

Abstract
Adenylate cyclase toxin (ACT) is an important B. pertussis virulence factor that is not included in current acellular pertussis vaccines. We previously demonstrated that immunization with the repeat-in-toxin (RTX) domain of ACT elicits neutralizing antibodies in mice and discovered the first two antibodies to neutralize ACT activities by occluding the receptor-binding site. Here, we fully characterized these antibodies and their epitopes. Both antibodies bind ACT with low nanomolar affinity and cross-react with ACT homologs produced by B. parapertussis and B. bronchiseptica. Antibody M1H5 binds B. pertussis RTX751 ~100-fold tighter than RTX751 from the other two species, while antibody M2B10 has similar affinity for all three variants. To initially map the antibody epitopes, we generated a series of ACT chimeras and truncation variants, which implicated the repeat blocks II–III. To identify individual epitope residues, we displayed randomly mutated RTX751 libraries on yeast and isolated clones with decreased antibody binding by flow cytometry. Next-generation sequencing identified candidate epitope residues based on enrichment of clones with mutations at specific positions. These epitopes form two adjacent surface patches on a model of the RTX751 domain, one for each antibody. Notably, the cellular receptor also binds within blocks II–III and shares at least one residue with the M1H5 epitope. A predicted structural model of RTX751 supports the notion that the antibody and receptor epitopes overlap. These data provide insight into mechanisms of ACT neutralization and guidance for engineering more stable RTX variants that may be more appropriate vaccine antigens.
Keywords: whooping cough, pertussis, epitope mapping, antigen engineering, yeast display, high-throughput sequencing
Graphical Abstract
Whooping cough is a highly contagious respiratory disease caused primarily by the gram-negative bacteria Bordetella pertussis, whose incidence has been steadily increasing over the past two decades. Accumulating evidence has implicated limitations in the current acellular pertussis vaccine as one of the major contributors to pertussis recurrence. This appears due to several factors, including the fact that vaccine-induced immune responses limit manifestations of clinical disease but have less effect on infection or transmission rates.1 As there is no clear serological correlate of protection, these acellular vaccines include between one and five different pertussis antigens. One proposal to improve vaccine efficacy is inclusion of additional highly conserved protective antigens.2–6
The adenylate cyclase toxin (ACT) is frequently cited as a leading candidate for inclusion in next generation vaccines,3, 7–13 but is not included in any current acellular vaccines due to it being unavailable at the time those vaccines were being developed and to poor understanding of its role in protection. ACT is a large, multi-domain protein that targets phagocytic leukocytes expressing the αMβ2 integrin (also known as CR3, Mac-1 and CD11b/CD18) through its C-terminal repeat-in-toxin (RTX) domain.14, 15 After the initial interaction with the cellular surface, the N-terminal cyclase domain translocates into the target cell cytoplasm16, where it associates with calmodulin and rapidly generates supraphysiological levels of cAMP17. These activities are thought to protect bacteria from innate immune responses during the early stages of infection by inhibiting bacterial phagocytosis.18, 19
We and others have previously demonstrated that the C-terminal RTX domain can recapitulate many of the responses resulting from immunization with ACT, while possessing superior biochemical characteristics.20, 21 Notably, intact ACT is poorly soluble and prone to aggregation as well as proteolytic degradation, while the cyclase domain bears homology to human adenylate cyclase. Antibodies induced by active vaccination with the Bordetella enzyme could cross-react with the intracellular mammalian homolog,22 thereby inducing undesirable auto-reactive responses. These points have suggested that an engineered variant of RTX may be preferable as a vaccine antigen. The RTX domain is intrinsically disordered in the absence of calcium and folds into β-roll motifs upon calcium binding,23, 24 mediated by ~40 calcium-binding, glycine- and aspartate-rich nonapeptide repeats. These repeats are distributed in five blocks between residues 1006–160023 which are entropically stabilized by the flanking region at the C-terminus of block V.25, 26 Receptor binding and subsequent intracellular delivery of the catalytic domain is dependent upon post-translational acylation (residues K983 when expressed in B. pertussis, residues K983 and K860 when expressed in E. coli) by the co-expressed enzyme CyaC27, 28 as well as calcium ion-mediated structural changes.29 Receptor binding has been suggested to directly involve RTX residues in the region 1166–1287.14, 15, 30 Recombinant RTX fragment spanning residues 751–1706 (RTX751) include the acylation sites, exhibit calcium-dependent conformational changes and retain the ability to bind purified receptor.21 This and related RTX fragments are potential surrogates for ACT in future vaccines.
Structure-function analyses of neutralizing antibody-antigen interactions can identify residues forming protective epitopes. This information is crucial to guide design of immunogens with increased epitope accessibility that elicit stronger neutralizing antibody responses than the native antigen. This approach has been employed to generate a single meningococcal factor H binding protein that provides protection against the high sequence variability observed among clinical Neisseria meningitidis B strains.31 It has also been used to stabilize the pre-fusion conformation of the fusion F protein from respiratory syncytial virus that presents all known neutralizing epitopes.32 Neutralizing epitopes can be mapped via structural and biochemical methods.33, 34 Since structural approaches can present technical challenges, especially for a large and poorly soluble protein such as ACT, mutagenic approaches employing high throughput selection and sequencing schemes are gaining popularity. In particular, fine epitope mapping using yeast surface display can comprehensively identify epitope residues.35–38
We previously discovered two neutralizing antibodies, M2B10 and M1H5, which bind non-overlapping epitopes within the RTX domain of ACT. Both antibodies neutralize toxin activities by blocking the interaction between ACT and its αMβ2 integrin cellular receptor but do not competitively inhibit each other. These were the first antibodies reported to neutralize ACT by binding the RTX domain or by inhibiting the ACT-receptor association step. In this study, we determined the epitopes for these two antibodies using biochemical approaches and a high-throughput yeast display approach coupled with fluorescence-activated cell sorting and next-generation sequencing. These data provide insight into the mechanisms of antibody-mediated ACT neutralization and will contribute to engineering an RTX immunogen better able to elicit protective immunity.
EXPERIMENTAL PROCEDURES
Molecular cloning
To generate plasmids expressing the soluble RTX N-terminal truncations (RTXII–V spanning repeat blocks II–V and residues 1132–1706, RTXIII–V spanning residues 1244–1706, RTXIV–V spanning residues 1381–1706, and RTXV spanning residues 1530–1706), DNA fragments encoding these regions were amplified with forward primers 5′-actattGCTAGCaatatcgagaatctgcacggctcccg-3′, 5′-actattGCTAGCtccgcgctgggcgtggactattac-3′, 5′-actattGCTAGCgtcgacaagctgggcgaggc-3′, 5′-actattGCTAGCagcgcgcgtgatgacgtgc-3′, and the common reverse primer 5′-actattGGATCCgcgccagttgacagccaggga-3′. To generate plasmids expressing C-terminally truncated variants (RTXI–II spanning residues 751–1242, RTXI–III spanning residues 751–1376, and RTXI–IV spanning residues 751–1529), the DNA fragments were amplified with common forward primer 5′-actattGCTAGCgccaattcggacggcctacgg-3′ and reverse primers 5′-actattGGATCCcttgcgcacccattccctggac-3′, 5′-actattGGATCCcaggtcagccaggatgcccag-3′, and 5′-actattGGATCCgcccacggcgttctcgatattgc-3′. To generate plasmids expressing the RTX751 domains of Bordetella parapertussis and Bordetella bronchiseptica, DNA fragments encoding these regions were amplified from respective colonies using primers 5′-actattGCTAGCgccaattcggacggcctacgg-3′ and 5′-actattGGATCCgcgccagttgacagccaggga-3′ with Q5 polymerase (NEB). The amplified fragments were gel purified, double digested with NheI and BamHI and ligated into similarly digested pET28a plasmid. Restriction sites are in uppercase and underlined.
To generate plasmids for yeast display of the RTX751 domain, a DNA fragment encoding residues 751–1706 of adenylate cyclase toxin was amplified with primers 5′-actattGCTAGCgccaattcggacggcctacgg-3′ and 5′-actattGGATCCgcgccagttgacagccaggga-3′, gel purified, double digested with NheI and BamHI and ligated into similarly digested pCTCON plasmid 39.
The Q5 Site-Directed Mutagenesis Kit (NEB) was used to alter codons in the B. pertussis RTX751 to those present in B. parapertussis. For the region 1140–1164 within Block II, a total of 5 residue changes were introduced (R1140S, R1144S, Q1150R, H1157D and R1164H) using primers 5′-cgagctctggggcgacgatggcaacgacacgatacacGGCCGGGGCGGCGACGAC-3′ and 5′-ttgtcccggtcgtcgccggcgatgctgtcgttcaggctGGAGCCGTGCAGATTCTCGATATTCTTGACATGGTC-3′. For the region 1237–1256 at the beginning of Block III, a total of nine residue changes were introduced (R1237E, E1238G, S1244A, A1245R, L1246R, V1248M, D1249G, N1253S and N1256S) using primers 5′-ggcatgggctactacgacagtgtccgcagtGTCGAAAACGTCATCGGTACG-3′ and 5′-gcgccgggccgccttgcgcacccacccttcGGACAGGTCCGCCTCGAT-3′. For the region 1357–1365 between Block III and IV, a total of three residue changes were introduced (T1357A, I1363V and A1365T) using primers 5′-cggcgttgccacgGGTCGCATCGGGCTGGGC-3′ and 5′-gcatgcgcgcccgcCTGGCTGTAATCGACGGTATCCAC-3′. Primers were designed using NEBaseChanger, in which uppercase nucleotides are identical or complementary to wild-type sequence and lowercase nucleotides contain the desired mutations.
Protein expression and purification
The different versions of RTX domains or truncations were expressed in E. coli strain BL21(DE3) and purified by immobilized metal affinity chromatography (IMAC) resin and size exclusion chromatography as described previously.21 Single chain antibody fragments (scAbs, comprised of the heavy and light chain variable regions joined by a flexible polypeptide linker and appended with a human kappa constant domain) and full-length chimeric IgG antibodies (mouse variable regions appended with human IgG1/κ constant regions) were expressed and purified as described previously.21
Antibody binding assessed by ELISA
To test the effect of calcium ions on antibody binding, ELISA plates were coated with 0.2 μg/mL of ACT diluted in PBS. After washing with HBST (50 mM Hepes, 150 mM NaCl, 0.05% Tween-20, pH 7.5), wells were blocked with HBST-1%BSA supplemented with 0, 0.2, 2, or 10 mM CaCl2. Then, M2B10 or M1H5 were serially diluted in respective blocking solutions with different calcium concentrations. After incubation and washing, bound antibodies were detected with anti-human Fc HRP antibody diluted in HBST-1%BSA. The plates were developed with TMB substrate (Pierce), quenched with 1M HCl, and read at 450nm.
To determine whether the formation of calcium-dependent epitopes is reversible, calcium was depleted in either the antigen coating or antibody incubation steps in ELISA. Purified RTX751 contains 2mM CaCl2 and was coated in HBS + 5mM EGTA to deplete calcium or in HBS + 2 mM CaCl2 to maintain calcium required for RTX folding. Full-length ACT, purified in the presence of 8 M urea without added calcium as described 21, was coated in HBS or HBS + 2 mM CaCl2. The wells were blocked with HBST-1% BSA ± 2 mM CaCl2 to maintain the calcium concentration in the coating buffer. Then, M2B10 and M1H5 diluted to 10 nM in HBST-1%BSA ± 2mM CaCl2 were added to the wells. Washing buffer was HBST for all samples. The diluents for the HRP-conjugated secondary antibodies were kept the same as for the primary antibody in respective wells. The plates were developed and read as above.
Measurement of antibody-RTX751 binding affinity and kinetics
Binding affinity experiments were performed on a KinExA 3000 (Sapidyne Instruments). RTX751 was coated on polymethylmethacrylate beads in HBSC (50 mM Hepes, 150 mM NaCl, 2 mM CaCl2, pH 7.5) at 4°C overnight, then washed and blocked with running buffer (50 mM Hepes, 150 mM NaCl, 0.05% Tween-20, 2 mM CaCl2, 1mg/mL BSA, 0.02% sodium azide, pH 7.5). Signal Test and Range Find procedures were performed to determine the fixed antibody concentration and the range of antigen concentration to be used in the titration. RTX751 serially diluted in running buffer to concentrations between 50 and 0.2 nM were incubated with 2 nM M1H5 or 3 nM M2B10 for 1 hour at room temperature before flowing through RTX751-coated beads followed by fluorescent antibody detection. The experiments were performed in duplicate, with non-specific binding signals subtracted. Data was fit to a five parameter logistic equation described by Ohmura et al.40
In vitro toxin neutralization assay
To compare the neutralizing activity of M2B10 and M1H5 against ACT produced by B. pertussis and B. bronchiseptica in the context of the whole bacteria, B. pertussis Tohama I and B. bronchiseptica were grown on Bordet-Gengou agar plate supplemented with 15% defibrinated sheep blood (BD) and inoculated into modified synthetic Stainer-Scholte medium (SSM). The optimal concentrations of each Bordetella species to cause cAMP elevation in J774A.1 cells roughly equivalent to 125 ng/mL B. pertussis ACT were determined empirically. Then, serially diluted M2B10 and M1H5 were pre-incubated with the bacteria before being added to J774A.1 cells. The intracellular cAMP levels of J774A.1 cells were measured and calculated as described.21
Western blot of whole cell lysates
To roughly determine the epitope locations of M2B10 and M1H5, 900 μL E. coli cultures expressing different RTX751 truncation variants were pelleted, resuspended in 50 μL 2× SDS-PAGE loading dye, and boiled for 10 min before loading 5 μL or a dilution thereof to each lane. Since the different RTX variants expressed at different levels and were recognized by the three antibodies with varying sensitivity, the culture volume resulting in a detectable band was empirically determined and was not the same for each lane/blot. Three identical gels were run and transferred to PVDF membranes, which were probed by 1:2500 anti His-tag HRP antibody or 5 nM M2B10 and 5 nM M1H5 followed by anti-human Fc HRP (1:4000), respectively. Membranes were developed using SuperSignal West Pico Chemiluminescent Substrate (Pierce) followed by X-ray film exposure.
Yeast culture and transformation
Saccharomyces cerevisiae EBY100 strain was used for yeast display. EBY100 cells were streaked on YPD agar plate supplemented with 1X penicillin/streptomycin (Life Technologies), and incubated at 28–30 °C for 2–3 days until colonies appear. Yeast cells carrying pCTCON vector were selected on YNB-CAA-glucose agar plates (0.67% yeast nitrogen base, 0.5% casamino acid, 2% glucose, 1.5% agar, 1X penicillin/streptomycin).
Heat shock competent cells were prepared using Frozen-EZ Yeast Transformation II kit (Zymo Research) to transform individual pCTCON vectors. Electrocompetent cells were prepared as described by Benatuil et al. 41 except without calcium in electroporation buffer, and were used to construct random mutagenesis libraries through homologous recombination. For surface display, yeast colonies harboring the pCTCON plasmids were inoculate into YNB-CAA-glucose liquid medium, grown at 30°C overnight until OD600 of 2.0–5.0, pelleted, resuspended in YNB-CAA-galactose (2%) medium to an OD600 of 0.75~1.0 and grown at room temperature for another 20–50 hrs, to induce the expression and display of Aga2-RTX fusion protein.
Random mutagenesis library construction
The RTX gene was cloned between unique NheI and BamHI sites in the pCTCON yeast display vector.39 A unique SphI site lies within the RTX gene, corresponding to ACT codons 1359–1361. The 1968 bp region between NheI and SphI sites was randomly mutated during amplification with primers 5′-ccatacgacgttccagactacgctctgcag-3′ and 5′-ggcagcagcgcctacgatac-3′ using Mutazyme II (Agilent). After gel purification, the product was re-amplified with nested primers 5′-tggtggttctggtggtggtgg-3′ and 5′-caggtcagccaggatgccc-3′ using Q5 Polymerase (NEB), resulting in a 40 bp overhang upstream of NheI and a 46bp overhang downstream of SphI. After gel purification, the DNA was precipitated with isopropanol, resuspended in water, and quantified by NanoDrop. To prepare the linearized vector, the pCTCON plasmid was digested with NheI and SphI, gel purified, isopropanol precipitated and resuspended in water.
To construct the library, 5 μg of insert DNA and 3 μg of linearized vector backbone were added into 300 μL freshly prepared yeast electrocompetent cells and electroporated in 2 mm cuvettes as described above. Three separate electroporations were performed and pooled. For control, linearized vector without insert DNA was electroporated. Aliquots of the library and control electroporations were serially diluted and plated on selection plates to determine the total number of yeast transformants. The pooled transformants were grown in liquid selection medium at 30 °C and passaged twice to minimize the chance of carrying more than one type of plasmid per cell.41
To sequence individual library clones, plasmid DNA was extracted from 0.1–0.2 OD of yeast cells using the Zymoprep Yeast Plasmid MiniPrep II kit (Zymo Research). Due to the low quantity and purity of DNA extracted from yeast cells, it was transformed into E. coli XL1-Blue competent cells for miniprep and sequencing. In order to cover the entire mutagenized region, three sequencing primers were used for each sample: 5′-CGATTGAAGGTAGATACCCATAC-3′, 5′-CAAGGTGGTGTCGCAACTGGTC-3′, and 5′-GCACGGCTCCCGCCTGAA-3′.
Yeast staining and flow cytometry
To stain the yeast cells for flow cytometric analysis, 0.1 OD of cells were stained with 100 μL of 67 nM M2B10 or M1H5 and 1H6 in HBSC-1% BSA, followed by detection with 1:200 dilution of the secondary antibodies (Alexa647 anti-human Fc and Alexa488 anti-mouse Fc). To determine whether the antibody binding was dependent on the RTX conformation, yeast cells were either stained in the absence of CaCl2 or heated at 85°C on a thermal cycler for 30 min before staining. Flow cytometric analysis was performed on a LSR Fortessa II (BD). All flow data were analyzed with Flowjo software.
For sorting, 5 OD of cells were stained with 5mL of 13.4 nM M2B10 or M1H5 and 1H6; 0.1 OD of positive and negative control were stained with 100 μL of antibodies at the same concentrations, followed by detection with 1:200 secondary antibodies (Alexa647 anti-human Fc and Alexa488 anti-mouse Fc), and sorted on a FACSAria (BD). Gates were drawn around populations with diminished binding (P4) and normal binding (P7) to respective antibodies. Another gate (P5) in between was drawn to circumvent the population with reduced binding. Sorted cells were collected in tubes containing YNB-CAA-glucose medium and grown at 30°C until OD600 >2.0.
Next-generation sequencing
Cells equivalent to 0.2 OD yeast cells from the sorted P5 and P7 populations as well as the starting library were pelleted for plasmid extraction, from which the mutagenized region (~1.9 kb) was amplified using Q5 polymerase and primers 5′-tggtggttctggtggtggtgg-3′ and 5′-caggtcagccaggatgccc-3′. After gel purification, the samples were processed by Genomic Sequencing and Analysis Facility at UT-Austin according to the protocol 42. In order to cover the 1.9 kb region, the samples were fragmented by sonication to an average size of 250bp and sequenced on an Illumina Hiseq 2500 in 2 × 125 bp (paired-end) mode. The minimal number of reads for P5 and P7 were set to 4 × 106 for P5 and P7 and 3.6 × 107 for the starting library. A PCR fragment of the wild-type sequence was included as a control for the sequencing error rate, with minimal reads of 106. The number of reads was sufficient to cover all sequences at a theoretical eight-fold excess, accounting for the length and diversity of each sample. About 40% of the read pairs contained only one nucleotide mutation as compared to the reference wild-type sequence, and these sequences were used to calculate the enrichment ratio of mutational rate at each position. Enrichment values for each mutation were calculated using the Enrich software package.43 Custom scripts (available on GitHub at https://github.com/jstapleton/Wang_et_al and https://github.com/JKlesmith/Deep_Sequencing_Analysis) converted the shotgun reads into a standardized format compatible with Enrich and processed the Enrich output. Epitope residues were identified by manual examination of enrichment values at each position.
RaptorX model generation
A computational model of the wild-type RTX751 domain was generated by RaptorX web server based on homologous RTX structures (pseudomonas alkaline protease PDB 1KAP and RTX block V PDB 5CVW and 5CXL structures).44
Results
Neutralizing antibodies recognize RTX epitopes with high affinity
We previously observed that the M1H5 and M2B10 antibodies bind non-overlapping epitopes within the RTX domain and neutralize ACT activities by blocking the ACT-αMβ2 receptor interaction. We wanted to better understand the biochemical basis of this interaction and, in particular, understand how antibody-RTX binding differs from receptor-RTX binding. For instance only calcium-complexed forms of RTX, including RTX751, bind the αMβ2 receptor while acylated forms of RTX have a higher apparent affinity for purified αMβ2 receptor and can more effectively intoxicate αMβ2 –bearing cells.14, 15, 21, 28 Both antibodies bind unacylated RTX751 while the M2B10 antibody also binds the truncated, unacylated RTX985 variant comprising residues 985–1706 with no apparent loss in affinity.21
Prior ELISA data suggested that both antibodies bind ACT and RTX751 with similar high affinities and ~100-fold more sensitively than the toxin variants bind soluble receptor.21 To gain a more quantitative measurement of the antibody binding affinities to guide our epitope mapping experiments, we turned to KinExA analysis, a method that measures equilibrium binding affinities of unmodified molecules in the solution phase. Purified RTX751 was immobilized on polymethylmethacrylate beads, after which the antibodies alone or antibodies incubated with varying concentrations of soluble RTX751 were flowed over the beads. Under these conditions, the solution affinity of M1H5 was measured as 1.6 ± 1.2 nM and M2B10 as 1.3 ± 0.7 nM (Figure 1). To measure the binding kinetics, we performed a similar set of experiments using SPR in which purified antibody was immobilized and RTX751 injected across the surface in concentrations from 0.1 to 50 nM. This yielded similar on-rates of ~2×105 M−1sec−1 for the two antibodies and off-rates of 3×10−4 and 1.5×10−3 sec−1, for M1H5 and M2B10, respectively. This yielded an equilibrium affinity for M1H5 of 1.3 nM, comparable to that measured by KinExA. The M2B10 measurements were compromised by reduced binding after regeneration, resulting in a faster off-rate and a calculated affinity of 9 nM (Table 1, Figure S1).
Figure 1. Antibody-RTX751 binding affinity measured by KinExA analysis.

Purified anti-RTX antibody (2 nM M1H5 or 3 nM M2B10) was incubated with soluble RTX751 at varying concentrations, then flowed over beads with immobilized RTX751. Free antibody was captured by the beads and subsequently detected with fluorescent anti-human-Fc antibody. A, antibody M1H5 had a measured affinity of 1.6 ± 1.2 nM; B, antibody M2B10 had a measured affinity of 1.3 ± 0.7 nM. Data was analyzed using KinExA Pro software based on equations described by Ohmura et al. 40
Table 1.
Antibody-RTX751 binding affinities and kinetics
| KinExA | Reichert SPR | ||||
|---|---|---|---|---|---|
| Kd (nM) | Chi2 (R2) | On-rate (sec−1M−1) | Off-rate (sec−1) | Kd (nM) | |
| M1H5 | 1.63 ± 1.21 | 0.001 (0.995) | 2.36×105 | 3.09×10−4 | 1.31 |
| M2B10 | 1.25 ± 0.68 | 0.00043 (0.998) | 1.74×105 | 1.53×10−3 | 8.78* |
Poor data fitting due to incomplete regeneration between cycles.
Neutralizing antibodies recognize calcium-sensitive RTX epitopes
Next, we aimed to better define the RTX conformations compatible with antibody binding. Since RTX undergoes a dramatic conformational change from disordered random coils to beta-roll structures upon calcium binding which is required for receptor binding,14, 26 we wanted to determine the effect of calcium ions on antibody binding. We performed ELISAs with immobilized ACT or RTX751 and varying amounts of CaCl2 in the buffers. While no binding was observed when CaCl2 concentrations were ≤0.2 mM, full binding was observed when CaCl2 concentrations were ≥2mM (Figure 2A, B). The presence of calcium during the antibody incubation step was sufficient to support binding, even if the ACT or RTX751 molecules had been previously exposed to EGTA sufficient to chelate available calcium ions (Figure 2C, D). In contrast, when calcium was not included during the antibody incubation step, no binding was observed even for antigens previously incubated with calcium. These data suggest that both antibodies bind the beta-roll form of RTX and that calcium-induced folding of RTX751 and ACT is a readily reversible process. This is distinct from a previously described panel of anti-ACT antibodies that do not require calcium for ELISA binding.45
Figure 2. Calcium is required for RTX751-neutralizing antibody binding.

Binding of ACT to A, M1H5 and B, M2B10 in the presence of varying amounts of CaCl2. ELISA plates were coated with 0.2 μg/mL of ACT diluted in PBS. After washing with HBST, wells were blocked with HBST-1%BSA supplemented with 0 mM CaCl2 (open circles), 0.2 mM CaCl2 (squares), 2 mM CaCl2 (triangle), or 10 mM CaCl2 (inverted triangle). Then, M1H5 or M2B10 was serially diluted in blocking solution with the same amount of CaCl2. Bound antibodies were detected with anti-human Fc HRP antibody diluted in HBST-1%BSA without calcium. The presence of CaCl2 during the antibody incubation step is necessary and sufficient for binding of C, ACT and D, RTX751 to the M1H5 and M2B10 antibodies. ACT and RTX751 were coated with or without calcium ions and then incubated with antibodies diluted in buffer with or without calcium ions prior to ELISA analysis. C, Purified ACT does not contain calcium and was coated in HBS + 2 mM CaCl2 (black bars) or just HBS (gray bars). D, RTX751 was purified with 2mM CaCl2 and thus coated in HBS + 2 mM CaCl2 to maintain calcium concentrations (black bars) or HBS + 5mM EGTA to deplete calcium (white bars). Subsequent steps used HBST-1% BSA + 2mM CaCl2 or HBST-1% BSA to match the calcium concentration in the coating buffer. Control wells (“no Ab”) omitted only the primary M1H5/M2B10 antibodies.
Neutralizing RTX epitopes are conserved across Bordetella species
While whooping cough is caused primarily by the gram-negative bacteria Bordetella pertussis, its close relatives B. bronchiseptica and B. parapertussis can also cause human disease, albeit less commonly.46 ACT is highly conserved among these species, with an amino acid sequence identity of ~97.7%. Most sequence variation occurs in the C-terminal RTX region (Figure S2). To determine whether M1H5 and M2B10 recognize conserved epitopes, the RTX751 regions from B. parapertussis and B. bronchiseptica were produced and purified as predominantly monomeric proteins (Figure 3A). Antibody M1H5 showed preferential binding to B. pertussis RTX751, with ~100-fold lower sensitivity for RTX751 from other species in an ELISA (Figure 3B). In contrast, M2B10 bound all three RTX751 versions with identical sensitivity (Figure 3C). Supporting this data, an in vitro toxin neutralization assay using live B. bronchiseptica bacteria and human J774A.1 cells showed that M2B10 was able to better suppress cAMP elevation than M1H5 (Figure 3D).
Figure 3. Antibody M2B10 but not M1H5 recognizes a highly conserved epitope.
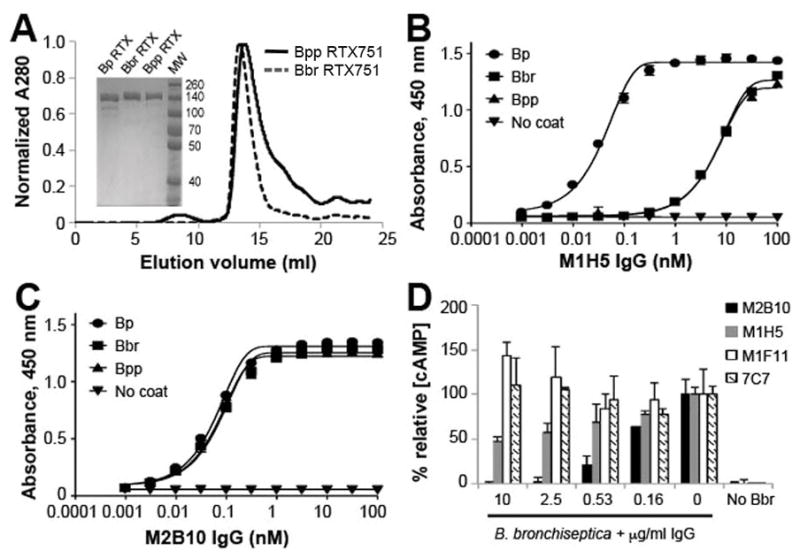
A, The RTX751 domains of B. bronchiseptica (Bbr) and B. parapertussis (Bpp) were cloned, expressed, and purified by IMAC and size exclusion chromatography. Purity was assessed by SDS-PAGE (inset). Purified RTX751 from the three species were coated on ELISA plates and compared for binding to the B, M2B10 and C, M1H5 antibodies by ELISA. Shown is the average of duplicates with error bars representing the data range; the assay was repeated twice. D, M2B10 and M1H5 IgG were tested for protection of J774A.1 cells from cAMP intoxication by live B. bronchiseptica. M1F11 and 7C7 are two non-neutralizing monoclonal antibodies, binding the catalytic and RTX domains, respectively. The % relative [cAMP] is calculated from the total cAMP concentration in the cellular lysate as determined by cAMP ELISA, divided by the protein concentration of the lysate normalized to control cells treated only with ACT. Error bars indicate range of duplicate assays; shown is the average ± range for duplicates. The assay was repeated twice.
Hypothesizing that the residues varying among species might help to define the epitope, we generated three chimeras in which the B. parapertussis mismatch region was substituted for the corresponding region in B. pertussis RTX751. B. bronchiseptica and B. parapertussis ACT sequences vary from each other by just three residues, one in the hydrophobic region and two within the RTX751 region (567A/V, 1249D/G and 1441D/E), and both share 36 additional amino acid differences from the B. pertussis ACT sequence (Figure S2). Closer inspection revealed three regions of greatest variation: residues 1139–1164 in RTX block II contains five mismatches; residues 1236–1256 at the beginning of block III contains nine mismatches; and residues 1357–1365 before block IV contains three mismatches.
Each of these regions was individually introduced into the B. pertussis RTX751 by site-directed mutagenesis and the resulting protein purified for use in ELISA (Figure 4). Notably, the block III chimera abolished binding to M1H5 but not to M2B10, suggesting some of these residues are involved, directly or indirectly, in the M1H5 epitope. For instance, non-homologous B. parapertussis residues outside block III may be required to form the M1H5 epitope in this ACT variant and thus contribute to species-specificity.
Figure 4. RTX751 block II chimera loses affinity for M1H5.

Several RTX751 chimeras were generated, in which regions of low homology were grafted from B. parapertussis into the B. pertussis RTX751 gene. A, Expression and purification of the three chimeras. Shown is a size exclusion chromatogram on an S200 column. Inset, SDS-PAGE gel with molecular weight markers indicated. B, Binding interactions between the chimeras and antibody M1H5, as measured by ELISA. The chimeras were used to coat the plate, followed by blocking and serial dilution of the antibody. Bound antibody was detected with anti-human-Fc-HRP. C, Binding between the chimeras and M2B10 antibody, evaluated as in panel B.
RTX truncations reveal linear components of the epitopes
We next aimed to localize the antibody epitopes by generating a series of truncated RTX751 variants. Variants were constructed by dividing the protein into five Gly-Asp blocks as described by Osicka et al. 47 and serially deleting blocks from the N- or C-terminus (Figure 5A). We used Western blot analysis to rapidly screen the resulting seven variants for antibody binding. Whole E. coli expressing the variants were lysed by boiling, and the resulting blots probed with an anti-hexahistidine control antibody and either the M1H5 or M2B10 antibody. The control antibody demonstrated the presence and approximate calculated molecular weight of each RTX variant. RTX constructs have previously been noted to run larger than expected, likely due to the large number of positive aspartic acid residues. The recognition patterns were similar for both M1H5 and M2B10 (Figure 5B). Of the four N-terminal truncations, both RTXII–V and RTXIII–V were recognized, while all three C-terminal truncations, RTXI–II, RTXI–III and RTXI–IV were recognized by both antibodies. Since the antibodies recognized RTX on a Western blot, the epitopes appear to include discontinuous linear components. The recognition patterns suggest that blocks II and III include residues important for both antibodies.
Figure 5. Repeat blocks II and III both contribute to antibody binding.

A, The RTX751 domain was serially truncated from the N- and C-termini to remove individual RTX repeats. Shaded blocks with numbers indicate the five blocks of repeats, “Mod” indicates the region prone to post-translational modification by the acylating enzyme CyaC. B, Western blot of whole cell lysates of the truncations, probed with M2B10, M1H5, and anti-His tag antibodies, respectively, in the presence of calcium. The amount of lysate loaded was empirically adjusted n order to clearly visualize the resulting bands on the blot. The M1H5 antibody appears to cross-react with an endogenous E. coli protein at ~50 kDa (marked with an arrow). C, SDS-PAGE of the purified preparations of the truncated RTX variants. Variants lacking block V were TCA precipitated due to very low yields.
To provide more quantitative binding information, we purified all truncated variants for use in ELISA. Variants with N-terminal truncations were produced in reasonable yields, except that Block II–V was heavily degraded. All variants with C-terminal truncations were poorly expressed and required TCA precipitation for visualization on a protein gel (Figure 5C). This is consistent with the previous report that Block V and its C-terminal flanking region up to residue 1681 are important for the proper folding of the RTX domain.23 Since the C-terminally truncated variants were detected by anti-hexahistidine antibody in whole cell lysates but not purified from the soluble fraction, it is likely that they formed aggregates or inclusion bodies.
The N-terminal truncations were further purified by size exclusion chromatography and the monomeric fractions assessed for antibody binding by ELISA. All variants bound rabbit polyclonal anti-ACT antibody strongly, but none showed significant binding to M1H5 or M2B10. (Figure S3). The discrepancy between western blot and ELISA result may be due to differences in the assay sensitivity.
RTX751 displayed on the yeast surface presents conformational epitopes
The relatively low-throughput nature of these approaches and large size of RTX751 (955 residues) led us to pursue high-throughput approaches to identify specific epitope residues. Yeast display with FACS-based selection and next-generation sequencing is emerging as a versatile tool for this effort.37, 38, 48 However, RTX751 is ~100 kDa while previous proteins used in yeast display have been <60kDa in size.49 Moreover, most proteins, including the Aga2 fusion proteins used in yeast display, include a secretion signal at the N-terminus, yet ACT is normally exported via a secretion signal at its C-terminus. We therefore needed to determine whether RTX could be displayed on yeast in a properly folded form.
Since all RTX truncations generated above compromised antibody binding measured by ELISA, the entire RTX751 gene (codons 751–1706) was cloned into the pCTCON yeast display vector as a C-terminal fusion to the yeast Aga2 protein.39 Yeast cells harboring the plasmid were induced and incubated with antibodies in the presence of 2 mM CaCl2 prior to flow cytometry analysis. Murine antibody 1H6, which binds a calcium-independent, linear epitope between residues 1589–1631 in the C-terminal RTX secretion sequence,45 was used to monitor display of full-length RTX on the yeast surface, while either M1H5 or M2B10 was used to monitor folding of the epitope of interest (Figure 6A). When the yeast cells displaying RTX751 were co-stained with 1H6 and M1H5 or M2B10, a double-positive population in the Q2 quadrant indicated a strong correlation between RTX751 display level and integrity of the M1H5 and M2B10 epitopes, while yeast with empty vector showed a tight double-negative population in Q4 (Figure 6B).
Figure 6. Yeast display intact RTX751 with conformational epitopes.
A, Diagramatic depiction of RTX751 yeast display and staining by two antibodies simultaneously. B, Yeast cells were grown and induced, then stained with antibodies 1H6 and either M1H5 or M2B10 in the presence of 2mM CaCl2, followed by AlexaFluor-488 anti-mouse IgG Fc-specific to detect 1H6 and AlexaFluor-647 anti-human IgG Fc-specific to detect M1H5 or M2B10 using a Fortessa flow cytometer. Controls included yeast cells grown and induced in the same manner but stained without CaCl2 or yeast cells that were heated at 85 °C for 30 min before staining in the presence of 2mM CaCl2. Control yeast harbored an empty pCTCON vector lacking the RTX751 gene.
We next aimed to determine whether the antibodies bound yeast-displayed RTX751 in a similar manner as soluble RTX751. To determine whether binding is calcium dependent, yeast displaying RTX751 were incubated with antibodies in the absence of calcium. Under this condition, neither M1H5 nor M2B10 bound the yeast but the calcium-independent 1H6 antibody retained binding, resulting in a cell population in quadrant Q3. To further assess the specificity of antibody binding, RTX751-displaying yeast were heated at 85°C for 30 min prior to antibody staining. In this case, no RTX-specific antibody binding was observed (Figure 6B). Taken together, these data suggest that yeast display is a feasible strategy to select variants of RTX751 with altered affinity for the M1H5 and M2B10 antibodies.
Selection of RTX751 variants with reduced antibody affinity by yeast display
For fine epitope mapping, our strategy was to generate a library in which the yeast displayed antigen was randomly mutated, followed by FACS to isolate variants with altered antibody binding properties.35, 37, 48 Since RTX751 is a large protein, encoded by ~2900 bp DNA, we used our biochemical data to focus on smaller RTX regions likely to include the entire epitope. The ACT species selectivity and truncation data suggested a role for RTX blocks II–III, while earlier work suggested that the receptor-binding region lies between RTX residues 1166–1287 that span blocks II–III.15, 30 We amplified codons 751–1358 (about two-thirds of RTX751) under error-prone conditions. A library with 2.1 × 107 clones was constructed using homologous recombination of NheI/SphI linearized vector backbone and error prone PCR product with overhangs on both ends.
To identify populations for sorting, the library and two control yeast populations, one expressing wild-type RTX751 and another with empty pCTCON vector, were induced prior to antibody staining. For the initial analysis, we used 67 nM M1H5/M2B10 for staining. This is much higher than the measured antibody affinities to ensure detection of RTX751 variants with modestly but not severely reduced binding. The library showed three populations: a double positive population similar to that observed for wild-type RTX751 (termed population P7), a population with lower Alexa647 fluorescence indicating reduced M2B10 binding (population P5), and a population with severely reduced M2B10 binding (population P4; Figure 7). The library stained with M1H5 and 1H6 showed similar populations (data not shown).
Figure 7. Antibody-binding populations isolated by flow cytometry.
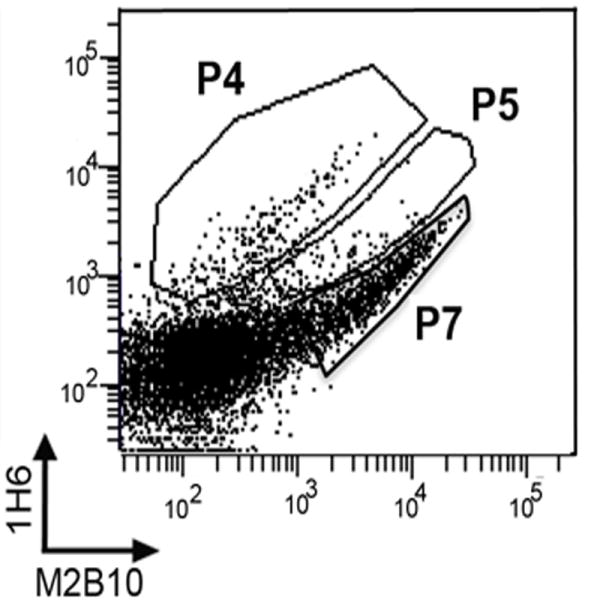
The RTX751 library was induced and stained with antibodies 1H6 and M2B10 or M1H5, followed by sorting on a FACS Aria cell sorter. DNA sequencing revealed that the P4 population, with severely reduced antibody binding, was dominated by clones with internal deletions. The P5 population, with moderately reduced antibody binding, contained full length RTX751 with random mutations.
All three populations (P4, P5 and P7) were isolated by FACS after incubation with either the M1H5 or M2B10 antibody. Since the initial screen showed a strong P7 population after incubation with 67 nM antibody, we reduced this to 13.4 nM to better detect clones with modestly reduced affinity. Sequencing of 12 random clones from each population showed that all P5 and P7 clones contained full length RTX751 gene with mutations. The P4 clones contained truncated RTX751 genes that lost internal portions of the randomized region and thus lost binding to M2B10 or M1H5 but not 1H6. This is likely a result of incorrect recombination events due to the highly similar repeats in RTX. To further characterize the isolated clones, four P5 clones were grown individually and stained with 1H6 and M2B10 (Figure S4). Compared to yeast displaying wild-type RTX751, all four clones showed a moderate to severe reduction of M2B10 binding, while the binding to 1H6 was not significantly affected, indicating isolation of the desired population. A total of ~50,000 cells were collected for the P5 and P7 populations after staining with each antibody.
Identification of RTX residues implicated in antibody epitopes
High-throughput sequencing of the isolated cell populations was used to identify individual residues affecting antibody binding. After sorting, the yeast clones were grown in selective medium, plasmids extracted and the mutagenized region amplified for sequencing with an Illumina Hiseq 2500. Every position was altered to at least four different residues. About 40% of the read pairs contained a single nucleotide mutation as compared to the wild-type B. pertussis sequence, and these reads were counted to determine the enrichment or depletion of each mutation at each position by the selective sort. Enrichment values for each mutation were calculated using the Enrich software package.43 Custom scripts (available on GitHub, see materials & methods) converted the shotgun reads into a standardized format compatible with Enrich and processed the Enrich output. Epitope residues were identified by manual examination of enrichment values at each position.
This analysis identified 20 residues implicated in M1H5 binding and 16 in M2B10 binding (Figure 8A). Most of these residues are not continuous in the primary sequence, suggesting they may be conformational. There is only one shared residue between the two sets (D1199), consistent with our prior observation that the two antibodies do not compete with each other. Interestingly, all the identified residues fall within the Gly-Asp rich blocks II to III for M1H5 and I–III for M2B10, despite similar mutational rates across codons 751–1358. A number of the identified residues are aspartic acids (seven for M1H5; two for M2B10). Given the known role of aspartic acid in calcium binding, these may play structural roles as opposed to antibody-antigen interactions.
Figure 8. Antibody epitope residues on RTX751 identified by next-generation sequencing.

A, The RTX region subjected to random mutagenesis is shown. Bold and red letters indicate candidate M1H5 epitope residues, while bold and blue letters identify potential M2B10 epitope residues. The sole shared residue is indicated by bold and purple. Bold, underlined and italized residues indicate those implicated in receptor binding 30. The boxes indicate the first three Gly-Asp rich repeat blocks. B, Six residues identified as potentially participating in each antibody epitope were individually altered to the most common enriched amino acid residues observed after sequencing of sorted libraries. Each variant, with a single residue change, was expressed and purified as soluble RTX751 protein. Binding affinity of M1H5 and M2B10 monovalent scAbs for each of the RTX751 variants was assessed by ELISA. Data is represented as relative affinity (EC50,wild-type/EC50,variant), so that a lower value indicates reduced binding. C, A predicted structure of RTX751 was generated using the RaptorX web server44 and the experimental structure of domain V (PDB 5CVW or 5CXL). Residues 1015–1679 encompassing the five repeat blocks are shown, with the individual block locations indicated. The putative epitope residues are highlighted in red for M1H5 and in blue for M2B10, the block II variable region is shown in green, the putative receptor binding site is shown in magenta and the block IV–V region in yellow.
Structural analysis and validation of epitope residues
To validate the library results, six residues predicted to be involved in each epitope were randomly chosen for further analysis. Soluble RTX751 variants containing a single residue change, from the wild-type residue to the residue most frequently observed in the reduced antibody binding P5 populations, were expressed and purified. Binding of antibodies M1H5 and M2B10 to the wild-type and variant RTX751 were compared by ELISA. Among the six M1H5 epitopic mutants, H1225D and F1229M showed significantly reduced binding to M1H5 but not to M2B10. The other four variants, D1161V, R1166P, D1179H and D1199H, demonstrated severely altered binding to both M1H5 and M2B10. Among the six variants encompassing the putative M2B10 epitope, W1074S, E1117D, K1118N, and P1120S showed significantly reduced binding to M2B10, but only minimally affected binding to M1H5 (Figure 8B). All variants bound 1H6 or a rabbit polyclonal anti-ACT antibody equally well (Figure S5), suggesting the difference in apparent affinity to M1H5 and M2B10 is not due to different coating densities on the ELISA surface.
To better understand the structural orientation of these putative epitope, we generated a computational model of the wild-type RTX751 domain with the RaptorX web server, based on homologous RTX structures (RTX block V PBD files 5CVW and 5CXL). The model shows beta roll domains with buried aspartic acid residues. The C-terminal secretion signal was predicted as an extended tail structure; since this does not seem reliable and this region is not predicted to be involved in antibody binding, all residues after D1680 were deleted. Blocks I–IV were modeled from a single experimental structure of block V, connected by linker regions. Since there is no experimental data supporting the interactions between the domains, the model may not accurately represent the quaternary RTX structure. Nevertheless, this model assumes the ratchet shape described by Bumba et al. 26
The majority of the identified residues formed two adjacent patches on the RTX model (Figure 8C). This further supports the idea that the antibodies do not compete with each other for ACT binding. While many of the epitope residues are predicted to be solvent exposed, several predicted residues are predicted as buried. In particular, residue G1262 is buried in the structure, and residue D1294 is located distal to the major epitope patch. For both variants we noticed significantly reduced expression level, supporting the idea that they affect RTX structure or stability. Aspartic acid residues in particular may mediate calcium binding and support beta roll formation as opposed to direct antibody interactions. Supporting this notion, expression level of the three variants analyzed (D1161V, D1179H and D1199H) was reduced to ~10% of wild-type RTX751 and size exclusion chromatography indicated that a large fraction of the protein eluted as higher molecular weight species (Figure S6). The candidate epitope residues identified by yeast display are consistent with our previous biochemical data.
Discussion
We previously discovered the first antibodies that neutralize ACT activities by binding the RTX domain of ACT and competitively inhibiting the RTX-receptor binding interaction.21 For other infectious diseases in which toxins play a prominent role, receptor blockade is a proven strategy for active and passive immunization: vaccination with tetanus toxoid primarily induces receptor-blocking antibodies;50 and antibodies using this mechanism are approved therapeutics to treat anthrax.51 These anti-RTX antibodies are broadly neutralizing and recognize conserved epitopes, as both bind RTX from the three major Bordetella strains, one with identical affinity. These epitopes are thus likely to be important for protection. Complementary epitope mapping approaches localized the antibody epitopes to the same vicinity as the receptor-binding site and suggest that at least one residue is shared by the M1H5 antibody and the receptor-binding site.30 These results shed light on mechanisms of antibody versus receptor binding to RTX as well as mechanisms of ACT neutralization and the potential for escape variants.
The M1H5 and M2B10 antibodies both recognize primarily conformational epitopes on RTX with low nanomolar affinity (Figure 1). No antibody binding was observed to the intact ACT or RTX751 in the absence of calcium, but the subsequent addition of 2 mM CaCl2 restored full binding (Figure 2). This indicates that the epitopes require the calcium-mediated beta roll conformation of RTX. Both antibodies detect RTX751 on a Western blot (Figure 5B), which likely reflects the very sensitive nature of the assay and the fact that conformational epitopes often include several short linear elements. Several lines of evidence localized the epitopes to repeat blocks II–III within RTX751. Of the three RTX chimeras we generated containing regions of sequence variation present in other Bordetella species, only the chimera with an altered block III lost affinity for the M1H5 antibody (Figure 4). Similarly, only RTX truncation variants retaining repeat blocks II or III retained antibody binding (Figure 5B).
Even this simple observation provides insight into the mechanism of antibody neutralization. ACT is known to fold upon encountering extracellular calcium ions while being secreted across the bacterial outer membrane.26 This results in locally high concentrations of ACT that can bind and intoxicate eukaryotic cells in close proximity to the bacteria.52 Considering that the antibodies primarily bind the folded RTX conformation and can neutralize ACT when produced by bacteria in vitro (Figure 3D), the antibodies may bind ACT as it is being secreted from the bacteria or shortly thereafter, and likely need to co-localize with the bacteria in order to effectively neutralize secreted ACT.
A strategy combining yeast display, cell sorting and high-throughput sequencing identified individual residues involved in the antibody epitopes. Despite the large size of RTX751 (~100 kDa) and the fact that it is normally secreted via a C-terminal secretion sequence, the full-length protein was presented on the yeast surface and recognized by our antibodies in a calcium-dependent manner (Figure 6). Interestingly, although our libraries retained the C-terminal secretion signal, removing this had no effect on RTX751 display or antibody binding (data not shown).
Since there is not yet a complete structure of RTX, we targeted random mutagenesis to the blocks II–III implicated by our biochemical analyses. To allow use of naturally occurring restriction sites, the mutated region spanned blocks I–III. Deep sequencing indicated that every residue in this region was altered to at least four different amino acids. Since cell populations with highly reduced M1H5/M2B10 binding were dominated by variants with internal deletions, we isolated and sequenced clones with modestly reduced antibody binding. This identified 20 residues in blocks II–III and 16 residues in blocks I–III that contribute to the M1H5 and M2B10 epitopes, respectively (Figure 8).
The residues identified by yeast display are largely consistent with our biochemical data. These residues form two adjacent regions on our homology model of RTX751 (Figure 8C), consistent with our observations that binding of one antibody does not inhibit binding of the other 21. Notably, only one residue (D1199) is shared between the two putative epitopes and we are unable to exclude a structural calcium-binding role for this or the other identified aspartic acid residues. Only one residue (R1164) was identified that falls within the variable regions used to generate RTX chimeras. A number of predicted epitope residues are present in the conserved regions directly adjacent to the block II and block III variable regions; variable residues could influence the orientation of these epitope residues, thus explaining the species specificity we observed for M1H5. The fact that both antibodies bind RTX751 from different species and that only one epitope residue varies among the three RTX751 alleles tested is evidence that the epitopes are highly conserved and that there is a low risk for escape variants.
There are a few caveats in our study. First, we mutated only 1.9 kb of the ~3 kb RTX751 gene. This region was implicated by our biochemical data and includes the putative receptor-binding site. However, there may be residues outside of this region that help to stabilize the epitope structure or are more directly involved in antibody binding. Second, we analyzed pairs of short (125 bp) sequence reads containing a single mutation, but we cannot exclude the possibility that additional mutations were present outside of the sequenced region. However, if a certain residue is enriched in the sorted library, this residue is unlikely to be a neutral mutation due to the large number of reads from different clones. Third, most of the residues we validated showed a modest decrease in antibody binding. By selecting the P5 population with modestly reduced antibody binding, we may have missed some of the hot spot residues critical for binding. Residues resulting in severely reduced antibody binding fell into the P4 gate which we did not deep sequence since most of these clones had large internal deletions due to aberrant homologous recombination events. Finally, RTX is a large protein that has been largely recalcitrant to crystallization and has little homology to other proteins with known structures. The X-ray structure of the block V repeat was recently reported and was used here to model the other blocks but the lack of an experimental RTX structure limits our ability to unambiguously distinguish between epitopic versus structural residues.
A key question is to what extent these antibodies mimic the receptor-RTX binding interaction. The receptor binding epitope was initially mapped by insertion of an octapeptide into convenient restriction sites to residues 1166–1287, spanning repeat blocks II–III.15 This region includes the block III variable region, with two additional species-specific residues (Figure S2). Our results show that the M1H5 epitope also spans blocks II–III, while the M2B10 epitope appears broader, with residues distributed over blocks I–III. M1H5 includes three epitope residues within and five more just upstream of 1166–1287, while M2B10 includes one. Regardless, the similar binding locales explain why both antibodies competitively inhibit receptor binding.
A recent study aiming to understand ACT-CR3 receptor interactions identified several ACT residues that impact receptor binding.30 Computational modeling suggested negatively charged residues within the 1166–1287 ACT region interact with positively charged residues between 614–682 on the CD11b chain of CR3. Two ACT variants were generated and observed to have strongly reduced cellular binding by flow cytometry and cellular penetration, as monitored by cAMP production. One variant included two (E1232A+D1234A) residue changes while the other included three changes (D1193A+D1194A+E1195A) within the putative receptor binding region. All five residues are conserved among the major Bordetella strains (Figure S2). The E1232A/D1234A residues are right before the block III variable region, suggesting they may have been preserved by a selective pressure. The triple-mutation variant includes D1194 that was also identified as an M1H5 epitope residue.
A striking difference is that neither these antibodies nor previously described anti-ACT monoclonal antibodies require palmitoylation for binding,16 while receptor binding and cellular are greatly enhanced by this post-translational modification.21, 27, 28 Palmitoylation has been suggested to stabilize the ACT-receptor interaction by interacting with the membrane. Considering the similarities in the antibody and receptor epitopes, the lipid may also directly interact with the receptor. There is precedence for such an interaction: the palmitoyl group on the Xenopus Wnt8 extends into a deep groove on its Frizzled receptor protein, contributing to the binding interface and stabilizing the interaction.53 It would be interesting to map receptor epitope on RTX domain using the approach used here, however co-expression of the acylating enzyme CyaC reduced RTX751 yeast-display levels and RTX751 on the surface did not bind to soluble receptor. The CyaC enzyme may not be compatible with the yeast palmitoyl-CoA:acyl carrier protein complex, or the C16 substrate may not be available in sufficient quantities.54
ACT, especially its RTX domain, is an attractive target for development as a vaccine antigen as it is an essential virulence factor that is also highly conserved.55 The M1H5 and M2B10 antibodies are broadly neutralizing and thus their epitopes are targets for protective immunity. The M1H5 epitope appears to more closely overlap with the receptor epitope, while M2B10 appears to less closely mimic the receptor epitope and may act by sterically inhibiting receptor binding as opposed to direct competition. As for other antigens, this detailed mapping of conserved neutralizing epitopes sets the stage for design of vaccine antigens that induce antibodies directed toward these epitopes. While yeast display is unlikely to facilitate a similar study to map the receptor binding epitope on RTX751, it could be used to engineer RTX751 variants, for instance smaller variants comprising blocks II–V or II-III–V, for increased solubility while retaining these neutralizing epitopes.
Supplementary Material
Acknowledgments
We thank Andrea M. diVenere for expert technical assistance and Richard A Salinas in Microscopy and Imaging Facility at The University of Texas at Austin for assistance with cell sorting.
Funding details: This work was support by the Welch Foundation under grant F-1767 and NIH grant RO1 AI122753 and 1S10RR028062 to JAM; NSF under grant CBET-1254238 to TAW and under NIH grant RO1 AI018000 to ELH.
Abbreviations
- ACT
adenylate cyclase toxin
- CR3
complement receptor 3
- ELISA
enzyme-linked immunosorbent assay
- IgG
immunoglobulin
- RTX
repeat in toxin
- RTX751
repeat in toxin fragment containing residues 751–1706
- scAb
single chain antibody
- SPR
surface plasmon resonance
Footnotes
Supporting information available Supplementary Table S1, Statistics for read coverage of the libraries; Figure S1, Antibody-RTX751 binding kinetics; Figure S2, Amino acid sequence alignment and RTX block regions for representative Bordetella species; Figure S3, Antibody binding to purified RTX C- and N- terminal truncations; Figure S4, Characterization of individual random mutation clones; Figure S5, ELISA validation of select candidate epitope residues; Figure S6, SEC elution profiles for wild-type RTX751 and variants.
References
- 1.Warfel JM, Zimmerman LI, Merkel TJ. Acellular pertussis vaccines protect against disease but fail to prevent infection and transmission in a nonhuman primate model. Proc Natl Acad Sci U S A. 2014;111:787–792. doi: 10.1073/pnas.1314688110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hewlett EL, Burns DL, Cotter PA, Harvill ET, Merkel TJ, Quinn CP, Stibitz ES. Pertussis pathogenesis--what we know and what we don’t know. J Infect Dis. 2014;209:982–985. doi: 10.1093/infdis/jit639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Plotkin SA. The pertussis problem. Clin Infect Dis. 2014;58:830–833. doi: 10.1093/cid/cit934. [DOI] [PubMed] [Google Scholar]
- 4.Guiso N. Bordetella pertussis: why is it still circulating? J Infect. 2014;68(Suppl 1):S119–124. doi: 10.1016/j.jinf.2013.09.022. [DOI] [PubMed] [Google Scholar]
- 5.Cherry JD. Why do pertussis vaccines fail? Pediatrics. 2012;129:968–970. doi: 10.1542/peds.2011-2594. [DOI] [PubMed] [Google Scholar]
- 6.Sheridan SL, Ware RS, Grimwood K, Lambert SB. Number and order of whole cell pertussis vaccines in infancy and disease protection. JAMA. 2012;308:454–456. doi: 10.1001/jama.2012.6364. [DOI] [PubMed] [Google Scholar]
- 7.Cherry JD, Heininger U, Richards DM, Storsaeter J, Gustafsson L, Ljungman M, Hallander HO. Antibody response patterns to Bordetella pertussis antigens in vaccinated (primed) and unvaccinated (unprimed) young children with pertussis. Clin Vaccine Immunol. 2010;17:741–747. doi: 10.1128/CVI.00469-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berbers GA, de Greeff SC, Mooi FR. Improving pertussis vaccination. Hum Vaccin. 2009;5:497–503. doi: 10.4161/hv.8112. [DOI] [PubMed] [Google Scholar]
- 9.Clark TA. Responding to pertussis. J Pediatr. 2012;161:980–982. doi: 10.1016/j.jpeds.2012.07.014. [DOI] [PubMed] [Google Scholar]
- 10.Melvin JA, Scheller EV, Miller JF, Cotter PA. Bordetella pertussis pathogenesis: current and future challenges. Nat Rev Microbiol. 2014;12:274–288. doi: 10.1038/nrmicro3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sebo P, Osicka R, Masin J. Adenylate cyclase toxin-hemolysin relevance for pertussis vaccines. Expert Rev Vaccines. 2014;13:1215–1227. doi: 10.1586/14760584.2014.944900. [DOI] [PubMed] [Google Scholar]
- 12.Guiso N, Hegerle N. Other Bordetellas, lessons for and from pertussis vaccines. Expert Rev Vaccines. 2014;13:1125–1133. doi: 10.1586/14760584.2014.942221. [DOI] [PubMed] [Google Scholar]
- 13.Edwards KM. Unraveling the challenges of pertussis. Proc Natl Acad Sci U S A. 2014;111:575–576. doi: 10.1073/pnas.1321360111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guermonprez P, Khelef N, Blouin E, Rieu P, Ricciardi-Castagnoli P, Guiso N, Ladant D, Leclerc C. The adenylate cyclase toxin of Bordetella pertussis binds to target cells via the alpha(M)beta(2) integrin (CD11b/CD18) J Exp Med. 2001;193:1035–1044. doi: 10.1084/jem.193.9.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.El-Azami-El-Idrissi M, Bauche C, Loucka J, Osicka R, Sebo P, Ladant D, Leclerc C. Interaction of Bordetella pertussis adenylate cyclase with CD11b/CD18: Role of toxin acylation and identification of the main integrin interaction domain. J Biol Chem. 2003;278:38514–38521. doi: 10.1074/jbc.M304387200. [DOI] [PubMed] [Google Scholar]
- 16.Gordon VM, Young WW, Jr, Lechler SM, Gray MC, Leppla SH, Hewlett EL. Adenylate cyclase toxins from Bacillus anthracis and Bordetella pertussis. Different processes for interaction with and entry into target cells. J Biol Chem. 1989;264:14792–14796. [PubMed] [Google Scholar]
- 17.Wolff J, Cook GH, Goldhammer AR, Berkowitz SA. Calmodulin activates prokaryotic adenylate cyclase. Proc Natl Acad Sci U S A. 1980;77:3841–3844. doi: 10.1073/pnas.77.7.3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Confer DL, Eaton JW. Phagocyte impotence caused by an invasive bacterial adenylate cyclase. Science. 1982;217:948–950. doi: 10.1126/science.6287574. [DOI] [PubMed] [Google Scholar]
- 19.Kamanova J, Kofronova O, Masin J, Genth H, Vojtova J, Linhartova I, Benada O, Just I, Sebo P. Adenylate cyclase toxin subverts phagocyte function by RhoA inhibition and unproductive ruffling. J Immunol. 2008;181:5587–5597. doi: 10.4049/jimmunol.181.8.5587. [DOI] [PubMed] [Google Scholar]
- 20.Betsou F, Sebo P, Guiso N. The C-terminal domain is essential for protective activity of the Bordetella pertussis adenylate cyclase-hemolysin. Infect Immun. 1995;63:3309–3315. doi: 10.1128/iai.63.9.3309-3315.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang X, Gray MC, Hewlett EL, Maynard JA. The Bordetella adenylate cyclase repeat-in-toxin (RTX) domain is immunodominant and elicits neutralizing antibodies. J Biol Chem. 2015;290:3576–3591. doi: 10.1074/jbc.M114.585281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Monneron A, Ladant D, d’Alayer J, Bellalou J, Barzu O, Ullmann A. Immunological relatedness between Bordetella pertussis and rat brain adenylyl cyclases. Biochemistry. 1988;27:536–539. doi: 10.1021/bi00402a005. [DOI] [PubMed] [Google Scholar]
- 23.Bauche C, Chenal A, Knapp O, Bodenreider C, Benz R, Chaffotte A, Ladant D. Structural and functional characterization of an essential RTX subdomain of Bordetella pertussis adenylate cyclase toxin. J Biol Chem. 2006;281:16914–16926. doi: 10.1074/jbc.M601594200. [DOI] [PubMed] [Google Scholar]
- 24.Angkawidjaja C, You DJ, Matsumura H, Kuwahara K, Koga Y, Takano K, Kanaya S. Crystal structure of a family I.3 lipase from Pseudomonas sp MIS38 in a closed conformation. FEBS Lett. 2007;581:5060–5064. doi: 10.1016/j.febslet.2007.09.048. [DOI] [PubMed] [Google Scholar]
- 25.Blenner MA, Shur O, Szilvay GR, Cropek DM, Banta S. Calcium-induced folding of a beta roll motif requires C-terminal entropic stabilization. J Mol Biol. 2010;400:244–256. doi: 10.1016/j.jmb.2010.04.056. [DOI] [PubMed] [Google Scholar]
- 26.Bumba L, Masin J, Macek P, Wald T, Motlova L, Bibova I, Klimova N, Bednarova L, Veverka V, Kachala M, Svergun DI, Barinka C, Sebo P. Calcium-Driven Folding of RTX Domain beta-Rolls Ratchets Translocation of RTX Proteins through Type I Secretion Ducts. Mol Cell. 2016;62:47–62. doi: 10.1016/j.molcel.2016.03.018. [DOI] [PubMed] [Google Scholar]
- 27.Hackett M, Guo L, Shabanowitz J, Hunt DF, Hewlett EL. Internal lysine palmitoylation in adenylate cyclase toxin from Bordetella pertussis. Science. 1994;266:433–435. doi: 10.1126/science.7939682. [DOI] [PubMed] [Google Scholar]
- 28.Basar T, Havlicek V, Bezouskova S, Hackett M, Sebo P. Acylation of lysine 983 is sufficient for toxin activity of Bordetella pertussis adenylate cyclase. Substitutions of alanine 140 modulate acylation site selectivity of the toxin acyltransferase CyaC. J Biol Chem. 2001;276:348–354. doi: 10.1074/jbc.M006463200. [DOI] [PubMed] [Google Scholar]
- 29.Hewlett EL, Gray L, Allietta M, Ehrmann I, Gordon VM, Gray MC. Adenylate cyclase toxin from Bordetella pertussis. Conformational change associated with toxin activity. J Biol Chem. 1991;266:17503–17508. [PubMed] [Google Scholar]
- 30.Osicka R, Osickova A, Hasan S, Bumba L, Cerny J, Sebo P. Bordetella adenylate cyclase toxin is a unique ligand of the integrin complement receptor 3. eLife. 2015;4:e10766. doi: 10.7554/eLife.10766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scarselli M, Arico B, Brunelli B, Savino S, Di Marcello F, Palumbo E, Veggi D, Ciucchi L, Cartocci E, Bottomley MJ, Malito E, Lo Surdo P, Comanducci M, Giuliani MM, Cantini F, Dragonetti S, Colaprico A, Doro F, Giannetti P, Pallaoro M, Brogioni B, Tontini M, Hilleringmann M, Nardi-Dei V, Banci L, Pizza M, Rappuoli R. Rational design of a meningococcal antigen inducing broad protective immunity. Sci Transl Med. 2011;3:91ra62. doi: 10.1126/scitranslmed.3002234. [DOI] [PubMed] [Google Scholar]
- 32.McLellan JS, Chen M, Joyce MG, Sastry M, Stewart-Jones GB, Yang Y, Zhang B, Chen L, Srivatsan S, Zheng A, Zhou T, Graepel KW, Kumar A, Moin S, Boyington JC, Chuang GY, Soto C, Baxa U, Bakker AQ, Spits H, Beaumont T, Zheng Z, Xia N, Ko SY, Todd JP, Rao S, Graham BS, Kwong PD. Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science. 2013;342:592–598. doi: 10.1126/science.1243283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cunningham BC, Wells JA. High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science. 1989;244:1081–1085. doi: 10.1126/science.2471267. [DOI] [PubMed] [Google Scholar]
- 34.Weiss GA, Watanabe CK, Zhong A, Goddard A, Sidhu SS. Rapid mapping of protein functional epitopes by combinatorial alanine scanning. Proc Natl Acad Sci U S A. 2000;97:8950–8954. doi: 10.1073/pnas.160252097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Levy R, Forsyth CM, LaPorte SL, Geren IN, Smith LA, Marks JD. Fine and domain-level epitope mapping of botulinum neurotoxin type A neutralizing antibodies by yeast surface display. J Mol Biol. 2007;365:196–210. doi: 10.1016/j.jmb.2006.09.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Doolan KM, Colby DW. Conformation-dependent epitopes recognized by prion protein antibodies probed using mutational scanning and deep sequencing. J Mol Biol. 2015;427:328–340. doi: 10.1016/j.jmb.2014.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van Blarcom T, Rossi A, Foletti D, Sundar P, Pitts S, Bee C, Melton Witt J, Melton Z, Hasa-Moreno A, Shaughnessy L, Telman D, Zhao L, Cheung WL, Berka J, Zhai W, Strop P, Chaparro-Riggers J, Shelton DL, Pons J, Rajpal A. Precise and efficient antibody epitope determination through library design, yeast display and next-generation sequencing. J Mol Biol. 2015;427:1513–1534. doi: 10.1016/j.jmb.2014.09.020. [DOI] [PubMed] [Google Scholar]
- 38.Kowalsky CA, Faber MS, Nath A, Dann HE, Kelly VW, Liu L, Shanker P, Wagner EK, Maynard JA, Chan C, Whitehead TA. Rapid fine conformational epitope mapping using comprehensive mutagenesis and deep sequencing. J Biol Chem. 2015;290:26457–26470. doi: 10.1074/jbc.M115.676635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feldhaus MJ, Siegel RW, Opresko LK, Coleman JR, Feldhaus JM, Yeung YA, Cochran JR, Heinzelman P, Colby D, Swers J, Graff C, Wiley HS, Wittrup KD. Flow-cytometric isolation of human antibodies from a nonimmune Saccharomyces cerevisiae surface display library. Nat Biotechnol. 2003;21:163–170. doi: 10.1038/nbt785. [DOI] [PubMed] [Google Scholar]
- 40.Ohmura N, Lackie SJ, Saiki H. An immunoassay for small analytes with theoretical detection limits. Anal Chem. 2001;73:3392–3399. doi: 10.1021/ac001328d. [DOI] [PubMed] [Google Scholar]
- 41.Benatuil L, Perez JM, Belk J, Hsieh CM. An improved yeast transformation method for the generation of very large human antibody libraries. Protein Eng Des Sel. 2010;23:155–159. doi: 10.1093/protein/gzq002. [DOI] [PubMed] [Google Scholar]
- 42.Podnar J, Deiderick H, Hunicke-Smith S. Next-generation sequencing fragment library construction. Curr Protoc Mol Biol. 2014;107:7 17 11–17 17 16. doi: 10.1002/0471142727.mb0717s107. [DOI] [PubMed] [Google Scholar]
- 43.Fowler DM, Araya CL, Gerard W, Fields S. Enrich: software for analysis of protein function by enrichment and depletion of variants. Bioinformatics. 2011;27:3430–3431. doi: 10.1093/bioinformatics/btr577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang S, Li W, Liu S, Xu J. RaptorX-Property: a web server for protein structure property prediction. Nucleic Acids Res. 2016;44:W430–435. doi: 10.1093/nar/gkw306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee SJ, Gray M, Guo L, Sebo P, Hewlett E. Epitope mapping of monoclonal antibodies against Bordetella pertussis adenylate cyclase toxin. Infection and immunity. 1999;67:2090. doi: 10.1128/iai.67.5.2090-2095.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mattoo S, Cherry JD. Molecular pathogenesis, epidemiology, and clinical manifestations of respiratory infections due to Bordetella pertussis and other Bordetella subspecies. Clin Microbiol Rev. 2005;18:326–382. doi: 10.1128/CMR.18.2.326-382.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Osicka R, Osickova A, Basar T, Guermonprez P, Rojas M, Leclerc C, Sebo P. Delivery of CD8(+) T-cell epitopes into major histocompatibility complex class I antigen presentation pathway by Bordetella pertussis adenylate cyclase: delineation of cell invasive structures and permissive insertion sites. Infect Immun. 2000;68:247–256. doi: 10.1128/iai.68.1.247-256.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chao G, Cochran JR, Wittrup KD. Fine epitope mapping of anti-epidermal growth factor receptor antibodies through random mutagenesis and yeast surface display. J Mol Biol. 2004;342:539–550. doi: 10.1016/j.jmb.2004.07.053. [DOI] [PubMed] [Google Scholar]
- 49.Kondo A, Ueda M. Yeast cell-surface display--applications of molecular display. Appl Microbiol Biotechnol. 2004;64:28–40. doi: 10.1007/s00253-003-1492-3. [DOI] [PubMed] [Google Scholar]
- 50.Lavinder JJ, Wine Y, Giesecke C, Ippolito GC, Horton AP, Lungu OI, Hoi KH, DeKosky BJ, Murrin EM, Wirth MM, Ellington AD, Dorner T, Marcotte EM, Boutz DR, Georgiou G. Identification and characterization of the constituent human serum antibodies elicited by vaccination. Proc Natl Acad Sci U S A. 2014;111:2259–2264. doi: 10.1073/pnas.1317793111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maynard JA, Maassen CB, Leppla SH, Brasky K, Patterson JL, Iverson BL, Georgiou G. Protection against anthrax toxin by recombinant antibody fragments correlates with antigen affinity. Nat Biotechnol. 2002;20:597–601. doi: 10.1038/nbt0602-597. [DOI] [PubMed] [Google Scholar]
- 52.Eby JC, Gray MC, Warfel JM, Paddock CD, Jones TF, Day SR, Bowden J, Poulter MD, Donato GM, Merkel TJ, Hewlett EL. Quantification of the adenylate cyclase toxin of Bordetella pertussis in vitro and during respiratory infection. Infect Immun. 2013;81:1390–1398. doi: 10.1128/IAI.00110-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Janda CY, Waghray D, Levin AM, Thomas C, Garcia KC. Structural basis of Wnt recognition by Frizzled. Science. 2012;337:59–64. doi: 10.1126/science.1222879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tehlivets O, Scheuringer K, Kohlwein SD. Fatty acid synthesis and elongation in yeast. Biochim Biophys Acta. 2007;1771:255–270. doi: 10.1016/j.bbalip.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 55.Weiss AA, Hewlett EL, Myers GA, Falkow S. Pertussis toxin and extracytoplasmic adenylate cyclase as virulence factors of Bordetella pertussis. J Infect Dis. 1984;150:219–222. doi: 10.1093/infdis/150.2.219. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.