

Abstract
Osteoprotegerin (OPG) is a secreted member of the tumor necrosis factor (TNF) receptor superfamily that binds to the ligand for receptor activator of nuclear factor κB (RANKL) and inhibits bone resorption. OPG can also bind and inhibit the activity of the TNF‐related apoptosis‐inducing ligand (Apo2L/TRAIL), raising the possibility that the anticancer efficacy of soluble Apo2L/TRAIL may be abrogated in the bone microenvironment where OPG expression is high. In this study we used a murine model of breast cancer growth in bone to evaluate the efficacy of recombinant soluble Apo2L/TRAIL against intratibial tumors that were engineered to overexpress native full‐length human OPG. In vitro, OPG‐overexpressing breast cancer cells were protected from Apo2L/TRAIL‐induced apoptosis, an effect that was reversed with the addition of soluble RANKL or neutralizing antibodies to OPG. In vivo, mice injected intratibially with cells containing the empty vector developed large osteolytic lesions. In contrast, OPG overexpression preserved the integrity of bone and prevented breast cancer–induced bone destruction. This effect was due primarily to the complete absence of osteoclasts in the tibias of mice inoculated with OPG‐transfected cells, confirming the biologic activity of the transfected OPG in vivo. Despite the secretion of supraphysiologic levels of OPG, treatment with Apo2L/TRAIL resulted in strong growth inhibition of both empty vector and OPG‐overexpressing intratibial tumors. While Apo2L/TRAIL‐induced apoptosis may be abrogated in vitro by OPG overexpression, the in vivo anticancer efficacy of recombinant soluble Apo2L/TRAIL is retained in the bone microenvironment even in the presence of biologically active OPG at supraphysiologic concentrations. © 2011 American Society for Bone and Mineral Research.
Keywords: APO2L/TRAIL, OPG, OSTEOLYSIS, BREAST CANCER, APOPTOSIS
Introduction
Recombinant soluble Apo2 ligand (Apo2L)/tumor necrosis factor–related apoptosis‐inducing ligand (TRAIL) is emerging as a promising new cancer therapeutic for the treatment of solid and hematologic malignancies. Apo2L/TRAIL induces apoptosis in a wide variety of cancer cell lines but not in most normal cells.1, 2 Induction of apoptosis by Apo2L/TRAIL is mediated by interactions with its death domain–containing cell surface receptors DR4 and DR5 to activate caspases that carry out the cell death program (reviewed in ref. 3). The potential of soluble Apo2L/TRAIL as an anticancer therapeutic agent has been well demonstrated in mouse xenograft models of human soft tissue cancers, including colorectal,4 breast,2 and lung cancers5 and multiple myeloma,6 and glioma.7 Apo2L/TRAIL is active alone and exhibits synergistic activity with chemotherapeutic agents or radiotherapy, causing marked regression or complete remission of tumors, with no evidence of toxicity to normal tissues and organs in animal experiments.7, 8 In light of these preclinical findings, recombinant soluble Apo2L/TRAIL now has progressed to the next stage of its development, with numerous early‐phase clinical trials initiated.9
While other apoptosis‐inducing members of the tumor necrosis factor (TNF) family, such as TNF‐α and FasL, initially showed great promise as anticancer agents, their severe toxicity toward normal tissues precluded their clinical use. In contrast, Apo2L/TRAIL is selectively toxic to cancer cells while sparing normal cells.1, 2 For example, we have found that normal human bone (NHB) cells, normal mammary epithelial cells, and human fibroblasts are resistant to Apo2L/TRAIL‐induced apoptosis.10, 11, 12 While many transformed cells are sensitive to Apo2L/TRAIL, some display resistance via mechanisms that remain poorly understood. There appear to be multiple mechanisms for this resistance, including increased expression of the cell surface “decoy” receptors DcR1 and DcR2. Unlike DR4 and DR5, the decoy receptors lack functional death domains, cannot mediate apoptosis, and may compete with death‐inducing receptors for Apo2L/TRAIL binding. We and others have not been able to demonstrate a consistent correlation between Apo2L/TRAIL receptor expression and sensitivity to Apo2L/TRAIL‐induced apoptosis. However, our previous work has implicated the decoy receptor DcR2 in the acquired loss of sensitivity of osteosarcoma cells.10 Additional mechanisms controlling Apo2L/TRAIL sensitivity/resistance have been proposed, including overexpression of intracellular inhibitory proteins such as FLIP or intracellular inhibitor of apoptosis molecules (IAPs) (reviewed in refs. 3 and 13).
Osteoprotegerin (OPG) is a widely expressed soluble member of the TNF receptor superfamily that binds to the ligand for receptor activator of nuclear factor κB (RANKL) and inhibits bone resorption.14, 15 However, OPG also can bind Apo2L/TRAIL, and although it has lower affinity for Apo2L/TRAIL at normal physiologic temperatures, it can block Apo2L/TRAIL‐induced apoptosis in vitro.16, 17, 18, 19 There is now mounting evidence supporting the ability of OPG to act as both an endocrine and paracrine survival factor for different tumor types. For example, human prostate cancer cell lines were shown to secrete OPG at concentrations capable of inhibiting Apo2L/TRAIL‐induced apoptosis in vitro.20 Similarly, multiple myeloma and prostate and breast cancer cells were protected from Apo2L/TRAIL‐induced apoptosis in the presence of OPG produced by osteoblast‐like cells or bone marrow stromal cells.17, 21, 22 Collectively, these data suggest that OPG production may represent an additional mechanism by which cancer cells escape host defense systems. Therapeutically, this raises the possibility that the activity of soluble Apo2L/TRAIL may be reduced in tumor environments where OPG expression is high. While there is supporting evidence for this hypothesis from in vitro studies, the potential for OPG to limit the efficacy of therapeutically relevant doses of recombinant soluble Apo2L/TRAIL in vivo has not been established.
In this study we used a murine intratibial model of breast cancer development and progression to investigate whether tumor‐derived OPG can limit the anticancer efficacy of recombinant soluble Apo2L/TRAIL in the bone microenvironment.
Materials and Methods
Cell lines and tissue culture
The MB‐231, MB‐453, MB‐468, ZR‐75, MCF‐7, and T47D human breast cancer cell lines and the MG‐63 human osteosarcoma cell line were obtained from ATCC (Manassas, VA, USA). The MB‐231 derivative cell line MB‐231‐TXSA was kindly provided by Dr Toshiyuki Yoneda (University of Texas Health Sciences Center, San Antonio, TX, USA). Murine monocytic RAW 264.7 cells, kindly supplied by Dr David Thomas (Peter McCallum Institute, Melbourne, Australia), were originally from the ATCC. Human bone marrow stromal cells (hBMSCs) were obtained from needle aspirates taken from the iliac crests of normal healthy donors, as described previously,23 and grown in α‐MEM (SAFC Biosciences, Victoria, Australia) containing 10% fetal bovine serum (Biosciences, Sydney, Australia) and ascorbic acid 2‐phosphate (NovaChem, Victoria, Australia). Human primary giant cell tumors (GCTs) of bone samples were processed as described previously.24 All other cell lines were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 2 mM glutamine, 100 IU/mL of penicillin, 160 µg/mL of gentamicin, Hepes (20 mM), and 10% fetal bovine serum (Biosciences, Sydney, Australia) in a 5% CO2–containing humidified atmosphere.
Reagents
Recombinant Apo2L/TRAIL was a kind gift from Dr Avi Ashkenazi, Genentech, Inc (South San Francisco, CA, USA). Anti‐human OPG MAB805 was obtained from R&D Systems (Minneapolis, MN, USA). Recombinant human soluble RANK ligand was purchased from Millipore (Billerica, MA, USA). An Acid Phosphatase, Leukocyte (TRACP) Kit was purchased from Sigma‐Aldrich (St Louis, MO, USA). The caspase inhibitor‐1 zVAD‐fmk was obtained from Calbiochem (La Jolla, CA, USA).
Generation of luciferase‐tagged MB‐231‐TXSA cells
Luciferase‐expressing MB‐231‐TXSA cells were generated using the retroviral expression vector SFG‐NES‐TGL, which gives rise to a single fusion protein encoding herpes simplex virus thymidine kinase (TK), green fluorescence protein (GFP), and firefly luciferase (Luc). Virus particle–containing supernatants were generated and filtered to remove any cellular debris and then used to infect cells, as described previously.12, 25, 26 The retrovirally transduced cells were grown as bulk cultures for 48 hours and subsequently sorted for positive GFP expression using fluorescence‐activated sorting (FACS) (Aria BD Biosciences, Franklin Lakes, NJ, USA). The cells were allowed to proliferate, and the 10% of cells expressing GFP most strongly were sorted by FACS to generate the subline MB‐231‐TXSA‐TGL.
Generation of luciferase‐tagged MB‐231‐TXSA‐TGL overexpressing native human OPG
The OPG coding sequence was excised from a full‐length cDNA clone (kindly provided by Amgen, Inc., Thousand Oaks, CA, USA). The OPG coding sequence was digested with NotI/XbaI, and nucleotide overhangs were removed using a Klenow fragment (no. M0212S, New England BioLabs, Inc., Beverly, MA, USA). In preparation for cloning of this fragment into pRuf‐IRES‐GFP,27 the vector was digested with XhoI, followed by removal of nucleotide overhands with a Klenow fragment. The OPG coding fragment was ligated subsequently into this vector, generating the retroviral construct pRuf‐IRES‐OPG‐GFP. Infectious retroviral particles were generated by transfecting HEK293T cells with retroviral constructs harboring cDNAs of interest (5 µg) along with retroviral packaging vectors pGP (4 µg) and pVSV‐G (4 µg) (Stratagene, La Jolla, CA, USA) using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA). Viral particle–containing supernatant was collected 48 hours after transfection and filtered through a 0.45‐µm low‐binding filter to remove any cellular debris, as described previously.25 Polybrene (4 µg/mL) was added to the viral particle–containing supernatant immediately prior to its addition to cells. Viral supernatant was removed 48 hours after infection, and cells were grown for a further 5 days in DMEM growth medium [DMEM supplemented with 10% (v/v) fetal calf serum (FCS), 2 mmol/L of l‐glumatine, 50 IU/mL of penicillin/streptomycin; JRH, Lenexa, KS, USA) at 37°C in a 5% CO2–containing humidified atmosphere. The cells were sorted (FACS; Aria BD Biosciences, Mountain View, CA, USA) for the top 30% of GFP‐expressing cells to generate pools of MB‐231‐TXSA‐TGL‐p‐RUF‐ and p‐OPG‐overexpressing cell lines.
RT‐PCR
Real‐time PCR reactions were performed using RT2 Real Time SYBR Green PCR Master Mix (SuperArray Biosciences Corp., Frederick, MD, USA) as per the manufacturer's instructions in a Rotor‐Gene (Corbett Research, New South Wales, Australia) with the following cycle parameters: 95°C for 15 seconds, 60°C for 26 seconds, and 72°C for 10 seconds, 40 × . The following RT‐PCR primers were used: OPG forward, 5'‐cgctcgtgtttctggacat ‐3'; reverse, 5'‐acacggtcttccactttgct‐3'. All specific products were expressed relative to their β‐actin or GAPDH internal control, as indicated.
Cell viability assays
To determine the cytotoxic effects of Apo2L/TRAIL on cell growth, 1 × 104 cells per well were seeded in 96‐well microtiter plates and allowed to adhere overnight. Cells were treated with increasing concentrations of Apo2L/TRAIL (10 to 100 ng/mL) for 24 hours. Cell viability was assessed using the Titer‐Blue Cell Viability Assay (Promega, Madison, WI, USA), as well as crystal violet staining, and optical density was measured at 570 nm (OD570). Experiments were performed in triplicate and repeated a minimum of three times. Results of representative experiments are presented as the mean ± SD.
DAPI staining of nuclei
MB‐231‐TXSA‐TGL cells were seeded on plastic chamber slides and treated with Apo2L/TRAIL at a concentration of 100 ng/mL. After two washes with PBS, cells were fixed in methanol for 5 minutes, washed again with PBS, and incubated with 0.8 mg/mL of 4',6‐diamidine‐2'‐phenylindole dihydrochloride (DAPI; Roche Diagnostics, Castle Hill, New South Wales, Australia) in PBS for 15 minutes at 37°C. After several washes in PBS, the coverslips were mounted on PBS/glycerol. DAPI staining was visualized by fluorescence microscopy.
Western blot analysis
MB‐231‐TXSA‐TGL cells were treated with Apo2L/TRAIL at 100 ng/mL in a time‐dependent manner (1, 4, 8, and 24 hours) and lysed in buffer containing 10 mM Tris‐HCl (pH 7.6), 150 mM NaCl, 1% Triton X‐100, 0.1% sodium dodecylsulfate (SDS), 2 mM sodium vanadate, and a protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany). Protein lysates from all the time points were stored at –70°C until ready to use. The amount of protein in each sample was quantified using the BCA Protein Assay Reagent (Rockford, IL, USA) according to the manufacturer's instructions. Prior to loading, protein extracts were mixed with an equal volume of running buffer containing 12 mM Tris‐HCL (pH 6.8), 6% SDS, 10% β‐mercaptoethanol, 20% glycerol, and 0.03% bromophenol blue. Protein samples then were heated at 70°C for 10 minutes and loaded into 4% to 20% polyacrylamide gels for electrophoresis under reducing conditions. Separated proteins were transferred electrophoretically to polyvinylidene difluoride (PVDF) transfer membranes (Novex, San Diego, CA, USA) and blocked in PBS containing 5% blocking reagent (Amersham, Castle Hill, New South Wales, Australia) for 1 hour at room temperature. Immunodetection was performed overnight at 4°C in PBS/blocking reagent containing 0.1% Tween 20 using the following primary antibodies at the dilutions suggested by the manufacturer: Anti‐caspase‐8 and anti‐caspase‐9 were purchased from Cell Signaling Technology (Beverly, MA, USA), mAb anti‐caspase‐10 from MBL (Naka‐ku, Nagoya, Japan). mAb anti‐caspase‐3 from Transduction Laboratories (Lexington, KY, USA), pAb anti‐Bid from Chemicon International (Temecula, CA, USA), and pAb anti–poly‐ADP‐ribose polymerase (anti‐PARP) from Roche Diagnostics (Mannheim, Germany). Anti‐DR5, anti‐DR4, anti‐DcR1, and anti DcR2 were purchased from R&D Systems (Minneapolis, MN, USA). Anti‐actin mAb (Sigma, St Louis, MO, USA) was used as a loading control. Membranes then were rinsed several times with PBS containing 0.1% Tween‐20 and incubated with 1:5000 dilution of anti‐mouse, anti‐goat, or anti‐rabbit alkaline phosphatase–conjugated secondary antibodies (Pierce, Rockford, IL, USA) for 1 hour. Visualization and quantification of protein bands were performed using the ECF Substrate Reagent Kit (GE Healthcare, Buckinghamshire, UK) on a FluorImager (Molecular Dynamics, Inc., Sunnyvale, CA, USA).
Preparation of conditioned medium and detection of OPG by ELISA
Cells were seeded at a density of 2.3 × 106 cells per T75 flask in 12 mL of standard medium as above. Medium was collected at 48 and 72 hours and cleared of cells by centrifugation (1200 rpm for 5 minutes) and filtration (0.2‐µm filter). The medium was stored at –80°C until required. The concentrations of OPG in the culture medium from each cell line and in blood serum collected from mice were determined using a commercial ELISA kit as per the manufacturer's instructions (KB 1011, Immunodiagnostics AG, Wien, Austria).
Detection of OPG by immunoprecipitation
Cell lysates or culture supernatants from MB‐231‐TXSA‐TGL‐p‐RUF and p‐OPG cell lines were prepared as described previously.25 Goat anti‐mouse Ig–coupled Dynabeads (Dynal, Oslo, Sweden) were washed twice in 1% (v/v) NP40–50 mM Tris‐HCl, 150 mM NaCl, and 1 mM EDTA prior to the addition of 3 µg of purified immunoglobulin (MAB805, R&D Systems, Inc, and the relevant isotype‐matched, nonbinding controls). This mixture then was incubated at 4°C for a minimum of 6 hours with rotation. The resulting prearmed Dynabeads were washed twice in 1% (v/v) NP40, 50 mM Tris‐HCL, 150 mM NaCl, and 1 mM EDTA, and the beads were collected using a magnetic particle collector (MPC‐1, Dynal). To these, 1.0‐mL aliquots of the appropriate NP40 cell lysate or 10 mL of culture supernatants was added. The samples were incubated for 2 hours at 4°C with rotation. The immunoprecipitates then were washed twice in 1% (v/v) NP40‐TSE, once in 0.1% (v/v) NP40‐TSE, and once in TSE (pH 8.0). Each immunoprecipitate represented the material from 5 × 106 cell equivalents or 10 mL of conditioned medium (CM). Immunoprecipitated proteins were detected by Western blotting using the monoclonal antibody to OPG (MAB8051, R&D Systems, Inc.)
Challenge with Apo2L/TRAIL in the presence of CM from MG‐63 cells and BMSCs
MB‐231‐TXSA‐TGL breast cancer cells were seeded in a 96‐well plate at a density of 1 × 104/well and allowed to adhere overnight. CM from MG‐63 cells and hBMSCs was collected as described earlier. Cells were treated with 25% CM from MG‐63 cells and human BMSCs in the presence of 20 ng/mL of Apo2L/TRAIL. In some experiments, breast cancer cells overexpressing OPG also were challenged with Apo2L/TRAIL in the presence of increasing concentrations of soluble RANKL (100 to 1000 ng/mL) or neutralizing antibodies to OPG (1 to 20 mg/mL). Cell viability was assessed using the Titer‐Blue Cell Viability Assay as well as crystal violet staining, and optical density was measured at 570 nm (OD570). Experiments were performed in triplicate and repeated a minimum of three times. Results of representative experiments are presented as the mean ± SD.
Osteoclast assays
Raw 264.7 murine monocytic cells were seeded in 96‐well plates and cultured in DMEM, as described earlier. When attached, the cells were treated with RANKL (100 ng/mL) and 25% CM from MB‐231‐TXSA‐TGL‐p‐RUF‐ and OPG‐overexpressing cells. Medium and treatments were replaced on day 3. Cells were fixed on day 5 and stained histochemically for tartrate resistant acid phosphatase (TRACP) according to the manufacturer's instructions (Sigma‐Aldrich, St Louis, MO, USA). TRACP+ cells were visualized by light microscopy. To assess bone resorption by mature osteoclasts, cells cultured from human GCT of bone were seeded onto dentine slices in 96‐well plates at a density of 4 × 105/well in the presence or absence of 25% CM from MB‐231‐TXSA‐TGL‐p‐RUF‐ and p‐OPG‐overexpressing cells for 5 days. CM was replaced on day 3. On day 5, the dentine slices were mounted on stubs, coated with carbon‐gold, and examined on a Philips XL‐20 scanning electron microscope (Eindhoven, Holland). Images then were analyzed and the number of resorption pits counted using ImageQuant software (GE Healthcare, Little Chalfont, United Kingdom; quadruplicate dentine slices for each treatment). Results are shown as average number of pits (± SEM), and the significant difference between treatments was determined using Student's t tests.
Animals
Female athymic nude mice at 4 to 6 weeks of age (Institute of Medical and Veterinary Services Division, Gilles Plains, SA, Australia) were acclimatized to the animal housing facility for a minimum period of 1 week prior to the commencement of experimentation. The general physical well‐being and weight of animals were monitored continuously throughout the experiments. All mice were housed under pathogen‐free conditions, and all experimental procedures on animals were carried out with strict adherence to the rules and guidelines for the ethical use of animals in research and were approved by the Animal Ethics Committees of the University of Adelaide and the Institute of Medical and Veterinary Science, Adelaide, SA, Australia.
Intratibial injection model
MB‐231‐TXSA‐TGL‐p‐RUF‐ and p‐OPG‐overexpressing human breast cancer cells were cultured as described earlier until they reached 70% to 80% confluency. Adherent cells were removed from flasks with 2 mM EDTA and resuspended in 1× PBS at 1 × 105 cells/10 µL and kept on ice in an Eppendorf tube. Mice were anesthetized with isoflurane (Faulding Pharmaceuticals, SA, Australia), the left tibia was wiped with 70% ethanol, a 27‐gauge needle coupled with a Hamilton syringe was inserted through the tibial plateau with the knee flexed, and 1 × 105 cells resuspended in 10 µL of PBS were injected into the marrow space. All animals were injected with PBS in the contralateral tibia as a control. Mice were randomly assigned into two groups for each cell line of five animals each, and treatment began 2 weeks after cancer cell transplantation. Mice in the vehicle group received an intraperitoneal (i.p.) injection of clear 1× PBS once weekly. Apo2L/TRAIL was administered at 30 mg/kg i.p for 5 consecutive days every second week.
Bioluminescence imaging (BLI) of tumor growth
Noninvasive whole‐body imaging for assessment of tumor growth was performed once weekly using the IVIS 100 Imaging system (Xenogen, Alameda, CA, USA). Mice were injected i.p. with 100 µL of the D‐Luciferin solution at final dose of 3 mg/20 g of mouse body weight (Xenogen) and then gas‐anesthetized with isoflurane (Faulding Pharmaceuticals). Images were acquired for 0.5 to 30 seconds (images are shown at 1 second) from the front angle, and the photon emission transmitted from the mice was captured and quantitated in photons/s/cm2/sr using Xenogen Living Image (Igor Pro Version 2.5) software (Alameda, CA, USA).
Micro–computed tomographic (µCT) analysis
Limbs for µCT analysis were surgically resected and scanned using a SkyScan‐1174 high‐resolution µCT Scanner (Skyscan, Kontich, Belgium). During µCT scanning, the tibias were placed vertically in tightly fitting plastic tubes. The µCT scanner was operated at 50 kV, 800 µA, rotation step 0.4, 0.25‐mm Al filter, and scan resolution of 7.78 µm per pixel. The cross sections were reconstructed using a cone‐beam algorithm (NRecon software, Skyscan). Files then were imported into CTAn software (Skyscan) for 3D analysis and 3D image generation. Using the 2D images obtained from the µCT scan, the growth plate was identified, and 500 sections were selected starting from the growth plate–tibia interface and moving down the tibia. All images are viewed and edited using CTvol visualization software (Skyscan). Histograms representing bone volume (mm3) from tumor‐bearing tibias were generated and compared with the contralateral non‐tumor‐bearing tibias. Tumor burdens, measured in cubic millimeters, were determined using the Skyscan software.
Histology
Tibias were fixed in 10% (v/v) buffered formalin (24 hours at 4°C), followed by 2 to 4 weeks of decalcification in 0.5 M EDTA–0.5% paraformaldehyde in PBS (pH 8.0) at 4°C. Complete decalcification was confirmed by radiography, and the tibias then were embedded in paraffin. Five‐micron longitudinal sections were prepared and stained with hematoxylin and eosine (H&E). Additional sections were used for TRACP (Sigma‐Aldrich) or TUNEL (Promega) staining following the manufacture's protocol. Analysis was performed on an Olympus CX41 (Hachioji‐shi, Tokyo, Japan) microscope, and images were taken using the NanoZoomer Digital Pathology (NDP‐Hamamatsu, Hamamatsu City, Shizuoka Pref., Japan). Tumor area measured in square millimeters was assessed using the NanoZoomer software.
Data analysis and statistics
Experiments were performed in triplicate, and data presented as mean ± SE. All statistical analysis was performed using SigmaStat for Windows Version 3.0 (Systat Software, Inc., Port Richmond, CA, USA) using the unpaired Student's t test. Measures of association between two variables were assessed using the Spearman rank correlation coefficient. Comparisons between groups were assessed using a one‐way ANOVA test. In all cases, p < .05 was considered statistically significant
Results
Apo2L/TRAIL induces apoptosis in breast cancer cell lines in vitro
Seven well‐established human breast cancer cell lines were examined for their sensitivity to the cytotoxic effects of recombinant soluble Apo2L/TRAIL in vitro. As shown in Fig. 1 A, culture of MB‐231‐TXSA, MB‐231, MB‐468, and T‐47D cells in increasing doses of Apo2L/TRAIL resulted in the induction of cell death after 24 hours of treatment, with 40% to 90% cell death occurring at 100 ng/mL of Apo2L/TRAIL. The remaining breast cancer cell lines were relatively resistant to the effects of Apo2L/TRAIL (Fig. 1 A). The mode of cell death induced by Apo2L/TRAIL was apoptosis, as shown in the MB‐231‐TXSA cells. Treatment with Apo2L/TRAIL induced the morphologic changes characteristic of apoptosis, including chromatin condensation and DNA fragmentation, as assessed by DAPI staining (Fig. 1 B). In addition, Apo2L/TRAIL activated the extrinsic apoptotic signaling pathway, including activation and processing of the initiator caspase‐8 and caspase‐10, leading to cleavage and activation of caspase‐3 that was concomitant with cleavage of the apoptosis target protein poly‐ADP‐ribose polymerase (PARP; Fig. 1 C). The presence of the pan‐caspase inhibitor zVAD‐fmk completely reversed the reduction in cell viability, providing further evidence that Apo2L/TRAIL‐induced apoptosis of MB‐231‐TXSA cells depends on caspase (Fig. 1 D). We also found evidence of activation of the “intrinsic” apoptotic pathway, which was associated with caspase‐8‐mediated cleavage of the Bcl2 protein family member Bid and activation of caspase‐9 (Fig. 1 C). Predictably, the Apo2L/TRAIL‐sensitive MB‐231, MB‐231‐TXSA, and MB‐468 cell lines expressed relatively higher levels of both DR4 and DR5 when compared with the resistant cell lines (Fig. 2 A). To assess whether OPG is a determinant of Apo2L/TRAIL sensitivity, an ELISA was used to measure OPG secreted into the medium of each cell line when cultured for 48 hours under the same conditions. MB‐231, MB‐231‐TXSA, and MCF‐7 cells produced varying levels of OPG ranging from 300 to 500 pg/mL of OPG, whereas the remaining cell lines produced OPG at levels near or below the detection limit of the system (Fig. 2 B). These levels did not correlate with sensitivity/resistance to the cytotoxic effects of Apo2L/TRAIL. In contrast, the levels of OPG produced by osteoblast‐like cells (MG‐63) or primary hBMSCs cultured from three donors were approximately 100‐fold higher than those produced by the panel of breast cancer cell lines. MG‐63 cells and hBMSCs secreted OPG at concentrations of 20 and 30 ng/mL, respectively, and CM from these cultures protected MB‐231‐TXSA‐sensitive breast cancer cells from Apo2L/TRAIL‐induced apoptosis (Fig. 2 C). These data demonstrate that levels of OPG secreted by breast cancer cells are relatively low and do not affect Apo2L/TRAIL‐induced apoptosis. However, the levels of OPG released by hBMSCs potentially could accumulate in the bone microenvironment at high enough levels to antagonize Apo2L/TRAIL‐induced apoptosis.
Figure 1.
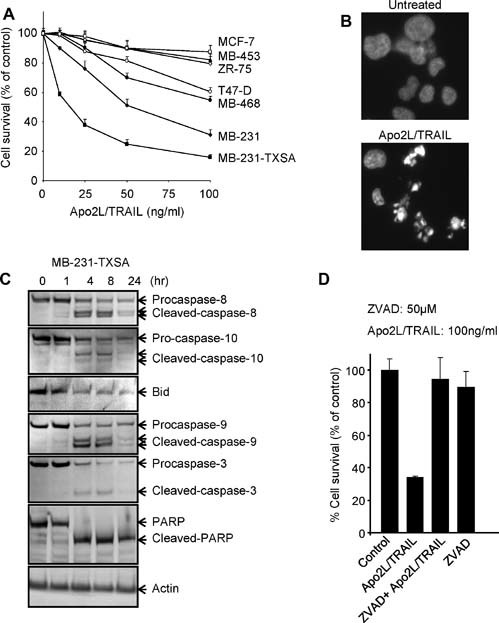
Apo2L/TRAIL induces apoptosis in breast cancer cell lines in vitro by activating both the intrinsic and the extrinsic apoptotic signaling pathways. (A) Seven well‐established human breast cancer cell lines were treated with increasing concentrations of Apo2L/TRAIL, as indicated. Cell viability was assessed by AlamarBlue staining 24 hours after treatment. (B) Fluorescence microscopy of DAPI‐stained MB‐231‐TXSA‐TGL cells treated for 24 hours with Apo2L/TRAIL at 100 ng/mL. (C) Western blot analysis of lysates isolated from MB‐231‐TXSA‐TGL cells treated with 100 ng/mL of Apo2L/TRAIl for 0, 1, 4, 8, and 24 hours. The caspase‐8, caspase‐9, caspase‐10, caspase‐3, poly‐ADP ribose polymerase (PARP), and Bid antibodies detect both full‐length and processed forms of the antigens. (D) MB‐231‐TXSA cells were treated for 12 hours with 100 ng/mL of Apo2L/TRAIL alone or were coincubated with the caspase inhibitor z‐VAD‐fmk (50 µM). To exclude possible toxic effects of the inhibitor, cells also were treated with the inhibitor alone. Cell viability was determined using the AlamarBlue assay and expressed as a percentage of control. Data points show means of quadruplicate results from a representative experiment repeated at least twice. Bars, ± SD.
Figure 2.
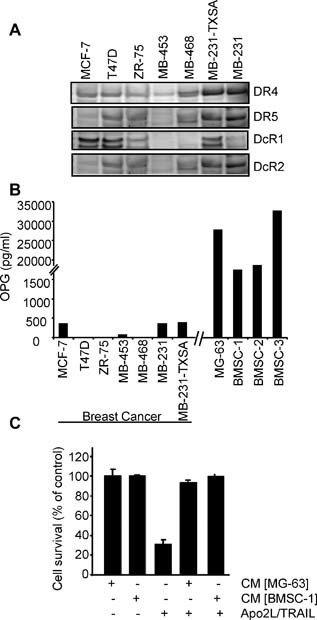
Differential sensitivity of breast cancer cell lines to soluble Apo2L/TRAIL is not correlated with basal levels of OPG. (A) Total cell lysates were collected for Western blot analysis to determine the basal expression of Apo2L/TRAIL receptors (B) OPG concentration in the cell supernatant from breast cancer cell lines, osteoblast‐like MG‐63 cells, and human bone marrow stromal cells (hBMSCs) from three independent donors were measured by OPG ELISA after culture of cells for 48 hours. (C) MB‐231‐TXSA cells were treated with 20 ng/mL of Apo2L/TRAIL in the presence or absence of 25% medium conditioned by MG‐63 cells or hBMSC‐1. Data points show means of quadruplicate results from a representative experiment repeated at least twice. Data are presented as the mean ± SD of quadruplicate wells and are expressed as a percentage of the number of control cells.
Generation and characterization of breast cancer cells overexpressing full‐length human OPG
To determine if OPG could accumulate at levels that would affect Apo2L/TRAIL‐induced apoptosis in vivo, we overexpressed full‐length human OPG in the MB‐231‐TXSA breast cancer cell line. This cell line was chosen for these studies because of its ability to form aggressive, rapidly growing tumors when injected into the orthotopic site of the mammary fat pad of nude mice. Furthermore, when injected into the tibial marrow cavity, osteolytic lesions arise within 3 to 4 weeks after cancer cell transplantation (see below). In addition, we have shown previously that these cells are highly sensitive to the apoptotic effects of Apo2L/TRAIL in vitro, and when tested in vivo, Apo2L/TRAIL reduced tumor burden in bone and prevented breast cancer–induced osteolysis in a mouse model.28 Quantitative RT‐PCR analysis showed a significant increase (1000‐fold) in OPG mRNA expression in the OPG‐transfected cell line (MB‐231‐TXSA‐p‐OPG) compared with the empty vector–transfected cells (MB‐231‐TXSA‐p‐RUF) (Fig. 3 A). Production of OPG protein also was confirmed by Western blot following immunoprecipitation of OPG from CM of the OPG‐infected cells. The Western blots revealed bands for OPG at 55 and 23 kDa, corresponding to dimeric and monomeric forms of OPG, respectively,15 that were present at high levels in the cell supernatant rather in than the total cell lysates (Fig. 3 B). Furthermore, ELISA analysis showed that transfection with full‐length OPG resulted in a substantial increase in OPG production, reaching 180 ng/mL, when compared with vector control cells, in which the OPG levels were only 0.3 ng/mL (Fig. 3 C). The in vitro growth kinetics of each cell line were identical, indicating that OPG overexpression had no effect on their growth rate (Fig. 3 D).
Figure 3.
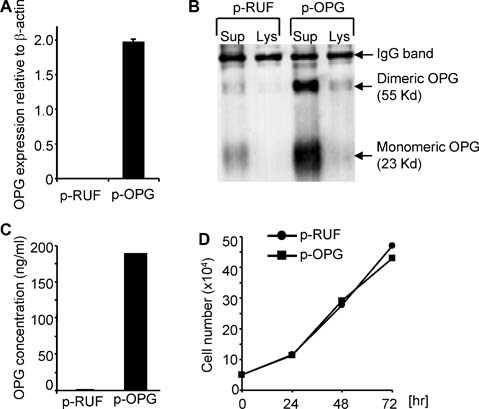
Characterization of MB‐231‐TXSA cells overexpressing full‐length human OPG. (A) Quantitative RT‐PCR analysis showing approximately 1000‐fold increase in OPG mRNA expression in the OPG‐transfected cells (MB‐231‐TXSA‐p‐OPG) compared with empty vector–transfected cells (MB‐231‐TXSA‐p‐RUF). (B) Conditioned medium (CM) from MB‐231‐TXSA‐p‐RUF and MB‐231‐TXSA‐p‐OPG cells was collected and tested for OPG production by Western blot analysis following immunoprecipitation of OPG. Each immunoprecipitate represented the material from 5 × 106 cell equivalents or 10 mL of CM. Immunoprecipitated proteins were detected by Western blotting using the monoclonal antibody to OPG MAB8051. (C) OPG concentration in the cell supernatant from each transfected cell line was measured by OPG ELISA after culture of cells for 48 hours. Transfection with full‐length human OPG resulted in a substantial increase in OPG production compared with vector‐infected cells. (D) MB‐231‐TXSA‐p‐RUF and MB‐231‐TXSA‐p‐OPG cells were seeded at 5 × 104/well in triplicate into 24‐well plates, and the number of cells was counted at 24, 48, and 72 hours. Data are presented as the mean ± SD of triplicate wells and are expressed as absolute number of cells.
Biologic activity of OPG secreted by the transfected cells
To determine whether the OPG secreted by the transfected breast cancer cells was biologically active, we performed in vitro osteoclast‐formation assays. Consistent with the role of OPG in inhibiting osteoclast differentiation and bone resorption, we found that when RAW264.7 murine monocytic cells were cultured with the receptor activator of nuclear factor κB ligand (RANKL), CM (25%) from p‐OPG‐overexpressing cells, but not from p‐RUF empty vector–transfected cells, inhibited the formation of TRACP+ multinucleated cells (Fig. 4 A). Similarly, the same CM inhibited bone resorption by mature osteoclasts that were isolated from human giant cell tumors (GCTs) of bone when cultured on bone matrix (Fig. 4 B).
Figure 4.
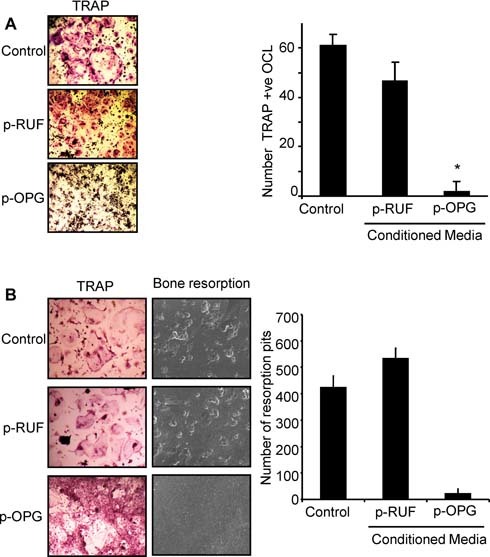
OPG secreted by MB‐231‐TXSA‐p‐OPG is biologically active. (A) RAW264.7 cells were seeded in 96‐well plates and treated with 100 ng/mL of RANKL in the absence (control) or presence of 25% CM prepared from MB‐231‐TXSA‐p‐RUF or MB‐231‐TXSA‐p‐OPG cells for 5 days. Shown are representative fields of the cell cultures as indicated after TRACP staining (left panel). The number of TRACP+ multinucleated osteoclasts (containing three or more nuclei) was scored (right panel). Data represent the means ± SD of three independent experiments. (B) Cells cultured from giant cell tumor of bone (GCT) samples known to contain abundant numbers of osteoclast‐like cells were seeded on plastic in 96‐well plates or were directly plated onto dentine slices in the presence or absence of CM for 5 days. Shown are representative fields of the cell cultures after TRACP staining (left panel). Bone slices were examined on a scanning electron microscope, and the number of resorption pits was counted using ImageQuant software (right panel). Results are shown as average number of pits (± SEM).
Effect of OPG overexpression on Apo2L/TRAIL‐induced apoptosis of MB‐231‐TXSA cells in vitro
Having established that OPG secreted by the transfected cells is biologically active, we next examined the effect of OPG overexpression on Apo2L/TRAIL‐induced apoptosis in vitro. Treatment of the empty vector–transfected cells with Apo2L/TRAIL for 24 hours resulted in a dose‐dependent loss of cell viability that reached a maximum of 80% at the highest dose of 100 ng/mL of Apo2L/TRAIL and with an IC50 value of 20 ng/mL. In contrast, the OPG‐transfected cells were relatively refractory to the cytotoxic effects of Apo2L/TRAIL, showing only a 20% loss of cell viability at the same dose (Fig. 5 A). These effects were mediated by OPG because the addition of soluble RANKL, which sequesters OPG, dose‐dependently reversed the protective effects conferred by the OPG‐overexpressing cells (Fig. 5 B). Similarly, when OPG‐overexpressing cells were preincubated with increasing concentrations of a neutralizing antibody to OPG, the level of Apo2L/TRAIL‐induced apoptosis was increased significantly, and cell survival was reduced correspondingly (Fig. 5 C).
Figure 5.
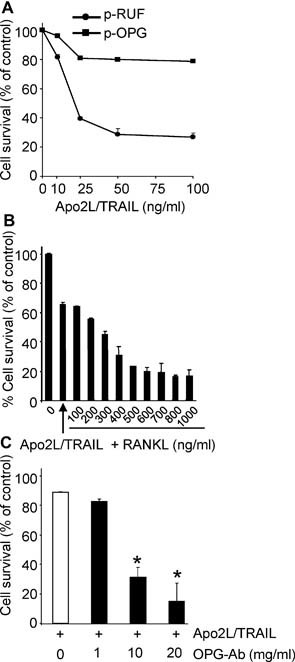
Overexpression of OPG by breast cancer cells protects against Apo2L/TRAIL‐induced apoptosis in vitro. (A) MB‐231‐TXSA‐p‐RUF and MB‐231‐TXSA‐p‐OPG cells were seeded in 96‐well plates and treated with increasing concentrations of Apo2L/TRAIL. Cell viability was assessed by AlamarBlue staining 24 hours after treatment. MB‐231‐TXSA‐p‐OPG cells were relatively resistant to the cytotoxic effects of Apo2L/TRAIL. (B, C) MB‐231‐TXSA‐p‐OPG cells were treated with Apo2L/TRAIL at 50 ng/mL in the presence of increasing concentrations of soluble RANKL (B) or OPG neutralizing antibody MAB805 (C). The addition of RANKL and OPG neutralizing antibodies dose‐dependently reversed Apo2L/TRAIL resistance of MB‐231‐TXSA‐p‐OPG cells. Data presented are the mean (n = 3) of representative experiments. Bars, ± SEM. *p < .05 compared with control.
Anticancer efficacy of Apo2L/TRAIL against OPG‐overexpressing intratibial tumors
To evaluate the efficacy of Apo2L/TRAIL against OPG‐overexpressing tumors within bone and its effects on cancer‐induced bone destruction, we used a xenogeneic tumor model in which the empty vector– or OPG‐overexpressing MB‐231‐TXSA breast cancer cells were transplanted directly into the tibial marrow cavity of athymic nude mice. We established noninvasive bioluminescence imaging approaches that provided sensitive real‐time in vivo assessment of breast cancer growth in bone. Development of breast cancer–induced bone destruction was assessed qualitatively and quantitatively using high‐resolution µCT analysis. For noninvasive bioluminescence imaging (BLI) of tumor growth, the vector‐ and OPG‐transfected cells were superinfected with a triple‐fusion protein reporter construct encoding herpes simplex virus thymidine kinase (TK), green fluorescent protein (GFP), and firefly luciferase (Luc).12, 26 After infection, cells were enriched for high‐level expression of GFP by two rounds of fluorescence‐activated cell sorting, thus generating the sublines MB‐231‐TXSA‐pRUF‐TGL and MB‐231‐TXSA‐pOPG‐TGL. In this model, we initiated Apo2L/TRAIL treatment on day 14 after cancer cell transplantation, following confirmation that breast cancer cells had established growth in the bone marrow cavity but displayed no evidence of osteolysis. All animals inoculated with the empty vector–transfected cells and treated with vehicle only showed an exponential increase of mean photon emission associated with an increase in tumor burden, which was clearly evident from day 14 onward (Fig. 6 A). By day 18, all animals in this group developed large intratibial tumors that penetrated the cortical bone, with the tumor mass invading the surrounding soft tissue. Reconstructed 3D µCT images of representative tumor‐bearing tibias demonstrated extensive osteolysis compared with the contralateral non‐tumor‐bearing tibias of vehicle‐treated animals (Fig. 6 B). To quantify the total bone volume (BV), we compared the tumor‐bearing with the contralateral non‐tumor‐bearing tibias of the vehicle‐treated animals at a selected region beginning at the growth plate and extending downward 750‐ × 5.2‐µm slices, which encompassed all the cancer lesions. As seen in Fig. 6 C, the amount of bone lost exceeded 40% in the tumor‐bearing tibias compared with the contralateral non‐tumor‐bearing tibias, confirming the qualitative assessment (Fig. 6 B). For ethical reasons, all vehicle‐treated animals were humanely killed on day 18 owing to high tumor load and extensive osteolysis. Treatment with Apo2L/TRAIL inhibited tumor growth in the tibias of all animals inoculated with the p‐RUF empty vector, as assessed by BLI (Fig. 6 A), whereas qualitative (Fig. 6 B) and quantitative (Fig. 6 C) µCT analysis demonstrated remarkable protection from breast cancer–induced osteolysis.
Figure 6.
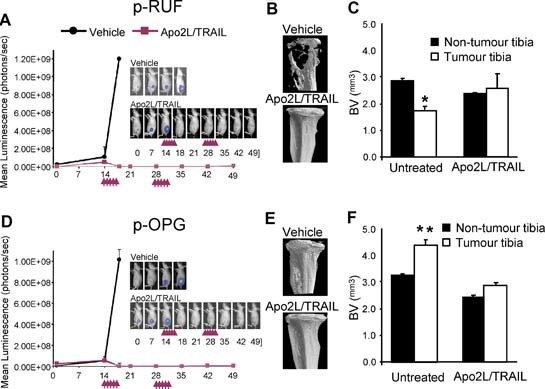
Antitumour efficacy of Apo2L/TRAIL is maintained in the bone microenvironment where biologically active OPG is present at high concentrations. (A, D) Representative whole‐body BLI of a single animal from each group of mice (n = 5 mice per group) bearing MB‐231‐TXSA‐TGL‐p‐RUF and MB‐231‐TXSA‐TGL‐p‐OPG tumor cells. The line graphs represent the average tumor signal over time of vehicle and Apo2l/TRAIL‐treated mice measured as mean photon counts per second. The arrows demonstrate the dosing schedule with Apo2L/TRAIL, which is described under “Materials and Methods.” All vehicle‐treated animals were killed humanely on day 18 for ethical reasons owing to high tumor load, whereas Apo2L/TRAIL‐treated mice, with either p‐RUF vector or p‐OPG‐overexpressing tumors, exhibited complete responses and remained free of tumor until day 49, when the experiment was terminated. (B, E) Qualitative 3D µCT images of representative animals from each group before and after treatment with Apo2L/TRAIL. (C, F) Quantitative assessment of bone volume (BV) in cubic millimeters in the tumor‐bearing tibias compared with the contralateral non‐tumor‐bearing tibias. Bars, ±SEM. *p < .01.
OPG overexpression by breast cancer cells in the absence of Apo2L/TRAIL had no effect on the total tumor burden in the tibias, as assessed by BLI, with the kinetics of tumor growth being identical to that of the tumors with the p‐RUF empty vector (compare Fig. 6 A with Fig. 6 D). Consistent with the role of OPG in inhibiting osteoclastic bone resorption, all animals inoculated with the p‐OPG‐transfected cells showed preservation of the integrity of bone around the tumors, as depicted in the representative reconstructed 3D µCT images (Fig. 6 E). OPG released by cancer cells translated to a significant increase in BV in the OPG‐bearing tibias compared with the tibias with tumors containing the empty vector (Fig. 6 F). It is interesting to note that OPG secretion by cancer cells not only protected against breast cancer–induced bone loss but also resulted in a significant increase in bone volume in the contralateral non‐tumor‐bearing tibias compared with the contralateral non‐tumor‐bearing tibias of the p‐RUF animals. While BLI demonstrated no significant differences in the total tumor burden, histologic inspection of the p‐OPG tibias showed that the distribution of the tumor was different in p‐OPG animals compared with vector‐only animals. Intraosseous tumor burden diminished considerably in p‐OPG animals, within the bone marrow space, which had now been significantly reduced and spatially constrained owing to the increase in trabecular bone density (Fig. 7 A). However, there was persistent growth of cancer cells in the extramedullary space, which was not affected by OPG overexpression, thus accounting for the failure to detect any differences in the overall tumor burden when assessed by BLI. The intra‐ and extraosseous tumor burden in the tibias, when quantified from histologic sections and expressed as an average tumor area per group, confirmed the qualitative histologic assessment (Fig. 7 B). Osteoclasts were abundantly present and attached to the bone surfaces in tumor lesions from the vector‐only‐transfected cells (Fig. 7 C). In contrast, the protective effect of OPG overexpression on bone destruction was primarily due to the complete absence of TRACP+ osteoclasts in tibia preparations of animals inoculated with p‐OPG‐transfected cells (Fig. 7 C). These data further confirm the biologic activity of the exogenous OPG in vivo. Importantly, the efficacy of Apo2L/TRAIL against p‐OPG‐expressing tumors was identical to that of the p‐RUF‐bearing tumors, showing similar kinetics of tumor growth inhibition (Fig. 6 D) and maintenance of bone integrity (Fig. 6 E, F). Histologic examination of representative sections from the intratibial p‐OPG‐expressing tumors 1 week after treatment indicated that Apo2L/TRAIL induced apoptosis in a substantial proportion of the tumor mass, with intense TUNEL+ staining of tumor cells compared with vehicle‐treated animals, thus confirming the biologic activity of Apo2L/TRAIL in vivo and the bioluminescence data (Fig. 7 D). Bloods were collected from representative animals at different times during the experiment, and the amount of circulating OPG was measured by ELISA. The data show that the basal level of circulating OPG in the non‐tumor‐bearing animals was approximately 30 pg/mL. These levels increased 40‐fold to 1200 pg/mL in the animals with established p‐OPG‐bearing tumors when measured on day 18 after cancer cell transplantation, clearly indicating that p‐OPG cells maintained expression of OPG over the entire days in vivo in the untreated animals (Fig. 7 E). Furthermore, treatment with Apo2L/TRAIL decreased the levels of circulating OPG to near‐basal levels owing to cancer cell apoptosis, further confirming the biologic activity and anticancer efficacy of Apo2L/TRAIL against OPG‐overexpressing tumors in vivo.
Figure 7.
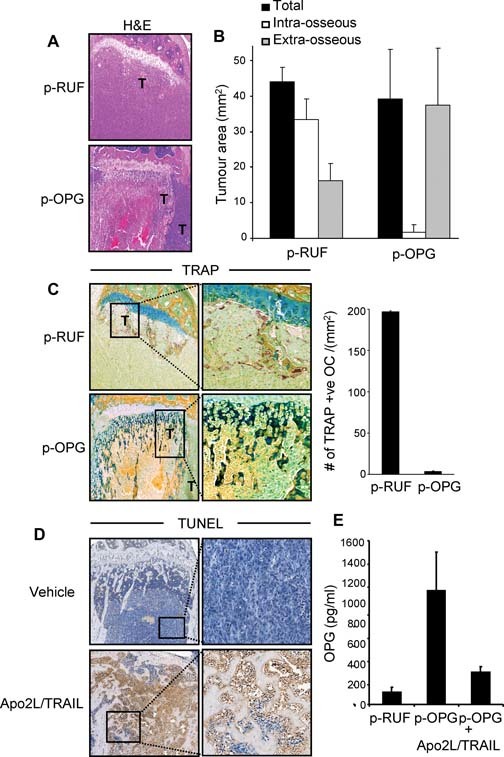
OPG overexpression maintains bone integrity but alters the intra‐ and extramedullary tumor distribution. (A) Representative H&E‐stained tibial sections showing the differential distribution of intra‐ and extramedullary tumor growth. (B) Quantitative assessment of intra‐ and extramedullary tumor area measured in square millimeters. (C) TRACP staining of histologic sections showing the absence of TRACP+ osteoclasts in tibia preparations of animals inoculated with OPG‐transfected cells compared with vector‐transfected cells in which osteoclasts were abundantly present and attached to the bone surfaces (left panel). Quantitative assessment of the number of TRACP+ osteoclasts (right panel). (D) Histologic examination of decalcified tibial sections from p‐OPG bearing tumors 1 week after treatment indicate that Apo2L/TRAIL‐induced apoptosis in a substantial proportion of intra‐ and extraosseous tumor mass with intense TUNEL+ staining of tumor cells compared with vehicle‐treated animals. (E) OPG concentration in the blood serum of representative mice (n = 3) collected at different time points during the experiment, as measured by ELISA. Bars, ±SD.
Discussion
In this study we tested the hypothesis that the anticancer efficacy of recombinant soluble Apo2L/TRAIL against breast cancer cell growth would be abrogated in the bone microenvironment, where OPG expression is high. Our in vitro results demonstrated that the basal level of OPG produced and secreted by a panel of breast cancer cell lines could not be correlated with Apo2L/TRAIL‐induced apoptosis. The levels varied considerably between breast cancer cell lines and were too low to counteract the apoptotic actions of Apo2L/TRAIL. Therefore, OPG production by breast cancer cells themselves is an unlikely mechanism by which invading breast cancer cells either evade the host defense system or may promote cell survival during Apo2L/TRAIL therapy. MB‐231‐TXSA cells were engineered to overexpress OPG at levels approximately 1000‐fold greater than the vector control cells. Apo2L/TRAIL‐induced apoptosis was significantly suppressed in these cells, indicating that accumulating levels of OPG can antagonize the apoptotic actions of Apo2L/TRAIL in vitro. The suppression of apoptosis by OPG overexpression in breast cancer cells was reversed by soluble RANKL, which sequestered the activity of OPG. Similarly, the addition of neutralizing OPG antibodies also reversed this protection, confirming that OPG produced by these cells was responsible for suppressing Apo2L/TRAIL‐induced apoptosis. These in vitro observations are in accordance with previously published studies on different types of tumor cells, all suggesting that OPG may be a survival factor for human cancer cells through inhibition of Apo2L/TRAIL‐induced apoptosis. In agreement with previously published data,17, 20, 21, 22 we have further shown that osteoblast‐like cells (MG‐63), or primary bone marrow stromal cells, secrete OPG at relatively high levels, and CM from these cells suppress Apo2L/TRAIL‐induced apoptosis of the sensitive MB‐231‐TXSA cells. These in vitro findings raise the possibility that the activity of recombinant soluble Apo2L/TRAIL could be reduced in the bone microenvironment, where the local concentrations of OPG secreted within the confines of the bone marrow is high. Therefore, in the context of cancer in bone, it has been suggested that recombinant soluble Apo2L/TRAIL may have limited use as a therapeutic agent. While the in vitro data support this hypothesis, the significance of the OPG‐Apo2L/TRAIL association in vivo and the potential antagonistic actions of OPG have not been established. In this study we have used a mouse model of intratibial injection of breast cancer cells overexpressing native full‐length human OPG. This in vivo model recapitulates the late stages of the bone metastatic process and is ideally suited to monitor the effects of Apo2L/TRAIL treatment on the growth of breast cancer cells in the bone microenvironment and on cancer‐induced bone destruction.
OPG plays a key role in inhibiting osteoclast formation and bone resorption.14 In our study, OPG overexpression decreased bone resorption markedly, protected the integrity of cortical and trabecular bone, and prevented the development of osteolytic lesions. We have shown that these effects were due to the inhibitory actions of OPG on osteoclast formation, which further confirmed the biologic activity of the transfected OPG in vivo. While OPG overexpression protected the bone from cancer‐induced osteolysis, there was no significant effect on the overall tumor burden (intraosseous and extramedullary), as measured using BLI. However, histologic inspection demonstrated that the intraosseous tumor burden in the p‐OPG tibias diminished considerably. In contrast, there was persistent growth in the extramedullary space. While the basis for the persistent growth of cancer cells outside the bone of mice with the p‐OPG‐expressing cells is not entirely clear, it should be noted that the trabecular bone density was considerably increased in the p‐OPG‐bearing tibias. This effect of p‐OPG translated to a significant reduction in bone marrow volume, contributing to the spatial constraints forcing cancer cells to escape through vascular spaces or through parts of the cortex that initially had been resorbed. The effects of p‐OPG on bone density were due to suppression of osteoclast formation and perhaps activity because p‐OPG expression dramatically decreased the number of TRACP+ osteoclasts lining the bone surfaces. Consistent with our findings, Buijs and colleagues have shown recently that osteoclast ablation by zoledronic acid or Fc‐OPG treatment following intratibial injection of highly osteolytic breast cancer cells protected against cancer‐induced osteolysis, and although the intraosseous tumor burden was diminished, the overall tumor burden was not affected significantly owing to the persistent growth in the extramedullary space.29 This effect is also consistent with our recently published data showing that osteoclast ablation with zoledronic acid had no significant effect on the overall tumor burden owing to the persistent growth of osteosarcoma cells in the extramedullary space, even though it protected against osteosarcoma‐induced bone destruction.30
Our in vivo results highlight a number of important points. First, we have shown that basal levels of OPG produced either by breast cancer cells or by bone marrow stromal cells and secreted locally within the confines of the bone marrow are insufficient to inhibit the anticancer efficacy of recombinant soluble Apo2L/TRAIL. These findings are consistent with our previously published observations showing that recombinant soluble Apo2L/TRAIL decreased tumor burden in bone and prevented cancer‐induced bone destruction in murine models of both breast cancer and multiple myeloma.12, 28 Second, we have shown that even in the presence of supraphysiologic levels of OPG secreted by OPG‐overexpressing breast cancer cells, the anticancer efficacy of Apo2L/TRAIL is adequately maintained. It should be noted that the circulating serum level of OPG in mice bearing tumors containing the empty vector, as measured by our ELISA assay, is approximately 0.08 ng/mL, whereas in mice with the OPG‐overexpressing tumors, the serum level is increased 15‐fold to 1.2 ng/mL prior to Apo2L/TRAIL treatment. However, the bioavailability of Apo2L/TRAIL when administered via the peritoneal cavity, as in this study, is approximately 30%, and the peak plasma levels expected with a 30 mg/kg i.p dose given over 5 days is approximately 7 µg/mL.12, 28 Therefore, on a molar‐ratio basis, the concentration of recombinant soluble Apo2L/TRAIL achieved in mice is in excess, nonlimiting, and unlikely to be antagonized by OPG. Similarly, in human patients with solid tumors, the serum concentration of Apo2L/TRAIL is approximately 36 mg/mL and is achieved at doses greater than 4 mg/kg, which is equivalent to the effective doses in preclinical models.31 Patients with solid tumors and metastatic disease to bone have a significantly higher level of circulating OPG of approximately 0.2 ng/mL,32 but this concentration does not approach the level that would be expected to counterbalance the level of recombinant soluble Apo2L/TRAIL achieved in vivo.32 It also should be noted that the binding affinity between Apo2L/TRAIL and OPG at normal physiologic temperature is much weaker than for Apo2L/TRAIL and its death‐inducing receptors DR4 and DR5.18 While it is conceivable that the low‐affinity associations between OPG and Apo2L/TRAIL may become significant under conditions where the local concentration of OPG is high, our study suggests that OPG is unlikely to play a significant role in modulating the therapeutic potential of recombinant soluble Apo2L/TRAIL in vivo.
Taken together, our data demonstrate that while Apo2L/TRAIL‐induced apoptosis may be abrogated in vitro by OPG overexpression, the in vivo anticancer efficacy of recombinant soluble Apo2L/TRAIL is retained in the bone microenvironment where biologically active OPG may be present at high concentrations.
Disclosures
All the authors state that they have no conflicts of interest.
Acknowledgements
This work was supported by grants from the National Health and Medical Research Council (NHMRC) of Australia, the Cancer Council of South Australia (TCCSA), and the National Breast Cancer Foundation (NBCF). We will like to acknowledge the University of Adelaide microscopy resources staff for their assistance and the IMVS animal facility staff for animal care assistance.
References
- 1. Ashkenazi A, Pai RC, Fong S, et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest. 1999; 104: 155–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Walczak H, Miller RE, Ariail K, et al. Tumoricidal activity of tumor necrosis factor–related apoptosis‐inducing ligand in vivo. Nat Med. 1999; 5: 157–163. [DOI] [PubMed] [Google Scholar]
- 3. Bouralexis S, Findlay DM, Evdokiou A. Death to the bad guys: targeting cancer via Apo2L/TRAIL. Apoptosis. 2005; 10: 35–51. [DOI] [PubMed] [Google Scholar]
- 4. Kelley SK, Harris LA, Xie D, et al. Preclinical studies to predict the disposition of Apo2L/tumor necrosis factor‐related apoptosis‐inducing ligand in humans: characterization of in vivo efficacy, pharmacokinetics, and safety. J Pharmacol Exp Ther. 2001; 299: 31–38. [PubMed] [Google Scholar]
- 5. Jin H, Yang R, Fong S, et al. Apo2 ligand/tumor necrosis factor–related apoptosis‐inducing ligand cooperates with chemotherapy to inhibit orthotopic lung tumor growth and improve survival. Cancer Res. 2004; 64: 4900–4905. [DOI] [PubMed] [Google Scholar]
- 6. Mitsiades CS, Treon SP, Mitsiades N, et al. TRAIL/Apo2L ligand selectively induces apoptosis and overcomes drug resistance in multiple myeloma: therapeutic applications. Blood. 2001; 98: 795–804. [DOI] [PubMed] [Google Scholar]
- 7. Gliniak B, Le T. Tumor necrosis factor‐related apoptosis‐inducing ligand' s antitumor activity in vivo is enhanced by the chemotherapeutic agent CPT‐11. Cancer Res. 1999; 59: 6153–6158. [PubMed] [Google Scholar]
- 8. Chinnaiyan AM, Prasad U, Shankar S, et al. Combined effect of tumor necrosis factor‐related apoptosis‐ inducing ligand and ionizing radiation in breast cancer therapy. Proc Natl Acad Sci U S A. 2000; 97: 1754–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pan Y. Application of pharmacodynamic assays in a phase 1a trial of Apo2L/TRAIL in pateients with advenced tumours. J Clin Oncol. 2007; (ASCO):Abstract 3535. [Google Scholar]
- 10. Bouralexis S, Findlay DM, Atkins GJ, Labrinidis A, Hay S, Evdokiou A. Progressive resistance of BTK‐143 osteosarcoma cells to Apo2L/TRAIL‐induced apoptosis is mediated by acquisition of DcR2/TRAIL‐R4 expression: resensitisation with chemotherapy. Br J Cancer. 2003; 89: 206–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Butler LM, Liapis V, Bouralexis S, et al. The histone deacetylase inhibitor, suberoylanilide hydroxamic acid, overcomes resistance of human breast cancer cells to Apo2L/TRAIL. Int J Cancer. 2006; 119: 944–954. [DOI] [PubMed] [Google Scholar]
- 12. Labrinidis A, Hay S, Liapis V, Ponomarev V, Findlay D, Evdokiou A. Zoledronic acid inhibits both the osteolytic and osteoblastic components of osteosarcoma lesions in a mouse model. Clin Cancer Res. 2009; 15: 3451–3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ashkenazi A. Targeting death and decoy receptors of the tumour‐necrosis factor superfamily. Nat Rev Cancer. 2002; 2: 420–430. [DOI] [PubMed] [Google Scholar]
- 14. Simonet WS, Lacey DL, Dunstan CR, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997; 89: 309–319. [DOI] [PubMed] [Google Scholar]
- 15. Tsuda E, Goto M, Mochizuki S, et al. Isolation of a novel cytokine from human fibroblasts that specifically inhibits osteoclastogenesis. Biochem Biophys Res Commun. 1997; 234: 137–142. [DOI] [PubMed] [Google Scholar]
- 16. Emery JG, McDonnell P, Burke MB, et al. Osteoprotegerin is a receptor for the cytotoxic ligand TRAIL. J Biol Chem. 1998; 273: 14363–14367. [DOI] [PubMed] [Google Scholar]
- 17. Shipman CM, Croucher PI. Osteoprotegerin is a soluble decoy receptor for tumor necrosis factor‐related apoptosis‐inducing ligand/Apo2 ligand and can function as a paracrine survival factor for human myeloma cells. Cancer Res. 2003; 63: 912–916. [PubMed] [Google Scholar]
- 18. Truneh A, Sharma S, Silverman C, et al. Temperature‐sensitive differential affinity of TRAIL for its receptors. DR5 is the highest affinity receptor. J Biol Chem. 2000; 275: 23319–23325. [DOI] [PubMed] [Google Scholar]
- 19. Vitovski S, Phillips JS, Sayers J, Croucher PI. Investigating the interaction between osteoprotegerin and receptor activator of NF‐kappaB or tumor necrosis factor‐related apoptosis‐inducing ligand: evidence for a pivotal role for osteoprotegerin in regulating two distinct pathways. J Biol Chem. 2007; 282: 31601–31609. [DOI] [PubMed] [Google Scholar]
- 20. Holen I, Croucher PI, Hamdy FC, Eaton CL. Osteoprotegerin (OPG) is a survival factor for human prostate cancer cells. Cancer Res. 2002; 62: 1619–1623. [PubMed] [Google Scholar]
- 21. Neville‐Webbe HL, Cross NA, Eaton CL, et al. Osteoprotegerin (OPG) produced by bone marrow stromal cells protects breast cancer cells from TRAIL‐induced apoptosis. Breast Cancer Res Treat. 2004; 86: 269–279. [DOI] [PubMed] [Google Scholar]
- 22. Nyambo R, Cross N, Lippitt J, et al. Human bone marrow stromal cells protect prostate cancer cells from TRAIL‐induced apoptosis. J Bone Miner Res. 2004; 19: 1712–1721. [DOI] [PubMed] [Google Scholar]
- 23. Atkins GJ, Bouralexis S, Evdokiou A, et al. Human osteoblasts are resistant to Apo2L/TRAIL‐mediated apoptosis. Bone. 2002; 31: 448–456. [DOI] [PubMed] [Google Scholar]
- 24. Evdokiou A, Atkins GJ, Bouralexis S, et al. Expression of alternatively‐spliced MDM2 transcripts in giant cell tumours of bone. Int J Oncol. 2001; 19: 625–632. [DOI] [PubMed] [Google Scholar]
- 25. Zannettino AC, Rayner JR, Ashman LK, Gonda TJ, Simmons PJ. A powerful new technique for isolating genes encoding cell surface antigens using retroviral expression cloning. J Immunol. 1996; 156: 611–620. [PubMed] [Google Scholar]
- 26. Ponomarev V, Doubrovin M, Serganova I, et al. A novel triple‐modality reporter gene for whole‐body fluorescent, bioluminescent, and nuclear noninvasive imaging. Eur J Nucl Med Mol Imaging. 2004; 31: 740–751. [DOI] [PubMed] [Google Scholar]
- 27. Diamond P, Labrinidis A, Martin SK, et al. Targeted disruption of the CXCL12/CXCR4 axis inhibits osteolysis in a murine model of myeloma‐associated bone loss. J Bone Miner Res. 2009; 24: 1150–1161. [DOI] [PubMed] [Google Scholar]
- 28. Thai le M, Labrinidis A, Hay S, et al. Apo2l/Tumor necrosis factor‐related apoptosis‐inducing ligand prevents breast cancer‐induced bone destruction in a mouse model. Cancer Res. 2006; 66: 5363–5370. [DOI] [PubMed] [Google Scholar]
- 29. Buijs JT, van der Pluijm G, Osteotropic cancers: from primary tumor to bone. Cancer Lett 2009; 273: 177–193. [DOI] [PubMed] [Google Scholar]
- 30. Labrinidis A, Hay S, Liapis V, Findlay DM, Evdokiou A. Zoledronic acid protects against osteosarcoma‐induced bone destruction but lacks efficacy against pulmonary metastases in a syngeneic rat model. Int J Cancer. 2009. [DOI] [PubMed] [Google Scholar]
- 31. Ling J, Herbst RS, Mendelson DS, et al. Apo2L/TRAIL pharmacokinetics in a phase 1a trial in advanced cancer and lymphoma. J Clin Oncol. 2006; 24: (ASCO Meeting Abstracts): 3047. [Google Scholar]
- 32. Lipton A, Ali SM, Leitzel K, et al. Serum osteoprotegerin levels in healthy controls and cancer patients. Clin Cancer Res. 2002; 8: 2306–2310. [PubMed] [Google Scholar]