

Abstract
The hedgehog signaling network regulates organogenesis, cell fate, proliferation, survival, and stem cell self-renewal in many mammalian tissues. Aberrant activation of the hedgehog signaling network is present in ∼25% of all cancers, including breast. Altered expression of hedgehog network genes in the mammary gland can elicit phenotypes at many stages of development. However, synthesizing a cohesive mechanistic model of signaling at different stages of development has been difficult. Emerging data suggest that this difficulty is due, in part, to non-canonical and tissue compartment-specific (i.e., epithelial, versus stromal, versus systemic) functions of hedgehog network components. With respect to systemic functions, hedgehog network genes regulate development of endocrine organs that impinge on mammary gland development extrinsically. These new observations offer insight into previously conflicting data, and have bearing on the potential for anti-hedgehog therapeutics in the treatment of breast cancer.
Keywords: hedgehog signaling, mammary gland, breast tumorigenesis, breast cancer metastasis, epithelial-stromal interactions
1. Introduction
The hedgehog signaling network is required for metazoan embryonic organogenesis, and for homeostasis of many adult tissues. In the mammary gland field, extensive effort has been exerted to dissect the roles of hedgehog network genes in development and breast cancer. In contrast to other organs, where data from hedgehog mutants and hedgehog-modulating pharmacological agents are largely consistent with canonical hedgehog signaling function, it has proven more difficult to synthesize a clear picture of hedgehog signaling network function in mammary gland development. Recent data from murine mammary gland development, human breast tumors, and the identification of non-canonical functions for hedgehog network genes have provided insight into these seemingly conflicting data. This review discusses the established roles of hedgehog network genes in mammary gland development and cancer, hedgehog network function in endocrine tissues that regulate the mammary gland, and the potential for hedgehog therapeutics in the clinic.
2. Brief Overview of Mammary Gland Development
Murine mammary gland development is initiated at approximately embryonic day 10.5 (E10.5) with the formation of the mammary ridges, or milk lines, two lines of thickened columnar epithelium displaced on either side of the ventral midline between the head and tail [1]. At ∼E11.5, five pairs of placodes form along the milk lines in the positions of the presumptive nipples [1]. Placode formation requires signaling events within the epithelium (e.g. activated Wnt/Lef1 signaling [1]), as well as paracrine signals originating from the underlying somites (e.g. GLI3 expression in the somites driving FGF10 expression) [1]. Placodes enlarge by ∼E13 to yield a mammary bud that invades an underlying condensed mammary mesenchyme [1]. By ∼E16, the mammary bud elongates to a mammary sprout that invades the mammary fat pad precursor mesenchyme. Thereafter, a small amount of branching morphogenesis is initiated, which produces a rudimentary ductal tree that fills only a small portion of the mammary fat pad at birth [1]. These phases of mammary gland growth and morphogenesis are entirely ovarian hormone independent.
The rudimentary ductal tree present at birth is largely growth quiescent until the onset of puberty at 3-4 weeks of age in most strains of laboratory mice. The systemic hormones present during puberty induce the formation of terminal end buds (TEBs), which are transient bulb-shaped structures positioned at the distal ends of the ductal tree during puberty [2]. Hormones required for pubertal mammary ductal elongation include ovary-derived estrogen and pituitary-derived growth hormone, which mediate TEB formation and ductal elongation [2,3].
The TEBs proliferate and invade rapidly to drive ductal outgrowth and fill the mammary fat pad stroma. The TEBs interact with stromal cell types, and require a particular context of extracellular matrix remodeling enzymes and ECM content for proper outgrowth [4]. Additionally, TEBs are the site of lumen formation, which is thought to be driven by apoptosis, anoikis (apoptosis due to loss of basement membrane contact), autophagy, and non-apoptotic cell death [5–7]. TEB-driven elongation and bifurcation continues until TEBs reach the edge of the mammary fat pad and regress to leave blunt or round-ended duct termini [8].
In most laboratory strains of mice, the mammary ductal tree reaches the edge of the mammary fat pad and side branching is complete by 8-10 weeks of age [3,9]. The mature virgin mammary duct consists of a single layer of luminal cells surrounded by a single layer of myoepithelial or basal cells, mammary stem cells, and luminal/basal progenitors [3,7]. The mammary epithelium exists within the mammary fat pad stroma consisting of diverse cell types, including fibroblasts, mature adipocytes, eosinophils, neutrophils, macrophages and other myeloid cells, endothelial cells, pericytes, and nervous tissue [4].
With pregnancy, a hormonal milieu including estrogen, progesterone, glucocorticoids, and prolactin drives production and differentiation of the alveolar cells, which are responsible for the production of milk during lactation [2]. This stage of development is characterized by dramatic stromal changes: ECM components are remodeled, adipocytes of the mammary fat pad transfer lipids to the alveolar cells causing the adipocytes to be depleted of lipid and to diminish in size, and the vasculature becomes augmented [9,10]. After lactation is complete and weaning of the pups induces milk stasis, mammary gland involution is initiated [9]. This dynamic phase of development is characterized by apoptosis and removal of a majority of the alveolar cells, epithelial remodeling, as well as reversal of the many stromal changes observed in pregnancy and lactation [11–13]. Thus, after involution, the mammary ductal tree resembles that of the adult virgin animal, but differs with respect to gene expression [9,14]. This cycle of production of alveolar cells, lactation, and involution can occur many times over the lifespan of a mammal, underscoring the extensive replicative and regenerative capacity of mammary stem/progenitor cells.
3. Overview of Canonical Hedgehog Signaling
The canonical mammalian hedgehog signaling cascade has two functional states depending on the presence or absence of hedgehog ligands. In the absence of the hedgehog ligands (Desert Hedgehog (DHH), Indian Hedgehog (IHH), and Sonic Hedgehog (SHH)), the Patched-1 (PTCH1) and Patched-2 (PTCH2) receptors, inhibit Smoothened (SMO), the main effector of signaling, which is located in vesicles in the cytoplasm [15,16] (Figure 1A). The mechanism by which PTCH1 (or PTCH2) inhibits SMO is not known, but recent data suggest that PTCH1 inhibits SMO both cell autonomously and non-autonomously by functioning as an efflux pump for an oxysterol [17]. PTCH1 inhibition of SMO may also be due to PTCH1-mediated inhibition of phosphatidylinositol-4-phosphate, which promotes the association of SMO with the cell surface membrane [15]. The conformation of SMO in the absence of hedgehog ligands is not compatible with cell surface accumulation of SMO [15]. SMO sequestered in vesicles can also be degraded, thereby attenuating signaling [18].
Figure 1. The Canonical Hedgehog Network in the Absence (A) or Presence (B) of Hedgehog Ligands.
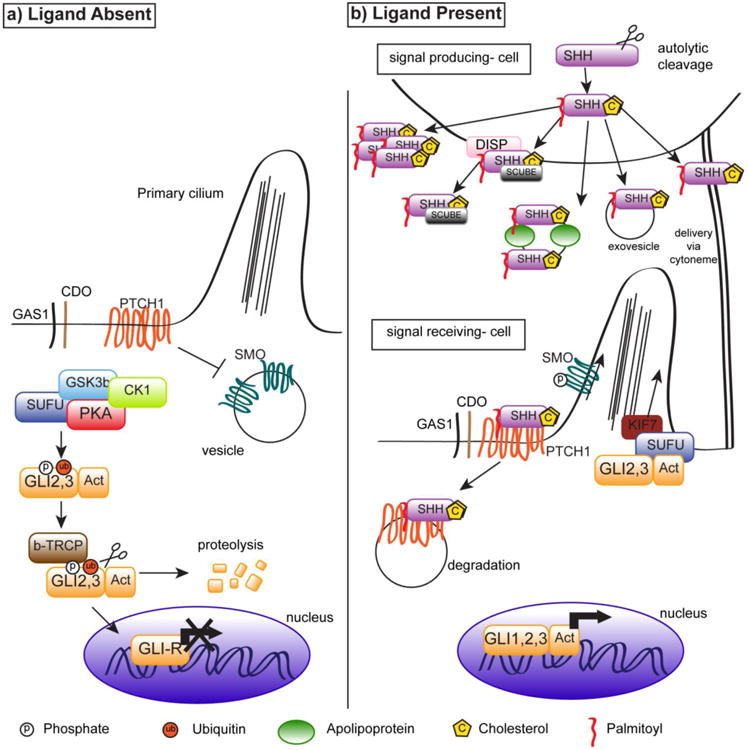
In the absence of HH, PTCH1 inhibits SMO, and SMO is sequestered in vesicles. A multiprotein complex (including SUFU, PKA, and GSK3β) phosphorylates and ubiquitinylates GLI transcription factors, which are cleaved to a repressor form (depicted as GLI-R in 1.a) lacking the activator domain, degraded, trafficked to the primary cilium, or translocate to the nucleus where they act as transcriptional repressors.
Secretion of hedgehog ligands (DHH, IHH, or SHH) by a signal- producing cell involves cleavage and post-translational modification, including addition of the cholesterol and palmitoyl groups to the hedgehog ligand, before secretion in various possible formats. HH binds PTCH1 or PTCH2 on a signal-receiving cell, inhibition of SMO by PTCH1/2 is alleviated, and SMO is phosphorylated. SMO then inhibits PKA, GSK3β, and SUFU, and is trafficked to the apical surface of the primary cilium due to interaction with KIF3a, a motor protein, and intraflagellar transport proteins. Due to inhibition of the multiprotein complex, GLI transcription factors retain their activator domain, (marked as ‘GLI1-Act), translocate to the nucleus, and activate transcription at start sites.
In the absence of active SMO, a multiprotein complex located in the cytoplasm regulates two GLI transcription factor family members (GLI2 and GLI3) by phosphorylating them, and promoting their cleavage into transcriptional repressor forms. The GLI transcription factors, GLI1, GLI2, and GLI3; are zinc finger proteins [15]. GLI1 is not subject to proteolytic cleavage (by virtue of lacking a cleavage site), and functions exclusively as a transcriptional activator [15], while GLI2 and GLI3 can act as activators or repressors of transcription, depending on the presence of the C-terminal activator domain.
The multiprotein complex regulating GLI activity consists of Suppressor of Fused (SUFU), Glycogen Synthase Kinase beta (GSK3β), Protein Kinase A (PKA), kinesin family member 7 (KIF7), and other proteins that modify GLI transcription factor function and localization through post-translational modifications and cleavage of GLIs. PKA phosphorylates GLI proteins at three sites, which primes GLIs for phosphorylation by GSK3β [19,20]. SUFU directly binds GLI proteins to retain full-length GLI proteins in the cytoplasm and thus inhibit transcriptional activation [21]. PKA-dependent phosphorylation targets GLIs for recognition by βTRCP (beta-transducin repeat-containing protein), which leads to ubiquitinylation of GLI transcription factors (and SKP-Cul-F box protein mediated degradation), proteolytic cleavage of the C-terminal activator domain of GLIs yielding transcriptional repressor forms that then translocate into the nucleus to mediate GLI-dependent transcriptional repression [22]. Aside from the multiprotein complex, HHIP, a membrane protein and transcriptional target of activated Hedgehog signaling, can also negatively regulate hedgehog network activation by binding Hh ligands.
Activation of the canonical hedgehog signaling cascade is initiated by secretion of ligands from a signal producing cell (Fig. 1B). In the endoplasmic reticulum of the signaling cell, ligands are autocatalytically cleaved by a transesterification reaction with cholesterol leading to a C-terminal lipid-modified species. This species is subsequently modified on the N-terminus by addition of the palmitoyl group to produce functional ligands [15]. Hedgehog ligands can be secreted in vesicles associated with lipoproteins and apolipoproteins, in vesicles without the additional apo- and lipoproteins, as a monomer via DISP (Dispatched) and SCUBE2 (Signal Peptide, CUB Domain, EGF-Like 2), or as a soluble multimer [15]. More targeted delivery of hedgehog ligands may be executed by transporting ligands through cytonemes, a cellular extension that can deliver ligand a distance of a few cell diameters away [15].
When Hh ligands (DHH, IHH, and SHH) bind to PTCH1/2 receptor complexes, PTCH1/2 inhibition of SMO is released. PTCH1/2 receptor complexes may include the CDO (Cell adhesion molecule, Down-regulated by Oncogenes), BOC (Brother of CDO), and IHOG (interference hedgehog) proteins [23]. These transmembrane proteins contain fibronectin type II domains for interaction with PTCH1 and HH ligands, and act as co-receptors to enhance hedgehog activation [23]. GAS1 (growth arrest specific 1) protein also acts in the receptor complex via a different domain and mechanism [23]. With the release of SMO inhibition, SMO is also phosphorylated by Casein Kinase 1 and G-protein coupled receptor regulatory Kinase 2 (GPRK2), which induces a conformational change, and trafficked to the cell surface on the primary cilium [23,24].
The trafficking of SMO to the apical surface of the cilium may be mediated by interactions with β-arrestin (BARR) and the KIF3a (kinesin family 3a) motor protein [15]. It is thought that intraflagellar transport, and the primary cilium, are required for hedgehog signaling activation [25]. SMO activation inhibits the GLI-modifying multiprotein complex containing GSK3β, PKA, and SUFU [15]. As a result, proteolytic cleavage of GLI transcription factors is inhibited. Thus, GLI proteins remain full-length and shuttle to the nucleus to function as transcriptional activators [15].
GLI transcription targets include factors that promote survival and proliferation [15]. GLI-dependent transcription also elicits autoregulatory negative feedback by upregulating mRNA levels of negative regulators of signaling, including Ptch1 and Hhip [15]. In opposition to the negative autoregulatory feedback, GLI transcription elicits positive autoregulatory feedback, for example of Gli1 and the Gas1 coreceptor [15].
In organogenesis, loss-of-function studies demonstrate that canonical hedgehog signaling regulates developmental events including branching morphogenesis of the lung, prostate, and pancreas [26–28], the specification of neuronal cell fate in the notochord [29], the self-renewal of adult neural stem cells, as well as many other organogenesis events [30–32]. Aside from organogenesis, canonical hedgehog signaling is important for the regulation of adult stem cell compartments, and is implicated in diseases including cancer [31,33].
Aberrant activation of canonical hedgehog signaling, due to inactivating mutations/heterozygosity of Ptch1, or activating mutations of Smo, induces medulloblastoma and basal cell carcinoma [34–36]. Somatic mutations in hedgehog network genes; such as Ptch1 heterozygosity, and mutations in Gli and Ihh; elicit diverse phenotypes in patients, including altered body size, increased tumor susceptibility, malformed phalanges, and holoprosencephaly (incomplete separation between hemispheres of the brain) [33,37].
Paracrine hedgehog signaling is critical in development and tumorigenesis. For example, inhibition of hedgehog signaling in the stroma appears to be important in prostate development [27], while activation of hedgehog signaling is correlated with increased prostate cancer metastasis [38].
4. Non-Canonical Signaling by Hedgehog Network Members
Aside from the canonical hedgehog signaling cascade described above, many hedgehog signaling network members participate in “non-canonical” signaling (Figure 2). For further reading and citations for this section (unless otherwise noted), refer to Jenkins 2009 [39] and Brennan 2012 [40]. Some examples of non-canonical functions of hedgehog network component are discussed below.
Figure 2. Non-Canonical Activities of Hedgehog Network Members.
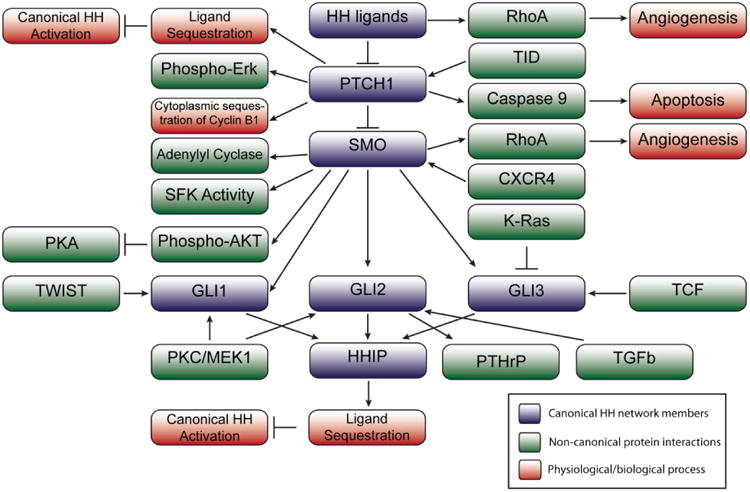
Members of the canonical hedgehog network and their interactions are shown in blue, with non-canonical protein interactions or functions shown in green, and physiological processes/results of a given signaling event shown in red.
With respect to ligand functions, all three hedgehog ligands can promote adoption of the activated conformation (i.e. GTP-bound) of RhoA in HUVECs (human umbilical vein endothelial cells). This activity of hedgehog ligands requires SMO, but does not require GLI-mediated transcription based on the rapid activation of RhoA, and the lack of GLI1-luciferase reporter activity with HH ligand treatment [40]. Hedgehog ligand stimulation of RhoA induced stress fiber formation and tubulogenesis of the HUVECs [40]. The promotion of angiogenesis may involve non-canonical functions of PTCH1, or more likely SMO, and SMO function as a G-protein coupled receptor, based on changes in cell survival, and response to pertussis toxin treatment to inhibit Gαi proteins [40].
At the receptor level, both PTCH1 and PTCH2 are 12 pass transmembrane proteins that resemble bacterial transport proteins that pump out toxins [15]. PTCH2 is not well studied, whereas PTCH1 has several non-canonical functions, including interactions with Cyclin B1, Caspase 9, GRB2, and TID proteins, as well as Hedgehog ligand sequestration [39–42]. PTCH1 can bind phosphorylated Cyclin B1 to sequester Cyclin B1 outside the nucleus in the absence of hedgehog ligand, which decreases proliferation in 293 T cells [39,40]. Tagged Xenopus PTCH1 and Cyclin B1 physically interact in 293T cells, and PTCH1 transfection decreases the proportion of nuclear Cyclin B1 [39,40]. SHH treatment increases the amount of nuclear Cyclin B1. Transfection experiments suggest that the intracellular loop of PTCH1 (residues 599-750) is required for this interaction [39,40].
PTCH1 can also promote apoptosis via a complex with Caspase 9 and downregulated in rhabdomyosarcoma LIM-domain protein (DRAL) in the absence of SHH according to Mille et al, 2009 [40]. A physical interaction is present between PTCH1 and DRAL in mammalian cells and the chick notochord [40]. Ptch1 overexpression increases apoptosis in the chick notochord, and recruits and activates Caspase 9, consistent with Thibert et al, 2003 [40]. This activity requires the intracellular C terminus domain of Ptch1 [40]. Thus, it seems PTCH1 acts as a dependence receptor in cultured mammalian cells and the chick notochord; in other words, the PTCH1 receptor must be bound for cell survival.
Another non-canonical function of PTCH1 observed in MCF10A and 293 cells is the direct interaction between the PTCH1 C terminus and the Src homology domain of GRB2 (growth factor receptor bound protein 2) promoting MEK phosphorylation of ERK [40]. In the presence of SHH, Ptch1 transfection increases phosphorylated ERK1/2 (extracellular related kinase 1), which is blocked by treatment with a function-blocking antibody to SHH (5E1) or a MEK inhibitor [40]. ERK1/2 phosphorylation was observed in the absence of detectable SMO [40]. Recently, genetic ablation of Ptch2 can induce PTCH1 phosphorylation of ERK (and canonical signaling) in the bone marrow niche [43].
PTCH1 may also participate in non-canonical signaling with tumorous imaginal disc (TID) proteins. In human basal cell carcinomas, Canamasas et al [39] observed concomitant loss of PTCH1 and TID proteins, and similar expression patterns by immunostaining. Further mechanistic/molecular data from mammalian systems is lacking. However, studies from Drosophila support a physical interaction between the PTCH1 C-terminus and TID proteins; additionally, Tid loss phenocopies loss of Ptch1 [39].
PTCH1 may also sequester hedgehog ligands to modulate activation of canonical hedgehog signaling. In Drosophila, Chen et al found that genetic loss of Ptch1 augments Hh expression in the wing disc, indicative of Ptch1 function in ligand sequestration to limit the range of ligands. In the mouse neural tube, low-level expression of Ptch1 can expand the SHH-dependent progenitor populations, which is distinct from the phenotype displayed by ablation of Ptch1 expression [41]. The phenotypes observed due to different combinations of Ptch1 and Hhip genetic ablation- specially, the stronger phenotypes with homozygous loss of both genes while expressing low-levels of Ptch1 suggest that Ptch1 and Hhip both function in ligand dependent antagonism opposing activation of canonical hedgehog signaling; however, Ptch1 works in ligand- independent antagonism as well [41]. Thus, ligand- dependent antagonism of activated hedgehog signaling by Ptch1 restricts the range and sharpens the morphogen gradient in the mammalian neural tube [41].
SMO, the main transducer of HH signaling, is a 7-pass transmembrane protein with a recently established non-canonical function as a G-protein coupled receptor (GPCR). In Sf9 cells, an insect cell line lacking expression of G proteins, co-transfection of Gi and Smo can increase GTP binding, indicative of G protein signaling [40]. Pertussis toxin treatment to inhibit G protein function decreases GLI-luciferase reporter activity with Shh or SmoM2 transfection in mammalian cells [40]. SmoM2 is constitutively activated form of SMO from a spontaneous human basal cell carcinoma, containing a point mutation resulting in the replacement of tryptophan with leucine. Transfection of mammalian cells with a Smo lacking its C terminus shows that Smo can act non-canonically as a GPCR, independent of GLI activation [40].
Data from the mammary gland further support a role of constitutively activated SMO (via the SmoM2 mutation) as a GPCR. While SmoM2-induced hyperproliferation is blocked by pertussis toxin treatment to inhibit Gαi proteins, treatment with GANT61 at concentrations able to block the uterine decidualization response does not block hyperproliferation, suggesting the hyperproliferation requires SMO but not GLI1/2 [44]. SmoM2 function as a GPCR is also supported by loss of the hyperproliferation phenotype when SmoM2 mice have conditional ablation of Gαi2, thus showing a phenotypic consequence of the putative function of SMO as a GPCR [44]. Importantly, the hyperproliferative phenotype is not blocked by disruption of either Gαi1 or Gαi3.
In addition to SMO function as a GPCR, Yam et al demonstrated that SMO, together with SHH and the BOC co-receptor, can rapidly stimulate Src family kinase activity of Src and Fyn in neurons independent of GLI-dependent transcription [40]. In some pancreatic cancer cell lines, stroma-derived stromal cell derived factor 1 (SDF-1) ligand activates chemokine receptor type 4 (CXCR4) to increase Smo and Gli1 mRNA and protein levels in the absence of hedgehog ligands [45], although the intermediate steps have not been elucidated.
GLI transcription factors can also function outside of the canonical signaling cascade. In MD-MBA-231 human breast cancer cells lacking SMO mRNA expression, GLI2 transcription is induced by treatment with TGFβ, but not in the presence of a dominant-negative TGFβ receptor [46]. TGFβ induction of Gli2 upregulates PTHrP mRNA and protein, and metastasis to bone in a xenograft model [46]. It is unknown whether these signaling events occur in other mammalian cells, and evidence of direct interactions is lacking. Riobo et al showed that GLI transcription is induced downstream of diacylglycerol analog treatment, acting via phorbol ester- responsive PKC and MEK-1 in NIH 3T3 cells [47]. This was observed with either endogenous or overexpressed GLI, and required the GLI N terminus (amino acids 1-30) to sense PKC activation [47].
Data from breast cancer cell lines indicate that the Twist transcription factor impinges on the hedgehog network [48]. Luciferase assays showed that Twist induces expression of the long non-coding RNA termed LncRNA-Hh; similarly, shRNA against Twist reduces GLI1 expression [48]. LncRNA-Hh may upregulate GLI1 and GAS1 transcription, since overexpression of LncRNA-Hh induced GLI1 and GAS1 protein levels; GLI1 and GAS1 were downregulated with shRNA against LncRNA-Hh [48].
Recent data suggests that SIX1 transcription factors can increase GLI1 expression non-cell autonomously via an unknown mechanism to induce and EMT (epithelial-mesenchymal transition) behaviors in breast cancer cell lines [49]. Treatment with a SMO inhibitor suggested that SMO was dispensable for Six1 dependent GLI1 expression [49].
K-Ras can influence hedgehog signaling in mammalian cell lines by increasing the ratio of repressor form of GLI3 relative to the activator form, increasing in SHH expression, and inhibiting Gli1-dependent transcription (with HH or SMO stimulation) independent of primary cilium presence [50]. K-RAS inhibition of canonical hedgehog signaling seems to be via DYRK1B (dual specificity tyrosine phosphorylated and regulated kinase 1B), since Dyrk1b siRNA abrogates the loss of Gli1 luciferase reporter signal [50]. Another type of non-canonical signaling shown to regulate Gli3 is Wnt1/3a; signaling of Wnt ligands via TCF (T cell factor) transcription factors induces Gli3 expression in the chick notochord; the Gli3 promoter contains TCF binding sites, and Gli3 can rescue phenotypes elicited by expression of a dominant-negative Tcf [51].
The multiprotein complex that modifies GLIs, which includes GSK3β and PKA, is a point of crosstalk with other signaling pathways, such as Wnt. As discussed in this section, hedgehog network proteins can elicit diverse canonical and non-canonical signaling events. Hedgehog network proteins can also have overlapping functions- for example, parallel pathways that impact proliferation. Non-canonical functions of hedgehog network members are an important consideration for contextualizing data, and necessitate the evaluation of multiple hedgehog network members to synthesize an accurate understanding of hedgehog network functions in a given biological context.
5. Hedgehog Signaling Network in Mammary Gland Development
5.1 The Hedgehog Network in Embryonic Development
Currently, no data indicate whether Dhh functions in embryonic development; however, data suggest that Shh and Ihh are dispensable. While in situ hybridization indicates that Shh and Ihh are expressed at E12.5-E16.5; transplanted glands with Shh or Ihh ablation suggested that neither ligand is essential in embryonic development, since transplants yield a comparable ductal tree capable of lactation [52,53]. It is unknown whether redundant hedgehog ligand expression masked potential phenotypes.
Looking downstream, in situ hybridization indicates that Ptch1 is expressed in the embryonic mammary bud and mesenchyme [54], however, based on studies of mutants later in development, Ptch1 loss may not grossly perturb embryonic mammary gland development [54,55]. True loss of function studies of Ptch1 in embryonic development have not been done; also, there are no data with respect to Ptch2. The Smo effector has not been studied in embryonic development.
Data from a Gli1-LacZ reporter mouse indicate that Gli1 is absent in the embryonic mammary bud [56]. A mutant homozygous for a Gli1- LacZ knock in allele (resulting in genetic ablation) showed that Gli1 is not required for embryonic development [56]. In the embryonic gland, Gli2 is expressed mostly in the mammary stroma and in a few basal cells at E16.5 by LacZ reporter [56,57]. Again, the adult phenotypes displayed with homozygous ablation of Gli2 suggest that Gli2 is not essential for embryonic development [57]. Thus, both Gli1 and Gli2 seem dispensable for embryonic mammary gland development.
In contrast with Gli1 and Gli2, Gli3 is essential in the somites adjacent to the embryonic mammary gland. Gli3 is expressed in somites, basal cells of the embryonic bud, and surrounding stroma [56]. Gli3xt/xt mice with loss of Gli3 (due to a spontaneous mutation, extra toes, resulting in intragenic deletion of Gli3) frequently showed loss of embryonic buds 3 and 5, as well as loss of TOPGal Wnt reporter activity characteristic of embryonic buds [56,58]. In vivo analysis of different combinations of Gli2/Gli3 mutants suggests the importance of hedgehog repression in embryonic development; heterozygous or homozygous loss of Gli3 reduces the number of buds, which is not observed with Gli2 loss [56]. The absence of mammary bud 3 in the Gli3xt/xt mutant was rescued in organ culture with an FGF10-containing pellet [58]. As Gli3 mRNA expression was not perturbed with in the Fgf10 homozygous null mutant, Gli3 acts upstream of Fgf10 [58]. The repressor functions of Gli3, and relative activation of the hedgehog network may distinguish embryonic specification of the mammary epithelium versus the hair follicle [52,59]. In brief, it seems that hedgehog network repression and Gli3 function in somites as a transcriptional repressor is critical for FGF10 expression, and embryonic mammary bud specification, while no roles have been defined for any other hedgehog network member.
5.2 Hedgehog Ligands in Postnatal Development
Although hedgehog ligands are expressed in the mammary gland, murine genetic models suggest that hedgehog ligands are individually dispensable for mammary gland organogenesis. SHH is expressed in the mammary epithelium of the virgin gland and during lactation [59]. Because homozygous Shh loss is embryonic lethal, embryonic mammary anlagen were transplanted to the kidney capsule to produce mammary gland outgrowths lacking SHH [59]. Resulting outgrowths suggested that Shh is not required for branching morphogenesis or lactation [59] (see Table 1 and Figure 3 for summary). Shh repression may be important, since mice overexpressing Shh under the whey acidic protein promoter (Wap) showed increased stromal condensation, Collagen 1 deposition, luminal to basal cell ratio, and ductal dysplasia after multiple pregnancies [60]. Ihh ligand, by in situ hybridization, is expressed in the body cells of TEBs and in the virgin mammary epithelium, pregnancy and lactation, and up until involution day 2 [54]. Ihh expression is undetectable at involution day 2, but expression returns in remodeled areas of the gland by involution day 14 [54]. Mutants lacking Ihh also did not show any mammary gland phenotypes when embryonic mammary anlagen was transplanted to cleared mammary fat pads[59]. Given that Ihh and Shh have similar expression patterns, these hedgehog ligands could compensate for each to mask potential phenotypes in these models. Data on DHH in mammary gland development are less extensive, however, Dhh was identified as a gene upregulated in TEBs compared to epithelium-free stroma by microarray analysis [61]. Dhh expression in the TEB epithelium was confirmed by in situ hybridization, with Dhh also present in some mature ducts [61]. Phenotypic analysis of Dhh loss or overexpression in the mammary gland has not been done. Thus, although Ihh and Shh are expressed extensively throughout development, loss-of-function studies have not yet identified functions of these molecules in mammary gland development, while data on Dhh are lacking.
Table 1.
Summary of mammary gland phenotypes displayed by animals with different mutant hedgehog network genes. Blue background indicates loss-of-function or hypomorphic alleles; orange indicates overexpression and/or conditionally activated alleles. (Tx) in the first column signifies that mammary epithelium was transplanted due to embryonic lethality of mutant animals. ND = No data. All references listed in far right column. See Figure 3 for cartoon summary.
| Mutation | Epithelial Histological Phenotype |
Stromal Phenotype |
Lactation Phenotype |
Stunted Ductal Outgrowth |
Epithelial Proliferation |
Other | Ref |
|---|---|---|---|---|---|---|---|
| Shh-/- (Tx) | No | No | No | No | ND | 49 | |
| Wap-Shh | Yes | Yes | Yes | No | Reduced in dysplasias | Dilated ducts, expanded basal population, hyperplasia | 49 |
| Ihh-/- (Tx) | No | No | No | No | ND | 49 | |
| Ptch1mes/mes | Yes | ND | ND | Yes | Sometimes increased | Epithelial, stromal, and systemic functions of Ptch1 (undefined) | 53 |
| PtchlΔ/+ | Yes | Yes | ND | Yes | Sometimes increased | Strain dependent hyperproliferation | 51 |
| MMTV-SmoM2 | Yes | Yes | ND | No | Increased | Incr. branching, decr. stem cell frequency | 57 |
| Ad-Cre; SmoM2 | Yes | Yes | ND | No | Increased | Incr branching, altered cell fate | 58 |
| Gli1 laczki/ki | ND | ND | No | ND | ND | No overt lactation or embryonic defects | 59 |
| MMTV-rtTA; TRE-Gli1 | Yes | Yes | Yes | No | Increased | Tumorigenesis | 60, 86 |
| Gli2-/- (Tx) | Yes | Yes | No | No | ND | Ductal dilation, focal dysplasia | 61 |
| Gli3xt/xt | ND | ND | ND | ND | ND | Loss of embryonic buds 3 and 5 | 59, 68 |
Figure 3. Overview of Hedgehog Network Functions in Mammary Gland Development.
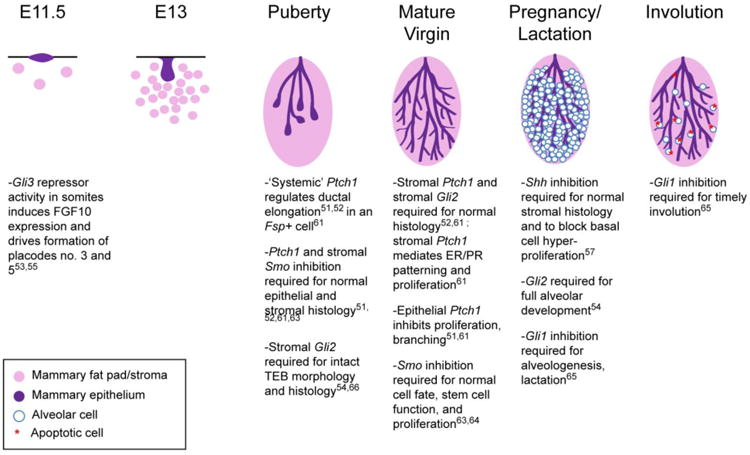
Above, the developmental stages of the murine mammary gland are listed proceeding from left to right. The critical hedgehog network member functions (or repression thereof) are briefly outlined in bullet points below. The legend (bottom left) describes which colors correspond to different cell types.
5.3 Patched Receptors in Postnatal Development
The Ptch1 receptor gene is expressed in the mammary gland throughout development as seen by in situ hybridization [54]. Animals heterozygous for the Ptch1 null allele (since homozygous loss of Ptch1 is embryonic lethal), showed filled-in ducts, dysmorphic TEBs, increased periductal stromal condensation, and loss of epithelial cell polarity [54]. Phalloidin staining indicated that luminal cells of different morphology filled ducts; interestingly, the occlusions resolved during pregnancy and lactation, but some were observed at involution day 14 [54]. Whole glands of Ptch1Δ/+ mice transplanted to a wild-type recipients retained some TEB and histological abnormalities, but transplants of epithelial fragments did not [54] minimally suggesting a role in mammary fat pad stroma. Subsequent studies employed the mesenchymal dysplasia (mes) allele of Ptch1, which arose spontaneously in mice, and encodes a protein where a 32 base pair deletion results in 220 amino acids of the Ptch1 C-terminus being replaced with 68 unrelated amino acids [55,62]. The Ptch1 C terminus has been shown to interact with TID proteins. Ptch1mes/mes animals displayed stunted ductal elongation in the adult virgin, and ductal hyperplasia and hyperproliferation in the DBA or B6D2F1 strains, but not C56B6 or FVB strains [55]. Epithelial fragment and whole gland transplantation experiments assessed the local, stromal, and systemic functions of Ptch1 contributing to these phenotypes. Whole gland or epithelial fragment transplantation of Ptch1mes/mes mutant tissues to wild type recipients rescued the stunted outgrowth phenotype, but morphological anomalies at the ductal termini were present in both cases, thus indicating both epithelial and stromal roles for Ptch1 [55]. Isografting of a wild type pituitary could rescue the stunted ducts, while estrogen and progesterone treatment did not, suggesting that Ptch1 may be required “systemically” in the pituitary for prolactin or growth hormone production to mediate ductal outgrowth [55]. Later, it was reported that Ptch1mes/mes stunted ducts could be rescued by MMTV (mouse mammary tumor virus) promoter-driven expression of an activated c-Src allele, albeit with a developmental delay, suggesting that c-Src activation downstream of Ptch1 may contribute to ductal elongation [63].
Recently, these studies were followed up with more precise tissue-compartment specific conditional ablation analyses of Ptch1 function. Adenovirus-Cre mediated ablation of Ptch1 with transplantation to wildtype hosts to achieve solely epithelial loss of Ptch1 demonstrates that Ptch1 in the mammary epithelium inhibits hyperproliferation and hyperbranching, but is dispensable for normal ductal histology in the mature adult [64]. The inability of IPI926 to inhibit hyperproliferation and hyperbranching in the mammary outgrowths lacking Ptch1 indicates that these functions of Ptch1 are SMO- independent, suggesting that Ptch1 may function primarily non-canonically to regulate branching and proliferation in the mammary epithelium.
Consistent with a stromal/systemic role for Ptch1 suggested by earlier studies, Fsp-Cre-mediated ablation of Ptch1 in fibroblasts and myeloid cells yields TEBs with altered histology, and mature ducts filled with luminal cells, with increased estrogen receptor positivity together with decreased progesterone receptor expression [64]. This model also displayed stunted ducts, and a loss of mammary epithelial cell proliferation. The stunted duct phenotype was rescued by whole gland transplantation to a wildtype recipient animal, while the filled-in ducts were not rescued [64]. Bone marrow transplantation did not rescue the ductal outgrowth or filled ducts [64]. Thus, Ptch1 appears to function in a mammary gland extrinsic Fsp+ cell to regulate pubertal ductal elongation, ER/PR patterning, and proliferation, while Ptch1 in mammary fat pad fibroblasts inhibits a DCIS-like phenotype [64].
In summary, Ptch1 has many functions in different tissue compartments to regulate mammary gland biology, including histology of TEBs and mature ducts, pubertal ductal outgrowth, proliferation, periductal stromal accumulation, and ER/PR expression patterns. These results show distinct systemic, stromal, and epithelial functions of Ptch1 [55,64]; which include non-canonical functions of Ptch1 as well, and suggest that that systemic and stromal functions of this gene may be of greater importance in regulation of the mammary gland elongation and histology compared to the mammary epithelium intrinsic role. In contrast, there are no data on the phenotypic consequences of Ptch2 disruption or overexpression in the mammary gland.
5.4 Smoothened Effector in Postnatal Development
Regulated expression of Smo, the primary effector of canonical hedgehog signaling, is important in mammary gland homeostasis. Murine models evaluating the role of Smo in the mammary gland have employed the conditional constitutively activated SmoM2 allele, which has a G-to-T transversion resulting in a tryptophan to leucine alteration identified in human basal cell carcinoma [65]. Mice with MMTV driven-SmoM2 (MMTV-SmoM2) expression displayed TEB dysmorphia and an increased number of TEBs persisting at 10 weeks of age, hyperproliferation at 10 weeks of age, hyperbranching/hyperbudding [66]. Other mouse models of conditional SmoM2 expression in the mammary gland (using MMTV-Cre, Adenovirus-Cre infected epithelial cells transplanted to a cleared fat pad, or intraductal Adenovirus- Cre injection) displayed similar phenotypes including hyperbudding, hyperbranching, and hyperproliferation [67]. The hyperbranching and hyperbudding phenotypes required a mixture of SmoM2 positive and SmoM2 negative cells [67]. Aberrant SmoM2 activation also produced phenotypes indicative of altered cell fate, namely loss of NCKK1 (Na–K–Cl co-transporter-1) in ducts, a protein which is normally lost during alveologenesis; and stromal changes, including an increase in the presence of periductal F4/80 positive macrophages and an increase in collagen deposition [67].
Smo has a stromal function in mammary gland development as well: mice with Fsp-Cre mediated SmoM2 expression show histological defects in the TEB- include cap cell layer detachment- and increased ductal filling [64]. The similarities to the Fsp-Cre; Ptch1fl/fl phenotypes suggest that Ptch1 inhibits Smo to block abnormal histology [64]. Thus, aberrant Smo activation in the mammary gland alters proliferation, cell fate, branching morphogenesis, and the periductal stroma, while regulated Smo expression in Fsp+ cells is important for normal histology. No published data address Smo complete loss-of-function in the mammary gland.
The downstream mechanism driving the phenotypes present due to Adenovirus-Cre-mediated SmoM2 expression was postulated to be Notch1 signaling, since Notch target genes were upregulated in SmoM2 positive cells relative to SmoM2 negative cells by qPCR [67]. Given data indicating that Smo could function as a G-protein coupled receptor (GPCR) in other systems [15], the hypothesis that a GPCR function of Smo mediates SmoM2-induced hyperproliferation was tested. Pertussis toxin treatment to block Gai activity, or conditional genetic ablation of Gai2 both blocked SmoM2-induced hyperproliferation, while ablation of neither Gai1 nor Gai3 blocked SmoM2-driven proliferation[44]. Further, SmoM2 induced hyperproliferation appeared to be GLI-independent, since treatment with a GLI1/2 inhibitor, GANT61, did not block hyperproliferation in SmoM2 animals [44]. Although SmoM2 may function differently than endogenous Smo, it is plausible that the paracrine signaling driving hyperproliferation, and perhaps hyperbudding/hyperbranching in these mouse models is non-canonical SMO action as a GPCR.
5.5 GLI Transcription Factors in Postnatal Development
Regulated expression of GLI transcription factors is also essential for normal mammary gland development. Gli1 expression, using a Gli1-LacZ reporter mouse, is absent in the mature virgin mammary epithelium; the only Gli1 reporter activity was in lymph vessels [56]. Pups born to mothers lacking Gli1 (homozygous for a Gli1-LacZ knock in allele) were viable, suggesting that lactation was not significantly perturbed [56]. Conditional overexpression of human Gli1 in the mammary epithelium delayed alveologenesis and impaired functional differentiation, in spite of increased proliferation during pregnancy [68]. The increased proliferation was present with an increase in TUNEL positive cells at lactation day 1 [68]. Involution was also delayed in Gli1 overexpression mutants, with more extensive mammary epithelium and F4/80 positive macrophages present relative to controls at involution day 14 [68]. There was an increase in periductal stroma as well [68]. Thus, Gli1 repression is required for normal lactation, a normal balance of proliferation/ cell death, stromal homeostasis, and involution.
In contrast to Gli1, Gli2 is essential for normal ductal morphogenesis. According to in situ hybridization and a Gli2-LacZ reporter, Gli2 is expressed mostly in the mammary stroma until pregnancy and lactation; during lactation, Gli2 is widely expressed in the mammary epithelium [56,57]. As homozygous Gli2 loss is perinatal lethal, homozygous Gli2 null glands were transplanted to evaluate loss-of-function phenotypes[57]. The Gli2 null outgrowths displayed ductal distension and aberrant micropapillary structures [57]. Transplantation data suggested that only stromal functions of Gli2 regulate mammary ductal histology [57]; which is consistent with the lack of Gli2 mRNA expression in the mammary epithelium until pregnancy. Mutants heterozygous for the Gli2 null allele displayed stunted ducts, radial branching, mammary lesions, and reduced alveolar development [57]. Whole gland transplantation of homozygous Gli2 null glands did not display any defects in alveologenesis [57], suggesting that the alveologenesis defect in heterozygotes is due to a systemic function of Gli2.
Recently, the tissue compartment specific roles of Gli2 have been further defined using Fsp-Cre;Gli2 conditional ablation animals [69]. This animal displays reduced proliferation, reduced stroma adjacent to mammary epithelium, hypoplasia, and reduced collagen deposition [69]. Transplantation experiments show that stromal Gli2 is required for normal mammary epithelial outgrowths [69]. Growth hormone and 17-β estradiol administration to these mutants did not elicit upregulation of respective target genes (i.e., Igf1 and Hgf, respectively); Wnt2 signaling is also altered [69]. Together with reduced colony formation in 3D culture, these results indicate that Gli2 in non-F4/80+ Fsp+ cells may regulate mammary stem cells and mammary epithelial responsiveness to hormone signaling. Thus, Gli2 is required for normal ductal histology, branching, and alveologenesis, and may also act as a tumor suppressor. Gli2 thus has tissue compartment specific functions, including mediating mammary epithelial proliferation, and regulating the mammary stem cell niche.
The Gli3 transcription factor is expressed in the mammary epithelium and stroma of pubertal and mid-pregnant animals [56]. While Gli3 has been studied in embryonic development (see 5.6), no published data define a postnatal function for Gli3 [56], although the ratio of activator to repressor forms of GLI3 increases the mammary repopulating cells (CD24+, CD29high) at pregnancy day 14, suggesting that hedgehog signaling is active in this subset of cells, and thus there could be a role in alveologenesis [70].
5.6 Primary Cilia and Mammary Gland Development
The primary cilium organelle may be required for canonical hedgehog signaling, and mammary gland development. Primary cilia have been observed by microscopy and immunostaining in both luminal and basal cells at 3-4 weeks of age [71,72], but only in 35% of the basal cells at 7 weeks of age [71]. Ablation of primary cilia throughout the mouse, via expression of a mutant form of Ift88 (intraflagellar transport protein 88) to disrupt primary cilium assembly, produced decreased branching [71]. The loss of branching in mutants in organ culture suggests this is an epithelium intrinsic defect [71]. Interestingly, while fat pad filling was reduced at 7 weeks, TEBs were present [71]. Alveologenesis was also perturbed. Ducts displayed reduced canonical hedgehog signaling and increased canonical Wnt signaling; it is not known which of these molecular changes produced the loss of branching [71].
Ablation of primary cilia in ovarian follicles (by conditional ablation of Ift88) yielded stunted mammary ductal outgrowths at an adult virgin timepoint [73] TEBs and pubertal ductal outgrowth was restored in these mutants with estrogen treatment, suggesting that defective estradiol production induced the stunted ducts and loss of TEBs in the mutant animals [72]. This hypothesis is consistent with the fact that the follicles, which produce estradiol, were genetically manipulated in this model [72]. It is unclear why these mutants did not have TEBs at the adult virgin stage, while the previous model did [71]; perhaps primary cilia have opposing functions in different cell types to regulate TEBs, or these models have different recombination efficiencies. Since primary cilia and Ptch1 have systemic roles in mammary gland development, we cannot exclude that other phenotypes from mice with global mutations- i.e. Gli2 heterozygotes- could be due to systemic functions of hedgehog network genes. The lack of data addressing the molecular drivers of these phenotypes makes it difficult to determine whether these phenotypes were due to altered hedgehog signaling. Tissue compartment-specific and more extensive molecular analysis would significantly elucidate the role of the primary cilium in mammary gland development.
5.7 Hedgehog Network and Mammary Stem Cells
Hedgehog signaling is important in the homeostasis of many adult stem cell compartments, such as the hair follicle bulge [74]. Hedgehog network members may also regulate stemness in the mammary gland. Mice heterozygous for a Ptch1 null allele showed increased proliferation, but decreased long-term label retention in a population enriched for mammary stem cells gland (CD24+CD29hi) [70]. The CD24+CD29hi population, with Ptch1 heterozygosity, showed increased Gata3 mRNA expression, which is associated with luminal progenitor cells [70]. In immortalized mammary epithelial cells, TAp63 ablation reduced Ihh mRNA levels in the stem cell enriched fraction, whereas expression of ΔNp63 increased Ihh mRNA [70]. Additionally, shIhh reduces the number of complex acini, whereas shGli3 increases complex acini formation. Together, these data suggest that Ptch1 loss pushes mammary stem cells into a progenitor state. A mechanism was postulated whereby Ihh drives altered TP63 promoter selection to forfeit mammary stem cell quiescence, and promote asymmetric division. A few caveats to this study include the fact that initial labeling in long-term label retention may have been different, which we may presume to be the case given data from other studies of Ptch1 heterozygosity [55]. Also, the conclusions with respect to stemness were based on correlations with the CD24+CD29hi markers rather than functional data.
SMO may also mediate stem cell function, as expression of the MMTV-SmoM2 transgene reduced stem cell frequency in limiting dilution transplantation assays, while mammosphere formation efficiency was increased and K6 expression (a marker of progenitor cells) was increased [66]. Thus, it appears that SmoM2 expression pushes stem cells into a progenitor state. Since SmoM2 acts as a GPCR in the mammary epithelium, SmoM2 function in mammary stem cells may be also be due to GPCR functions of SmoM2 rather than activation of canonical hedgehog signaling [44].
Animals with Wap promoter-driven Shh overexpression and a Ptch1-LacZ reporter showed basal/stem-cell related phenotypes after multiple pregnancies to induce Shh [60]. A slow-cycling subset of basal cells were positive for the Ptch1-LacZ reporter, and displayed Integrin β3, K15, and P63 expression and primary cilia (thought to be required for canonical hedgehog signaling) by immunostaining [60]. Basal cell hyperplasia in the Wap-Shh mice this suggests that hedgehog activation by epithelial Shh ligands may regulate stemness and proliferation of basal cells [60]. The association of K14+ cells with primary cilia is supported by data from normal human breast tissue; co-staining of primary cilia and K14 was observed, while primary cilia were rare in luminal cells [75]. It is unknown whether cells with this phenotype exist in the absence of pregnancy, or in the presence of physiological levels of SHH. Also, the conclusions with regard to stemness in this report were based on co-immunofluorescence for p63, K15 (a luminal progenitor marker), and integrin β3, rather than functional assays.
Another report using normal human breast tissue suggests canonical hedgehog signaling activity promotes stemness or a less-differentiated state. Experiments with normal human breast tissue from reduction mammoplasties indicated that PTCH1, SMO, GLI1, and GLI2 mRNAs were upregulated in mammospheres compared to differentiated cells [76]. SHH treatment increased primary and secondary mammosphere formation, which was blocked by Cyclopamine, suggesting this effect was SMO-dependent [76]. Additionally, GLI2 overexpression increased mammosphere formation [76]. GLI1 and GLI2 overexpressing mammospheres induced BMI-1 transcription, and BMI-1 siRNA significantly decreased mammosphere formation efficiency, suggesting that BMI-1 may mediate stemness downstream of GLI in the mammary gland [76]. Thus, autocrine hedgehog signaling may control mammary stem cell self-renewal, however, this conclusion is based solely on mammosphere formation assays.
Aside from these studies examining epithelial cell autonomous functions of hedgehog network genes, recent data shows that mice with Fsp-Cre mediated Gli2 conditional ablation display reduced colony formation and reduced mammary epithelial cell proliferation, while the number of basal cells was unchanged by flow cytometry [69]. Most convincingly, transplantation of CD24+CD49hi stem cell enriched cells transplanted to Fsp-Cre;Gli2fl/fl mutants display reduced fat pad filling; additionally, mammary epithelial cells from Fsp-Cre;Gli2fl/fl mutants are consistently outcompeted when transplanted with wildtype cells in a competitive repopulation transplantation assay [69]. Thus, stromal Gli2 may be critical for the mammary epithelial stem cell niche.
Together, these data indicate that members of the hedgehog signaling network could control cell fate and stem cell self-renewal in the normal mammary gland, by potentially acting upstream of alternative p63 promoter selection or inducing Bmi-1 transcription [76]. Additionally, Gli2 in Fsp+ non- macrophage stromal cells may regulate regenerative potential of mammary stem cells via an unknown paracrine mechanism [69]. Functional data from physiologically relevant systems are needed to fully understand the role of the hedgehog network in mammary stem cell self-renewal and differentiation.
6. Hedgehog Signaling Network in Breast Cancer
Aberrant activation of hedgehog signaling is found in multiple cancer types. For example, in basal cell carcinoma, mutations in hedgehog network genes resulting in hedgehog network activation are sufficient to induce, and required to maintain carcinomas [34]. Mutations that activate canonical hedgehog signaling drive medulloblastoma tumorigenesis as well [35,77]. In contrast to these explicit data in basal cell carcinoma and medulloblastoma, the data with respect to hedgehog network activation and breast tumorigenesis are much less definitive- but do suggest misregulation of the hedgehog network.
6.1 Changes in Hedgehog Network DNA, and Expression in Breast Cancer
Hedgehog network genes are often misregulated at the DNA level, including point mutations and copy number variations, in breast tumors. SHH is amplified in 1-2% of breast cancers, while DHH and IHH are less frequently altered at the DNA level (see Table 2). PTCH1 mutations and deletions are present in 1-2% of breast cancers [78]. SMO is also mutated (missense point mutations) or amplified in about 1% of breast tumors; and, consistently, GLI1, GLI2, and GLI3 are mutated (missense point mutations) in 1-2.5% of breast cancers [78]. While the TCGA dataset [19] (consisting of fewer samples) reported amplifications together with less frequent missense mutations and deep deletions in hedgehog network members, the larger METABRIC dataset [79,80] only shows amplifications in hedgehog network genes (Table 2). In a screen for promoter methylation in 6 breast cancer cell lines, the PTCH1 promoter was frequently methylated [81]. Comparative hybridization of 47 human breast tumors indicated that a region of chromosome 9 containing PTCH1 is lost in 26% of tumors, while chromosome 12q13-15 containing GLI is frequently amplified [82]. Taken together, the hedgehog signaling network member genes and methylation patterns are perturbed in a subset of human breast cancers.
Table 2.
Here, the percent of breast tumors from the TCGA dataset [1] (left columns) with mutations of different hedgehog network members is listed, together with information on what type of mutation is present. The right hand columns show this information for the METABRIC datasets [2,3]. The TCGA dataset includes 816 samples [1], and the METABRIC datasets include 2051 samples [2,3]. Data obtained with CBioPortal [4,5].
| TCGA | TCGA | METABRIC | METABRIC | |
|---|---|---|---|---|
| Gene | % Breast Cancers with Mutations | Types of Mutations | % Breast Cancers with Mutations | Types of Mutations |
| Shh | 1.5% | Amplifications | 2.1% | Amplifications |
| Ihh | 1% | Amplifications, deep deletions | 0.2% | Amplifications |
| Dhh | 0.2% | Amplifications | 0.3% | Amplifications |
| Ptch1 | 1.8% | Missense, with a few amplifications and deep deletions | 0.6% | Amplifications |
| Ptch2 | 1.8% | Amplifications, with a few deletions, truncations, and missense | 1.3% | Amplifications |
| Smo | 1.2% | Amplifications | 1.4% | Amplifications |
| Hhip | 0.7% | Amplifications | 0.7% | Amplifications |
| Gli1 | 1.5% | Amplifications and missense | 0.6% | Amplifications |
| Gli2 | 1.5% | Missense | 0.3% | Amplifications |
| Gli3 | 2.6% | Amplifications and missense | 2.4% | Amplifications |
Since the mammary gland is an ectodermally-derived tissue, it was hypothesized that SMO polymorphisms present in basal cell carcinomas may be present in breast cancers. This hypothesis was refuted in a study of 128 breast tumors [83]. Missense coding mutations in PTCH1 were also absent in a panel of 45 breast cancers [83]. This finding with respect to PTCH1 mutations conflicts with the new analysis of TCGA data presented here (Figure 4) [78]. The divergence of hedgehog signaling in skin and mammary tumors is consistent with divergent embryonic functions of hedgehog signaling in these tissues [59].
Figure 4. Comparison of Paired Breast Tumor-Normal mRNA Levels.
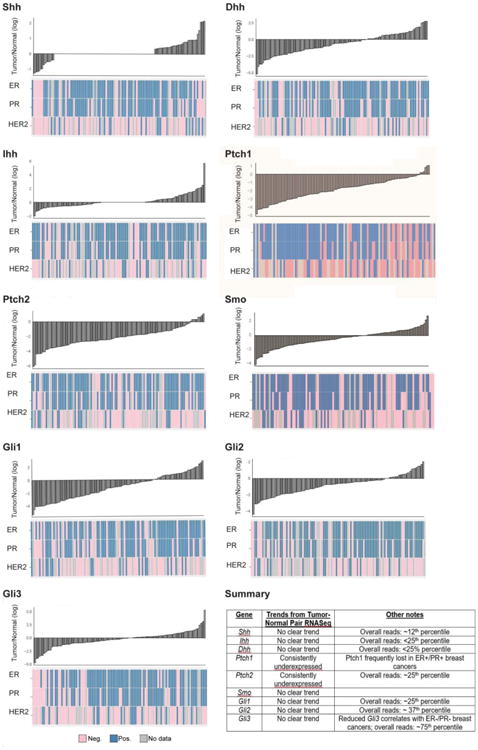
There is consistent downregulation of Ptch1 and Ptch2 mRNA compared to paired normal tissue, as seen in waterfall plots for 109 tumor-normal pairs.See below for ER/PR/HER2 status as determined by immunohistochemistry (IHC). On the other hand, Shh, Ihh, Dhh, Smo, Gli1, Gli2, and Gli3 mRNAs do not display a consistent trend in breast tumors compared to normal tissue. Waterfall plots display fold change for 109 tumor-normal pairs [19] on a log2 scale, with normal expression as baseline. Graphs produced using ggplot2 in R version 3.0.1.
Reports also show misregulation of the hedgehog network at the mRNA level. SHH mRNA is enriched in invasive breast cancers (IBC) that relapse compared to non-invasive breast cancers [84]. PTCH mRNA is lower in breast tumors relative to normal samples, as observed by qPCR, which strongly correlated with lower GLI1 and GLI2 mRNA levels (but not HIP or GLI3 mRNA), suggesting activated canonical hedgehog signaling in breast tumors [81]. In 10 paired samples, SHH or DHH mRNA was higher, PTCH1 mRNA was lower, and GLI1 mRNA was higher in breast tumors relative to normal tissue [85]. Similar results were observed in breast cancer cell lines compared to cells from reduction mammoplasties, except there was no significant difference in PTCH1 mRNA [85]. In a different panel, GLI1 and SHH mRNA levels were highest in IBC, less high in DCIS, and at lowest expression in normal breast tissue; additionally, GLI1 and SHH mRNA levels showed a positive correlation[86]. A new isoform of GLI1 (tGLI1), containing a deletion of part of exons 3 and 4, has been identified exclusively in breast cancer cell lines but not normal breast tissue [87]. It is not known if this variant is common in breast tumors.
Given that the studies comparing tumor-normal pairs had small sample sizes, we analyzed PTCH1 and SMO mRNA levels in tumor-normal pairs using the updated TCGA dataset [19]. Comparison of PTCH1 and PTCH2 tumor mRNA levels relative to 109 paired normal tissues revealed consistent downregulation of PTCH1 and PTCH2 mRNA in tumors (see Figure 4). In contrast, other hedgehog network members, including SHH, IHH, DHH, SMO, GLI1, GLI2, and GLI3 mRNAs do not follow a consistent trend in tumors relative to normal tissue (Figure 4).
Hedgehog network members may also be misregulated at the protein level in breast cancer. SHH, PTCH1 and GLI1 protein expression is increased in breast tumors relative to non-paired normal tissue [88]. However, the specificity of the antibodies used was not demonstrated (a persistent problem in the field). Similar to mRNA results, SHH and GLI1 expression were correlated in breast carcinomas, and nuclear GLI1 expression was highest in IBC, next highest in DCIS, and lowest in normal tissue [86]. In tumor-normal paired samples, SHH, PTCH1, and GLI1 were all upregulated in breast cancer versus paired normal tissue [85] from 10 patients. It should be noted that the sample size of this study is small, the samples with immune infiltrate were excluded (possibly biasing the data), and most importantly, the PTCH1 and SHH antibodies used have very weak, diffuse staining patterns, including some unexpected nuclear staining, as seen in the papers cited as a positive control. The authors allude to a positive control experiment for the SHH and GLI1 antibodies, but these data are not shown. This paper [85] presents the staining immunoscore without primary images. Consistent with aberrant hedgehog network activation, another report showed SMO protein was undetectable in the normal human breast (or normal mouse mammary gland), but was expressed in 70% of DCIS and 30% of IBCs [66], as well as in mice expressing MMTV-SmoM2 to indicate antibody specificity.
While the data with respect to other hedgehog network members in breast cancer are more consistent, PTCH1 expression data have been confused by the use of poor antibodies. In addition to the above report using the unverified PTCH1 antibody [85], two other studies have used this unverified antibody (and report upregulation of PTCH1 in breast cancer relative to normal tissue) [88,89]. On the other hand, other studies have used a rigorously validated PTCH1 antibody [66,81]. These studies showed that PTCH1 protein expression is detectable in both the epithelium and stroma of the normal human breast [66,81]. The high expression of PTCH1 in the normal mammary gland is consistent with the in situ hybridization data from the normal murine mammary gland [54]; further, increased signal was present in MMTV-SmoM2 positive cells [66]. In contrast to the ubiquitous expression in the normal breast, PTCH1 expression is reduced or lost in 50% of cases of ductal carcinoma in situ (DCIS) (a precursor of malignant carcinoma) and 50% of invasive breast cancers (IBC) [66]. Similarly, PTCH1 was reduced in 40% of DCIS samples and 52% of IBCs [81] using the same PTCH1 antibody on a panel of 105 IBCs, 104 DCIS samples, and 175 adjacent normal tissue samples. Although there are reports to the contrary, given the more extensive validation of the antibody used in [66,81] and the large samples sizes in these studies, it seems more likely that PTCH1 is frequently underexpressed in pre-malignant lesions and breast cancer, while SMO expression increases in breast disease.
6.2 Hedgehog Network and Tumorigenesis
The hypothesized role of hedgehog network activation in breast tumorigenesis contrasts with negative data from mouse models and patients. In genetically engineered mice, neither Ptch1 heterozygosity nor MMTV-driven expression of SmoM2 induced mammary tumorigenesis [55,66], although ectopic SMO may be correlated with proliferation of adjacent cells in breast cancer [66]. Gli2 loss enhanced dysplasia formation in mice [57]. Neither Gorlin syndrome patients that are haploinsufficient for PTCH1, nor patients with other mutations in the hedgehog network (i.e. IHH, GLI) are reported to be at an elevated risk for breast cancer [35].
The most data exist with respect to GLI1 in tumorigenesis. GLI1, 2, and 3 proteins were all upregulated in breast tumors relative to normal tissue [89]. Transgenic expression of Gli1 induced tumorigenesis, with Gli1 expression required for tumor survival in a mouse model [90]. Gli1 has been implicated in breast cancer cell survival, proliferation, and metastasis in xenografts and cell line models [46,89,91–93]. The tGLI1 isoform of GLI1, lacking exon 3 and part of exon 4, has been identified in a few breast cancer cell lines. TGLI1 increases anchorage-independent cell growth in MDA-MB-231 cells, and induces VEGF-a transcription to promote angiogenesis [87]. Additionally, many reports correlate GLI1 expression with poorer patient survival (see section 6.6).
Data suggest a connection between Shh and Gli1 expression in breast cancer, consistent with a role for activated canonical hedgehog signaling in tumorigenesis. SHH expression is correlated with GLI1 expression in patient samples [86,88,94], which correlated with higher tumor grade [86]. Shh overexpression increased angiogenesis and metastasis in tumors independent of VEGF via Gli1 transcription of Cyr61 (Cysteine-rich angiogenic inducer 61) in xenografted cells [93]. Thus, Shh-Gli1 signaling may control critical aspects of breast tumorigenesis; for further information, see 6.3 for discussion of the associations between Shh and Gli1 in ER+ breast cancer.
While there are limited data connecting growth factor, TGFβ, and/or Wnt1 signaling with hedgehog activation in breast cancer, this area bears further investigation. It is well-established that Wnt1, EGF, FGF, and TGFβ misregulation contribute to breast tumorigenesis and proliferation. Hedgehog signaling has non-canonical interactions downstream of Wnt1 [51]; additionally, MMTV-Wnt1 mice display Gli1 reporter activity [95]. While FGF10 is induced by Gli3 in embryonic mammary gland development [58], and Gli2 transcription is promoted by TGFβ signaling in the vicious cycle- a feed-forward signaling cascade that promotes breast tumor metastasis to bone [46], these signaling events have not been studied in tumorigenesis. Also supporting connections between these signaling pathways, LncHH expression in breast cancer cells (which upregulated hedgehog signaling) was associated with upregulated Wnt, ErbB, and TGF beta signaling by microarray and gene ontology analysis [48]. Survival and/or proliferation are promoted by an interaction between the cytoplasmic domain of ErbB2 (EGF) and TID1 in breast cancer cells [96]. While Ptch1 involvement has not been addressed, it seems likely since TID1 binds a PTCH1 domain lost in the Ptch1Fvb mutant allele associated with squamous cell carcinoma (see [39] for further discussion).
6.3 Hedgehog Network in ER+ Breast Cancer
Recent data suggest a connection between the hedgehog network and estrogen signaling in ER+ breast cancer. An interaction between a PTCH1 polymorphism in the C-terminus, Pro1315Leu, is associated with increased breast cancer risk with oral contraceptive use [97]. PTCH protein expression showed a positive correlation with ER positive, but not PR positive breast cancers [81]. Gli1 (nuclear GLI1 protein) and ERα mRNA and protein expression correlate in many patient tumors and breast cancer cell lines [86,98–100]. Estradiol treatment can induce GLI1 mRNA, and nuclear localization of GLI1 in ER positive breast cancer cell lines to promote proliferation, survival, and invasiveness [86,98,100]. On the other hand, a different study reported that Gli1 attenuated the mitogenic response of breast cancer cells to estradiol [92]. Gli1 also induces invasiveness in ER negative cancers via upregulation of MMP-11 in MDA-MB-231 cells [91]. Si- and shRNA to GLI1 and SHH treatment suggests that SHH signaling via Gli1 in ER positive cells induces migration in vitro [86]. Conversely, estrogen depletion in cell culture reduced SHH and GLI1 expression; additionally, SMO inhibitors decreased ERα-luciferase activity and proliferation of ER positive cells [94]. It would interesting to evaluate GLI1 expression and localization in tumors of patients treated with anti-estrogen therapies, and elucidate estrogen-GLI1 signaling mechanisms and functions in ER positive cancers.
6.4 Hedgehog Signaling in TICs, EMT, and Metastasis
Hedgehog signaling may also function in the tumor-initiating cells in breast cancer. Tumor initiating cells (TICs), also referred to as cancer stem cells, are a subset of the tumor population with the capacity to regenerate the tumor bulk and self-renew. TIC are of interest since TICs may be particularly resistant to conventional cytotoxic chemotherapy and radiation [101]. One subset of cells with enhanced tumor initiating capacity is the CD44+CD24-Lin- population. These cells display higher expression of PTCH1, GLI1, and GLI2 mRNA [76]. Similarly, other data showed that two populations enriched for stemness, CD44+CD24-Lin- and Hoechst Dye excluding cells, display higher Shh and Gli1 mRNA and protein levels relative to the tumor bulk [102]. Cyclopamine treatment or Gli1 depletion reduced the size of this population of cells [102]. ΔNP63 regulates mammosphere formation in MMTV-ErbB2 tumor cells, and p63 shRNA decreased SHH, PTCH1, GLI2, and BMI-1 mRNA levels [103]. These hedgehog target genes may be direct transcriptional targets of p63 [103]. An in vitro experiment suggested that estrogen stimulation of TIC survival depends on GLI activity [98]. Additionally, the MMTV-Wnt-1 transgenic mouse model displayed Gli1-LacZ reporter activity in p63+, K14+ basal cells, thus correlating Gli-dependent transcription with basal cell marker expression [95]. Consistently, primary cilia were detected primarily in basal cells but rarely luminal cells of normal breast epithelium [75]. Thus, canonical hedgehog signaling may be activated in TICs.
Hedgehog signaling may also activated in EMT (epithelial mesenchymal transition), which is the concomitant loss of epithelial traits and gain of mesenchymal traits by a cell, which enables invasion and metastasis [48]. The LncRNA-Hh long non-coding RNA is upregulated in Twist-expressing MCF-7 or MCF10A cells (with induced EMT), and data suggest that lncRNA-Hh regulates GLI1 and GAS1 expression [48]. Overexpression of LncRNA-Hh stimulates primary tumor growth in xenografted cell lines, while LncRNA-Hh shRNA reduces tumorigenesis [48]. LncRNA-Hh also positively regulates mammosphere formation efficiency and size, thus a connection between Twist-LncRNA-Hh-Gli1/Gas1 signaling and stemness [48]. It is unknown whether LncRNA-Hh is expressed in patient tumors or normal tissue. The induction of hedgehog signaling by an established EMT transcription factor could fit with patient data suggesting that Gli-dependent signaling positively regulates invasiveness.
The tGLI1 isoform of GLI drives a number of characteristics associated with increased invasive and migratory behavior of MDA-MB-231 cells [87]. TGLI1 accelerates gap closure in the scratch assay and transwell migration, and increases CD24 and MMP expression [87]. It would be interesting to test whether tGLI1 is expressed in human breast cancers.
Bridging metastasis and EMT, recent data implicated GLI1 in EMT behavior. Data from an upcoming report suggests that the EMT- promoting properties of TWIST1 and SNAILl1 acting via the SIX1 transcription factor drives metastasis by stimulating GLI1 expression in non-EMT HMLER cells by a paracrine mechanism [49]. Paracrine activation of GLI1 via Twist1, Snail1, or Six1 was sufficient to increase invasiveness, EMT characteristics, and reduce anoikis of the non-EMT MCF7 cells, which was blocked with GANT61 treatment to inhibit GLI1/2 [49]. In patient derived xenografts expressing hedgehog network members, GANT61 treatment, but not pharmacological inhibition of SMO, inhibited tumor growth [49]. Expression data from human breast cancer data sets suggests that EMT transcription factor (TWIST1, SNAIL1, SIX1) activation of GLI1 can be Hh- and Smo- dependent or independent, and thus either canonical or non-canonical (or both) [49].
Hedgehog network members may enable breast cancer metastasis to bone. Ptch1-dependent signaling, or blocking SMO by LDE225 treatment, attenuated pro-metastatic osteoclast activity in vitro [104]. Thus, activated canonical hedgehog signaling may accelerate the ‘vicious cycle’ [104]. The vicious cycle is a process in breast tumor metastasis to bone involving signaling which increases osteoclast activity and enables metastasis growth in a feed-forward response. These results are consistent with findings that tumor-derived Shh and/or Ihh induces osteoclast activity [105,106]. Specifically, Ihh may be required for Runx2 induction of PTHrP mRNA (parathyroid hormone related protein) to increase osteoclast activity [105]. Hedgehog ligands may also promote metastasis by stimulating expansion of pre-osteoclasts via osteopontin [106]. Transfection of Gli2 can induce PTHrP-luciferase activity and PTHrP mRNA expression, osteoclast activity, and the vicious cycle, but in this study, osteoclast activity did not require hedgehog ligands or SMO activation [46,107]. While these data suggest a role for paracrine hedgehog signaling to bone in the vicious cycle, it should be noted that these studies used a single cell line; additional studies to determine whether Hh signaling acts on osteoclasts in vivo would be informative.
6.5 Stromal Misregulation of the Hedgehog Network in Breast Cancer
Recent data suggest that hedgehog network misregulation in the stroma rather than in the tumor itself may be important for breast tumorigenesis. HH overexpressing xenografted tumors grew faster, and were more proliferative and invasive than tumors without HH ligand overexpression [108]. QPCR and staining indicated that tumor-derived SHH ligands activated canonical hedgehog signaling in stromal cells rather than signaling to the tumor itself [108]. this signaling pattern fits with Wap-Shh, Gli1 overexpression, or SmoM2 expression in the mouse eliciting stromal phenotypes including hyperplasia and increased collagen deposition [60,67,90]. Treatment of animals bearing xenografted cells with 5E1, a HH function blocking antibody, reduced lung metastasis [108]. Data from human cell lines showed differential expression of hedgehog network genes, and different responses to cyclopamine in epithelial versus stromal cells [85].
Consistent with paracrine activation of hedgehog signaling in the cancer-associated stroma, GLI1 mRNA was higher in the cancer stroma than in normal stroma; however PTCH1 and SMO mRNA levels were higher in normal stroma (than cancer associated stroma) in a small study [85]. The upregulation of GLI1 in cancer-associated stroma fits with mouse data showing strong Gli1-LacZ reporter activity in the stroma of MMTV-Wnt1 transgenic mice [95]. Primary cilia are widely expressed in breast tumor-associated fibroblasts [75]. Activated hedgehog signaling in the breast tumor-associated stroma fits with similar expression in other epithelial tumors, including prostate and pancreatic cancer.
6.6 Hedgehog Network and Breast Cancer Patient Clinical Outcomes
Data associate higher expression of hedgehog network genes with poorer clinical outcome. Comparison of non-IBC (invasive breast cancer) and IBC breast tumors showed that higher SHH mRNA levels predicted for relapse [84]. High SHH and GLI1 expression correlated with higher tumor grade in patient samples [86]. Additionally, nuclear GLI1 protein was correlated with poorer patient disease-free and overall survival, higher tumor stage, and increased lymph node positivity [109,110]. GLI1 also predicted reduced pathological complete response in hormone receptor negative breast cancers [100,110]. However, in a different report, nuclear GLI1 correlates with triple negative breast cancer, and does not predict worse patient survival in ER positive cancer [100]. An association between stromal Gli1 expression and poorer patient outcome has also been reported: high hedgehog ligand expression in breast tumors present with nuclear GLI1 in the adjacent stroma predicted for invasive breast cancers with worse patient outcomes higher grade tumors [108]. This finding [108] raises the possibility that datasets associating Shh expression with poor patient outcome could have also displayed higher stromal GLI1 expression to elicit worse patient outcome [84].
Gli1 expression may also correlate with therapeutic resistance. GLI1 protein may mediate tamoxifen resistance in a model of parental and tamoxifen resistant MCF-7 xenografted cells [109], which fits with the putative interaction between Gli1 and estrogen signaling (see 6.3). Nuclear GLI1 protein and resistance to neoadjuvant therapy are correlated in HER2 positive tumors, as well as nuclear GLI1 and lower pathological complete response [110]. Taken together, Gli1, potentially with Shh, could be a biomarker of worse overall patient survival, disease- free survival, and pathologic complete response, or tamoxifen or anti-HER2 therapy resistance.
7. Hedgehog Network in Endocrine Tissues
The mammary gland is exquisitely sensitive to, and exhibits extensive physiological changes in response to hormones throughout development, and in breast cancer. Normal development of, and crosstalk between the hypothalamus, pituitary, adrenal glands, and ovary is required for the production of steroid hormones, peptides, and proteins involved in pubertal outgrowth and lactation. The factors regulating postnatal mammary gland development include estradiol and progesterone from the ovary; gonadotropic releasing hormone and growth hormone releasing hormone from the hypothalamus; prolactin, growth hormone, and oxytocin from the pituitary; and glucocorticoids from the adrenal gland. The hypothalamus-pituitary-ovary signaling axis, involving extensive neuro-endocrine crosstalk, cyclically regulates these factors and the steroids and peptides required for their release. In addition to developmental roles for hormone signaling to the mammary gland, hormone signaling is of critical importance for understanding breast tumorigenesis and disease progression, since 70% of breast tumors are receptor positive and receive anti-estrogen therapies.
Genetic manipulation of hedgehog network genes elicits developmental phenotypes in these endocrine organs. These data are important given that mammary gland extrinsic functions of hedgehog network genes (i.e. Ptch1 and Gli2) impinge on mammary gland development, and the data connecting hedgehog network members and estrogen signaling in ER positive breast cancer. Here, we summarize findings the data with respect to hedgehog network function in mammary-gland relevant endocrine functions. For this section, please refer to [111] and the references therein.
In the hypothalamus, Shh mediates proliferation, and anterior-posterior and mediolateral patterning. Gli2 and Gli3 are required for different progenitor domains and functions. Currently, no data directly connect hedgehog network genes with the production of the hypothalamic hormones required for mammary gland development; however, we cannot exclude this possibility.
In the pituitary, data suggest that hedgehog network activation mediates hormone secretion. In Dario rerio, Gli1 and 2 are required for the formation of cells secreting growth hormone, cortisol, and prolactin; cyclopamine treatment blocked the formation of some of these lineages. In mammalian systems, SHH stimulates cortisol production in rats, as well as growth hormone and prolactin production in cell lines. PTCH1, PTCH2, and GLI1 protein are expressed in the anterior pituitary, which contains cells producing growth hormone and prolactin. Additionally, pituitary isograft of a wildtype pituitary into Ptch1mes/mes mutants rescues a stunted mammary gland phenotype, suggesting that expression of Ptch1 in the pituitary regulates pubertal estrogen signaling [55]. Patients with pituitary adenomas with altered hormone levels display changes in PTCH2, GLI2, GLI3, and HHIP protein expression. Thus, regulation of the hedgehog network may be critical for the normal function of many cell types and hormone production in the pituitary.
In the adrenal gland, hedgehog network members including Shh, Gli1, Gli2, and Gli3 mRNAs are expressed in the peripheral cortex. Conditional ablation of Shh produced abnormal organization of the gland, and loss of proliferation. Expression patterns suggest paracrine hedgehog signaling in the adrenal gland, with ligand-producing cells in the undifferentiated areas inducing canonical hedgehog signaling in the adjacent mesenchyme. A LacZ reporter showed Shh co-localizes with adrenocortical progenitor cells. At this time, there is no direct evidence of hedgehog signaling required for the production of glucocorticoids in the adrenal cortex, but it seems that regulated hedgehog expression is important for adrenal gland development and homeostasis.
The ovaries produce hormones regulating mammary gland development and a large proportion of breast cancers. All hedgehog ligands, Ptch1, Ptch2, Gli1, and Hhip are expressed in the adult murine ovary. In situ hybridization suggests paracrine signaling from the granulosa cells producing Ihh and Dhh signaling to the adjacent theca layers expressing Ptch1 and Gli1 to regulate theca lineages. Hedgehog ligands can regulate granulosa and theca cell proliferation. SmoM2 expression in the ovary altered follicular development, ovulation, and expression of steroidogenic enzymes by microarray. Loss of the primary cilia by genetic ablation of the intraflagellar transport protein (Ift88) produced abrogated estrous cycling and ovulation, and loss of the corpus luteum- the progesterone producing structure of the ovary. Recently, it was observed that fertility was abrogated with Fsp-Cre mediated ablation of Ptch1; this finding is consistent with previous data suggesting the importance of paracrine hedgehog signaling in the ovary [64]. It is unknown whether the abrogated fertility was due to a function of Ptch1 in the ovary and/or other organs of the hypothalamus-gonadal axis. While direct evidence showing that hedgehog network activity regulates hormone production in the ovary are not established, the defects in ovaries due to altered hedgehog signaling, loss of steroidogenic structures such as the corpus luteum, and altered expression of steroidogenic enzymes strongly suggests that the hedgehog network may be essential for the production of ovarian hormones.
These data provide insight into the connection between hedgehog network expression and hormone receptor positive breast cancer, and the ‘systemic’ roles of hedgehog network genes in mammary gland development. Further studies are necessary to determine the requirement for hedgehog network genes and canonical (or non-canonical) hedgehog activation in specific hormone- producing cell populations, and to understand the ‘systemic’ functions of the hedgehog network impinging on mammary gland development and tumorigenesis.
8. Summary of Hedgehog Network Function in Mammary Gland Development and Breast Cancer
The Hedgehog Network in Mammary Gland Development
With regard to embryonic mammary gland development, the data indicate that inhibition of hedgehog signaling is critical, as is Gli3 function as a repressor in somites. In contrast, synthesizing a model for the hedgehog signaling network in postnatal mammary gland development has been more complex, since data seemed to conflict. One previously published model [112] postulated the following: in the TEB, DHH/IHH ligands abrogate PTCH1 inhibition of SMO, leading to SMO functioning possibly canonically, possibly non-canonically as a GPCR coupling with Gαi proteins, whereas in the mature duct, hedgehog signaling is inactive, PTCH1 inhibits SMO, and GLI3 repressor is expressed. In the mature stroma, it was hypothesized that PTCH1 inhibits SMO, while GLI2 and GLI3 act as repressors.
This model was created prior to the publication of many tissue-specific knockout mutants. Yet, many aspects of this model are supported by newer experimental data. The postulated stromal PTCH1 inhibition of SMO, and GLI2 repressor function supporting normal development still fit with the experimental data. Given the histological data from Fsp-Cre;Ptch1fl/fl, Fsp-Cre;Gli2fl/fl, and Fsp-Cre;SmoM2 mice [64,69], we may still hypothesize that Gli2 functions as a repressor, or non-canonically downstream of TGFβ in the stroma.
The previous model should be revised in the mature epithelium. While the data still indicate that hedgehog signaling is not active, it is now established that Ptch1 functions non-canonically independent of SMO to suppress proliferation and branching [64]. Experimental data support the importance of Smo inhibition in morphogenesis and normal branching; however, rather than inhibition of canonical functions of Smo, it is likely non-canonical function of Smo as a GPCR coupled to Gαi2 [44] that must be inhibited in the normal mature duct. Similar to the embryonic bud, there is no evidence for a function for active canonical hedgehog signaling in the mature mammary gland.
Modulation of hedgehog network members (stromal Ptch1 and SmoM2) perturbs TEBs [64], as does Gli2 loss [57]; however, it remains difficult to define local, stromal functions of these genes in TEB morphogenesis given the ‘systemic’ functions of hedgehog network genes. Returning to the previous model, we should add that stromal Smo inhibition is important for TEB homeostasis, given the disrupted histology of Fsp-Cre;SmoM2 mutants [64]. Whether Smo regulation of TEBs is also via Gαi2 is unknown.
While the data with respect to stemness are not definitive given largely correlative rather than functional data, reports from MMTV-SmoM2, Wap-Shh, and heterozygous Ptch1 animals [60,67,70] suggest that in the murine mammary gland, activated canonical hedgehog signaling promotes stem cell differentiation into a progenitor state. The correlation of activated hedgehog signaling with a progenitor state contrasts with data from human mammospheres, which suggest that autocrine hedgehog signaling regulates stemness. The Fsp-Cre;Gli2fl/fl data introduce the possibility that stromal hedgehog network members impinge on stemness as well; signaling downstream of this Gli2 function is unknown [69].
It is important to consider these data in the context of hedgehog network function in mammotrophic neuro-endocrine organs, which can shape the interpretation of data from mutants where the whole animal has been genetically altered. New data [64,69] show that stromal Gli2 and stromal Ptch1 regulate hormone responsiveness and hormone receptor positivity in the mammary gland, respectively. These functions may both be non-canonical, since there was no evidence of perturbed hormone signaling in Fsp-Cre;SmoM2 mice [64]. The fertility defect displayed by Fsp-Cre;Ptch1fl/fl mice [64], together with previous studies of SmoM2 in the ovary [113,114] and conditional ablation of primary cilia [72] suggest that hedgehog network members regulate mature ovarian biology, in addition to regulating development of the pituitary, adrenal glands, and possibly the hypothalamus [55,111]. Experimentation to clearly define hedgehog network gene function, and the downstream signaling, in hormone production would inform our understanding of these genes in mammary ductal morphogenesis.
Data also suggest that hedgehog network members control stromal homeostasis. Overexpressing positive regulators of hedgehog signaling (including in epithelium) can alter stromal composition; Gli1 and epithelial SmoM2 overexpression increases periductal F4/80+ macrophages [67,68]. Ptch1 loss and Wap-Shh expression elicit stromal hyperplasia [54,60]; in contrast, Fsp-Cre mediated Gli2 ablation causes stromal hypoplasia [69]. SmoM2 or Wap-Shh expression increase ECM deposition [60,67]. These data are important to consider since the stromal compartment is critical for postnatal mammary morphogenesis (as discussed earlier).
Hedgehog Signaling in Breast Cancer
Hedgehog network members are associated with many aspects of breast cancer biology, both tumor-intrinsic and extrinsic (Figure 5). With respect to tumor-intrinsic biology, hedgehog network activation [increased SHH, GLI1, with Ptch1/2 mRNA and PTCH1 protein loss] may be present in breast tumors and involved in tumor initiation; however, genetic activation is generally insufficient to drive tumorigenesis in mouse models (aside from Gli1 overexpression [90]). A subset of breast tumors displays genetic mutations or aberrant methylation of hedgehog network genes (Fig. 4) [78,81]. Upregulated SHH and GLI1 is associated with ER positive tumors in particular, and this expression is associated with proliferation, invasiveness, and anti-estrogen therapy resistance [86,89,92,98,100]. Hedgehog signaling activation may also promote TIC self-renewal [102,103,110]. Gli1 may also be a key factor promoting EMT behaviors via an unknown paracrine factor [49]. While data suggest that hedgehog misregulation may govern critical features of breast tumor biology, further investigation is needed to define the role of hedgehog network members. Additional studies might address whether hedgehog network members function non-canonically in breast tumors, which is a distinct possibility since Smo and Ptch1 function primarily non-canonically in the normal mammary gland [42,44], and Gli1 activation can promote EMT independent of ligands [49]. For instance, a non-canonical role for Smo is consistent with immunostaining from breast tumors where SMO expression correlated with proliferation of neighboring cells via an unknown paracrine mechanism [49].
Figure 5. Putative Functions of Hedgehog Signaling Activation in the Context of Breast Cancer.
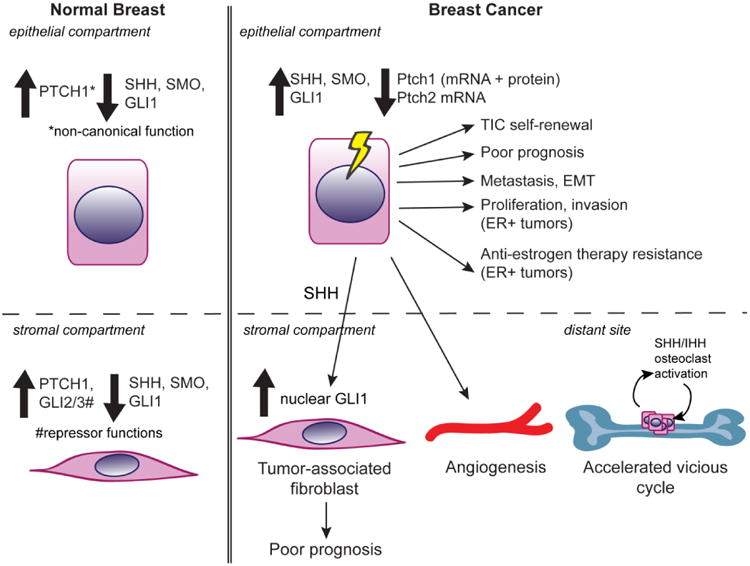
This figure provides an overview of some tumor-intrinsic and tumor-extrinsic roles of hedgehog network members- and hedgehog network activation- related to breast tumorigenesis and patient prognosis, in contrast to hedgehog network expression patterns in the normal breast. SHH, SMO, and GLI1 protein levels are increased in breast tumor cells, while PTCH1 levels are decreased, leading to increased invasion and poorer prognosis, particularly in ER+ breast tumors. Additionally, paracrine activation of Gli1 in breast cancer cells increases invasion and metastasis. Hedgehog misregulation may also significantly impact tumor stroma biology; tumor production of SHH ligands may stimulate angiogenesis and stromal accumulation of nuclear GLI1. Shh/Ihh may drive the vicious cycle in bone metastasis by stimulating osteoclasts.
Newer data suggest that hedgehog network activation in the tumor-adjacent stroma mediates breast tumor progression, invasion, and metastasis [108]. Thus, stromal nuclear GLI1 has potential as a biomarker for predicting patient survival or response. It has not been determined how signaling downstream of hedgehog ligand production by the tumor impacts the stromal compartment to potentially drive more aggressive phenotypes [87,93,115], aside from data indicating that SHH/IHH production by tumor cells can drive the vicious cycle and metastasis to bone [46,105]. Additionally, GLI1/2 transcription factor activation may promote breast cancer metastasis via activation by EMT transcription factor expressing cells [49]. Stromal misregulation of hedgehog network members altering the tumor microenvironment is consistent with the data from postnatal development, where altering expression of hedgehog network members is sufficient to alter stromal constituents.
In brief, data continue to suggest that hedgehog inhibition in breast cancer may be clinically beneficial. Newer data indicate that hedgehog network inhibition may need to target non-canonical functions of hedgehog network members for breast cancer treatment. Thus, testing whether various inhibitors block non-canonical functions of hedgehog network members may be informative. Recent data also suggest that hedgehog inhibition in breast cancer may be beneficial by targeting the stroma, TICs, and/or EMT properties. Functional data with respect to hedgehog signaling and tumor initiation would be informative, given that killing TICs may be critical for enhancing patient survival. Additionally, the association between Shh/ Gli1 expression, ER positive cancer, and tamoxifen resistance suggests that inhibiting estrogen signaling together with hedgehog signaling may be beneficial to patients and bear further investigation. Finally, we should continue to explore the basic biology of hedgehog network member function in the breast tumor associated stroma, perhaps coupled with animal models with tissue-compartment specific manipulation to evaluate whether hedgehog network expression in the stroma increases DCIS incidence, drives tumorigenesis, or could serve as a useful biomarker.
Highlights.
Properly regulated hedgehog network gene function in the mammary epithelium, mammary fat pad stroma, and endocrine organs, is critical for normal mammary ductal morphogenesis and normal proliferation rates.
Upregulated expression of hedgehog network members, including SHH, SMO, and GLI1, together with reduced PTCH1/2 mRNA and PTCH1 protein expression, is associated with tumorigenesis, invasion/metastasis, and TIC (tumor initiating cell) function
SHH/GLI1 upregulation may be particularly associated with ER+ breast tumors.
Data show roles for hedgehog network members in the neuroendocrine signaling axis, but specific roles with respect to mammary gland development and breast cancer are undefined.
Acknowledgments
We thank Dr. Chad Shaw, Lukas M. Simon, and David Henke at Baylor College of Medicine for bioinformatics assistance. We also thank Dr. JoAnne Richards and Dr. Yi Athena Ren at Baylor College of Medicine for feedback on a portion of the manuscript. This work was supported by the National Institutes of Health and National Cancer Institute (RO1 CA127857 to M.T.L. and P30 CA125123 grant for the Dan L. Duncan Cancer Center to C. Kent Osborne).
Abbreviations
- Shh
Sonic Hedgehog
- Ihh
Indian Hedgehog
- Dhh
Desert Hedgehog
- Patched-1
Patched-1
- Smo
Smoothened
- PKA
Protein Kinase A
- SUFU
Suppressor of Fused
- Hhip
Hedgehog Interacting Protein
- Gas1
Growth Arrest Specific protein 1
- TID
Tumorous Imaginal Disc
- GPCR
G Protein-Coupled Receptor
- PTHrP
Parathyroid Related Protein
- TEB
Terminal End Bud
- DCIS
Ductal Carcinoma in situ
- IBC
Invasive Breast Cancer
- TIC
Tumor Initiating Cell
- ER
Estrogen Receptor, EMT, Epithelial-Mesenchymal Transition
- TGFβ
Transforming Growth Factor beta
- Wnt
Wingless
- SFK
Src Family Kinase
- pERK
phospho- Extracellular signal- Regulated Kinase
- TCF
T Cell Factor
- MMTV
mouse mammary tumor virus
- PKC
Protein Kinase C
- MEK-1
Mitogen-Activated Protein Kinase kinase
- CXCR4
Chemokine associated receptor 4
Footnotes
Disclosures: Michael T. Lewis is a founder of, and Limited Partner in StemMed Ltd., and is a Manager of StemMed LLP, its General Partner.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hens JR, Wysolmerski JJ. Key stages of mammary gland development: molecular mechanisms involved in the formation of the embryonic mammary gland. Breast Cancer Res. 2005;7:220–224. doi: 10.1186/bcr1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brisken C, O'Malley B. Hormone action in the mammary gland. Cold Spring Harb Perspect Biol. 2010;2:a003178. doi: 10.1101/cshperspect.a003178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hennighausen L, Robinson GW. Information networks in the mammary gland. Nat Rev Mol Cell Biol. 2005;6:715–725. doi: 10.1038/nrm1714. [DOI] [PubMed] [Google Scholar]
- 4.Wiseman BS, Werb Z. Stromal Effects on Mammary Gland Development and Breast Cancer. Science (80-) 2002;296:1046–1049. doi: 10.1126/science.1067431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Humphreys RC, Krajewska M, Krnacik S, Jaeger R, Weiher H, Krajewski S, Reed JC, Rosen JM. Apoptosis in the terminal endbud of the murine mammary gland: a mechanism of ductal morphogenesis. Development. 1996;122:4013–4022. doi: 10.1242/dev.122.12.4013. [DOI] [PubMed] [Google Scholar]
- 6.Wen HC, Avivar-Valderas A, Sosa MS, Girnius N, Farias EF, Davis RJ, Aguirre-Ghiso Ja. p38α Signaling Induces Anoikis and Lumen Formation During Mammary Morphogenesis. Sci Signal. 2011;4:ra34. doi: 10.1126/scisignal.2001684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Macias H, Hinck L. Mammary gland development. Wiley Interdiscip Rev Dev Biol. 2012;1:533–557. doi: 10.1002/wdev.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hinck L, Silberstein GB. Key stages in mammary gland development: the mammary end bud as a motile organ. Breast Cancer Res. 2005;7:245–51. doi: 10.1186/bcr1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richert MM, Schwertfeger KL, Ryder J, Anderson SM. An Atlas of Mouse Mammary Gland Development. J Mammary Gland Biol Neoplasia. 2000;5:227–241. doi: 10.1023/a:1026499523505. [DOI] [PubMed] [Google Scholar]
- 10.Seagroves TN, Hadsell D, McManaman J, Palmer C, Liao D, McNulty W, Welm B, Wagner KU, Neville M, Johnson RS. HIF1alpha is a critical regulator of secretory differentiation and activation, but not vascular expansion, in the mouse mammary gland. Development. 2003;130:1713–1724. doi: 10.1242/dev.00403. [DOI] [PubMed] [Google Scholar]
- 11.Monks J, Rosner D, Jon Geske F, Lehman L, Hanson L, Neville MC, Fadok VA. Epithelial cells as phagocytes: apoptotic epithelial cells are engulfed by mammary alveolar epithelial cells and repress inflammatory mediator release. Cell Death Differ. 2005;12:107–114. doi: 10.1038/sj.cdd.4401517. [DOI] [PubMed] [Google Scholar]
- 12.O'Brien J, Lyons T, Monks J, Lucia MS, Wilson RS, Hines L, Man YG, Borges V, Schedin P. Alternatively activated macrophages and collagen remodeling characterize the postpartum involuting mammary gland across species. Am J Pathol. 2010;176:1241–1255. doi: 10.2353/ajpath.2010.090735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li M, Liu X, Robinson G, Bar-Peled U, Wagner KU, Young WS, Hennighausen L, Furth Pa. Mammary-derived signals activate programmed cell death during the first stage of mammary gland involution. Proc Natl Acad Sci U S A. 1997;94:3425–3430. doi: 10.1073/pnas.94.7.3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.D'Cruz CM, Moody SE, Master SR, Hartman JL, Keiper EA, Imielinski MB, Cox JD, Wang JY, Ha SI, Keister BA, Chodosh LA. Persistent Parity-Induced Changes in Growth Factors, TGF-β3, and Differentiation in the Rodent Mammary Gland. Mol Endocrinol. 2002;16:2034–2051. doi: 10.1210/me.2002-0073. [DOI] [PubMed] [Google Scholar]
- 15.Briscoe J, Thérond PP. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol. 2013;14:416–29. doi: 10.1038/nrm3598. [DOI] [PubMed] [Google Scholar]
- 16.Zhulyn O, Nieuwenhuis E, Liu YC, Angers S, chung Hui C. Ptch2 shares overlapping functions with Ptch1 in Smo regulation and limb development. Dev Biol. 2015;397:191–202. doi: 10.1016/j.ydbio.2014.10.023. [DOI] [PubMed] [Google Scholar]
- 17.Roberts B, Casillas C, Alfaro AC, Jägers C, Roelink H. Patched1 and Patched2 inhibit Smoothened non-cell autonomously. Elife. 2016;5:e17634. doi: 10.7554/eLife.17634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martín V, Carrillo G, Torroja C, Guerrero I. The sterol-sensing domain of patched protein seems to control smoothened activity through patched vesicular trafficking. Curr Biol. 2001;11:601–607. doi: 10.1016/s0960-9822(01)00178-6. [DOI] [PubMed] [Google Scholar]
- 19.Chang K, Creighton CJ, Davis C, Donehower L, Drummond J, Wheeler D, Ally A, Balasundaram M, Birol I, Butterfield YSN, Chu A, Chuah E, Chun HJE, Dhalla N, Guin R, Hirst M, Hirst C, Holt RA, Jones SJM, Lee D, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Price MA, Kalderon D. Proteolysis of the Hedgehog Signaling Effector Cubitus interruptus Requires Phosphorylation by Glycogen Synthase Kinase 3 and Casein Kinase 1. Cell. 2002;108:823–835. doi: 10.1016/s0092-8674(02)00664-5. [DOI] [PubMed] [Google Scholar]
- 21.Kogerman P, Grimm T, Kogerman L, Krause D, Undén aB, Sandstedt B, Toftgård R, Zaphiropoulos PG. Mammalian suppressor-of-fused modulates nuclear-cytoplasmic shuttling of Gli-1. Nat Cell Biol. 1999;1:312–319. doi: 10.1038/13031. [DOI] [PubMed] [Google Scholar]
- 22.Huntzicker EG, Estay IS, Zhen H, Lokteva La, Jackson PK, Oro AE. Dual degradation signals control Gli protein stability and tumor formation Email alerting service Dual degradation signals control Gli protein stability and tumor formation. Genes Dev. 2006;20:276–281. doi: 10.1101/gad.1380906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robbins DJ, Fei DL, Riobo Na. The Hedgehog signal transduction network. Sci Signal. 2012;5:1–13. doi: 10.1126/scisignal.2002906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Izzi L, Lévesque M, Morin S, Laniel D, Wilkes BC, Mille F, Krauss RS, McMahon AP, Allen BL, Charron F. Boc and gas1 each form distinct shh receptor complexes with ptch1 and are required for shh-mediated cell proliferation. Dev Cell. 2011;20:788–801. doi: 10.1016/j.devcel.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu A, Wang B, Niswander LA. Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development. 2005;132:3103–3111. doi: 10.1242/dev.01894. [DOI] [PubMed] [Google Scholar]
- 26.Hebrok M, Kim SK, St Jacques B, McMahon AP, Melton DA. Regulation of pancreas development by hedgehog signaling. Development. 2000;127:4905–4913. doi: 10.1242/dev.127.22.4905. [DOI] [PubMed] [Google Scholar]
- 27.Wang BE, Shou J, Ross S, Koeppen H, De Sauvage FJ, Gao WQ. Inhibition of epithelial ductal branching in the prostate by sonic hedgehog is indirectly mediated by stromal cells. J Biol Chem. 2003;278:18506–18513. doi: 10.1074/jbc.M300968200. [DOI] [PubMed] [Google Scholar]
- 28.Bellusci S, Furuta Y, Rush MG, Henderson R, Winnier G, Hogan BL. Involvement of Sonic hedgehog (Shh) in mouse embryonic lung growth and morphogenesis. Development. 1997;124:53–63. doi: 10.1242/dev.124.1.53. [DOI] [PubMed] [Google Scholar]
- 29.Jessell TM. Neuronal specification in the spinal cord: inductive signals and transcriptional codes. Nat Rev Genet. 2000;1:20–29. doi: 10.1038/35049541. [DOI] [PubMed] [Google Scholar]
- 30.Adolphe C, Hetherington R, Ellis T, Wainwright B. Patched1 functions as a gatekeeper by promoting cell cycle progression. Cancer Res. 2006;66:2081–8. doi: 10.1158/0008-5472.CAN-05-2146. [DOI] [PubMed] [Google Scholar]
- 31.Lai K, Kaspar BK, Gage FH, Schaffer DV. Sonic hedgehog regulates adult neural progenitor proliferation in vitro and in vivo. Nat Neurosci. 2003;6:21–27. doi: 10.1038/nn983. [DOI] [PubMed] [Google Scholar]
- 32.Petrova R, Joyner La. Roles for Hedgehog signaling in adult organ homeostasis and repair. Development. 2014;141:3445–3457. doi: 10.1242/dev.083691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gorlin RJ. Nevoid Basal Cell Carcinoma Syndrome. Medicine (Baltimore) 1987;66:98–113. doi: 10.1097/00005792-198703000-00002. [DOI] [PubMed] [Google Scholar]
- 34.Hutchin ME, Kariapper MS, Grachtchouk M, Wang A, Wei L, Cummings D, Liu J, Michael LE, Glick A, Dlugosz AA. Sustained Hedgehog signaling is required for basal cell carcinoma proliferation and survival: conditional skin tumorigenesis recapitulates the hair growth cycle. Genes Dev. 2005;19:214–223. doi: 10.1101/gad.1258705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Evans DG, Ladusans EJ, Rimmer S, Burnell LD, Thakker N, Farndon Pa. Complications of the naevoid basal cell carcinoma syndrome: results of a population based study. J Med Genet. 1993;30:460–464. doi: 10.1136/jmg.30.6.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goodrich LV, Milenković L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- 37.Kalff-Suske M, Wild a, Topp J, Wessling M, Jacobsen EM, Bornholdt D, Engel H, Heuer H, Aalfs CM, Ausems MG, Barone R, Herzog a, Heutink P, Homfray T, Gillessen-Kaesbach G, König R, Kunze J, Meinecke P, Müller D, Rizzo R, et al. Point mutations throughout the GLI3 gene cause Greig cephalopolysyndactyly syndrome. Hum Mol Genet. 1999;8:1769–77. doi: 10.1093/hmg/8.9.1769. [DOI] [PubMed] [Google Scholar]
- 38.Fan L, Pepicelli CV, Dibble CC, Catbagan W, Zarycki JL, Laciak R, Gipp J, Shaw A, Lamm MLG, Munoz A, Lipinski R, Thrasher JB, Bushman W. Hedgehog signaling promotes prostate xenograft tumor growth. Endocrinology. 2004;145:3961–70. doi: 10.1210/en.2004-0079. [DOI] [PubMed] [Google Scholar]
- 39.Jenkins D. Hedgehog signalling: Emerging evidence for non-canonical pathways. Cell Signal. 2009;21:1023–1034. doi: 10.1016/j.cellsig.2009.01.033. [DOI] [PubMed] [Google Scholar]
- 40.Brennan D, Chen X, Cheng L, Mahoney M, Riobo N. Noncanonical Hedgehog Siganling. Vitam Horm. 2012;88:55–72. doi: 10.1016/B978-0-12-394622-5.00003-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jeong J, McMahon AP. Growth and pattern of the mammalian neural tube are governed by partially overlapping feedback activities of the hedgehog antagonists patched 1 and Hhip1. Development. 2005;132:143–154. doi: 10.1242/dev.01566. [DOI] [PubMed] [Google Scholar]
- 42.Chang H, Li Q, Moraes RC, Lewis MT, Hamel Pa. Activation of Erk by sonic hedgehog independent of canonical hedgehog signalling. Int J Biochem Cell Biol. 2010;42:1462–1471. doi: 10.1016/j.biocel.2010.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klein C, Zwick A, Kissel S, Forster CU, Pfeifer D, Follo M, Illert AL, Decker S, Benkler T, Pahl H, Oostendorp RAJ, Aumann K, Duyster J, Dierks C. Ptch2 loss drives myeloproliferation and myeloproliferative neoplasm progression. J Exp Med. 2016;213:273–90. doi: 10.1084/jem.20150556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Villanueva H, Visbal AP, Obeid NF, Ta AQ, Faruki AA, Wu M, Hilsenbeck SG, Shaw CA, Yu P, Plummer NW, Birnbaumer L, Lewis MT. An essential role for G a i2 in Smoothened-stimulated epithelial cell proliferation in the mammary gland. Sci Signal. 2015;8:1–11. doi: 10.1126/scisignal.aaa7355. [DOI] [PubMed] [Google Scholar]
- 45.Li X, Ma Q, Xu Q, Liu H, Lei J, Duan W, Bhat K, Wang F, Wu E, Wang Z. SDF-1/CXCR4 signaling induces pancreatic cancer cell invasion and epithelial-mesenchymal transition in vitro through non-canonical activation of the Hedgehog pathway. Cancer Lett. 2012;322:169–176. doi: 10.1016/j.canlet.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johnson RW, Nguyen MP, Padalecki SS, Grubbs BG, Merkel AR, Oyajobi BO, Matrisian LM, Mundy GR, Sterling JA. TGF-beta promotion of Gli2-induced expression of parathyroid hormone-related protein, an important osteolytic factor in bone metastasis, is independent of canonical Hedgehog signaling. Cancer Res. 2011;71:822–831. doi: 10.1158/0008-5472.CAN-10-2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Riobo NA, Haines GM, Emerson CP. Protein kinase C-?? and mitogen-activated protein/extracellular signal-regulated kinase-1 control GLI activation in hedgehog signaling. Cancer Res. 2006;66:839–845. doi: 10.1158/0008-5472.CAN-05-2539. [DOI] [PubMed] [Google Scholar]
- 48.Zhou M, Hou Y, Yang G, Zhang H, Tu G, Du Y, Wen S, Xu L, Tang X, Tang S, Yang L, Cui X, Liu M. LncRNA-Hh Strengthen Cancer Stem Cells Generation in Twist-Positive Breast Cancer via Activation of Hedgehog Signaling Pathway. Stem Cells. 2015:n/a–n/a. doi: 10.1002/stem.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Neelakantan D, Zhou H, Oliphant MU, Zhang X, Simon LM, Henke DM, Shaw CA, Wu M, Hilsenbeck SG, White LD, Lewis MT, Ford H. Cooperativity between EMT and non-EMT cells promotes breast cancer metastasis via paracrine GLI activation. Nat Commun. 2017 doi: 10.1038/ncomms15773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lauth M, Bergström Å, Shimokawa T, Tostar U, Jin Q, Fendrich V, Guerra C, Barbacid M, Toftgård R. DYRK1B-dependent autocrine-to-paracrine shift of Hedgehog signaling by mutant RAS. Nat Struct Mol Biol. 2010;17:718–725. doi: 10.1038/nsmb.1833. [DOI] [PubMed] [Google Scholar]
- 51.Alvarez-Medina R, Cayuso J, Okubo T, Takada S, Martí E. Wnt canonical pathway restricts graded Shh/Gli patterning activity through the regulation of Gli3 expression. Development. 2008;135:237–247. doi: 10.1242/dev.012054. [DOI] [PubMed] [Google Scholar]
- 52.Michno K, Boras-Granic K, Mill P, Hui CC, Hamel Pa. Shh expression is required for embryonic hair follicle but not mammary gland development. Dev Biol. 2003;264:153–165. doi: 10.1016/s0012-1606(03)00401-9. [DOI] [PubMed] [Google Scholar]
- 53.Gallego MI, Binart N, Robinson GW, Okagaki R, Coschigano KT, Perry J, Kopchick JJ, Oka T, Kelly PA, Hennighausen L. Prolactin, growth hormone, and epidermal growth factor activate Stat5 in different compartments of mammary tissue and exert different and overlapping developmental effects. Dev Biol. 2001;229:163–175. doi: 10.1006/dbio.2000.9961. [DOI] [PubMed] [Google Scholar]
- 54.Lewis MT, Ross S, Strickland PA, Sugnet CW, Jimenez E, Scott MP, Daniel CW. Defects in mouse mammary gland development caused by conditional haploinsufficiency of Patched-1. Development. 1999;126:5181–5193. doi: 10.1242/dev.126.22.5181. [DOI] [PubMed] [Google Scholar]
- 55.Moraes RC, Chang H, Harrington N, Landua JD, Prigge JT, Lane TF, Wainwright BJ, Hamel PA, Lewis MT. Ptch1 is required locally for mammary gland morphogenesis and systemically for ductal elongation. Development. 2009;136:1423–1432. doi: 10.1242/dev.023994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hatsell SJ, Cowin P. Gli3-mediated repression of Hedgehog targets is required for normal mammary development. Development. 2006;133:3661–3670. doi: 10.1242/dev.02542. [DOI] [PubMed] [Google Scholar]
- 57.Lewis MT, Ross S, Strickland Pa, Sugnet CW, Jimenez E, Hui C, Daniel CW. The Gli2 transcription factor is required for normal mouse mammary gland development. Dev Biol. 2001;238:133–44. doi: 10.1006/dbio.2001.0410. [DOI] [PubMed] [Google Scholar]
- 58.Veltmaat JM, Relaix F, Le LT, Kratochwil K, Sala FG, van Veelen W, Rice R, Spencer-Dene B, Mailleux Aa, Rice DP, Thiery JP, Bellusci S. Gli3-mediated somitic Fgf10 expression gradients are required for the induction and patterning of mammary epithelium along the embryonic axes. Development. 2006;133:2325–2335. doi: 10.1242/dev.02394. [DOI] [PubMed] [Google Scholar]
- 59.Gallego MI, Beachy Pa, Hennighausen L, Robinson GW. Differential requirements for shh in mammary tissue and hair follicle morphogenesis. Dev Biol. 2002;249:131–139. doi: 10.1006/dbio.2002.0761. [DOI] [PubMed] [Google Scholar]
- 60.García-Zaragoza E, Pérez-Tavarez R, Ballester A, Lafarga V, Jiménez-Reinoso A, Ramírez Á, Murillas R, Gallego MI. Intraepithelial paracrine Hedgehog signaling induces the expansion of ciliated cells that express diverse progenitor cell markers in the basal epithelium of the mouse mammary gland. Dev Biol. 2012;372:28–44. doi: 10.1016/j.ydbio.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 61.Kouros-Mehr H, Werb Z. Candidate regulators of mammary branching morphogenesis identified by genome-wide transcript analysis. Dev Dyn. 2006;235:3404–3412. doi: 10.1002/dvdy.20978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Makino S, Masuya H, Ishijima J, Yada Y, Shiroishi T. A spontaneous mouse mutation, mesenchymal dysplasia (mes), is caused by a deletion of the most C-terminal cytoplasmic domain of patched (ptc) Dev Biol. 2001;239:95–106. doi: 10.1006/dbio.2001.0419. [DOI] [PubMed] [Google Scholar]
- 63.Chang H, Balenci L, Okolowsky N, Muller WJ, Hamel PA. Mammary epithelial-restricted expression of activated c-src rescues the block to mammary gland morphogenesis due to the deletion of the C-terminus of Patched-1. Dev Biol. 2012;370:187–197. doi: 10.1016/j.ydbio.2012.07.027. [DOI] [PubMed] [Google Scholar]
- 64.Monkkonen T, Landua JD, Visbal AP, Lewis MT. Epithelial and non-epithelial Patched-1 (Ptch1) play opposing roles to regulate proliferation and morphogenesis of the mouse mammary gland. Development. 2017;144:1317–1327. doi: 10.1242/dev.140434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mao J, Ligon KL, Rakhlin EY, Thayer SP, Bronson RT, Rowitch D, McMahon AP. A novel somatic mouse model to survey tumorigenic potential applied to the Hedgehog pathway. Cancer Res. 2006;66:10171–10178. doi: 10.1158/0008-5472.CAN-06-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Moraes RC, Zhang X, Harrington N, Fung JY, Wu MF, Hilsenbeck SG, Allred DC, Lewis MT. Constitutive activation of smoothened (SMO) in mammary glands of transgenic mice leads to increased proliferation, altered differentiation and ductal dysplasia. Development. 2007;134:1231–1242. doi: 10.1242/dev.02797. [DOI] [PubMed] [Google Scholar]
- 67.Visbal AP, LaMarca HL, Villanueva H, Toneff MJ, Li Y, Rosen JM, Lewis MT. Altered differentiation and paracrine stimulation of mammary epithelial cell proliferation by conditionally activated Smoothened. Dev Biol. 2011;352:116–127. doi: 10.1016/j.ydbio.2011.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fiaschi M, Rozell B, Bergstrom A, Toftgard R, Kleman MI. Targeted expression of GLI1 in the mammary gland disrupts pregnancy-induced maturation and causes lactation failure. J Biol Chem. 2007;282:36090–36101. doi: 10.1074/jbc.M704280200. [DOI] [PubMed] [Google Scholar]
- 69.Zhao C, Cai S, Shin K, Lim A, Kalisky T, Lu WJ, Clarke MF, Beachy PA. Stromal Gli2 activity coordinates a niche signaling program for mammary epithelial stem cells. Science (80-) 2017;3485 doi: 10.1126/science.aal3485. [DOI] [PubMed] [Google Scholar]
- 70.Li N, Singh S, Cherukuri P, Li H, Yuan Z, Ellisen LW, Wang B, Robbins D, DiRenzo J. Reciprocal intraepithelial interactions between TP63 and hedgehog signaling regulate quiescence and activation of progenitor elaboration by mammary stem cells. Stem Cells. 2008;26:1253–1264. doi: 10.1634/stemcells.2007-0691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McDermott KM, Liu BY, Tlsty TD, Pazour GJ. Primary Cilia Regulate Branching Morphogenesis during Mammary Gland Development. Curr Biol. 2010;20:731–737. doi: 10.1016/j.cub.2010.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Johnson ET, Nicola T, Roarty K, Yoder BK, Haycraft CJ, Serra R. Role for primary cilia in the regulation of mouse ovarian function. Dev Dyn. 2008;237:2053–2060. doi: 10.1002/dvdy.21612. [DOI] [PubMed] [Google Scholar]
- 73.Xie J, Johnson RL, Zhang X, Bare JW, Waldman FM, Cogen PH, Menon AG, Warren RS, Chen LC, Scott MP, Epstein EH. Mutations of the PATCHED gene in several types of sporadic extracutaneous tumors. Cancer Res. 1997;57:2369–2372. [PubMed] [Google Scholar]
- 74.Adolphe C, Narang M, Ellis T, Wicking C, Kaur P, Wainwright B. An in vivo comparative study of sonic, desert and Indian hedgehog reveals that hedgehog pathway activity regulates epidermal stem cell homeostasis. Development. 2004;131:5009–5019. doi: 10.1242/dev.01367. [DOI] [PubMed] [Google Scholar]
- 75.Yuan K, Frolova N, Xie Y, Wang D, Cook L, Kwon YJ, Steg AD, Serra R, Frost AR. Primary cilia are decreased in breast cancer: analysis of a collection of human breast cancer cell lines and tissues. J Histochem Cytochem. 2010;58:857–870. doi: 10.1369/jhc.2010.955856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu S, Dontu G, Mantle ID, Patel S, Ahn NS, Jackson KW, Suri P, Wicha MS. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006;66:6063–6071. doi: 10.1158/0008-5472.CAN-06-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Goodrich LV, Milenković L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science (80-) 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- 78.Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, Zhang H, McLellan M, Yau C, Kandoth C, Bowlby R, Shen H, Hayat S, Fieldhouse R, Lester SC, Tse GMK, Factor RE, Collins LC, Allison KH, Chen YY, et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell. 2015;163:506–519. doi: 10.1016/j.cell.2015.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, Gräf S, Ha G, Haffari G, Bashashati A, Russell R, McKinney S, Langerød A, Green A, Provenzano E, Wishart G, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–52. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pereira B, Chin SF, Rueda OM, Vollan HKM, Provenzano E, Bardwell HA, Pugh M, Jones L, Russell R, Sammut SJ, Tsui DWY, Liu B, Dawson SJ, Abraham J, Northen H, Peden JF, Mukherjee A, Turashvili G, Green AR, McKinney S, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 2016;7:11479. doi: 10.1038/ncomms11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wolf I, Bose S, Desmond JC, Lin BT, Williamson Ea, Karlan BY, Koeffler HP. Unmasking of epigenetically silenced genes reveals DNA promoter methylation and reduced expression of PTCH in breast cancer. Breast Cancer Res. 2007;105:139–55. doi: 10.1007/s10549-006-9440-4. [DOI] [PubMed] [Google Scholar]
- 82.Nessling M, Richter K, Schwaenen C, Roerig P, Wrobel G, Wessendorf S, Fritz B, Bentz M, Sinn HP, Radlwimmer B, Lichter P. Candidate genes in breast cancer revealed by microarray-based comparative genomic hybridization of archived tissue. Cancer Res. 2005;65:439–447. [PubMed] [Google Scholar]
- 83.Vorechovsky I, Benediktsson KP, Toftgard R. The patched/hedgehog/smoothened signalling pathway in human breast cancer: no evidence for H133Y SHH, PTCH and SMO mutations. Eur J Cancer. 1999;35:711–713. doi: 10.1016/s0959-8049(99)00017-9. [DOI] [PubMed] [Google Scholar]
- 84.Bieche I, Lerebours F, Espie M, Marty M, Lidereau R. Molecular Profiling of Inflammatory Breast Cancer : Identification of a Poor-Prognosis Gene Expression Signature. Clin Cancer Res. 2004;10:6789–6795. doi: 10.1158/1078-0432.CCR-04-0306. [DOI] [PubMed] [Google Scholar]
- 85.Mukherjee S, Frolova N. Hedgehog signaling and response to cyclopamine differs in epithelial and stromal cells in benign breast and breast cancer. Cancer Biol …. 2006;5:674–683. doi: 10.4161/cbt.5.6.2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Souzaki M, Kubo M, Kai M, Kameda C, Tanaka H, Taguchi T, Tanaka M, Onishi H, Katano M. Hedgehog signaling pathway mediates the progression of non-invasive breast cancer to invasive breast cancer. Cancer Sci. 2011;102:373–81. doi: 10.1111/j.1349-7006.2010.01779.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cao X, Geradts J, Dewhirst M, Lo HW. Upregulation of VEGF-A and CD24 Gene Expression by the tGLI1 Transcription Factor Contributes to the Aggressive Behavior of Breast Cancer Cells. Oncogene. 2012;31:104–115. doi: 10.1038/onc.2011.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kubo M, Nakamura M, Tasaki A, Yamanaka N, Nakashima H, Nomura M, Kuroki S, Katano M. Hedgehog signaling pathway is a New Therapeutic Target for Patients with Breast Cancer. Cancer Res. 2004;64:6071–6074. doi: 10.1158/0008-5472.CAN-04-0416. [DOI] [PubMed] [Google Scholar]
- 89.Xuan Y, Lin Z. Expression of Indian Hedgehog signaling molecules in breast cancer. J Cancer Res Clin Oncol. 2009;135:235–40. doi: 10.1007/s00432-008-0451-x. [DOI] [PubMed] [Google Scholar]
- 90.Fiaschi M, Rozell B, Bergström A, Toftgård R. Development of mammary tumors by conditional expression of GLI1. Cancer Res. 2009;69:4810–7. doi: 10.1158/0008-5472.CAN-08-3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kwon YJ, Hurst DR, Steg AD, Yuan K, Vaidya KS, Welch DR, Frost AR. Gli1 enhances migration and invasion via up-regulation of MMP-11 and promotes metastasis in ERα negative breast cancer cell lines. Clin Exp Metastasis. 2011;28:437–449. doi: 10.1007/s10585-011-9382-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhao J, Chen G, Cao D, Li Y, Diao F, Cai H, Jin Y, Lu J. Expression of Gli1 correlates with the transition of breast cancer cells to estrogen-independent growth. Breast Cancer Res Treat. 2010;119:39–51. doi: 10.1007/s10549-009-0323-3. [DOI] [PubMed] [Google Scholar]
- 93.Harris LG, Pannell LK, Singh S, Samant RS, Shevde LA. Increased vascularity and spontaneous metastasis of breast cancer by hedgehog signaling mediated upregulation of cyr61. Oncogene. 2012;31:3370–3380. doi: 10.1038/onc.2011.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Koga K, Nakamura M, Nakashimja H, Akiyoshi T, Kubo M, Sato H, Kuroki S, Nomura M, Tanaka M, Katano M. Novel Link between Estrogen Receptor α and Hedgehog Pathway in Breast Cancer. Anticancer Res. 2008;28:731–739. [PubMed] [Google Scholar]
- 95.Teissedre B, Pinderhughes A, Incassati A, Hatsell SJ, Hiremath M, Cowin P. MMTV-Wnt1 and -ΔN89β-catenin induce canonical signaling in distinct progenitors and differentially activate hedgehog signaling within mammary tumors. PLoS One. 2009;4 doi: 10.1371/journal.pone.0004537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kim SW, Chao TH, Xiang R, Lo JF, Campbell MJ, Fearns C, Lee JD. Tid1, the human homologue of a Drosophila tumor suppressor, reduces the malignant activity of ErbB-2 in carcinoma cells. Cancer Res. 2004;64:7732–7739. doi: 10.1158/0008-5472.CAN-04-1323. [DOI] [PubMed] [Google Scholar]
- 97.Chang-Claude J, Dunning A, Schnitzbauer U, Galmbacher P, Tee L, Wjst M, Chalmers J, Zemzoum I, Harbeck N, Pharoah PD, Hahn H. The patched polymorphism Pro1315Leu (C3944T) may modulate the association between use of oral contraceptives and breast cancer risk. Int J Cancer. 2003;103:779–783. doi: 10.1002/ijc.10889. [DOI] [PubMed] [Google Scholar]
- 98.Sun Y, Wang Y, Fan C, Gao P, Wang X, Wei G, Wei J. Estrogen promotes stemness and invasiveness of ER-positive breast cancer cells through Gli1 activation. Mol Cancer. 2014;13:137. doi: 10.1186/1476-4598-13-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Xu L, Kwon YJ, Frolova N, Steg AD, Yuan K, Johnson MR, Grizzle WE, Desmond RA, Frost AR. Gli1 promotes cell survival and is predictive of a poor outcome in ERalpha-negative breast cancer. Breast Cancer Res Treat. 2010;123:59–71. doi: 10.1007/s10549-009-0617-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Xu L, Kwon YJ, Frolova N, Steg AD, Yuan K. Gli1 promotes cell survival and is predictive of a poor outcome in ERa-negative breast cancer. Breast Cancer Res Treat. 2010;123:59–71. doi: 10.1007/s10549-009-0617-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, Rimm DL, Wong H, Rodriguez A, Herschkowitz JI, Fan C, Zhang X, He X, Pavlick A, Gutierrez MC, Renshaw L, Larionov Aa, Faratian D, Hilsenbeck SG, Perou CM, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci U S A. 2009;106:13820–13825. doi: 10.1073/pnas.0905718106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tanaka H, Nakamura M, Kameda C, Kubo M, Sato N, Kuroki S, Tanaka M, Katano M. The Hedgehog signaling pathway plays an essential role in maintaining the CD44+CD24-/low subpopulation and the side population of breast cancer cells. Anticancer Res. 2009;29:2147–2157. [PubMed] [Google Scholar]
- 103.Memmi EM, Sanarico AG, Giacobbe A, Peschiaroli A, Frezza V, Cicalese A, Pisati F, Tosoni D, Zhou H, Tonon G, Antonov A, Melino G, Pelicci PG, Bernassola F. p63 Sustains self-renewal of mammary cancer stem cells through regulation of Sonic Hedgehog signaling. Proc Natl Acad Sci U S A. 2015;112:3499–504. doi: 10.1073/pnas.1500762112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Heller E, Hurchla Ma, Xiang J, Su X, Chen S, Schneider J, Joeng KS, Vidal M, Goldberg L, Deng H, Hornick MC, Prior JL, Piwnica-Worms D, Long F, Cagan R, Weilbaecher KN. Hedgehog signaling inhibition blocks growth of resistant tumors through effects on tumor microenvironment. Cancer Res. 2012;72:897–907. doi: 10.1158/0008-5472.CAN-11-2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pratap J, Wixted JJ, Gaur T, Zaidi SK, Dobson J, Gokul KD, Hussain S, van Wijnen AJ, Stein JL, Stein GS, Lian JB. Runx2 transcriptional activation of Indian Hedgehog and a downstream bone metastatic pathway in breast cancer cells. Cancer Res. 2008;68:7795–802. doi: 10.1158/0008-5472.CAN-08-1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Das S, Samant RS, Shevde La. Hedgehog signaling induced by breast cancer cells promotes osteoclastogenesis and osteolysis. J Biol Chem. 2011;286:9612–9622. doi: 10.1074/jbc.M110.174920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sterling JA. The Hedgehog Signaling Molecule Gli2 Induces Parathyroid Hormone-Related Peptide Expression and Osteolysis in Metastatic Human Breast Cancer Cells. Cancer Res. 2006;66:7548–7553. doi: 10.1158/0008-5472.CAN-06-0452. [DOI] [PubMed] [Google Scholar]
- 108.O'Toole SA, Machalek DA, Shearer RF, Millar EK, Nair R, Schofield P, McLeod D, Cooper CL, McNeil CM, McFarland A, Nguyen A, Ormandy CJ, Qiu MR, Rabinovich B, Martelotto LG, Vu D, Hannigan GE, Musgrove EA, Christ D, Sutherland RL, et al. Hedgehog overexpression is associated with stromal interactions and predicts for poor outcome in breast cancer. Cancer Res. 2011;71:4002–4014. doi: 10.1158/0008-5472.CAN-10-3738. [DOI] [PubMed] [Google Scholar]
- 109.Ramaswamy B, Lu Y, Teng KY, Nuovo G, Li X, Shapiro CL, Majumder S. Hedgehog signaling is a novel therapeutic target in tamoxifen-resistant breast cancer aberrantly activated by PI3K/AKT pathway. Cancer Res. 2012;72:5048–5059. doi: 10.1158/0008-5472.CAN-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Liu S, Duan X, Xu L, Ye J, Cheng Y, Liu Q, Zhang H, Zhang S, Zhu S, Li T, Liu Y. Nuclear Gli1 expression is associated with pathological complete response and event-free survival in HER2-positive breast cancer treated with trastuzumab-based neoadjuvant therapy. Tumor Biol. 2015 doi: 10.1007/s13277-015-4325-y. [DOI] [PubMed] [Google Scholar]
- 111.Huang CC, Yao HHC. Diverse Functions of Hedgehog Signaling in Formation and Phyisology of Steroidogenic Organs. Mol Reprod Dev. 2010;77:489–496. doi: 10.1002/mrd.21174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Visbal AP, Lewis MT. Hedgehog Signaling in Mammary Gland Development and Breast Cancer. Springer; London: 2011. [Google Scholar]
- 113.Ren Y, Cowan RG, Harman RM, Quirk SM. Dominant activation of the hedgehog signaling pathway in the ovary alters theca development and prevents ovulation. Mol Endocrinol. 2009;23:711–723. doi: 10.1210/me.2008-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ren Y, Cowan RG, Migone FF, Quirk SM. Overactivation of hedgehog signaling alters development of the ovarian vasculature in mice. Biol Reprod. 2012;86:174. doi: 10.1095/biolreprod.112.099176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chinchilla P, Xiao L, Kazanietz MG, Riobo Na. Hedgehog proteins activate pro-angiogenic responses in endothelial cells through non-canonical signaling pathways. Cell Cycle. 2014;9:570–579. doi: 10.4161/cc.9.3.10591. [DOI] [PubMed] [Google Scholar]