plate was shaken for 10 min. Cell lysates were transferred to eppendorf tubes and spun at 16,000g for 5 min at 4°C. The supernatants (10 µl) were mixed with an equal volume of 2×SDS loading buffer (without reducing agents) for 5 min at room temperature, and loaded onto a protein gel for Western blot analysis. Horseradish peroxidase-conjugated anti-Flag (Sigma M2) and anti-HA (Sigma) were used to probe for free and cross-linked arrestin and rhodopsin proteins.
Cell-based Tango assay for GPCR-arrestin interaction
Rhodopsin-arrestin interaction Tango assay was performed as described previously (Kang et al., 2015). Briefly, rhodopsin or β2AR coding sequence was fused to a tobacco etch virus (TEV) protease cleavage site (TEV site), and followed by the transcriptional activator tTA. The rhodopsin 4M was used in this experiment. To avoid the interference of the long tail of β2AR, the flexible region (341–388) of the receptor was deleted from the fusion. Visual arrestin or β-arrestin-1 was fused to TEV protease (β-arrestin1-TEV protease). HTL cells were a gift from G. Barnea and R. Axel (Brown University and Columbia University). Cell transfection, harvest and luciferase assay were performed as described (Kang et al., 2015). GPCR-arrestin binding activity was measured via tTA activated luciferase signal that was normalized to Renilla luciferase activity (Luc/Ren) used as transfection control.
Molecular dynamics
All-atom atmospheric simulations were performed using GROMACS5.0.6 (Abraham et al., 2015) in the isothermal–isobaric (NPT) ensemble with periodic boundary conditions and the CHARMM36 force field (Huang and MacKerell, 2013). Chain A of the rhodopsin arrestin complex reported in this manuscript was prepared by removing the N-acetylglucosamine and aligned for membrane insertion using the Orientations of Proteins in Membranes database (Lomize et al., 2006). Missing residues 324–330 in the C-tail were modelled and subjected to 500 rounds of very_slow loop refinement assed by DOPE scoring using Modeller9.13 (Eswar et al., 2006). The final simulation model was chosen based on favorable molpdf and DOPE energy scores as well as visual inspection of hydrophobic residue placements and potential hydrogen bonding partners. Optimal hydrogen bonding networks and side chain protonation states were determined at pH 7.0 by PROPKA included in Schrödinger Release 2016-1. The resulting complex was capped with neutral acetyl and methylamine groups and embedded into a palmitoyl-oleoyl-phosphatidylcholine (POPC) lipid bilayer solvated in a 115×115×135 Å box of TIP3P waters with 0.150 mM NaCl (neutralized by removing 6 chloride ions; approximately 156,000 atoms in total). Prior to production simulations, 50,000 steps of energy minimization were performed, followed by equilibration in the canonical (NVT) and NPT ensembles for 10 and 50 ns respectively with positional restraints (1000 kJ mol−1 nm−2) placed on backbone atoms. Temperature was maintained at 310 K using the v-rescale method with a coupling time of 0.1 ps and pressure was maintained at 1 bar using the Berendsen barostat with a coupling time (tp) of 1.0 ps and compressibility of 4.5 × 10−5 bar−1. Two independent 3 µs production simulations were performed for a combined 6 µs of simulation. To assess dynamics of the C-terminal tail, production simulations were aligned using the backbone atoms of the arrestin molecule taken from the last frame of NPT equilibration. Root mean squared deviations (RMSDs) were calculated for heavy atoms within select residues in the C-terminal tail. For root mean squared fluctuation (RMSF) calculations, the first 1 µs was omitted to exclude initial structure relaxation and only alpha carbons within the C-terminal tail were used. Simulation analysis was performed using MDTraj 1.7.2 (McGibbon et al., 2015) and VMD 1.9.2 (Humphrey et al., 1996) and CPPTRAJ (Roe and Cheatham, 2013). Plots were generated using the R statistical package (https://stat.ethz.ch/pipermail/r-help/2008-May/161481.html).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis of reporter assays
All reporter assays (Tango assays) were repeated at least 3 times in triplicate. Error bars show S.D. Data were analyzed by two-tailed, unpaired t-test using GraphPad Prism 5 or Excel software. Statistical significance of the differences is shown, as follows: *, p<0.05; **, p<0.01; ***, p<0.001.
DATA AND SOFTWARE AVAILABILITY
Data Resources
The XFEL diffraction images for T4L-rhodopsin-arrestin complex structure determination were deposited in Coherent X-ray Imaging Data Bank http://dx.doi.org/10.11577/1241101, and are accessible via web service at http://www.cxidb.org with CXIDB ID 32.
The coordinates and structures factors of T4L-rhodopsin-visual arrestin complex have been submitted to the Protein Data Bank under accession number 5W0P.
Software availability
All software used in this paper are reported in Method Details and indicated in the Key Resources Table.
Supplementary Material
Figure S1. Electron density maps for the rhodopsin-arrestin complex. Related to Figures 1 and 2. (A–B) Overall model of T4L-rhodopsin-arrestin complex A with a 2Fo-Fc map (A) and a 2Fo-Fc composite omit map (B), both of which were contoured at 1 σ. (C) A 2Fo-Fc density map of the arrestin C domain with the putative membrane-touching tip loop (F339-L343) shown as a stick model. (D) A 2Fo-Fc density map of the NAG molecules at N15 of the rhodopsin extracellular side. (E–F) Fo-Fc omit maps contoured at 3 σ (E) or 2 σ (F), respectively, showing the density of the rhodopsin C-tail in the rhodopsin-arrestin complex.
Figure S2. Liquid-chromatography-tandem mass spectrometry analysis (LS-MS/MS) of T4L-rhodopsin-arrestin samples expressed in mammalian system. Related to Figure 1 and 2.
(A–C) Mass spectra of T4L-rhodopsin-arrestin fusion protein indicated that residues S334 (A), T336 (B), and S338 (C) were phosphorylated.
Figure S3. Validation of the rhodopsin-arrestin interface. Related to Figures 3 and 4.
(A) Summary of disulfide crosslinking mapping of the interface between the rhodopsin C-tail and the N-domain of the arrestin. (B) HDX mass spectrometry perturbation map derived from the difference in the HDX rate between rhodopsin-bound arrestin and free arrestin. The bars below the arrestin sequence represent the peptide fragments resolved by mass spectrometry and the colors of the bars indicate the relative decrease in deuterium exchange (color code at bottom). (C) HDX differences mapped on the crystal structure. Rhodopsin is in red and arrestin is colored based on the exchange rate differences between the free and rhodopsin-bound arrestin as shown in (B).
Figure S4. Interaction network and range between the rhodopsin C-tail and arrestin. Related to Figure 5. (A) Detailed analysis of the bonding network between pT336, pS338 and E341 and key residues comprising arrestin’s three phosphate binding pockets for both simulations. Only interactions with frequencies greater than 5% are shown. Residues marked with * indicated backbone interactions (B) Simulation B-factors (left) derived from Cα RMSFs agree with room temperature XFEL structure B-factors (right).
Figure S5. Receptor C-tail-arrestin interfaces are conserved. Related to Figure 6. (A) Binding of V2R C-tail (magenta) to the β-arrestin N-terminal surface (PDB code: 4JQI). Blue indicates positive charges, and red negative charges at the arrestin surface. The phospho-residues binding to the arrestin positive pockets (A, B and C) are labeled in white. Among the residues that contribute to the positively charged density of the pocket C is R7, corresponding to V12 in visual arrestin (see panel B). (B) A comparison of the receptor binding surface of the visual arrestin N-domain (brown) with that of β-arrestin-1 (gray). Labeled are the receptor interfacre residues that differ between the two arrestins. (C) Sequence alignment of mouse visual arrestin, and human β-arrestins and visual arrestins. Residues in red boxes form the charged pockets (denoted as A, B, and C) at the arrestin surfaces for binding to phosphorylated residues in the rhodopsin C-tail.
Figure S6. Phosphorylation code frequency within C-tails of GPCR subfamilies identified using PhosCoFinder (http://tools.vai.org/phoscofinder/). Related to Figure 5.
(A) Presence of C-tail phosphorylation codes in different receptor classes suggests a common mechanism of arrestin recruitment. For all classes, the majority of receptors contain complete or partial C-tail codes with codeless receptors representing the minority. (B) Frequency of complete short and long phosphorylation codes per receptor in class A GPCRs. For receptors lacking either short or long phosphorylation codes, the frequency of partial codes is significantly increased (p=2.2e-16; data not shown). (C) The classification of GPCRs based on their arrestin binding affinities (”class A” and “class B”, by Lohse and Hoffmann, 2014) strongly correlates with the occurrence of complete phosphorylation codes in their C-terminal tails. Left panel shows that “Class A” GPCRs (α1- and β2-adrenergic, µ-opioid, endothelin ETA and dopamine D1A receptors) with a preference for binding β-arrestin-2 over β-arrestin-1 and visual arrestin, contain no or only one phosphorylation code, while “class B” GPCRs (angiotensin II type 1A receptor, neurotensin receptor 1, vasopressin V2 receptor, thyrotropin-releasing hormone receptor, and substance P receptor) which bind both β-arrestins and visual arrestin, contain two or more phosphorylation codes in their C-tails. Right panel is a list showing the number of short, long and partial phosphorylation codes for each of the two classes of GPCRs.
Figure S7. Dose response curves of phosphorylation code mutants of the β2-AR or β2-AR-V2R chimera in Tango assay. Related to Figures 6 and 7. (A) Representative dose response of the phosphorylation code 6A mutant (S357A/T359A/T360A/S362A/S363A/S364A) of the β2AR-V2R chimera. EC50 of wild type β2-AR-V2R chimera and 6A mutant is 0.82µM and 0.63µM, respectively. (B) Representative dose response curves of the phosphorylation code mutants of the β2AR C-tail cluster 1 (341–365). EC50 of wild type β2AR C-tail cluster 1, 4A (S355A/S356A/T360A/E362A) and N359S are 1.76µM, 0.69µM and 1.97 µM, respectively. (C) Representative dose response curves of the phosphorylation code mutants of the β2AR C-tail cluster 2 (389–413). EC50 of wild type β2AR C-tail cluster 2 (389–413) and mutant I399S/N405S are 1.31µM and 1.52µM, respectively. Data were from triplicated experiments with error bars indicating S.D.
Highlights.
A rhodopsin–arrestin complex structure with phosphorylated rhodopsin C-terminus
Structural mechanism for recognition of phosphorylated rhodopsin by visual arrestin
Phosphorylation codes are a common mechanism of arrestin recruitment by GPCRs
Acknowledgments
This work was supported in part by NIH grant DK071662, American Asthma Foundation, Jay and Betty Van Andel Foundation, Ministry of Science and Technology (China) grants 2012ZX09301001, 2012CB910403, 2013CB910600, XDB08020303, 2013ZX09507001 to H.E.X., the Canada Excellence Research Chairs program, the Canadian Institute for Advanced Research and the Anne and Max Tanenbaum Chair in Neuroscience to O.P.E., NIH PO1 GM108635 to V.C., NIH RO1 EY011500 and R35 GM122491 to V.V.G., RO1 EY05216 and the Jules Stein Professorship Endowment to W.L.H., and the Van Andel Research Institute Cryo-EM core facility. The original XFEL data was collected at the Linac Coherent Light Source at SLAC National Laboratory, and was assisted by Sébastien Boutet, Garth Williams, and members of the Petra Fromme, Uwe Weierstall, and John Spence groups from Arizona State University, which are funded in part by NSF Science and Technology Center award 1231306 (J.S., P.F. and U.W). All computations were performed on the Van Andel Research Institute compute cluster.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Conceptualization, X.E.Z. and H.E.X.; Methodology, X.E.Z., Y.H., X.G., P.W.dW., Y.K., N.V.E., Y.Y., K.P., D.G., T.A.W., A.B., N.R.L., H.N.C., W.L.H., R.C.S., P.R.G. and O.P.E.; Resources, H.N.C., O.P.E., R.C.S., P.R.G., K.M. and H.E.X.; Writing, X.E.Z., P.W.dW., V.V.G., R.O.D., V.C., K.M. and H.E.X. Funding Acquisition, H.E.X., K.M., P.R.G., O.P.E., V.V.G., H.N.C., R.C.S., V.C. and W.L.H.
References
- Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1–2:19–25. [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta crystallographica Section D, Biological crystallography. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altenbach C, Kusnetzow AK, Ernst OP, Hofmann KP, Hubbell WL. High-resolution distance mapping in rhodopsin reveals the pattern of helix movement due to activation. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:7439–7444. doi: 10.1073/pnas.0802515105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14 Azevedo AW, Doan T, Moaven H, Sokal I, Baameur F, Vishnivetskiy SA, Homan KT, Tesmer JJ, Gurevich VV, Chen J, et al. C-terminal threonines and serines play distinct roles in the desensitization of rhodopsin, a G protein-coupled receptor. eLife. 2015;4 doi: 10.7554/eLife.05981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnea G, Strapps W, Herrada G, Berman Y, Ong J, Kloss B, Axel R, Lee KJ. The genetic design of signaling cascades to record receptor activation. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:64–69. doi: 10.1073/pnas.0710487105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barty A, Kirian RA, Maia FR, Hantke M, Yoon CH, White TA, Chapman H. software for high-throughput reduction and analysis of serial femtosecond X-ray diffraction data. J Appl Crystallogr. 2014;47:1118–1131. doi: 10.1107/S1600576714007626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battye TG, Kontogiannis L, Johnson O, Powell HR, Leslie AG. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta crystallographica Section D, Biological crystallography. 2011;67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao W, Luttrell LM, Medvedev AV, Pierce KL, Daniel KW, Dixon TM, Lefkowitz RJ, Collins S. Direct binding of activated c-Src to the beta 3-adrenergic receptor is required for MAP kinase activation. The Journal of biological chemistry. 2000;275:38131–38134. doi: 10.1074/jbc.C000592200. [DOI] [PubMed] [Google Scholar]
- Chen W, Kirkbride KC, How T, Nelson CD, Mo J, Frederick JP, Wang XF, Lefkowitz RJ, Blobe GC. Beta-arrestin 2 mediates endocytosis of type III TGF-beta receptor and down-regulation of its signaling. Science. 2003;301:1394–1397. doi: 10.1126/science.1083195. [DOI] [PubMed] [Google Scholar]
- Duisenberg AJM. Indexing in Single-Crystal Diffractometry with an Obstinate List of Reflections. J Appl Crystallogr. 1992;25:92–96. [Google Scholar]
- Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. Comparative protein structure modeling using Modeller. Current protocols in bioinformatics. 2006;(Unit 5 6) doi: 10.1002/0471250953.bi0506s15. Chapter 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Arenas E, Calleja E, Martinez-Martin N, Gharbi SI, Navajas R, Garcia-Medel N, Penela P, Alcami A, Mayor F, Jr, Albar JP, et al. beta-Arrestin-1 mediates the TCR-triggered re-routing of distal receptors to the immunological synapse by a PKC-mediated mechanism. The EMBO journal. 2014;33:559–577. doi: 10.1002/embj.201386022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleissner MR, Cascio D, Hubbell WL. Structural origin of weakly ordered nitroxide motion in spin-labeled proteins. Protein Sci. 2009;18:893–908. doi: 10.1002/pro.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimenez LE, Kook S, Vishnivetskiy SA, Ahmed MR, Gurevich EV, Gurevich VV. Role of receptor-attached phosphates in binding of visual and non-visual arrestins to G protein-coupled receptors. The Journal of biological chemistry. 2012;287:9028–9040. doi: 10.1074/jbc.M111.311803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girnita L, Shenoy SK, Sehat B, Vasilcanu R, Girnita A, Lefkowitz RJ, Larsson O. {beta}-Arrestin is crucial for ubiquitination and down-regulation of the insulin-like growth factor-1 receptor by acting as adaptor for the MDM2 E3 ligase. The Journal of biological chemistry. 2005;280:24412–24419. doi: 10.1074/jbc.M501129200. [DOI] [PubMed] [Google Scholar]
- Goswami D, Devarakonda S, Chalmers MJ, Pascal BD, Spiegelman BM, Griffin PR. Time window expansion for HDX analysis of an intrinsically disordered protein. J Am Soc Mass Spectrom. 2013;24:1584–1592. doi: 10.1007/s13361-013-0669-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich EV, Gurevich VV. Arrestins: ubiquitous regulators of cellular signaling pathways. Genome biology. 2006a;7:236. doi: 10.1186/gb-2006-7-9-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV, Gurevich EV. The structural basis of arrestin-mediated regulation of G-protein-coupled receptors. Pharmacol Ther. 2006b;110:465–502. doi: 10.1016/j.pharmthera.2005.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV, Gurevich EV. Structural determinants of arrestin functions. Prog Mol Biol Transl Sci. 2013;118:57–92. doi: 10.1016/B978-0-12-394440-5.00003-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebda JK, Leclair HM, Azzi S, Roussel C, Scott MG, Bidere N, Gavard J. The C-terminus region of beta-arrestin1 modulates VE-cadherin expression and endothelial cell permeability. Cell communication and signaling : CCS. 2013;11:37. doi: 10.1186/1478-811X-11-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, MacKerell AD., Jr CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J Comput Chem. 2013;34:2135–2145. doi: 10.1002/jcc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Kabsch W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr D. 2010;66:133–144. doi: 10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Zhou XE, Gao X, He Y, Liu W, Ishchenko A, Barty A, White TA, Yefanov O, Han GW, et al. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature. 2015;523:561–567. doi: 10.1038/nature14656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Vishnivetskiy SA, Van Eps N, Alexander NS, Cleghorn WM, Zhan X, Hanson SM, Morizumi T, Ernst OP, Meiler J, et al. Conformation of receptor-bound visual arrestin. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:18407–18412. doi: 10.1073/pnas.1216304109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YJ, Hofmann KP, Ernst OP, Scheerer P, Choe HW, Sommer ME. Crystal structure of pre-activated arrestin p44. Nature. 2013;497:142–146. doi: 10.1038/nature12133. [DOI] [PubMed] [Google Scholar]
- Krasel C, Zabel U, Lorenz K, Reiner S, Al-Sabah S, Lohse MJ. Dual role of the beta2-adrenergic receptor C terminus for the binding of beta-arrestin and receptor internalization. The Journal of biological chemistry. 2008;283:31840–31848. doi: 10.1074/jbc.M806086200. [DOI] [PubMed] [Google Scholar]
- Lally CC, Bauer B, Selent J, Sommer ME. C-edge loops of arrestin function as a membrane anchor. Nature communications. 2017;8:14258. doi: 10.1038/ncomms14258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- Liggett SB. Phosphorylation barcoding as a mechanism of directing GPCR signaling. Sci Signal. 2011;4:pe36. doi: 10.1126/scisignal.2002331. [DOI] [PubMed] [Google Scholar]
- Lohse MJ, Hoffmann C. Arrestin interactions with G protein-coupled receptors. Handbook of experimental pharmacology. 2014;219:15–56. doi: 10.1007/978-3-642-41199-1_2. [DOI] [PubMed] [Google Scholar]
- Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI. OPM: orientations of proteins in membranes database. Bioinformatics. 2006;22:623–625. doi: 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- Mashukova A, Spehr M, Hatt H, Neuhaus EM. Beta-arrestin2-mediated internalization of mammalian odorant receptors. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2006;26:9902–9912. doi: 10.1523/JNEUROSCI.2897-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGibbon RT, Beauchamp KA, Harrigan MP, Klein C, Swails JM, Hernandez CX, Schwantes CR, Wang LP, Lane TJ, Pande VS. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys J. 2015;109:1528–1532. doi: 10.1016/j.bpj.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez A, Burns ME, Roca A, Lem J, Wu LW, Simon MI, Baylor DA, Chen J. Rapid and reproducible deactivation of rhodopsin requires multiple phosphorylation sites. Neuron. 2000;28:153–164. doi: 10.1016/s0896-6273(00)00093-3. [DOI] [PubMed] [Google Scholar]
- Nobles KN, Xiao K, Ahn S, Shukla AK, Lam CM, Rajagopal S, Strachan RT, Huang TY, Bressler EA, Hara MR, et al. Distinct phosphorylation sites on the beta(2)-adrenergic receptor establish a barcode that encodes differential functions of beta-arrestin. Sci Signal. 2011;4:ra51. doi: 10.1126/scisignal.2001707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS. Differential affinities of visual arrestin, beta arrestin1, and beta arrestin2 for G protein-coupled receptors delineate two major classes of receptors. The Journal of biological chemistry. 2000;275:17201–17210. doi: 10.1074/jbc.M910348199. [DOI] [PubMed] [Google Scholar]
- Ostermaier MK, Peterhans C, Jaussi R, Deupi X, Standfuss J. Functional map of arrestin-1 at single amino acid resolution. Proc Natl Acad Sci U S A. 2014a;111:1825–1830. doi: 10.1073/pnas.1319402111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostermaier MK, Schertler GF, Standfuss J. Molecular mechanism of phosphorylation-dependent arrestin activation. Current opinion in structural biology. 2014b;29:143–151. doi: 10.1016/j.sbi.2014.07.006. [DOI] [PubMed] [Google Scholar]
- Pascal BD, Willis S, Lauer JL, Landgraf RR, West GM, Marciano D, Novick S, Goswami D, Chalmers MJ, Griffin PR. HDX Workbench: Software for the Analysis of H/D Exchange MS Data. J Am Soc Mass Spectr. 2012;23:1512–1521. doi: 10.1007/s13361-012-0419-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nature reviews Molecular cell biology. 2002;3:639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- Pitcher JA, Freedman NJ, Lefkowitz RJ. G protein-coupled receptor kinases. Annual review of biochemistry. 1998;67:653–692. doi: 10.1146/annurev.biochem.67.1.653. [DOI] [PubMed] [Google Scholar]
- Por ED, Bierbower SM, Berg KA, Gomez R, Akopian AN, Wetsel WC, Jeske NA. beta-Arrestin-2 desensitizes the transient receptor potential vanilloid 1 (TRPV1) channel. The Journal of biological chemistry. 2012;287:37552–37563. doi: 10.1074/jbc.M112.391847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Por ED, Gomez R, Akopian AN, Jeske NA. Phosphorylation regulates TRPV1 association with beta-arrestin-2. The Biochemical journal. 2013;451:101–109. doi: 10.1042/BJ20121637. [DOI] [PubMed] [Google Scholar]
- Reiter E, Ahn S, Shukla AK, Lefkowitz RJ. Molecular mechanism of beta-arrestin-biased agonism at seven-transmembrane receptors. Annual review of pharmacology and toxicology. 2012;52:179–197. doi: 10.1146/annurev.pharmtox.010909.105800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe DR, Cheatham TE., 3rd PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J Chem Theory Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- Seibold A, Williams B, Huang ZF, Friedman J, Moore RH, Knoll BJ, Clark RB. Localization of the sites mediating desensitization of the beta(2)-adrenergic receptor by the GRK pathway. Molecular pharmacology. 2000;58:1162–1173. doi: 10.1124/mol.58.5.1162. [DOI] [PubMed] [Google Scholar]
- Shukla AK, Manglik A, Kruse AC, Xiao K, Reis RI, Tseng WC, Staus DP, Hilger D, Uysal S, Huang LY, et al. Structure of active beta-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature. 2013;497:137–141. doi: 10.1038/nature12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla AK, Westfield GH, Xiao K, Reis RI, Huang LY, Tripathi-Shukla P, Qian J, Li S, Blanc A, Oleskie AN, et al. Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature. 2014;512:218–222. doi: 10.1038/nature13430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla AK, Xiao K, Lefkowitz RJ. Emerging paradigms of beta-arrestin-dependent seven transmembrane receptor signaling. Trends in biochemical sciences. 2011;36:457–469. doi: 10.1016/j.tibs.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JS, Rajagopal S. The beta-Arrestins: Multifunctional Regulators of G Protein-coupled Receptors. The Journal of biological chemistry. 2016;291:8969–8977. doi: 10.1074/jbc.R115.713313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton RB, Vishnivetskiy SA, Robert J, Hanson SM, Raman D, Knox BE, Kono M, Navarro J, Gurevich VV. Crystal Structure of Cone Arrestin at 2.3Å: Evolution of Receptor Specificity. J Mol Biol. 2005;354:1069–1080. doi: 10.1016/j.jmb.2005.10.023. [DOI] [PubMed] [Google Scholar]
- Thomsen AR, Plouffe B, Cahill TJ, 3rd, Shukla AK, Tarrasch JT, Dosey AM, Kahsai AW, Strachan RT, Pani B, Mahoney JP, et al. GPCR-G Protein-beta-Arrestin Super-Complex Mediates Sustained G Protein Signaling. Cell. 2016;166:907–919. doi: 10.1016/j.cell.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan DJ, Millman EE, Godines V, Friedman J, Tran TM, Dai W, Knoll BJ, Clark RB, Moore RH. Role of the G protein-coupled receptor kinase site serine cluster in beta2-adrenergic receptor internalization, desensitization, and beta-arrestin translocation. The Journal of biological chemistry. 2006;281:7684–7692. doi: 10.1074/jbc.M500328200. [DOI] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Raman D, Wei J, Kennedy MJ, Hurley JB, Gurevich VV. Regulation of arrestin binding by rhodopsin phosphorylation level. J Biol Chem. 2007;282:32075–32083. doi: 10.1074/jbc.M706057200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehbi VL, Stevenson HP, Feinstein TN, Calero G, Romero G, Vilardaga JP. Noncanonical GPCR signaling arising from a PTH receptor-arrestin-Gbetagamma complex. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:1530–1535. doi: 10.1073/pnas.1205756110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West GM, Pascal BD, Ng LM, Soon FF, Melcher K, Xu HE, Chalmers MJ, Griffin PR. Protein conformation ensembles monitored by HDX reveal a structural rationale for abscisic acid signaling protein affinities and activities. Structure. 2013;21:229–235. doi: 10.1016/j.str.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White TA, Mariani V, Brehm W, Yefanov O, Barty A, Beyerlein KR, Chervinskii F, Galli L, Gati C, Nakane T, et al. Recent developments in CrystFEL. J Appl Crystallogr. 2016;49:680–689. doi: 10.1107/S1600576716004751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisler JW, DeWire SM, Whalen EJ, Violin JD, Drake MT, Ahn S, Shenoy SK, Lefkowitz RJ. A unique mechanism of beta-blocker action: carvedilol stimulates beta-arrestin signaling. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:16657–16662. doi: 10.1073/pnas.0707936104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao K, McClatchy DB, Shukla AK, Zhao Y, Chen M, Shenoy SK, Yates JR, 3rd, Lefkowitz RJ. Functional specialization of beta-arrestin interactions revealed by proteomic analysis. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:12011–12016. doi: 10.1073/pnas.0704849104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Yu X, Liu C, Qu CX, Gong Z, Liu HD, Li FH, Wang HM, He DF, Yi F, et al. Phospho-selective mechanisms of arrestin conformations and functions revealed by unnatural amino acid incorporation and (19)F-NMR. Nature communications. 2015;6:8202. doi: 10.1038/ncomms9202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Yang F, Zhang D, Liu Z, Lin A, Liu C, Xiao P, Yu X, Sun JP. Phosphorylation of G protein-coupled receptors: from the barcode hypothesis to the flute model. Molecular pharmacology. 2017 doi: 10.1124/mol.116.107839. [DOI] [PubMed] [Google Scholar]
- Yu MC, Su LL, Zou L, Liu Y, Wu N, Kong L, Zhuang ZH, Sun L, Liu HP, Hu JH, et al. An essential function for beta-arrestin 2 in the inhibitory signaling of natural killer cells. Nature immunology. 2008;9:898– 907. doi: 10.1038/ni.1635. [DOI] [PubMed] [Google Scholar]
- Zhou XE, Gao X, Barty A, Kang Y, He Y, Liu W, Ishchenko A, White TA, Yefanov O, Han GW, et al. X-ray laser diffraction for structure determination of the rhodopsin-arrestin complex. Scientific data. 2016;3:160021. doi: 10.1038/sdata.2016.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zindel D, Butcher AJ, Al-Sabah S, Lanzerstorfer P, Weghuber J, Tobin AB, Bunemann M, Krasel C. Engineered hyperphosphorylation of the beta2-adrenoceptor prolongs arrestin-3 binding and induces arrestin internalization. Molecular pharmacology. 2015;87:349–362. doi: 10.1124/mol.114.095422. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Electron density maps for the rhodopsin-arrestin complex. Related to Figures 1 and 2. (A–B) Overall model of T4L-rhodopsin-arrestin complex A with a 2Fo-Fc map (A) and a 2Fo-Fc composite omit map (B), both of which were contoured at 1 σ. (C) A 2Fo-Fc density map of the arrestin C domain with the putative membrane-touching tip loop (F339-L343) shown as a stick model. (D) A 2Fo-Fc density map of the NAG molecules at N15 of the rhodopsin extracellular side. (E–F) Fo-Fc omit maps contoured at 3 σ (E) or 2 σ (F), respectively, showing the density of the rhodopsin C-tail in the rhodopsin-arrestin complex.
Figure S2. Liquid-chromatography-tandem mass spectrometry analysis (LS-MS/MS) of T4L-rhodopsin-arrestin samples expressed in mammalian system. Related to Figure 1 and 2.
(A–C) Mass spectra of T4L-rhodopsin-arrestin fusion protein indicated that residues S334 (A), T336 (B), and S338 (C) were phosphorylated.
Figure S3. Validation of the rhodopsin-arrestin interface. Related to Figures 3 and 4.
(A) Summary of disulfide crosslinking mapping of the interface between the rhodopsin C-tail and the N-domain of the arrestin. (B) HDX mass spectrometry perturbation map derived from the difference in the HDX rate between rhodopsin-bound arrestin and free arrestin. The bars below the arrestin sequence represent the peptide fragments resolved by mass spectrometry and the colors of the bars indicate the relative decrease in deuterium exchange (color code at bottom). (C) HDX differences mapped on the crystal structure. Rhodopsin is in red and arrestin is colored based on the exchange rate differences between the free and rhodopsin-bound arrestin as shown in (B).
Figure S4. Interaction network and range between the rhodopsin C-tail and arrestin. Related to Figure 5. (A) Detailed analysis of the bonding network between pT336, pS338 and E341 and key residues comprising arrestin’s three phosphate binding pockets for both simulations. Only interactions with frequencies greater than 5% are shown. Residues marked with * indicated backbone interactions (B) Simulation B-factors (left) derived from Cα RMSFs agree with room temperature XFEL structure B-factors (right).
Figure S5. Receptor C-tail-arrestin interfaces are conserved. Related to Figure 6. (A) Binding of V2R C-tail (magenta) to the β-arrestin N-terminal surface (PDB code: 4JQI). Blue indicates positive charges, and red negative charges at the arrestin surface. The phospho-residues binding to the arrestin positive pockets (A, B and C) are labeled in white. Among the residues that contribute to the positively charged density of the pocket C is R7, corresponding to V12 in visual arrestin (see panel B). (B) A comparison of the receptor binding surface of the visual arrestin N-domain (brown) with that of β-arrestin-1 (gray). Labeled are the receptor interfacre residues that differ between the two arrestins. (C) Sequence alignment of mouse visual arrestin, and human β-arrestins and visual arrestins. Residues in red boxes form the charged pockets (denoted as A, B, and C) at the arrestin surfaces for binding to phosphorylated residues in the rhodopsin C-tail.
Figure S6. Phosphorylation code frequency within C-tails of GPCR subfamilies identified using PhosCoFinder (http://tools.vai.org/phoscofinder/). Related to Figure 5.
(A) Presence of C-tail phosphorylation codes in different receptor classes suggests a common mechanism of arrestin recruitment. For all classes, the majority of receptors contain complete or partial C-tail codes with codeless receptors representing the minority. (B) Frequency of complete short and long phosphorylation codes per receptor in class A GPCRs. For receptors lacking either short or long phosphorylation codes, the frequency of partial codes is significantly increased (p=2.2e-16; data not shown). (C) The classification of GPCRs based on their arrestin binding affinities (”class A” and “class B”, by Lohse and Hoffmann, 2014) strongly correlates with the occurrence of complete phosphorylation codes in their C-terminal tails. Left panel shows that “Class A” GPCRs (α1- and β2-adrenergic, µ-opioid, endothelin ETA and dopamine D1A receptors) with a preference for binding β-arrestin-2 over β-arrestin-1 and visual arrestin, contain no or only one phosphorylation code, while “class B” GPCRs (angiotensin II type 1A receptor, neurotensin receptor 1, vasopressin V2 receptor, thyrotropin-releasing hormone receptor, and substance P receptor) which bind both β-arrestins and visual arrestin, contain two or more phosphorylation codes in their C-tails. Right panel is a list showing the number of short, long and partial phosphorylation codes for each of the two classes of GPCRs.
Figure S7. Dose response curves of phosphorylation code mutants of the β2-AR or β2-AR-V2R chimera in Tango assay. Related to Figures 6 and 7. (A) Representative dose response of the phosphorylation code 6A mutant (S357A/T359A/T360A/S362A/S363A/S364A) of the β2AR-V2R chimera. EC50 of wild type β2-AR-V2R chimera and 6A mutant is 0.82µM and 0.63µM, respectively. (B) Representative dose response curves of the phosphorylation code mutants of the β2AR C-tail cluster 1 (341–365). EC50 of wild type β2AR C-tail cluster 1, 4A (S355A/S356A/T360A/E362A) and N359S are 1.76µM, 0.69µM and 1.97 µM, respectively. (C) Representative dose response curves of the phosphorylation code mutants of the β2AR C-tail cluster 2 (389–413). EC50 of wild type β2AR C-tail cluster 2 (389–413) and mutant I399S/N405S are 1.31µM and 1.52µM, respectively. Data were from triplicated experiments with error bars indicating S.D.