

Abstract
During the last decade, the use of small molecule (MW< 500 Dalton) compounds that modulate (inhibit or activate) important proteins of different biological pathways became widespread. Recently, the homologous recombination (HR) pathway emerged as a target for such modulators. Development of small molecule modulators pursues two distinct but not mutually exclusive purposes: to create a research tool to study the activities or functions of proteins of interest and to produce drugs targeting specific pathologies. Here, we review the progress of small molecule development in the area of HR.
HR plays a critical role in repair of the most harmful types of DNA lesions, DNA double-stranded breaks (DSBs) and interstrand cross-links (ICLs), in faithful segregation of homologous chromosomes during meiosis, and in telomere maintenance 1. However, the mechanisms of HR, especially in mammals, remain to be understood. Traditional genetic analysis of the HR pathway in mammals is complicated because most of HR proteins are essential for viability. However this impediment also provides an impetus for development of small molecule modulators as an alternative approach to investigate the mechanisms of HR. Since many anticancer chemotherapies or radiotherapy act by inducing DSBs or ICLs the modulators of HR may also have important clinical applications. They may either sensitize cancer cells to anticancer therapies by inhibiting HR, or provide protection to normal cells against these therapies by stimulating HR. Because of the intrinsic genome instability of cancer cells they often accumulate mutations in various DNA repair pathways. The loss of alternative DNA repair pathways could make HR essential for cancer cell viability rendering them vulnerable for HR inhibitors even in the absence of therapeutic agents. Conversely, in cancers that are deficient in HR the alternative DNA repair pathways could become essential for cell viability. Thus, Poly-ADP-ribose polymerase 1 (PARP1), which plays a signaling role in DNA Base Excision Repair pathway, is essential for viability of familial breast cancer cells deficient in HR proteins BRCA1&2 2. As a result, these cells are sensitive to inhibitors of PARP1.
HR performs DNA repair through an essentially error-free mechanism by employing homologous DNA as a template (Fig. 1)3. The initial step of HR involves exonucleolytic processing of the DNA ends into a resected DNA duplex with protruding 3′-ssDNA tails (Fig. 1)4. Then, RAD51 recombinase loads onto the ssDNA to form a contiguous helical nucleoprotein filament that promotes a search for the homologous dsDNA and exchange of DNA strands that results in formation of joint molecules 5 Joint molecules, key intermediates of HR, provide both a template and a primer for the DNA synthesis to generate an overlap with another end of broken DNA for the subsequent re-joining of DSB ends. The joint molecules are processed through either of the two mechanisms, synthesis-dependent strand annealing (SDSA) and DNA double-strand break repair (DSBR) 3. In the SDSA mechanism, the joint molecules dissociate after DNA synthesis (Fig. 1a). After their dissociation the released 3′-ssDNA end rejoins with the second end of the broken chromosome. It is thought that this mechanism takes place in both mitotically and meiotically dividing cells and produces non-crossover recombinants. The DSBR mechanism that operates in meiosis involves the second end capture, formation of double Holliday junctions that are resolved at a later step by structure-specific endonucleases leading to crossing over which is crucial for proper segregation of homologous chromosomes in meiotic prophase I (Fig. 1b) 6.
Figure 1. Two major HR pathways.
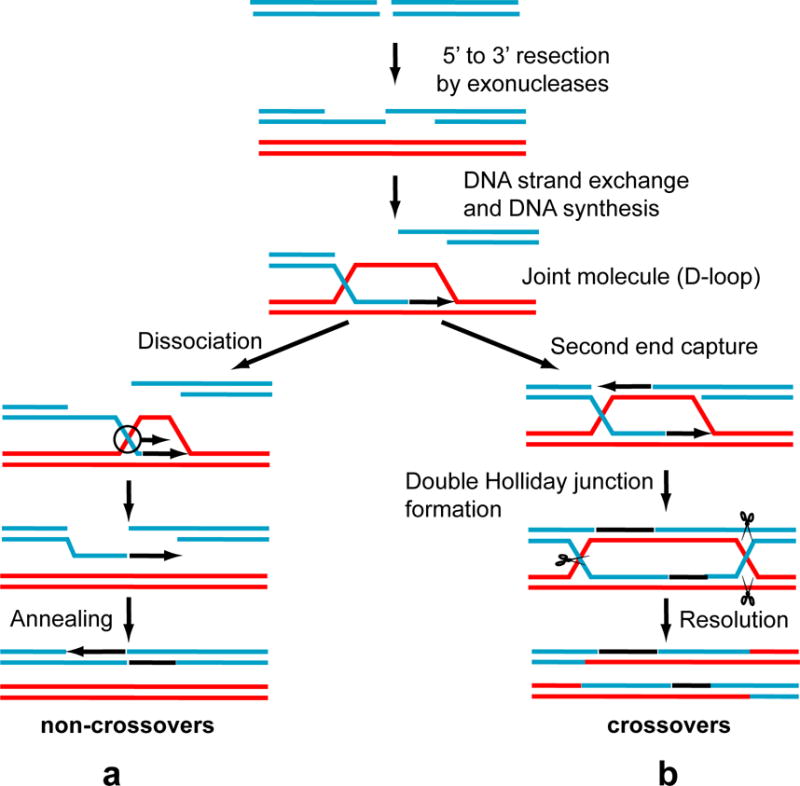
Formation of non-crossovers (a) and crossovers (b) occurring in mitotically and in meiotically dividing cells.
In eukaryotes, the HR genes were discovered by genetic screens of Saccharomyces cerevisiae for mutants conferring strong ionizing radiation sensitivity and moderate ultraviolet (UV) light sensitivity 7. In mammals, the core of the HR pathway includes RAD51, RAD51 paralogs (XRCC2, XRCC3, Rad51B, Rad51C, and Rad51D), RAD52, RAD54, RAD54 paralog RAD54B, BRCA1 and BRCA2, breast cancer susceptibility proteins 1 and 2; RAD50, MRE11, and NBS1 which form a complex (MRN) 4. Rad51 is highly evolutionarily conserved and ubiquitous in all kingdoms of life 8. Most other members of the HR core are also evolutionarily conserved in eukaryotes, but have no obvious bacterial homologues. In addition to the core HR proteins, a significant number of other proteins participate in specialized HR events, particularly the proteins of the Fanconi anemia (FA) group 9 and the RecQ family 10, 11. Also, there are a number of HR proteins specifically expressed during meiosis 6. Below we review the recent progress in development of small molecule modulators that target several important HR proteins.
Targeting RAD51 protein with small molecule compounds
RAD51, the central protein of HR, was found to be overexpressed in many tumors and transformed cell lines 12–16. High level of RAD51 expression was associated with increased resistance to chemotherapies and radiotherapies in several experimental tumor systems 17–19. In contrast, depletion of RAD51 by antisense RNAs attenuated radioresistance in vitro and in vivo 20, 21 and enhanced killing of cancer (HeLa) cells by cisplatin 22. Consequently, RAD51 was recognized as an important target for development of small molecule inhibitors to supplement anticancer therapies. Small molecule inhibitors may also provide a tool for investigation of the mechanism of DNA strand exchange and the cellular functions of RAD51.
Biochemical and cell-based assays have been developed for high throughput screening (HTS) for RAD51 inhibitors. The active form of RAD51 is a nucleoprotein filament which RAD51 forms with ssDNA in the presence of ATP and divalent metal cofactors5. Consequently, the assay was developed to screen for compounds that disrupt (or stimulate) binding of purified RAD51 to ssDNA in vitro23. The effect of compounds on ssDNA binding by RAD51 was measured using fluorescently labeled ssDNA substrates using fluorescence polarization.
In vivo, RAD51 competes with Replication Protein A (RPA), a ubiquitous ssDNA binding protein, for ssDNA binding. It was found that BRCA2 stimulates RAD51 loading on ssDNA in the presence of RPA24. This loading is mediated by RAD51 interaction with eight conserved BRC domains of BRCA2. Consequently, a cell-based assay to screen for small molecule compounds that disrupt RAD51 interaction with BRC peptide was developed using yeast two-hybrid system25.
DNA strand exchange, the salient activity of RAD51, can be reconstituted in vitro with purified RAD51, ATP, divalent metal ions, and homologous ssDNA and dsDNA substrates. Three different DNA strand exchange assays were adapted for screening RAD51 inhibitors (Fig. 2). The first, known as three-strand reaction, employs circular ssDNA and linear dsDNA of bacteriophages, like M13 or φX174. In this assay, the RAD51 filament formed on circular ssDNA promotes DNA strand exchange with homologous linear dsDNA. The DNA products of DNA strand exchange are analyzed by gel electrophoresis in agarose gels and visualized by staining with fluorescent DNA intercalating dyes. The second, known as D-loop (displacement loop) assay, utilizes supercoiled covalently closed plasmid dsDNA and synthetic 32P-labeled ssDNA substrates. In this assay, the RAD51 filament formed on ssDNA invades the plasmid dsDNA to produce a D-loop; D-loop formation is analyzed by electrophoresis in agarose gels. The third assay is based on the use of synthetic DNA substrates. In this assay, synthetic dsDNA carries a fluorophore group on one strand that is opposed by a quencher group attached to the complementary strand. DNA strand exchange catalyzed by the RAD51 filament formed on unlabeled homologous ssDNA leads to separation of dsDNA strands carrying a fluorophore and a quencher and to an increase in the fluorescence intensity. This assay is well suited for HTS. In a version of this assay, radiolabeled DNA substrates are used and the DNA products are analyzed by gel-electrophoresis in polyacrylamide gels.
Figure 2. The assays for screening of inhibitors of the RAD51 DNA strand exchange activity.
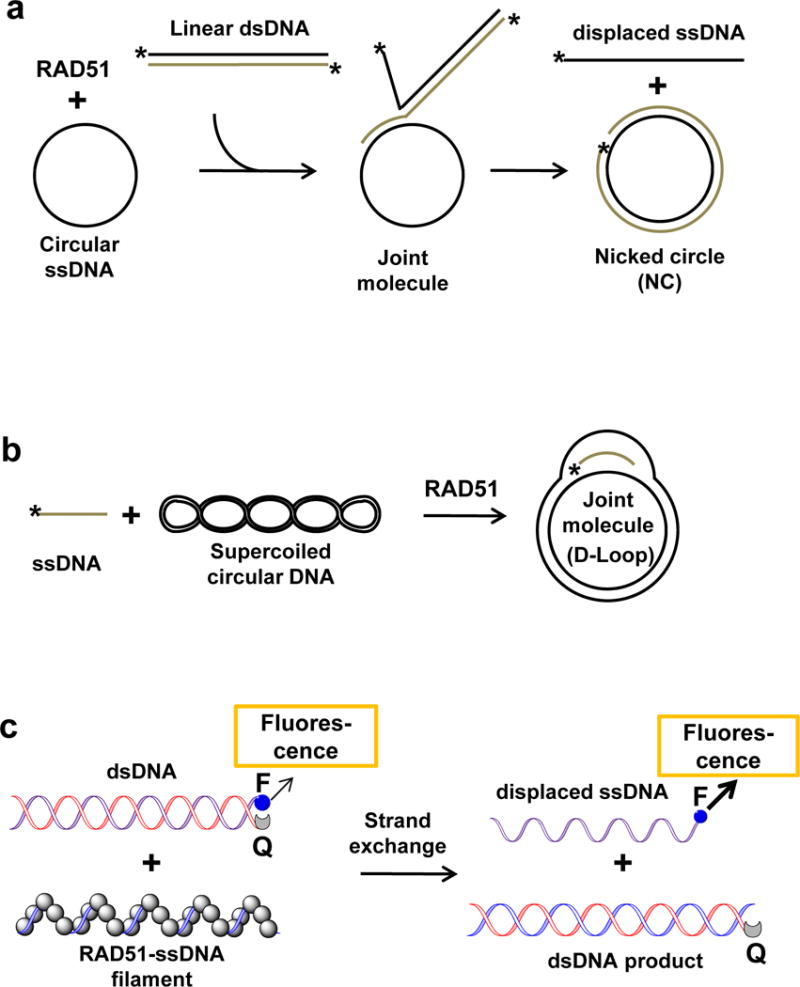
The three-strand (a), D-loop (b), and synthetic DNA-based assay (c). The asterisk denotes 32P-label; “F” and “Q” indicate fluorophore and quencher groups, respectively.
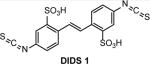
While DNA strand exchange promoted by RAD51 is a basic HR activity, the mechanism of this activity remains to be fully understood. Small molecule inhibitors may present a valuable tool for analysis of the mechanism of DNA strand exchange in vitro, e.g., by disrupting it into separate steps or by stabilizing transient reaction intermediates. Two compounds, DIDS (1) and metatungstate (2) (Table 1), were used specifically for the analysis of DNA strand exchange in vitro26, 27. DIDS was identified by screening a library of 185 potential antitumor agents for inhibitors of the RAD51 DNA26. strand exchange activity The screen was performed using the three-strand assay using φX174 ssDNA and linearized dsDNA as substrates. The IC50 of DIDS is in 1–10 μM range (at 6 μM RAD51). DIDS acts by disrupting RAD51 binding to ssDNA. Using the surface plasmon resonance (SPR) method it was shown that DIDS binds directly to RAD51 with Kd = 2 μM. It was suggested that DIDS binds to a RAD51 region close to the DNA binding site(s), and directly competes with DNA for RAD51 binding. To test this hypothesis it would be important to solve the structure of the RAD51-DIDS complex. Because of high cell toxicity the effect of DIDS on the DNA repair and HR was not evaluated in human cells28. DIDS is also known for its ability to inhibit ion channels and membrane transporters29.
Table 1.
Inhibitors and stimulators of HR proteins
| Small molecule compound | Target protein | Mechanism | Potency (EC50) in vitro | Specificity in vitro | Cellular effect |
|---|---|---|---|---|---|
|
RAD51 | Inhibits RAD51 binding to ssDNA | 1–2 μM | N/A | N/A |
|
Metatungstate (Na6H2W12O40) 2 |
MvRadA | Inhibits MvRadA binding to ssDNA and dsDNA | ~2.5 μM | N/A | N/A |
|
RAD51 | Stabilizes the RAD51-ssDNA filament | N/A | Yes | Induces resistance of human fibroblasts to cisplatin. |
|
RAD51 | Disrupts RAD51 oligomerization |
4:5–30 μM/(0.2–0.4 μM RAD51) 5 : 44 μM/(0.2–0.4 μM RAD51) |
Yes yes |
Sensitizes human cancer cells to mitomycin C |
|
RAD51 | Disrupts RAD51 binding to BRCA2 and RAD51 oligomerization | N/A | yes | Inhibits of HR in human cells; increases the sensitivity of MCF7 to IR. |
|
RAD51 | Disrupts RAD51 binding to DNA | 27μM/1μM RAD51 | yes | Increases cell sensitivity to cisplatin and mitomycin C |
|
RAD54 | Binds to RAD54 and inactivates its ATPase by generating reactive oxygen species | 16.5μM/(0.1μM RAD54) | yes | N/A |
|
BLM | Disrupts BLM binding to DNA | Helicase activity: 3μM/10nM; BM activity:50μM/10nM |
No (also inhibits WRN in vitro) | Enhances the toxicity of aphidicolin and exerts antiproliferative effect in cells expressing BLM. |
|
WRN | Inhibits the helicase activity with a milder inhibition of the ATPase and exonuclease activity | 10: 20 μM/1.2nM; 11:N/A | Yes | NSC617145 causes sensitization of FA and NHEJ-deficient cells to mitomycin C |
|
MRN | Inhibits Mre11 exonuclease activity and prevents MRN-dependent activation of ATM | 12 μM | yes | Inhibits HR-mediated DNA repair in HEK293 cells |
Note: MvRadA = Methanococcus voltae RadA
It was found fortuitously that a member of the polyoxometalate family sodium metatungstate inhibited DNA strand exchange activity of Methanococcus voltae RadA (MvRadA), an archaeal Rad51 homologue 27. In the oligonucleotide-based DNA strand exchange assay, the IC50 of metatungstate was approximately 2.5 μM (at 5 μM MvRadA). The structure of the RadA-ADP-metatungstate complex was solved. Metatungstate binds MvRadA between the DNA-binding loops L1 and L2 locking the protein in an inactive conformation. The binding was mediated by electrostatic interactions between the anionic groups of metatungstate and the cationic surface of the DNA-binding region of the RadA. These structural data may potentially provide an insight to the mechanism of DNA strand exchange. Consistent with the known role of the L1 and L2 loops in DNA binding, metatungstate inhibited ssDNA and dsDNA binding by MvRadA. The inhibitor lacked specificity for MvRadA, it also inhibited EcRecA, an Escherichia coli homologue. The effect of metatungstate was not tested on cells, and due to its high charge metatungstate is unlikely to permeate the cell membrane.
While the structure of RAD51 is highly conserved, its functions may vary in different species. In all organisms, RAD51 is important for DNA damage repair, but in vertebrates it is also essential for viability 30. Currently, it is not clear whether RAD51 acquires additional essential functions in vertebrates, or the HR mechanism of DNA repair became essential in organisms with high genome complexity. Because RAD51 mutants are often unstable in the cell, small molecule inhibitors that target different domains or activities of RAD51 may represent an attractive option to investigate RAD51 cellular functions.
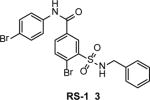
While most studies with small molecule compounds are focused on development of RAD51 inhibitors, a unique RAD51-stimulatory compound 1 (RS-1) (3) (Table 1) was also reported23. RS-1 was identified by screening a 10,000-compound library (Chembridge DIVERSet) for stimulators of RAD51 binding to ssDNA using fluorescence polarization23. RS-1 (20 μM) increases the apparent affinity of RAD51 (0.3 μM) for ssDNA by a factor of two and stimulates RAD51 strand exchange. RS-1 has no effect on S. serevisiae Rad51 and Dmc1, or EcRecA, indicating its specific interaction with human RAD51. The mechanism of this stimulation appears to be unique. It is known that formation of an active nucleoprotein filament requires ATP binding, but not ATP hydrolysis by RAD51. Moreover, ATP hydrolysis leads to accumulation of RAD51-ADP complexes that inhibit DNA strand exchange31. Previously, it was shown that inhibition of the RAD51 ATPase by Ca2+ stimulates DNA strand exchange31. However, unlike Ca2+, RS-1 does not to inhibit the RAD51 ATPase activity. Instead, the authors suggested that RS-1 allosterically “locks” RAD51 in an active conformation overriding the inhibitory effect of RAD51-ADP-complexes23. Future structural studies on the RAD51-RS-1 complex might provide an important insight into the mechanisms of regulation of the RAD51 DNA strand exchange activity. Docking simulation of RS-1 binding to RAD51 would also be useful. Though only a partial structure of the human RAD51 is currently available32, the structures of several RAD51 orthologs27, 33 may help to model the RAD51-RS-1 complex in silico. In neonatal human dermal fibroblasts, RS-1 (7.5 μM) showed a 1.5–2.0-fold increase in survival after cisplatin treatment indicating biological activity of RS-1 in cells. The authors proposed that RS-1 can be used as a protector against DNA damage or as an enhancer of gene targeting.
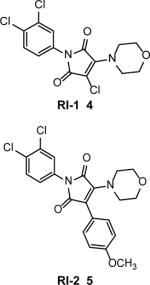
RAD51 inhibitory compound 1 (RI-1) (4) (Table 1), was identified by screening a 10,000-compound library (Chembridge DIVERSet) for inhibitors of RAD51 binding to ssDNA using fluorescence polarization34. RI-1 covalently binds through its chloromaleimide group to the C319 thiol group in RAD51. RI-1 may disrupt nucleoprotein filament formation on ssDNA because C319 lies on the protein interface between protein subunits. The IC50 of RI-1 inhibition of ssDNA binding was 5–30 μM (at 0.2–0.4 μM RAD51). Consistent with inhibition of RAD51 in cells, RI-1 (5–20 μM) reduces gene conversion in the DR-GFP assay35 in U2OS cells and stimulates single strand annealing in the SA-GFP assay36 in HEK293 cells. Also, RI-1 (20 μM) inhibited the formation of DNA damage-induced RAD51 foci in immortalized human fibroblasts, consistent with targeting RAD51. RI-1 (25 μM) also sensitizes human cancer cells HeLa, MCF-7 and U2OS to mitomycin C (MMC). Surprisingly, inhibition of the RAD51 (0.4 μM) DNA strand exchange activity in vitro requires relatively high RI-1 concentration (80–160 μM). The apparent discrepancy between the relatively low potency of RI-1 in the DNA strand exchange assay in vitro and the much more potent inhibition of HR in cells requires further investigation. It is possible that sub-optimal conditions for DNA strand exchange used in this work (e.g., 5 mM Mg2+ and no Ca2+ or other stimulatory agents) diminished the apparent inhibitory effect of RI-1 on DNA strand exchange.
In order to reduce the likelihood of off-target covalent interactions and improve compound stability in the cell, RI-2 (5) (Table 1) was synthesized. It lacks the chloromaleimide group but retains RAD51 inhibitory activity37. RI-2 was shown to bind reversibly to the same site on the RAD51 protein as does RI-1. In biochemical and cellular assays, RI-2 showed inhibitory activities similar to RI-1, but with somewhat lower potency. The IC50 of RI-2 inhibition of RAD51 ssDNA binding was 44 μM. In HEK293 cells, inhibition of the DR-GFP gene conversion and sensitization to MMC was observed at 60 μM and 150 μM RI-2, respectively.
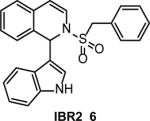
A yeast reversed two-hybrid system was used to identify IBR2 (6) (Table 1) by screening of a 24,000-compound library for inhibitors that disrupt RAD51 interaction with BRC, a conserved domain of BRCA2 25. The disruption of the RAD51-BRC interaction by IBR2 was confirmed with SPR. Because the RAD51 hydrophobic pocket involved in BRC binding is also responsible for protein multimerization, IBR2 also disrupts RAD51 multimerization. In cells, it was found, somewhat unexpectedly, that IBR2 promotes proteasome-mediated degradation of RAD51, presumably because of disruption of RAD51 multimers that are resistant to degradation. IBR2 inhibits HR in human cells; at 20 μM it causes an approximately 10-fold decrease in DSB-induced HR in the DR-GFP construct in HeLa cells and a 2-fold decrease in IR-induced RAD51 foci formation in MCF7 cells. IBR2 (5 μM) also caused a slight, but statistically significant increase, in MCF17 sensitivity to IR. By decreasing the level of RAD51, IBR2 caused an antitumor effect by prolonging survival of imatinib-resistant CML model mice bearing the T315I Bcr-abl mutation. IBR2 suppressed proliferation of CD34+ progenitor cells from CML patients by inducing apoptosis without significantly affecting the viability of normal cells. These results indicate that cancer cells require a higher level of RAD51 for their survival than normal cells.
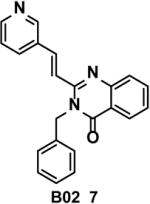
B02 (7) (Table 1) was identified by screening of a 202,556-compound library for inhibitors of the RAD51 DNA strand exchange activity in fluorescence quenching assay with oligonucleotide DNA substrates 38. In the D-loop assay, the IC50 of RAD51 (1 μM) inhibition by B02 was 27 μM, and inhibition was specific, as no inhibition of EcRecA or human RAD54 was observed. SPR showed that B02 binds to RAD51 directly with Kd = 5.6 μM39. The mechanism of B02 inhibition of RAD51 includes disruption of RAD51 binding to ssDNA and also inhibition of dsDNA binding by the RAD51-ssDNA filament 39. Importantly, B02 shows substantial inhibition of HR and DNA repair in human and mouse cells. In human HEK293 cells, B02 (5–20 μM) reduces gene conversion in the DR-GFP construct 35 up to 8-fold. In HEK293 cells, B02 (25–50 μM) suppresses IR-induced RAD51 foci formation, approximately 4-fold. In mouse embryonic fibroblasts (MEF), B02 (5 μM) increases cell sensitivity to cross-linking agents cisplatin and MMC 17- and 5-fold, respectively. Similar sensitization to cisplatin by B02 was observed in human HEK293 cells. In MEF, B02 (5 μM) potentiates (3–4-fold) the effect of the PARP1 inhibitor olaparib 40 on cell sensitivity to alkylating agent MMS 39. In contrast to IBR2, B02 does not cause a decrease in the RAD51 expression, consistent with direct binding to RAD51 in the cell.
Inhibitors of helicases and helicase-like proteins involved in HR
Three helicase-like HR proteins, RAD54, WRN, and BLM, important for maintenance of genome stability have been targeted by small molecule inhibitors. These proteins belong to the Helicase Superfamily 2 of ATPase-dependent DNA translocases. Inhibitors of these proteins may be used to sensitize cancer cells to therapeutics and for studies of protein activities and cellular functions 10, 11, 41.
The fluorescence quenching assays with synthetic DNA substrates were used to identify inhibitors of RAD54 and BLM (Fig. 3) 42, 43. A fluorophore and a quencher group were attached to DNA substrates opposing each other so that dissociation of these substrates due to RAD54 branch migration or BLM DNA unwinding (helicase) activities resulted in an increase of the fluorescence intensity. A version of the helicase assay with radioactively-labeled DNA substrates was used to identify inhibitors of the WRN helicase activity; in this case, the product formation was analyzed by electrophoresis in polyacrylamide gels 44.
Figure 3. The assays for screening of RAD54 and BLM inhibitors.
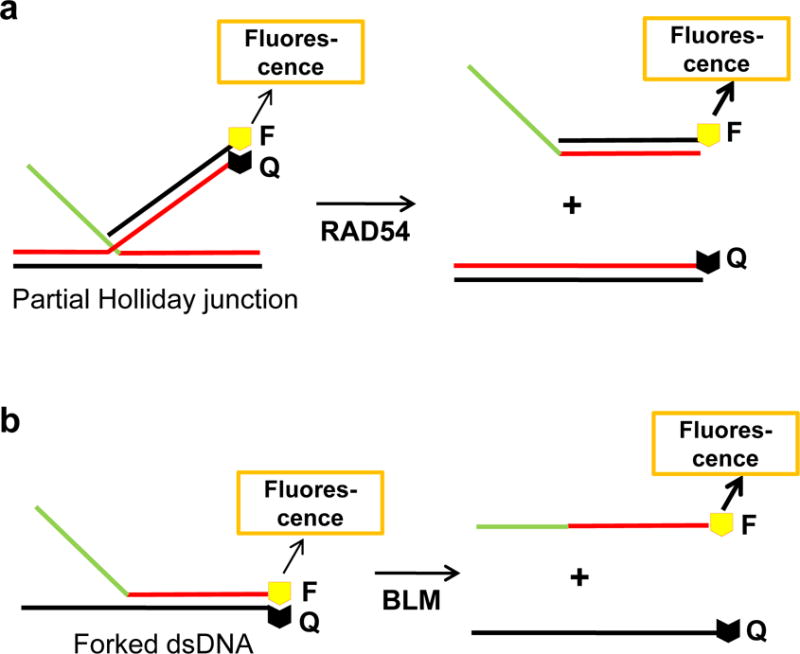
Branch migration (a) and DNA helicase (b) assays. “F” and “Q” indicate fluorophore and quencher groups, respectively.
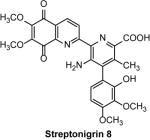
RAD54, a member of the Swi2/Snf2 family of ATP-dependent DNA translocases, is conserved in all eukaryotes 41. In S. cerevisiae, the Rad54−/− mutant is among the three most IR-sensitive single mutants, along with Rad51−/− and Rad52−/− mutants. In mammals, in contrast to most other HR proteins, RAD54 is not essential for cell viability, but important for repair of DNA damage, especially ICLs. RAD54 is a multifunctional protein. In vitro, it stimulates the DNA strand exchange activity of RAD51, promotes branch migration of Holliday junctions, and catalyzes remodeling of chromatin and nonchromatin nucleoprotein complexes. Streptonigrin (SN) (8) (Table 1), an aminoquinone compound 45, was identified as an inhibitor of the RAD54 branch migration activity by screening a 2000-compound library using fluorescence-quenching assay with synthetic Holliday junction substrates (Fig. 3) 42. The IC50 of RAD54 (0.1 μM) inhibition by SN is16.5 μM; inhibition was specific as SN does not affect the branch migration activity of EcRuvAB or DNA strand exchange activity of human RAD51. Interestingly, SN does not affect RAD54 DNA binding and targets specifically the RAD54 ATPase activity. The mechanism of inhibition includes direct binding of SN to RAD54 in the proximity of ATPase center and generation of reactive oxygen species during cycles of reduction/autoxidation of the SN quinone groups. Cu2+ potentiated the inhibitory effect of SN by ~2-fold by increasing the rate of generation of reactive oxygen species through stimulation of reduction of the SN quinone group. SN was especially instrumental for the analysis of the RAD54 activities in vitro. Using SN, it was found that RAD54 ATPase plays a relatively minor role in stimulation of DNA strand exchange activity of RAD51, contrary to common expectations. A new mechanism was proposed in which stimulation of DNA strand exchange is mediated by protein:protein interactions between RAD51 and RAD54. The effect of SN on HR was not tested in cells due to its high toxicity.
BLM is a member of the conserved RecQ helicase family 10. In humans, BLM mutations are responsible for Bloom’s syndrome, a cancer predisposition abnormality. On the cellular level, BLM mutations increase the frequency of sister chromatid exchanges (SCEs), a diagnostic phenotype of Bloom’s syndrome, and increase cell sensitivity to DNA damage. In vitro, BLM has a DNA helicase activity, DNA branch migration activity, and G-quadruplex DNA disruption activity.
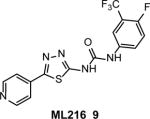
A fluorescence-quenching assay (Fig. 3) was used to identify ML216 (9) (Table 1), an inhibitor of the BLM helicase activity, through HTS of a ~350,000 compound library and medicinal chemistry optimization 43, 46. ML216 inhibited the BLM helicase (IC50 = 3.0 μM, at 10 nM BLM) stronger than its branch migration activity (IC50 = 50 μM) with only minimal effect on the G-quadruplex disrupting activity, consistent with different mechanisms underlying these two BLM activities 47, 48. In controls, ML216 did not significantly inhibit the helicase activity of EcUvrD or human RECQ1 and RECQ5, but substantially inhibited the WRN helicase (IC50 = 5.0 μM). In vitro, ML216 acts through competitive inhibition of DNA binding by BLM.
In cell-based assays, ML216 (12.5–50 μM) inhibits cell proliferation and increases the frequency of SCE in BLM-proficient fibroblast cells (PSNF5) while having only minimal effects on BLM-deficient fibroblast cells (PSNG13), indicating on-target activity in cells. ML216 (50 uM) sensitizes PSNF5 cells to aphidicolin, an inhibitor of replicative DNA polymerases, and suppresses γ-H2AX focus formation in MMC-treated PSNF5, but not in PSNG13 cells, also consistent with targeting BLM. In contrast, ML216 similarly inhibited cell proliferation of WRN− and WRN+ cells and sensitized both cell lines to aphidicolin. This result indicates that WRN is not a primary cellular target for ML216, despite the inhibition of the WRN helicase in vitro. BLM inhibitors may help define the mechanisms of BLM activities in vitro and provide a basis for development of cancer therapeutics by sensitizing cells to DNA damage.
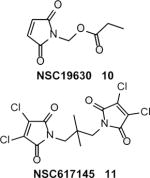
WRN is another member of the RECQ helicase family10. WRN functions are not redundant with functions of BLM or other RECQ family members and currently are under intense investigation. In humans, WRN mutations are responsible for Werner’s syndrome, a chromosomal instability disorder with clinical symptoms of premature aging. In vitro, the WRN helicase has DNA helicase and exonuclease activities. NSC19630 (10) (Table 1), an inhibitor of the WRN helicase was identified by HTS of a 500-compound library using an in vitro radiometric assay with a 19-bp forked duplex DNA substrate 44. NSC19630 inhibited the WRN helicase (IC50 = 20 μM; at WRN = 1.2 nM), but not human BLM, FANCJ, RECQ1, and E. coli RecQ, UvrD, or DnaB helicases. In contrast to the WRN helicase activity, NSC19630 caused only a modest inhibition of the WRN ATPase and exonuclease activity. NSC19630 (3 uM) inhibits proliferation of HeLa (and U2OS), but not WRN-depleted HeLa cells, indicating specific targeting of WRN. NSC19630 (2 μM) also caused an elevated proliferating cell nuclear antigen (PCNA) foci and delayed S-phase progression, consistent with the accumulation of stalled replication forks and other DNA lesions in a WRN-dependent manner. NSC19630 sensitized U2OS cells to the G-quadruplex-binding compound telomestatin or a PARP1 inhibitor KU0058948. NSC 19630 (1 uM) and the chemotherapy drug topotecan (0.1 uM) acted synergistically to inhibit HeLa proliferation. Based on structural similarity with NSC19630, another inhibitor of WRN, NSC617145 (11) (Table 1) was identified 49. NSC617145 causes sensitization of human glioblastoma cells deficient in both the Fanconi anemia (FA) and Non-homologous End-joining (NHEJ) pathways to DNA cross-linking agent MMC indicating a functional link between WRN and these two pathways. Experiments with cells derived from patients with Fanconi anemia (SV-40 transformed fibroblasts FA-D2−/− and FA-A−/−) showed that WRN inhibition perturbs the function of the HR pathway and channels DSBs formed during ICL repair into the NHEJ pathway 48. Thus, the use of small molecule inhibitors provided new insights into the role of WRN in DNA damage response.
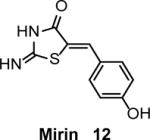
MRE11-RAD50-NBS1 (MRN) protein complex acts at the initial steps of DNA damage response including recognition and resection of DSBs, and activation of ATM 50, 51. Mirin (12) (Table 1), a small molecule inhibitor of MRE11 was identified by screening a 10,000-compound DIVERSet library (Chembridge Corporation) using ATM-dependent phosphorylation in cell free extracts from Xenopus laevis eggs as the readout 52. The IC50 of inhibition of MRN-dependent ATM activation by mirin is 66 μM in cells free extracts and 12 μM in vitro with purified recombinant proteins. Mirin (100 μM) specifically inhibits the 3′ to 5′ exonuclease activity of MRN. In U2OS cells mirin (25–100 μM) abolished the IR-induced G2/M checkpoint, consistent with inhibition of ATM signaling. In HEK293 cells, mirin (25 μM) decreased DSB-induced recombination in the DR-GFP construct by 70%.
Inhibitors that downregulate HR proteins
Several small molecule inhibitors of protein kinases were reported to inhibit HR by downregulation of HR proteins. PI3K inhibitor, BKM120, caused downregulation of BRCA1 reducing the efficiency of HR repair53. Gleevec (imatinib), an inhibitor of BRC-Abl tyrosin kinase, was reported to reduce the RAD51 level increasing radiosensitivity of the glioma or bladder cancer cells54–56. Similarly, gefitinib (IressaR, ZD1839) an inhibitor of epidermal growth factor receptor (EGFR) tyrosine kinase decreased the level of RAD51expression increasing cytotoxic effect of MMC, cisplatin and other drugs in non-small-cell lung cancer cells57. The receptor tyrosine kinase inhibitor amuvatinib (MP470) also was shown to inhibit the RAD51 expression sensitizing H1299 lung carcinoma cells to IR and MMC and suppressing HR in the DR-GFP assay58. Even though protein kinases have numerous cellular targets these inhibitors may help to investigate the mechanisms of posttranslational regulation of HR which are currently poorly understood.
Conclusions
Several of the above examples demonstrate that small molecule inhibitors can be used as tools to study the activities of HR proteins. For instance, metatungstate and RS-1 that lock RAD51 in an inactive and active state 23, 27, respectively, likely reveal physiologically relevant mechanisms of RAD51 regulation. Experiments with SN provided important insight to the role of RAD54 ATPase 42. The study with metatungstate showed the importance of structural approaches for the analysis of biochemical activities using small molecule inhibitors 27. Several inhibitors showed biological activity in mammalian cells. Some of them, like NSC19630 or ML216, were used as a tool to investigate functions of their target proteins WRN and BLM in DNA repair 43, 44, others like B02, IBR2, and RI-1 25, 34, 39 were helpful in justification of new targets for cancer therapies. The main challenge in cellular studies remains the specificity of chemical probes. The approaches to exam this specificity include the use of cell lines lacking the target protein and cellular assays for specific activities of target proteins. There is no doubt that further work based on structural analysis, medicinal chemistry, and cell-based approaches will lead to more potent and specific inhibitors for study the mechanisms of HR in the cells and development of novel combination chemotherapies.
Acknowledgments
The authors are thankful to Dr. Olga Mazina and Mr. Nadish Goyal for helpful discussion. This work was supported by the NIH grants CA100839, DA033981, MH097512 (to AVM).
Footnotes
The authors declare no conflict of interest.
References
- 1.Moynahan ME, Jasin M. Nat Rev Mol Cell Biol. 2010;11:196. doi: 10.1038/nrm2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Helleday T. Carcinogenesis. 2010;31:955. doi: 10.1093/carcin/bgq064. [DOI] [PubMed] [Google Scholar]
- 3.Pâques F, Haber JE. Microbiol Mol Biol Rev. 1999;63:349. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Symington LS, Gautier J. Annu Rev Genet. 2011;45:247. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- 5.Kowalczykowski SC. Nature. 2008;453:463. doi: 10.1038/453463a. [DOI] [PubMed] [Google Scholar]
- 6.Neale MJ, Keeney S. Nature. 2006;442:153. doi: 10.1038/nature04885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Game JC. Mutat Res. 2000;451:277. doi: 10.1016/s0027-5107(00)00055-5. [DOI] [PubMed] [Google Scholar]
- 8.Lin Z, Kong H, Nei M, Ma H. Proc Natl Acad Sci USA. 2006;103:10328. doi: 10.1073/pnas.0604232103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wrighton KH. Nat Rev Mol Cell Biol. 2010;11:603. doi: 10.1038/nrm2958. [DOI] [PubMed] [Google Scholar]
- 10.Chu WK, Hickson ID. Nat Rev Cancer. 2009;9:644. doi: 10.1038/nrc2682. [DOI] [PubMed] [Google Scholar]
- 11.Brosh RM., Jr Nat Rev Cancer. 2013;13:542. doi: 10.1038/nrc3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raderschall E, Stout K, Freier S, Suckow V, Schweiger S, Haaf T. Cancer Res. 2002;62:219. [PubMed] [Google Scholar]
- 13.Maacke H, Opitz S, Jost K, Hamdorf W, Henning W, Kruger S, Feller AC, Lopens A, Diedrich K, Schwinger E, Sturzbecher HW. International Journal of Cancer. 2000;88:907. doi: 10.1002/1097-0215(20001215)88:6<907::aid-ijc11>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 14.Hannay JA, Liu J, Zhu QS, Bolshakov SV, Li L, Pisters PW, Lazar AJ, Yu D, Pollock RE, Lev D. Molecular cancer therapeutics. 2007;6:1650. doi: 10.1158/1535-7163.MCT-06-0636. [DOI] [PubMed] [Google Scholar]
- 15.Thacker J. Cancer Lett. 2005;219:125. doi: 10.1016/j.canlet.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 16.Hine CM, Seluanov A, Gorbunova V. Proc Natl Acad Sci USA. 2008;105:20810. doi: 10.1073/pnas.0807990106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Raderschall E, Bazarov A, Cao J, Lurz R, Smith A, Mann W, Ropers HH, Sedivy JM, Golub EI, Fritz E, Haaf T. J Cell Sci. 2002;115:153. doi: 10.1242/jcs.115.1.153. [DOI] [PubMed] [Google Scholar]
- 18.Vispe S, Cazaux C, Lesca C, Defais M. Nucleic Acids Res. 1998;26:2859. doi: 10.1093/nar/26.12.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu ZY, Loignon M, Han FY, Panasci L, Aloyz R. The Journal of pharmacology and experimental therapeutics. 2005;314:495. doi: 10.1124/jpet.105.084053. [DOI] [PubMed] [Google Scholar]
- 20.Taki T, Ohnishi T, Yamamoto A, Hiraga S, Arita N, Izumoto S, Hayakawa T, Morita T. Biochem Biophys Res Commun. 1996;223:434. doi: 10.1006/bbrc.1996.0911. [DOI] [PubMed] [Google Scholar]
- 21.Ohnishi T, Taki T, Hiraga S, Arita N, Morita T. Biochemical & Biophysical Research Communications. 1998;245:319. doi: 10.1006/bbrc.1998.8440. [DOI] [PubMed] [Google Scholar]
- 22.Ito M, Yamamoto S, Nimura K, Hiraoka K, Tamai K, Kaneda Y. The journal of gene medicine. 2005;7:1044. doi: 10.1002/jgm.753. [DOI] [PubMed] [Google Scholar]
- 23.Jayathilaka K, Sheridan SD, Bold TD, Bochenska K, Logan HL, Weichselbaum RR, Bishop DK, Connell PP. Proc Natl Acad Sci USA. 2008;105:15848. doi: 10.1073/pnas.0808046105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holloman WK. Nat Struct Mol Biol. 2011;18:748. doi: 10.1038/nsmb.2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu J, Zhou L, Wu G, Konig H, Lin X, Li G, Qiu XL, Chen CF, Hu CM, Goldblatt E, Bhatia R, Chamberlin AR, Chen PL, Lee WH. EMBO Mol Med. 2013;5:353. doi: 10.1002/emmm.201201760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ishida T, Takizawa Y, Kainuma T, Inoue J, Mikawa T, Shibata T, Suzuki H, Tashiro S, Kurumizaka H. Nucleic Acids Res. 2009;37:3367. doi: 10.1093/nar/gkp200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y, He Y, Luo Y. Biochemistry. 2009;48:6805. doi: 10.1021/bi900832t. [DOI] [PubMed] [Google Scholar]
- 28.Takaku M, Kainuma T, Ishida-Takaku T, Ishigami S, Suzuki H, Tashiro S, van Soest RW, Nakao Y, Kurumizaka H. Genes Cells. 2011;16:427. doi: 10.1111/j.1365-2443.2011.01494.x. [DOI] [PubMed] [Google Scholar]
- 29.Wulff H. ACS chemical biology. 2008;3:399. doi: 10.1021/cb800140m. [DOI] [PubMed] [Google Scholar]
- 30.Lim DS, Hasty P. Mol Cell Biol. 1996;16:7133. doi: 10.1128/mcb.16.12.7133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bugreev DV, Mazin AV. Proc Natl Acad Sci USA. 2004;101:9988. doi: 10.1073/pnas.0402105101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pellegrini L, Yu DS, Lo T, Anand S, Lee M, Blundell TL, Venkitaraman AR. Nature. 2002;420:287. doi: 10.1038/nature01230. [DOI] [PubMed] [Google Scholar]
- 33.Conway AB, Lynch TW, Zhang Y, Fortin GS, Fung CW, Symington LS, Rice PA. Nat Struct Mol Biol. 2004;11:791. doi: 10.1038/nsmb795. [DOI] [PubMed] [Google Scholar]
- 34.Budke B, Logan HL, Kalin JH, Zelivianskaia AS, Cameron McGuire W, Miller LL, Stark JM, Kozikowski AP, Bishop DK, Connell PP. Nucleic Acids Res. 2012 doi: 10.1093/nar/gks353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pierce AJ, Johnson RD, Thompson LH, Jasin M. Genes Dev. 1999;13:2633. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bennardo N, Cheng A, Huang N, Stark JM. PLoS Genet. 2008;4:e1000110. doi: 10.1371/journal.pgen.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Budke B, Kalin JH, Pawlowski M, Zelivianskaia AS, Wu M, Kozikowski AP, Connell PP. J Med Chem. 2013;56:254. doi: 10.1021/jm301565b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang F, Motlekar NA, Burgwin CM, Napper AD, Diamond SL, Mazin AV. ACS chemical biology. 2011;6:628. doi: 10.1021/cb100428c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang F, Mazina OM, Zentner IJ, Cocklin S, Mazin AV. J Med Chem. 2012;55:3011. doi: 10.1021/jm201173g. [DOI] [PubMed] [Google Scholar]
- 40.Menear KA, Adcock C, Boulter R, Cockcroft XL, Copsey L, Cranston A, Dillon KJ, Drzewiecki J, Garman S, Gomez S, Javaid H, Kerrigan F, Knights C, Lau A, Loh VM, Jr, Matthews IT, Moore S, O’Connor MJ, Smith GC, Martin NM. J Med Chem. 2008;51:6581. doi: 10.1021/jm8001263. [DOI] [PubMed] [Google Scholar]
- 41.Mazin AV, Mazina OM, Bugreev DV, Rossi MJ. DNA Repair (Amst) 2010;9:286. doi: 10.1016/j.dnarep.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deakyne JS, Huang F, Negri J, Tolliday N, Cocklin S, Mazin AV. J Biol Chem. 2013;288:31567. doi: 10.1074/jbc.M113.502195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nguyen GH, Dexheimer TS, Rosenthal AS, Chu WK, Singh DK, Mosedale G, Bachrati CZ, Schultz L, Sakurai M, Savitsky P, Abu M, McHugh PJ, Bohr VA, Harris CC, Jadhav A, Gileadi O, Maloney DJ, Simeonov A, Hickson ID. Chem Biol. 2013;20:55. doi: 10.1016/j.chembiol.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aggarwal M, Sommers JA, Shoemaker RH, Brosh RM., Jr Proc Natl Acad Sci USA. 2011;108:1525. doi: 10.1073/pnas.1006423108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bolzan AD, Bianchi MS. Mutat Res. 2001;488:25. doi: 10.1016/s1383-5742(00)00062-4. [DOI] [PubMed] [Google Scholar]
- 46.Rosenthal AS, Dexheimer TS, Gileadi O, Nguyen GH, Chu WK, Hickson ID, Jadhav A, Simeonov A, Maloney DJ. Bioorg Med Chem Lett. 2013;23:5660. doi: 10.1016/j.bmcl.2013.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mazina OM, Rossi MJ, Deakyne JS, Huang F, Mazin AV. J Biol Chem. 2012;287:11820. doi: 10.1074/jbc.M112.341347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aggarwal M, Banerjee T, Sommers JA, Iannascoli C, Pichierri P, Shoemaker RH, Brosh RM., Jr Cancer Res. 2013;73:5497. doi: 10.1158/0008-5472.CAN-12-2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aggarwal M, Banerjee T, Sommers JA, Brosh RM., Jr Cell Cycle. 2013;12:3329. doi: 10.4161/cc.26320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee JH, Paull TT. Science. 2005;308:551. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 51.Stracker TH, Petrini JH. Nat Rev Mol Cell Biol. 2011;12:90. doi: 10.1038/nrm3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dupre A, Boyer-Chatenet L, Sattler RM, Modi AP, Lee JH, Nicolette ML, Kopelovich L, Jasin M, Baer R, Paull TT, Gautier J. Nature chemical biology. 2008;4:119. doi: 10.1038/nchembio.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ibrahim YH, Garcia-Garcia C, Serra V, He L, Torres-Lockhart K, Prat A, Anton P, Cozar P, Guzman M, Grueso J, Rodriguez O, Calvo MT, Aura C, Diez O, Rubio IT, Perez J, Rodon J, Cortes J, Ellisen LW, Scaltriti M, Baselga J. Cancer Discov. 2012;2:1036. doi: 10.1158/2159-8290.CD-11-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Russell JS, Brady K, Burgan WE, Cerra MA, Oswald KA, Camphausen K, Tofilon PJ. Cancer Res. 2003;63:7377. [PubMed] [Google Scholar]
- 55.Qiao B, Kerr M, Groselj B, Teo MT, Knowles MA, Bristow RG, Phillips RM, Kiltie AE. Cancer Res. 2013;73:1611. doi: 10.1158/0008-5472.CAN-12-1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Choudhury A, Zhao H, Jalali F, Al Rashid S, Ran J, Supiot S, Kiltie AE, Bristow RG. Molecular cancer therapeutics. 2009;8:203. doi: 10.1158/1535-7163.MCT-08-0959. [DOI] [PubMed] [Google Scholar]
- 57.Ko JC, Hong JH, Wang LH, Lin YW. Exp Cell Res. 2008;314:1881. doi: 10.1016/j.yexcr.2008.02.013. [DOI] [PubMed] [Google Scholar]
- 58.Zhao H, Luoto KR, Meng AX, Bristow RG. Radiother Oncol. 2011;101:59. doi: 10.1016/j.radonc.2011.08.013. [DOI] [PubMed] [Google Scholar]