

Abstract
IFN-γ is essential for control of Mycobacterium tuberculosis infection in vitro and in vivo. However, the mechanisms by which IFN-γ controls infection remain only partially understood. One of the crucial IFN-γ target genes required for control of M. tuberculosis is Inducible nitric oxide synthase (iNOS). While nitric oxide (NO) produced by iNOS is thought to have direct bactericidal activity against M. tuberculosis, the role of NO as a signaling molecule has been poorly characterized in the context M. tuberculosis infection. Here, we find that iNOS broadly regulates the macrophage transcriptome during M. tuberculosis infection, activating antimicrobial pathways while also limiting inflammatory cytokine production. The transcription factor Hypoxia inducible factor-1α (HIF-1α) was recently shown to be critical for IFN-γ mediated control of M. tuberculosis infection. We find that HIF-1α function requires NO production, and that HIF-1α and iNOS are linked by a positive feedback loop that amplifies macrophage activation. Furthermore, we find that NO inhibits NF-kB activity to prevent hyper-inflammatory responses. Thus, NO activates robust microbicidal programs while at the same time limiting damaging inflammation. IFN-γ signaling must carefully calibrate an effective immune response that does not cause excessive tissue damage, and this work identifies NO as a key player in establishing this balance during M. tuberculosis infection.
Keywords: Mycobacterium tuberculosis, inflammation, nitric oxide, HIF-1α, Interferon-γ, macrophages
Introduction
Mycobacterium tuberculosis infection is responsible for an enormous burden of morbidity and mortality worldwide, causing ~1.5 million deaths annually (1). Successful immunity to M. tuberculosis infection requires the cytokine Interferon-γ (IFN-γ) (2, 3). The dependence on IFN-γ for control of M. tuberculosis infection can be seen both in cell culture and mouse models of infection, and patients lacking components of the IFN-γ signaling pathway are extremely susceptible to mycobacterial infections (4). While the essentiality of IFN-γ activation to successful control of M. tuberculosis infection is well established, the mechanisms through which this occur remain poorly understood. IFN-γ activation of macrophages is thought to control infection by transforming infected macrophages into a microbicidal environment, while also limiting damaging inflammation caused by recruitment of neutrophils (5–7).
Multiple IFN-γ dependent mechanisms of cell intrinsic control of M. tuberculosis have been proposed, including restriction of nutrient availability, enhanced production of antimicrobial peptides, induction of autophagy, and the production of nitric oxide (NO) by Inducible nitric oxide synthase (iNOS) (8–12). Mice deficient for iNOS are extremely susceptible to M. tuberculosis infection (12), accounting for a substantial portion of the susceptibility of IFN-γ deficient mice. While the role of NO as a second messenger that regulates mammalian signaling is well known, in the context of M. tuberculosis infection the possible regulatory roles of NO have been mostly overlooked. Instead, the susceptibility of iNOS deficient mice has long been attributed to direct bactericidal activity of NO (5, 13). However, several lines of evidence suggest that this model is incomplete. First, a study using microarrays to examine the transcriptional response to M. tuberculosis infection in the context of IFN-γ activation demonstrated that a large proportion of the gene expression changes observed under these conditions require expression of iNOS and/or phagosome oxidase (phox) (14). Although this study made the important point that NO likely modulates signaling during M. tuberculosis infection, a mechanistic basis for the impact of NO on the macrophage transcriptome was not identified. More recently, it was shown that the ability of NO to suppress inflammasome dependent production of IL-1 is important to prevent excessive inflammation that contributes to immune pathology independent of bacterial growth in mice infected with M. tuberculosis (15).
In this study, we demonstrate that NO has a broad impact on the macrophage transcriptome during M. tuberculosis infection and identify key regulatory pathways through which this occurs. We find that during M. tuberculosis infection NO is required for IFN-γ dependent stabilization of HIF-1α, a transcription factor we have recently shown to be critical for control of M. tuberculosis infection (16). Interestingly, we find that HIF-1α and iNOS are linked in a positive feedback loop during IFN-γ activation of M. tuberculosis infected macrophages. RNAseq profiling reveals that HIF-1α and iNOS regulate a largely overlapping set of genes, suggesting that iNOS plays an unexpectedly broad role in mediating antibacterial responses of macrophages, via the activation of HIF-1α. Surprisingly, we also find that HIF-1α and iNOS antagonistically regulate a small but critical set of inflammatory cytokines and chemokines that are positively regulated by HIF-1α but negatively regulated by iNOS. We find that iNOS suppresses prolonged nuclear localization of the NF-kB family member RelA and that this activity at least partially explains the divergent functions of iNOS that allow both an enhancement of antimicrobial responses and suppression of excessive inflammatory cytokine and chemokine production. These findings fit with an emerging understanding that IFN-γ signaling must carefully calibrate an effective immune response that does not cause excessive tissue damage, and provide further support for the hypothesis that NO is a key player in establishing this balance.
Materials and Methods
Ethics Statement
All procedures involving the use of mice were approved by the University of California, Berkeley IACUC, the Animal Care and Use Committee (protocol number R353-1113B). All protocols conform to federal regulations, the National Research Council’s Guide for the Care and Use of Laboratory Animals and the Public Health Service’s (PHS’s) Policy on Humane Care and Use of Laboratory Animals.
Reagents
Recombinant mouse IFN-γ (485 MI/CF) was obtained from R&D systems. 1400W, and Dimethyloxalylglycine (DMOG) were obtained from Cayman Chemical. Pam3CysK4 (PAM) was obtained from EMC Microcollections. Ascorbate, citrulline, and S-Nitroso-N-acetylpenicillamine (SNAP) were obtained from Sigma-Aldrich. The following primary antibodies were used: HIF-1α (NB100-479, Novus Biologicals), IL-1b (AF-401-NA, R&D systems), and the following Cell Signaling Technology antibodies: HIF-1α (D2U3T), RelA (D14E12), RelB (C1E4), NF-kB1 (D4P4D), IkBa (L35A5), α/β-Tubulin (2148), Histone H3 (D1H2), and β-actin (13E5).
Mice
Wildtype mice were C57BL/6, were obtained from Jackson Laboratory, and then bred in house. All knockout mice are on the C57BL/6 background. B6.129-Hif1atm3Rsjo/J (HIF1αflox) mice were obtained from the Jackson Laboratory and were crossed with B6.129P2-Lyz2tm1(cre)Ifo/J (LysMcre) to generate Hif1aflox/flox, LysMcre+/+ mice that had Hif1a deletion targeted to the myeloid lineage. B6.129P2-Nos2tm1Lau/J (iNOS-) mice were obtained from the Jackson Laboratory and were bred in house, and also crossed to Hif1aflox/flox, LysMcre+/+ mice to generate Hif1aflox/flox, LysMcre+/+, Nos2−/− mice. NFkB-RE-luc mice (10499, Taconic) on the BALB/c background were obtained and BMDM were prepared from these mice for NFkB-luciferase experiments.
Cell Culture
Macrophages were derived from bone marrow of wildtype, Nos2−/−, Hif1aflox/flox, LysMcre+/+, and Hif1aflox/flox, LysMcre+/+, Nos2−/− by culturing in DMEM with 10% FBS, 2 mM L-glutamine and 10% supernatant from 3T3-M-CSF cells for 6 days with feeding on day 3. After differentiation, macrophages were cultured in DMEM supplemented with 10% FBS, 2mM GlutaMAX, and 10% supernatant from 3T3-M-CSF cells (BMDM media).
Bacterial culture
The M. tuberculosis strain Erdman was used for all experiments. M. tuberculosis was grown in Middlebrook 7H9 liquid media supplemented with 10% ADS (albumin-dextrose-saline), 0.4% glycerol, and 0.05% Tween-80. The fluorescent mCherry strain used for microscopy was derived from an Erdman strain, with episomal PMV261 containing Zeo-mCherry under the GroEL promoter.
In vitro infections
BMDM were plated into 96-well or 24-well plates with 5×104 and 3×105 macrophages per well respectively, and were allowed to adhere and rest for 24 hours. For all experiments where IFN-γ was used, BMDM were pretreated with IFN-γ overnight (at 6.25ng/ml unless otherwise indicated) and then infected in DMEM supplemented with 5% horse serum and 5% FBS at MOI=5. After a 4-hour phagocytosis period, infected BMDM were washed with PBS before replacing with BMDM media. For experiments with 1400W or DMOG, these reagents were added to the BMDM media after the 4-hour phagocytosis. For IFN-γ pretreated wells, IFN-γ was also added after infection at the same concentration. For enumeration of CFU, infected BMDM were washed with PBS, lysed in water with 0.5% Triton-X for ten minutes at 37C, and serial dilutions were prepared in PBS with 0.05% Tween-80 and plated onto 7H10 plates. CFU per well values reported refer to the total number of bacteria per well of the assay plate averaged across 4 or 5 replicate wells.
For experiments using the RAW-NF-κB luciferase reporter cells (a kind gift from Greg Barton), cells were plated and infections were performed once cells reached ~75% confluence. Complete RPMI media was used, and infections were performed as described above with the following difference: IFN-γ was only added post-infection. For conditions with 1400W, it was added the same time as IFN-γ, immediately following the 4-hour phagocytosis. To measure luciferase reporter activity, the Dual-Luciferase Reporter Assay System (E1910, Promega) was used as per the manufacturer’s protocol.
Glycolysis assays
Glucose depletion from the media was measured using the Glucose (HK) assay kit (GAHK20, Sigma Aldrich). The protocol was modified to perform the assays in 96 well plates with 100uL reactions instead of 1mL reactions in cuvettes as described in the manufacturer’s protocol. Glucose consumption was calculated by measuring glucose levels in the media after infection and subtracting from glucose measured in cell-free media. Lactate measurements were made using LC MS/MS as previously described (16)
ELISAs
For IL-1β ELISAs, supernatants from BMDM in 24 well plates were used. A mouse IL-1 β ELISA kit (DY401, R&D systems) was used as per the manufacturer’s protocol.
Western blots
BMDM were washed with PBS, lysed in 1x SDS-PAGE buffer on ice, and heat sterilized for 20 min at 100°C before removal from the BSL3 facility. Total protein lysates were analyzed by SDS-PAGE using pre-cast Tris-HCl criterion gels (Bio-Rad). HRP conjugated secondary antibodies were used. Western Lightning Plus-ECL chemiluminescence substrate (PerkinElmer) was used and blots were developed on film or using a ChemiDoc MP System (Bio-Rad).
qRT-PCR and RNA-seq
For q-RT-PCR, 3×105 BMDM were seeded in 24-well dishes and infected as described. At 24h post-infection, cells were washed with room temperature PBS and lysed in 1 mL TRIzol (Invitrogen Life Technologies). Total RNA was extracted using chloroform (200 μL) and the aqueous layer was further purified using RNeasy spin columns (Qiagen). For qPCR, cDNA was generated from 1 μg of RNA using Superscript III (Invitrogen Life Technologies) and Oligo-dT primers. Select genes were analyzed using Maxima SYBR green qPCR master mix (Thermo Scientific). Each sample was analyzed in triplicate on a CFX96 Real-time PCR detection system (Bio-Rad). CQ values were normalized to values obtained for actin and relative changes in gene expression were calculated using the ΔΔCQ method. All methods for iNOS RNA-seq were performed as previously described (16). Although the iNOS dataset was not previously reported, the data was collected simultaneously with the wildtype and HIF-1α datasets. Data for selected genes from the wildtype and HIF-1α deficient macrophages was depicted in a previous publication (16). RNAseq data is from three independent infections, and all error bars represent the SD between the three independent infections. RNA-seq was performed at the Genome Center and Bioinformatics Core Facility at the University of California, Davis. Data was deposited into SRA: http://www.ncbi.nlm.nih.gov/sra/SRP108696.
Microscopy
BMDM were plated on glass coverslips in 24 well plates at 3×105 per coverslip, and infected with M. tuberculosis Erdman-mCherry. 24h post-infection, coverslips were fixed in formalin, and stained for RelA. Microscopy was performed on a Carl Zeiss LSM710 confocal microscope. Images shown were taken with a 63x objective. For quantification, larger fields were taken with a 20x objective. Nuclear fluorescence from RelA staining was quantified from a minimum of 800 cells for each condition. Microscopy was performed at The CNR Biological Imaging Facility at The University of California, Berkeley. Research reported in this publication was supported in part by the National Institutes of Health S10 program under award number 1S10RR026866-01.
Transcription factor prediction
oPOSSUM, a tool for identification of over-represented transcription factor binding sites in co-expressed genes (17) was used to predict transcription factors regulated by NO. To identify transcription factors activated by NO, oPOSSUM analysis was run on all genes found to be lower in the Nos2−/− BMDM during M. tuberculosis infection with IFN-γ activation in our RNAseq data. To identify transcription factors inhibited by NO, oPPOSSUM analysis was run all genes found to be 4-fold or more elevated in the Nos2−/− BMDM during M. tuberculosis infection with IFN-γ activation. Ingenuity IPA analysis, which utilizes curated experimental data to predict transcription factor regulation was also performed. To predict transcription factors both activated and inhibited by NO, analysis was performed on all genes 2fold or more upregulated or downregulated and statistically significant (p<.05) in the Nos2−/− BMDM during M. tuberculosis infection with IFN-γ activation in our RNAseq data.
In vivo infections
Cohorts of age and sex matched wild-type and Nos2−/− mice were infected by aerosol route with M. tuberculosis strain Erdman at a dose of ~300 CFU. All mice were on the C57BL/6 background, and were 7–12 weeks of age when infected. Aerosol infection was done using a Nebulizer and Full Body Inhalation System (Glas-Col). Mice were weighed the day of infection, and weights were followed until a humane 15% weight loss cutoff was reach at which point the mice were euthanized. Neutrophil depletions were performed as described in (7), using the anti-Ly6G mAb (clone 1A8, BioXcell) or an isotype control (clone 2A3, BioXcell). 200ug antibody was injected every other day starting 10 days post-infection. Alternatively, the anti-Ly6G/Ly6C mAb (clone RB6-8C5, BioXcell) was used, with 250ug antibody or isotype control injected every other day starting 10 days post-infection. To confirmation depletion of neutrophils, eye-bleeds were performed d15 after infection (after 3 antibody injections), and cells were stained for CD45, CD11b, Ly6G, and Ly6C. Data shown in figure 4H is gated on CD45+ cells.
Figure 4. iNOS and HIF-1α antagonistically regulate inflammatory cytokine production.
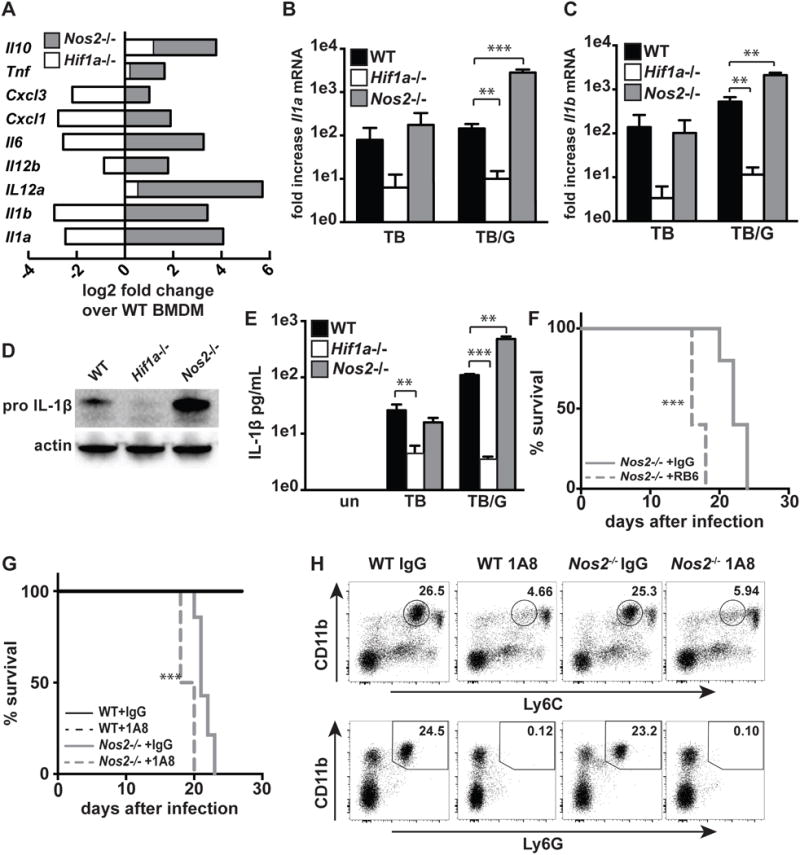
(A) RNAseq data showing fold difference in cytokines and chemokines in Nos2−/− and Hif1a−/− BMDM relative to wildtype during M. tuberculosis infection with IFN-γ activation. (B,C) RNAseq data from wildtype, Hif1a−/−, and Nos2−/− BMDM showing fold change over uninfected for Il1a and Il1b with either M. tuberculosis infection [TB] or M. tuberculosis infection with IFN-γ activation [TB/G]. (D) Western blot for pro-IL-1B from wildtype, Hif1a−/− and Nos2−/− BMDM 24h post-infection with M. tuberculosis and IFN-γ activation. (E) ELISA for IL1B from supernatants of wildtype, Hif1a−/−and Nos2−/− BMDM 36h post-infection with and without IFN-γ activation. (F,G) Survival of Nos2−/− mice following aerosol infection with the virulent M. tuberculosis strain Erdman, with antibody mediated neutrophil depletion or with control IgG treatment. Mice injected with RB6 (F) or 1A8 (G) antibody every other day beginning 10 days post-infection. (H) Flow cytometry confirming neutrophil depletion from mice treated with 1A8 antibody. Data is from peripheral blood, after 3 antibody injections (15 days post-infection). RNAseq data is from 3 independent experiments. (D,E,F,G) are representative of 2 or more experiments. The p values were determined using an unpaired t test for (B,C,E), and using the Log-rank Mantel-Cox test for (F and G). **p<.01, ***p<.001
Results
Nitric oxide shapes the macrophage transcriptome during infection
To identify potential signaling roles for nitric oxide (NO) during M. tuberculosis infection, we performed RNAseq on wildtype and iNOS deficient (Nos2−/−) bone marrow derived macrophages (BMDM). RNAseq data was deposited into SRA (http://www.ncbi.nlm.nih.gov/sra/SRP108696). We found that upon infection of IFN-γ activated BMDM with M. tuberculosis, over 1500 genes were differentially expressed in Nos2−/− BMDM compared with wildtype (Figure 1A). A much smaller number of genes were differentially expressed upon infection with M. tuberculosis in the absence of IFN-γ activation, and the transcriptome of resting wildtype and Nos2−/− BMDM were essentially identical. This correlates with NO production, which is undetectable in resting BMDM, very low with M. tuberculosis infection and robust with IFN-γ activation combined with M. tuberculosis infection (Figure 1B). Similarly, Nos2 transcript levels are not substantially elevated with either M. tuberculosis infection alone or IFN-γ activation alone, but are robustly induced by the combination of the two stimuli (Figure S1A). The NO dependent alterations to the macrophage transcriptome are extensive, and comprise 32% of all IFN-γ regulated genes during M. tuberculosis infection (Figure 1C).
Figure 1. NO has large effects on the macrophage transcriptome.
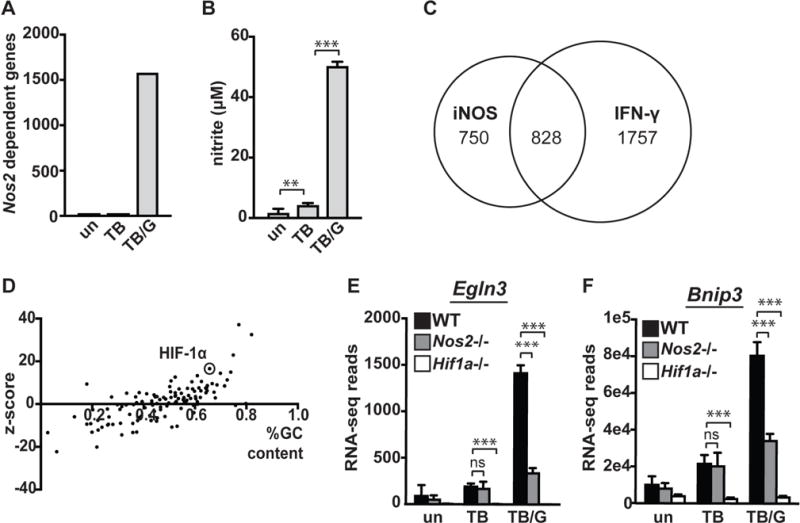
(A) Total number of differentially expressed genes between wildtype and Nos2−/− BMDM in untreated [un], M. tuberculosis infected [TB], and M. tuberculosis infected and IFN-γ treated [TB/G] BMDM 24h post-infection. For this analysis, statistical significance (p<.05) and a 2-fold or greater difference in expression was used as a cutoff. For RNAseq data and all other in vitro infections, the M. tuberculosis strain Erdman was used at an MOI of 5. (B) Griess assay measuring NO production 24h post-infection of wildtype BMDM with and without IFN-γ. (C) Venn diagram showing overlap of iNOS regulated genes during M. tuberculosis infection with IFN-γ treatment, and genes differentially expressed between M. tuberculosis infected wildtype macrophages with and without IFN-γ (D) Bioinformatic prediction (using oPOSSUM) of transcription factors responsible for regulation of all genes found to be expressed at a lower level in Nos2−/− BMDM during M. tuberculosis infection with IFN-γ treatment. (E) RNAseq reads 24h post-infection in wildtype, Nos2−/−, and Hif1a−/− BMDM in untreated [un], M. tuberculosis infected [TB], and M. tuberculosis infected with IFN-γ activation [TB/G] for HIF-1α target genes Egln3 and (F) Bnip3. RNAseq data is from 3 independent infections. (B) is representative of >5 experiments. The p values were determined using an unpaired t test. **p<.01, ***p<.001
The large number of genes dependent on NO production for expression suggests that NO might impact the function of global transcriptional regulators. To identify candidate transcription factors regulated by NO during M. tuberculosis infection, we applied two bioinformatic methods to our RNA-seq dataset. First, we used Ingenuity IPA analysis, which utilizes curated experimental data to predict transcription factor regulation. Transcription factors predicted to be activated by NO, and transcription factors predicted to be inactivated by NO are listed in (Table SI). Second, we used oPOSSUM, a tool for identification of over-represented transcription factor binding sites in co-expressed genes (17). Analysis of genes positively regulated by NO (downregulated in Nos2−/− BMDM) using both IPA and oPOSSUM identified HIF-1α (Figure 1D, Table SI), which we have recently identified as a key mediator of IFN-γ dependent control of M. tuberculosis infection (16).
iNOS activates HIF-1α signaling
As HIF-1α was identified in our analysis as a potential NO regulated transcription factor, and has been shown to be important for control of M. tuberculosis infection, we selected this transcription factor for further analysis. The genes Egln3 and Bnip3 are canonical HIF-1α dependent target genes (18). In M. tuberculosis infected BMDM where production of NO is low, we observed low levels of Egln3 and Bnip3 expression that are HIF-1α dependent but NO independent (Figure 1E, 1F). However, in IFN-γ activated BMDM infected with M. tuberculosis, where NO production is high, we observed an upregulation of Egln3 and Bnip3 that is strongly dependent on both HIF-1α and iNOS (Figure 1E, 1F).
In resting macrophages, HIF-1α is constitutively transcribed and translated, but kept at low levels by constitutive proteasomal degradation (19, 20). While HIF-1α protein levels are primarily regulated at the level of protein stability, HIF-1α can also be transcriptionally regulated (21). We first tested whether NO impacts transcription of the Hif1a gene, and found that transcript levels were equivalent or higher in Nos2−/− BMDM relative to wildtype (Figure 2A), indicating that NO dependent upregulation of HIF-1α target genes is unlikely to be due to NO dependent upregulation of Hif1a transcript. Next, HIF-1α protein accumulation during M. tuberculosis infection with IFN-γ activation was assessed in wildtype and Nos2−/− BMDM. In wildtype BMDM HIF-1α protein levels rise, peaking at 12h post infection (Figure 2B). In contrast, iNOS deficient macrophages have a weak and transient accumulation of HIF-1α (Figure 2B), indicating that the large accumulation of HIF-1α that occurs after 4h is dependent on the production of NO. The addition of ascorbate, a reducing agent that reverses S-nitrosylation (15), inhibited HIF-1α stabilization in wildtype BMDM, suggesting the possibility that protein S-nitrosylation by NO contributes to HIF-1α stabilization (Figure 2C).
Figure 2. iNOS is required for HIF-1α stabilization and transcriptional activity.
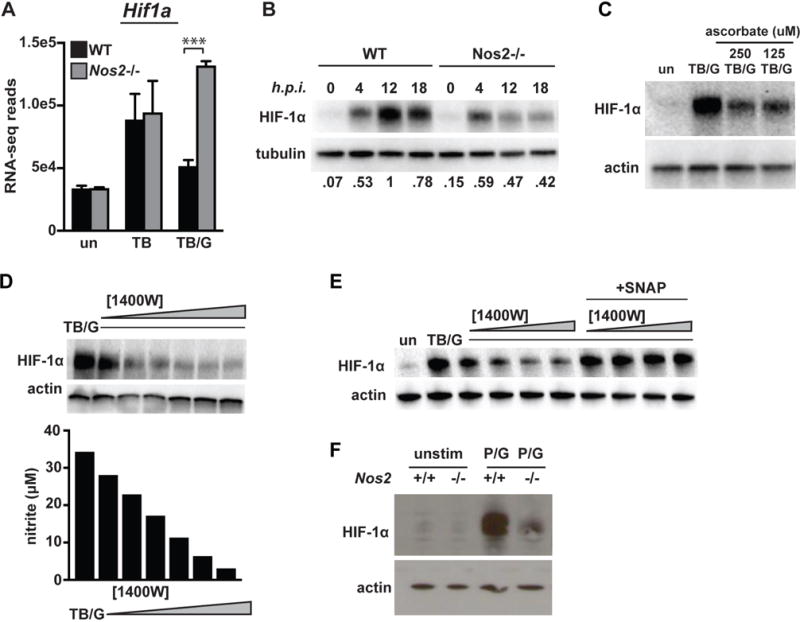
(A) RNAseq reads for Hif1a 24h post-infection in wildtype and Nos2 BMDM in untreated [un], M. tuberculosis infected [TB], and M. tuberculosis infected with IFN-γ activation [TB/G]. (B) Western blot for HIF-1α in M. tuberculosis infected, IFN-γ activated BMDM at 0,4,12 and 18h post-infection in wildtype and Nos2−/− BMDM. Values for quantification of HIF-1α/tubulin ratios are normalized to lane 3 (maximal HIF-1α levels) and are shown below the image. (C) Western blot for HIF-1α during infection with M. tuberculosis with IFN-γ activation in the presence or absence of ascorbate (D) Western blot for HIF-1α in M. tuberculosis infected, IFN-γ activated BMDM with a dose response of the iNOS inhibitor 1400W [.625, 1.25, 2.5, 5, 10, 25uM]. Griess assay for NO production was done on the supernatant of the cells used for the western blot. (E) Western blot for HIF-1α in M. tuberculosis infected, IFN-γ activated BMDM with a dose response of 1400W [.625, 1.25, 2.5, 5uM] with and with addition of the NO donor SNAP [250uM]. (F) HIF-1α levels were measured by western blot at 12h after stimulation with PAM [50ng/ml] and IFN-γ [6.25 ng/ml]. RNAseq data is from 3 independent experiments. All other experiments are representative of 2 or more experiments. Error bars represent the SD and p values were determined using an unpaired t test. ***p<.001
To identify a threshold of NO production required for HIF-1α stabilization, BMDM were treated with a dose response of the iNOS inhibitor 1400W following infection with M. tuberculosis and activation with IFN-γ. Even a modest inhibition (~50%) of NO production completely reverses NO dependent HIF-1α stabilization (Figure 2D). Additionally, the addition of the exogenous NO donor S-Nitroso-N-acetylpenicillamine (SNAP) is able to rescue this 1400W dependent abrogation of HIF-1α stabilization (Figure 2E), confirming that NO itself is responsible for the defect in HIF-1α stabilization in Nos2−/− BMDM. Finally, we tested whether this signaling role for NO is specific to M. tuberculosis infection of IFN-γ activated macrophages, or whether it is a more general feature of IFN-γ activation. Wildtype and Nos2−/− BMDM were treated with the TLR1/2 agonist PAMCysK4 and IFN-γ, and probed for HIF-1α by Western blot 12h post-treatment. We observed a similar result in the context of sterile stimulation of BMDM as with M. tuberculosis infection, with Nos2−/− BMDM exhibiting a strong defect in HIF-1α stabilization (Figure 2F). This suggests that NO stabilizing HIF-1α is a general feature of IFN-γ activation of macrophages.
iNOS and HIF-1α are linked in a positive feedback loop, and regulate aerobic glycolysis
We have previously shown that HIF-1α is required for maximal production of NO in BMDM activated with IFN-γ and infected with M. tuberculosis, and that this metabolic transition requires both stimulation by M. tuberculosis and IFN-γ (16). We confirmed that these HIF-1α dependent phenotypes result specifically from HIF-1α deletion and not from the LysMcre+/+ phenotype (Figure S1B–S1E). The dependence of NO production on HIF-1α is observable across a wide range of IFN-γ concentrations (Figure 3A). Furthermore, treatment with the HIF-1α stabilizer dimethyloxalylglycine (DMOG) enhances NO production in wildtype but not HIF-1α deficient (Hif1a−/−) BMDM (Figure 3A). Thus, iNOS and HIF-1α positively regulate each other, with NO being required for IFN-γ dependent HIF-1α stabilization, and HIF-1α being required for maximal NO production. We next compared genes differentially expressed in Hif1a −/− BMDM with genes differentially expressed in Nos2−/− BMDM. During M. tuberculosis infection with IFN-γ, we identified ~1700 genes in Hif1a−/− BMDM and ~1600 genes in Nos2−/− BMDM that had altered expression levels compared to wildtype BMDM. Approximately 40% of these regulated genes overlapped (Figure 3B), indicating that NO dependent regulation of HIF-1α activity accounts for a substantial portion of the HIF-1α dependent transcriptional response in IFN-γ activated macrophages during M. tuberculosis infection.
Figure 3. iNOS and HIF-1α are linked by positive feedback, and regulate aerobic glycolysis.
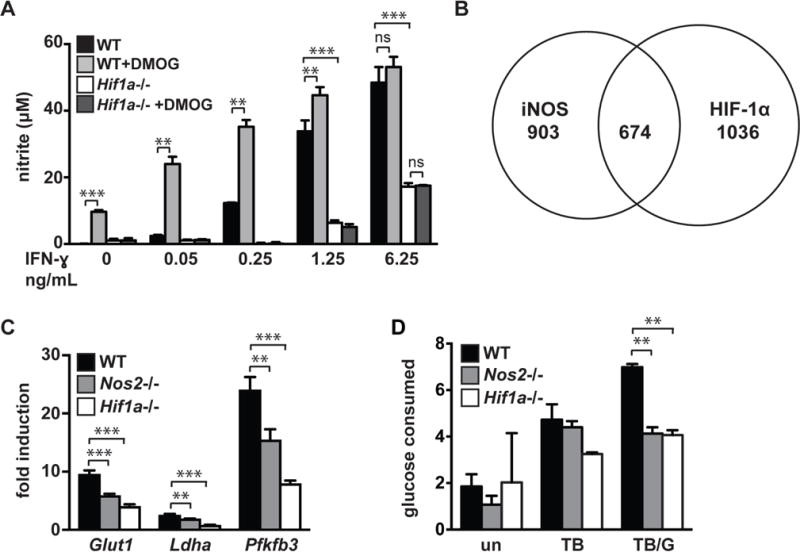
(A) Griess assay measuring NO production 24h post-infection of M. tuberculosis infected wildtype and Hif1a−/− BMDM, with a dose response of IFN-γ. The HIF-1α stabilizer DMOG [200uM] increases NO production in a HIF-1α dependent manner. (B) Venn diagram of RNAseq data showing overlap of genes differentially expressed in Nos2−/− vs wildtype and Hif1a−/− vs wildtype BMDM during M. tuberculosis infection with IFN-γ activation. (C) RNAseq data showing fold induction of the glycolytic genes Glut1, Ldha, and Pfkfb3 during M. tuberculosis infection with IFN-γ activation in wildtype, Nos2−/−, Hif1a−/− BMDM relative to untreated. (D) Measurement of glucose uptake in wildtype, Nos2−/− and Hif1a−/− BMDM either untreated [un], M. tuberculosis infected [TB], or M. tuberculosis infected and IFN-γ activated [TB/G]. RNAseq data is from 3 independent experiments. (A,D) are representative of 2 or more experiments. Error bars represent SD from 3 or more wells (A,D) or 3 independent experiments (C). The p values were determined using an unpaired t test. **p<.01, ***p<.001
This overlapping gene regulation includes regulation of aerobic glycolysis, which we and others have previously shown to be a key function required for control of M. tuberculosis in macrophages (16, 22, 23). RNAseq data indicates that Nos2−/− and Hif1a −/− BMDM have a defect in transcription of genes that are induced as a part of the program of aerobic glycolysis, including glut1, ldha, and pfkfb3 (Figure 3C). Consistent with this observation, glucose assays indicate that both Nos2−/− and Hif1a −/− BMDM are deficient for glucose uptake during M. tuberculosis infection with IFN-γ activation (Figure 3D). We observed the same NO dependence of aerobic glycolysis in cells stimulated with PAM and IFN-γ using lactate levels as the readout (Figure S1F).
iNOS and HIF-1α have opposing roles in the regulation of inflammation
Our finding that NO positively regulates HIF-1α stabilization suggests a role for NO in positively regulating inflammatory gene expression, as we and others have shown HIF-1α to be a key transcriptional regulator of inflammatory cytokines during bacterial infection (16, 24, 25). However, an excessive inflammatory response has been suggested to contribute to the susceptibility of both Nos2−/− and IFN-γ deficient mice, suggesting that in fact NO acts as an anti-inflammatory mediator (7, 15).
Analysis of our RNAseq data indicated that of the nearly 700 genes that are regulated by both iNOS and HIF-1α, only ~5% are regulated in opposite directions (increased expression in one genotype and decreased expression in the other genotype for a single gene). Interestingly, this subset is highly enriched for inflammatory cytokines and chemokines. Surprisingly, given the positive feedback relationship between HIF-1α and NO, HIF-1α broadly upregulates inflammatory cytokines and chemokines, while NO strongly suppresses mRNA abundance of a key subset of cytokines and chemokines (Figure 4A). We examined the Il1a and Il1b transcripts in more detail, as IL-1 is essential for control of M. tuberculosis infection (26). Levels of IL-1 produced during M. tuberculosis infection must be carefully balanced to protect the host; IL-1 is required for the protective immune response to M. tuberculosis but excessive IL-1 leads to host pathology and death (15). For Il1a and Il1b, a nearly identical transcriptional phenotype was observed, with Hif1a −/− BMDM having substantially lower levels during M. tuberculosis infection both in the presence and absence of IFN-γ, while Nos2−/− BMDM have much higher transcript levels than WT, but only in the context of IFN-γ activation (Figure 4B, 4C). Analysis of intracellular pro-IL-1β by western blot indicates that during M. tuberculosis infection with IFN-γ activation, Nos2−/− BMDM have markedly higher levels than wildtype BMDM, while pro-IL-1β is barely detectable in Hif1a −/− BMDM (Figure 4D). Similarly, ELISAs for IL-1β from supernatants of M. tuberculosis infected BMDM recapitulate the transcriptional phenotype, with markedly higher levels of IL-1β detected in Nos2−/− BMDM and dramatically lower levels detected in Hif1a −/− BMDM (Figure 4E). Thus while HIF-1α and iNOS are regulated by positive feedback, and coordinately regulate hundreds of genes, they have antagonistic roles with regard to inflammatory cytokine production. Interestingly, mRNA levels of IL-1 and CXCL1, which both play a role in recruitment of neutrophils, are elevated in Nos2−/− BMDM (Figure 4A). This raises the possibility that NO is the IFN-γ dependent effector responsible for restricting destructive inflammation resulting from excessive neutrophil recruitment. Depleting neutrophils from IFN-γ deficient mice is reported to extend their survival (7). We tested whether depleting neutrophils from Nos2−/− mice is also protective. We independently tested two antibodies for neutrophil depletion, and confirmed neutrophil depletion in mice treated with the antibody 1A8, which targets Ly6G (Figure 4H). We found that under these experimental conditions, depleting neutrophils did not extend the life of Nos2−/− mice, but instead slightly exacerbated infection (Figure 4F,G).
iNOS suppresses NF-kB activity during M. tuberculosis infection
NO has been demonstrated to regulate NF-kB signaling via S-nitrosylation of NF-kB family members, including p50 and p65 (27, 28). However, depending on the context, NO can either inhibit or enhance NF-kB dependent transcriptional responses (29). Indeed, increased activation of NF-kB by NO has been observed in multiple inflammatory settings (30, 31). Our bioinformatic analyses suggest that in macrophages infected with M. tuberculosis and activated with IFN-γ, NO suppresses NF-kB dependent signaling. Indeed, both IPA and oPOSSUM transcription factor prediction analysis identify NF-kB family members as top hits for transcription factors inhibited by NO (Figure 5A, Table SI, Table SI). We hypothesized that this activity of NO might explain the hyper-inflammatory phenotype of Nos2−/− BMDM, and the divergent phenotypes of Hif1a −/− and Nos2−/− BMDM for a subset of inflammatory cytokines (Figure 4A).
Figure 5. iNOS suppresses NF-kB activity.
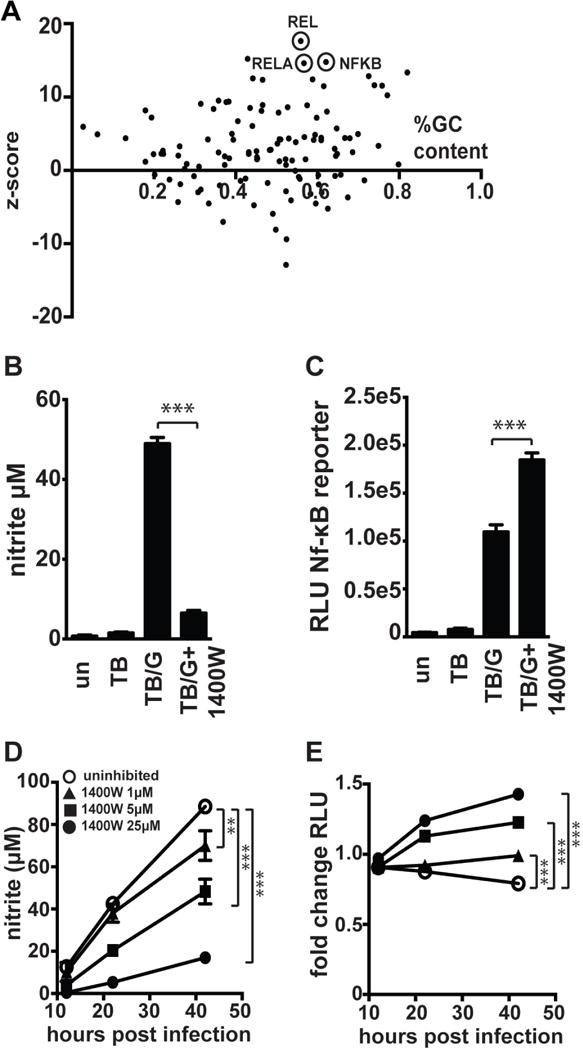
(A) Bioinformatic analysis of RNAseq data using oPOSSUM to predict transcription factors regulating the subset of genes found to be 4-fold or more upregulated in Nos2−/− BMDM relative to wildtype BMDM during M. tuberculosis infection with IFN-γ activation. (B) Griess assay measuring NO production 24h post-infection in RAW macrophages carrying an NF-kB luciferase reporter, with M. tuberculosis infection without IFN-γ [TB], with IFN-γ activation [TB/G], and with IFN-γ activation and addition of 25uM 1400W [TB/G+1400W]. (C) Luciferase assay 24h post-infection for the same cells as in (B) to measure NF-kB activity. (D) Griess assays measuring NO production over a timecourse in M. tuberculosis infected, IFN-γ activated BMDM (carrying an NF-kB luciferase reporter) with a dose response of 1400W. (E) Luciferase assay for NF-kB activity from the same cells as in (D). (B,C) representative of 3 experiments, (D,E) representative of 2 experiments. The p values were determined using an unpaired t test. **p<.01, ***p<.001
To experimentally determine whether NO inhibits NF-kB activity in IFN-γ activated and M. tuberculosis infected macrophages we first examined the effect of NO on a NF-kB dependent firefly luciferase reporter gene in both the RAW cell line and primary BMDM. Inhibition of NO production with 25uM 1400W (Figure 5B) led to an increase in reporter activity in RAW cells at 36h after infection (Figure 5C), suggesting that NO may suppress NF-kB dependent signaling at late timepoints after macrophage activation. However, it was surprising that even without inhibition of NO production, there were high levels of activation of NF-kB in M. tuberculosis infected and IFN-γ activated macrophages 36h after infection. We therefore sought to test whether a similar effect was observed in primary cells, and utlilized BMDM prepared from a mouse expressing an NF-kB luciferase reporter in all cells (32). We found that 12h after infection NF-kB signaling in M. tuberculosis infected and IFN-γ activated BMDM was comparable to that found in resting macrophages. However, using a dose response of 1400W to titrate NO levels, we found that inhibition of NO production caused prolonged NF-kB activity at later time-points in a dose dependent manner (Figure 5D, 5E). We confirmed that 1400W treatment had no effect on BMDM NF-kB luciferase activity in the absence of IFN-γ (Figure S2A). We next sought to identify which NF-kB family members contribute to the prolonged NF-kB activity observed. Increased mRNA levels were observed for Rel, Relb, Nfkb1, and Nfkb2 in Nos2−/− BMDM relative to wildtype BMDM infected with M. tuberculosis and activated with IFN-γ (Figure S2C–G). To determine whether the enhanced levels of mRNA translated to increased protein levels and increased activation of NF-kB family members, western blotting was performed in whole cell lysates and nuclear extracts for all NF-kB family members 24h post-infection. While whole cell and nuclear levels of RelB increased with M. tuberculosis infection of IFN-γ treated BMDM, there was no clear difference between wildtype and Nos2−/− BMDM (Figure 6A). There was also no clear difference between wildtype and Nos2−/− BMDM for NF-kB1 (Figure 6A). We were unable to detect nuclear c-Rel or any NF-kB2 protein under the conditions examined (data not shown). Also, as expected, IkBa levels were reduced with infection, but there was no obvious difference between wildtype and Nos2−/− BMDM (Figure 6A). The only NF-kB family member for which there was a clear difference in Nos2−/− BMDM was the RelA/p65 subunit. While total levels of RelA remained unchanged across all conditions, there was markedly more nuclear RelA in Nos2−/− BMDM than in wildtype BMDM after infection (Figure 6A). This result was confirmed by confocal immunofluorescence microscopy for RelA/p65. In resting BMDM, staining is predominantly cytoplasmic, but upon M. tuberculosis infection of IFN-γ activated BMDM there is a relocalization of RelA to the nucleus in iNOS deficient BMDM that is observable at 24h after infection (Figure 6B). Quantification of RelA positive nuclei shows that ~3x as many nuclei are RelA positive in Nos2−/− BMDM relative to wildtype (Figure 6C). Taken together, this supports the hypothesis that NO deficiency causes prolonged nuclear localization of RelA with a concomitant increase in the expression of specific inflammatory cytokines, including IL-1.
Figure 6. iNOS deficiency leads to aberrantly high nuclear RelA.
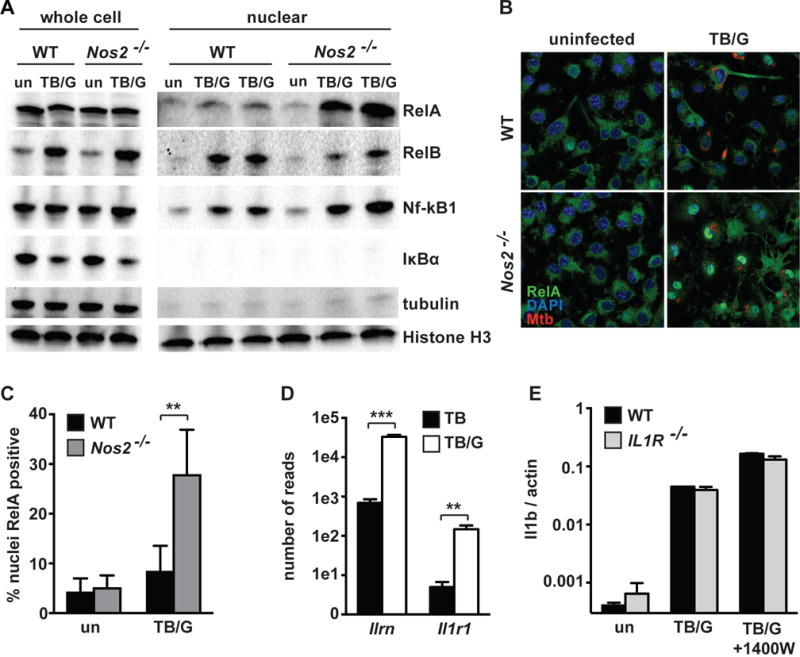
(A) Western blots 24h post-infection from whole cell lysates and nuclear extracts for RelA, RelB, NF-kB1, and IkBa, with tubulin as a cytoplasmic marker and Histone H3 as a nuclear marker. Western blots with wildtype and Nos2−/− BMDM either uninfected [un] or M. tuberculosis infected with IFN-γ activation [TB/G]. For nuclear extracts, [TB/G] performed in biological duplicate for each repeat. (B) Confocal microscopy at 63x for RelA 24h post-infection in wildtype and Nos2−/− BMDM, uninfected or during M. tuberculosis infection with IFN-γ activation [TB/G]. For microscopy, the M. tuberculosis strain Erdman, carrying a fluorescent mCherry was utilized. (C) Quantification of p65 positive nuclei from the same samples as in (B), but from 20x images. >800 nuclei were analyzed for each condition, error bars represent SD between 5 fields. (D) RNAseq data showing number of reads for the IL1R antagonist (Ilrn) and IL1R (Il1r1) in wildtype BMDM infected with M. tuberculosis [TB] or M. tuberculosis with IFN-γ activation [TB/G]. (E) qPCR for Il1b transcript in wildtype and Il1r−/− BMDM, either untreated [un], during M. tuberculosis infection with IFN-γ activation [TB/G], or during M. tuberculosis infection with IFN-γ activation and 1400W treatment at 25uM [TB/G+1400W]. All data representative of 2 or more experiments. The p values were determined using an unpaired t test. **p<.01, ***p<.001
As NO is reported to play an anti-inflammatory role during M. tuberculosis infection by inhibiting NLRP3 dependent IL1b processing (15), we sought to test whether the upregulation of NF-kB activity in NO deficient BMDM that we observed might be a secondary effect following IL-1 autocrine or paracrine signaling between macrophages. RNAseq data suggests that this is unlikely, as the IL1 receptor antagonist (Il1rn) is ~100x more highly expressed than then IL1 receptor (Il1r1) during M. tuberculosis infection with and without IFN-γ activation (Figure 6D). To functionally assess whether IL-1 autocrine/paracrine signaling plays a role, the iNOS inhibitor 1400W was used in wildtype and IL1R−/− BMDM infected with M. tuberculosis and activated with IFN-γ. As expected, 1400W treatment increased Il1b expression in wildtype macrophages, recapitulating a Nos2−/− phenotype (Figure 6E). However, In IL1R−/− BMDM, 1400W treatment enhanced Il1b transcription to the same level as in wildtype BMDM (Figure 6E), indicating that signaling through the IL-1 receptor is not required for Il1b upregulation, and suggesting that the broad transcriptional repression of cytokines by NO occurs by more direct modulation of NF-kB. We also tested whether the absence of citrulline accumulation in Nos2−/− BMDM might be responsible for the increased NF-kB activity observed. Addition of citrulline (a byproduct of NO production) to Nos2−/− BMDM during M. tuberculosis infection with IFN-γ activation did not reverse the aberrantly high Il1b transcript levels observed in Nos2−/− BMDM (Figure S2B), again indicating that it is NO that is directly modulating NF-kB activity.
Simultaneous deletion of HIF-1α and iNOS from macrophages balances inflammatory cytokine production, but does not restore control of infection
To further characterize the role of iNOS and its interactions with HIF-1α in the regulation of IFN-γ dependent transcriptional responses and cell intrinsic control of M. tuberculosis, we examined BMDM lacking both HIF-1α and iNOS. Nos2−/− BMDM are known to be defective in IFN-γ dependent control of M. tuberculosis infection, and we have shown that Hif1a −/− BMDM are also defective in control of M. tuberculosis specifically in the context of IFN-γ activation (16). First, we compared Nos2−/−, Hif1a −/−, and Hif1a−/− Nos2−/− double knockout BMDM for their ability to control infection. We observed no differences in bacterial growth in the absence of IFN-γ between any of the genotypes tested (Figure 7A). In the context of IFN-γ activation we found that Nos2−/−, Hif1a −/−, and Hif1a−/− Nos2−/− BMDM all had significantly elevated CFU relative to wildtype BMDM (Figure 7B). Interestingly, all three mutant genotypes had a remarkably similar defect in IFN-γ dependent restriction of bacterial growth, and the Hif1a−/− Nos2−/− double knockout BMDM had no further loss of control relative to the individual knockouts (Figure 7B). This result suggests that iNOS and HIF-1α are acting through the same cell intrinsic pathway for control of M. tuberculosis infection. We next compared cytokine expression by qPCR in Nos2−/−, Hif1a −/−, and Hif1a−/− Nos2−/− BMDM following M. tuberculosis infection and IFN-γ activation. Hif1a −/− BMDM have a strong defect in Il1a, Il1b and Il6 transcript levels while Nos2−/− BMDM have highly increased levels (Figure 7C, 7D, 7E). Interestingly, the Hif1a−/− Nos2−/− BMDM balance out these hyper-inflammatory and hypo-inflammatory phenotypes and have intermediate Il1a, Il1b and Il6 transcript levels, similar to that of wildtype BMDM (Figure 7C, 7D, 7E). Thus, NO appears to be required for cell intrinsic control of M. tuberculosis infection via activation of HIF-1α, while at the same time iNOS and HIF-1α antagonistically regulate inflammatory cytokine production to produce a properly calibrated inflammatory response.
Figure 7. Double knockout of iNOS and HIF-1α balances inflammation.
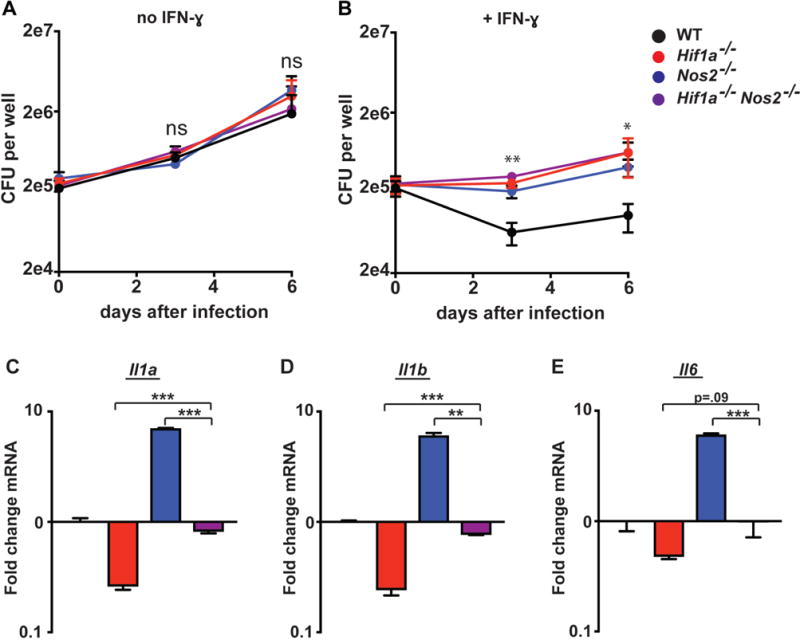
Wildtype, Nos2−/−, Hif1a−/−, and Nos2−/− Hif1a−/− BMDM were infected with M. tuberculosis without IFN-γ (A) and with IFN-γ (B). CFU was enumerated immediately following phagocytosis of bacteria, and again 3d and 6d post-infection. (C,D,E) wildtype, Nos2−/−, Hif1a−/−, and Nos2−/− Hif1a−/− BMDM were treated with IFN-γ and infected with M. tuberculosis. qPCR for Il1a, Il1b, and Il6 was performed on RNA isolated from BMDM 24h post-infection. (A–E) representative of 3 or more independent experiments. The p values were determined using an unpaired t test. *p<.05, **p<.01, ***p<.001
Discussion
In this report, we identify two key signaling roles for iNOS during M. tuberculosis infection. First, we find that NO mediates stabilization of HIF-1α, a transcription factor that is required for optimal expression of both pro-inflammatory cytokines and antimicrobial effectors. Second, we find that NO represses NF-kB signaling, and prevents prolonged nuclear RelA localization. Additionally, we find that iNOS and HIF-1α are linked by a positive feedback loop while at the same time they antagonistically regulate inflammatory cytokine production. Thus, we find that iNOS promotes the microbicidal functions of the pro-inflammatory transcription factor HIF-1α while simultaneously preventing excessive inflammatory responses via suppression of the NF-kB family member RelA/p65 (Figure S3).
NO is produced in macrophages in response to a diverse array of microbial pathogens and has been found to be protective in most infection models (33). In the case of M. tuberculosis infection, NO is produced following the onset of adaptive immunity and IFN-γ signaling (5). It has long been known that Nos2−/− mice are extremely susceptible to M. tuberculosis infection, accounting for much of the susceptibility of IFN-γ−/− mice (12). It has been assumed that the predominant mechanism by which NO controls infection in macrophages is via direct killing of M. tuberculosis. However, the need for re-examination of this hypothesis has been raised by several key observations. First, the isolation of M. tuberculosis mutants that are dramatically more susceptible to nitrosative stress can be interpreted as evidence that M. tuberculosis has evolved resistance mechanisms to direct NO dependent killing (13). Second, part of the susceptibility phenotype of IFN-γ deficient mice has been attributed to an excessive inflammatory response caused by a failure to suppress the recruitment of neutrophils to sites of infection (7). Finally, an intriguing paper demonstrated that NO prevents immunopathology during M. tuberculosis infection independent of bacterial replication, providing the first concrete evidence that NO contributes to control of M. tuberculosis infection through a mechanism other than direct bacterial toxicity (15).
While the role of NO as a second messenger that regulates mammalian signaling is well known (34), in the context of M. tuberculosis infection the potential regulatory roles of NO have not been well defined. In addition to its well-studied role in regulating cyclic GMP (cGMP) dependent kinases, NO has broad effects on signaling via NO-induced post translational modifications including S-nitrosylation, S-glutathionylation and tyrosine nitration (35). In addition, NO can inhibit the function of iron dependent proteins (36). The role of NO in regulating inflammation is complex, and can either be pro-inflammatory or anti-inflammatory depending on the context (29, 31).
We identified HIF-1α as a key transcription factor that is activated by NO production during M. tuberculosis infection. RNAseq profiling of Nos2−/− and Hif1a −/− macrophages indicates that there is a large overlap of genes regulated by both HIF-1α and NO. We have recently shown that HIF-1α is a key mediator of IFN-γ mediated control of M. tuberculosis infection in vitro and in vivo. This implies that NO may have a broader role in mediating IFN-γ dependent antibacterial defense against M. tuberculosis than was previously appreciated, and may do so in large part through the activation of HIF-1α dependent bactericidal effectors.
Aerobic glycolysis is associated with M1 polarized bactericidal macrophages (37) and we have recently shown that in the context that M. tuberculosis infection, aerobic glycolysis is required for IFN-γ dependent control of infection in macrophages (16). We find that the metabolic program of aerobic glycolysis in M. tuberculosis infected and IFN-γ activated macrophages requires both iNOS and HIF-1α. Thus, NO may contribute to control of M. tuberculosis infection by regulating aerobic glycolysis. Although NO positively regulates both HIF-1α and aerobic glycolysis during M. tuberculosis infection, the mechanism by which this occurs remains an open question. The observation that ascorbate treatment abrogates HIF-1α stabilization under these conditions suggests that S-nitrosylation of a protein involved in HIF-1α regulation may be involved. Direct S-nitrosylation and activation of HIF-1α has been reported to lead to PHD independent stabilization of HIF1 (38). Similarly, NO could interfere with PHD function either by nitrosylation or via disruption of the catalytic site via iron binding (39). An additional possibility is that the NO dependent upregulation of HIF-1α is indirect, and downstream of an upregulation of aerobic glycolysis, which we have shown to be required for HIF-1α stabilization during M. tuberculosis infection (16). Finally, because the reaction catalyzed by iNOS requires the use of molecular oxygen, it is possible that consumption of O2 by iNOS creates a locally hypoxic environment that results in stabilization of HIF-1α.
Interestingly, while there is broad overlap in HIF-1α and iNOS regulated genes, there is a small but critical set of inflammatory cytokines and chemokines that are dramatically upregulated in Nos2−/− macrophages and downregulated in Hif1a −/− macrophages. This suggests that in addition to enhancing antimicrobial and inflammatory activity via HIF-1α, NO production is also profoundly anti-inflammatory in the context of M. tuberculosis infection. This is a somewhat surprising result given the positive feedback between HIF-1α and iNOS, but implies that NO serves as an important braking mechanism to prevent excessive inflammation at the transcriptional level. It has been demonstrated that NO specifically nitrosylates and inactivates the NLRP3 inflammasome, thus limiting IL-1β processing, and that Nos2−/− mice have a striking hyper-inflammatory response to M. tuberculosis infection in vivo (15). We find that the immunosuppressive functions of NO are broader than just the inactivation of the NLRP3 inflammasome. Our RNA-seq data reveals that in M. tuberculosis infected, IFN-γ activated macrophages, NO represses not only IL-1α and IL-1β at the transcriptional level, but also many other inflammatory cytokines and chemokines including CXCL1. Interestingly, IL1 and CXCL1 both promote the recruitment of neutrophils, suggesting that NO might function to suppress the recruitment of neutrophils to the site of infection thereby preventing the excessive inflammatory response that can be mediated by neutrophils. Although we find that NO negatively regulates the transcription of neutrophil attractant chemokines in vitro, we did not observe an increased survival time in Nos2−/− mice following neutrophil depletion in vivo (Figure 4F–H). However, the dose we used for infection is 2–4 fold higher than the dose used to observe the protective effect in IFN-γ−/− mice upon neutrophil depletion (7). It is possible that the aerosol infection model we use results in a hyperinflammation via multiple mechanisms, thereby masking a protective effect of neutrophil depletion. While this manuscript was under review, a new study highlighting the role of NO in suppressing damaging neutrophilic infiltration in M. tuberculosis infected mouse lungs was published (40). In their study, the authors demonstrate that depleting neutrophils using the 1A8 antibody decreases CFU in the lungs of Nos2−/− mice. However, they did not report survival data so it is unclear how this result relates to our observation that this treatment does not prolong survival. Nevertheless, this intriguing study suggests that in their experimental system, the bactericidal activity of NO is not important in vivo. However, NO clearly has an important role in cell intrinsic control in macrophages infected in vitro. Thus, it will be important to determine whether the bactericidal vs immune suppressive activities of NO are context dependent, or whether the dominant in vivo role of NO during infection is to dampen inflammatory responses.
To identify effectors of the broad transcriptional upregulation of inflammatory cytokines and chemokines we observe in Nos2−/− macrophages, we performed a bioinformatic analysis of genes found to be upregulated in Nos2−/− macrophages, and identified NF-kB family members as the best candidates. We experimentally validated this bioinformatic prediction, and found that the p65/RelA subunit of NF-kB is inhibited by NO production during IFN-γ activation of M. tuberculosis infected macrophages. This result fits with the known role of NO in nitrosylating and inhibiting multiple NF-kB family members including both the p50 and p65 NF-kB monomers (27, 28). Thus, while iNOS activates HIF-1α dependent cell intrinsic bactericidal mechanisms, it also counterbalances HIF-1α dependent upregulation of inflammatory mediators by inhibiting pro-inflammatory cytokine transcription by NF-kB. We propose that this dual role for NO allows for robust activation of IFN-γ dependent cell intrinsic bactericidal effectors via HIF-1α while also suppressing excessive production of inflammatory cytokines that would result from activation of both NF-kB and HIF-1α, presumably to preclude excessive tissue damage during an effective IFN-γ mediated response to infection. Indeed, several lines of evidence suggest that excessive production of either IFN-γ or IFN-γ dependent effectors results in significant loss of control of infection, as both PD-1 deficient and ARE-del mice have increased IFN-γ and increased susceptibility to infection (41, 42).
To further characterize role of iNOS in mediating both control of bacterial growth and preventing excessive inflammatory cytokine production, we utilized Hif1a−/− Nos2−/− macrophages. We assessed their ability to regulate inflammatory cytokine production and their ability to control M. tuberculosis infection relative to Nos2−/− macrophages and Hif1a−/− macrophages. We found that Hif1a−/− Nos2−/− macrophages have inflammatory cytokine transcription that is intermediate between the hyperinflammatory Nos2−/− macrophages and hypoinflammatory Hif1a−/− macrophages, closely matching wildtype cytokine transcription. This result indicates that NO is both activating cytokine production via HIF-1α while also potently inhibiting cytokine production by inactivating NF-kB activity. This result also indicates that although HIF-1α protein levels and transcriptional activity are reduced in Nos2−/− macrophages, there is still some HIF-1α activity in the Nos2−/− macrophages that further contributes to the hyperinflammatory phenotype caused by NO deficiency.
Although we find that Hif1a−/− Nos2−/− macrophages have wildtype levels of cytokine transcript, this does not lead to effective control of M. tuberculosis in macrophages, implying that NO is required for cell intrinsic control independent of cytokine production. Indeed we find that Nos2−/− macrophages, Hif1a−/− macrophages, and Hif1a−/− Nos2−/− macrophages have an identical defect in IFN-γ dependent killing of M. tuberculosis. This result implies that HIF-1α and iNOS act through the same pathway to effect cell intrinsic control of M. tuberculosis infection. Although we have recently identified a variety of effector functions that are impaired in HIF-1α deficient macrophages during M. tuberculosis infection (16), it remains an open question as to what specifically mediates HIF-1α dependent killing of M. tuberculosis in macrophages. Autophagy and LRG-47, previously thought to explain the NO independent IFN-γ dependent cell intrinsic control, have recently been shown to play no role in control of M. tuberculosis infection (43, 44). There are clearly important and undiscovered regulators of IFN-γ dependent, cell intrinsic control of M. tuberculosis infection.
Whether NO plays a role in human macrophages remains controversial. Production of NO by primary human macrophages derived from peripheral blood mononuclear cells is not consistently observed, with the majority of reports suggesting that these human cells produce far less NO than do mouse macrophages (45). Because we have observed neither NO production nor control of M. tuberculosis infection with IFN-γ addition to primary human monocyte derived macrophages, we are unable to evaluate the relavance of NO signaling in primary human cells. However, there is ample evidence that NO is produced during M. tuberculosis infection of human patients; NO can be detected in the breath of human TB patients, and macrophages isolated from lungs of human TB patients have been shown to produce NO in mycobactericidal amounts (46, 47). It is therefore possible that the iNOS promoter is silenced in human monocyte derived macrophages, and that human macrophages require tissue and inflammation specific cues to express iNOS and produce NO.
In conclusion, we demonstrate that iNOS dependent NO production is responsible for broad regulation of the macrophage transcriptome during M. tuberculosis infection, and we identify two key transcription factors that are regulated by NO production in the context of IFN-γ activation. We identify HIF-1α as a transcription factor that is activated by NO and supports cell intrinsic bacterial restriction, and we identify NF-kB as a key transcription factor inhibited by NO that prevents excessive inflammatory cytokine production during M. tuberculosis infection.
Supplementary Material
Acknowledgments
The authors thank Lutz Froenicke (DNA Technologies and Expression Analysis Core, UC Davis) and Monica Britton, Joseph Fass and Blythe Durbin (Genome Center and Bioinformatics Core Facility, UC Davis), for RNA-seq and data analysis; Russell Vance, Jeffrey Cox, Bennett Penn, Kim Sogi and Katie Lien for helpful discussions. The authors also thank Erik Van Dis for help with mouse procedures and flow cytometry.
References
- 1.Floyd K. World Health Organizationed. WHO Press; 2016. pp. 1–100. [Google Scholar]
- 2.Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russell DG, Orme IM. Disseminated tuberculosis in interferon gamma gene-disrupted mice. J Exp Med. 1993;178:2243–2247. doi: 10.1084/jem.178.6.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, Bloom BR. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. 1993;178:2249–2254. doi: 10.1084/jem.178.6.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abel L, Casanova J-L. Genetic predisposition to clinical tuberculosis: bridging the gap between simple and complex inheritance. American journal of human genetics. 67:274. doi: 10.1086/303033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan J, Xing Y, Magliozzo RS, Bloom BR. Killing of virulent Mycobacterium tuberculosis by reactive nitrogen intermediates produced by activated murine macrophages. J Exp Med. 175:1111–1122. doi: 10.1084/jem.175.4.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Desvignes L, Ernst JD. Interferon-gamma-responsive nonhematopoietic cells regulate the immune response to Mycobacterium tuberculosis. 2009;31:974–985. doi: 10.1016/j.immuni.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nandi B, Behar SM. Regulation of neutrophils by interferon-γ limits lung inflammation during tuberculosis infection. Journal of Experimental Medicine. 208:2251–2262. doi: 10.1084/jem.20110919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang YJ, Reddy MC, Ioerger TR, Rothchild AC, Dartois V, Schuster BM, Trauner A, Wallis D, Galaviz S, Huttenhower C, Sacchettini JC, Behar SM, Rubin EJ. Tryptophan Biosynthesis Protects Mycobacteria from CD4 T-Cell-Mediated Killing. 2013;155:1296–1308. doi: 10.1016/j.cell.2013.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alonso S, Pethe K, Russell DG, Purdy GE. Lysosomal killing of Mycobacterium mediated by ubiquitin-derived peptides is enhanced by autophagy. 2007;104:6031–6036. doi: 10.1073/pnas.0700036104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.MacMicking JD. Immune Control of Tuberculosis by IFN- -Inducible LRG-47. Science. 302:654–659. doi: 10.1126/science.1088063. [DOI] [PubMed] [Google Scholar]
- 11.Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. 313:1438–1441. doi: 10.1126/science.1129577. [DOI] [PubMed] [Google Scholar]
- 12.MacMicking JD, North RJ, LaCourse R, Mudgett JS, Shah SK, Nathan CF. Identification of nitric oxide synthase as a protective locus against tuberculosis. 94:5243–5248. doi: 10.1073/pnas.94.10.5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Darwin KH. The Proteasome of Mycobacterium tuberculosis Is Required for Resistance to Nitric Oxide. Science. 2003;302:1963–1966. doi: 10.1126/science.1091176. [DOI] [PubMed] [Google Scholar]
- 14.Ehrt S, Schnappinger D, Bekiranov S, Drenkow J, Shi S, Gingeras TR, Gaasterland T, Schoolnik G, Nathan C. Reprogramming of the Macrophage Transcriptome in Response to Interferon-γ and Mycobacterium tuberculosis. 2001;194:1123–1140. doi: 10.1084/jem.194.8.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mishra BB, Rathinam VAK, Martens GW, Martinot AJ, Kornfeld H, Fitzgerald KA, Sassetti CM. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome–dependent processing of IL-1β. 2012;14:52–60. doi: 10.1038/ni.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Braverman J, Sogi KM, Benjamin D, Nomura DK, Stanley SA. HIF-1α Is an Essential Mediator of IFN-γ-Dependent Immunity to Mycobacterium tuberculosis. 2016;197:1287–1297. doi: 10.4049/jimmunol.1600266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ho Sui SJ. oPOSSUM: identification of over-represented transcription factor binding sites in co-expressed genes. 2005;33:3154–3164. doi: 10.1093/nar/gki624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Benita Y, Kikuchi H, Smith AD, Zhang MQ, Chung DC, Xavier RJ. An integrative genomics approach identifies Hypoxia Inducible Factor-1 (HIF-1)-target genes that form the core response to hypoxia. Nucleic Acids Research. 2009;37:4587–4602. doi: 10.1093/nar/gkp425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salceda S, Caro J. Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. Journal of Biological Chemistry. 1997;272:22642–22647. doi: 10.1074/jbc.272.36.22642. [DOI] [PubMed] [Google Scholar]
- 20.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 21.Bárdos JI, Ashcroft M. Negative and positive regulation of HIF-1: A complex network. 2005;1755:107–120. doi: 10.1016/j.bbcan.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 22.Gleeson LE, Sheedy FJ, Palsson-McDermott EM, Triglia D, O’Leary SM, O’Sullivan MP, O’Neill LAJ, Keane J. Cutting Edge: Mycobacterium tuberculosis Induces Aerobic Glycolysis in Human Alveolar Macrophages That Is Required for Control of Intracellular Bacillary Replication. J Immunol. 2016;196:2444–2449. doi: 10.4049/jimmunol.1501612. [DOI] [PubMed] [Google Scholar]
- 23.Shi L, Salamon H, Eugenin EA, Pine R, Cooper A, Gennaro ML. Infection with Mycobacterium tuberculosis induces the Warburg effect in mouse lungs. Sci Rep. 2015;5:18176. doi: 10.1038/srep18176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, Zheng L, Gardet A, Tong Z, Jany SS, Corr SC, Haneklaus M, Caffrey BE, Pierce K, Walmsley S, Beasley FC, Cummins E, Nizet V, Whyte M, Taylor CT, Lin H, Masters SL, Gottlieb E, Kelly VP, Clish C, Auron PE, Xavier RJ, O’Neill LAJ. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. 2013;496:238–242. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, Hurtado-Ziola N, Nizet V, Johnson RS. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. 115:1806–1815. doi: 10.1172/JCI23865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mayer-Barber KD, Barber DL, Shenderov K, White SD, Wilson MS, Cheever A, Kugler D, Hieny S, Caspar P, Nunez G, Schlueter D, Flavell RA, Sutterwala FS, Sher A. Cutting Edge: Caspase-1 Independent IL-1 Production Is Critical for Host Resistance to Mycobacterium tuberculosis and Does Not Require TLR Signaling In Vivo. 184:3326–3330. doi: 10.4049/jimmunol.0904189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.delaTorre A, Schroeder RA, Punzalan C, Kuo PC. Endotoxin-mediated S-nitrosylation of p50 alters NF-kappa B-dependent gene transcription in ANA-1 murine macrophages. The Journal of Immunology. 1999;162:4101–4108. [PubMed] [Google Scholar]
- 28.Kelleher ZT, Matsumoto A, Stamler JS, Marshall HE. NOS2 regulation of NF-kappaB by S-nitrosylation of p65. Journal of Biological Chemistry. 2007;282:30667–30672. doi: 10.1074/jbc.M705929200. [DOI] [PubMed] [Google Scholar]
- 29.Marshall HE, Hess DT, Stamler JS. S-nitrosylation: physiological regulation of NF-kappaB. Proc Natl Acad Sci USA. 101:8841–8842. doi: 10.1073/pnas.0403034101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yakovlev VA, Barani IJ, Rabender CS, Black SM, Leach JK, Graves PR, Kellogg GE, Mikkelsen RB. Tyrosine nitration of IkappaBalpha: a novel mechanism for NF-kappaB activation. Biochemistry. 2007;46:11671–11683. doi: 10.1021/bi701107z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Connelly L, Palacios-Callender M, Ameixa C, Moncada S, Hobbs AJ. Biphasic regulation of NF-kappa B activity underlies the pro- and anti-inflammatory actions of nitric oxide. The Journal of Immunology. 2001;166:3873–3881. doi: 10.4049/jimmunol.166.6.3873. [DOI] [PubMed] [Google Scholar]
- 32.Carlsen H, Ø Moskaug J, Fromm SH, Blomhoff R. In vivo imaging of NF-kappa B activity. The Journal of Immunology. 2002;168:1441–1446. doi: 10.4049/jimmunol.168.3.1441. [DOI] [PubMed] [Google Scholar]
- 33.MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol. 1997;15:323–350. doi: 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- 34.Francis SH, Busch JL, Corbin JD, Sibley D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol Rev. 2010;62:525–563. doi: 10.1124/pr.110.002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martínez-Ruiz A, Lamas S. Two decades of new concepts in nitric oxide signaling: from the discovery of a gas messenger to the mediation of nonenzymatic posttranslational modifications. IUBMB Life. 2009;61:91–98. doi: 10.1002/iub.144. [DOI] [PubMed] [Google Scholar]
- 36.Cooper CE. Nitric oxide and iron proteins. Biochimica et Biophysica Acta. 1999;1411:290–309. doi: 10.1016/s0005-2728(99)00021-3. [DOI] [PubMed] [Google Scholar]
- 37.Galván-Peña S, O’Neill LAJ. Metabolic reprograming in macrophage polarization. Front Immunol. 2014;5:420. doi: 10.3389/fimmu.2014.00420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li F, Sonveaux P, Rabbani ZN, Liu S, Yan B, Huang Q, Vujaskovic Z, Dewhirst MW, Li C-Y. Regulation of HIF-1α Stability through S-Nitrosylation. 2007;26:63–74. doi: 10.1016/j.molcel.2007.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Metzen E, Zhou J, Jelkmann W, Fandrey J, Brune B. Nitric oxide impairs normoxic degradation of HIF-1alpha by inhibition of prolyl hydroxylases. Molecular Biology of the Cell. 2003;14:3470–3481. doi: 10.1091/mbc.E02-12-0791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mishra BB, Lovewell RR, Olive AJ, Zhang G, Wang W, Eugenin E, Smith CM, Phuah JY, Long JE, Dubuke ML, Palace SG, Goguen JD, Baker RE, Nambi S, Mishra R, Booty MG, Baer CE, Shaffer SA, Dartois V, McCormick BA, Chen X, Sassetti CM. Nitric oxide prevents a pathogen-permissive granulocytic inflammation during tuberculosis. Nat Microbiol. 2017;2:17072. doi: 10.1038/nmicrobiol.2017.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sakai Shunsuke, K KD, S MA, S AH, Y HA, G VV, B DL. CD4 T Cell-Derived IFN-γ Plays a MinimalRole in Control of Pulmonary Mycobacteriumtuberculosis Infection and Must Be ActivelyRepressed by PD-1 to Prevent Lethal Disease. PLOS Pathogens. 2016;12:1–22. doi: 10.1371/journal.ppat.1005667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lázár-Molnár E, Chen B, Sweeney KA, Wang EJ, Liu W, Lin J, Porcelli SA, Almo SC, Nathenson SG, Jacobs WR. Programmed death-1 (PD-1)-deficient mice are extraordinarily sensitive to tuberculosis. Proc Natl Acad Sci USA. 2010;107:13402–13407. doi: 10.1073/pnas.1007394107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kimmey JM, Huynh JP, Weiss LA, Park S, Kambal A, Debnath J, Virgin HW, Stallings CL. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature. 2015;528:565–569. doi: 10.1038/nature16451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hunn JP, Howard JC. The mouse resistance protein Irgm1 (LRG-47): a regulator or an effector of pathogen defense? PLOS Pathogens. 2010;6:e1001008. doi: 10.1371/journal.ppat.1001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weinberg JB. Nitric oxide production and nitric oxide synthase type 2 expression by human mononuclear phagocytes: a review. Mol Med. 1998;4:557–591. doi: 10.1007/BF03401758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nathan C, Shiloh MU. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. PNAS. 2000;97:8841–8848. doi: 10.1073/pnas.97.16.8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang CS, Yuk J-M, Jo E-K. The Role of Nitric Oxide in Mycobacterial Infections. Immune Netw. 2009;9:46. doi: 10.4110/in.2009.9.2.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.