

Abstract
As part of an ongoing drug development effort aimed at selective opioid receptor ligands based on the pawhuskin natural products we have synthesized a small set of amide isosteres. These amides were centered on lead compounds which are selective antagonists for the delta and kappa opioid receptors. The amide isomers revealed here show dramatically different activity from the parent stilbene compounds. Three of the isomers synthesized showed antagonist activity for the opioid growth factor (OGF)/opioid growth factor receptor (OGFR) axis which is involved in cellular and organ growth control. This cellular signaling mechanism is targeted by “low-dose” naltrexone therapy which is being tested clinically for multiple sclerosis, Crohn’s disease, cancer, and wound healing disorders. The compounds described here are the first selective small molecule ligands for the OGF/OGFR system and will serve as important leads and probes for further study.
Keywords: Opioid receptor, Opioid growth factor, Low-dose naltrexone, Pawhuskin, Stilbene isostere
Graphical Abstract
1. Introduction
Opiates are some of the most well studied and highly used medicines in history.[1] The use of morphine for pain control dates back centuries[2] and the development of more potent opiates, both agonists[3] and antagonists,[4, 5] has been one of the great stories of modern pharmacology and chemistry. It is not however without its downside, for the use of opiates to ameliorate pain can lead to tolerance and addiction ending in severe substance use disorder.[6] Since the middle of the 20th century it has been understood that subtle changes in the structure of these compounds can lead to large differences in activity.[7–9] In the latter half of the century the cloning and characterization of specific opioid receptors showed them to be members of the 7-transmembrane domain G-protein coupled receptor family.[10–12] This was followed recently by crystallization and determination of the structures of these proteins by X-ray diffraction,[13–16] which in turn has led to a new era of drug design with the aim of reducing or eliminating the unwanted effects.[17–23]
The four traditional opioid receptor subtypes are the mu (MOR), delta (DOR), kappa (KOR) and nociceptin receptors (NOR).[24] The NOR is related to the others, but it does not respond to the same ligands.[25, 26] The MOR is the subtype which has been traditionally associated with the analgesic properties of the opioid ligands morphine and codeine, as well as the other clinically used synthetic opioid agonists.[24] Functional studies of the KOR have shown its importance to analgesia,[27] but also demonstrate that this receptor subtype has significant additional roles, including mediating the dysphoria induced by stress responses.[28]
Recently KOR antagonists have elicited some interest for the treatment of depression and substance use disorder, particularly in the context of the conjunction between stress and relapse to drug seeking and taking behaviors.[28–30] KOR agonism is thought to be of key importance to the development of the cycle of substance abuse where the dysphoric effects of this receptor encourage the further use of the substances to relieve negative feelings. This was called the “dark-side” of addiction by Koob et al.[31, 32] and is operative in amphetamine, alcohol, and narcotic abuse as evidenced by the ability of KOR antagonists to ameliorate the development of chronic dose escalation in models of addiction using such agents.[33, 34]
Another opioid binding protein known as the opioid growth factor receptor (OGFR) has also been of interest lately in the context of immune modulation, wound healing, and potential for cancer treatment.[35] The OGFR is not a member of the 7-transmembrane G-protein coupled receptor family and bears no structural homology to the other classic opioid receptor subtypes.[36] The OGFR binds the natural ligand [Met5]-enkephalin, a ligand which also has selectivity for the classical DOR.[36] The natural ligand agonist acts to reduce growth of several human cancer cell lines in vitro and in vivo, and the disruption of signaling through OGFR causes increased growth.[37–39] The non-selective antagonist naltrexone blocks signaling through the OGFR in another overlap in receptor pharmacology.[40] Blockade of OGFR is being pursued as a therapeutic strategy for wound healing.[36, 41–44] Interestingly, short term blockade of the OGFR by low doses of naltrexone results in upregulation of the OGFR signaling axis and causes a reduction in tumor growth.[45, 46] This is being explored for the treatment of several cancers,[47–51] as well as other diseases[52–56] and is termed “low-dose naltrexone therapy”.[57]
Our interest in opioid receptor modulators dates to the isolation of the natural product pawhuskin A (1, Figure 1) from Dalea purpurea by Belofsky et al.[58] This prenylated resveratrol analogue was shown to have opioid receptor binding activity by displacement of radioactive naloxone from animal tissues. Because of a long running interest in the synthesis and biology of prenylated stilbenes,[59, 60] we undertook the total synthesis and testing of this compound and demonstrated that it is a selective KOR antagonist.[61, 62] Subsequently, a medicinal chemistry program was established which has resulted in the synthesis of both DOR (2)[63, 64] and KOR (3)[63, 64] selective antagonists that are analogues of the natural product. These compounds are structurally unrelated to the classical opioids, and do not have the basic nitrogen found in morphine and other clinically used compounds such as meperidine, fentanyl or the antagonist naltrexone. Thus the pawhuskin core provides a potential lead for the development of novel compounds which could have a different side effect-profile from other structural classes under development.
Figure 1.
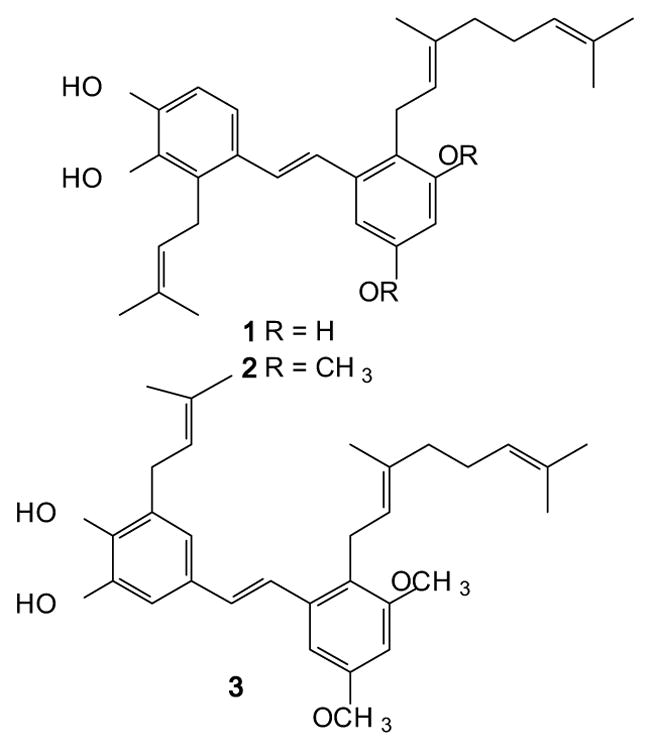
Structures of Pawhuskin A (1) and related DOR and KOR selective antagonist analogues 2 and 3.
Initial studies of the prenylated isomers 2 and 3 showed them to have very low water solubility. It is our intention to test the hypothesis that a compound derived from this class has effects in an animal model, and so it was desirable to design analogues with improved solubility as a first goal for the drug development effort. Docking studies of the KOR selective antagonist 3 indicated that key interactions could be with the free phenols of the catechol substructure, with the prenyl and geranyl chains providing additional hydrophobic interactions (Figure 2). This led to the hypothesis that the stilbene olefin would be buried within the binding motif and would be amendable to changes with the goal of improving drug-likeness.
Figure 2.
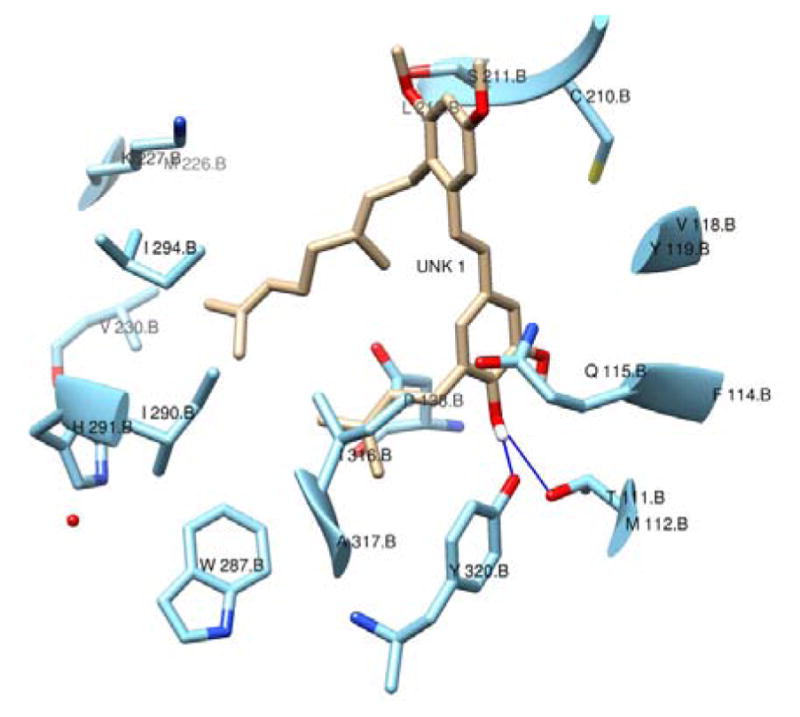
Compound 3 docked into the KOR ligand binding site. H-bonds with protein are shown in blue.
As a first foray into trans-stilbene isosteric replacements the amides 4 and 5 (Figure 3) were viewed as a natural starting point, with the isomeric pair 6 and 7 bearing the opposite amide orientation also being reasonable targets. It is expected that these compounds will be more hydrophilic. Calculated partition coefficients (CLogP) for the stilbenes 2 and 3 are 9.56 vs. 7.04 for the amide isosteres, which should translate into increased water solubility. A study of the amide isosteres of the related compound resveratrol as quinone reductase 2 inhibitors showed that this substitution was tolerated in that model although the activity was slightly reduced.[65]
Figure 3.

Structure of the amide compounds designed as stilbene isosteres.
2. Chemistry
At the outset of this effort, the more accessible amides appeared to be those derived from an amine “right half” and a “left half” carboxylic acid. Geranyl-substituted benzaldehydes that have been used to prepare other pawhuskin analogues[66] could serve as potential starting materials for the desired amine. However, a more concise approach might be based on an electrophilic aromatic substitution given that Friedel-Crafts acylation of the acetanilide 8 is known to afford the ortho-substituted isomer,[67] as long as a parallel alkylation could be conducted with an isoprenoid alcohol. Accordingly acetanilide 8 was prepared from commercial 3,5-dimethoxyaniline and acetic anhydride.[68] The subsequent BF3·OEt2 mediated geranylation of compound 8 preserved the E-olefin stereochemistry, as determined by the 13C NMR spectrum of the product,[69, 70] but gave a mixture of the ortho-and para-substituted products. The desired product 9 was the major isomer, it was readily isolated by flash chromatography, and the regiochemistry was easily identified by comparison to the symmetrical para isomer. Standard basic hydrolysis of the amide 9 gave the necessary amine 10.
Preparation of the first amide then required a left half carboxylic acid, and compound 12 was readily prepared via Pinnick oxidation of the known aldehyde 11.[61] Treatment of the resulting carboxylic acid 12 with thionyl chloride followed by reaction with the amine 10 gave the desired amide 13, but the same amide was obtained in better yield via an EDC-mediated condensation. Final hydrolysis of the two MOM protecting groups gave the targeted amide 4.
The isomeric A-ring amide 5 was prepared via a parallel series of reactions. For this target, oxidation of the known aldehyde 14[61, 71] gave the expected carboxylic acid 15 in good conversion and modest isolated yield. Treatment of this carboxylic acid with thionyl chloride followed by the amine 10 afforded the desired amide 16 but only in low yield (16%). The EDC-mediated condensation of acid 15 with the amine 10 gave the amide 16 in better yield (56%), and sufficient material was obtained to complete the reaction sequence. Final hydrolysis of the MOM groups then gave the target amide 5.
Synthesis of the isomeric amides derived from a carboxylic acid right half and an amine left half began with preparation of an appropriate carboxylic acid. In this case, regiocontrolled introduction of the geranyl group could be achieved through the aryl bromide 17, which was readily prepared by bromination of the corresponding methyl benzoate with NBS.[72, 73] Although it may not be common, halogen-metal exchange in the presence of an ester group is known,[74] and the presence of adjacent polar functionality can facilitate metalation.[75] In this specific case, treatment of the aryl bromide 17 with n-BuLi and CuBr·DMS followed by reaction with geranyl bromide gave the desired alkylation product 18 in good yield. A final basic ester hydrolysis then gave the carboxylic acid 19.
The carboxylic acids 12 and 15 were viewed as attractive starting materials for the aromatic amines needed for the target amides, as long as decarboxylation could be achieved without reaction at the isoprenoid olefin. Potential side reactions could include isomerization of the olefin into conjugation with the aromatic ring and cyclization of the isoprenoid chain to an ortho oxygen.[60, 76] Fortunately, treatment of the carboxylic acid 12 with diphenyl phosphoryl azide followed by hydrolysis of the presumed intermediate gave the desired amine 20. Once this amine was in hand, an EDC-mediated coupling with the acid 19 gave the amide 21, and a final hydrolysis of the MOM groups gave the amide 6. In a parallel series of reactions, the isomeric carboxylic acid 15 was converted to the amine 22, coupled with the carboxylic acid 19 to give the amide 23, and then deprotected to afford the desired target 7.
3. Biological Evaluation
We first tested compounds 4 and 5 for agonism and antagonism of the KOR, DOR, and MOR using a GTPχS functional assay. Neither of these amides showed any activity in this assay up to 10 μM concentrations indicating that they are not acting as traditional antagonists or agonists (data not shown). It was apparent that the amides 6 and 7 were also unlikely to show any agonist or antagonist activity at the traditional opioid receptors, but they were tested for intrinsic agonist activity (Figure 4). As suspected, based on the very similar compounds 4 and 5, neither amide 6 nor 7 showed any activity in recruitment of β-arrestin, an assay of agonist activity at the MOR, DOR, or KOR.
Figure 4.

Concentration response curves for β-arrestin recruitment assays of agonism in cells transfected with the human MOR, DOR and KOR. Positive controls for each receptor are shown as well.
The four amide isomers also were tested for activity at the OGFR. The COS-7 cell line was chosen for this experiment because earlier work had shown that OGFR and OGF are expressed in this line, while it fails to express the MOR, KOR, and DOR.[77] Activity at the OGFR receptor might be expected because the stilbene 2 showed selective antagonist activity at the DOR, and natural ligands for DOR and OGFR are closely related. In fact, in a standard model of cell growth in the COS-7 cell line, the DOR-selective stilbene 2 shows growth enhancement whereas the KOR selective ligand 3 does not (Figure 5). Likewise the comparable amide 4, an isostere of compound 2, shows activity in this assay while the amide 5, which is structurally parallel to the KOR-selective stilbene 3, does not. Indeed the structurally related stilbene 2 and amide 4 demonstrate activity as potent as naltrexone in this assay.
Figure 5.
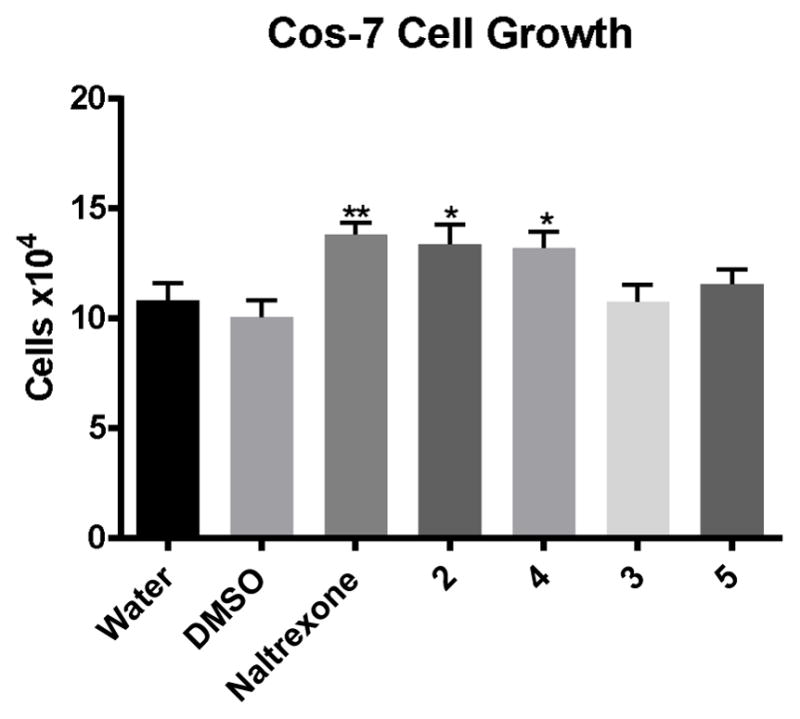
Cell growth in COS-7 cells at 72 hours. Cells were treated with 1 μM concentration of the indicated compounds. * p < 0.05, ** p <0.005
The second pair of amide isomers, compounds 6 and 7, showed differential activity in terms of COS-7 cell growth using an assay based on incorporation of 5-bromo-2’-deoxyuridine (BrdU) into DNA. The BrDU assay was employed to demonstrate that cell growth enhancement was due to DNA synthesis, and compound 2 was included as a positive control for OGFR enhanced growth. In this assay, amide 7, which is an isostere of the KOR-selective stilbene 3, showed increased cell growth (Figure 6). In contrast, amide 6, an isostere of the DOR-selective stilbene 2, did not demonstrate significant cell growth promotion. This finding suggests that ligand binding to the OGFR and DOR is not a simple relationship.
Figure 6.
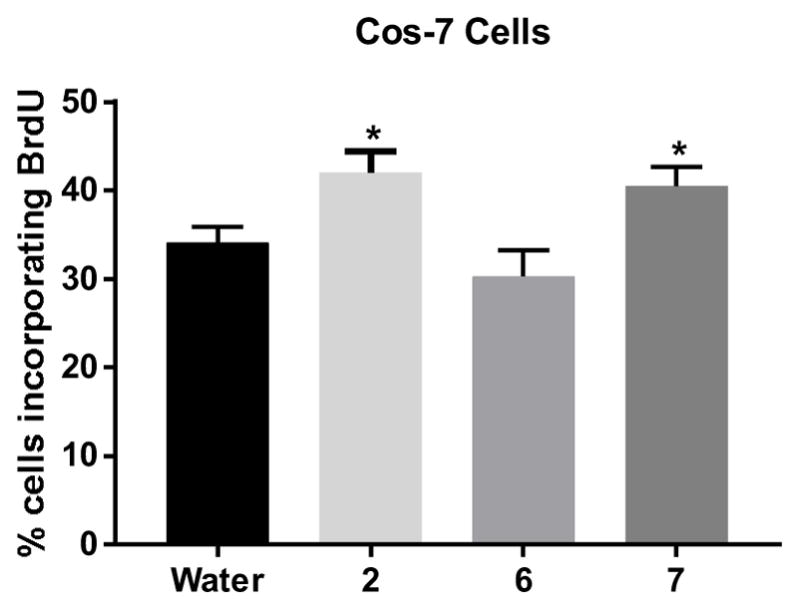
Cell growth for COS-7 cells at 72 hours. Cells were treated with 1 μM concentration of compounds. Cell growth was assessed by incorporation of BrdU. * P < 0.05
To assess further this pro-growth effect some of the compounds were tested in an additional cell line model. The OGF-OGFR axis is active in all human cancer cell lines examined.[35] A specific cell line that has received extensive testing in this regard is the human ovarian cancer cell line SKOV-3. Treatment of SKOV-3 with numerous opioid agonists specific for the MOR, DOR and KOR show that these ligands do not have growth effects in this cell line.[78] Cell growth by OGF-OGFR signaling can be induced by knock-down of the OGFR.[77, 79] Treatment with naltrexone in the presence of OGFR siRNA shows no additional growth enhancement in SKOV-3 cells indicating that the interaction with OGFR is the sole mechanism of growth induction.[78]
Treatment of SKOV-3 cells with these new amides revealed that compounds 4, 6 and 7 resulted in significantly enhanced growth in this model (Figure 7, left panel) at 2 time points in the logarithmic growth phase. Growth was only enhanced when cells were continuously treated with compound. In “low-dose” naltrexone regimens, a small dose of naltrexone is taken once a day leading to pulsed target engagement. Consistent with the effects of this pulsed naltrexone on the OGFR, treatment of SKOV-3 cells with pulsed dose of 6 hours of compound every other day led to significant decreases in cell growth with compounds 4, 6, and 7. The decrease in cell growth is, presumably, the result of an adaptive response to short duration target engagement leading to increased OGF and OGFR which when released leads to increased signaling leading to reduced cellular growth.[47]
Figure 7.

Treatment of SKOV-3 human derived ovarian cancer cells with compound 4, 6, and 7 at 10 μM concentration showed enhanced cell growth at 96, and 120 hour time points with 24 hour (constant) drug treatment (Left Panels), and reduced growth at the same time points when the “low-dose naltrexone” regimen of pulsed therapy is used (Right Panels). Here the “low-dose” therapy is achieved by pulsing drug on for 6 hours every other day.
Finally, a dose response experiment was conducted with the two compound 6 and 7, to further support that this is indeed a pharmacological effect. Treatment of SKOV-3 cells under both pulsed and continuous conditions do show that these compounds have a tendency to a dose response for both growth inhibitory and growth enhancing effects as would be expected of selective OGFR antagonists (Figure 8). In this experiment compound 7 shows significant activity at both 5 and 10 μM concentrations for the growth inhibition effects, while the amide 6 only inhibits growth at 10 μM indicating that 7 is the more potent compound of the two.
Figure 8.

Treatment of SKOV-3 human derived ovarian cancer cells with compounds 6 and 7 at four concentration showed enhanced cell growth at the 120 hour time point with 24 hour (constant) drug treatment ( Upper Panel), and reduced growth at the same time point when the “low-dose naltrexone” regimen of pulsed therapy is used (Lower Panels).
4. Conclusions
Here the first attempts to improve the “drug-likeness” of opioid receptor modulators derived from the pawhuskin family of natural products is described. We sought to synthesize analogues of our two lead structures, stilbenes 2 and 3, which demonstrate selectivity for the DOR and KOR respectively, through synthesis of analogues containing an amide linkage in place of the central stilbene connection. The synthesis of these compounds proved quite efficient and gave all four of the isomers desired, which should facilitate the preparation of additional analogues. Furthermore, the biological activity displayed by these compounds has been very interesting. Our hypothesis, based on molecular docking studies, was that the change to the amide substructure should not have large effects on the interactions of these compounds with the traditional receptors. This turned out to be incorrect: there was no activity of any of the amides (4–7) in any assays for agonism or antagonism at MOR, DOR, or KOR.
Because one of the endogenous ligands for the DOR is Met-enkephalin, also known as OGF, and because some stilbenes have DOR activity, the new compounds were tested for their effects at this receptor. Interestingly, three of the new amides, compounds 4, 6, and 7, display significant activity in this regard. While compound 6 showed activities in the SKOV-3 ovarian cancer assay of cell growth as potent as that of the other compounds it failed to show activity in the COS-7 cells. This could reflect the difference in concentration between the two assays. The dose response experiment as shown in Figure 8 provides some evidence to support this view indicating that the compound 7 is slightly more potent than 6 in the 1 – 10 μM range.
These results demonstrate the first selective antagonists of OGFR. Naltrexone is somewhat selective for DOR, and OGFR, but has significant effects at all the opioid receptors. This could be a significant disadvantage for the use of naltrexone-based OGFR antagonism to treat cancers where many patients are being treated for chronic pain. In such a setting naltrexone could interfere with pain management. Naltrexone also can cause acute gastrointestinal disturbances due to blockade of the MOR in the intestines, and it carries a label warning contraindicating use in patients with liver function problems. For these reasons, compounds 4, 6 and 7 can be used as leads to develop new drugs for the indications currently being treated with “low-dose” naltrexone therapy, including multiple sclerosis,[80] cancer,[81–83] and Crohn’s disease.[57] Further work on these lead compounds and OGFR signaling will be reported in due course.
5. Experimental section
5.1 Chemical Synthesis
5.1.1 (E)-N-(2-(3,7-Dimethylocta-2,6-dienyl)-3,5 dimethoxyphenyl)acetamide (9)
An oven-dried flask was charged with N-(3,5-dimethoxyphenyl)acetamide[68] (4.61 g, 23.6 mmol) in anhydrous dioxane (120 mL). To this solution was added BF3·OEt2 (2.33 mL, 18.9 mmol) dropwise via syringe. The flask was heated to 50 °C and then geraniol (2.07 mL, 11.8 mmol) in dioxane (20 mL) was added over 1 hour using a syringe pump. After the addition was complete, the flask was stirred at 50 °C overnight. The solution was then washed with H2O, the layers were separated, and the aqueous layer was extracted with CH2Cl2 (3x). The organic layers were combined, washed with brine and dried (Na2SO4). The mixture was filtered and the filtrate was concentrated in vacuo. Final purification of the resulting oil via flash chromatography (40% EtOAc in hexanes) afforded amide 9, 1.60 g, 40%) as a white solid along with a smaller amount of the symmetrical para substituted isomer. For 9: 1H NMR (400 MHz, CDCl3) δ 7.32 (s, 1H), 7.27 (d, J = 1.9 Hz, 1H), 6.29 (d, J = 2.0 Hz, 1H), 5.07 (m, 2H), 3.79 (s, 3H), 3.78 (s, 3H), 3.33 (d, J = 6.7 Hz, 2H), 2.10 (s, 3H), 2.09 – 1.98 (m, 4H), 1.82 (s, 3H), 1.66 (s, 3H), 1.58 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 168.2, 159.1, 157.9, 138.0, 136.6, 132.1, 123.9, 122.9, 112.0, 99.4, 95.8, 55.9, 55.5, 39.7, 26.7, 25.8, 24.7, 22.8, 17.8, 16.3; HRMS (ESI) m/z calcd for C20H30NO3 (M + H)+ 332.2226, found 332.2212.
5.1.2 (E)-2-(3,7-Dimethylocta-2,6-dienyl)-3,5-dimethoxyaniline (10).
To a stirred solution of amide 9 (400 mg, 1.21 mmol) in MeOH (5 mL) was added NaOH (386 mg, 9.66 mmol). The flask was fitted with a reflux condenser and the solution was heated at reflux for 24 hours. After the mixture was diluted with EtOAc and washed with saturated aqueous NH4Cl, the layers were separated and the aqueous layer was extracted with EtOAc (3x). The organic extracts were combined, washed with brine, and dried (Na2SO4). The mixture was filtered and the filtrate was concentrated in vacuo. Final purification using an ISCO Combiflash Rf chromatography gradient (0 to 20% EtOAc in hexanes) afforded amine 10 (250 mg, 86%) as a beige solid: 1H NMR (400 MHz, CDCl3) δ 5.98 (d, J = 2.1 Hz, 1H), 5.91 (d, J = 2.2 Hz, 1H), 5.11 – 5.02 (m, 2H), 3.77 (s, 3H), 3.75 (s, 3H), 3.67 (s, 2H), 3.26 (d, J = 5.8 Hz, 2H), 2.12 – 2.04 (m, 2H), 2.04 – 1.96 (m, 2H), 1.77 (s, 3H), 1.66 (s, 3H), 1.58 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 159.4, 158.8, 146.7, 136.2, 131.6, 124.4, 122.9, 107.2, 93.7, 89.7, 55.8, 55.3, 39.8, 26.8, 25.9, 22.6, 17.8, 16.2. HRMS (ESI) m/z calcd for C18H28NO2 (M + H)+ 290.2120, found 290.2101.
5.1.3 3,4-Bis(methoxymethoxy)-2-(3-methylbut-2-enyl)benzoic acid (12)
To a stirred solution of the corresponding aldehyde[61] (43 mg, 0.1 mmol) in 2–methyl–2-butene (4.2 mL, 39.7 mmol) and t–BuOH (1.2 mL) was added NaClO2 (137 mg, 1.5 mmol) and NaH2PO4 (112 mg, 0.9 mmol) in H2O (0.5 mL) over 11 min.[84] The reaction was allowed to stir for 3.5 h, and then was extracted with Et2O. The combined organic layers were dried (MgSO4), filtered, and concentrated in vacuo to afford acid 12 (54 mg, 100% by NMR) as a white solid: 1H NMR (400 MHz, CDCl3) δ 7.81 (d, J = 8.8 Hz, 1H), 7.05 (d, J = 8.8 Hz, 1H), 5.26 (s, 2H), 5.19 – 5.13 (m, 1H), 5.10 (s, Hz, 2H), 3.89 (d, J = 7.2 Hz, 2H), 3.62 (s, 3H), 3.51 (s, 3H), 1.77 (s, 3H), 1.67 (d, J = 1.0 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 172.6, 153.9, 145.0, 139.8, 131.9, 129.0, 123.4, 122.9, 112.7, 99.3, 94.8, 57.9, 56.6, 26.4, 25.9, 18.2.; HRMS (ESI) m/z calcd for C16H22O6Na (M + Na)+ 333.1314, found 333.1323.
5.1.4 (E)-N-(2-(3,7-Dimethylocta-2,6-dienyl)-3,5-dimethoxyphenyl)-3,4-bis(methoxymethoxy)-2-(3-methylbut-2-enyl)benzamide (13)
To a stirred solution of the acid 12 (98 mg, 0.32 mmol) and the amine 10 (41 mg, 0.14 mmol) in anhydrous CH2Cl2 (1.5 mL) was added 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC, 121 mg, 0.632 mmol) and 4-(dimethylamino)pyridine (DMAP, 4 mg, 0.03 mmol). The resulting solution was stirred at room temperature overnight and then washed with saturated aqueous NH4Cl. The layers were separated and the aqueous layer was extracted with CH2Cl2 (3x). The combined organic extracts were washed with brine and dried (Na2SO4). The mixture was filtered and the filtrate was concentrated in vacuo. Final purification using an ISCO Combiflash Rf chromatography gradient (0 to 30% EtOAc in hexanes) afforded amide 13 (55 mg, 67%) as a yellow oil: For 13: 1H NMR (400 MHz, CDCl3) δ 7.60 (s, 1H), 7.47 (s, 1H), 7.10 (d, J = 8.3 Hz, 1H), 7.00 (d, J = 8.4 Hz, 1H), 6.31 (d, J = 2.1 Hz, 1H), 5.24 – 5.18 (m, 3H), 5.12 (s, 2H), 5.06 – 4.98 (m, 2H), 3.83 (s, 3H), 3.80 (s, 3H), 3.67 (d, J = 6.6 Hz, 2H), 3.60 (s, 3H), 3.50 (s, 3H), 3.29 (d, J = 6.8 Hz, 2H), 1.98 – 1.91 (m, 2H), 1.91 – 1.83 (m, 2H), 1.64 (s, 3H), 1.62 (s, 6H), 1.55 (s, 3H), 1.41 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 167.7, 159.1, 157.9, 151.5, 145.3, 138.4, 137.2, 135.3, 132.4, 131.8, 124.1, 123.2, 122.1, 113.4, 111.7, 99.4, 98.9, 95.8, 95.1, 57.8, 56.4, 55.9, 55.6, 39.8, 26.8, 26.2, 25.9, 25.8, 22.7, 18.2, 17.8, 15.9; HRMS (ESI) m/z calcd for C34H48NO7 (M + H)+ 582.3431, found 582.3429.
5.1.5 (E)-N-(2-(3,7-Dimethylocta-2,6-dienyl)-3,5-dimethoxyphenyl)-3,4-dihydroxy-2-(3-methylbut-2-enyl)benzamide (4)
To a stirred solution of amide 13 (25 mg, 43 μmol) in MeOH (4.3 mL) was added p-TsOH·H2O (31 mg, 0.16 mmol). The flask was sealed and stirred overnight at room temperature. The solution was diluted with EtOAc and then washed with saturated aqueous NaHCO3. The layers were separated and the aqueous layer was extracted with EtOAc (3x). The combined organic extracts were washed with brine and dried (Na2SO4). After the mixture was filtered and the filtrate was concentrated in vacuo, final purification using an ISCO Combiflash Rf chromatography gradient (20 to 60% EtOAc in hexanes) afforded amide 4 (8 mg, 38%) as an off-white solid: 1H NMR (400 MHz, CDCl3) δ 7.74 (s, 1H), 7.47 (s, 1H), 6.89 (d, J = 8.2 Hz, 1H), 6.73 (d, J = 8.2 Hz, 1H), 6.32 (d, J = 2.3 Hz, 1H), 6.10 (s, 1H), 5.78 (s, 1H), 5.31 – 5.23 (m, 1H), 5.08 – 4.98 (m, 2H), 3.82 (s, 3H), 3.80 (s, 3H), 3.60 (d, J = 6.7 Hz, 2H), 3.31 (d, J = 6.7 Hz, 2H), 1.99 – 1.85 (m, 4H), 1.75 (s, 3H), 1.73 (s, 3H), 1.64 (s, 3H), 1.54 (s, 3H), 1.46 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 168.1, 159.2, 158.0, 146.5, 143.0, 138.4, 137.5, 135.7, 131.9, 129.5, 126.7, 124.0, 122.1, 122.0, 119.9, 112.8, 111.8, 98.6, 95.8, 55.9, 55.6, 39.7, 26.8, 26.7, 25.9, 25.8, 22.8, 18.1, 17.8, 16.2; HRMS (ESI) m/z calcd for C30H40NO5 (M + H)+ 494.2906, found 494.2912, ; HRMS (ESI) m/z calcd for C30H40NO5 (M + H)+ 494.2906, found 494.2912.
5.1.6 3,4-Bis(methoxymethoxy)-5-(3-methylbut-2-enyl)benzaldehyde (14)
To a stirred solution of the corresponding alcohol[61, 71] (368 mg, 1.24 mmol) and pyridine (0.300 mL, 3.72 mmol) in CH2Cl2 (4 mL) was added Dess-Martin periodinane (683 mg, 1.61 mmol). The resulting solution was stirred at room temperature for 1 hour and then washed with 2M NaOH. The layers were separated and the aqueous layer was extracted with CH2Cl2 (3x). The organic extracts were combined, washed with brine, and dried (Na2SO4). After the mixture was filtered and the filtrate was concentrated in vacuo, final purification using an ISCO Combiflash Rf chromatography gradient (0 to 40% EtOAc in hexanes) afforded aldehyde 14 (315 mg, 86%) as a yellow solid:[85] 1H NMR (400 MHz, CDCl3) δ 9.86 (s, 1H), 7.52 (d, J = 1.9 Hz, 1H), 7.39 (d, J = 1.9 Hz, 1H), 5.32 (m, 1H), 5.25 (s, 2H), 5.23 (s, 2H), 3.60 (s, 3H), 3.51 (s, 3H), 3.47 (d, J = 7.3 Hz, 2H), 1.77 (s, 3H), 1.73 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 191.4, 150.3, 150.2, 136.9, 133.9, 132.7, 126.0, 121.7, 114.1, 99.2, 95.2, 57.8, 56.6, 28.6, 25.9, 18.0; HRMS (TOF MS EI) m/z calcd for C16H22O5 (M+) 294.1467, found 294.1472.
5.1.7 3,4-Bis(methoxymethoxy)-5-(3-methylbut-2-enyl)benzoic acid (15)
To a stirred solution of aldehyde 14 (306 mg, 1.04 mmol) and 2-methyl-2-butene (2.20 mL, 20.8 mmol) in t-BuOH (10.4 mL) was added NaH2PO4 (437 mg, 3.64 mmol) and NaClO2 (564 mg, 6.24 mmol) in H2O (3.5 mL) via syringe. The solution was stirred vigorously at room temperature for 3.5 hours and then washed with saturated aqueous NH4Cl. The mixture was extracted with EtOAc (3x) and the combined organic extracts were washed with brine and dried (Na2SO4). The mixture was filtered and filtrate was concentrated in vacuo. Final purification using an ISCO Combiflash Rf chromatography gradient (10 to 30% EtOAc in hexanes) afforded the carboxylic acid 15 (104 mg, 69%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 7.72 (d, J = 2.0 Hz, 1H), 7.64 (d, J = 2.0 Hz, 1H), 5.31 (m, 1H), 5.24 (s, 2H), 5.22 (s, 2H), 3.60 (s, 3H), 3.52 (s, 3H), 3.45 (d, J = 7.2 Hz, 2H), 1.76 (d, J = 0.8 Hz, 3H), 1.73 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 171.7, 149.7, 149.3, 136.2, 133.5, 125.6, 124.9, 121.9, 115.8, 99.0, 95.2, 57.6, 56.4, 28.6, 25.8, 17.9; HRMS (TOF MS EI) m/z calcd for C16H22O6Na (M + Na)+ 333.1314, found 333.1309.
5.1.8 (E)-N-(2-(3,7-Dimethylocta-2,6-dienyl)-3,5-dimethoxyphenyl)-3,4-bis(methoxymethoxy)-5-(3-methylbut-2-enyl)benzamide (16)
To a stirred solution of the carboxylic acid 15 (18 mg, 58 μmol) and amine 10 (20 mg, 70 μmol) in anhydrous CH2Cl2 (1 mL) was added EDC (33 mg, 0.17 mmol) and DMAP (1.5 mg, 12 μmol). The solution was stirred at room temperature for 1.5 days and then washed with saturated aqueous NH4Cl. The layers were separated and the aqueous layer was extracted with CH2Cl2 (3x). The combined organic extracts were washed with brine, dried over Na2SO4, and filtered, and the filtrate was concentrated in vacuo. Final purification using an ISCO Combiflash Rf chromatography gradient (0 to 80% EtOAc in hexanes) afforded 16 (18.8 mg, 56%) as a yellow oil: 1H NMR (400 MHz, CDCl3) δ 7.88 (s, 1H), 7.52 (d, J = 2.1 Hz, 1H), 7.44 (d, J = 1.8 Hz, 1H), 7.22 (d, J = 2.1 Hz, 1H), 6.33 (d, J = 2.4 Hz, 1H), 5.30 – 5.24 (m, 2H), 5.22 (s, 2H), 5.18 (s, 2H), 5.15 – 5.09 (m, 1H), 5.05 – 4.99 (m, 1H), 3.82 (s, 3H), 3.80 (s, 3H), 3.59 (s, 3H), 3.50 (s, 3H), 3.45 (d, J = 6.9 Hz, 2H), 3.37 (d, J = 6.3 Hz, 2H), 2.02 – 1.96 (m, 4H), 1.73 (d, J = 1.1 Hz, 3H), 1.72 (s, 3H), 1.66 (d, J = 0.9 Hz, 3H), 1.61 (s, 3H), 1.52 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 165.4, 159.1, 158.0, 150.0, 148.1, 137.9, 137.5, 136.5, 133.3, 131.3, 124.0 (2C), 122.3, 122.1, 113.8, 112.7, 99.2 (2C), 96.1, 95.5, 57.7, 56.6, 55.9, 55.6, 39.7, 29.0, 26.7, 25.8, 25.7, 22.8, 18.1, 17.8, 16.6; HRMS (TOF MS ES+) m/z calculated for C34H48NO7 (M+H)+ 582.3431, found 582.3437.
5.1.9 (E)-N-(2-(3,7-Dimethylocta-2,6-dienyl)-3,5-dimethoxyphenyl)-3,4-dihydroxy-5-(3-methylbut-2-enyl)benzamide (5)
To a stirred solution of amide 16 (24.6 mg, 42 μmol) in MeOH (4.2 mL) was added p-TsOH·H2O (31 mg, 0.16 mmol). The flask was sealed and stirred for 48 hours at room temperature. The solution was diluted with EtOAc and then washed with saturated aqueous NaHCO3. The layers were separated and the aqueous layer was extracted with EtOAc (3x). The combined organic extracts were washed with brine and dried (Na2SO4). After the mixture was filtered and the filtrate was concentrated in vacuo, final purification using an ISCO Combiflash Rf chromatography gradient (20 to 60% EtOAc in hexanes) afforded amide 5 (10.3 mg, 49%) as an off-white solid: 1H NMR (500 MHz, CDCl3) δ 8.04 (s, 1H), 7.82 (s, 1H), 7.64 (d, J = 1.6 Hz, 1H), 7.31 (s, 1H), 7.01 (s, 1H), 6.34 (d, J = 2.0 Hz, 1H), 6.01 (s, 1H), 5.27 (t, J = 6.5 Hz, 1H), 5.13 (t, J = 5.8 Hz, 1H), 5.08 – 4.99 (m, 1H), 3.83 (s, 3H), 3.81 (s, 3H), 3.37 (d, J = 6.5 Hz, 4H), 2.04 (m, 4H), 1.74 (s, 6H), 1.67 (s, 3H), 1.62 (s, 3H), 1.53 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 166.4, 159.1, 158.1, 146.6, 144.3, 137.9, 137.8, 134.2, 131.9, 127.6, 126.3, 124.1, 122.3, 121.8, 119.8, 113.6, 113.3, 99.6, 96.3, 56.0, 55.6, 39.7, 29.3, 26.7, 25.9, 25.7, 22.9, 18.1, 17.8, 16.7; HRMS (ESI) m/z calcd for C30H40NO5 (M + H)+ 494.2906, found 494.2905.
5.1.10 Methyl 2-bromo-3,5-dimethoxybenzoate (17)
According to the published procedures,[72, 73, 86] to a stirred solution of methyl 3,5-dimethoxybenzoate (2.0 g, 10 mmol) in CH3CN (100 mL) at 0 °C was added N-bromosuccinimide (2.0 g, 11.2 mmol). After the addition was complete, the solution was allowed to equilibrate to room temperature and stirred for an additional 3.5 hours. The reaction was then quenched by the addition of saturated aqueous Na2S2O3. A majority of the CH3CN was removed under reduced pressure and then the mixture was washed with saturated aqueous NH4Cl. The mixture was extracted with EtOAc (3x) and the combined organic extracts were washed with brine, dried (Na2SO4), and filtered. After the filtrate was concentrated in vacuo, final purification via flash chromatography (25% EtOAc in hexanes) afforded bromide 17 as a yellow solid (2.29 g, 82%): 1H NMR (400 MHz, CDCl3) δ 6.80 (d, J = 2.8 Hz, 1H), 6.58 (d, J = 2.8 Hz, 1H), 3.93 (s, 3H), 3.89 (s, 3H), 3.82 (s, 3H).[86]
5.1.11 (E)-Methyl 2-(3,7-dimethylocta-2,6-dienyl)-3,5-dimethoxybenzoate (18)
An oven-dried flask was charged with ester 17 (1.0 g, 3.64 mmol) in anhydrous THF (25 mL) at −78 °C under argon. To this solution was added n-BuLi (2.5 M in hexanes, 1.6 mL, 4.00 mmol) dropwise via syringe. The solution was stirred at −78 °C for 15 minutes and then CuBr·DMS (0.820 g, 4.00 mmol) was added. After this solution was stirred an additional 15 minutes at −78 °C, geranyl bromide (0.870 g, 4.00 mmol) was added dropwise via syringe. The solution was allowed to equilibrate to room temperature overnight and then the reaction was quenched by the addition of saturated aqueous NH4Cl. The layers were separated and the aqueous layer was extracted with EtOAc (3x). The organic extracts were combined, washed with brine, and dried (Na2SO4). The mixture was filtered and the filtrate was concentrated in vacuo. Final purification using an ISCO Combiflash Rf chromatography gradient (0 to 20% EtOAc in hexanes) afforded ester 18 (847 mg, 70%) as a yellow oil: 1H NMR (400 MHz, CDCl3) δ 6.88 (d, J = 2.5 Hz, 1H), 6.60 (d, J = 2.5 Hz, 1H), 5.18 – 5.12 (m, 1H), 5.11 – 5.01 (m, 1H), 3.89 (s, 3H), 3.84 (s, 3H), 3.83 (s, 3H), 3.59 (d, J = 6.7 Hz, 2H), 2.10 – 2.03 (m, 2H), 2.02 – 1.94 (m, 2H), 1.75 (s, 3H), 1.67 (d, J = 1.0 Hz, 3H), 1.60 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 168.9, 159.0, 158.3, 134.9, 132.1, 131.3, 124.6, 124.4, 123.5, 105.1, 102.3, 56.0, 55.6, 52.2, 39.9, 26.9, 25.8, 25.2, 17.8, 16.3; HRMS (TOF MS ES+) m/z calcd for C20H28O4 (M + Na)+ 355.1885, found 355.1888.
5.1.12 (E)-2-(3,7-Dimethylocta-2,6-dienyl)-3,5-dimethoxybenzoic acid (19)
To a stirred solution of ester 18 (520 mg, 1.56 mmol) in CH2Cl2 (9 mL) and MeOH (1 mL) was added NaOH (1.2 g, 30 mmol). The solution was stirred vigorously at room temperature overnight and then diluted with saturated aqueous NH4Cl. The layers were separated and the aqueous layer was extracted with CH2Cl2 (3x). The combined organic extracts were washed with brine, dried (Na2SO4), and filtered, and the filtrate was concentrated in vacuo. Final purification using an ISCO Combiflash Rf chromatography gradient (20 to 40% EtOAc in hexanes) afforded the carboxylic acid 19 (239 mg, 48%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 7.05 (d, J = 2.5 Hz, 1H), 6.64 (d, J = 2.5 Hz, 1H), 5.19 – 5.12 (m, 1H), 5.08 – 5.02 (m, 1H), 3.83 (s, 3H), 3.82 (s, 3H), 3.68 (d, J = 6.7 Hz, 2H), 2.08 – 2.00 (m, 2H), 1.99 – 1.91 (m, 2H), 1.75 (d, J = 1.0 Hz, 3H), 1.63 (d, J = 0.9 Hz, 3H), 1.56 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 173.8, 159.1, 158.3, 135.0, 131.3, 130.5, 125.9, 124.6, 123.4, 105.8, 103.4, 56.0, 55.6, 40.0, 26.9, 25.8, 25.2, 17.8, 16.3; HRMS (TOF MS ES+) m/z calcd for C19H26O4Na (M + Na)+ 341.1729, found 341.1732.
5.1.13 3,4-Bis(methoxymethoxy)-2-(3-methylbut-2-enyl)aniline (20)
An oven-dried flask was charged with the carboxylic acid 12 (66 mg, 0.21 mmol) in anhydrous benzene. To this stirred solution was added triethylamine (0.300 mL, 2.13 mmol) followed by the dropwise addition of diphenyl phosphoryl azide (0.460 mL, 2.13 mmol) via syringe. The solution was stirred at room temperature for 15 minutes, and then the flask was fitted with a reflux condenser and heated at reflux overnight. The benzene was removed under reduced pressure and the residue was dissolved in THF (2.1 mL). To this solution was added aqueous 4N LiOH (1.1 mL, 4.5 mmol) and the resulting mixture was stirred vigorously at room temperature for 1 hour. The mixture was diluted with H2O and the layers were separated. The aqueous layer was extracted with EtOAc (3x) and the combined organic extracts were washed with brine and dried (Na2SO4). After the mixture was filtered and the filtrate was concentrated in vacuo, final purification using an ISCO Combiflash Rf chromatography gradient (20 to 40% EtOAc in hexanes) afforded amine 20 (44 mg, 74%) as a yellow oil: 1H NMR (400 MHz, CDCl3) δ 6.85 (d, J = 8.6 Hz, 1H), 6.39 (d, J = 8.6 Hz, 1H), 5.11 – 5.09 (m, 1H), 5.09 (s, 2H), 5.08 (s, 2H), 3.57 (s, 3H), 3.50 (s, 3H), 3.39 (d, J = 6.5 Hz, 2H), 1.80 (d, J = 0.8 Hz, 3H), 1.72 (d, J = 1.3 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 146.0, 142.7, 141.2, 133.4, 122.1, 121.5, 116.5, 111.3, 99.5, 96.5, 57.7, 56.2, 25.8, 24.5, 18.1.; HRMS (TOF MS ES+) m/z calcd for C15H23NO4 (M + H)+ 282.1699, found 282.1705.
5.1.14 (E)-N-(3,4-Bis(methoxymethoxy)-2-(3-methylbut-2-enyl)phenyl)-2-(3,7-dimethylocta-2,6-dienyl)-3,5-dimethoxybenzamide (21)
To a stirred solution of amine 20 (34 mg, 0.12 mmol) and carboxylic acid 19 (42 mg, 0.13 mmol) in anhydrous CH2Cl2 (1 mL) was added EDC (69 mg, 0.36 mmol) and DMAP (3 mg, 24 μmol). The solution was stirred at room temperature overnight and then was washed with saturated aqueous NH4Cl. The layers were separated and the aqueous layer was extracted with CH2Cl2 (3x). The combined organic extracts were washed with brine, dried (Na2SO4), and filtered, and the filtrate was concentrated in vacuo. Final purification using an ISCO Combiflash Rf chromatography gradient (10 to 40% EtOAc in hexanes) afforded amide 21 (44 mg, 63%) as a yellow oil: 1H NMR (400 MHz, CDCl3) δ 7.80 (d, J = 8.9 Hz, 1H), 7.53 (s, 1H), 7.07 (d, J = 9.0 Hz, 1H), 6.52 (s, 2H), 5.20 – 5.14 (m, 3H), 5.09 (s, 2H), 5.08 – 5.01 (m, 2H), 3.83 (s, 3H), 3.79 (s, 3H), 3.56 (s, 3H), 3.51 (s, 3H), 3.45 (d, J = 6.8 Hz, 2H), 3.41 (d, J = 6.6 Hz, 2H), 2.02 – 1.96 (m, 2H), 1.95 – 1.87 (m, 2H), 1.63 (s, 3H), 1.59 (s, 6H), 1.55 (s, 3H), 1.37 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 168.0, 159.0, 158.9, 147.1, 145.0, 138.8, 135.4, 134.2, 131.8, 131.4, 126.7, 124.5, 123.3, 121.8, 120.9, 119.0, 114.9, 102.8, 100.2, 99.6, 95.6, 57.8, 56.4, 55.9, 55.6, 40.0, 27.0, 25.8, 25.7, 25.3, 24.8, 17.8, 17.6, 16.3; HRMS (TOF MS ES+) m/z calcd for C34H48NO7 (M + H)+ 582.3431, found 582.3425.
5.1.15 (E)-N-(3,4-Dihydroxy-2-(3-methylbut-2-enyl)phenyl)-2-(3,7-dimethylocta-2,6-dienyl)-3,5-dimethoxybenzamide (6)
To a stirred solution of amide 21 (28 mg, 48 μmol) in MeOH (4.8 mL) was added p-TsOH·H2O (35 mg, 0.18 mmol). The flask was sealed and stirred overnight at room temperature. The solution was diluted with EtOAc and then washed with saturated aqueous NaHCO3. The layers were separated and the aqueous layer was extracted with EtOAc (3x). The combined organic extracts were washed with brine, dried (Na2SO4), and filtered, and the filtrate was concentrated in vacuo. Final purification using an ISCO Combiflash Rf chromatography gradient (0 to 40% EtOAc in hexanes) afforded amide 6 (7 mg, 30%) as an off-white solid: 1H NMR (400 MHz, CDCl3) δ 7.46 (s, 1H), 6.97 (d, J = 8.5 Hz, 1H), 6.64 (d, J = 2.4 Hz, 1H), 6.58 (d, J = 8.6 Hz, 1H), 6.56 (d, J = 2.4 Hz, 1H), 5.17 (t, J = 6.1 Hz, 1H), 5.12 (t, J = 7.0 Hz, 1H), 5.03 (t, J = 6.7 Hz, 1H), 3.84 (s, 3H), 3.82 (s, 3H), 3.50 (d, J = 6.7 Hz, 2H), 3.33 (d, J = 6.6 Hz, 2H), 2.02 – 1.96 (m, 2H), 1.96 – 1.90 (m, 2H), 1.63 (s, 6H), 1.63 (s, 3H), 1.55 (s, 3H), 1.54 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 169.6, 159.1, 159.0, 143.1, 142.5, 137.8, 136.1, 134.1, 131.5, 128.2, 124.4, 123.3, 122.3, 121.7, 121.0, 117.1, 113.5, 103.3, 100.7, 56.0, 55.7, 39.9, 29.9, 26.9, 25.8 (2C), 25.6, 24.8, 17.8, 16.4; HRMS (TOF MS ES+) m/z calcd for C30H40NO5 (M + H)+ 494.2906, found 494.2905.
5.1.16 3,4-Bis(methoxymethoxy)-5-(3-methylbut-2-enyl)aniline (22)
An oven-dried flask was charged with carboxylic acid 15 (77 mg, 0.25 mmol) in anhydrous benzene (25 mL). To this stirred solution was added triethylamine (0.345 mL, 2.50 mmol) followed by the dropwise addition of diphenyl phosphoryl azide (0.536 mL, 2.50 mmol) via syringe. The solution was stirred at room temperature for 15 minutes, and then the flask was fitted with a reflux condenser and heated at reflux overnight. The benzene was removed under reduced pressure and the residue was dissolved in THF (2 mL). To this solution was added aqueous 4N LiOH (1.25 mL, 5.00 mmol) and the resulting mixture was stirred vigorously at room temperature for 2 hours. The mixture was diluted with H2O and the layers were separated. The aqueous layer was extracted with EtOAc (3x) and the organic extracts were combined, washed with brine, and dried (Na2SO4). After the mixture was filtered and the filtrate was concentrated in vacuo, final purification using an ISCO Combiflash Rf chromatography gradient (0 to 40% EtOAc in hexanes) afforded amine 22 (55 mg, 79%) as a yellow oil: 1H NMR (400 MHz, CDCl3) δ 6.38 (d, J = 2.7 Hz, 1H), 6.16 (d, J = 2.7 Hz, 1H), 5.31 – 5.24 (m, 1H), 5.14 (s, 2H), 4.99 (s, 2H), 3.59 (s, 3H), 3.49 (s, 3H), 3.32 (d, J = 7.2 Hz, 2H), 1.74 (d, J = 1.1 Hz, 3H), 1.70 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 150.5, 143.0, 137.1, 136.7, 132.5, 122.8, 109.3, 101.7, 99.3, 95.1, 57.4, 56.1, 28.5, 25.8, 17.8; HRMS (TOF MS ES+) m/z calcd for C15H23NO4 (M + H)+ 282.1704, found 282.1705.
5.1.17 (E)-N-(3,4-Bis(methoxymethoxy)-5-(3-methylbut-2-enyl)phenyl)-2-(3,7-dimethylocta-2,6-dienyl)-3,5-dimethoxybenzamide (23)
To a stirred solution of amine 22 (43 mg, 0.15 mmol) and carboxylic acid 19 (58 mg, 0.18 mmol) in anhydrous CH2Cl2 (1 mL) was added EDC (88 mg, 0.36 mmol) and DMAP (4 mg, 31 μmol). The solution was stirred at room temperature overnight and then washed with saturated aqueous NH4Cl. The layers were separated and the aqueous layer was extracted with CH2Cl2 (3x). The combined organic extracts were washed with brine, dried (Na2SO4), filtered, and the filtrate was concentrated in vacuo. Final purification using an ISCO Combiflash Rf chromatography gradient (10 to 40% EtOAc in hexanes) afforded amide 23 (49 mg, 55%) as a yellow oil: 1H NMR (400 MHz, CDCl3) δ 7.47 (d, J = 2.5 Hz, 1H), 7.44 (s, 1H), 6.87 (d, J = 2.5 Hz, 1H), 6.65 (d, J = 2.4 Hz, 1H), 6.53 (d, J = 2.3 Hz, 1H), 5.31 – 5.25 (m, 1H), 5.22 – 5.17 (m, 3H), 5.09 – 5.03 (m, 3H), 3.83 (s, 3H), 3.80 (s, 3H), 3.59 (s, 3H), 3.51 (s, 3H), 3.44 (d, J = 6.2 Hz, 3H), 3.40 (d, J = 7.1 Hz, 2H), 2.08-2.01 (m, 2H), 2.01 – 1.95 (m, 2H), 1.73 (s, 3H), 1.71 (s, 3H), 1.64 (s, 3H), 1.60 (s, 3H), 1.55 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 167.7, 159.1, 158.9, 150.0, 141.5, 138.5, 136.5, 134.5, 133.1, 131.5, 124.4, 124.2, 123.4, 122.5, 120.1, 114.2, 106.6, 103.3, 100.8, 99.3, 95.4, 57.6, 56.5, 55.9, 55.7, 39.7, 28.7, 28.2, 26.8, 25.9, 25.8, 18.0, 17.8, 16.6; HRMS (TOF MS ES+) m/z calcd for C34H48NO7 (M + H)+ 582.3431, found 582.3431.
5.1.18 (E)-N-(3,4-Dihydroxy-2-(3-methylbut-2-enyl)phenyl)-2-(3,7-dimethylocta-2,6-dienyl)-3,5-dimethoxybenzamide (7)
To a stirred solution of amide 23 (17.4 mg, 30 μmol) in MeOH (3 mL) was added p-TsOH·H2O (22 mg, 0.11 mmol). The flask was sealed and stirred overnight at room temperature. The solution was diluted with EtOAc and then washed with saturated aqueous NaHCO3. The layers were separated and the aqueous layer was extracted with EtOAc (3x). The combined organic extracts were washed with brine, dried (Na2SO4), and filtered, and the filtrate was concentrated in vacuo. Final purification using an ISCO Combiflash Rf chromatography gradient (0 to 40% EtOAc in hexanes) afforded amide 7 (14.8 mg, 53%) as a yellow oil: 1H NMR (400 MHz, CDCl3) δ 8.14 (d, J = 2.4 Hz, 1H), 7.55 (s, 1H), 6.68 (d, J = 2.4 Hz, 1H), 6.58 (d, J = 2.4 Hz, 1H), 6.17 (d, J = 2.4 Hz, 1H), 5.30 – 5.23 (m, 1H), 5.19 (t, J = 5.9 Hz, 1H), 5.06 (t, J = 6.7 Hz, 1H), 3.85 (s, 3H), 3.82 (s, 3H), 3.44 (d, J = 6.1 Hz, 2H), 3.30 (d, J = 7.1 Hz, 2H), 2.09 – 2.02 (m, 2H), 2.02 – 1.96 (m, 2H), 1.73 (s, 3H), 1.69 (s, 3H), 1.64 (s, 3H), 1.56 (s, 3H), 1.55 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 168.6, 159.2, 159.0, 144.3, 140.0, 138.0, 137.2, 133.4, 131.7, 130.1, 127.4, 124.3, 123.2, 122.2, 119.9, 111.3, 105.7, 103.3, 101.1, 56.0, 55.7, 39.7, 28.3, 26.8, 26.0, 25.9, 25.8, 17.9, 17.8, 16.8; HRMS (ESI) m/z calcd for C30H40NO5 (M + H)+ 494.2906, found 494.2906.
5.2 Biological assays
5.2.1 Cell Culture
Two cell lines were used in these studies SKOV3 COS-7 cells were obtained from the American Type Culture Collection (Manassas, VA, USA). SKOV-3 cells were grown in a humidified atmosphere of 5% CO295% air at 37°C in RPMI 1640 supplemented with 10% fetal calf serum and antibiotics (5000 units/mL penicillin, 5 μg/mL streptomycin, and 10mg/mL neomycin) unless otherwise noted. COS-7 cells were grown in under the same general conditions in DMEM.
5.2.2 Cell Growth
SKOV3 and COS-7 cells were plated at 20,000 cells/well in 24 well plates and counted 24 hours later for time 0 to determine seeding efficiency. Cells were then treated with Naltrexone (Tocris Biosciences, Bristol, United Kingdom), various different combinations of compounds 2–7 or sterile water at 10−6 mol/L for 24 hours daily or 6 hours every 48 hours. At the end of the 6 hours treatment, the media containing the drugs were removed and replaced with fresh media containing no drugs. Media with and without drugs were changed daily. At time points 0, 24, 48, 72, 96, and 120 hours of treatment, cells were harvested with trypsin and counted with a hemacytometer.
5.2.3 β-Arrestin Assay
β-Arrestin express GPCR assay kits were obtained from DiscoverX (Fremont, CA, USA) and protocol provided was followed. Briefly, cells expressing MOR, DOR, or KOR were plated at 100μL per well in the provided 96well plate in cell plating reagent. The cells were incubated in a humidified atmosphere of 5% CO2, 95% air at 37°C for 48 hours. Cells were then treated with 10μL of 11x of known agonist (DADLE for DOR, Met5-Enkephalin for MOR, and Dynorphin A for KOR (Tocris Biosciences, Bristol, United Kingdom)), compound 6, or 7 for 180 minutes at concentration starting at 5μM and decreasing at a 1:3 ratio titration and incubated in a humidified atmosphere of 5% CO295% air at 37°C. Working solution was added after the 180 minutes and the plates were allowed to incubate at room temperature in the dark for 1 hour, then read for chemiluminescent signal.
5.2.4 5-Bromo-2=-deoxyuridine incorporation study
To determine DNA synthesis, 5-bromo-2-deoxyuridine (BrdU) was utilized. COS-7 cells growing on 22-mm coverslips were incubated compounds (naltrexone, 6 and 7) for 24 h. Fresh, complete DMEM medium with 30 M BrdU (Sigma) was added to each culture for 3 h, and cells were stained according to our prior procedures.[78] Cells were counterstained with DAPI (1 g/ml) and sealed onto glass slides. At least 100 cells/treatment were counted in a masked fashion for each experiment
Supplementary Material
Scheme 1.

Synthesis of amides 4 and 5.
Scheme 2.

Synthesis of compounds 6 and 7.
Acknowledgments
We thank the Center for Biocatalysis and Bioprocessing for a fellowship (to D. P. S.) through the predoctoral Training Program in Biotechnology (T32 GM008365). Financial support from the Roy J. Carver Charitable Trust (01-224 to D. F. W.) is gratefully acknowledged.
Footnotes
The 1H and 13C NMR spectra for all new compounds.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Trescot AM, Datta S, Lee M, Hansen H. Opioid Pharmacology. Pain Physician. 2008;11:S133–S153. [PubMed] [Google Scholar]
- 2.Ramoutsaki IA, Askitopoulou H, Konsolaki E. Pain relief and sedation in Roman Byzantine texts: Mandragoras officinarum, Hyoscyamos niger and Atropa belladonna. In: Diz JC, Franco A, Bacon DR, Rupreht J, Alvarez J, editors. History of Anesthesia. 2002. pp. 43–50. [Google Scholar]
- 3.Inturrisi CE. Clinical pharmacology of opioids for pain. Clinical Journal of Pain. 2002;18:S3–S13. doi: 10.1097/00002508-200207001-00002. [DOI] [PubMed] [Google Scholar]
- 4.Dhawan BN, Cesselin F, Raghubir R, Reisine T, Bradley PB, Portoghese PS, Hamon M International union of pharmacology. 12. Classification of opioid receptors. Pharmacological Reviews. 1996;48:567–592. [PubMed] [Google Scholar]
- 5.Portoghese PS, Sultana M, Takemori AE. Naltrindole, a highly selective and potent non-peptide-delta opioid receptor antagonist. European Journal of Pharmacology. 1988;146:185–186. doi: 10.1016/0014-2999(88)90502-x. [DOI] [PubMed] [Google Scholar]
- 6.Koob GF, LeMoal M. Drug abuse: Hedonic homeostatic dysregulation. Science. 1997;278:52–58. doi: 10.1126/science.278.5335.52. [DOI] [PubMed] [Google Scholar]
- 7.Portoghese PS. From models to molecules: Opioid receptor dimers, bivalent ligands, and selective opioid receptor probes. J Med Chem. 2001;44:2259–2269. doi: 10.1021/jm010158+. [DOI] [PubMed] [Google Scholar]
- 8.Portoghese PS, Sultana M, Nagase H, Takemori AE. Application of the message adress concept in the design of highly potent and selective non-peptide delta-opioid receptor antagonists. J Med Chem. 1988;31:281–282. doi: 10.1021/jm00397a001. [DOI] [PubMed] [Google Scholar]
- 9.Eguchi M. Recent advances in selective opioid receptor agonists and antagonists. Medicinal Research Reviews. 2004;24:182–212. doi: 10.1002/med.10059. [DOI] [PubMed] [Google Scholar]
- 10.Kieffer BL, Befort K, Gaveriauxruff C, Hirth CG. The delta-opioid receptor - Isolation of a CDNA by expression cloning and pharmacological characterization. Proc Natl Acad Sci U S A. 1992;89:12048–12052. doi: 10.1073/pnas.89.24.12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Y, Mestek A, Liu J, Hurley JA, Yu L. Molecular cloning and functiional expression of a mu-opioid receptor from rat brain. Mol Pharmacol. 1993;44:8–12. [PubMed] [Google Scholar]
- 12.Raynor K, Kong HY, Chen Y, Yasuda K, Yu L, Bell GI, Reisine T. Pharmacological characterization of the cloned kappa-opioid, delta-opioid and mu-opioid receptors. Mol Pharmacol. 1994;45:330–334. [PubMed] [Google Scholar]
- 13.Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S. Crystal structure of the mu-opioid receptor bound to a morphinan antagonist. Nature. 2012;485:321–U170. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Granier S, Manglik A, Kruse AC, Kobilka TS, Thian FS, Weis WI, Kobilka BK. Structure of the delta-opioid receptor bound to naltrindole. Nature. 2012;485:400–U171. doi: 10.1038/nature11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu HX, Wacker D, Mileni M, Katritch V, Han GW, Vardy E, Liu W, Thompson AA, Huang XP, Carroll FI, Mascarella SW, Westkaemper RB, Mosier PD, Roth BL, Cherezov V, Stevens RC. Structure of the human kappa-opioid receptor in complex with JDTic. Nature. 2012;485:327–U369. doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thompson AA, Liu W, Chun E, Katritch V, Wu HX, Vardy E, Huang XP, Trapella C, Guerrini R, Calo G, Roth BL, Cherezov V, Stevens RC. Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature. 2012;485:395–U150. doi: 10.1038/nature11085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shang Y, LeRouzic V, Schneider S, Bisignano P, Pasternak GW, Filizola M. Mechanistic Insights into the Allosteric Modulation of Opioid Receptors by Sodium Ions. Biochemistry. 2014;53:5140–5149. doi: 10.1021/bi5006915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shang Y, Filizola M. Opioid receptors: Structural and mechanistic insights into pharmacology and signaling. European Journal of Pharmacology. 2015;763:206–213. doi: 10.1016/j.ejphar.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaserer T, Lantero A, Schmidhammer H, Spetea M, Schuster D. mu Opioid receptor: novel antagonists and structural modeling. Scientific Reports. 2016;6 doi: 10.1038/srep21548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perlikowska R, Piekielna J, Gentilucci L, De Marco R, Cerlesi MC, Calo G, Artali R, Tomboly C, Kluczyk A, Janecka A. Synthesis of mixed MOR/KOR efficacy cyclic opioid peptide analogs with antinociceptive activity after systemic administration. European Journal of Medicinal Chemistry. 2016;109:276–286. doi: 10.1016/j.ejmech.2015.12.012. [DOI] [PubMed] [Google Scholar]
- 21.Shang Y, Yeatman HR, Provasi D, Alt A, Christopoulos A, Canals M, Filizola M. Proposed Mode of Binding and Action of Positive Allosteric Modulators at Opioid Receptors. ACS Chem Biol. 2016;11:1220–1229. doi: 10.1021/acschembio.5b00712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen Q, Qian YY, Huang XQ, Xu XJ, Li W, Liu JG, Fu W. Discovery of Potent and Selective Agonists of delta Opioid Receptor by Revisiting the "Message-Address" Concept. Acs Medicinal Chemistry Letters. 2016;7:391–396. doi: 10.1021/acsmedchemlett.5b00423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Teramoto H, Yamauchi T, Terado Y, Odagiri S, Sasaki S, Higashiyama K. Design and Synthesis of a Piperidinone Scaffold as an Analgesic through Kappa-Opioid Receptor: Structure-Activity Relationship Study of Matrine Alkaloids. Chem Pharm Bull. 2016;64:410–419. doi: 10.1248/cpb.c15-00962. [DOI] [PubMed] [Google Scholar]
- 24.Waldhoer M, Bartlett SE, Whistler JL. Opioid receptors. Annu Rev Biochem. 2004;73:953–990. doi: 10.1146/annurev.biochem.73.011303.073940. [DOI] [PubMed] [Google Scholar]
- 25.Meunier JC. Nociceptin/orphanin FQ and the opioid receptor-like ORL1 receptor. European Journal of Pharmacology. 1997;340:1–15. doi: 10.1016/s0014-2999(97)01411-8. [DOI] [PubMed] [Google Scholar]
- 26.Calo G, Guerrini R, Rizzi A, Salvadori S, Regoli D. Pharmacology of nociceptin and its receptor: a novel therapeutic target. Br J Pharmacol. 2000;129:1261–1283. doi: 10.1038/sj.bjp.0703219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pasternak GW. Pharmacological mechanisms of opioid analgesics. Clinical Neuropharmacology. 1993;16:1–18. doi: 10.1097/00002826-199302000-00001. [DOI] [PubMed] [Google Scholar]
- 28.Land BB, Bruchas MR, Lemos JC, Xu M, Melief EJ, Chavkin C. The dysphoric component of stress is encoded by activation of the dynorphin kappa-opioid system. J Neurosci. 2008;28:407–414. doi: 10.1523/JNEUROSCI.4458-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bruchas MR, Land BB, Chavkin C. The dynorphin/kappa opioid system as a modulator of stress-induced and pro-addictive behaviors. Brain Research. 2010;1314:44–55. doi: 10.1016/j.brainres.2009.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Land BB, Bruchas MR, Schattauer S, Giardino WJ, Aita M, Messinger D, Hnasko TS, Palmiter RD, Chavkin C. Activation of the kappa opioid receptor in the dorsal raphe nucleus mediates the aversive effects of stress and reinstates drug seeking. Proc Natl Acad Sci U S A. 2009;106:19168–19173. doi: 10.1073/pnas.0910705106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.George O, Koob GF, Vendruscolo LF. Negative reinforcement via motivational withdrawal is the driving force behind the transition to addiction. Psychopharmacology. 2014;231:3911–3917. doi: 10.1007/s00213-014-3623-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koob GF, Buck CL, Cohen A, Edwards S, Park PE, Schlosburg JE, Schmeichel B, Vendruscolo LF, Wade CL, Whitfield TW, George O. Addiction as a stress surfeit disorder. Neuropharmacology. 2014;76:370–382. doi: 10.1016/j.neuropharm.2013.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schlosburg JE, Whitfield TW, Park PE, Crawford EF, George O, Vendruscolo LF, Koob GF. Long-Term Antagonism of kappa Opioid Receptors Prevents Escalation of and Increased Motivation for Heroin Intake. J Neurosci. 2013;33:19384–19392. doi: 10.1523/JNEUROSCI.1979-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Whitfield TW, Schlosburg JE, Wee S, Gould A, George O, Grant Y, Zamora-Martinez ER, Edwards S, Crawford E, Vendruscolo LF, Koob GF. kappa Opioid Receptors in the Nucleus Accumbens Shell Mediate Escalation of Methamphetamine Intake. J Neurosci. 2015;35:4296–4305. doi: 10.1523/JNEUROSCI.1978-13.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zagon IS, Donahue RN, McLaughlin PJ. Opioid growth factor-opioid growth factor receptor axis is a physiological determinant of cell proliferation in diverse human cancers. American Journal of Physiology-Regulatory Integrative and Comparative Physiology. 2009;297:R1154–R1161. doi: 10.1152/ajpregu.00414.2009. [DOI] [PubMed] [Google Scholar]
- 36.Zagon IS, Verderame MF, McLaughlin PJ. The biology of the opioid growth factor receptor (OGFr) Brain Research Reviews. 2002;38:351–376. doi: 10.1016/s0165-0173(01)00160-6. [DOI] [PubMed] [Google Scholar]
- 37.Avella DM, Kimchi ET, Donahue RN, Tagaram HRS, McLaughlin PJ, Zagon IS, Staveley-O'Carroll KF. The opioid growth factor-opioid growth factor receptor axis regulates cell proliferation of human hepatocellular cancer. American Journal of Physiology-Regulatory Integrative and Comparative Physiology. 2010;298:R459–R466. doi: 10.1152/ajpregu.00646.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hytrek SD, Lang CM, Smith JP, McLaughlin PJ, Zagon IS. Opioid growth-factor modulates tumorigenesis of human colon carcinomas grown in nude mice. Gastroenterology. 1995;108:A483–A483. [Google Scholar]
- 39.Zagon IS, Hytrek SD, Smith JP, McLaughlin PJ. Opioid growth factor (OGF) inhibits human pancreatic cancer transplanted into nude mice. Cancer Lett. 1997;112:167–175. doi: 10.1016/s0304-3835(96)04566-1. [DOI] [PubMed] [Google Scholar]
- 40.Hytrek SD, Smith JP, McGarrity TJ, McLaughlin PJ, Lang CM, Zagon IS. Identification and characterization of zeta-opioid receptor in human colon cancer. American Journal of Physiology-Regulatory Integrative and Comparative Physiology. 1996;271:R115–R121. doi: 10.1152/ajpregu.1996.271.1.R115. [DOI] [PubMed] [Google Scholar]
- 41.Zagon IS, Jenkins JB, Sassani JW, Wylie JD, Ruth TB, Fry JL, Lang CM, McLaughlin PJ. Naltrexone an opioid antagonist facilitates reepithelialization of the cornea in diabetic rat. Diabetes. 2002;51:3055–3062. doi: 10.2337/diabetes.51.10.3055. [DOI] [PubMed] [Google Scholar]
- 42.Zagon IS, Sassani JW, McLaughlin PJ. Opioid growth-factor modulates corneal epithelial outgrowth in tissue-culture. American Journal of Physiology-Regulatory Integrative and Comparative Physiology. 1995;268:R942–R950. doi: 10.1152/ajpregu.1995.268.4.R942. [DOI] [PubMed] [Google Scholar]
- 43.Zagon IS, Sassani JW, McLaughlin PJ. Re-epithelialization of the rabbit cornea is regulated by opioid growth factor. Brain Research. 1998;803:61–68. doi: 10.1016/s0006-8993(98)00610-6. [DOI] [PubMed] [Google Scholar]
- 44.Zagon IS, Sassani JW, McLaughlin PJ. Reepithelialization of the human cornea is regulated by endogenous opioids. Investigative Ophthalmology & Visual Science. 2000;41:73–81. [PubMed] [Google Scholar]
- 45.Hytrek SD, Smith JP, McLaughlin PJ, Lang CM, Zagon IS. Inhibition of human colon cancer by naltrexone. Gastroenterology. 1996;110:A532–A532. doi: 10.1016/0304-3835(96)04119-5. [DOI] [PubMed] [Google Scholar]
- 46.McLaughlin PJ, Zagon IS. Duration of opioid receptor blockade determines biotherapeutic response. Biochem Pharmacol. 2015;97:236–246. doi: 10.1016/j.bcp.2015.06.016. [DOI] [PubMed] [Google Scholar]
- 47.Donahue RN, McLaughlin PJ, Zagon IS. Low-dose naltrexone targets the opioid growth factor-opioid growth factor receptor pathway to inhibit cell proliferation: mechanistic evidence from a tissue culture model. Experimental Biology and Medicine. 2011;236:1036–1050. doi: 10.1258/ebm.2011.011121. [DOI] [PubMed] [Google Scholar]
- 48.Donahue RN, McLaughlin PJ, Zagon IS. The opioid growth factor (OGF) and low dose naltrexone (LDN) suppress human ovarian cancer progression in mice. Gynecologic Oncology. 2011;122:382–388. doi: 10.1016/j.ygyno.2011.04.009. [DOI] [PubMed] [Google Scholar]
- 49.Donahue RN, McLaughlin PJ, Zagon IS. Low-dose naltrexone suppresses ovarian cancer and exhibits enhanced inhibition in combination with cisplatin. Experimental Biology and Medicine. 2011;236:883–895. doi: 10.1258/ebm.2011.011096. [DOI] [PubMed] [Google Scholar]
- 50.McLaughlin PJ, Stucki JK, Zagon IS. Modulation of the opioid growth factor ( Met5 -enkephalin)-opioid growth factor receptor axis: Novel therapies for squamous cell carcinoma of the head and neck. Head and Neck-Journal for the Sciences and Specialties of the Head and Neck. 2012;34:513–519. doi: 10.1002/hed.21759. [DOI] [PubMed] [Google Scholar]
- 51.Rogosnitzky M, Finegold MJ, McLaughlin PJ, Zagon IS. Opioid growth factor (OGF) for hepatoblastoma: a novel non-toxic treatment. Invest New Drug. 2013;31:1066–1070. doi: 10.1007/s10637-012-9918-3. [DOI] [PubMed] [Google Scholar]
- 52.Hammer LA, Waldner H, Zagon IS, McLaughlin PJ. Opioid growth factor and low-dose naltrexone impair central nervous system infiltration by CD4+T lymphocytes in established experimental autoimmune encephalomyelitis, a model of multiple sclerosis. Experimental Biology and Medicine. 2016;241:71–78. doi: 10.1177/1535370215596384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McLaughlin PJ, Immonen JA, Zagon IS. Topical naltrexone accelerates full-thickness wound closure in type 1 diabetic rats by stimulating angiogenesis. Experimental Biology and Medicine. 2013;238:733–743. doi: 10.1177/1535370213492688. [DOI] [PubMed] [Google Scholar]
- 54.McLaughlin PJ, Sassani JW, Klocek MS, Zagon IS. Diabetic keratopathy and treatment by modulation of the opioid growth factor (OGF)-OGF receptor (OGFr) axis with naltrexone: A review. Brain Research Bulletin. 2010;81:236–247. doi: 10.1016/j.brainresbull.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McLaughlin PJ, Zagon IS. The opioid growth factor-opioid growth factor receptor axis: Homeostatic regulator of cell proliferation and its implications for health and disease. Biochem Pharmacol. 2012;84:746–755. doi: 10.1016/j.bcp.2012.05.018. [DOI] [PubMed] [Google Scholar]
- 56.Turel AP, Oh KH, Zagon IS, McLaughlin PJ. Low Dose Naltrexone for Treatment of Multiple Sclerosis A Retrospective Chart Review of Safety and Tolerability. Journal of Clinical Psychopharmacology. 2015;35:609–611. doi: 10.1097/JCP.0000000000000373. [DOI] [PubMed] [Google Scholar]
- 57.Smith JP, Stock H, Bingaman S, Mauger D, Rogosnitzky M, Zagon IS. Low-dose naltrexone therapy improves active Crohn's disease. American Journal of Gastroenterology. 2007;102:820–828. doi: 10.1111/j.1572-0241.2007.01045.x. [DOI] [PubMed] [Google Scholar]
- 58.Belofsky G, French AN, Wallace DR, Dodson SL. New geranyl stilbenes from Dalea purpurea with in vitro opioid receptor affinity. J Nat Prod. 2004;67:26–30. doi: 10.1021/np030258d. [DOI] [PubMed] [Google Scholar]
- 59.Neighbors JD, Beutler JA, Wiemer DF. Synthesis of nonracemic 3-deoxyschweinfurthin B. J Org Chem. 2005;70:925–931. doi: 10.1021/jo048444r. [DOI] [PubMed] [Google Scholar]
- 60.Topczewski JJ, Neighbors JD, Wiemer DF. Total Synthesis of (+)-Schweinfurthins B and E. J Org Chem. 2009;74:6965–6972. doi: 10.1021/jo901161m. [DOI] [PubMed] [Google Scholar]
- 61.Neighbors JD, Buller MJ, Boss KD, Wiemer DF. A Concise Synthesis of Pawhuskin A. J Nat Prod. 2008;71:1949–1952. doi: 10.1021/np800351c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Neighbors JD, Salnikova MS, Wiemer DF. Total synthesis of pawhuskin C: a directed ortho metalation approach. Tetrahedron Lett. 2005;46:1321–1324. [Google Scholar]
- 63.Hartung AM, Navarro HA, Wiemer DF, Neighbors JD. A selective delta opioid receptor antagonist based on a stilbene core. Bioorg Med Chem Lett. 2015;25:5532–5535. doi: 10.1016/j.bmcl.2015.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hartung AM, Beutler JA, Navarro HA, Wiemer DF, Neighbors JD. Stilbenes as kappa-Selective, Non-nitrogenous Opioid Receptor Antagonists. J Nat Prod. 2014;77:311–319. doi: 10.1021/np4009046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.St John SE, Jensen KC, Kang S, Chen YF, Calamini B, Mesecar AD, Lipton MA. Design, synthesis, biological and structural evaluation of functionalized resveratrol analogues as inhibitors of quinone reductase 2. Bioorg Med Chem. 2013;21:6022–6037. doi: 10.1016/j.bmc.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hartung AM. University of Iowa; 2014. [Google Scholar]
- 67.Gaslonde T, Covello F, Velazquez-Alonso L, Leonce S, Pierre A, Pfeiffer B, Michel S, Tillequin F. Synthesis and cytotoxic activity of benzo a acronycine and benzo b acronycine substituted on the A ring. Eur J Med Chem. 2011;46:1861–1873. doi: 10.1016/j.ejmech.2011.02.050. [DOI] [PubMed] [Google Scholar]
- 68.Duan HL, Zheng J, Lai QL, Liu Z, Tian GH, Wang Z, Li JF, Shen JS. 2-Phenylquinazolin-4(3H)-one, a class of potent PDE5 inhibitors with high selectivity versus PDE6. Bioorg Med Chem Lett. 2009;19:2777–2779. doi: 10.1016/j.bmcl.2009.03.125. [DOI] [PubMed] [Google Scholar]
- 69.Bohlmann F, Zeisberg R, Klein E. C-13 NMR-Spectra of Monoterpenes. Organic Magnetic Resonance. 1975;7:426–432. [Google Scholar]
- 70.Matthiesen RA, Wills VS, Metzger JI, Holstein SA, Wiemer DF. Stereoselective Synthesis of Homoneryl and Homogeranyl Triazole Bisphosphonates. J Org Chem. 2016;81:9438–9442. doi: 10.1021/acs.joc.6b01693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hartung AM. Potential Opioid Receptor Modulators Derived From Novel Stilbenes. Department of Chemistry, University of Iowa; 2014. [Google Scholar]
- 72.Bringmann G, Hinrichs J, Henschel P, Kraus J, Peters K, Peters EM. Novel concepts in directed biaryl synthesis. Part 97. Atropo-enantioselective synthesis of the natural bicoumarin (+)-isokotanin A via a configurationally stable biaryl lactone. European Journal of Organic Chemistry. 2002:1096–1106. [Google Scholar]
- 73.Wang PF, Hu HY, Wang Y. Application of the excited state meta effect in photolabile protecting group design. Organic Letters. 2007;9:2831–2833. doi: 10.1021/ol071085c. [DOI] [PubMed] [Google Scholar]
- 74.Ohba K, Koga Y, Nomura S, Nakata M. Functionalized aryl-beta-C-glycoside synthesis by Barbier-type reaction using 2,4, 6-triisopropylphenyllithium. Tetrahedron Lett. 2015;56:1007–1010. [Google Scholar]
- 75.Klatt T, Markiewicz JT, Samann C, Knochel P. Strategies To Prepare and Use Functionalized Organometallic Reagents. J Org Chem. 2014;79:4253–4269. doi: 10.1021/jo500297r. [DOI] [PubMed] [Google Scholar]
- 76.Topczewski JJ, Callahan MP, Neighbors JD, Wiemer DF. A Tandem Cascade Cyclization-Electrophilic Aromatic Substitution: Application in the Total Synthesis of (+)-Angelichalcone. J Am Chem Soc. 2009;131:14630–14631. doi: 10.1021/ja906468v. [DOI] [PubMed] [Google Scholar]
- 77.Zagon IS, Verderame MF, McLaughlin PJ. The expression and function of the OGF-OGFr axis -a tonically active negative regulator of growth -in COS cells. Neuropeptides. 2003;37:290–297. doi: 10.1016/j.npep.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 78.Donahue RN, McLaughlin PJ, Zagon IS. Cell proliferation of human ovarian cancer is regulated by the opioid growth factor-opioid growth factor receptor axis. American Journal of Physiology-Regulatory Integrative and Comparative Physiology. 2009;296:R1716–R1725. doi: 10.1152/ajpregu.00075.2009. [DOI] [PubMed] [Google Scholar]
- 79.Zagon IS, Donahue RN, Rogosnitzky M, McLaughlin PJ. Imiquimod upregulates the opioid growth factor receptor to inhibit cell proliferation independent of immune function. Experimental Biology and Medicine. 2008;233:968–979. doi: 10.3181/0802-RM-58. [DOI] [PubMed] [Google Scholar]
- 80.Cree BAC, Kornyeyeva E, Goodin DS. Pilot Trial of Low-Dose Naltrexone and Quality of Life in Multiple Sclerosis. Annals of Neurology. 2010;68:145–150. doi: 10.1002/ana.22006. [DOI] [PubMed] [Google Scholar]
- 81.Berkson BM, Rubin DM, Berkson AJ. The long-term survival of a patient with pancreatic cancer with metastases to the liver after treatment with the intravenous alpha-lipoic acid/low-dose naltrexone protocol. Integrative Cancer Therapies. 2006;5:83–89. doi: 10.1177/1534735405285901. [DOI] [PubMed] [Google Scholar]
- 82.Berkson BM, Rubin DM, Berkson AJ. Reversal of signs and symptoms of a B-Cell lymphoma in a patient using only low-dose naltrexone. Integrative Cancer Therapies. 2007;6:293–296. doi: 10.1177/1534735407306358. [DOI] [PubMed] [Google Scholar]
- 83.Berkson BM, Rubin DM, Berkson AJ. Revisiting the ALA/N (alpha-Lipoic Acid/Low-Dose Naltrexone) Protocol for People With Metastatic and Nonmetastatic Pancreatic Cancer: A Report of 3 New Cases. Integrative Cancer Therapies. 2009;8:416–422. doi: 10.1177/1534735409352082. [DOI] [PubMed] [Google Scholar]
- 84.Arima Y, Kinoshita M, Akita H. Natural product synthesis from (8aR)-and (8aS)-bicyclofarnesols: synthesis of (+)-wiedendiol A, (+)-norsesterterpene diene ester and (−)-subersic acid. Tetrahedron-Asymmetry. 2007;18:1701–1711. [Google Scholar]
- 85.Li YL, Zhao YL. Studies on the prenylflavonoids. Part XI: The total synthesis of amorisin and sigmoidin-B. Chin Chem Lett. 1994;5:1009–1012. [Google Scholar]
- 86.Haseltine JN, Cabal MP, Mantlo NB, Iwasawa N, Yamashita DS, Coleman RS, Danishefsky SJ, Schulte GK. Total synthesis of calicheamicinone - New arrangements for the actuation of the reductive cycloaromatization of taglycon congeners. J Am Chem Soc. 1991;113:3850–3866. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.