

Abstract
The transcription factor C/EBP-homologous protein (CHOP) is a key component of the terminal unfolded protein response (UPR) that mediates unresolvable endoplasmic reticulum stress-induced apoptosis. CHOP induction is known to cause cancer cell death. Chemicals that induce CHOP expression would thus be valuable as potential cancer therapeutics and as research tools. Here, we identified 5-nitrofuran-2-amide derivatives as small molecule activators of CHOP expression that induced apoptosis in triple negative breast cancer (TNBC) cells. Our preliminary structure-activity relationship studies indicated that compounds with an N-phenyl-5-nitrofuran-2-carboxamide skeleton were particularly potent inducers of TNBC cell apoptosis. The compounds activate CHOP expression via the PERK–eIF2α–ATF4 branch of the UPR. These results indicate that small molecule activators of CHOP expression may have therapeutic potential for TNBC.
1. Introduction
The endoplasmic reticulum (ER) is the major cellular organelle responsible for protein folding and secretion, calcium storage and release, and lipid biogenesis. Disturbance in the ER environment leads to the accumulation of misfolded or unfolded proteins in the ER, a condition termed ER stress. Under ER stress, cells activate the unfolded protein response (UPR), a signaling pathway mediated by three ER transmembrane protein sensors: inositol-requiring enzyme 1α (IRE1α), protein kinase RNA-like ER kinase (PERK), and activating transcription factor 6 (ATF6), in an attempt to restore homeostasis.1–3 The UPR is initiated as an adaptive mechanism to alleviate the accumulation of misfolded- or unfolded proteins in the ER by altering protein translation, folding, and post-translational modifications.3–5 However, if these adaptive responses fail to re-establish homeostasis because ER stress is excessive or prolonged, a terminal UPR becomes activated and induces cell death.
ER stress and UPR activation have been implicated in the pathogenesis of human cancers.6–9 Cancer cells utilize the adaptive branch of the UPR pathway to survive and progress in a stressful microenvironment. For example, the B-cell neoplasm multiple myeloma (MM) displays chronic ER stress and is dependent on the adaptive Ire1α –X-box binding protein 1 (XBP1, an Ire1α substrate) branch of the UPR pathway for survival.9 Similarly, in triple negative breast cancer (TNBC), which is defined by the absence of the estrogen receptor, progesterone receptor, and human epidermal growth factor receptor-2 and is among the most aggressive and treatment-resistant forms of breast cancer10, XBP1 is highly activated and plays a pivotal role in the tumorigenicity and progression.8 Accordingly, inhibiting the adaptive Ire1α -XBP1 pathway has been proposed as a promising strategy for the development of anticancer therapy.8, 11, 12 Indeed, blockade of XBP1 activation by small molecule Ire1α inhibitors has been shown to cause significant growth inhibition of MM cells.12, 13 On the other hand, another recently proposed therapeutic rationale is to augment the terminal UPR in cancer cells whose adaptive UPR is active so as to tip the balance to apoptosis instead of survival.6, 14, 15 However, this strategy has not been experimentally proven, nor has any chemicals been identified to serve this purpose.
The transcription factor C/EBP-homologous protein (CHOP) is a key component of the ER stress-induced terminal UPR5, 16, 17 and is activated mainly by the PERK pathway, although the IRE1α and ATF6 pathways also contribute.5, 18 CHOP deletion has been shown to increase tumorigenesis in mouse models of lung and liver cancers.19, 20 Therefore, we reasoned that if the postulation that activation of the terminal UPR triggers cancer cell death is correct, compounds that enhance the expression or activity of CHOP in cancer cells could tip the scale in favor of apoptosis.
In this study, we sought to identify small molecule inducers of CHOP expression and revealed that these molecules are capable of inducing apoptosis of TNBC cells. Using a high- throughput screening assay with HEK293 cells expressing a CHOP promoter-luciferase (CHOP-Luc) reporter, we identified several 5-nitrofuran-2-amide derivatives that induced CHOP-Luc activity. These compounds induced apoptosis in multiple TNBC cell lines by inducing CHOP gene expression. Our preliminary structure-activity relationship (SAR) studies indicated that compounds with an N-phenyl-5-nitrofuran-2-carboxamide skeleton were particularly potent inducers of TNBC cell apoptosis. We further showed that these compounds preferentially activate the eukaryotic initiation factor-2α (eIF2α)- activating transcription factor 4 (ATF4) pathway to induce CHOP expression. Our results provide first demonstration that augmentation of the terminal UPR pathway may serve as a promising therapy for cancers with adaptive UPR activation.
2. Results and Discussion
To identify compounds that activate the expression of CHOP, a HEK293 cell line stably expressing a CHOP-Luc reporter construct that faithfully reflects endogenous CHOP gene expression21 was used to screen approximately 50,000 structurally diverse small molecules. A novel compound 1, N-(4-iodophenyl)-5-nitrofuran-2-carboxamide was identified that increased the activity of the CHOP-Luc reporter by 24-fold at the concentration of 10 µM (Figure 1A, B). We further determined whether compound 1 affects the expression of the endogenous CHOP gene. As shown in Figure 1C, compound 1 significantly increased the expression of mRNA level of the endogenous CHOP gene in HEK293 cells, by up to 30- fold increase, as measured by quantitative RT-PCR (Figure 1C).
Figure 1.

Structure of compound 1 and induction of CHOP mRNA. (A) Structure of compound 1. (B) Cells were treated with DMSO (control) or 1 (10 µM) for 24 h, and luciferase activity was determined using the Bright-Glo assay. (C) HEK293 cells were treated with DMSO or 1 (10 µM) for 24 h and CHOP mRNA levels were analyzed by qRT-PCR. In B and C, the results are the means of 3 replicate wells and are representative of 3 independent experiments. **P < 0.01 and ***P < 0.001 by Student’s t-test compared with control cells.
Given the known functions of CHOP in ER stress-induced apoptosis16, 17 and in regulating cancer cell death,6, 19, 20 we investigated whether compound 1 affects the viability of TNBC cells. Three human TNBC cell lines, HCC-1806, HCC-1143, and HCC-38, were treated with doses of 1 ranging from 0.125 µM to 20 µM and their viability was assessed using the CellTiter-Glo assay, which measures intracellular ATP levels. The viability of the three TNBC cell lines was significantly reduced by 1 in a dose-dependent manner, and the IC50 values were similar on all three cell lines; 6.2 ± 0.8 µM, 9.5 ± 1.9 µM, 8.7 ± 1.5 µM (mean±SD), for HCC-1806, HCC-1143, and HCC-38 cells, respectively. These results indicate that 1 exhibits antitumor activity in TNBC cells.
To identify more potent 5-nitrofuran-2-amide derivatives, we performed SAR analysis on a series of 1 analogues and assessed their antitumor activity using the HCC-1806 cell line. We first analyzed the effects on efficacy of various substituted groups introduced to the phenyl ring. Replacement of the 4-iodine group with methyl, ethyl, or chlorine appeared to have no effect on or slightly improved the antitumor activity, as indicated by the similar IC50s of the respective compounds 2a, 2b, and 2c (Table 1) compared to that of 1. In contrast, 2,6-di-Me and 2-iBu replacement resulted in inactive analogues 2k and 2l, suggesting that steric hindrance reduces the potency. In addition, introduction of 2,5-dimethyl (2f) or 2-ethyl (2i) to the phenyl ring in 1 moderately improved the potency (Table 1).
Table 1.
Anticancer effect of 2a–2l in HCC-1806 cells.
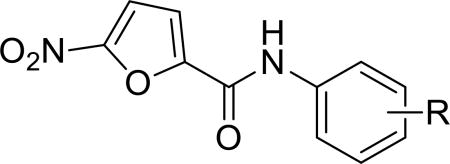
| ||
|---|---|---|
| Compd | R | HCC-1806 IC50 (µM) a |
| 2a | 4-Me | 6.7 |
| 2b | 4-Et | 5.0 |
| 2c | 4-Cl | 5.1 |
| 2d | 3-OCF3 | 19.6 |
| 2e | 2,5-di-Cl | 8.6 |
| 2f | 2,5-di-Me-4-I | 3.0 |
| 2g | 2-Cl-5-CF3 | 5.1 |
| 2h | 2-OMe-5-Cl | 7.0 |
| 2i | 2-Et-4-I | 5.5 |
| 2j | 2,3-di-Me | 9.7 |
| 2k | 2,6-di-Me | >40 |
| 2l | 2-iBu | >40 |
IC50 value for cancer cell viability calculated with GraphPad Prism.
Given that the substituents at the para position of the phenyl ring were well tolerated, various six-ring substituents were introduced to the phenyl ring, and the compounds were tested for their anti-TNBC activity. We observed that all of the six-ring derivatives tested, including morpholine (3a, 3b), piperidine (3c, 3d), and piperazine (3e), exhibited substantially improved IC50 values for inhibition of HCC-1806 viability (Table 2). Compounds 3a-e all also showed significant activity on HCC-1143 and HCC-38 (Table 2). Moreover, further SAR analysis indicated that the nitro group on the left furan ring is critical for the anticancer activity, since its deletion in compounds 4a–4d eliminated or severely inhibited their activity (Table 3). Overall, our preliminary SAR studies indicated that compounds with an N-phenyl-5-nitrofuran-2-carboxamide skeleton were potent inhibitors of TNBC cell viability.
Table 2.
Anticancer effect of 3a–3e in HCC-1806, HCC-1143 and HCC-38 cells.
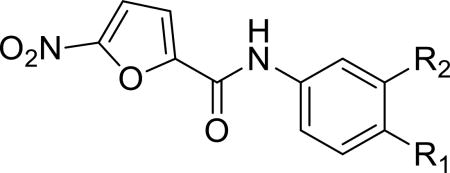
| |||||
|---|---|---|---|---|---|
| Compd | R1 | R2 | HCC-1806 IC50 (µM) a |
HCC-1143 IC50 (µM) a |
HCC-38 IC50 (µM) a |
| 3a |
|
H | 1.5 | 2.3 | 3.1 |
| 3b |
|
Cl | 2.6 | 2.5 | 2.0 |
| 3c |
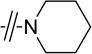
|
Cl | 3.5 | 4.5 | 3.8 |
| 3d |
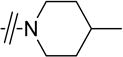
|
H | 2.8 | 2.1 | 2.5 |
| 3e |
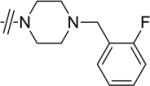
|
H | 3.0 | 3.8 | 1.9 |
IC50 value for cancer cell viability calculated with GraphPad Prism.
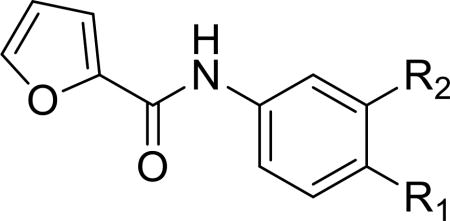
Table 3.
Anticancer effect of 4a–4d in HCC-1806 cells.
| |||
|---|---|---|---|
| Compd | R1 | R2 | HCC-1806 IC50 (µM) a |
| 4a |
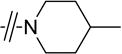
|
H | >40 |
| 4b |
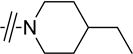
|
Cl | >40 |
| 4c |
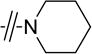
|
F | >40 |
| 4d |
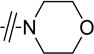
|
Br | >40 |
IC50 value for cancer cell viability calculated with GraphPad Prism.
To determine whether the reduction in TNBC cell viability by the 5-nitrofuran-2-amide derivatives was due to the induction of apoptosis, we analyzed cleavage of caspase-3, a critical executioner of apoptosis, in HCC-1806 cells treated with compound 3d. Indeed, treatment of HCC-1806 cells with 3d increased cleaved caspase-3 protein levels at 8 h and 24 h, indicating that 3d activated apoptosis in the TNBC cells (Figure 2A). To confirm this, HCC-1806 cells were cultured to near confluence and treated with 10 µM 3d or DMSO for 24 h and then imaged by live-cell phase-contrast microscopy. Whereas the DMSO-treated cells remained confluent, few cells were observed in the 3d-treated culture, indicative of significant cell death and thus detachment from the culture dish, rather than a reduction in proliferation, (Figure 2B, 2C).
Figure 2.

Compound 3d induces apoptosis of HCC-1806 cells. (A) Cells were treated with 3d (10 µM) for the indicated times, and cleavage of caspase-3 was determined by Western blotting. α-Tubulin was used as a loading control. The data shown are representative of 3 independent experiments. (B, C) Cells were treated with DMSO (B) or 3d (10 µM) (C) for 24 h, and live-cell phase-contrast images were acquired (magnification 10×).
We next investigated the effect of 3d on CHOP gene expression in HCC-1806 cells using quantitative real-time PCR. Treatment of cells with 3d significantly increased CHOP mRNA levels in a dose-dependent manner (Figure 3A). Similarly, a kinetic analysis revealed that treatment with 10 µM 3d increased CHOP transcription in a time-dependent fashion (Figure 3B). In both dose- and time-dependent studies, 3d-induced CHOP mRNA levels were up to 9-fold higher than in DMSO-treated cells. In agreement with its effects on CHOP transcription, 3d treatment of HCC-1086 cells also increased CHOP protein levels, with significant increases detected between 4 h and 24 h (Figure 3C). These results demonstrate that 3d activates the expression of the CHOP gene in TNBC cells.
Figure 3.

Compound 3d induces CHOP expression in HCC-1806 cells. (A) Cells were treated with 3d at the indicated concentrations for 8 h, and CHOP mRNA levels were analyzed by qRT-PCR. (B) Cells were treated with 3d (10 µM) for the indicated times, and CHOP mRNA levels were analyzed by qRT-PCR. The results in (A) and (B) are the means of 4 replicate wells and are representative of 3 independent experiments. *P < 0.05 and **P < 0.01 by Student’s t-test compared with cells treated with DMSO (in A) or with 3d for 0 h (in B). (C) Cells were treated with 3d (10 µM) for the indicated times, and CHOP protein levels were analyzed by Western blotting. α-Tubulin was used as a loading control. The data shown are representative of 3 independent experiments. (D). HCC-1806 and MCF7 cells were treated with 3d (10 µM) for 3d. Cell viability was assessed by CellTiter-Glo kit. The results are the means of 4 replicate wells and are representative of 3 independent experiments.
TNBC cells display a highly active Ire1α-XBP1 an adaptive branch of the UPR pathway that plays a pivotal role in the tumorigenicity and progression in a stressful microenvironment.8 We therefore hypothesize that augmentation of the terminal UPR (such as activation of CHOP) in fast growing TNBC cells could disrupt the ER environment and hence drive these cells to apoptosis instead of survival. This suggests that TNBC cells may be more vulnerable to CHOP activation than cells that are not known for the XBP1 activation (such as estrogen receptor positive adenocarcinoma cell line MCF7).22, 23 To test this idea, we compared the viabilities of HCC-1086 cells and MCF7 cells treated with 3d. As shown in Figure 3D, treatment of 3d caused the death in both cell lines in a dose-dependent manner. However, HCC-1086 cells showed a significantly more cell death than MCF7 cells at the concentrations of 2.5, 5, and 10 µM, indicating that HCC-1086 cells are more vulnerable than MCF7 cells to 3d treatment.
Prolonged or severe ER stress activates CHOP expression primarily through the PERK branch of the UPR, although the IRE1α and ATF6 branches also contribute.5, 18 The 5-nitrofuran-2-amide derivatives could induce CHOP expression by acting as an ER stressor that activates all three branches of UPR or by preferentially activating a select branch of UPR. To distinguish between these possibilities, we investigated which branches of the UPR were affected by 3d. Activation of the PERK pathway by ER stress leads to phosphorylation of eIF2α, followed by activation of the transcription and translation of the transcription factor ATF4, a 5′-upstream ORF-containing gene, and ATF4-mediated CHOP expression.5, 24 To determine whether the PERK pathway is involved in 3d-mediated CHOP induction, we analyzed ATF4 expression and eIF2α phosphorylation in HCC-1806 cells by qRT-PCR and Western blotting. Compound 3d significantly increased ATF4 expression at both the mRNA (Figure 4A) and protein (Figure 4B) levels, and substantially increased the phosphorylation of eIF2α (Figure 4B). Of note, the 3d-induced increase in eIF2α phosphorylation preceded the increase in ATF4 mRNA, and both effects peaked within several hours of 3d treatment; a pattern consistent with a typical ER stress-mediated response.6 PERK–eIF2α–ATF4–CHOP axis is regulated in a complex manner. First, ATF4 and CHOP are also known to induce the expression of the growth arrest and DNA damage-inducible protein (GADD34), a regulatory subunit of protein phosphatase 1 that participates in a feedback loop to dephosphorylate eIF2α.6 GADD34 expression would be expected to increase following 3d-triggered CHOP induction. Indeed, we observed that GADD34 mRNA level increased significantly in HCC-1806 cell treated with 3d over time (Figure 4C). The increase in GADD34 level after 3d treatment is consistent with our observation on the gradual decline of eIF2α phosphorylation and subsequent decline of ATF4 translation (protein level) over time (Figure 4B) as GADD34 is expected to dephosphorylate eIF2α. Second, although it is well known that eIF2α phosphorylation preferentially activates the translation of ATF4 under ER stress, Dey et al recently reported that ER stress activates the transcription of ATF4 mRNA.24 These may explain our observation that while 3d treatment activated ATF4 transcription (Figure 4A); its continued treatment subsequently down-regulated ATF4 translation (Figure 4B) due to the GADD34-mediated negative feedback on ATF4 translation. Taken together, these results support a role for the PERK–eIF2α–ATF4–CHOP branch of the UPR in 3d-mediated TNBC cancer cell death.
Figure 4.

Compound 3d selectively activates eIF2α-ATF4 pathway. (A) HCC-1806 cells were treated with 3d (10 µM) for the indicated times, and ATF4 mRNA levels were analyzed by qRT-PCR. Results are the means of 4 replicate wells and are representative of 3 independent experiments. *P < 0.05 by Student’s t-test compared with DMSO-treated cells. (B) HCC-1806 cells were treated with 3d (10 µM) for the indicated times, and ATF4 and p-eIF2α protein levels were analyzed by Western blotting. α-Tubulin was used as a loading control. The data shown are representative of 3 independent experiments. (C) HCC-1806 cells were treated with 3d (10 µM) for the indicated times, and GADD34 mRNA levels were analyzed by qRT-PCR. Results are the means of 4 replicate wells and are representative of 3 independent experiments. *P < 0.05 by Student’s t-test compared with DMSO-treated cells. (D) HCC-1806 cells were treated with 3d (10 µM) or tunicamycin (Tm, 1 µg/mL) for the indicated times. XBP1 mRNA levels were analyzed by RT-PCR and the products were resolved by agarose gel electrophoresis. The full-length (unspliced, XBP1u) and spliced (XBP1s) forms of XBP1 mRNA are indicated. GAPDH mRNA was used as an internal control. (D’) Quantification of data shown in (A) by densitometry. The percentage of XBP1s relative to total XBP1 was calculated as: (XBP1s/[XBP1s+XBP1u]) × 100%. The data shown are representative of 3 independent experiments. (E) HEK293 cells stably expressing CHOP-Luc, ERSE-Luc, or UPRE-luc reporters were treated with DMSO, 3d (10 µM), or Tm (1 µg/mL) for 24 h, and luciferase activity was measured using the Bright-Glo assay. Results are the means of 4 replicate wells and are representative of 3 independent experiments.
We next asked whether the IRE1α and ATF6 branches of the UPR were also activated by 3d treatment of TNBC cells. Under ER stress, activated IRE1α cleaves X-box binding protein-1 (XBP1) mRNA to generate a spliced form of XBP1 that is translated into a potent transcription factor XBP1s (for spliced XBP1).14 XBP1s increases transcription of UPR genes encoding factors involved in ER protein folding and degradation by binding to the unfolded protein response element (UPRE) in the gene promoters, either as a homodimer or as an XBP1s–ATF6 heterodimer.25, 26 We used XBP1 mRNA splicing and a UPRE-Luciferase (UPRE-Luc) reporter as markers for activation of IRE1α pathway. We observed that XBP1 mRNA splicing, as measured by XBP1s levels, increased only slightly in the first 24 h after 3d treatment of HCC-1806 cells (from 11% of total XBP1 at 0 h to 15~20% at 2~24 h) (Figure 4D, D’). This compared with the dramatic increase of XBP1s (~87% at 8 h) after treatment with tunicamycin, a well-characterized ER stressor (Figure 4D, D’). We also analyzed the effect of 3d treatment on the IRE1α pathway using a HEK293 UPRE-Luc reporter cell line, and found that luciferase activity was increased less than 2-fold by 3d, compared with >25-fold by tunicamycin (Figure 4E). These results suggest that activation of the IRE1α pathway is likely to contribute marginally, if at all, to the anticancer activity of 3d. We next determined whether 3d activates the ATF6 pathway by evaluating its effect on the activity of an ER stress response elements (ERSE)-Luc reporter stably established in HEK293 cell line. Under ER stress, activated ATF6 functions as a nuclear transcription factor and activates the expression of genes encoding ER chaperones by binding to ERSEs in their promoters.26, 27 Whereas ERSE-driven luciferase activity was increased by ~12-fold upon treatment with tunicamycin, treatment with 3d for 24 h had no significant effect (Figure 4E). Taken together, these results indicate that 3d does not behave as a general ER stressor to activate all three branches of the UPR (e.g., tunicamycin), but instead, induces CHOP expression by selectively activating the PERK- eIF2α–ATF4 branch of the UPR.
To confirm that CHOP induction plays a role in 3d-mediated TNBC cell death, we asked whether siRNA-mediated knockdown of CHOP mitigated 3d-induced cell death. For this, HCC-1806 cells were transfected with different concentrations of CHOP siRNA, and knockdown efficiency was assessed by RT-PCR. CHOP mRNA levels were reduced by >90% at 48 h after transfection with 20 nM CHOP-specific siRNA compared with control siRNA (Figure 5A), and neither siRNA had a significant effect on cell viability under control conditions (Figure 5B). As expected, 3d caused a marked reduction in the viability of cells transfected with the control siRNA; however, CHOP siRNA significantly attenuated the effects of 3d on HCC-1806 cell death (Figure 5B). We note that the rescue of CHOP siRNA knockdown on 3d-induced apoptosis is incomplete, albeit significant. One possibility for this partial rescue could be continued induction of CHOP expression in the presence of 3d. We therefore evaluated the CHOP gene expression in HCC-1806 cells after compound treatment in the presence of CHOP or control siRNA transfection. As shown in Figure 5C, we observed that CHOP mRNA level increased significantly in 3d-treated HCC-1806 cells under both CHOP siRNA and control siRNA conditions, indicating the enhanced transcription of CHOP mRNA could contribute to the aforementioned incomplete rescue. These results indicate that CHOP is critical for 3d-induced TNBC cell death.
Figure 5.

Compound 3d induces CHOP-dependent apoptosis in HCC-1806 cells. (A) HCC-1806 cells were transfected with indicated concentrations of CHOP siRNA or scrambled siRNA (indicated as 0 nM) for 48 h, and CHOP mRNA levels were analyzed by RT-PCR. GAPDH mRNA was used as an internal control. CHOP knockdown efficiency was quantified with the indicated concentrations of CHOP siRNA relative to control siRNA. The data shown are representative of 3 independent experiments. (B) Control or CHOP siRNA (20 nM) was transfected into HCC-1806 cells, and 6 h later, cells were treated with 3d (5 µM) or DMSO for 48 h. Cell viability was measured using the CellTiter-Glo assay. The data shown are representative of 3 independent experiments. *P < 0.05 by Student’s t-test compared with DMSO-treated cells. (C) HCC-1806 cells were transfected with indicated concentrations of CHOP siRNA or scrambled siRNA (indicated as 0 nM), and 6 h later, cells were treated with 3d (5 µM) or DMSO for 48 h, and CHOP mRNA levels were analyzed by RT-PCR. GAPDH mRNA was used as an internal control. The data shown are representative of 3 independent experiments. (D) HCC-1806 cells were treated with 3d (10 µM) for the indicated times, and BCL2 and cleaved caspase 4 protein levels were analyzed by Western blotting. α-Tubulin was used as a loading control. The data shown are representative of 3 independent experiments. (E) HCC-1806 cells were treated with 3d (10 µM) for the indicated times, and Bim mRNA levels were analyzed by qRT-PCR. Results are the means of 4 replicate wells and are representative of 3 independent experiments. *P < 0.05 and ** P < 0.01 by Student’s t-test compared with DMSO-treated cells.
Finally we investigated how 3d-induced CHOP could lead to TNBC cell death. CHOP activation under ER stress inhibits expression of the gene encoding anti-apoptotic BCL-2 while inducing the transcription of the gene encoding pro-apoptotic BH3-only protein BIM.28, 29 Once activated, BH3-only proteins triggers the oligomerization of the multi-domain proapoptotic proteins Bax and/or Bak, leading to mitochondrial permeabilization and caspase-mediated apoptosis.4 If 3d induces apoptosis via activation of CHOP, it will be predicted to affect its downstream targets BCL-2 and Bim. We therefore assessed whether BCL-2 and BIM were affected by 3d treatment. We observed that BCL2 protein decreased drastically in HCC-1806 cells from 2 h of 10 µM 3d treatment onwards (Figure 5D). In contrast, Bim mRNA level increased significantly in HCC-1806 cells treated with 10 µM 3d in a time-dependent manner (Figure 5E). Caspases play essential roles in initiating apoptotic signaling and executing the final stages of cell death. The executioner caspase 3 is known to be critical for ER stress-induced apoptosis and indeed, we have shown that caspase 3 was induced in HCC-1806 cells by 3d treatment (shown in Figure 2A). On the other hand, a subfamily of caspases [caspase 4 (human) and caspase 12 (rodents)] has been proposed to be the initiator caspases in ER stress-triggered apoptosis. However, caspase 12 was later shown to be downstream of BAX/BAK-dependent mitochondrial permeabilization, indicating a role in amplifying rather than initiating ER stress-induced apoptosis.30 Irregardless this controversy, caspase 4/12 appears to be activated during ER stress-mediated apoptosis. We therefore investigated whether 3d triggered the activation of caspase 4. Indeed, as shown in Figure 5D, cleaved caspase 4 was induced in HCC-1806 cells treated with10 µM 3d from 8 h onwards. Taken together, these results support a CHOP-dependent role in 3d-induced TNBC cell apoptosis.
3. Conclusion
In this study, we identified a number of 5-nitrofuran-2-amide derivatives that induced expression of CHOP, a key component of the pro-apoptotic arm of the UPR. We showed that the derivatives induce CHOP expression by preferentially activating the PERK–eIF2α–ATF4 branch of the UPR; an observation that suggests a highly selective mode of action. 5-nitrofuran-2-amide derivatives were previously reported to possess anti-microbial and immunomodulatory activities.31, 32 Our identification of similar small molecules as novel activators of CHOP expression may lead to the development of new classes of therapeutics for drug-resistant TNBCs.
4. EXPERIMENTAL SECTION
4.1. Chemicals
The chemical libraries were obtained from ChemBridge (San Diego, CA, USA), Maybridge (Cornwall, UK), and MicroSource (Ann Arbor, MI, USA). The compounds were supplied as 10 mM solutions in DMSO. All 5-nitrofuran-2-amide derivatives were obtained from ChemBridge. The structures and purities were confirmed by the suppliers using NMR and HPLC. Tunicamycin was obtained from Sigma (St Louis, MO, USA). All chemicals were dissolved in DMSO and used at the indicated concentrations. Bright-Glo and CellTiter-Glo kits were purchased from Promega (Madison, WI, USA).
4.2. Cell culture
HEK293T cells were cultured in DMEM medium (Corning, NY, USA) supplemented with 10% fetal bovine serum (FBS; Atlanta Biologicals, Norcross, GA), and antibiotics (100 UI/mL penicillin and 100 µg/mL streptomycin; Corning) and maintained in a humidified 5% CO2 atmosphere at 37°C. HCC-1806, HCC-1143, and HCC-38 cells (from ATCC) were cultured in RPMI 1640 medium (Corning) with 10% FBS (Atlanta Biologicals) and antibiotics (100 UI/mL penicillin and 100 µg/mL streptomycin; Corning) and maintained in a humidified 5% CO2 atmosphere at 37°C.
4.3. CHOP, ERSE, and UPRE reporter cell lines
The HEK293T CHOP reporter cell line (CHOP-Luc) was previously described.21, 33, 34 HEK293T cells were stably transfected with ERSE-Luciferase27 and UPRE-Luciferase25, 35 reporters to generate ERSE-Luc and UPRE-Luc reporter cell lines, respectively. Reporter cells were plated at 7 × 103 cells/well in a 384-well plate and incubated for 16 h. Test compounds or Tm at 1 µg/mL were then added. Luciferase activity was measured with a Bright-Glo kit 24 h later.
4.4. High-throughput chemical screening
HEK293T CHOP-Luc cells were seeded at 7 × 103 cells/well in 384-well plates and treated with 10 µM of the library compounds the next day. After 24 h treatment, the medium was aspirated and 20 µL/well of Bright-Glo luciferase assay reagent was added. Luminescence was measured with an EnVision multilabel plate reader (PerkinElmer, Waltham, MA, USA). Hit selection was based on standard scores. The mean and standard deviation (SD) of luminescence for each compound was determined, and the standard score for each compound was then calculated as (raw measurement of a compound − mean)/SD of the plate. Compounds that increased ATP levels >3 SD compared with control wells (standard score >3) were considered hits.
4.5. Cell survival assay
HCC-1806, HCC-1143, or HCC-38 cells were seeded at 3 × 103 cells/well in a 384-well plate overnight and treated with compounds at the indicated concentrations the next day. After 3 d treatment, the medium was aspirated and 20 µL/well of CellTiter-Glo reagent was added. Cell viability was measured with an EnVision multilabel plate reader. The IC50 value for cell viability of each compound was calculated with GraphPad Prism6 (La Jolla, CA, USA).
4.6. RNA isolation and qRT-PCR
HCC-1806 cells were seeded at 4 × 105 cells/well in 6-well plates and treated with compounds for the indicated times. Total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol, and 2 µg of total RNA was reverse transcribed using a Superscript kit (Invitrogen). Real-time PCR was performed in 96-well format using SYBR Select Master Mix (Applied Biosystems, Foster City, CA) with an ABI 7500 PCR system (Applied Biosystems). The primer sequences used were: Human CHOP: F, 5′-GCCTTTCTCTTCG-3′ and R, 5′-TGTGACCTCTGCTGGTTCTG-3′. Human ATF4: F, 5′-TTCTCCAGCGACAAGGCTAAGG-3′ and R, 5′-CTCCAACATCCAATCTGTCCCG-3′. Human XBP1: F, 5′-GCTTGTGATTGAGAACCAGG-3′ and R, 5′-GAAAGG GAGGCTGGTAAGGAAC-3′. Human Cyclophilin A: F, 5′-GCCTCTCCCTAGCTTTGGTT-3′ and R, 5′-GGTCTGTTAAGGTGGGCAGA-3′. Human GAPDH: F, 5′-CACAGTCCATGCCATCACTG-3′ and R, 5′-TACTCCTTGGAGGCCATGTG-3′.
4.7. Western blotting
HCC-1806 cells were seeded in 60-mm dishes at 8 × 105 cells/dish and treated for the indicated times. Cells were then washed with PBS and lysed with lysis buffer (Cell Signaling Technology, Danvers, MA, USA) containing EDTA and phosphatase inhibitors. Aliquots of 20 µg total protein were separated on 7% SDS-PAGE gels (Life Technologies, Carlsbad, CA, USA) and transferred to PVDF membranes. The membranes were probed with primary antibodies followed by the appropriate HRP-conjugated secondary antibodies (goat anti-rabbit IgG and goat anti-mouse IgG, 1:3000; Santa Cruz Biotechnology, Santa Cruz, CA, USA). Blots were then developed. The primary antibodies and dilutions used were: CHOP (1:1000 no. MA1-250; Thermo, IL, USA), cleaved caspase 3 (1:1000 no. 9661; Cell Signaling Technology), ATF4 (1:1000 no. 10835-1-AP; ProteinTech Group, IL, USA), p-eIF2α (Ser51) (1:1000 no. 9721; Cell Signaling Technology), and α-tubulin (1:2000 no. SC-8035; Santa Cruz Biotechnology).
4.8. Transfection of CHOP siRNA
HCC-1806 cells incubated with serum-free RPMI 1640 medium (Corning) were transfected with scrambled control or CHOP siRNA (E-004819-00; Dharmacon/Thermo Scientific, IL, USA) using LipofectAMINE reagent (Invitrogen). After 6 h, the medium was replaced with RPMI 1640 medium supplemented with 10% FBS and compound 3d was added. After 48 h, the medium was aspirated and 60 µL/well of CellTiter-Glo reagent was added in 96-w format. Cell viability was measured with an EnVision multilabel plate reader.
4.9. Statistical analysis
Data are presented as means ± SD unless specified. Comparisons were performed by two-tailed paired Student’s t-test. A P value of <0.05 was considered statistically significant.
Supplementary Material
References
- 1.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nature reviews. Molecular cell biology. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 2.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 3.Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J. Cell Biol. 2012;197:857–867. doi: 10.1083/jcb.201110131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shore GC, Papa FR, Oakes SA. Signaling cell death from the endoplasmic reticulum stress response. Curr. Opin. Cell Biol. 2011;23:143–149. doi: 10.1016/j.ceb.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011;13:184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer. 2014;14:581–597. doi: 10.1038/nrc3800. [DOI] [PubMed] [Google Scholar]
- 7.Verfaillie T, Salazar M, Velasco G, Agostinis P. Linking ER Stress to Autophagy: Potential Implications for Cancer Therapy. International journal of cell biology. 2010;2010:930509. doi: 10.1155/2010/930509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen X, Iliopoulos D, Zhang Q, Tang Q, Greenblatt MB, Hatziapostolou M, Lim E, Tam WL, Ni M, Chen Y, Mai J, Shen H, Hu DZ, Adoro S, Hu B, Song M, Tan C, Landis MD, Ferrari M, Shin SJ, Brown M, Chang JC, Liu XS, Glimcher LH. XBP1 promotes triple-negative breast cancer by controlling the HIF1alpha pathway. Nature. 2014;508:103–107. doi: 10.1038/nature13119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davenport EL, Moore HE, Dunlop AS, Sharp SY, Workman P, Morgan GJ, Davies FE. Heat shock protein inhibition is associated with activation of the unfolded protein response pathway in myeloma plasma cells. Blood. 2007;110:2641–2649. doi: 10.1182/blood-2006-11-053728. [DOI] [PubMed] [Google Scholar]
- 10.Bosch A, Eroles P, Zaragoza R, Vina JR, Lluch A. Triple-negative breast cancer: molecular features, pathogenesis, treatment and current lines of research. Cancer treatment reviews. 2010;36:206–215. doi: 10.1016/j.ctrv.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 11.Petrocca F, Altschuler G, Tan SM, Mendillo ML, Yan H, Jerry DJ, Kung AL, Hide W, Ince TA, Lieberman J. A genome-wide siRNA screen identifies proteasome addiction as a vulnerability of basal-like triple-negative breast cancer cells. Cancer Cell. 2013;24:182–196. doi: 10.1016/j.ccr.2013.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Papandreou I, Denko NC, Olson M, Van Melckebeke H, Lust S, Tam A, Solow-Cordero DE, Bouley DM, Offner F, Niwa M, Koong AC. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood. 2011;117:1311–1314. doi: 10.1182/blood-2010-08-303099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mimura N, Fulciniti M, Gorgun G, Tai YT, Cirstea D, Santo L, Hu Y, Fabre C, Minami J, Ohguchi H, Kiziltepe T, Ikeda H, Kawano Y, French M, Blumenthal M, Tam V, Kertesz NL, Malyankar UM, Hokenson M, Pham T, Zeng Q, Patterson JB, Richardson PG, Munshi NC, Anderson KC. Blockade of XBP1 splicing by inhibition of IRE1alpha is a promising therapeutic option in multiple myeloma. Blood. 2012;119:5772–5781. doi: 10.1182/blood-2011-07-366633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hetz C, Chevet E, Harding HP. Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 2013;12:703–719. doi: 10.1038/nrd3976. [DOI] [PubMed] [Google Scholar]
- 15.Wang Y, Xiao J, Zhou H, Yang S, Wu X, Jiang C, Zhao Y, Liang D, Li X, Liang G. A novel monocarbonyl analogue of curcumin, (1E,4E)-1,5-bis(2,3-dimethoxyphenyl)penta-1,4-dien-3-one, induced cancer cell H460 apoptosis via activation of endoplasmic reticulum stress signaling pathway. J. Med. Chem. 2011;54:3768–3778. doi: 10.1021/jm200017g. [DOI] [PubMed] [Google Scholar]
- 16.Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, Harding HP, Ron D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma Y, Brewer JW, Diehl JA, Hendershot LM. Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J. Mol. Biol. 2002;318:1351–1365. doi: 10.1016/s0022-2836(02)00234-6. [DOI] [PubMed] [Google Scholar]
- 19.Huber AL, Lebeau J, Guillaumot P, Petrilli V, Malek M, Chilloux J, Fauvet F, Payen L, Kfoury A, Renno T, Chevet E, Manie SN. p58(IPK)-mediated attenuation of the proapoptotic PERK-CHOP pathway allows malignant progression upon low glucose. Molecular cell. 2013;49:1049–1059. doi: 10.1016/j.molcel.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 20.Nakagawa H, Umemura A, Taniguchi K, Font-Burgada J, Dhar D, Ogata H, Zhong Z, Valasek MA, Seki E, Hidalgo J, Koike K, Kaufman RJ, Karin M. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell. 2014;26:331–343. doi: 10.1016/j.ccr.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tran K, Li Y, Duan H, Arora D, Lim HY, Wang W. Identification of Small Molecules That Protect Pancreatic beta Cells against Endoplasmic Reticulum Stress-Induced Cell Death. ACS chemical biology. 2014 doi: 10.1021/cb500740d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singha PK, Pandeswara S, Venkatachalam MA, Saikumar P. Manumycin A inhibits triple-negative breast cancer growth through LC3-mediated cytoplasmic vacuolation death. Cell death & disease. 2013;4:e457. doi: 10.1038/cddis.2012.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghosh S, Adhikary A, Chakraborty S, Bhattacharjee P, Mazumder M, Putatunda S, Gorain M, Chakraborty A, Kundu GC, Das T, Sen PC. Cross-talk between endoplasmic reticulum (ER) stress and the MEK/ERK pathway potentiates apoptosis in human triple negative breast carcinoma cells: role of a dihydropyrimidone, nifetepimine. J. Biol. Chem. 2015;290:3936–3949. doi: 10.1074/jbc.M114.594028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dey S, Baird TD, Zhou D, Palam LR, Spandau DF, Wek RC. Both transcriptional regulation and translational control of ATF4 are central to the integrated stress response. J. Biol. Chem. 2010;285:33165–33174. doi: 10.1074/jbc.M110.167213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 26.Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, Harada A, Mori K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell. 2007;13:365–376. doi: 10.1016/j.devcel.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 27.Yoshida H, Okada T, Haze K, Yanagi H, Yura T, Negishi M, Mori K. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell. Biol. 2000;20:6755–6767. doi: 10.1128/mcb.20.18.6755-6767.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol. 2001;21:1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Puthalakath H, O'Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, Hughes PD, Michalak EM, McKimm-Breschkin J, Motoyama N, Gotoh T, Akira S, Bouillet P, Strasser A. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129:1337–1349. doi: 10.1016/j.cell.2007.04.027. [DOI] [PubMed] [Google Scholar]
- 30.Ruiz-Vela A, Opferman JT, Cheng EH, Korsmeyer SJ. Proapoptotic BAX and BAK control multiple initiator caspases. EMBO reports. 2005;6:379–385. doi: 10.1038/sj.embor.7400375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De La Fuente R, Sonawane ND, Arumainayagam D, Verkman AS. Small molecules with antimicrobial activity against E. coli and P. aeruginosa identified by high-throughput screening. Br. J. Pharmacol. 2006;149:551–559. doi: 10.1038/sj.bjp.0706873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Horrigan S, Zong Q, Soppet D, Castaneda J, Chen B, Cibotti R, Audoly L, Coyle A, Kiener P. PCT Application. 2006. Compounds and methods for treating or preventing autoimmune diseases. WO2008144011. [Google Scholar]
- 33.Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J. Cell Biol. 2001;153:1011–1022. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harding HP, Zhang Y, Khersonsky S, Marciniak S, Scheuner D, Kaufman RJ, Javitt N, Chang YT, Ron D. Bioactive small molecules reveal antagonism between the integrated stress response and sterol-regulated gene expression. Cell Metab. 2005;2:361–371. doi: 10.1016/j.cmet.2005.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y, Shen J, Arenzana N, Tirasophon W, Kaufman RJ, Prywes R. Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic reticulum stress response. J. Biol. Chem. 2000;275:27013–27020. doi: 10.1074/jbc.M003322200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.