

Abstract
The zebrafish has emerged as an important model for whole-organism small-molecule screening. However, most zebrafish-based chemical screens have achieved only mid-throughput rates. Here we describe a versatile whole-organism drug discovery platform that can achieve true high-throughput screening (HTS) capacities. This system combines our automated reporter quantification in vivo (ARQiv) system with customized robotics, and is termed ‘ARQiv-HTS’. We detail the process of establishing and implementing ARQiv-HTS: (i) assay design and optimization, (ii) calculation of sample size and hit criteria, (iii) large-scale egg production, (iv) automated compound titration, (v) dispensing of embryos into microtiter plates, and (vi) reporter quantification. We also outline what we see as best practice strategies for leveraging the power of ARQiv-HTS for zebrafish-based drug discovery, and address technical challenges of applying zebrafish to large-scale chemical screens. Finally, we provide a detailed protocol for a recently completed inaugural ARQiv-HTS effort, which involved the identification of compounds that elevate insulin reporter activity. Compounds that increased the number of insulin-producing pancreatic beta cells represent potential new therapeutics for diabetic patients. For this effort, individual screening sessions took 1 week to conclude, and sessions were performed iteratively approximately every other day to increase throughput. At the conclusion of the screen, more than a half million drug-treated larvae had been evaluated. Beyond this initial example, however, the ARQiv-HTS platform is adaptable to almost any reporter-based assay designed to evaluate the effects of chemical compounds in living small-animal models. ARQiv-HTS thus enables large-scale whole-organism drug discovery for a variety of model species and from numerous disease-oriented perspectives.
INTRODUCTION
In the effort to bring new and useful therapeutics to patients, whole-organism drug discovery represents a complementary approach to in vitro HTS. Whole-organism chemical screening typically involves a visual phenotyping assay, which limits this approach to mid-throughput capacities. Nevertheless, a number of first-in-class drugs have emerged from phenotypic screening1,2. It stands to reason that modern drug discovery could benefit from a methodology enabling whole-organism phenotyping at high-throughput rates—i.e., large-scale drug tests directly in small animal models. To develop such a system, we sought to adapt existing HTS methods to in vivo contexts. We emphasized approaches for quantifying light-emitting reporters (e.g., microtiter plate readers) and demonstrated a potential for this strategy to accelerate whole-organism drug discovery (i.e., tens of thousands of organisms evaluated per day) using several different reporter-based assays3. One of our main goals behind the drive to increase throughput was to facilitate optimal screening practices, such as titration-based chemical screening4, for whole-organism drug discovery. We recently completed the first large-scale, titration-based, whole-organism screen with a fully automated iteration of this methodology, evaluating >0.5 million transgenic zebrafish larvae to identify and validate 24 Food and Drug Administration (FDA)-approved drugs that increased the number of insulin-producing beta cells in the pancreas5. In this first iteration of the ARQiv-HTS platform, we encountered emergent throughput issues that caused the entire screen to take a little over a year to complete. In an effort to streamline future in vivo drug discovery efforts, we discuss the below methods for addressing the bottlenecks that we encountered in this first screen. Importantly, as the approach is based on quantification of reporters, a near-limitless number of whole-organism assays can be applied to this screening platform.
HTS-based drug discovery provides a powerful methodology for identifying potential therapeutics. Over the past two decades, HTS has been focused almost exclusively on target-based screens— i.e., identifying compounds that bind a specific molecular moiety. Although greatly advancing understanding of molecular biology, target-based screening has not been as successful for drug discovery as initially hoped6. It has been proposed that the absence of disease complexity, or in vivo context, in reductionist HTS approaches leads to high failure rates during drug discovery7–9. Thus, one advantage of whole-organism screening is that it more fully accounts for biological complexity—i.e., evaluating compound effects within intact disease models. In addition, whole-organism drug discovery is typically performed from a target-agnostic perspective and is therefore well suited to identifying new druggable targets. Subsequently, identification of the molecular mechanism of action of ‘hit’ compounds can be pursued to identify the molecular target(s) and/or signaling pathway(s) eliciting the desired effect (e.g., absence of pathology)10.
Over the past 15 years, the zebrafish (Danio rerio) has emerged as a powerful vertebrate model system for whole-organism chemical screening11. In 2000, the first large-scale chemical screen of zebrafish embryos arrayed in multiwell plates was reported by Peterson et al.12. Since then, >60 successful zebrafish chemical screens have been published. These assays have covered a diverse array of research areas11,13–27. Importantly, a high degree of ‘pharmacological conservation’ has been observed between fish and humans. For example, a study of cardiac drugs that alter the cardiac cycle (e.g., slow heart rate) in humans found that 22 of 23 compounds had similar effects in zebrafish28,29. In addition, drugs that affect lipid metabolism, anticoagulants, and narcotics all have remarkably similar physiological effects in zebrafish30. Critically, a drug initially discovered to increase hematopoietic stem cell (HSC) proliferation rates in zebrafish is now entering phase II trials as a means of improving engraftment in patients; ProHema, a prostaglandin E2 derivative, is used to expand the number of umbilical cord blood-derived HSCs before transplantation, thus further demonstrating conservation of drug action across phyla. This work clearly demonstrates the potential for rapidly translating findings from fish tank to bedside31. Collectively, these studies (i) have demonstrated the utility and power of this animal model for rapid in vivo assessment of drug activities, and (ii) strongly suggest that drugs discovered in zebrafish can act as potent therapeutics for human patients.
Whole-organism drug discovery efforts have recently benefitted from the development of sophisticated high-content screening (HCS) methods that facilitate rapid imaging of phenotypes of interest. Automated phenotyping methods range from relatively straightforward morphological image acquisition systems to more complex platforms, such as rapid microscopic imaging with automated feature detection32 and automated tracking and quantification of behaviors25. Although extremely powerful, HCS methods are by nature slower than classic HTS approaches, typically achieving mid-throughput rates at best. Moreover, to counterbalance the lower throughput rates of HCS approaches, compromises are often required in key assay parameters—e.g., low sample numbers, screening of smaller drug libraries, and/or testing of drugs at a single concentration. This limits the capacity to apply optimal screening strategies known to reduce false-hit rates, such as the titration-based screening (e.g., quantitative HTS (qHTS)) strategy that we apply for ARQiv-HTS, which can enhance the value of whole-organism drug discovery immensely4. Reviews comparing throughput capacities and other key features of recently developed HCS systems with each other, and with ARQiv, are available for those interested in further details33,34.
Development of the protocol
To overcome this limitation, we sought an existing HTS methodology for rapidly and accurately quantifying phenotypes in living small animal models—e.g., zebrafish. Because of the prevalence of transgenic lines expressing fluorescent reporters in worms, flies, and fish, we reasoned that a fluorescence detection system would be quite powerful. Eventually, we identified a fluorescence plate reader that provided the necessary parameters for accurate signal-to-noise detection levels in vivo. We termed this methodology ‘automated reporter quantification in vivo’3. ARQiv differs from in vivo HCS assay platforms by providing only quantitative data. This limitation in content is offset by marked improvements in throughput. At maximal rates, a single plate reader can process >100,000 1-to 2-d-old zebrafish embryos per 12-h screening session3. However, this level of throughput creates bottlenecks in terms of the basic mechanics of in vivo assays such as egg/larvae production, dispensing into microtiter plates, and liquid and plate handling. To address these issues, we developed several methods aimed at surmounting impediments to attaining true in vivo HTS.
To better facilitate HTS-paced in vivo screening, we coupled ARQiv with a custom-designed robotics workstation and termed the fully automated platform ‘ARQiv-HTS’. The robotized system circumvents many of the bottlenecks attending large-scale in vivo assays, alleviates repetitively mundane aspects of the screening process, and removes much of the potential for user error. The first full-scale deployment of the ARQiv-HTS system was recently completed. Applying qHTS principles, >0.5 million zebrafish larvae were screened to identify potential new therapeutics for diabetic patients. Notably, this screen also highlighted the sensitivity of plate reader-based in vivo reporter quantification in that hit compounds increased the number of pancreatic beta cells by only 10 cells: from ~30 to ~40 (ref. 5). Here we provide a protocol that is based on a series of methods we developed during this first iteration of the ARQiv-HTS platform to overcome obstacles encountered in our effort to attain true in vivo HTS rates. Although some of the emergent bottlenecks and caveats have yet to be fully resolved, we found solutions for the majority of the issues hampering the attainment of HTS-paced drug discovery in zebrafish. We also outline a generalized process for applying ARQiv-HTS to drug discovery that encompasses assay design, mass egg and larvae production, automated drug dilutions, dispensing of embryos or larvae into microtiter plates, quantitative reporter detection, and real-time data outputs. Finally, we provide the specific ARQiv-HTS protocol that we used to identify drugs that elevate insulin reporter activity, thus potentially increasing pancreatic beta-cell mass5. The ARQiv-HTS platform has the potential to greatly expand the types of large-scale chemical screens that can be performed in vivo, and it provides a means to attain the throughput that is necessary to apply optimal screening practices to whole-organism drug discovery.
Overview of the procedure
The protocol includes two parts (see Fig. 1 for an overview). The first part, ‘Prescreen assay optimization’, is used to establish, test, and optimize ARQiv-HTS assays using the following components: (i) fluorescence detection in vivo—optimizing plate reader settings for distinguishing ‘signal’ from ‘noise’ in transgenic larvae; (ii) assay quality tests—assessing whether an assay is HTS-ready; (iii) calculation of sample size—using power calculations to establish sample number across a range of error rates; and (iv) choice of hit selection criteria—a method for simulating assay performance with a control reference compound is used to calculate statistical values for hit criteria (i.e., ‘effect size’). The second part, ‘Large-scale in vivo drug discovery’, details the cyclic screening process and includes the following: (i) large-scale egg production and larvae maintenance, (ii) preparation of drug plates, (iii) dispensing larvae into 96-well plates, (iv) plate reader scans, and (v) real-time HTS data analysis. Notably, we have established an R-based ARQiv package that simplifies much of the assay design and implementation procedures (e.g., automating key labor-intensive steps), which adds substantial value to the screening process. For instance, converting raw data into graphical outputs in real time facilitates near-immediate compound performance evaluation and thus same-day visual inspection of plates of interest.
Figure 1.
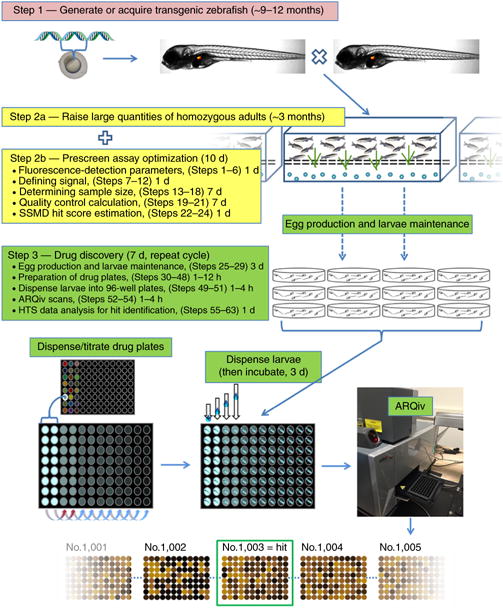
Development and implementation of HTS-ARQiv. Step 1 (pink box): Generate or acquire a transgenic zebrafish. In-cross and identify homozygous adults. Step 2a (yellow box): rapidly expand this population to generate several thousand homozygous transgenic adults within ~3 months. Step 2b (yellow box, complete concurrent with Step 2a). Optimize assay parameters before large-scale screen (i.e., maximize assay Signal:Background ratio, determine the sample size, choose hit selection method). If necessary, repeat Step 2b multiple times while expanding adult transgenic population. Step 3: Drug discovery phase can be performed iteratively (e.g., offset matings by 2 d) to complete multiple 7-d cycles in the course of a single week. This substantial work cycle contains both manual (egg production, larvae maintenance) and semiautonomous/autonomous (preparation of drug plates, larvae dispensing, ARQiv scans, data analysis, and hit selection) components.
Experimental design—in vivo HTS assay development
Several practical considerations need to be resolved during the process of designing whole-organism HTS assays. Below, we focus on issues that impinge on maximizing in vivo detection of the light-emitting reporter. Careful consideration of these parameters will help ensure that optimal Signal-to-Background (S:B) ratios are achieved.
Age
The relevance of the developmental stage (e.g., embryonic versus larval) to the biology being tested and temporal limits, with regard to maintaining fish in microplate wells for extended periods of time, must be factored in.
Pigmentation
Blocking pigmentation to create more ‘transparent’ fish generally facilitates increased S:B ratios. However, transparency is associated with concomitant increases in autofluorescence3. Therefore, the choice of pigmentation mutant(s) or a pigmentation-blocking agent needs to be evaluated for each assay. Available mutants include those with reduced melanophore pigmentation (e.g., albino (alb) and nacre (nac)), reduced iridophore pigmentation (e.g., roy orbison (roy) and transparent (tra)), and reduced xanthophores (e.g., pfeffer (pfe)). Double and triple mutants have also been derived (e.g., roy;alb). Chemical inhibition (e.g., 1-phenyl 2-thiourea (PTU); a tyrosinase inhibitor) can also be used to block melanization. For instance, application of 200 nM PTU at 24 h post fertilization (h.p.f.) is sufficient to block pigmentation in tissues other than the eye, whereas treatment at 16 h.p.f. also inhibits retinal pigment epithelium melanization. To date, PTU treatment of roy mutant fish has provided the best S:B ratios for ARQiv assays.
Fluorophore(s)
To maximize S:B ratios, wavelengths exhibiting low autofluorescent background in unlabeled controls should be emphasized. For this reason, we tend to favor yellow to red emission ranges when conducting fish larvae assays3. A second, complementary reporter, used as a fiducial marker, can act as a baseline indicator around which the experimental reporter fluctuates. This strategy can facilitate ratiometric assessments of effect size, confirm that animals were scanned in cases in which experimental signal can decrease to background levels (e.g., complete loss of fluorescent signal following ablation of reporter-expressing cells in regeneration assays), and control for differences in sample number per well (e.g., a recently developed ultra-HTS Caenorhabditis elegans assay system)35.
Signal variability
Cell culture and in vivo HTS assays tend to generate higher signal variability than in silico HTS assays. This issue can be addressed in the assay design or through data processing. For instance, individually tracking reporter levels before and after compound exposures serves to normalize all observed changes in fluorescence to initial signal levels. Signal variability across individuals is therefore controlled by expressing signal changes as a percentage of each individual’s initial values3. However, the need to anesthetize fish before plate reader scans complicates the use of this approach for large-scale screens. We are currently developing methods to overcome this limitation. Alternatively, increased variability, unequal variance, and/or outliers associated with in vivo HTS are well accounted for by preprocessing signal data via log transformation, a common HTS practice (Supplementary Fig. 1)36. This can result in an increased dynamic range of statistical metrics used for quality control (QC) assessments and/or ‘hit’ calls. For instance, compare strictly standardized mean difference scores (SSMD and robust SSMD*; see below for further discussion) achieved before and after data transformation (Supplementary Fig. 1a,b, upper right). In turn, increased dynamic ranges can enable finer delineation of compound effect size during primary screens.
Plate reader detection of in vivo reporters
A microplate reader that allows z dimension focus and multiple ‘regional’ scans per well, with values reported per each individual region rather than averaged across regions, is required for reliable quantification of fluorescent reporters in zebrafish larvae (e.g., the Tecan Infinite M1000)3. Monochromator-based readers allow maximal flexibility with regard to excitation and emission settings. In that regard, a 5-nm bandwidth minimum can be critical for eliminating autofluorescent background and achieving robust S:B ratios for most of the commonly applied fluorophores. It is essential that regional scans be reported individually rather than averaged across all regions. This allows ‘noise’ from regions that do not contain fish to be eliminated from signal calculations; for further details, see Walker et al.3.
QC tests
HTS assay QC needs to be assessed before initiating a full-scale screen. Statistical methods have been developed for this that account for both S:B ratios and variance, the most common being the Z′ factor37. However, we have found that methods that account for non-normal distributions, unequal variance, and/or outliers (e.g., SSMD QC) are better suited to assessing quality and establishing ‘hit’ selection criteria for in vivo HTS assays37–40. In addition, log transformation of signal data can also be used to control for high variability. If initial analyses indicate that assay improvements are necessary, the Z′ factor and/or SSMD QC scores can be used to compare assay quality following adjustments made to key assay parameters.
Preparation of drug plates
To limit potential circadian effects on signal strength, we perform ARQiv scans only during subjective daytime hours. Therefore, to maximize daytime availability of robotics units for screening, all drugs are dispensed overnight on the evening before placement of embryos into drug plates. A software package that facilitates interfacing between robotics elements and all other automated assay components (e.g., sorter/dispenser and plate reader) is required (e.g., SoftLinx application, Hudson Robotics).
Dispensing larvae in 96-well plates
To dispense individual zebrafish embryos or larvae into single wells of microtiter plates, we use an organismal fluorescence-gated sorting system developed for this purpose—the Complex Object Parametric Analytic Sorter (e.g., COPAS-XL, Union Biometrica). The COPAS-XL can sort and dispense large batches of embryos or larvae at a rate of 0.5–1 per s. Therefore, it is capable of dispensing as many as 50,000 fish over a 14-to 16-h period. The dispensing rate depends on fish density (1–2 fish per ml is recommended), whether anesthetic is used, and the stringency of sorting parameters. Standard dispense rates for live/dead sorting range from 0.9 to 1.25 s per fish. However, care must be taken in setting up the sorting parameters to avoid dispensing errors (e.g., blank wells) that can otherwise compromise assay quality. The COPAS-XL can also be used to sort transgenic from nontransgenic fish, or to ‘gate’ transgenic populations according to a user-defined expression cutoff level (as with standard fluorescence-activated sorting). In theory, this could be used to circumvent transgene expression variability issues3. However, it would reduce the number of fish available for screening per round, and thereby decrease throughput.
Real-time HTS data analysis and hit selection
Real-time data analysis facilitates immediate visual follow-up of potential ‘hit’ compounds for toxic/teratological effects, autofluorescence, and/or confirmation of desired biological outcome. ARQiv data are limited to purely quantitative outputs and therefore prone to potential false-hit calls (e.g., nonspecific increases in fluorescence due to compound autofluorescence/staining, toxicity, detritus, and so on) that could be eliminated using imaging-based screening approaches. Conversely, imaging all plates at the throughput rates possible with ARQiv is not feasible. In an effort to eliminate false-hit calls, end users can visually scan only the subset of plates deemed of interest with standard fluorescence microscopy. To facilitate this approach, we developed an R-based ARQiv package to generate real-time graphical data outputs, which are useful for flagging potential hit plates immediately following ARQiv scans4. We use three different graphical output formats to represent separate aspects of the data: (i) standard box plots to visualize dose-response curves; (ii) SSMD analysis to generate a numerical metric for comparing compound efficacy; and (iii) heat maps to facilitate visual follow-up of specific wells. Collectively, these analyses facilitate identification and prioritization of ‘hit’ compounds based on relative activity levels and whether a reasonable dose-response is evident. See the PROCEDURE for further details.
Limitations
Here we discuss some of the outstanding issues and current limitations that impact the ability to fully leverage ARQiv-HTS for high-throughput drug discovery in vivo.
A key obstacle in the development of fluorescent-reporter-based whole-organism assays that we identified previously is the inherent variability of transgene expression, even when transgene copy number is controlled for3. This can be addressed experimentally, for instance, by targeting transgenes to a defined locus resistant to silencing, or by presorting for desired reporter expression levels. Alternatively, this issue can be addressed statistically during the assay development/design stages by establishing appropriate sample sizes and using robust statistical methods that replace mean and standard deviation with median and median absolute deviation, and/or logarithmic data transformations. Serendipitously, the transgenic line used for the Wang et al. screen exhibited a low level of variability as compared with the lines that we evaluated previously3 (perhaps because of the small number of cells labeled). Ongoing efforts in our lab are directed at circumventing this issue by facilitating tracking of reporter levels longitudinally over time (e.g., before and after treatment) in individually arrayed subjects. More specifically, we are exploring various multiwell ‘insert’ designs that would facilitate media exchanges and help position anesthetized fish in a defined orientation to eliminate this variable. Importantly, a successful insert system will enable changes in reporter levels to be expressed as a percentage of initial signal values per each condition/individual, thus internally controlling for variation across conditions/individuals and thereby negating any innate differences in reporter expression3.
Another bottleneck encountered during our effort to implement in vivo HTS with zebrafish larvae is the extent of egg production required to obtain sufficient sample numbers. Zebrafish HTS assays require tens of thousands of developmentally synchronized eggs per week; this can be accomplished by collecting embryos at defined intervals (e.g., every hour). This challenge can be surmounted using commercial (MEPS, Aquatic Habitats; iSpawn, Techniplast) or custom-made (Supplementary Fig. 2, assembled from plastic storage units nested in sequence) large-scale embryo production systems. Alternatively, submersible ‘drop-in’ mating chambers (Supplementary Fig. 2d–e) may be used when facility bench space is limited. In our experience, mass mating of 100–200 pairs of fish consistently yields ~10,000 embryos per session. However, if a daily supply of this magnitude is needed, we recommend rotating through ~1,000 adult breeders on a weekly basis to avoid stress-related issues incurred by frequent group matings. In addition, age, health, and genotype can markedly affect the number of breeders needed to achieve sufficient productivity levels.
To accommodate HTS in small-animal models, there are established robotics solutions for drug dispensing and dilutions (e.g., Solo, Hudson Robotics; Echo, Labcyte), as well as plate handling (e.g., PlateCrane EX, Hudson Robotics). However, only one current option is available for rapid sorting of small organisms into multiwell plates in a known volume (Biosorter and COPAS, Union Biometrica). A small table-top fish egg sorting system named ‘ZebraFactor’41 (Swiss Center for Electronics and Microtechnology), if successfully coupled to a multiwell dispensing system, may also be useful for the latter purpose. The dispense rate of the COPAS-XL system currently represents the rate-limiting step in the ARQiv-HTS process, maximally allowing ~50,000 embryos/larvae to be dispensed individually into single wells of a 96-well plate over an ~16-h period. In addition, dispensing errors (e.g., blank wells, multiple organisms per well) need to be minimized by carefully determining optimal sorting parameters.
Another important consideration for large-scale chemical screening is the timing of chemical exposure. Developmental issues may affect treatment window options for the assayed phenotype and need to be considered carefully. For instance, early-stage treatments (e.g., before 4 h.p.f.) can cause high rates of developmental defects or death. Treatments at 24 h.p.f. or later can help reduce developmental-related effects and/or toxicity due to the slower rate of cell cycling16. A positive-control chemical treatment known to elicit the desired phenotype should be used to establish exposure times and to verify the assay during screening, when available. Alternatively, for certain types of assays, nontreated transgenic controls or mutant lines can be used as a proxy for a control compound-induced, or control compound-nullified, phenotype.
If required, food sources need to be considered carefully; feeding in microtiter plates incurs unique challenges, and conditions for promoting optimal survival under small-volume conditions are not well defined for some species. For instance, zebrafish larvae are self-sustaining until 7 d post fertilization (7 d.p.f.), after which they need an added food source. For ARQiv-HTS, it is important that food sources do not interfere with reporter signal detection (e.g., autofluorescence) both before and after ingestion3. As a workaround, multiwell mesh inserts could facilitate daily media exchanges and/or feeding regimens, thereby improving long-term survival in microtiter plates.
MATERIALS
REAGENTS
Zebrafish (strain choice depends on the experimental design; for the experiments outlined in this protocol, AB wild-type (Zebrafish International Resource Center, cat. no. ZL1) and transgenic Tg(ins:PhiYFP-Eco.NfsB,sst2:TagRFP)lmc01 lines (available upon request3) were used) ▲ CRITICAL All zebrafish experiments should be performed in accordance with the relevant guidelines and regulations and approved by the applicable institutional animal care and use committees. Permission was obtained from the Johns Hopkins University Animal Care and Use Committee (ACUC), and all animal studies were carried out in accordance with onsite ACUC protocols using methods to minimize and/or eliminate procedures that have the potential to induce discomfort or pain in subjects.
DMSO (Fisher, cat. no. BP231-100) ! CAUTION DMSO is flammable and readily penetrates skin, thereby carrying other chemicals into the body. Wear appropriate personal protective equipment (PPE; includes protective gloves, protective clothing, eye protection, and face protection).
1-Phenyl-2-thiourea (PTU; Acros Organics, cat. no. 103-85-5) ! CAUTION PTU is highly toxic; wear appropriate PPE when handling the compound.
Eugenol (clove oil; Sigma-Aldrich, cat. no. C8392) ! CAUTION Eugenol is toxic when inhaled, and it may cause skin/eye irritation; wear appropriate PPE when handling the compound.
NaCl (Sigma-Aldrich, cat. no. S7653)
KCl (Sigma-Aldrich, cat. no. P9541)
CaCl2 (Sigma-Aldrich, cat. no. C3881) ! CAUTION CaCl2 causes serious eye irritation; wear appropriate PPE when handling the compound.
MgSO4 (Fisher, cat. no. M63)
DAPT (N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester, a cell-permeable γ-secretase inhibitor; Sigma-Aldrich, cat. no. 208255-80-5)
ddH2O
COPAS Z-Sheath Concentrate: solution A: 100× salts (Union Biometrica, cat. no. 370-5070-100)
COPAS Z-Sheath Concentrate: solution B: 100× surfactant (Union Biometrica, cat. no. 370-5070-100)
COPAS cleaning solution (Union Biometrica, cat. no. 300-5072-000)
Tris base (Fischer, cat. no. BP152-1)
NaOH (Fisher, cat. no. S318-100) ! CAUTION NaOH is hygroscopic and causes severe burns by all exposure routes. Use the compound only under a fume hood; wear appropriate PPE when handling the compound.
HCl, 12.1 N (Fisher, cat. no. A144S-500) ! CAUTION HCl is highly toxic and causes severe burns by all exposure routes. It causes respiratory irritation and may damage organs through prolonged or repeated exposure. Use only under a fume hood; wear appropriate PPE when handling the compound.
-
Tricaine methanesulfonate (MS-222, Argent Laboratories)
! CAUTION MS-222 causes skin irritation and serious eye damage. Prepare stock solutions under a fume hood; wear appropriate PPE when handling the compound.
Johns Hopkins Drug Library (source: J. Liu, Department of Pharmacology and Molecular Science, Johns Hopkins School of Medicine)
EQUIPMENT
Incubator set at 28 °C
96-Well microtiter plate sealing tape (Fisher Scientific, cat. no. 12-565-71)
U-shaped 96-well microtiter plate, black (Greiner Bio-One, cat. no. 650209)
32-liter storage bin (Hefty, cat. no. 71030101110446)
Darice plastic canvas, 7 count 10 × 13 inch (black; Darice, cat. no. 33900-20)
250-μM Nylon woven mesh sheet (Small Parts, cat. no. CMN-0250-C)
Mating chambers (Aquatic Habitats, cat. no. SBTANK2)
Economy wash bottle, low-density polyethylene (1,000 ml; Nalgene, cat. no. 2401-1000)
Plastic strainer, 3.5-inch (Cultures for Health, cat. no. 4045)
Petri dishes (150 × 15 mm; Fisherbrand, cat. no. FB0875714)
Petri dishes (100 × 20 mm; Fisherbrand, cat. no. FB0875711Z)
Corning square bioassay dishes (245 × 245 mm; Fisherbrand, cat. no. 07-200-600)
Plastic transfer pipettes (Fisherbrand, cat. no. 13-711-7M)
COPAS-XL (Union Biometrica, cat. no. 370-5000-000)
Software for data processing (R v3.3.1, R Studio v0.99.903, and the ARQiv package; all available at https://github.com/mummlab/ARQiv)
Windows 7 Pro-capable computer for robotics automation and ARQiv-HTS data acquisition (e.g., 64-bit OS, 3.4 GHz processor, 16 GB RAM)
Windows 7 Pro-capable computer for ARQiv-HTS data processing (e.g., 64-bit OS, 3.4 GHz processor, 16 GB RAM)
Microplate reader
Tecan Infinite M1000 microplate reader (Tecan, cat. no. 30036489)
Excitation module (Tecan, cat. no 30066074)
Emission module (Tecan, cat. no. 30069022)
Fluorescence top module (Tecan, cat. no. 30034121)
i-Control software package (included with Infinite M1000)
Liquid/plate handler
Solo automated pipettor (Hudson Robotics, cat. no. 800230)
SOLOsoft Software (Hudson Robotics, included with Solo automated pipettor)
Micro 10× robotic dispenser (×2, Hudson Robotics, cat. no. 500110)
4-Way valve (Hudson Robotics, cat. no. 500205)
PlateCrane EX plate handler (Hudson Robotics, cat. no. 280200)
PA1000 Barcode Print and Apply (Hudson Robotics, cat. no. 224100)
Barcode Reader (Hudson Robotics, cat. no. 200209)
SoftLinx Software (Hudson Robotics, cat. no. 720500)
200-μl Clear presterilized pipette tip boxes (Hudson Robotics, cat. no. 800352-S)
REAGENT SETUP
100× E3 medium
Combine 29.22 g of NaCl, 1.27 g of KCl, 3.33 g of CaCl2, and 3.97 g of MgSO4 to produce 1 liter of 100× E3 medium. Place the medium at room temperature (RT: ~21°C) for 1 h on a stir plate.
Adjust the pH to 7.4 with NaOH. Large stocks of 100× E3 may be stored at RT for 3 months or at 4°C for 6 months.
1× E3 medium
Dilute 100× E3 medium (above) to 1× in water.
The final 1× E3 solute concentration is as follows: NaCl (5 mM), KCl (0.17 mM), CaCl2 (0.3 mM), and MgSO4 (0.33 mM). Large stocks of 1× E3 may be stored at RT for 3 months.
50× PTU stock solution
Add 1.520 g of PTU to 1 liter of 1× E3 medium. Mix the solution on a hotplate at 50 °C for ≥2 h (protect from light). Let the solution cool to RT, and then prepare aliquots in a fume hood. Aliquotted PTU stock solution can be stored at −20 °C for 1 year (protected from light).
1× PTU solution
Dilute 50× PTU stock solution (above) to 1× with 1× E3. This solution can be stored at RT for 3 months.
Negative-control solution
Prepare a 0.1% (vol/vol) DMSO solution in 1× PTU solution. The negative-control solution should be freshly prepared during automated preparation of drug plates.
Positive-control solution
If solubility allows, prepare a 10,000× stock solution of the positive-control compound in 100% (vol/vol) DMSO. This facilitates automated serial dilution such that fish are treated at 4×, 2×, 1×, 0.5×, 0.25×, and 0.125× concentrations (in 0.1% (vol/vol) DMSO). For the Wang et al. screen, this was not possible; instead, a twofold dilution series from 200 to 6.25 μM of DAPT was prepared manually. DAPT is a γ-secretase inhibitor that was previously identified as inducing precocious secondary islet formation and thereby increasing insulin reporter activity42.
Dilution buffer 1 (for 1:2 serial dilution)
Prepare the buffer by adding 0.9 ml of 100% (vol/vol) DMSO to 1 liter of 1× PTU solution (0.09% (vol/vol) DMSO final). This buffer can be stored at RT for 3 months.
Dilution buffer 2
Prepare the buffer by adding 1.3 ml of 100% (vol/vol) DMSO to 1 liter of 1× PTU solution (0.13% (vol/vol) DMSO final). This buffer can be stored at RT for 3 months.
34× Eugenol anesthetic solution
Prepare a 0.68% (vol/vol) Eugenol solution in negative-control solution. This is diluted to a 0.02% working solution in negative-control solution. The stock and working solutions should be freshly made for each assay.
20× Tricaine anesthetic solution
Add 336 mg of tricaine methanesulfonate (MS-222) to 97.8 ml of water and 2.2 ml of 1 M Tris (pH9). Dilute to a 1× working stock with negative-control solution. The 20× stock can be stored at −20 °C for 1 year.
1 M Tris pH9
Add 12.114 g of Tris base to 100 ml of water, and lower the pH to 9 with 1 M HCl. This solution can be stored at RT for 6 months.
5 N NaOH
Add 20.0g of NaOH to 80 ml of water. When cooled, bring the final volume to 100 ml by adding water. This solution can be stored at RT for 6 months.
1 M HCl
Add 16.53 ml of 12.1 N HCl slowly to 83.47 ml of water in a fume hood. This solution can be stored at RT for 6 months.
EQUIPMENT SETUP
COPAS-XL parameters
Established COPAS parameters for sorting Tg(ins:PhiYFP-Eco.NfsB,sst2:TagRFP)lmc01 larvae are as follows:
Sort:/Well = 1; Delay = 60 (ms); Width = 40 (ms); Thresholds: Signal = 15; TOF Minimum = 2000 (ms); Gains: Signal (Red) = 5; Multiplier (Red) = 900 (v); Pressure: Sheath = 4.00 (psi); Sample = 0.3 (psi); Sorter = 2.75 (psi); Clean = 10.00 (psi). These parameters (along with a COPAS ‘top-cup’ larvae density of 1–2 fish per ml) avail consistent filling of 96-well plates in 2–2.25 min. ▲ CRITICAL Care must be taken to optimize sorting parameters to minimize dispensing errors (e.g., blank wells, multiple organisms per well). Using the parameters above, our error rate was 1.67 per plate. In an effort to account for this, increases in sample size per condition can be applied (e.g., we used a sample size of 16 for Wang et al., despite requiring a sample size of only 14). In addition, an R-based code used to evaluate ARQiv data in real time (see below) flags ‘outlier’ wells exhibiting fluorescence Signal levels above Background in five or more regions within a single well (indicating multiple larvae), and it eliminates such wells from the analysis.
Plate reader parameters
Established parameters for Tg(ins:PhiYFP-Eco. NfsB,sst2:TagRFP)lmc01:
Yellow fluorescent protein (YFP)
Excitation: 520 nm; Bandwidth: 5 nm; Emission: 546 nm; Bandwidth: 5 nm; Mode = Top; Gain (Manual) = 236; Settle time = 30 ms; Z-Position (Manual) = 22,040 μm; Multiple reads per well, Type: Square (filled), Size: 3 × 3; Border = 200 μm; Flashes: Mode 2 [100Hz] = 10; Integration time = 20 μs.
RFP
Excitation: 555 nm; Bandwidth 5 nm; Emission: 585 nm; Bandwidth 10 nm; Mode = Top; Gain (Manual) = 212; Settle time = 30 ms; Z-Position (Manual) = 22,040 μm; Multiple reads per well, Type: Square (filled), Size: 3 × 3; Border = 200 μm; Flashes: Mode 2 [100Hz] = 10; Integration time = 20 μs.
ARQiv software setup
We have created an R-based software package, ‘ARQiv’, containing functions for developing and optimizing ARQiv-HTS assays and for processing ARQiv-HTS data in real time during full-scale screens. To simplify the analysis process, a graphical user interface (GUI)43 was created, which allows the user to easily input files and obtain relevant data outputs (Supplementary Fig. 3)44. The ARQiv software package, including download and installation instructions, has been placed on the GitHub open-source website (https://github.com/mummlab/ARQiv).
PROCEDURE
Prescreen assay optimization—fluorescence detection parameters ● TIMING 1 d
▲ CRITICAL Generation of zebrafish transgenic lines for quantitative reporter-based assays typically takes 9 months to a year, and additional time is required to generate large breeding stocks (Fig. 1). The following protocol uses the dual-reporter homozygous transgenic line Tg(ins:PhiYFP-Eco.NfsB,sst2:TagRFP)lmc01, in which the insulin promoter drives the expression of YFP (PhiYFP, Evrogen) in beta cells, whereas the somatostatin 2 promoter drives RFP (TagRFP, Evrogen) in adjacent delta cells in pancreatic islets. The process of drug-induced precocious pancreatic beta-cell formation45 can be visualized using fluorescence microscopy45 or it can be quantified via ARQiv3,5,46. Here we outline the ARQiv-HTS drug discovery assay used to identify compounds that increased the number of insulin-producing beta cells in the pancreas.
-
1|
At the age(s) when fluorescence will be assessed, dispense ~12 each transgenic (or labeled) and nontransgenic (or unlabeled) larvae individually into single wells of a black U-shaped 96-well microtiter plate containing 330 μl of negative-control solution per well. These are used in the following steps to optimize all basic assay parameters: Excitation, Emission, Bandwidth, Z-Position, and Gain. Note that U-shaped well plates are optimal for ARQiv detection of zebrafish embryos and larvae3.
▲ CRITICAL STEP To maximize transgene expression intensity and to ensure maximal numbers of transgenically equivalent larvae, maintain only homozygous breeder stocks.
-
2|
Anesthetize fish by diluting a 34× stock solution of 0.68% (vol/vol) Eugenol to a final concentration of 0.02% (i.e., add 10 μl of 34× Eugenol to 330 μl of negative-control solution that is already in the well). Wait ~15 min for the anesthetic to take effect.
-
3|
Place the plate into a fluorescence plate reader, allowing Z-dimension focus and regionally reported scans (e.g., Tecan Infinite M1000).
-
4|
Establish an optimal Z-Position setting by running a Z-dimension focus assay on approximately four to six wells containing transgenic fish. Use the published Ex/Em maxima specific to your fluorophore for initial z-depth optimization. For example, the published Ex/Em maxima for Tg(ins:PhiYFP-2a-nsfB, sst2:tagRFP)lmc01 larvae are 525/537 (PhiYFP) and 555/584 (tagRFP). Use the average of the Z-Positions providing maximal signal detection between representative wells for all subsequent scans.
▲ CRITICAL STEP Ensure that the fish are centered in each well if regional scans are not an option with the Z-Position function.
-
5|
Establish optimal Excitation, Emission, and Bandwidth settings. Excitation and emission scan functions are useful for this, whereby a range of excitation wavelengths can be evaluated at a specific emission wavelength and vice versa. Keep both bandwidths at the minimal value to maximize differences between wavelengths. It is important to assess Ex/Em spectra for both transgenic (or labeled) and nontransgenic (or unlabeled) fish to account for potential signal detection interference from autofluorescence—i.e., to establish settings for which signal detection is maximal and autofluorescence is minimal. Optimal Bandwidth settings are defined empirically and are best determined after establishing optimal excitation and emission wavelengths. In our experience, narrower bandwidths (e.g., 5 nm) tend to minimize autofluorescence, thereby maximizing true signal detection; however, some fluorophore ranges tolerate wider bandwidths (e.g., 20 nm) without compromising signal detection.
-
6|
Establish Optimal Gain. It is critical that the photomultiplier ‘Gain’ setting remains consistent throughout the screen, as it is not possible to normalize the data across scans if this parameter fluctuates. Accordingly, the Gain should be set to a value that allows maximal effect ranges to be resolved. An ‘Optimal Gain’ setting can be obtained for the transgenic fish to establish a reasonable starting value. Assuming that these values are near the upper limit of signal to be detected, subtract ~20–50% from the Optimal Gain setting to allow for the possibility of test compound performances exceeding that of the control.
Prescreen assay optimization—defining the signal ● TIMING 1 d
-
7|
Establish a large Background data set using nontransgenic (or unlabeled) fish to determine autofluorescent signal levels. Dispense 96 nontransgenic (or unlabeled) fish individually into wells of a new black U-shaped 96-well microtiter plate containing 330 μl of negative-control solution per well.
-
8|
Repeat Steps 2 and 3, and then use the optimized reporter acquisition parameters defined in Steps 4–6 to scan all wells in a region-by-region manner (as depicted in Walker et al.3). Select the ‘Multiple Reads per Well’ check box to ensure that the entire area of each well is scanned. Select ‘Square (filled)’, ‘3 × 3’, and ‘300’ (μm) in the ‘Type’, ‘Size’, and ‘Border’ pull-down menus, respectively.
▲ CRITICAL STEP A plate reader that reports regionalized scan values individually per each region, rather than averaged across all regions (e.g., the Tecan Infinite M1000), is necessary to account for variability in larvae orientation, thus facilitating elimination of noise (e.g., blank regions) from the fluorescent signal.
? TROUBLESHOOTING
-
9|
Save the data from the microplate reader in .xml format.
-
10|Calculate Background (B) to establish a cutoff value for delineating fluorescent signal from nonfluorescent background. A standard detection limit equation is used for this—i.e., the average plus three standard deviations of the maximal regional values from the nontransgenic data:
where μn and sn are the mean and standard deviation of the negative control, respectively.
Background calculations are performed in this protocol using the ARQiv package, but they can also be done manually using the equation above. To use the ARQiv package, open R Studio and load the GUI by typing the following at the prompt in the Console window command line (see Supplementary Fig. 3 for an overview of the ARQiv GUI):
> library(gWidgets2RGtk2) > library(ARQiv) > GUI()
▲ CRITICAL STEP The GUI has two sections: the top section is used for all assay optimization functions and the lower section will be used later for analyzing test compounds (note that there are similarly named windows in each section; be sure to input data only into the appropriate section for each type of analysis; see Supplementary Fig. 3).
-
11|
Upload the background file (.xml format) by clicking ‘bkgd’ button in the ‘INPUT’ section and browsing for and selecting the appropriate file. Upon selection, the name of the file should appear in the adjacent window.
-
12|Click the ‘BACKGROUND’ button in the ‘OUTPUT’ section to reveal the calculated Background value cutoff. Once ‘Background’ is established, the R-based code will use that cutoff value to automatically calculate ‘Signal’ levels for each well. ‘Signal’ is defined as the sum of all regional values per well from the transgenic data greater than the Background limit (B). That is, if more than one region of the well produces signal, these values are summed to obtain the total signal for that well. The formula for signal calculation is as follows:
If calculating Background values manually in Step 10, insert the manually calculated Background cutoff by selecting ‘VALUE’ and inputting the value in the adjacent window. When doing this, ensure that the acquisition parameters defined in Steps 4–6 have been maintained. If these are adjusted for any reason, a new Background value must be calculated.
Prescreen assay optimization—determination of sample size ● TIMING 7 d
▲ CRITICAL To establish an appropriate sample size for detecting compounds that exhibit a desired effect, the signal from transgenic fish treated with a positive-control compound (ideally one producing the maximal desired effect) is compared with that from transgenic fish treated with vehicle alone (negative control). Using large data sets from positive and negative controls, ‘power calculations’ are then used to determine a minimum sample number required to identify compounds of interest (i.e., ‘hit’ drugs). For HTS, false-positive and false-negative rates (i.e., type I and type II errors, respectively) need to be minimized as much as possible while also adjusting for the desired compound effect size (i.e., percentage of effect relative to the positive control) and accounting for practical limitations of the screening process itself.
-
13|
Establish large positive- and negative-control data sets. To do this, dispense transgenic (or labeled) fish individually into each well of two black U-shaped 96-well microtiter plates. One plate will serve as the positive control (condition producing maximal signal, e.g., containing a positive-control compound) and the other as the negative control (condition producing minimal signal, e.g., containing vehicle).
-
14|
Treat for the amount of time required for the desired outcome to be achieved. Next, perform scans as described in Step 8.
-
15|
Save the data from each plate in .xml format.
-
16|
Access the GUI per Step 10, and upload the positive- and negative-control files (.xml format) by clicking the ‘+’ and ‘−’ buttons in the ‘INPUT’ section to browse for and select the appropriate files. Upon selection, the name of the file should appear in the adjacent window.
▲ CRITICAL STEP If control data sets have been manually compiled into a single .csv format (i.e., two columns, one positive and one negative), select the ‘CSV’ file type and browse for the file using the ‘+/−’ button in the ‘INPUT’ window. An example control data set is provided in the Supplementary Data for testing ‘Assay Optimization’ functions (Supplementary Data file named “Excel.Pos.Neg.Control”; .csv format).
-
17|
Click the ‘ASSAY DIRECTORY’ button in the ‘OUTPUT’ section to select a destination for the sample size file.
-
18|
Click the ‘SAMPLE SIZE’ button in the ‘OUTPUT’ section. This produces a ‘SampleSize.csv’ file that is automatically saved. The R-based code that we developed calculates sample size across a range of error rates and effect sizes for both raw and log2-transformed data. Note that ‘DEFAULT’ effect sizes are calculated at 25, 50, 75, and 100% of the positive control. Alternatively, a ‘CUSTOM’ window is available for entering a user-defined effect size. Table 1 provides an example of the type of data that can be obtained. Alternatively, sample size can be manually calculated using the control data sets established in Steps 13–15 and the equation provided in Box 1.
TABLE 1.
Sample size calculation.
| Drug effect (% of positive control) |
Normalized scaling factor | Minimum sample no | Type I (α) error | Type II (β) error | ||
|---|---|---|---|---|---|---|
|
| ||||||
| P | Zα | P | Zβ | |||
| Raw signal | ||||||
| 100% | 1.000 | 15.14 | 0.05 | 1.96 | 0.2 (80% power) | 0.84 |
| 75% | 1.249 | 17.25 | 0.05 | 1.96 | 0.2 (80% power) | 0.84 |
| 50% | 1.663 | 21.89 | 0.05 | 1.96 | 0.2 (80% power) | 0.84 |
| 25% | 2.489 | 39.10 | 0.05 | 1.96 | 0.2 (80% power) | 0.84 |
| 100% | 1.000 | 25.03 | 0.05 | 1.96 | 0.05 (95% power) | 1.64 |
| 75% | 1.249 | 28.52 | 0.05 | 1.96 | 0.05 (95% power) | 1.64 |
| 50% | 1.663 | 36.18 | 0.05 | 1.96 | 0.05 (95% power) | 1.64 |
| 25% | 2.489 | 64.64 | 0.05 | 1.96 | 0.05 (95% power) | 1.64 |
| Log2 signal | ||||||
| 100% | 1.000 | 7.05 | 0.05 | 1.96 | 0.05 (95% power) | 1.64 |
| 75% | 1.249 | 9.41 | 0.05 | 1.96 | 0.05 (95% power) | 1.64 |
| 50% | 1.663 | 14.73 | 0.05 | 1.96 | 0.05 (95% power) | 1.64 |
| 25% | 2.489 | 35.19 | 0.05 | 1.96 | 0.05 (95% power) | 1.64 |
| 100% | 1.000 | 10.00 | 0.05 | 1.96 | 0.01 (99% power) | 2.33 |
| 75% | 1.249 | 13.36 | 0.05 | 1.96 | 0.01 (99% power) | 2.33 |
| 50% | 1.663 | 20.91 | 0.05 | 1.96 | 0.01 (99% power) | 2.33 |
| 25% | 2.489 | 49.97 | 0.05 | 1.96 | 0.01 (99% power) | 2.33 |
Box 1. Calculation of sample number.
Formula for sample size calculation (where, Zα, Zβ, σ, μp, μn represent the desired level of statistical significance, desired power, standard deviation (of the control sample with the greatest variance), mean of the positive control, and mean of the negative control, respectively):
For example, the sample number calculation (P = 0.05/power = 95.0%) showed that an n of ~14 is sufficient to detect a drug effect at 50% of the strength of the positive control (Table 1). However, an n of 16 will be used to account for occasional automation errors (blank wells), and to adjust for the layout of a standard 96-well plate.
Formula for scaling effect size (where, μp, μn, and R, represent the mean of the positive control, mean of the negative control, and effect size ratio, respectively:
For example, for a desired drug effect size of 50% (R = 0.5), where μp = 8,000 and μn = 4,000, the normalized scaling factor is 1.33. Sample size or predicted SSMD score (below) would be calculated by dividing the positive-control value by 1.33 (instead of 2.00) to produce a minimum sample size or SSMD score corresponding to a predicted drug effect of 50% (i.e., 6,000, halfway between 8,000 and 4,000).
▲ CRITICAL STEP Performing a log2-transformation of the signal data can be used to account for variability, thereby increasing signal resolution. The ARQiv R package will do this automatically and produce results from both raw and log-transformed data to allow users to determine whether transformation is useful for their assay.
▲ CRITICAL STEP If calculating the sample size manually, the final sample number should be set at an integer convenient for microplate assay design (e.g., 6, 8, 12, and 16) that is greater than the calculated minimum sample size. Rounding up on the sample number serves to negate automation errors (e.g., blank wells).
Prescreen assay optimization—QC calculations ● TIMING 7 d
-
19|
To assess assay quality, use the positive- and negative-control data sets to calculate Z′ factor and SSMD QC scores. The control data sets were uploaded in Step 16; therefore, simply access the GUI per Step 10.
-
20|
Calculate the Z′ factor. This can be automatically calculated by simply clicking the ‘Z′-FACTOR’ button in the ‘OUTPUT’ section. The calculated score will appear in the adjacent window. Alternatively, Z′ factor can be manually calculated using the control data sets established in Steps 13–15 and the equation described in Box 2.
Box 2. Statistical assay QC tests.
Z′ factor
This is the most popular measure of HTS quality; it compares signals from positive- and negative-control data, taking into account the range and variability of both to determine assay quality. Minimally, for an HTS-ready assay a Z′ factor ≥0 is required; optimally the Z′ factor should range from 0.5 to 1 (ref. 52). Formula for Z′ factor calculation (where μp, μn, σp, and σn, are the sample means and sample standard deviations of the positive and negative controls, respectively):
SSMD QC
The Z′ factor can be problematic because of the assumption of normal distribution and sensitivity to unequal variance and/or outliers, elements that typically arise during in vivo HTS. An alternative assay quality measure that is less susceptible to these issues is the SSMD (formula presented below)37,39,40,52. Moreover, variants of this equation can be applied to both QC and hit selection in HTS applications (see below). The use of the ‘robust’ SSMD (SSMD*) is preferred for QC (SSMD QC), as variance distorts the sample mean more than the sample median40. Formula for the robust SSMD* estimate under unequal variance, when outliers are evident (where , , , and are the sample medians and sample median absolute deviations in the positive and negative controls, respectively):
For example, the S:B of 7 d.p.f. Tg(ins:PhiYFP-Eco.NfsB,sst2:TagRFP)lmc01 compared with nontransgenic age-matched siblings is ~5.5:1 (for YFP)5. DAPT-treated positive-control fish produced a robust SSMD* of 1.06 (2.25 for log2-transformed data) relative to 0.1% (vol/vol) DMSO-treated negative controls, suggesting that the assay is of sufficient quality (Supplementary Fig. 1).
▲ CRITICAL STEP As an example, inputting the .csv control file (‘Excel.Pos.Neg.Control’) and a Background cutoff value of 3,970 will produce a Z′ factor of −1.36.
▲ CRITICAL STEP Assay quality metrics can be used for assay optimization and to balance practical considerations (e.g., keeping the sample size as small as possible) with desirable assay quality metrics (e.g., Z′ factor >0.5).
-
21|
Calculate the SSMD QC score. This can be automatically calculated by simply clicking the ‘SSMD QC’ button in the ‘OUTPUT’ section. The calculated score will appear in the adjacent window. Alternatively, the SSMD QC score can be manually calculated using the control data sets established in Steps 13–15 and the equation described in Box 1.
▲ CRITICAL STEP As an example, inputting the .csv control file (‘Excel.Pos.Neg.Control’) and a Background cutoff value of 3,970 will produce an SSMD QC score of 1.06.
▲ CRITICAL STEP Although the Z′ factor is considered an ‘industry standard’ for HTS, this test was developed for highly reductionist HTS assays that typically exhibit less variance than cell-based or whole-organism assays. The Z′ factor may therefore be too stringent for most ARQiv-HTS applications. Accordingly, because of the increased tolerance of compressed signal windows and higher variance, we recommend using the SSMD QC for ARQiv-HTS assay quality tests.
Prescreen assay optimization—SSMD hit score estimation ● TIMING 1 d
▲ CRITICAL A metric for identifying compounds of interest (i.e., ‘hits’) will need to be established by calculating an estimated SSMD score cutoff. The R code that we developed uses computational iterations to randomly sample the positive-and negative-control data sets in subsets defined by the sample size. The resulting set of ‘virtual assays’ approximates the positive-control compound’s performance in a large-scale screen. As for the sample size, ‘DEFAULT’ effect sizes are calculated at 25, 50, 75, and 100% of the positive control. However, the effect size can be scaled to any percentage of interest relative to the positive control to set an SSMD score cutoff for flagging hit compounds.
-
22|
The control data sets were uploaded in Step 16; therefore, simply access the GUI per Step 10.
-
23|
Set the desired effect size, sample size, and number of iterations (e.g., 10,000) in the corresponding windows provided.
-
24|
Click the ‘SSMD ESTIMATE’ button in the ‘OUTPUT’ section to produce an ‘ssmd_est.csv’ file that is automatically saved. See Table 2 and Figure 2 as an example of the data output. Alternatively, the SSMD hit score cutoff can be manually calculated using the equation shown in Box 3.
TABLE 2.
Predicted SSMD scores.
| Effect Size | Predicted SSMD (raw) | Predicted SSMD (log2) |
|---|---|---|
| 100% | 1.21 | 2.27 |
| 75% | 1.17 | 2.01 |
| 50% | 1.11 | 1.67 |
| 25% | 0.97 | 1.17 |
Figure 2.
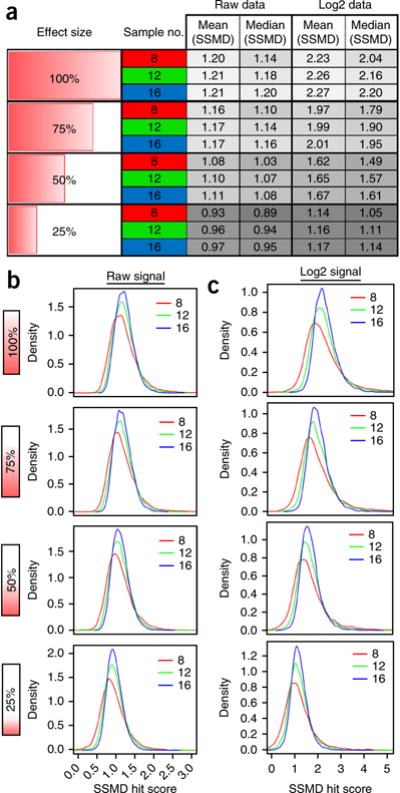
Predicted SSMD scores using bootstrapping. (a and b) Computational random sampling with replacement (‘bootstrapping’) of positive- and negative-control data sets provides a means of estimating HTS assay performance via a series of ‘virtual’ experiments at a defined sample size. This strategy is useful for establishing predicted SSMD scores relative to effect size, particularly for in vivo HTS assays in which same-day follow-up is desirable (e.g., visual phenotyping of ‘hit’ plates following ARQiv). Real-time data outputs (Fig. 5) facilitate same-day follow-up, but bootstrapping is necessary to establish reasonable approximations of hit criteria before initiating the full-scale screen. (a) Predicted SSMD scores (gray columns, right) were generated via 10,000 bootstrap iterations of raw and log-transformed control data. SSMD hit estimates were calculated relative to effect size (left column) and at three different sample numbers (8, 12, and 16; red, green, and blue, respectively, middle column). The results show SSMD estimates decreasing with smaller effect sizes and demonstrate the expanded dynamic range that log-transforming data provide. Note that for both raw and log-transformed data sets, lowering the sample number decreases the SSMD estimate (mean/median). (b) Density plots of bootstrap iterations using raw data show how increasing sample size reduces SSMD variance. Comparisons across effect size show a gradual decrease in predicted SSMD average as effect size is reduced. (c) Density plots of bootstrap iterations using log-transformed data also show reduced variance with larger sample sizes. Comparisons across effect size show the expanded SSMD score range following log transformation.
Box 3. SSMD-based quantification of effect size—hit identification.
The formula for SSMD hit score estimation is provided below (this equation is also used for real-time ARQiv-HTS data analysis for hit identification).
Formula for calculating the SSMD score of the ith drug compound with replicates n (where n represents sample number; and Si, are the sample mean and standard deviation of dij, where dij is the paired difference between the jth measured value of the ith compound and the median value of the negative-control data; and Γ() is a gamma function):
For example, after treatment with DAPT (positive-control compound) for 4 d, the 7 d.p.f. Tg(ins:PhiYFP-Eco.NfsB,sst2:TagRFP)lmc01 larvae produce a characteristic SSMD score. All other drug library compounds are compared with this positive control with regard to modulation of insulin reporter activity and potentially pancreatic beta-cell mass.
▲ CRITICAL STEP SSMD calculations are better suited to evaluating drug performance for in vivo HTS. Calculating the SSMD hit criteria with the appropriate sample number is critical because sample number affects variance, thus significantly impacting SSMD score estimates and associated confidence intervals.
As an example, the predicted SSMD hit score derived from Tg(ins:PhiYFP-Eco.NfsB,sst2:TagRFP)lmc01 control data sets—calculated from 10,000 iterations of the positive- and negative-control data at a 50% effect size and sample size of 16—suggests a hit selection cutoff of 1.11 (raw data) or 1.67 (log transformed data); see Table 2 and Figure 2. Lower sample numbers decrease predicted SSMD averages, particularly for log-transformed data (Fig. 2). However, lower sample numbers also result in higher variance in the range of SSMD scores, thus leading to less desirable confidence intervals47. The full implications of sample number, effect size, statistical power, and confidence intervals have recently been elucidated eloquently by Halsey and colleagues39.
Large-scale in vivo drug discovery—egg production and larvae maintenance ● TIMING 1–3 d
▲ CRITICAL The optimal breeding age for zebrafish is considered to be between 6 and 12 months48. Ensure that your large mating stock(s) of fish are cycled every ~3–6 months to provide consistent fecundity throughout the drug discovery process. We have found that a 1:2 (male to female) ratio is ideal for maximal production of viable embryos.
-
25|
Use customized mass fish-breeding chambers to collect large quantities of Tg(ins:PhiYFP-Eco.NfsB,sst2:TagRFP)lmc01 eggs. Gridded plastic canvases are used to replace the base of upper ‘mating’ chambers, allowing fertilized eggs to descend to a middle ‘collection’ chamber that is ported with 250 μM nylon woven mesh to allow water to flow through while retaining eggs. The mating and collection chambers are nested inside an intact bottom ‘water’ chamber (Supplementary Fig. 2b).
▲ CRITICAL STEP Keeping the water height in the upper chamber relatively low (4–6 cm) and the density of fish relatively high (1–2 adult fish per 10 cm2 or 100–200 adult fish per large-scale grouped breeding unit) ensures maximal interaction and abundant mating. Nested breeding units can also be angled to facilitate shallow ‘ramps’ within the mating chamber to further facilitate breeding49.
? TROUBLESHOOTING
-
26|
Collect eggs every hour for 1–2 h after light onset, and maintain as timed subpopulations. Pour the contents of the egg collection chamber through a fine-mesh strainer, rinse with E3 medium to remove detritus, and dispense into large (>500-ml capacity) containers.
▲ CRITICAL STEP Remove any eggs dropped overnight (prior to light onset) that would otherwise confound the first timed subpopulation. This is easily achieved by moving the mating chamber to a fresh nested collection chamber and water unit at light onset.
-
27|
Estimate the number of eggs using a graduated cylinder (~600 eggs per ml, Supplementary Fig. 2c).
-
28|
Maintain eggs in E3 medium at a volume density of ≤2 eggs per ml and a surface area density of ~1 egg per 3 cm2, with standard lighting and temperature conditions (14:10 h (light:dark) cycle, 28–29°C). We prefer Corning square bioassay dishes (245 × 245 mm), which have a capacity to hold ~400 embryos. At ~16–24 h.p.f., transfer the eggs to 200 nM PTU in E3 medium (i.e., 1× PTU solution).
▲ CRITICAL STEP To accurately detect fluorophores in the eye of the zebrafish, the transfer to 1× PTU must occur at ~16 h.p.f. to avoid early pigmentation. Alternatively, the eggs can be maintained overnight at 25 °C to delay development. This allows PTU to be added early the next morning, once the embryos have reached a developmental stage equivalent to 16 h.p.f. at 28–29°C (i.e., 14 somite stage). This can be preferable to transferring the eggs to 1× PTU at 2–4 AM. For detecting fluorophores in other regions, it is sufficient to transfer to 1× PTU at ~24 h.p.f.; maintain the eggs at 28–29 °C overnight in this case before transferring to 1× PTU.
-
29|
Maintain the eggs at a media volume density of ≤2 eggs per ml and a surface area density ~1egg per 3 cm2, with standard lighting and temperature conditions (14:10 h (light:dark) cycle, 28–29 °C). To ensure maximal viability, remove all detritus daily (waste matter, unfertilized eggs, chorions, and so on), and replace the medium with fresh 1× PTU throughout the incubation period. Continue to monitor the developmental progress of the embryos using a stereomicroscope until 3 d.p.f.
▲ CRITICAL STEP This ensures sustained action of PTU, which is photodegradable, while also removing potential autofluorescent contaminants. The chorion debris is much lighter than the hatched embryos and easily decanted via several rinses.
▲ CRITICAL STEP Remove unhealthy eggs or larvae daily until drug treatments commence. The morphological parameters that we use to assess larval zebrafish health include cardiac edema, curvature of the spine, and small eye.
? TROUBLESHOOTING
Large-scale in vivo drug discovery—preparation of drug plates ● TIMING 1–12 h
▲ CRITICAL Here we detail the compound titration process for the qHTS assays performed in Wang et al. We used a robotics platform (Fig. 3) and software provided by Hudson Robotics (SoftLinx) for integration of work cells to automate this process with liquid handlers. Drugs (Johns Hopkins Drug Library) were supplied as 5 μl of a 10 mM solution in 100% (vol/vol) DMSO. For the demonstration, a 1:2 dilution series is used to produce six different concentrations (4–0.125 μM) of a single compound on a 96-well plate with 16 fish assessed per concentration (96 fish assayed per compound). In addition, drug stock plates used the interior 10 columns only (thus 80 drugs). Therefore, we placed positive- and negative-control diluent and/or compounds in the outer columns of the stock plate to facilitate automated ‘bookending’, whereby control plates border each set of ten drug plates (Supplementary Fig. 4). This strategy allows assay quality assessments to be temporally synchronized to each subset of ten drugs, controlling for reporter changes due to extended assay timelines (e.g., 16 h) while enabling qHTS at relatively high sample numbers (for flowchart, see Fig. 4).
Figure 3.
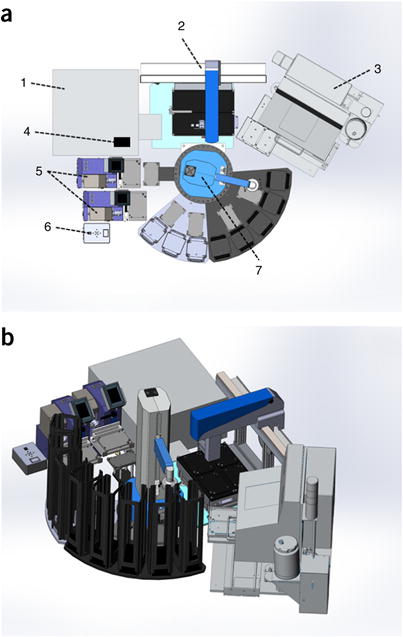
Robotics platform schematic. (a) Top-down schematic of ARQiv-HTS workstation. (1) Tecan Infinite M1000Pro microplate reader performs ARQiv scans, (2) Solo automated pipettor dispenses and serially dilutes drug stocks, (3) the COPAS XL sorts/dispenses fish into microtiter plates, (4) the barcode reader scans barcode-labeled working stock plates and drug treatment plates, (5) the Micro 10× robotic dispenser dispenses E3/anesthetic/metronidazole stock solutions, (6) the 4-Way Valve automates transition between stock solutions for the robotic dispenser, (7) the PlateCrane plate handler, with eight stacks and five auxiliary nests, moves microtiter plates to various workstation coordinates. (b) Isometric projection of the ARQiv-HTS workstation shown in a (rotated ~60 degrees clockwise). Diagrams courtesy of Hudson Robotics.
Figure 4.
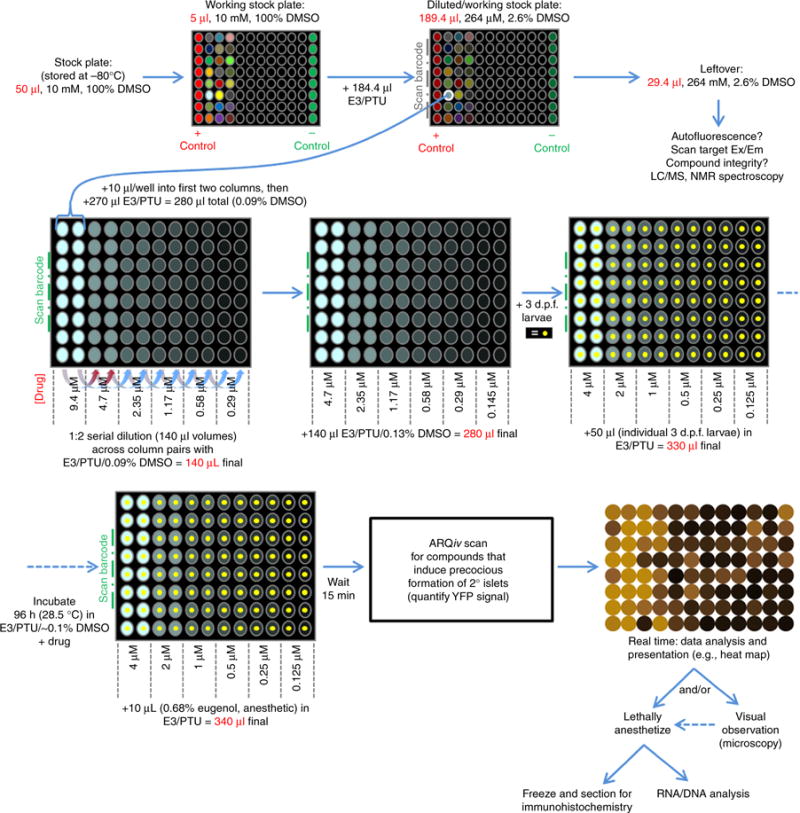
Example protocol flowchart. Diagram of ‘pancreatic beta-cell’ ARQiv-HTS assay process involving (clockwise from top left): tested compounds (stock plates); predilute for serial dilution; serially diluted per qHTS principles; dispensing larvae; incubation period; ARQiv scans; and real-time data process.
-
30|
Use a barcode print and apply unit (e.g., PA1000 Barcode Print and Apply, Hudson Robotics) to adhere a unique label and barcode to all multiwell plates used during the drug screening process, including stock plates (unless already labeled appropriately), drug treatment plates, and control plates. Note that serialized labels for drug treatment and control plates corresponding to drug stock plate, column, and row are particularly useful (e.g., P7A5, for plate 7, column A, row 5).
-
31|
Remove drug stock plate(s) from storage at −80 °C and thaw at RT, protected from light. Chemical libraries will thaw at a rate dependent on the solvent being used. For example, a library maintained at −80 °C in 100% (vol/vol) DMSO will fully thaw in 3 h at RT.
! CAUTION Store thawing plate(s) in a safe location, such as a fume hood, with hazmat labeling and sealing tape. Wear appropriate PPE—eyewear, two sets of gloves, and a lab coat—when handling chemicals.
-
32|
Prepare a fresh positive-control solution for automated preparation of drug plates. Add 5 μl of a 10,000× positive-control solution (in 100% (vol/vol) DMSO) to the first column (no. 1) of the drug stock plate, and 5 μl of negative-control solution (100% (vol/vol) DMSO) to the last column (no. 12).
▲ CRITICAL STEP Preparing a 10,000× stock solution facilitates automated serial dilution of the positive control such that fish are treated at 4×, 2×, 1×, 0.5×, 0.25×, and 0.125× concentrations (in a 0.1% (vol/vol) DMSO final)— a strategy that helps negate lot variability and thus ensures that maximal effects are observed in the control condition. However, for the Wang et al. screen this was not possible because of solubility issues; instead, a twofold dilution series from 200 μM to 6.25 μM DAPT was prepared manually. Accordingly, if a single dilution protocol cannot be used for test compounds and the positive control, it will be necessary to prepare positive-control plates manually (or to develop an independent dilution series strategy for this aspect of the screen).
-
33|
Drug dilution series. To facilitate overnight preparation of drug plates, use a robotics-integrated plate handling system (e.g., Hudson Robotics, PlateCrane EX, Fig. 3) and liquid handlers (e.g., Micro 10× and Solo, Hudson Robotics) to automate the drug dilution process. First, determine how many drugs in the stock plate(s) can be screened according to the number of viable larvae, and add 184.4 μl of 1× PTU medium to only the drugs that will be screened in this round; mix well. Each test drug is now at 264 μM in 2.6% (vol/vol) DMSO (Fig. 4).
-
34|
If you are unable to dilute all compounds in the drug stock plate, reseal the remainder with microtiter plate sealing tape and store it at 4°C until the next screening session (ideally, use within 1 week) to avoid additional freeze/thaw cycles.
-
35|
Manually place the drug treatment plates with barcodes corresponding to the drugs that will be screened in sequence (top to bottom) in PlateCrane stack(s).
▲ CRITICAL STEP Ensuring that labeled plates are in sequence will facilitate drug processing later. However, if a discrepancy arises, the SoftLinx software maintains a record linking drug stock plate positions to each drug treatment plate.
-
36|
Use the PlateCrane to move individual plates in sequence to a Micro 10×.
-
37|
Use one Micro 10× platform to add 270 μl of 1× PTU to each well of the first two columns (1 and 2) of each drug treatment plate, use the PlateCrane to return to a new stack, and repeat until all plates are processed.
-
38|
Use the PlateCrane to move individual plates (now in reverse sequence) to a second Micro 10×.
-
39|
Use the second Micro 10× to add 140 μl of dilution buffer 1 (PTU/E3/0.09% (vol/vol) DMSO) to each well of the remaining 10 columns (3–12) of the drug treatment plate, use the PlateCrane to return to a new stack (such that top to bottom sequence is restored), and repeat until all plates are processed.
-
40|
Use the PlateCrane to move the drug stock plate to the barcode reader to link the plate to the SoftLinx software, and then move the plate to the Solo platform for serial dilution.
-
41|
Use the PlateCrane to move two 200-μl tip boxes to the Solo platform.
-
42|
Use the PlateCrane to move the first drug treatment plate to the barcode reader, and then to the Solo platform. Note that the stock and treatment plate barcodes are now linked in the SoftLinx software.
-
43|
Use the Solo automated pipettor to dispense 10 μl of a 264 μM drug stock solution into each well of the first two columns of the drug treatment plate. The test drug is now at 9.4 μM in 0.09% (vol/vol) DMSO (Fig. 4).
-
44|
Use the Solo automated pipettor to perform a 1:2 dilution series across the drug treatment plate. Transfer 140 μl of the solution in the first column to the third column (i.e., into 140 μl of dilution buffer 1); repeat this process for the second and fourth columns. Continue the process between paired columns per Figure 4. Discard the final 140 μl of diluted drug solution. Each well should now contain 140 μl of diluted drug solution in PTU/E3/0.09% (vol/vol) DMSO, ranging from 9.4 μM to 290 nM (Fig. 4).
▲ CRITICAL STEP For qHTS assays, we use a 1:2 dilution series, testing all compounds at either six or eight different concentrations (i.e., in rows or columns of a 96-well microtiter plate, depending on the sample number required to detect the desired effect size). Drug dilution protocols need to ensure consistent diluent concentrations. Diluting from the desired diluent concentration into the same condition (e.g., 0.1% (vol/vol) DMSO) across the dilution series is a simple method for achieving this. We favor the use of 0.1% (vol/vol) DMSO rather than 1% (vol/vol) DMSO in order to avoid deleterious effects of this diluent on embryonic/larval zebrafish development50. Ongoing experiments are assessing whether 0.01% (vol/vol) DMSO is sufficient for compound penetration in hatched embryos/larvae, as this would avoid documented additional effects of this diluent on locomotion and induction of stress proteins, and deleterious effects on xenobiotic metabolism50,51.
-
45|
Use the PlateCrane to move each diluted drug plate to a new stack after the dilution series is complete; repeat this step until all plates are processed.
-
46|
Use the PlateCrane to move individual plates (now in reverse sequence) to a Micro 10×.
-
47|
Use the Micro 10× to add an additional 140 μl of dilution buffer 2 (PTU/E3/0.13% (vol/vol) DMSO) to all wells at the end to bring the volume to 280 μl per well. Use the PlateCrane to return to a new stack (such that top to bottom sequence is restored), and repeat until all plates are processed. Concentrations across the plate will range from 4.7 to 0.145 μM (Fig. 4).
■ PAUSE POINT Drug plates can be kept at RT overnight.
-
48|
Store leftover compound not applied to serial dilutions (in this example, ~29 μl at 264 μM) at 4°C for up to 1 week. Leftover compounds can be used to determine compound autofluorescence at established assay parameters, for subsequent confirmatory assays and/or to verify compound identity/integrity (e.g., via NMR spectroscopy).
Large-scale in vivo drug discovery—dispensing larvae in 96-well plates ● TIMING 1 h to several hours
-
49|
After preparation of drug dilution plates, use a COPAS-XL unit (Fig. 3) to sort and dispense individual Tg(ins:PhiYFP-Eco. NfsB,sst2:TagRFP)lmc01 larvae in 50-μl droplets (1× PTU) into each well. The volume will now be 330 μl per well, and drug concentration will range from ~ 4 μM to ~125 nM in 0.1% (vol/vol) DMSO. See Equipment Setup for COPAS-XL sorting parameters. Use the PlateCrane to move the drug dilution plates in sequence from the stack(s) to the COPAS XL platform and back to a new stack.
▲ CRITICAL STEP The dilution factor associated with dispensing fish into wells must be accounted for. Assuming a standard droplet size of 50 μl and assay incubation volume of 330 μl per well, fish dispensing incurs an additional 15.25% reduction in drug and diluent concentration. In practice, droplet size can vary between dispensed larvae—our tests show a range of 48.9–53.3 μl (50.7 ± 1.1 μl). Such variation could, in theory, complicate titration schemes. We recommend dispensing into a large volume to reduce relative differences between wells intended to represent like conditions. However, any variation in dispense volume will be applied randomly without any specific bias and thus will be accounted for during assay QC tests. Optimal system pressure settings may vary between systems, impacting the size of the dispensed liquid droplet (for additional details see Supplementary Fig. 5).
▲ CRITICAL STEP In our experience, 3-d-old larvae do not exhibit a level of motility necessitating the use of anesthetic for sorting via COPAS-XL. However, for older larvae (>5 d.p.f.), 1× tricaine can be used for sorting into 96-well plates. The final concentration of tricaine will be 0.15×: an approximately sixfold dilution (50-μl droplet in a 280-μl volume).
? TROUBLESHOOTING
-
50|
Cover the plates with clear plastic lids and incubate them at 28.5 °C for 4 d until ARQiv-based reporter quantification at 7 d.p.f.
▲ CRITICAL STEP Store fish-dispensed drug plates under standard lighting/temperature conditions such that aberrant effects on established circadian cycles are prevented (e.g., avoid stacking nontransparent microtiter plates).
-
51|
After 4 d of drug treatment, use the PlateCrane to bring individual plates to a Micro 10× for anesthetic treatments. 15 min before ARQiv scanning, add 10 μl of 0.68% (vol/vol) Eugenol (Sigma-Aldrich) in drug solvent to each well to anesthetize larvae. The final volume will be 340 μl per well, and the Eugenol concentration will be 0.02%. A final volume of ~340 μl per well provides a relatively flat liquid surface near the top of the well, thus avoiding light scatter due to a concave/convex liquid-air interface. For longer drug incubations, evaporation should also be accounted for.
▲ CRITICAL STEP Zebrafish embryos anesthetized with Eugenol show fewer light-induced startle movements as compared with those treated with tricaine methanesulfonate (tricaine). Therefore, in our experience, Eugenol has provided more consistent readings. The orientation of larvae within the wells has a major impact on the quality of readout and signal strength produced by Tecan. We observed that longer periods of incubation of larvae in 96-well plates help to center the larvae within the wells. Use of the Tecan orbital shaking function before scanning also helps to normalize fish placement.
? TROUBLESHOOTING
Large-scale in vivo drug discovery—ARQiv scans ● TIMING 1 h to several hours
-
52|
Use the PlateCrane to bring the anesthetized plates to the barcode reader to link the plate to the SoftLinx software, and then move the plate to the plate reader for scanning.
-
53|
Using optimized reporter-detection parameters (Equipment Setup), scan each plate using ARQiv (plate reader) to quantify reporter levels, and then add the plates to a new stack.
-
54|
Use the SoftLinx software to automatically save ARQiv data files in .xml format with a file name matching the label of the barcode on each plate.
■ PAUSE POINT If you are keeping zebrafish embryos after the scans, they need to be removed from the anesthetic immediately after the scans and kept in E3 medium. Otherwise, lethally anesthetize the larvae with an overdose of Eugenol and/or fix as desired to use for secondary assays (e.g., immunohistochemistry and RNAseq).
Large-scale in vivo drug discovery—real-time HTS data analysis for hit identification ● TIMING 1 d
▲ CRITICAL Use the ‘ANALYSIS’ function in the ARQiv package to generate three graphical representations of test compound performance in real time (Fig. 5). Real-time data analysis is used to identify potential hit plates immediately following ARQiv scans4, enabling immediate visual follow-up detection of potential false positives (nonspecific increases in fluorescence, toxicity, detritus, and so on). This also facilitates prioritization of ‘hit’ compounds based on relative activity levels and whether a reasonable dose-response is evident. The process for producing graphical outputs manually is described below. To automate real-time data processing, it will be necessary to set up networked file transfer between the SoftLinx data acquisition computer and a data analysis computer.
Figure 5.
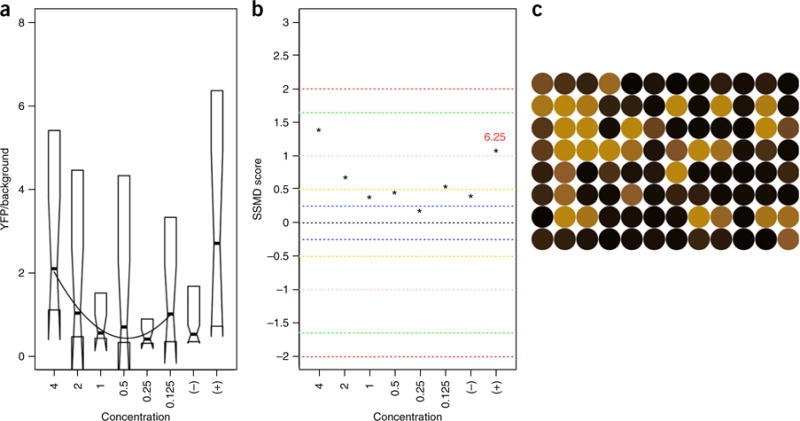
Real-time ARQiv data processing. (a–c) Graphs/maps produced via R-based code for plate parsing, extraction algorithms, and processing of ARQiv data. Representative assay data were collected from transgenic zebrafish Tg(ins:PhiYFP-2A-nfsB;sst2:TagRFP)lmc01 larvae exposed to N-acetylaspartic acid for 96 hrs, from 3 to 7 d.p.f. N-acetylaspartic acid was tested across six different concentrations (twofold dilution series ranging from 4 to 0.125 μM) for the capacity to increase beta-cell reporter activity (i.e., induce precocious formation of insulin-producing beta cells). ARQiv was used to quantify YFP reporter levels across conditions, and the data were downloaded and processed in real time using the R-based code to produce near-instant feedback on compound performance. We chose the following data plots to highlight the indicated aspects of the data: (a) boxplots—to show dose-response relationships and an indication of variability; (b) SSMD (strictly standardized mean difference) scores—a relative measure of effect size (blue, yellow, pink, green, and red dashed bounded ranges indicate, very weak, weak, weakly moderate, moderate and fairly strong effects, respectively); and (c) a heat map of signal intensity in a 96-well plate—to facilitate visual follow-up of select rows, columns, or wells to assess toxic/teratological effects, autofluorescence, and/or confirmation of biological effect. Toxicity includes but is not limited to observations of cardiac edema, curvature of the spine, small eyes, and startle response. However, to conserve time, toxicity may be applied simply as a measure of lethality (often associated with an increase in autofluorescence). Note that all graphs maintain a consistent orientation with the drug plates, and thus higher concentrations are on the left.
-
55|
Open the ARQiv package GUI per Step 10.
-
56|
Upload the background file (.xml format) by clicking the ‘bkgd’ button in the ‘COMPOUND ANALYSIS INPUT’ section (lower half) and browsing for and selecting the appropriate file. Upon selection, the name of the file should appear in the adjacent window. The Background cutoff value will be automatically calculated but does not appear in an output window. Alternatively, a manually calculated Background value can be inserted by selecting ‘VALUE’ and inputting the value in the adjacent window.
-
57|
Upload the positive- and negative-control files (.xml format) corresponding to the drug plates to be analyzed by clicking the ‘+’ and ‘−’ buttons in the ‘INPUT’ section to browse for and select the appropriate files. For instance, for Wang et al., control plates bookended each set of ten drug treatment plates. Upon selection, the name of the files should appear in the adjacent window. For an example, open the positive- and negative-control files provided in the Supplementary Data file (See example files named “Positive Control.XML” and “Negative Control.XML”).
-
58|
Create a ‘well label’ file (.csv format) that describes the layout of the positive- and negative-control plates, and the compound plates. Input the concentrations tested (in micromoles) in the appropriate wells. For an example, see the Supplementary Data (See example file named “Well label.CSV”). The file shows the layout of the plates used for the Wang et al. screen; the negative-control plate is composed of a single condition (0.1% (vol/vol) DMSO, denoted as ‘−’), the positive-control plate included six different concentrations of DAPT (200–6.25 μM)—with each condition arrayed in two columns of 8—and the positive-control plate was similarly arrayed but with concentrations ranging from 4 to 0.125 μM. Upload the well label file by clicking the ‘.CSV’ button in the ‘INPUT’ section to browse for and select the appropriate file. Upon selection, the name of the file should appear in the adjacent window.
-
59|
Upload the drug treatment plate files (.xml format) to be analyzed by clicking the ‘DRUG FILES(.XML)’ button in the ‘INPUT’ section to browse for and select the appropriate files. For an example, see the Supplementary Data (see example file named “DRUG FILES(.XML)”.
-
60|
Click the ‘COMPOUND DIRECTORY’ button in the ‘OUTPUT’ section to select a destination for the compound analysis graphical output files.
-
61|
Click the ‘ANALYSIS’ button in the ‘OUTPUT’ section. This produces a .png file that is automatically saved, referencing the name of the input drug treatment plate. The R-based code that we developed diagrams the compound analysis data in three graphical formats that represent separate aspects of the data: standard box plots, which allow visualization of dose-response curves; SSMD analysis, which provides a numerical metric for comparing compound activities; and heat maps, which facilitate visual follow-up of specific wells. As an example, inputting the control and drug files referenced above should produce a graphical output file identical to Figure 5.
▲ CRITICAL STEP To reduce false positives and to rescue false negatives arising from outlier control plates, we improved our R-based script to flag outlier negative-control plates by comparing each to the median of all previous negative-control plates (i.e., ‘pooled’ controls). Drug plates associated with outlier controls are re-evaluated using the pooled control values. In addition, once the screen is complete, all data are reprocessed with pooled control values. Compounds producing hit SSMD scores with both approaches, same-day and pooled control values, are deemed top priority for validation steps going forward. We found this method to be helpful in eliminating the outliers from further analysis.
-
62|
Analyze SSMD scores to measure compound effect size between groups (serial dilution, ± control).
? TROUBLESHOOTING
-
63|
Perform visual follow-up on plates of interest via upright epifluorescence microscope, and then lethally anesthetize zebrafish larvae in 0.68% (vol/vol) (34×) Eugenol stock solution.
▲ CRITICAL STEP Toxicity is simply a measure of lethality in drug screening. It can be detected by performing visual follow-up on plates or it can be detected by the ARQiv package (region-based threshold, labeled as autofluorescent ‘A’ in the heat map). Given enough separation between the transgenic negative-control and nontransgenic background populations, this same methodology could be applied to identify blank ‘B’ wells, accounting for occasional COPAS error.
? TROUBLESHOOTING
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 3.
TABLE 3.
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 8 | Fish larvae are not reliably detected by the Tecan microplate reader (well appears empty) | Convex/concave shape of the liquid:air interface refracts both excitation and emission light, decreasing acquired signal | Check the total volume of wells and if necessary adjust to ~330 μl to provide a consistent flat liquid surface before the scan. Make sure that U-shaped 96-well plates were used |
| Irregular placement of zebrafish within wells—i.e., organ or tissue of interest is irregularly placed and potentially missed in regional scans | Consider using the Tecan orbital shaking function before the scan for a more uniform larva orientation in each well. Alternatively, determine which type of 96-well plate (U-bottom, V-bottom, flat-bottom) permits the most uniform placement of your larvae (age- and tissue-specific) | ||
| Fish larvae are not fully anesthetized | Ensure that the final concentration of clove oil is accurate (0.02% final concentration of clove oil). Wait for 15 min before the scan; verify the lack of a startle response by tap-ping the edge of the plate, and check for movement of larvae | ||
| ‘Z-Position’ setting (μm) of the 96-well-plate scan is not optimal | Complete a ‘Z-Position’ scan to re-establish/verify optimal settings. Take the average of the depths from each well that provided maximal signal; this is your new ‘Manual Z-Position’ | ||
| Inappropriate parameter settings | Settings may need to be adjusted for maximal signal detection | ||
| Suboptimal Tecan Excitation, Emission and/or Bandwidth settings are used | Perform Excitation and Emission ‘Fluorescence Intensity Scans’ (±10 nM) centered at published Ex/Em maxima for your fluoro-phore (±10 nm) to maximize Signal:Background ratio | ||
| The fluorescent protein is not expressed at high-enough levels | Use an alternative transgenic line or promoter that drives higher transgene expression. Consider CRIPSR knock-in versus Tol2-mediated transgenesis, or a bi-partite system to enhance expression levels | ||
| PTU (photo-labile) is degraded and embryos have become pigmented | Protect PTU stock solution from light and transfer the eggs to PTU/E3 medium before 16 hpf. Ensure that 96-well plates are uniformly exposed to light—i.e., avoid stacking plates | ||
| 25 | Insufficient egg quantity (low fecundity) | Aquaculture system conditions are not favorable for fish health | Check the levels of the following: pH, nitrate, nitrite, ammonia, salinity, water temperature, and daily exchange rate of system water; verify consistent light:dark cycle. Check with your aquaculture system manufacturer for recommended settings |
| Fish are aged or their age is not optimal for mating | Maintain replacement breeder stocks to rotate new fish into the screen every 6 months (3-to 6-month-old adults provide highest fecundity) | ||
| 29 | Insufficient egg quality | Unfertilized eggs (opaque/white) | Increase the male:female ratio or consider replacing aged males (9+ months) with younger (3+ months) males |
| Robust mycotic (fungal) growth in plates | Thoroughly rinse the eggs during collection and remove detritus. Use embryo medium that includes methylene blue (anti-mycotic) for newly fertilized eggs during the first 16–24 h | ||
| 49 | Splashing of drug compound (of varying concentrations) between wells when the fish droplet is dispensed from COPAS | Droplet size is too large | Adjust COPAS ‘width’ metric (in milliseconds) to obtain an approximately 50-μl droplet size. Dispense larvae into a well that is 1/3 full. For details, see Supplementary Figure 5 |
| Droplet placement is not centered in the well | Recalibrate COPAS plate coordinates to recenter the droplets. In addition, verify that COPAS ‘Sorter’ pressure (psi) is optimized for your other component pressures (‘Sheath’, ‘Sample’, ‘Clean’) | ||
| No fish in the wells | Component pressures (‘Sheath’, ‘Sample’, ‘Clean’, ‘Sorter’) and/or gating/sorting parameters (Supplementary Fig. 5) are not optimized | Pass several hundred nontransgenic, and then transgenic, larvae through the COPAS to separate fish from nonfish (gating) and transgenic from nontransgenic (sorting). Adjust component pressures and/or gating/sorting windows, and repeat this process until optimal | |
| Multiple fish in the wells | Fish concentration in ‘Top Cup’ of COPAS (where large quantities of fresh larvae are placed) is too high | Decrease the concentration of fish in ‘Top Cup’; 1–2 fish per milliliter is ideal | |
| Embryo dispension rate is slow | Not enough sample pressure in COPAS | Increase the ‘Top Cup’ and/or ‘Sample Cup’ pressure in COPAS | |
| Fish concentration in ‘Top Cup’ of COPAS (where large quantities of fresh larvae are placed) is too low | Increase the concentration of fish in ‘Top Cup’, 1–2 fish per milliliter is ideal | ||
| 51 | Evaporation proximal to the edges of the plate | There is not enough humidity where the plates are stored. This will lead to an ‘edge-effect’ in which signal acquired from the edges of the plate is decreased as compared with that from more central wells | Check the humidity level and consider storing plates in a humidified incubator |
| Condensation on lids of plates, leading to mixed concentrations of serially titrated drug compounds | Frequent temperature fluctuations (e.g., incubator door opened frequently) | Consider storing the plates in a humidified incubator, and make sure that the incubator door seals well | |
| 62 | R GUI—‘ARQiv package graphical user interface (GUI)’ is not functional | ‘well.label.csv’ file is incorrectly formatted | This file identifies the location (96-well plate format) of positive/negative controls, as well as that of drug compounds. Verify the accuracy of this template file |
| Software needs to be updated and/or the package is missing | Ensure that R and R Studio are up to date. Re-download the ARQiv package from Github | ||
| 63 | Dead embryos after treatment | Drug concentration is too high or drug is toxic to zebrafish | Adjust the drug serial dilution concentration(s) to potentially retest this compound |
| Timing of chemical exposure is wrong | Establish appropriate exposure times with a positive control drug (i.e., length of exposure, or specific to developmental window) |
● TIMING
The time required to perform ARQiv-HTS-based drug screening will vary depending on the transgenic line used. Here, we provide an estimate of the time required for a qHTS-based chemical screening using larval-stage zebrafish. The first part, pre-screen assay optimization (Steps 1–24), takes 10 d; the second part, large-scale in vivo drug discovery (Steps 25–63), takes 7 d.
Steps 1–6, fluorescent detection parameters: 1 d
Steps 7–12, defining the signal, 1 d
Steps 13–18, determination of sample size, 7 d
Steps 19–21, QC calculations, 7 d, concurrent with sample size determination
Steps 22–24, SSMD hit score estimation, 1 d
Steps 25–29, egg production and larvae maintenance: 1–3 d
Steps 30–48, preparation of drug plates: 1–12h, depending on the number of drugs to be tested in a day
Step 49, dispensing of larvae into 96-well plates: 1 h to several hours, depending on the number of embryos to be assayed
Step 50, incubation of larvae in drugs: several hours to several days, depending on the assay
Step 51, anesthetization of larvae: 15–30 min
Steps 52–54, ARQiv scans: 1 h to several hours, depending on the number of plates to be assayed
Steps 55–63, real-time HTS data analysis for hit identification: 1 d
ANTICIPATED RESULTS
We recently completed a full-scale ARQiv-HTS drug discovery project in which we achieved near high-throughput metrics while applying qHTS-based methods—a first for vertebrate-based whole-organism screening. Transgenic reporter signals were quantified and compared between more than a half million transgenic zebrafish larvae to identify compounds that increased pancreatic beta-cell mass—i.e., increased the number of YFP-labeled insulin-producing cells (Fig. 6)4. We tested the Johns Hopkins Drug Library (JHDL, courtesy of J. Liu)— a collection of 3,348 drugs, most of which are approved for use in humans (2,290 approved by the FDA or international counterparts; 775 at various stages in clinical trials)—at an average rate of 60–120 drugs per week (72–144 plates per week, when accounting for control plates).
Figure 6.
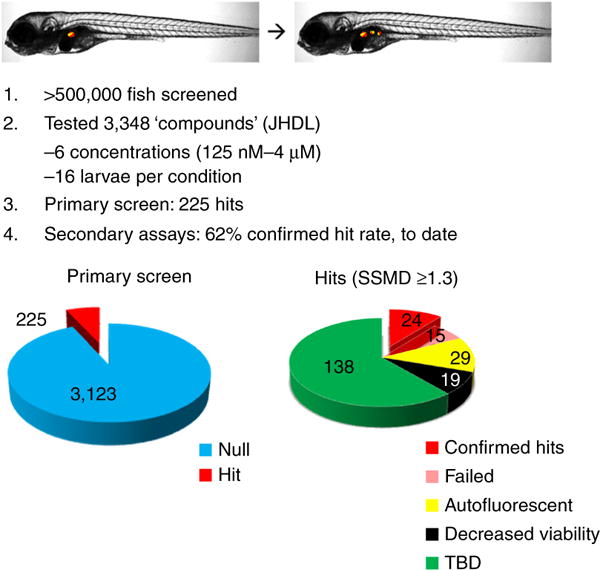
Summary of beta-cell neogenesis screen. Graphic representation of first ARQiv-HTS drug discovery with results summarized. Top: depiction of zebrafish larvae wherein precocious formation of pancreatic beta and delta cells is induced (i.e., increased beta-cell mass). Middle, 1–4: key points of screen; >0.5 million larvae were used to screen the Johns Hopkins Drug Library (JHDL, 3,348 compounds) using a twofold titration series (4–0.125 μM) with a 62% confirmed hit rate. Bottom: 225 ‘hit’ compounds produced an SSMD of ≥1.3 (left chart). Of the 87 compounds with the highest SSMD scores, 48 were either autofluorescent or negatively impacted fish health (yellow and black wedges, right chart). 24 of the remaining 38 compounds were confirmed to increase beta-cell mass (solid red wedge, right chart). For further description, see ‘ANTICIPATED RESULTS’.
For the screen, each JHDL compound was evaluated at six different concentrations (4 μM–125 nM) at a sample number of 16 per condition, with negative- and positive-control plates flanking every ten drug plates (per Supplementary Fig. 4). 3-d-old transgenic larvae, Tg(ins:PhiYFP-Eco.NfsB,sst2:TagRFP)lmc013, were treated for 4 d and then anesthetized, and reporter levels were quantified by ARQiv.
Compound performance was evaluated using real-time data readouts (per Fig. 5), and hit plates (producing SSMDs of ≥1.3) were visually evaluated to assess fish health, to flag possible nonspecific fluorescence, and to perform an initial visual assessment of the desired effect: increased beta-cell (YFP) signal.
Of 3,348 compounds tested, 225 (6.7%) produced an SSMD of ≥1.3 (Fig. 6, red wedge, left chart). However, 29 proved to be autofluorescent or caused increased larval fluorescence (Fig. 6, yellow wedge) and 19 negatively impacted fish health (black wedge), eliminating these 48 compounds from further evaluation. The remaining 177 ‘hits’ are being tested by our collaborator, M. Parsons (Department of Surgery, Johns Hopkins School of Medicine), using a series of secondary confirmation assays. To date, 24 of 39 hit compounds (61.5%) have been confirmed as being capable of increasing beta-cell mass in larval zebrafish (red wedge, right chart). The remaining 138 compounds (green wedge) await further evaluation.
Confirmed lead compounds represent potential therapeutics for diabetic patients. Moreover, ‘repurposing’ existing drugs could accelerate translation to the clinic. Another advantage of screening pre-existing drugs is that probable molecular mechanisms of action are known. This also provided a straightforward pathway to drug optimization and simplified secondary follow-up assay design, allowing us to quickly delineate novel roles for key signaling pathways in regulating beta-cell biology.
Supplementary Material
Acknowledgments
We are grateful to R. L. Daily, 3rd (integration designer, Hudson Robotics) for creating the ARQiv-HTS workstation schematics. A huge thanks goes to our in vivo drug discovery collaborators J. Liu (Department of Pharmacology and Molecular Science, Johns Hopkins School of Medicine) and M. Parsons (Department of Surgery, Johns Hopkins School of Medicine) and their research teams. We also thank M. Saxena for her editorial assistance, as well as other past and present Mumm and Luminomics laboratory members for helpful discussions. This work was supported by grants to J.S.M. from the NIH (RC4DK090816, R01EY022810, R41TR000945, and F31EY021713), DoD (MR130301), and the Foundation Fighting Blindness (TA-NMT-0614-0643-JHU-WG).
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS J.S.M. conceived of the ARQiv-HTS platform, designed reporter-based assay systems, and supervised the project. D.T.W., A.U.E., L.Z., S.S., S.K.R., and S.L.W. accomplished large-scale implementation of the ARQiv-HTS system and analyzed data. G.W., D.D., S.L.W., H.J., and J.Q. developed the software packages for assay optimization and real-time data analysis. D.T.W., A.U.E., and J.S.M. wrote the manuscript with input from all authors.
COMPETING FINANCIAL INTERESTS The authors declare competing financial interests: details are available in the online version of the paper.
References
- 1.Swinney DC, Anthony J. How were new medicines discovered? Nat Rev Drug Discov. 2011;10:507–519. doi: 10.1038/nrd3480. [DOI] [PubMed] [Google Scholar]
- 2.Eder A, et al. Effects of proarrhythmic drugs on relaxation time and beating pattern in rat engineered heart tissue. Basic Res Cardiol. 2014;109:436. doi: 10.1007/s00395-014-0436-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walker SL, et al. Automated reporter quantification in vivo: high-throughput screening method for reporter-based assays in zebrafish. PLoS One. 2012;7:e29916. doi: 10.1371/journal.pone.0029916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Inglese J, et al. Quantitative high-throughput screening: a titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc Natl Acad Sci USA. 2006;103:11473–11478. doi: 10.1073/pnas.0604348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang G, et al. First quantitative high-throughput screen in zebrafish identifies novel pathways for increasing pancreatic β-cell mass. Elife. 2015;4 doi: 10.7554/eLife.08261. http://dx.doi.org/10.7554/eLife.08261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scannell JW, Blanckley A, Boldon H, Warrington B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat Rev Drug Discov. 2012;11:191–200. doi: 10.1038/nrd3681. [DOI] [PubMed] [Google Scholar]
- 7.Rishton GM. Nonleadlikeness and leadlikeness in biochemical screening. Drug Discov Today. 2003;8:86–96. doi: 10.1016/s1359644602025722. [DOI] [PubMed] [Google Scholar]
- 8.Medina O, Estrada JC, Servin M. Robust adaptive phase-shifting demodulation for testing moving wavefronts. Opt Express. 2013;21:29687–29694. doi: 10.1364/OE.21.029687. [DOI] [PubMed] [Google Scholar]
- 9.Walker MJA, Barrett T, Guppy LJ. Functional pharmacology: the drug discovery bottleneck? Drug Discov Today TARGETS. 2004;3:208–215. [Google Scholar]
- 10.Rennekamp AJ, Peterson RT. From phenotype to mechanism after zebrafish small molecule screens. Drug Discov Today Dis Models. 2013;10:e51–e55. doi: 10.1016/j.ddmod.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rennekamp AJ, Peterson RT. 15 years of zebrafish chemical screening. Curr Opin Chem Biol. 2015;24:58–70. doi: 10.1016/j.cbpa.2014.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peterson RT, Link BA, Dowling JE, Schreiber SL. Small molecule developmental screens reveal the logic and timing of vertebrate development. Proc Natl Acad Sci USA. 2000;97:12965–12969. doi: 10.1073/pnas.97.24.12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peterson RT, et al. Chemical suppression of a genetic mutation in a zebrafish model of aortic coarctation. Nat Biotechnol. 2004;22:595–599. doi: 10.1038/nbt963. [DOI] [PubMed] [Google Scholar]
- 14.Burns CG, et al. High-throughput assay for small molecules that modulate zebrafish embryonic heart rate. Nat Chem Biol. 2005;1:263–264. doi: 10.1038/nchembio732. [DOI] [PubMed] [Google Scholar]
- 15.Stern HM, et al. Small molecules that delay S phase suppress a zebrafish bmyb mutant. Nat Chem Biol. 2005;1:366–370. doi: 10.1038/nchembio749. [DOI] [PubMed] [Google Scholar]
- 16.Murphey RD, Zon LI. Small molecule screening in the zebrafish. Methods. 2006;39:255–261. doi: 10.1016/j.ymeth.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 17.Anderson C, et al. Chemical genetics suggests a critical role for lysyl oxidase in zebrafish notochord morphogenesis. Mol Biosyst. 2007;3:51–59. doi: 10.1039/b613673g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Molina GA, Watkins SC, Tsang M. Generation of FGF reporter transgenic zebrafish and their utility in chemical screens. BMC Dev Biol. 2007;7:62. doi: 10.1186/1471-213X-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Owens KN, et al. Identification of genetic and chemical modulators of zebrafish mechanosensory hair cell death. PLoS Genet. 2008;4:e1000020. doi: 10.1371/journal.pgen.1000020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cao Y, et al. Chemical modifier screen identifies HDAC inhibitors as suppressors of PKD models. Proc Natl Acad Sci USA. 2009;106:21819–21824. doi: 10.1073/pnas.0911987106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Molina G, et al. Zebrafish chemical screening reveals an inhibitor of Dusp6 that expands cardiac cell lineages. Nat Chem Biol. 2009;5:680–687. doi: 10.1038/nchembio.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yeh HH, et al. Ha-ras oncogene-induced Stat3 phosphorylation enhances oncogenicity of the cell. DNA Cell Biol. 2009;28:131–139. doi: 10.1089/dna.2008.0762. [DOI] [PubMed] [Google Scholar]
- 23.Coffin AB, et al. Chemical screening for hair cell loss and protection in the zebrafish lateral line. Zebrafish. 2010;7:3–11. doi: 10.1089/zeb.2009.0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Groh ED, et al. Inhibition of histone deacetylase expands the renal progenitor cell population. J Am Soc Nephrol. 2010;21:794–802. doi: 10.1681/ASN.2009080851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kokel D, et al. Rapid behavior-based identification of neuroactive small molecules in the zebrafish. Nat Chem Biol. 2010;6:231–237. doi: 10.1038/nchembio.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paik EJ, de Jong JLO, Pugach E, Opara P, Zon LI. A chemical genetic screen in zebrafish for pathways interacting with cdx4 in primitive hematopoiesis. Zebrafish. 2010;7:61–68. doi: 10.1089/zeb.2009.0643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rihel J, et al. Zebrafish behavioral profiling links drugs to biological targets and rest/wake regulation. Science. 2010;327:348–351. doi: 10.1126/science.1183090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Milan DJ, Peterson TA, Ruskin JN, Peterson RT, MacRae CA. Drugs that induce repolarization abnormalities cause bradycardia in zebrafish. Circulation. 2003;107:1355–1358. doi: 10.1161/01.cir.0000061912.88753.87. [DOI] [PubMed] [Google Scholar]
- 29.Ali S, van Mil HGJ, Richardson MK. Large-scale assessment of the zebrafish embryo as a possible predictive model in toxicity testing. PLoS One. 2011;6:e21076. doi: 10.1371/journal.pone.0021076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Langheinrich U. Zebrafish: a new model on the pharmaceutical catwalk. Bioessays. 2003;25:904–912. doi: 10.1002/bies.10326. [DOI] [PubMed] [Google Scholar]
- 31.North TE, et al. Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature. 2007;447:1007–1011. doi: 10.1038/nature05883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peravali R, et al. Automated feature detection and imaging for high-resolution screening of zebrafish embryos. Biotechniques. 2011;50:319–324. doi: 10.2144/000113669. [DOI] [PubMed] [Google Scholar]
- 33.MacRae CA, Peterson RT. Zebrafish as tools for drug discovery. Nat Rev Drug Discov. 2015;14:721–731. doi: 10.1038/nrd4627. [DOI] [PubMed] [Google Scholar]
- 34.Mathias JR, Saxena MT, Mumm JS. Advances in zebrafish chemical screening technologies. Future Med Chem. 2012;4:1811–1822. doi: 10.4155/fmc.12.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leung CK, et al. An ultra high-throughput, whole-animal screen for small molecule modulators of a specific genetic pathway in Caenorhabditis elegans. PLoS One. 2013;8:e62166. doi: 10.1371/journal.pone.0062166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goktug AN, Chai SC, Chen T. Data analysis approaches in high throughput screening. In: El-Shemy HA, editor. Drug Discovery. InTech; 2013. [Google Scholar]
- 37.Zhang XD, et al. Integrating experimental and analytic approaches to improve data quality in genome-wide RNAi screens. J Biomol Screen. 2008;13:378–389. doi: 10.1177/1087057108317145. [DOI] [PubMed] [Google Scholar]
- 38.Zhang J, Chung T, Oldenburg K. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 39.Zhang XD. Novel analytic criteria and effective plate designs for quality control in genome-scale RNAi screens. J Biomol Screen. 2008;13:363–377. doi: 10.1177/1087057108317062. [DOI] [PubMed] [Google Scholar]
- 40.Zhang XD. Illustration of SSMD, z score, SSMD*, z* score, and t statistic for hit selection in RNAi high-throughput screens. J Biomol Screen. 2011;16:775–785. doi: 10.1177/1087057111405851. [DOI] [PubMed] [Google Scholar]
- 41.Graf SF, Hötzel S, Liebel U, Stemmer A, Knapp HF. Image-based fluidic sorting system for automated zebrafish egg sorting into multiwell plates. J Lab Autom. 2011;16:105–111. doi: 10.1016/j.jala.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 42.Parsons MJ, et al. Notch-responsive cells initiate the secondary transition in larval zebrafish pancreas. Mech Dev. 2009;126:898–912. doi: 10.1016/j.mod.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lawrence M, Lang D. RGtk2: a graphical user interface toolkit for R. J Stat Softw. 2010;37:1–52. [Google Scholar]
- 44.Borchers HW. pracma: Practical Numerical Math Functions (2015) 2016 https://rdrr.io/rforge/pracma/
- 45.Rovira M, et al. Chemical screen identifies FDA-approved drugs and target pathways that induce precocious pancreatic endocrine differentiation. Proc Natl Acad Sci USA. 2011;108:19264–19269. doi: 10.1073/pnas.1113081108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ariga J, Walker SL, Mumm JS. Multicolor time-lapse imaging of transgenic zebrafish: visualizing retinal stem cells activated by targeted neuronal cell ablation. J Vis Exp. 2010;43:e2093. doi: 10.3791/2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Halsey LG, Curran-Everett D, Vowler SL, Drummond GB. The fickle P value generates irreproducible results. Nat Methods. 2015;12:179–185. doi: 10.1038/nmeth.3288. [DOI] [PubMed] [Google Scholar]
- 48.Westerfield M. A guide for the laboratory use of zebrafish (Danio rerio) Eugene, OR: University of Oregon Press; 2007. [Google Scholar]
- 49.Adatto I, Lawrence C, Thompson M, Zon LI. A new system for the rapid collection of large numbers of developmentally staged zebrafish embryos. PLoS One. 2011;6:e21715. doi: 10.1371/journal.pone.0021715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hallare A, Nagel K, Köhler HR, Triebskorn R. Comparative embryotoxicity and proteotoxicity of three carrier solvents to zebrafish (Danio rerio) embryos. Ecotoxicol Environ Saf. 2006;63:378–388. doi: 10.1016/j.ecoenv.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 51.David RM, et al. Interference with xenobiotic metabolic activity by the commonly used vehicle solvents dimethylsulfoxide and methanol in zebrafish (Danio rerio) larvae but not Daphnia magna. Chemosphere. 2012;88:912–917. doi: 10.1016/j.chemosphere.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang XD. A pair of new statistical parameters for quality control in RNA interference high-throughput screening assays. Genomics. 2007;89:552–561. doi: 10.1016/j.ygeno.2006.12.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.