

Abstract
KATP channel openers have emerged as potential therapeutics for the treatment of glaucoma, lowering intraocular pressure in animal models and cultured human anterior segments. We have prepared water soluble phosphate and dipeptide derivatives of the KATP channel opener cromakalim and evaluated their IOP lowering capabilities in vivo. In general, the phosphate derivatives proved to be more chemically robust and efficacious at lowering IOP with once daily dosing in a normotensive mouse model. Two of these phosphate derivatives were further evaluated in a normotensive rabbit model, with a significant difference in activity observed. No toxic effects on cell structure or alterations in morphology of the aqueous humor outflow pathway were observed after treatment with the most efficacious compound (3S,4R)−2, suggesting that it is a strong candidate for development as an ocular hypotensive agent.
Table of Contents Graphic
Introduction
Glaucoma is the leading cause of irreversible blindness, affecting an estimated 64 million people worldwide.1 This number is expected to rise to 112 million by 2040 as the global population of the elderly increases. The most common form of the disease is primary open-angle glaucoma (POAG) which is responsible for approximately 74% of glaucoma cases.2 Elevated intraocular pressure (IOP) is the most prevalent and only treatable risk factor for the disease.3–5 All therapies for glaucoma include administration of ocular hypotensive agents to the eye or surgically modifying the outflow pathway to lower IOP. Several long-term clinical trials have demonstrated that lowering IOP slows the progression of glaucoma clinically, even in patients who have normal IOP.6–8
Aqueous humor is produced in the eye by the non-pigmented ciliary body epithelium and supplies nutrients to the avascular tissues of the anterior chamber of the eye. In essence, it serves the same function to the front of the eye that blood does to the rest of the body. Aqueous humor exits the eye by one of two outflow routes: the conventional (trabecular) pathway or the uveoscleral pathway. IOP is generated by the balance between the amount of aqueous humor produced and the amount of aqueous humor that is removed from the front part of the eye. 9–11 In adult humans, the conventional outflow pathway, comprising the trabecular meshwork, Schlemm’s canal and the associated collector channels is responsible for the drainage of 80% or more of the secreted aqueous humor from the anterior chamber.5, 12–13 Most cases of POAG are believed to be caused by an increase in resistance to the drainage of aqueous humor through the conventional pathway, leading to a build-up of fluid in the eye and a corresponding rise in IOP, which in turn damages the optic nerve.10
Existing classes of therapeutics lower IOP by either reducing the production of aqueous humor (β-adrenergic receptor blockers and carbonic anhydrase inhibitors), by increasing the outflow of the fluid from the eye (prostaglandin agonists and cholinergic drugs) or by a combination of the two mechanisms (α-adrenergic agonists).14–15 These therapeutics often need to be used in combination to achieve an IOP-lowering effect of the desired magnitude. While there are now several existing classes of therapeutics to lower IOP, each class comes with its own set of side effects such as ocular adverse events for prostaglandins, cardiac and respiratory side effects for β -blockers, and local allergic reactions with α-adrenergic agonists.15–16 Additionally, none of the current classes of ocular hypotensive agents target the trabecular meshwork, which is the site of increased outflow resistance in glaucoma. As a result, the search for new drugs to treat glaucoma is still a very active area of research.14,17–19
Recently, we identified functional ATP-sensitive potassium (KATP) channels in the human and murine outflow pathways and demonstrated that KATP channel openers including diazoxide and cromakalim (Figure 1) lower IOP in cultured human anterior segments and in vivo in mice.20–22 KATP channels are hetero-octameric complexes comprised of four regulatory sulfonylurea (SUR) receptor subunits and four pore forming inwardly rectifying potassium (Kir) channel subunits. There are three SUR subtypes (SUR1, SUR2A, and SUR2B) as well as two Kir subunits (Kir6.1 and Kir6.2). The subunits that form KATP channels vary by tissue; for instance KATP channels are comprised primarily of SUR2A and Kir6.2 in ventricular myocytes,23 SUR2B and Kir6.1 in adipose tissue,24 and SUR1 and Kir6.2 in pancreatic beta-cells.25 Utilizing pharmacologic KATP channel openers with selective subunit specificity, and a combination of immunohistochemistry and treatment of Kir6.2(−/−) mice, we determined that the IOP lowering effect of KATP channel openers was due to their action on KATP channels comprised of SUR2B and Kir6.2 subunits.21 This combination of SUR and Kir subunits has also been identified in other cell types such as cardiomyocytes.26
Figure 1.

Structures of KATP channel openers diazoxide and cromakalim.
One of the most promising aspects of our results with diazoxide and cromakalim was the strong ocular hypotensive effect in vivo of these drugs with only once daily dosing. This is important because patient non-compliance is one of the major causes of treatment failure for glaucoma and once a day dosing can help increase compliance.16, 27 In addition, cromakalim showed a significant additive effect when used in combination with latanoprost,22 a prostaglandin analog that is one of the first-line drugs used to lower IOP in glaucoma. However, cromakalim and diazoxide have limited aqueous solubility and in our in vivo studies we used DMSO and Cremophor EL to solubilize the KATP channel openers and permeabilize the cornea so that they could be administered topically.20–22 Because the toxicity of DMSO makes its clinical use in eye drops undesirable, 28–29 we synthesized water soluble prodrugs of KATP channel openers and evaluated whether they could be administered topically to lower IOP. Having aqueous solubility is advantageous for topical ophthalmic drug delivery because it allows the minimum amount of fluid to be administered to the eye, which in turn reduces the amount of drug lost to lacrimal drainage.30 The synthesis of novel prodrugs also allows for the generation of potentially patentable new chemical entities, easing the path to clinical development.31 We chose to focus our derivatization efforts on cromakalim as it was significantly more potent than diazoxide in reducing IOP at lower concentrations in our human anterior segments,20, 22 consistent with the relative potencies of the two drugs in activating recombinant SUR2B/Kir6.2 subunit containing KATP channels (3 µM vs. 200 µM).32 In addition, the secondary alcohol of cromakalim lends itself to chemical conjugations that can be used to improve solubility. Herein we report the synthesis of a series of phosphate and dipeptide derivatives of cromakalim and examine their ability to lower IOP in vivo.
Results and Discussion
Synthesis of Phosphate Analogs of Cromakalim
Our initial efforts were focused on synthesizing phosphate prodrugs of cromakalim because typically these types of prodrugs are relatively easy to prepare, add significantly to the aqueous solubility of the parent drug, and display good chemical stability.33–34 In addition, a phosphate prodrug (prednisolone sodium phosphate) is already marketed for ophthalmic use. 35–36
There are two main classes of phosphate ester prodrugs that are commonly used: one in which the phosphate ester is directly attached to the parent compound through an alcohol and one in which the phosphate ester is attached through an oxymethyl spacer group (known as oxymethylphosphate or OMP prodrugs). Both types of prodrugs are activated by alkaline phosphatase, but OMP prodrugs are typically activated more rapidly for steric reasons and release an equivalent of formaldehyde in addition to the parent drug upon bio-activation.33–34 Alkaline phosphatase is present in the cornea37 and previous investigations into phosphate-based prodrugs for ophthalmic use have found complete release of the active parent compounds prior to or during corneal absorption.30, 36 Juntunen and coworkers determined that a phosphate ester of a lipophilic cannabinoid analog had significantly higher flux across rabbit corneas than the parent compound administered as a suspension.30
Cromakalim is a racemic compound, with its activity derived almost entirely from its (3S,4R)-enantiomer levcromakalim.38–39 We initially prepared both the directly-linked and OMP prodrugs of cromakalim in racemic form (compounds 2 and 4, respectively), and based on the results of our initial animal studies, we proceeded to synthesize the individual enantiomers of 2.
To obtain the directly-linked phosphate ester prodrug (3S,4R)-2, levcromakalim was phosphitylated with dibenzyl N,N-dimethylphosphoramidite40 in the presence of tetrazole. The resulting phosphite triester was oxidized in situ with H2O2 to furnish phosphate triester (3S,4R)-1 (Scheme 1). Because attempts to debenzylate 1 using catalytic Pd/C and hydrogen led to extensive decomposition, 1 was instead deprotected using TMSBr.41 The resulting material was purified by reverse-phase chromatography and converted to a sodium salt by ion-exchange column using Dowex resin.42 The (3R,4S)-enantiomer of 2 was prepared by the same synthetic route using dexcromakalim43 instead of levcromakalim as the starting material. Compound 2 was readily soluble in water at 10 mg/mL and was stable in D2O solution for extended periods of time (> 6 months). Addition of alkaline phosphatase (in a buffer containing ZnCl2, MgCl2 and sodium glycine) to an aqueous solution of (3S,4R)-2 led to the release of levcromakalim, confirming that (3S,4R)-2 can act as a prodrug of levcromakalim.
Scheme 1. Synthesis of Directly-Linked Phosphate Cromakalim Analog (3S,4R)-2a.

aReagents and conditions: (a) dibenzyl N,N-dimethylphosphoramidite, tetrazole, CH2Cl2, CH3CN; (b) 30% H2O2, THF, 0 °C; (c) TMSBr, CH2Cl2; (d) ion exchange using Na+-Dowex resin.
The synthesis of OMP prodrug (±)-4 began with conversion of cromakalim into the methylthiomethyl derivative (±)-3 via a Pummerer rearrangement using DMSO, acetic acid and acetic anhydride42 Next, (±)-3 was stirred with N-iodosuccinimide (NIS) and orthophosphoric acid44 to furnish (±)-4, which was obtained as a sodium salt after ion-exchange filtration through Dowex resin. As some OMP prodrugs can be chemically unstable in aqueous solution,42 the stability of compound 4 was monitored by NMR spectroscopy. No signs of decomposition were observed after one week in D2O solution by either 1H or 31P NMR.
We also prepared the more lipophilic CF3-substituted analog (3S,4R)-9, as previous studies have suggested that moderately lipophilic compounds may penetrate the cornea better than highly hydrophilic compounds 45–46 Compound 9 is a prodrug of compound 7 (Scheme 3), which as a racemate was reported to have more potent activity than cromakalim at relaxing airway smooth muscle in guinea pigs.47 The enantioselective synthesis of (3S,4R)-9 began with 4-(trifluoromethyl)phenol,48 which was condensed with 1,1-diethoxy-3-methyl-2-butene in the presence of catalytic base49 to furnish 6-trifluoromethyl-2,2-dimethylchromene (compound 5). Jacobsen epoxidation yielded (S,S)-6, which was purified by silica gel chromatography and then recrystallized to increase the enantiomeric purity of the compound.43 The epoxide was reacted with 2-pyrrolidone and NaH to furnish (3S,4R)-7. HPLC analysis determined the enantiomeric excess of (3S,4R)-7 to be 99%. The synthesis of (3S,4R)-9 was completed using the same sequence of steps used to obtain 2 from levcromakalim.
Scheme 3. Enantioselective Synthesis of CF3-Analog (3S,4R)-9a.

aReagents and conditions: (a) 3-picoline, p-xylene, Δ; (b) (S,S)-Jacobsen’s catalyst, NaOCl, CH2Cl2, H2O, pH ≈ 11.3, 0 °C; (c) 2-pyrrolidone, NaH, DMSO; (d) dibenzyl N,N-dimethylphosphoramidite, tetrazole, CH2Cl2, CH3CN; (e) 30% H2O2, THF, 0 °C; (f) TMSBr, CH2Cl2; (g) ion exchange using Na+-Dowex resin.
Synthesis of Dipeptide-Conjugated Analogs
In addition to the phosphate-based prodrugs described above, we also prepared a set of analogs in which levcromakalim was conjugated to a dipeptide. These dipeptide analogs offer several advantages with regards to ocular bioavailability that make their preparation attractive. First, they have the potential to be actively transported into the eye by oligopeptide transporters located within the cornea45, 50 Second, they are less polar than the highly hydrophilic phosphate prodrugs, and thus would be expected to better penetrate the eye by passive transport mechanisms.
In our initial efforts, we attempted to synthesize Gly-Sar and Gly-Leu conjugates of levcromakalim, as these dipeptides are known to be good substrates for the oligopeptide transporter PepT1.51 However, the synthesis of these compounds proved to be challenging, likely due to the crowded steric environment near the alcohol of levcromakalim, and unfortunately we were unable to obtain significant amounts of the Gly-Sar conjugate. The best conditions identified for preparing the Gly-Leu conjugate started by coupling Boc-protected Gly-Leu to levcromakalim using several equivalents of HATU and DMAP (Scheme 4). This procedure led to a mixture of epimers (10 and 12). After separation by silica gel chromatography, epimers 10 and 12 were individually Boc-deprotected and purified by reverse-phase chromatography, yielding compounds 11 and 13, respectively, as formic acid salts. Our initial effort to assign the stereochemistry of the two epimers consisted of coupling Boc-Gly-D-Leu-OH with levcromakalim. However, this led to a mixture of 10 and 12 in the same ratio that was obtained with Boc-Gly-Leu-OH, meaning that it was not possible to assume the major product of each reaction contained an unepimerized leucine residue. Instead, X-ray crystallography was used to definitively assign stereochemistry. Since we were unable to obtain suitable X-ray quality single crystals from compounds 10–13, a more crystalline product was synthesized by coupling compound 11 to 4-bromobenzoic acid (Scheme 4). Analysis of an X-ray crystal structure of the resulting amide 14 determined that 11 was the conjugate of levcromakalim and Gly-D-Leu-OH (see Supporting Information).
Scheme 4. Synthesis of Levcromakalim Analogs Directly Conjugated to Dipeptidesa.

aReagents and conditions: (a) Boc-Gly-Leu-OH, HATU, DMAP, CH2Cl2; (b) 4M HCl in dioxane; (c) 4-bromobenzoic acid, HATU, DMAP, CH2Cl2.
Because the in vivo release of levcromakalim from prodrugs 11 and 13 by esterases could potentially be hindered by the sterically crowded environment near the ester in these prodrugs, we also prepared compound (3S,4R)-15, in which the Gly-Leu dipeptide was conjugated to levcromakalim via an acetalester.52–53 Analogous to OMP prodrug 4, this compound would release the parent drug and an equivalent of formaldehyde upon bioactivation by esterases. To synthesize this compound, (3S,4R)-3 was prepared as a single enantiomer from levcromakalim by the same method that was used to obtain racemic 3 from cromakalim (Scheme 2). Next, this methylthiomethyl derivative was treated with sulfuryl chloride followed by addition of Boc-Gly-Leu-OH to obtain the Boc-protected conjugate (Scheme 5). Deprotection with HCl yielded the desired (3S,4R)-15 as an HCl salt.
Scheme 2. Synthesis of Cromakalim Oxymethylphosphate Analog (±)-4a.

aReagents and conditions: (a) DMSO, Ac2O, AcOH; (b) H3PO4N-iodosuccinimide, molecular sieves, THF; (c) ion exchange using Na+-Dowex resin.
Scheme 5. Synthesis of a Levcromakalim Analog Conjugated to a Dipeptide through an Acetalester Linkera.

aReagents and conditions: (a) sulfuryl chloride, DCE, Boc-Gly-Leu-OH, DIEA; (b) 4M HCl in dioxane.
Intraocular Pressure Lowering Effects of Cromakalim Prodrugs in Normotensive Mice
We have previously reported that cromakalim reduces IOP in normotensive C57BL/6 wild type mice by 3.19 ± 0.41 mm Hg compared to vehicle treated contralateral eyes over a 5 day treatment period with once daily topical administration of 5 mM cromakalim (5 µL) dissolved in a mixture of DMSO, Cremophor EL and phosphate-buffered saline (PBS).22 To evaluate the cromakalim phosphate-based prodrugs, we treated >7 month old C57BL/6 retired breeders daily for 7 consecutive days with (±)-2 or (±)-4 dissolved in PBS. Compounds were administered topically once daily as a 5 µl bolus to one eye while vehicle (PBS) was administered to the contralateral eye. IOP was measured and recorded at times corresponding to 1, 4 and 23 hours after treatment in conscious, non-anesthetized mice using a hand held rebound tonometer (Icare Tonolab; Colonial Medical Supply, Franconia, NH). The average of the 1, 4 and 23 hour readings was used as the daily IOP and expressed as change in IOP (ΔIOP=IOP of treated – IOP of control eye in mm Hg) with respect to the vehicle treated contralateral eye. Treatment with (±)-2 (2.5 mM) resulted in a significant reduction in IOP (2.50 ± 0.16 mm Hg reduction, n=5, p<0.001) following a once daily topical administration for 7 consecutive days (Figure 2A, purple line). In contrast, OMP prodrug (±)-4 used at a concentration of 2.5 mM (1.81 ± 0.85 mm Hg reduction, n=5, p<0.001) (Figure 2A, green line) was both less potent than (±)-2 at lowering IOP and took a greater number of administrations to reach its maximum effect. In both experiments contralateral eyes treated with vehicle (PBS) showed a slight, but not statistically significant, increase in pressure compared to pre-treatment baseline measurements (see Table S1 in Supporting Information).
Figure 2.

(A) Comparison of IOP lowering effects of cromakalim prodrugs following once daily treatment for 7 consecutive days. (±)-2, 2.5 mM in PBS, n=5 (days 1–8) and n=3 (days 9–13), purple line; (±)-4, 2.5 mM in PBS, n=5 (all days), green line; (3S,4R)-2, 2.5 mM in PBS, n=10 (days 1–10) and n=6 (days 11–13), red line. Note that several animals were sacrificed prior to post-treatment to examine ocular histology and morphology. (B) Representative images of the aqueous outflow pathway in mice following treatment with (3S,4R)-2 or vehicle control. Hematoxylin and eosin stained sections show that tissue morphology is maintained in (3S,4R)-2 treated eyes. Transmission electron micrographs show that no observable changes occurred to cell structure or number in the aqueous outflow pathway following treatment with (3S,4R)-2. TM, trabecular meshwork; SC, Schlemm’s canal; AC, anterior chamber.
Based on the ocular hypotensive effects of (±)-2 in mice, we synthesized and tested its individual enantiomers. Treatment of C57BL/6 mice with (3S,4R)-2, the enantiomer of 2 derived from levcromakalim, resulted in a significant reduction in IOP (3.14 ± 0.42 mm Hg, n=10, p<0.001) following a topical once daily administration of a 5 µl bolus of a 2.5 mM solution in PBS for 7 consecutive days (Figure 2A, red line). Comparison of IOP at 1, 4, and 23 hours after daily administration of (3S,4R)-2 showed no significant difference, suggesting that the compound may be suitable for once daily dosing (Figure 3). Additionally, cessation of treatment resulted in IOP returning to baseline within 48 hours. Notably, the robust IOP-lowering ability of an aqueous solution of (3S,4R)-2 in the absence of DMSO and Cremophor EL shows that these permeabilizing agents are not necessary for a strong in vivo effect. Furthermore, treatment with (3S,4R)-2 did not alter morphology of the aqueous humor outflow pathway and did not show any toxic effects on cell structure (Figure 2B). Taken together, these results suggest that (3S,4R)-2 is a strong candidate as an ocular hypotensive agent due to its aqueous solubility, once daily dosing, IOP reduction comparable to the parent compound cromakalim22, and no observable toxicity following treatment. Additionally, (3S,4R)-2 shows similar IOP reduction to other clinically approved drugs that are currently used in first-line treatment of glaucoma (e.g. latanoprost).54
Figure 3.

C57BL/6 mice treated with (3S,4R)-2 (2.5 mM in PBS) at 1, 4, and 23 h post-treatment, n=10 (for days 1–10) and n=6 (for days 11–13)
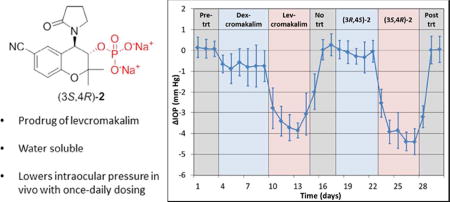
We next examined and compared the IOP lowering ability of levcromakalim and (3S,4R)-2 to their respective enantiomers dexcromakalim and (3R,4S)-2 (Figure 4A). Following establishment of baseline IOP, mice were treated once daily with a topical application of dexcromakalim (initially dissolved in DMSO at a 100 mM concentration and diluted to a 5mM solution with 10% cremophor EL in PBS) for 6 days, followed by 5 days of treatment with levcromakalim (5 mM solution prepared the same as dexcromakalim). Subsequently, IOP was allowed to return to baseline and 5 mM (3R,4S)-2 (dissolved in PBS) was added once daily for 5 days followed by 5 mM (3S,4R)-2 (dissolved in PBS) for another 5 days. Results show a significant but weak reduction of IOP with dexcromakalim (0.73 ± 0.42 mm Hg reduction, n=10, p<0.001) compared to a robust IOP reduction with levcromakalim (3.37 ± 0.44 mm Hg, n=10, p<0.001). Similarly, treatment with (3R,4S)-2 showed minimal IOP reduction (0.14 ± 0.26 mm Hg reduction, n=10, p=0.12) whereas (3S,4R)-2 lowered IOP by 3.82 ± 0.35 (n=10, p<0.001) which was similar in magnitude to that observed with its parent compound levcromakalim. Analysis of the aqueous outflow pathways in mouse eyes following treatment did not show any observable changes to cell structure or number when compared to vehicle treated control (Figure 4B).
Figure 4.

(A) Comparison of IOP-lowering ability of individual enantiomers of cromakalim and (±)-2: n=10 (days 1–27) and n=6 (days 28–30). Dexcromakalim and levcromakalim were administered in a mixture of DMSO, cremophor EL, and PBS. (3R,4S)-2 and (3S,4R)-2 were dissolved in PBS. All drugs were used at 5 mM and added as a 5 µl bolus. Note: four mice were sacrificed on day 27 to examine ocular histology and morphology. (B) Histologic analysis showed no difference in cellular morphology of the aqueous outflow pathway between vehicle control and treated eyes.
The more lipophilic CF3-substituted analog (3S,4R)-9 also showed very good IOP reduction when administered topically at 5 mM to mouse eyes, lowering IOP by 3.91 ± 0.29 mmHg compared to vehicle treated contralateral eyes over a 6 day treatment period (Figure 5, purple line). Like (3S,4R)-2, the CF3-substituted analog showed very little difference between IOP reduction measured at 1, 4, and 23 hours after each dose which is an ideal profile for once daily dosing. However, after treatment was stopped it took more than 72 h for IOP to return to baseline with (3S,4R)-9, which was longer than that observed with (3S,4R)-2.
Figure 5.

C57BL/6 mice treated with once daily 5 mM solution of (3S,4R)-9 in PBS at 1, 4, and 23 h post-treatment, n=10 (for days 1–9) and n=6 (for days 10–12).
In contrast to the phosphate-based cromakalim analogs, the dipeptide derivatives 11 (Figure 6, blue line), 13 (red line), and 15 (green line) administered once daily at 5 mM in PBS all performed poorly at lowering IOP in vivo in mice. Although IOP changes were statistically significant when compared to the contralateral eye, all dipeptide compounds exhibited less efficacy and took longer to produce a maximum IOP reduction in comparison to the phosphate-based analogs (11, 1.57 ± 0.55 mm Hg, n=5, p=0.003; 13, 2.45 ± 0.27 mm Hg, n=9, p<0.001; 15, 1.93 ± 0.39 mm Hg, n=5, p<0.001). In addition, compound 13 showed inconsistent IOP reduction throughout the course of the treatment regiment, suggesting it may be unstable in solution. This is not unexpected, as previously synthesized dipeptide analogs have shown pH-dependent chemical stability profiles, with good stability observed only under moderately acidic conditions (pH 4–6).50 Because of these less than optimal results, the development of the dipeptide derivatives was discontinued and the phosphate-based analogs advanced into further testing.
Figure 6.

C57BL/6 mice treated with dipeptides 11 (n=5); 13 (n=9, for days 1–11, n=5 for days 12–14), 15 (n=5). All were dissolved in PBS and treated at 5 mM dose. *No drug added to mice treated with 13 on day 11.
Intraocular Pressure Lowering Effects of Compounds in Normotensive Rabbits
Mice have significantly thinner corneas than humans (approximately 130 µM vs. 535 µM),55 making it easier for topically applied therapeutics to enter the eye. In contrast, rabbits have a thicker cornea than mice (approximately 380 µM)56 and thus may serve as a better model for evaluating the efficacy of the phosphate-based analogs. Accordingly, both (3S,4R)-2 and (3S,4R)-9 dissolved in PBS were applied topically to normotensive Dutch-belted pigmented rabbits at a 2.5 mM concentration in a 50 µL bolus once daily for four days, followed by daily dosing at 5 mM for an additional four days (Figure 7). While both compounds showed similar efficacy in the normotensive mouse model, (3S,4R)-2 showed better IOP lowering potency than (3S,4R)-9 in the rabbit model at 2.5 mM (2.02 ± 0.63 mm Hg versus 1.06 ± 0.39 mm Hg) and at 5 mM (2.26 ± 0.57 mm Hg versus 1.60 ± 0.52 mm Hg for (3S,4R)-9).
Figure 7.

Both (3S,4R)-2 (red line; n=5) and (3S,4R)-9 (blue line; n=5) dosed at 2.5 and 5 mM lowered IOP in normotensive Dutch-belted pigmented rabbits, but (3S,4R)-2 produced a greater and more consistent reduction over the treatment period. Compounds were administered topically once daily at 2.5 mM for 4 days followed by 5 mM for 4 days. Data points are an average of IOP measurements taken at 1, 4, and 23 hours after each dose and are expressed as the average absolute difference in IOP (in mmHg) between the treated and control eyes.
Summary
KATP channels have emerged as a new therapeutic target for the treatment of glaucoma.20–22 We have synthesized a set of phosphate and dipeptide derivatives of the KATP channel opener cromakalim and evaluated their IOP lowering capabilities in vivo. In general, the phosphate derivatives proved to be more chemically robust and efficacious at lowering IOP in a normotensive mouse model. These compounds also had high aqueous solubility, which should facilitate their clinical development. Two of these derivatives, (3S,4R)-2 and (3S,4R)-9 were further evaluated in a normotensive rabbit model, with a significant difference in activity observed. Because of the efficacy of compound (3S,4R)-2 at lowering IOP in both mouse and rabbit models, we are continuing its development as a potential clinical candidate for the treatment of glaucoma.
Experimental Protocols
Chemistry
NMR spectra were recorded using a Bruker 400 spectrometer. 1H NMR data are reported as follows: chemical shift in parts per million downfield of tetramethylsilane (TMS), multiplicity (s = singlet, bs = broad singlet, d = doublet, t = triplet, q = quartet, quint = quintet and m = multiplet), coupling constant (Hz), and integrated value. Coupling constants listed as J31P disappeared when 1H NMR spectra were obtained with 31P decoupling. 31P NMR spectra were taken with complete proton decoupling and were referenced to 85% phosphoric acid, which was added to the NMR tube in a sealed capillary tube. Cromakalim was synthesized by the method of Ashwood38 and levcromakalim by the method of Jacobsen.43 All compounds tested in vivo were found to have > 95% purity by HPLC or LC/MS analysis.
Dibenzyl ((3S,4R)-6-cyano-2,2-dimethyl-4-(2-oxopyrrolidin-1-yl)chroman-3-yl) phosphate (1)
To a stirred suspension of levcromakalim (150 mg, 0.175 mmol) in CH2Cl2 (15 mL) was added 0.45 M tetrazole in acetonitrile (11.7 mL, 5.28 mmol) followed by dibenzyl N,N-dimethylphosphoramidite (0.600 mL, 2.3 mmol). The reaction mixture was stirred at rt for 2.5 h, then cooled to 0 °C. THF (15 mL) was added, followed by dropwise addition of 30% H2O2 (3 mL). After stirring for an additional 15 min., saturated aqueous Na2S2O3 (60 mL) was added slowly. The mixture was diluted with water (100 mL) and extracted with CH2Cl2 (3 × 100 mL). The combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. The crude material was purified by flash column chromatography on silica gel (5–30% acetone in hexanes as eluent). Impure fractions were repurified as above and combined with the pure material to obtain the title compound as a white foam (0.272 g, 86% yield). 1H NMR (400 MHz, CDCl3): 7.46 (d, J = 8.5 Hz, 1H), 7.40–7.30 (m, 10H), 7.26 (s, 1H), 6.91 (d, J = 8.5 Hz, 1H), 5.55 (d, J = 9.9 Hz, 1H), 5.10–4.96 (m, 4H), 4.60 (dd, J = 9.9 Hz, J31P = 10 Hz, 1H), 3.56–3.46 (m, 1H), 2.94–2.83 (m, 1H), 2.51–2.39 (m, 1H), 2.35–2.23 (m, 1H), 2.02–1.88 (m, 2H), 1.51 (s, 3H), 1.28 (s, 3H). 13C NMR (100 MHz, CDCl3): 18.0, 18.8, 26.7, 31.0, 42.3, 50.2, 69.7 (d, J = 5.7 Hz), 69.8 (d, J = 5.9 Hz), 75.1 (d, J = 6.2 Hz), 79.1 (d, J = 5.0 Hz), 104.9, 118.7, 118.8, 120.4, 127.9, 128.2, 128.65, 128.67, 128.74, 128.75, 131.8, 133.3, 135.3 (d, J = 4.3 Hz), 135.4 (d, J = 5.3 Hz), 156.8, 177.2. 31P NMR (162 MHz, CDCl3): −0.86. HRMS m/z calcd for C30H31N2NaO6P [M + Na]+ 569.1812, found 569.1831. [α]D22 −8.1 (c 1.0, CHCl3).
Sodium (3S,4R)-6-cyano-2,2-dimethyl-4-(2-oxopyrrolidin-1-yl)chroman-3-yl phosphate [(+)-2]
To a solution of (−)-1 (122.3 mg, 0.224 mmol) in dry CH2Cl2 (6 mL) was added TMSBr (100 µL, 0.758 mmol) by syringe. After stirring for overnight, the reaction mixture was concentrated under reduced pressure. The resulting residue was purified by chromatography (0% acetonitrile/40 mM triethylammonium acetate buffer to 100% acetonitrile, C18 column) to yield a white solid after lyophilization. To prepare the sodium salt of (+)-2, a 1 cm wide column was filled with 12 cm of DOWEX 50W2 (50–100 mesh, strongly acidic) ion exchange resin. The column was prepared by sequentially washing with 1:1 acetonitrile/water, 1M aqueous NaHCO3 (lots of gas evolution), water, and then finally 1:1 acetonitrile/water. The reaction product was dissolved in 1:1 acetonitrile/water and loaded onto the column, which was eluted with 1:1 acetonitrile/water. The product containing fractions were lyophilized to furnish the title compound as a white solid (63.5 mg, 69% yield). 1H NMR (400 MHz, D2O): 7.63 (dd, J = 8.6, 1.8 Hz, 1H), 7.46 (bs, 1H), 7.00 (d, J = 8.6 Hz, 1H), 5.26 (d, J = 9.8 Hz, 1H), 4.46 (dd, J = 10.0, J31P = 10 Hz, 1H), 3.66–3.56 (m, 1H), 3.21–3.07 (m, 1H), 2.67–2.51 (m, 2H), 2.20–1.99 (m, 2H), 1.58 (s, 3H), 1.32 (s, 3H). 13C NMR (100 MHz, D2O): 17.9, 18.6, 26.7, 31.9, 52.1, 72.8 (d, J = 6.4 Hz), 81.1 (d, J = 4.3 Hz), 104.2, 119.1, 120.3, 121.1, 132.8, 134.4, 157.5, 181.3. 31P NMR (162 MHz, D2O): 0.30. HRMS m/z calcd for C16H18N2Na2O6P [M + H]+ 411.0692, found 411.0686. [α]D22 +22.9 (c 1.0, H2O). Racemic 2 was prepared starting from cromakalim by the same set of procedures used to synthesize (+)-2 from levcromakalim (see Supporting Information).
(±)-(3S,4R)-2,2-Dimethyl-3-((methylthio)methoxy)-4-(2-oxopyrrolidin-1-yl)chromane-6-carbonitrile (3)
To a solution of racemic cromakalim (188.2 mg, 0.657 mmol) in DMSO (10 mL) was added acetic anhydride (10 mL) and acetic acid (6 mL). After stirring at rt for 24 hours, the reaction mixture was diluted with water (400 mL) and carefully neutralized with solid NaHCO3. The mixture was extracted with ethyl acetate (3 × 400 mL). The organic layers were then each further extracted with water (400 mL), combined, dried (Na2SO4), filtered, and concentrated under reduced pressure. Purification by flash chromatography (30% to 100% ethyl acetate/hexanes) on silica gel furnished 202.5 mg of (±)-3 (89% yield) as a white solid. Use of levcromakalim as the starting material in this procedure yielded (+)-3 as product [optical rotation [α]D22 +21.0 (c 1.0, CHCl3); mp 152–154 °C]. 1H NMR (400 MHz, CDCl3): 7.45 (d, J = 8.5 Hz, 1H), 7.25 (bs, 1H), 6.89 (d, J = 8.5 Hz, 1H), 5.40 (d, J = 9.5 Hz, 1H), 4.83 (d, J = 12.0 Hz, 1H), 4.69 (d, J = 12.0 Hz, 1H), 3.80 (d, J = 9.5 Hz, 1H), 3.53–3.44 (m, 1H), 3.00–2.90 (m, 1H), 2.65–2.44 (m, 2H), 2.18 (s, 3H), 2.16–1.98 (m, 2H), 1.54 (s, 3H), 1.28 (s, 3H). 13C NMR (100 MHz, CDCl3): 14.3, 17.9, 19.1, 27.1, 31.2, 43.0, 50.7, 74.7, 76.4, 80.0, 104.6, 118.8, 118.9, 121.5, 132.0, 133.0, 157.2, 176.5. HRMS m/z calcd for C18H22N2NaO3S [M + Na]+ 369.1243, found 369.1233.
(±)-Sodium (((3S,4R)-6-cyano-2,2-dimethyl-4-(2-oxopyrrolidin-1-yl)chroman-3-yl)oxy)methyl phosphate (4)
To a stirred suspension of (±)-3 (30.8 mg, 0.089 mmol), phosphoric acid (74.1 mg, 0.756 mmol) and 4 Å molecular sieves (239 mg) in THF (3 mL) at 0 °C, was added a solution of N-iodosuccinimide (32.9 mg, 0.146 mmol) in THF (1 mL). After warming to rt over 2 h, a TLC showed starting material remaining, so additional NIS was added (37 mg, 0.164 mmol). An hour later, the mixture was decanted to remove the sieves. Aqueous sodium thiosulfate was added until the color disappeared and then 0.5 mL of 1 M triethylammonium acetate buffer was added. The THF was removed under reduced pressure and the resulting residue purified by chromatography (0% acetonitrile/20 mM triethylammonium acetate buffer to 100% acetonitrile, C18 column) to yield 13.4 mg of brown solid after lyophilization. The sodium salt of 4 was prepared by the same procedure described above for 2. White solid (7.7 mg, 20% yield from 3). 1H NMR (400 MHz, D2O): 7.64 (d, J = 8.6 Hz, 1H), 7.49 (s, 1H), 7.01 (d, J = 8.6 Hz, 1H), 5.26 (d, J = 9 Hz, 1H), 5.07 (dd, J = 5.3, J31P = 6 Hz, 1H), 5.02 (dd, J = 5.3, J31P = 6 Hz, 1H), 4.14 (d, J = 10.0 Hz, 1H), 3.72–3.60 (m, 1H), 3.26–3.08 (m, 1H), 2.82–2.68 (m, 1H), 2.66–2.54 (m, 1H), 2.32–2.17 (m, 1H), 2.15–1.99 (m, 1H), 1.61 (s, 3H), 1.30 (s, 3H). LC/MS calculated for (C17H21N2O7P-H)−, 395.1; observed, 395.5.
6-Trifluoromethyl-2,2-dimethylchromene (5)
To a 100 mL 3-necked round bottom flask equipped with a distillation head was added 5,5-diethoxy-2-methylpent-2-ene (5.9 mL, 29.0 mmol) in p-xylene (60 mL), followed by 4-(trifluoromethyl)phenol (3.0 g, 18.5 mmol) and 3-picoline (0.60 mL, 6.2 mmol). The reaction flask was heated to an internal temperature of 95 °C for 12 h., then heated at an internal temperature of 115 °C for 7 h., and cooled to room temperature. The reaction mixture was diluted with EtOAc (30 mL) and washed with 1M HCl (3 × 20 mL). The organic layer was washed with saturated aqueous NaHCO3 (2 × 20 mL) and brine (20 mL). The organic layer was dried over MgSO4, filtered, and concentrated under reduced pressure. The crude material was purified by flash column chromatography on silica gel (0–10% EtOAc in hexanes as eluent) to obtain the title compound (1.03 g, 24% yield) as a clear, colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.35 (dd, J = 8.4, 2.2 Hz, 1H), 7.22 (d, J = 2.2 Hz, 1H), 6.83 (d, J = 8.4 Hz, 1H), 6.33 (d, J = 9.8 Hz, 1H), 5.69 (d, J = 9.9 Hz, 1H), 1.46 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 28.4, 77.4, 116.6, 121.3, 121.6, 123.0 (q, J = 32.4 Hz), 123.6 (q, J = 3.8 Hz), 124.6 (q, J = 269.5 Hz), 126.4 (q, J = 3.7 Hz), 132.0, 155.8.
(1aS,7bS)-2,2-dimethyl-6-(trifluoromethyl)-1a,7b–dihydro-2H–oxireno[2,3-c]chromene (6)
A solution of bleach (NaOCl) buffered to pH ≈ 11.3 was prepared by addition of 0.05 M Na2HPO4 and 1 N NaOH. To 30 mL of this solution cooled to 0 °C was added a solution of 5 (1.26 g, 5.52 mmol) and (S,S)-Jacobsen’s catalyst (500 mg, 0.79 mmol) in CH2Cl2 (20 mL). The mixture was stirred vigorously at 0 °C for 6 hours, after which it was diluted with EtOAc (250 mL) and extracted with water (200 mL) followed by brine (200 mL). The organic layer was dried (Na2SO4), filtered, and concentrated under reduced pressure. Purification by flash chromatography (0–30% ethyl acetate/hexanes) on silica gel furnished 1.008 g product as a yellow solid. The solid was dissolved in hot ethanol (11 mL) and then water (8 mL) was added slowly to the solution. After cooling to rt, 747.9 mg of 6 (55%) was obtained as a yellow solid. 1H NMR (400 MHz, CDCl3): 7.61 (d, J = 2.0 Hz, 1H), 7.49 (dd, J = 8.5, 1.8 Hz, 1H), 6.88 (d, J = 8.5 Hz, 1H), 3.93 (d, J = 4.3 Hz, 1H), 3.52 (d, J = 4.4 Hz, 1H), 1.60 (s, 3H), 1.28 (s, 3H). 13C NMR (100 MHz, CDCl3): 22.8, 25.6, 50.4, 62.5, 74.0, 118.3, 120.3, 123.3 (q, J = 32.7 Hz), 124.2 (q, J = 268.3 Hz), 127.0 (q, J = 3.9 Hz), 127.5 (q, J = 3.7 Hz), 155.4.
1-((3S,4R)-3-Hydroxy-2,2-dimethyl-6-(trifluoromethyl)chroman-4-yl)pyrrolidin-2-one (7)
To a stirred solution of (S,S)-6 (735 mg, 3.01 mmol) and 2-pyrrolidinone (0.350 mL, 4.61 mmol) in DMSO (5 mL) was added NaH (60% dispersion in mineral oil, 190 mg, 4.75 mmol). After 2 hours, the mixture was diluted with water (125 mL) and extracted with EtOAc (3 × 150 mL). Each organic extract was then further extracted with water (2 × 125 mL). The organic extracts were combined, dried (Na2SO4), filtered, and concentrated under reduced pressure. Purification by flash chromatography (30–100% ethyl acetate/hexanes) on silica gel furnished 7 as a white solid (483.8 mg, 48.8%). The enantiomeric excess of 7 was determined to be 98.9% by HPLC analysis using a Chiralcel OD column (90% hexanes/10% IPA, 1 ml/min., 220 nM). 1H NMR (400 MHz, CDCl3): 7.42 (bd, J = 8.7 Hz, 1H), 7.17 (bs, 1H), 6.90 (d, J = 8.6 Hz, 1H), 5.31 (d, J = 10.3 Hz, 1H), 3.83 (d, J = 6.6 Hz, 1H), 3.76 (dd, J = 10.1, 6.6 Hz, 1H), 3.39–3.29 (m, 1H), 3.11–2.99 (m, 1H), 2.65–2.48 (m, 2H), 2.21–1.97 (m, 2H), 1.54 (s, 3H), 1.27 (s, 3H). 13C NMR (100 MHz, CDCl3): 18.2, 18.4, 26.7, 31.4, 42.7, 52.2, 70.7, 79.9, 118.1, 119.6, 123.1 (q, J = 32.9 Hz), 124.2 (q, J = 271.5 Hz), 124.5 (q, J = 3.8 Hz), 126.4 (q, J = 3.6 Hz), 156.4, 178.2. [α]D22 −17.1 (c 1.0, CHCl3). Mp 219–222 °C. HRMS m/z calcd for C16H18F3N1NaO3 [M+Na]+ 352.1136, found 352.1144.
Dibenzyl ((3S,4R)-2,2-dimethyl-4-(2-oxopyrrolidin-1-yl)-6-(trifluoromethyl)chroman-3-yl) phosphate (8)
Compound (3S,4R)-8 was prepared from (3S,4R)-7 by the same procedure described above for the synthesis of 1 from levcromakalim. Clear colorless oil, 63% yield. 1H NMR (400 MHz, CDCl3): 7.43 (d, J = 8.6 Hz, 1H), 7.40–7.30 (m, 10H), 7.19 (s, 1H), 6.91 (d, J = 8.6 Hz, 1H), 5.58 (d, J = 9.9 Hz, 1H), 5.10–4.96 (m, 4H), 4.60 (dd, J = 9.9 Hz, J31P = 10 Hz, 1H), 3.54–3.44 (m, 1H), 2.93–2.84 (m, 1H), 2.51–2.39 (m, 1H), 2.36–2.24 (m, 1H), 2.03–1.83 (m, 2H), 1.51 (s, 3H), 1.28 (s, 3H). 13C NMR (100 MHz, CDCl3): 18.2, 18.7, 26.8, 31.2, 42.3, 50.5, 69.6 (d, J = 5.5 Hz), 69.7 (d, J = 5.9 Hz), 75.6 (d, J = 6.4 Hz), 78.5 (d, J = 4.9 Hz), 118.2, 119.4, 123.8 (q, J = 33.0 Hz), 124.1 (q, J = 271.1 Hz), 124.6 (q, J = 3.6 Hz), 126.6 (q, J = 3.6 Hz), 127.9, 128.3, 128.63, 128.66, 128.69, 128.70, 135.46 (d, J = 2.7 Hz), 135.54 (d, J = 1.4 Hz), 155.8, 177.2. 31P NMR (162 MHz, CDCl3): −0.75. HRMS m/z calcd for C30H31F3N1NaO6P [M + Na]+ 612.1733, found 612.1745.
Sodium (3S,4R)-2,2-dimethyl-4-(2-oxopyrrolidin-1-yl)-6-(trifluoromethyl)chroman-3-yl phosphate (9)
Compound (3S,4R)-9 was prepared from (3S,4R)-8 by the same procedure described above for the synthesis of 2 from 1. White solid, 80% yield. 1H NMR (400 MHz, D2O): 7.58 (dd, J = 8.6, 1.4 Hz, 1H), 7.30 (bs, 1H), 7.00 (d, J = 8.6 Hz, 1H), 5.22 (d, J = 9.9 Hz, 1H), 4.41 (dd, J = 10.0, J31P = 10 Hz, 1H), 3.77–3.65 (m, 1H), 3.17–3.03 (m, 1H), 2.74–2.61 (m, 1H), 2.60–2.48 (m, 1H), 2.21–2.08 (m, 1H), 2.08–1.94 (m, 1H), 1.60 (s, 3H), 1.30 (s, 3H). 13C NMR (126 MHz, D2O): 17.4, 17.9, 23.2, 26.4, 31.5, 44.0, 52.1, 71.5 (d, J = 6.1 Hz), 80.6 (d, J = 3.5 Hz), 117.7, 120.2, 123.0 (q, J = 33.1 Hz), 124.2 (q, J = 270 Hz), 124.5 (q, J = 3.8 Hz), 126.6 (q, J = 3.6 Hz), 155.6, 180.7. 31P NMR (162 MHz, D2O): 2.09. HRMS m/z calcd for C16H18F3NNa2O6P [M + H]+ 454.0614, found 454.0604.
(3S,4R)-6-Cyano-2,2-dimethyl-4-(2-oxopyrrolidin-1-yl)chroman-3-yl (tert-butoxycarbonyl)glycyl-D-leucinate (10) and (3S,4R)-6-Cyano-2,2-dimethyl-4-(2-oxopyrrolidin-1-yl)chroman-3-yl (tert-butoxycarbonyl)glycyl-L-leucinate (12)
A suspension of levcromakalim (80.6 mg, 0.281 mmol), DMAP (308.9 mg, 2.53 mmol), and Boc-Gly-Leu-OH (334.7 mg, 1.16 mmol) was stirred for 10 min. in DCM (5 mL). HATU (473.7 mg, 1.25 mmol) was added and the mixture stirred at rt. After 24 h., the solvent was removed under reduced pressure and the resulting residue was purified by chromatography (25% to 75% ethyl acetate/hexanes) on silica gel to furnish 103 mg of 10, followed by 31.9 mg of 12, both as waxy white solids. Compound 10: 1H NMR (400 MHz, CDCl3): 7.48 (dd, J = 8.6, 1.6 Hz, 1H), 7.31 (bs, 1H), 6.93 (d, J = 8.6 Hz, 1H), 6.57 (d, J = 7.4 Hz, 1H), 5.76 (bs, 1H), 5.44 (d, J = 10.5 Hz, 1H), 5.22 (d, J = 10.5 Hz, 1H), 4.56–4.43 (m, 1H), 3.93 (dd, J = 16.7, 6.4 Hz, 1H), 3.75 (dd, J = 16.6, 5.9 Hz, 1H), 3.35–3.25 (m, 1H), 3.00–2.89 (m, 1H), 2.62–2.43 (m, 2H), 2.12–1.95 (m, 2H), 1.74–1.48 (m, 3H), 1.46 (s, 9H), 1.42 (s, 3H), 1.34 (s, 3H), 0.99 (d, J = 6.4 Hz, 3H), 0.95 (d, J = 6.4 Hz, 3H). 13C NMR (100 MHz, CDCl3): 18.2, 19.6, 21.7, 22.7, 24.9, 26.2, 28.3, 31.0, 40.5, 42.4, 44.4, 48.9, 51.6, 69.7, 78.5, 80.1, 104.9, 118.8, 119.0, 120.0, 131.8, 133.3, 156.6, 156.9, 170.3, 171.1, 177.4. LC/MS calculated for C29H40N4O7 + H+, 557.3; observed, 557.5. Compound 12: 1H NMR (400 MHz, CDCl3): 7.48 (dd, J = 8.3, 1.5 Hz, 1H), 7.25 (s, 1H), 6.94 (d, J = 8.6 Hz, 1H), 6.65–6.53 (m, 1H), 5.46 (d, J = 10.0 Hz, 1H), 5.33–5.21 (m, 1H), 5.14 (d, J = 10.1 Hz, 1H), 4.67–4.57 (m, 1H), 3.91–3.81 (m, 2H), 3.36–3.26 (m, 1H), 3.00–2.90 (m, 1H), 2.60–2.38 (m, 2H), 2.09–1.97 (m, 2H), 1.74–1.50 (m, 3H), 1.45 (s, 9H), 1.41 (s, 3H), 1.33 (s, 3H), 0.96 (d, J = 6.0 Hz, 3H), 0.94 (d, J = 6.0 Hz, 3H). LC/MS calculated for C29H40N4O7 + H+, 557.3; observed, 557.5.
(3S,4R)-6-Cyano-2,2-dimethyl-4-(2-oxopyrrolidin-1-yl)chroman-3-yl glycyl-D-leucinate (11)
Compound 10 (26 mg, 0.047 mmol) was dissolved in 4 M HCl in dioxane (5 mL) and stirred for 30 min. The reaction mixture was concentrated under reduced pressure. The resulting residue was purified by chromatography (10% acetonitrile/water with 0.1% formic acid to 100% acetonitrile, C18 column) to yield 5.0 mg after lyophilization (21% yield) as a formic acid salt. 1H NMR (400 MHz, DMSO-d6): 8.41 (d, J = 6.5 Hz, 1H), 8.18 (s, 1H), 7.69 (dd, J = 8.6, 1.8 Hz, 1H), 7.49 (bs, 1H), 7.04 (d, J = 8.6 Hz, 1H), 5.30 (d, J = 10.8 Hz, 1H), 5.18 (d, J = 10.5 Hz, 1H), 4.31–4.19 (m, 1H), 3.34–3.29 (m, 2H), 3.20–3.10 (m, 1H), 2.99–2.89 (m, 1H), 2.51–2.46 (m, 1H), 2.28–2.16 (m, 1H), 2.05–1.92 (m, 1H), 1.88–1.74 (m, 1H), 1.71–1.59 (m, 1H), 1.59–1.42 (m, 2H), 1.37 (s, 3H), 1.32 (s, 3H), 0.91 (d, J = 6.6 Hz, 3H), 0.86 (d, J = 6.5 Hz, 3H). LC/MS calculated for C24H32N4O5 + H+, 457.2; observed, 457.4.
(3S,4R)-6-cyano-2,2-dimethyl-4-(2-oxopyrrolidin-1-yl)chroman-3-yl glycyl-L-leucinate (13)
Compound 12 (18.5 mg, 0.033 mmol) was dissolved in 4 M HCl in dioxane (5 mL) and stirred for 30 min. The reaction mixture was concentrated under reduced pressure. The resulting residue was purified by chromatography (10% acetonitrile/water with 0.1% formic acid to 100% acetonitrile, C18 column) to yield 4.1 mg after lyophilization (25% yield) as a formic acid salt. 1H NMR (400 MHz, DMSO-d6): 8.32 (d, J = 6.6 Hz, 1H), 8.26 (s, 1H), 7.68 (dd, J = 8.5, 1.9 Hz, 1H), 7.49 (bs, 1H), 7.03 (d, J = 8.6 Hz, 1H), 5.27 (d, J = 10.8 Hz, 1H), 5.17 (d, J = 10.3 Hz, 1H), 4.26–4.16 (m, 1H), 3.24–3.12 (m, 1H), 3.00–2.89 (m, 1H), 2.51–2.41 (m, 1H), 2.31–2.19 (m, 1H), 2.02–1.90 (m, 1H), 1.89–1.78 (m, 1H), 1.70–1.56 (m, 2H), 1.53–1.40 (m, 1H), 1.33 (s, 3H), 1.27 (s, 3H), 0.92 (d, J = 6.5 Hz, 3H), 0.87 (d, J = 6.3 Hz, 3H). LC/MS calculated for C24H32N4O5 + H+, 457.2; observed, 457.4.
(3S,4R)-6-cyano-2,2-dimethyl-4-(2-oxopyrrolidin-1-yl)chroman-3-yl (4-bromobenzoyl)glycyl-D-leucinate (14)
Compound 10 (29.9 mg, 0.054 mmol) was dissolved in 4 M HCl in dioxane (2.5 mL) and stirred for 30 min. The reaction mixture was concentrated under reduced pressure. The residue was dissolved in DCM (5 mL) and 4-bromobenzoic acid (11.4 mg, 0.057 mmol) was added followed by DMAP (134 mg, 1.10 mmol) and HATU (92.1 mg, 0.242 mmol). After 24 h., the solvent was removed under reduced pressure and the resulting residue was purified by chromatography (10% to 100% ethyl acetate/hexanes) on silica gel to furnish 23.1 mg of 14 (67% yield) as a white solid. Crystals suitable for X-Ray crystallography were obtained from DCM/pentane. 1H NMR (400 MHz, CDCl3): 8.30 (t, J = 6.5 Hz, 1H), 7.92 (d, J = 8.5 Hz, 2H), 7.55 (d, J = 8.5 Hz, 2H), 7.49 (dd, J = 8.5, 1.7 Hz, 1H), 7.21 (d, J = 1.3 Hz, 1H), 6.89 (d, J = 8.5 Hz, 1H), 6.72 (d, J = 6.8 Hz, 1H), 5.19 (ABq, J = 11 Hz, 2H), 4.51–4.39 (m, 2H), 3.77 (dd, J = 15.2, 5.1 Hz, 1H), 3.37–3.27 (m, 1H), 2.99–2.91 (m, 1H), 2.82–2.71 (m, 1H), 2.54–2.44 (m, 1H), 2.22–1.94 (m, 2H), 1.79–1.67 (m, 1H), 1.62–1.42 (m, 2H), 1.30 (s, 3H), 1.00 (d, J = 6.6 Hz, 3H), 0.95 (d, J = 6.5 Hz, 3H), 0.89 (s, 3H). Mp 240–241 °C.
(((3S,4R)-6-cyano-2,2-dimethyl-4-(2-oxopyrrolidin-1-yl)chroman-3-yl)oxy)methyl glycyl-L-leucinate (15)
To a solution of (3S,4R)-3 (57.0 mg, 0.165 mmol) in DCE (1.5 mL) was added sulfuryl chloride (0.95 mL of 1 M in DCM solution, 0.95 mmol). After stirring for 90 min., the solvent was removed under reduced pressure and the resulting residue was left under vacuum for 10 min. The residue was then dissolved in acetonitrile (1 mL) and a solution of Boc-Gly-Leu-OH (110.8 mg, 0.384 mmol) in acetonitrile (3 mL) and DIEA (300 µL, 1.72 mmol) added by syringe. After 3 h., the solvent was removed under reduced pressure and the resulting residue was purified by chromatography (30% to 100% ethyl acetate/hexanes) on silica gel to furnish 96.1 mg of (((3S,4R)-6-cyano-2,2-dimethyl-4-(2-oxopyrrolidin-1-yl)chroman-3-yl)oxy)methyl (tert-butoxycarbonyl)glycyl-L-leucinate as a white solid (99% yield). 1H NMR (400 MHz, CDCl3): 7.46 (d, J = 8.3, 1H), 7.23 (bs, 1H), 6.89 (d, J = 8.5 Hz, 1H), 6.47 (d, J = 6.4 Hz, 1H), 5.46–5.32 (m, 3H), 5.17–5.04 (m, 1H), 4.65–4.54 (m, 1H), 3.89 (d, J = 9.6 Hz, 1H), 3.87–3.73 (m, 2H), 3.45–3.34 (m, 1H), 3.08–2.96 (m, 1H), 2.67–2.49 (m, 2H), 2.21–2.06 (m, 2H), 1.73–1.48 (m, 3H), 1.48 (s, 3H), 1.45 (s, 9H), 1.27 (s, 3H), 0.95 (d, J = 4.5 Hz, 6H). 13C NMR (100 MHz, CDCl3): 18.2, 19.0, 21.5, 23.0, 24.8, 26.7, 28.3, 31.1, 41.0, 43.0, 44.4, 50.7, 51.0, 76.4, 79.2, 80.5, 89.4, 104.8, 118.8, 118.9, 120.9, 131.7, 133.2, 156.1, 157.0, 169.6, 172.5, 176.5. LC/MS calculated for C30H42N4O8 + H+, 587.3; observed, 587.3.
The Boc-protected conjugate (42.4 mg, 0.072 mmol) was dissolved in 4 M HCl in dioxane (2 mL) and stirred for 30 min. The reaction mixture was concentrated under reduced pressure. The resulting residue was washed with diethyl ether, which was then discarded. The remaining material was purified by chromatography (10% acetonitrile/water to 100% acetonitrile, C18 column). The product containing fractions were lyophilized after a small amount of dilute HCl was added to them. 8.8 mg white solid (23% yield) as an HCl salt. 1H NMR (400 MHz, DMSO-d6): 8.81 (d, J = 7.3, 1H), 8.04 (bs, 3H), 7.65 (d, J = 8.7, 1H), 7.45 (s, 1H), 6.99 (d, J = 8.4 Hz, 1H), 5.44 (d, J = 5.8 Hz, 1H), 5.21 (d, J = 6.2 Hz, 1H), 5.13 (d, J = 9.2 Hz, 1H), 4.44–4.33 (m, 1H), 4.07 (d, J = 9.6 Hz, 1H), 3.64 (d, J = 16.3 Hz, 1H), 3.57 (d, J = 16.4 Hz, 1H), 3.46–3.33 (m, 1H), 3.05–2.92 (m, 1H), 2.60–2.44 (m, 1H), 2.42–2.30 (m, 1H), 2.11–1.92 (m, 2H), 1.75–1.62 (m, 1H), 1.62–1.51 (m, 2H), 1.47 (s, 3H), 1.18 (s, 3H), 0.92 (d, J = 6.5 Hz, 3H), 0.87 (d, J = 5.8 Hz, 3H). LC/MS calculated for C25H34N4O6 + H+, 487.3; observed, 487.4.
In Vivo Experiments
All animal studies adhered to the tenets of the Association for Research in Vision and Ophthalmology (ARVO) Statement for the use of animals in ophthalmic and vision research. All protocols utilizing animals were reviewed and approved by the Mayo Clinic Institutional Animal Care and Use Committee (IACUC A43414, A65613, A67713, A42713).
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the National Eye Institute (EY21727; MPF), Research to Prevent Blindness (MPF), Mayo Foundation (MPF), and the Minnesota Partnership for Biotechnology (MNP #12.06 & TPDF #15.01; PID, MPF) and the National Center for Advancing Translational Sciences of the National Institutes of Health Award Number UL1TR000114 (PID). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We gratefully acknowledge Drs. Michael A. Walters, Vadim Gurvich, and Gunda Georg for helpful discussions about this research. We thank Nicholas Bleeker and Andrew Goode for performing HPLC analysis of products and Dr. Victor G. Young, Jr, Director of the X-Ray Crystallographic Facility at the University of Minnesota, for obtaining the X-ray crystal structure of 14.
Abbreviations Used
- DCE
1,2-dichloroethane
- DCM
dichloromethane
- DIEA
N,N-diisopropylethylamine
- DMAP
4-(dimethylamino)pyridine
- DMSO
dimethyl sulfoxide
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- IOP
intraocular pressure
- KATP channel
ATP-sensitive potassium channel
- Kir channel
inwardly rectifying potassium channel
- NIS
N-iodosuccinimide
- OMP
oxymethylphosphate
- PBS
phosphate-buffered saline
- POAG
primary open angle glaucoma
- SUR
sulfonylurea
- THF
tetrahydrofuran
- TMSBr
trimethylsilyl bromide
- Sar
sarcosine
Footnotes
Supporting Information
Synthesis of compounds (±)-2 and (−)-2. Table S1, effect of compounds (3S,4R)-2, (±)-2, and (±)-4 on IOP in mice. X-ray crystal structure of compound 14.
Crystallographic Information
A CIF file with the X-ray crystal structure of 14 has been deposited with the Cambridge Crystallographic Data Centre (CCDC 1476916).
Author Contributions
The laboratories of Michael P. Fautsch and Peter I. Dosa contributed equally to work described in this manuscript.
References
- 1.Tham Y-C, Li X, Wong TY, Quigley HA, Aung T, Cheng C-Y. Global Prevalence of Glaucoma and Projections of Glaucoma Burden through 2040: A Systematic Review and Meta-Analysis. Ophthalmology. 2014;121:2081–2090. doi: 10.1016/j.ophtha.2014.05.013. [DOI] [PubMed] [Google Scholar]
- 2.Quigley HA, Broman AT. The Number of People with Glaucoma Worldwide in 2010 and 2020. Br. J. Ophthalmol. 2006;90:262–267. doi: 10.1136/bjo.2005.081224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kwon YH, Fingert JH, Kuehn MH, Alward WLM. Primary Open-Angle Glaucoma. N. Engl. J. Med. 2009;360:1113–1124. doi: 10.1056/NEJMra0804630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stamer WD, Acott TS. Current Understanding of Conventional Outflow Dysfunction in Glaucoma. Curr. Opin. Ophthalmol. 2012;23:135–143. doi: 10.1097/ICU.0b013e32834ff23e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Winkler NS, Fautsch MP. Effects of Prostaglandin Analogues on Aqueous Humor Outflow Pathways. J. Ocul. Pharmacol. Ther. 2013;30:102–109. doi: 10.1089/jop.2013.0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kass MA, Heuer DK, Higginbotham EJ, et al. The Ocular Hypertension Treatment Study: A Randomized Trial Determines that Topical Ocular Hypotensive Medication Delays or Prevents the Onset of Primary Open-angle Glaucoma. Arch. Ophthalmol. 2002;120:701–713. doi: 10.1001/archopht.120.6.701. [DOI] [PubMed] [Google Scholar]
- 7.Heijl A, Leske M, Bengtsson B, et al. Reduction of Intraocular Pressure and Glaucoma Progression: Results from the Early Manifest Glaucoma Trial. Arch. Ophthalmol. 2002;120:1268–1279. doi: 10.1001/archopht.120.10.1268. [DOI] [PubMed] [Google Scholar]
- 8.Collaborative Normal-Tension Glaucoma Study Group. Comparison of Glaucomatous Progression between Untreated Patients with Normal-tension Glaucoma and Patients with Therapeutically Reduced Intraocular Pressures. Am. J. Ophthalmol. 1998;126:487–497. doi: 10.1016/s0002-9394(98)00223-2. [DOI] [PubMed] [Google Scholar]
- 9.To CH, Kong CW, Chan CY, Shahidullah M, Do CW. The Mechanism of Aqueous Humour Formation. Clin. Exp. Optom. 2002;85:335–349. [PubMed] [Google Scholar]
- 10.Llobet A, Gasull X, Gual A. Understanding Trabecular Meshwork Physiology: A Key to the Control of Intraocular Pressure? News Physiol. Sci. 2003;18:205–209. doi: 10.1152/nips.01443.2003. [DOI] [PubMed] [Google Scholar]
- 11.Fitt AD, Gonzalez G. Fluid Mechanics of the Human Eye: Aqueous Humour Flow in The Anterior Chamber. Bull. Math. Biol. 2006;68:53–71. doi: 10.1007/s11538-005-9015-2. [DOI] [PubMed] [Google Scholar]
- 12.Toris CB, Yablonski ME, Wang Y-L, Camras CB. Aqueous Humor Dynamics in the Aging Human Eye. Am. J. Ophthalmol. 1999;127:407–412. doi: 10.1016/s0002-9394(98)00436-x. [DOI] [PubMed] [Google Scholar]
- 13.Gabelt BAT, Kaufman PL. Changes in Aqueous Humor Dynamics with Age and Glaucoma. Prog. Retinal Eye Res. 2005;24:612–637. doi: 10.1016/j.preteyeres.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 14.Zhang K, Zhang L, Weinreb RN. Ophthalmic Drug Discovery: Novel Targets and Mechanisms for Retinal Diseases and Glaucoma. Nat. Rev. Drug Discovery. 2012;11:541–559. doi: 10.1038/nrd3745. [DOI] [PubMed] [Google Scholar]
- 15.Roy Chowdhury U, Hann CR, Stamer WD, Fautsch MP. Aqueous Humor Outflow: Dynamics and DiseaseAqueous Humor Outflow. Invest. Ophthalmol. Visual Sci. 2015;56:2993–3003. doi: 10.1167/iovs.15-16744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bettin P, Di Matteo F. Glaucoma: Present Challenges and Future Trends. Ophthalmic Res. 2013;50:197–208. doi: 10.1159/000348736. [DOI] [PubMed] [Google Scholar]
- 17.Mainolfi N, Powers J, Amin J, Long D, Lee W, McLaughlin ME, Jaffee B, Brain C, Elliott J, Sivak JM. An Effective Prodrug Strategy to Selectively Enhance Ocular Exposure of a Cannabinoid Receptor (CB1/2) Agonist. J. Med. Chem. 2013;56:5464–5472. doi: 10.1021/jm4004939. [DOI] [PubMed] [Google Scholar]
- 18.Harrison BA, Almstead ZY, Burgoon H, Gardyan M, Goodwin NC, Healy J, Liu Y, Mabon R, Marinelli B, Samala L, Zhang Y, Stouch TR, Whitlock NA, Gopinathan S, McKnight B, Wang S, Patel N, Wilson AGE, Hamman BD, Rice DS, Rawlins DB. Discovery and Development of LX7101, a Dual LIM-Kinase and ROCK Inhibitor for the Treatment of Glaucoma. ACS Med. Chem. Lett. 2015;6:84–88. doi: 10.1021/ml500367g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Donegan RK, Lieberman RL. Discovery of Molecular Therapeutics for Glaucoma: Challenges, Successes, and Promising Directions. J. Med. Chem. 2016;59:788–809. doi: 10.1021/acs.jmedchem.5b00828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chowdhury UR, Bahler CK, Hann CR, Chang M, Resch ZT, Romero MF, Fautsch MP. ATP-Sensitive Potassium (KATP) Channel Activation Decreases Intraocular Pressure in the Anterior Chamber of the Eye. Invest. Ophthalmol. Visual Sci. 2011;52:6435–6442. doi: 10.1167/iovs.11-7523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chowdhury UR, Holman BH, Fautsch MP. ATP-Sensitive Potassium (KATP) Channel Openers Diazoxide and Nicorandil Lower Intraocular Pressure In Vivo. Invest. Ophthalmol. Visual Sci. 2013;54:4892–4899. doi: 10.1167/iovs.13-11872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roy Chowdhury U, Bahler CK, Holman BH, Dosa PI, Fautsch MP. Ocular Hypotensive Effects of the ATP-Sensitive Potassium Channel Opener Cromakalim in Human and Murine Experimental Model Systems. PLoS ONE. 2015;10:e0141783. doi: 10.1371/journal.pone.0141783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flagg TP, Enkvetchakul D, Koster JC, Nichols CG. Muscle KATP Channels: Recent Insights to Energy Sensing and Myoprotection. Physiol. Rev. 2010;90:799–829. doi: 10.1152/physrev.00027.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gabrielsson B, Karlsson AC, Lönn M, Olofsson L, Johansson J, Torgerson J, Sjöström L, Carlsson B, Edén S, Carlsson LS. Molecular Characterization of a Local Sulfonylurea System in Human Adipose Tissue. Mol. Cell. Biochem. 2004;258:65–71. doi: 10.1023/b:mcbi.0000012837.11847.c8. [DOI] [PubMed] [Google Scholar]
- 25.Gloyn AL, Siddiqui J, Ellard S. Mutations in the Genes Encoding the Pancreatic Beta-cell KATP Channel Subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) in Diabetes Mellitus and Hyperinsulinism. Hum. Mutat. 2006;27:220–231. doi: 10.1002/humu.20292. [DOI] [PubMed] [Google Scholar]
- 26.Jovanović S, Ballantyne T, Du Q, Blagojević M, Jovanović A. Phenylephrine Preconditioning in Embryonic Heart H9c2 Cells is Mediated by Up-Regulation of SUR2B/Kir6.2: A First Evidence for Functional Role of SUR2B in Sarcolemmal KATP Channels and Cardioprotection. Int. J. Biochem. Cell Biol. 2016;70:23–28. doi: 10.1016/j.biocel.2015.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jin J, Sklar GE, Min Sen Oh V, Chuen Li S. Factors Affecting Therapeutic Compliance: A Review from the Patient’s Perspective. Ther. Clin. Risk Manage. 2008;4:269–286. doi: 10.2147/tcrm.s1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Galvao J, Davis B, Tilley M, Normando E, Duchen MR, Cordeiro MF. Unexpected Low-dose Toxicity of the Universal Solvent DMSO. FASEB J. 2014;28:1317–1330. doi: 10.1096/fj.13-235440. [DOI] [PubMed] [Google Scholar]
- 29.Windrum P, Morris TCM, Drake MB, Niederwieser D, Ruutu T. Variation in Dimethyl Sulfoxide Use in Stem Cell Transplantation: a Survey of EBMT Centres. Bone Marrow Transplant. 2005;36:601–603. doi: 10.1038/sj.bmt.1705100. [DOI] [PubMed] [Google Scholar]
- 30.Juntunen J, Järvinen T, Niemi R. In-vitro Corneal Permeation of Cannabinoids and their Water-soluble Phosphate Ester Prodrugs. J. Pharm. Pharmacol. 2005;57:1153–1157. doi: 10.1211/jpp.57.9.0009. [DOI] [PubMed] [Google Scholar]
- 31.Huttunen KM, Raunio H, Rautio J. Prodrugs—from Serendipity to Rational Design. Pharmacol. Rev. 2011;63:750–771. doi: 10.1124/pr.110.003459. [DOI] [PubMed] [Google Scholar]
- 32.Vivaudou M, Moreau C, Terzic A. ATP-sensitive K+ Channels. In: Kew JN, Davies CH, editors. Ion Channels: from Structure to function. Oxford University Press; Oxford: 2010. pp. 454–473. [Google Scholar]
- 33.Heimbach T, Fleisher D, Kaddoumi A. Overcoming Poor Aqueous Solubility of Drugs for Oral Delivery. In: Stella VJ, Borchardt RT, Hageman MJ, Oliyai R, Maag H, Tilley JW, editors. Prodrugs: Challenges and Rewards Part 1. Springer New York; New York, NY: 2007. pp. 157–215. [Google Scholar]
- 34.Rautio J, Kumpulainen H, Heimbach T, Oliyai R, Oh D, Jarvinen T, Savolainen J. Prodrugs: Design and Clinical Applications. Nat. Rev. Drug Discovery. 2008;7:255–270. doi: 10.1038/nrd2468. [DOI] [PubMed] [Google Scholar]
- 35.Olejnik O, Weisbecker CA. Ocular Bioavailability of Topical Prednisolone Preparations. Clin. Ther. 1990;12:2–11. [PubMed] [Google Scholar]
- 36.McGhee CNJ, Noble MJ, Watson DG, Dutton GN, Fern AI, Healey TM, Midgley JM. Penetration of Topically Applied Prednisolone Sodium Phosphate into Human Aqueous Humour. Eye. 1989;3:463–467. doi: 10.1038/eye.1989.69. [DOI] [PubMed] [Google Scholar]
- 37.Čejková J, Bolková A. A Study on Alkaline Phosphatase in Cornea of Various Animals with Special Regard to Keratocytes. Graefe’s Arch. Clin. Exp. Ophthalmol. 1977;204:209–214. doi: 10.1007/BF00414848. [DOI] [PubMed] [Google Scholar]
- 38.Ashwood VA, Buckingham RE, Cassidy F, Evans JM, Faruk EA, Hamilton TC, Nash DJ, Stemp G, Willcocks K. Synthesis and Antihypertensive Activity of 4-(Cyclic Amido)-2H-1-benzopyrans. J. Med. Chem. 1986;29:2194–2201. doi: 10.1021/jm00161a011. [DOI] [PubMed] [Google Scholar]
- 39.Attwood MR, Brown BS, Dunsdon RM, Hurst DN, Jones PS, Kay PB. Synthesis of Homochiral Potassium Channel Openers: Role of the Benzopyranyl 3-Hydroxyl Group in Cromakalim and Pyridine N-Oxides in Determining the Biological Activities of Enantiomers. Bioorg. Med. Chem. Lett. 1992;2:229–234. [Google Scholar]
- 40.Chow CP, Berkman CE. Synthesis of N-Phosphoryl Amino Acids via Phosphoramidite Amine-exchange. Tetrahedron Lett. 1998;39:7471–7474. [Google Scholar]
- 41.Maruoka H, Jayasekara MPS, Barrett MO, Franklin DA, de Castro S, Kim N, Costanzi S, Harden TK, Jacobson KA. Pyrimidine Nucleotides with 4-Alkyloxyimino and Terminal Tetraphosphate δ-Ester Modifications as Selective Agonists of the P2Y4 Receptor. J. Med. Chem. 2011;54:4018–4033. doi: 10.1021/jm101591j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dosa PI, Ward T, Castro RE, Rodrigues CMP, Steer CJ. Synthesis and Evaluation of Water-Soluble Prodrugs of Ursodeoxycholic Acid (UDCA), an Anti-apoptotic Bile Acid. ChemMedChem. 2013;8:1002–1011. doi: 10.1002/cmdc.201300059. [DOI] [PubMed] [Google Scholar]
- 43.Lee NH, Muci AR, Jacobsen EN. Enantiomerically Pure Epoxychromans via Asymmetric Catalysis. Tetrahedron Lett. 1991;32:5055–5058. [Google Scholar]
- 44.DeGoey DA, Grampovnik DJ, Flosi WJ, Marsh KC, Wang XC, Klein LL, McDaniel KF, Liu Y, Long MA, Kati WM, Molla A, Kempf DJ. Water-Soluble Prodrugs of the Human Immunodeficiency Virus Protease Inhibitors Lopinavir and Ritonavir. J. Med. Chem. 2009;52:2964–2970. doi: 10.1021/jm900080g. [DOI] [PubMed] [Google Scholar]
- 45.Järvinen T, Niemi R. Prodrug Approaches to Ophthalmic Drug Delivery. In: Stella V, Borchardt R, Hageman M, Oliyai R, Maag H, Tilley J, editors. Prodrugs. V. Springer; New York: 2007. pp. 125–155. [Google Scholar]
- 46.Kwatra D, Vaishya R, Gaudana R, Jwala J, Mitra AK. Prodrugs and Targeted Delivery. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim, Germany: 2010. Ocular Delivery Using Prodrugs; pp. 181–205. [Google Scholar]
- 47.Buckle DR, Arch JRS, Fenwick AE, Houge-Frydrych CSV, Pinto IL, Smith DG, Taylor SG, Tedder JM. Relaxant Activity of 4-Amido-3,4-dihydro-2H-1-benzopyran-3-ols and 4-Amido-2H-1-benzopyrans on Guinea Pig Isolated Trachealis. J. Med. Chem. 1990;33:3028–3034. doi: 10.1021/jm00173a019. [DOI] [PubMed] [Google Scholar]
- 48.A two step synthesis of compound 5 has been previously reported. Evans JM, Stemp G, Tedder JM. Preparation of aminotrifluoromethylbenzopyrans as bronchodilators and antihypertensives. PCT. Int. Appl. WO 9109031. 1991 [Google Scholar]
- 49.North JT, Kronenthal DR, Pullockaran AJ, Real SD, Chen HY. Synthesis of 6-Cyano-2,2-dimethyl-2H-1-benzopyran and Other Substituted 2,2-Dimethyl-2H-1-benzopyrans. J. Org. Chem. 1995;60:3397–3400. [Google Scholar]
- 50.Anand BS, Nashed YE, Mitra AK. Novel Dipeptide Prodrugs of Acyclovir for Ocular Herpes Infections: Bioreversion, Antiviral Activity and Transport across Rabbit Cornea. Curr. Eye Res. 2003;26:151–163. doi: 10.1076/ceyr.26.3.151.14893. [DOI] [PubMed] [Google Scholar]
- 51.Fei Y-J, Kanai Y, Nussberger S, Ganapathy V, Leibach FH, Romero MF, Singh SK, Boron WF, Hediger MA. Expression Cloning of a Mammalian Proton-coupled Oligopeptide Transporter. Nature. 1994;368:563–566. doi: 10.1038/368563a0. [DOI] [PubMed] [Google Scholar]
- 52.Martin AR, Lavergne T, Vasseur J-J, Debart F. Assessment of New 2′-O-Acetalester Protecting Groups for Regular RNA Synthesis and Original 2′-Modified proRNA. Bioorg. Med. Chem. Lett. 2009;19:4046–4049. doi: 10.1016/j.bmcl.2009.06.015. [DOI] [PubMed] [Google Scholar]
- 53.Biscans A, Rouanet S, Bertrand J-R, Vasseur J-J, Dupouy C, Debart F. Synthesis, Binding, Nuclease Resistance and Cellular Uptake Properties of 2′-O-Acetalester-modified Oligonucleotides Containing Cationic Groups. Bioorg. Med. Chem. 2015;23:5360–5368. doi: 10.1016/j.bmc.2015.07.054. [DOI] [PubMed] [Google Scholar]
- 54.Whitlock NA, Harrison B, Mixon T, Yu X-Q, Wilson A, Gerhardt B, Eberhart DE, Abuin A, Rice DS. Decreased Intraocular Pressure in Mice Following Either Pharmacological or Genetic Inhibition of ROCK. J. Ocul. Pharmacol. Ther. 2009;25:187–194. doi: 10.1089/jop.2008.0142. [DOI] [PubMed] [Google Scholar]
- 55.Henriksson JT, McDermott AM, Bergmanson JPG. Dimensions and Morphology of the Cornea in Three Strains of Mice. Invest. Ophthalmol. Visual Sci. 2009;50:3648–3654. doi: 10.1167/iovs.08-2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li HF, Petroll WM, Moller-Pedersen T, Maurer JK, Cavanagh HD, Jester JV. Epithelial and Corneal Thickness Measurements by in vivo Confocal Microscopy through Focusing (CMTF) Curr. Eye Res. 1997;16:214–221. doi: 10.1076/ceyr.16.3.214.15412. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.