

Abstract
Background
Equine influenza is a major health problem of equines worldwide. The polymerase genes of influenza virus have key roles in virus replication, transcription, transmission between hosts and pathogenesis. Hence, the comprehensive genetic and codon usage bias of polymerase genes of equine influenza virus (EIV) were analyzed to elucidate the genetic and evolutionary relationships in a novel perspective.
Results
The group - specific consensus amino acid substitutions were identified in all polymerase genes of EIVs that led to divergence of EIVs into various clades. The consistent amino acid changes were also detected in the Florida clade 2 EIVs circulating in Europe and Asia since 2007. To study the codon usage patterns, a total of 281,324 codons of polymerase genes of EIV H3N8 isolates from 1963 to 2015 were systemically analyzed. The polymerase genes of EIVs exhibit a weak codon usage bias. The ENc-GC3s and Neutrality plots indicated that natural selection is the major influencing factor of codon usage bias, and that the impact of mutation pressure is comparatively minor. The methods for estimating host imposed translation pressure suggested that the polymerase acidic (PA) gene seems to be under less translational pressure compared to polymerase basic 1 (PB1) and polymerase basic 2 (PB2) genes. The multivariate statistical analysis of polymerase genes divided EIVs into four evolutionary diverged clusters - Pre-divergent, Eurasian, Florida sub-lineage 1 and 2.
Conclusions
Various lineage specific amino acid substitutions observed in all polymerase genes of EIVs and especially, clade 2 EIVs underwent major variations which led to the emergence of a phylogenetically distinct group of EIVs originating from Richmond/1/07. The codon usage bias was low in all the polymerase genes of EIVs that was influenced by the multiple factors such as the nucleotide compositions, mutation pressure, aromaticity and hydropathicity. However, natural selection was the major influencing factor in defining the codon usage patterns and evolution of polymerase genes of EIVs.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-4063-1) contains supplementary material, which is available to authorized users.
Keywords: Equine influenza virus, H3N8, Polymerase genes, Codon usage bias, Evolution
Background
Equine influenza outbreaks are of major concern throughout the world. The vaccines often fail to protect horses, especially in the case of newly emerged equine influenza A (H3N8) viruses owing to antigenic variation of haemagglutinin (HA) protein [1, 2]. Thus due to its importance for vaccine strain selection [3, 4] most evolutionary dynamic studies have been focused on HA gene [5–7]. Since their first isolation in 1963 (Miami/63), EIV H3N8 have diverged due to variation in the HA gene viz., Pre-divergent, Eurasian and American (evolved into currently circulating Florida sub-lineage clade1 & 2) [5, 7, 8]. The evolution of internal genes of EIV H3N8 in Greek outbreak [9] and evidence of species-jumping of H3N8 from horse to canine [10–12]; and pigs [13] emphasizes the role of internal genes, especially polymerase genes in replication, transcription and host adaptation.
The three largest genome segments of EIVs encode polymerase heterotrimeric proteins namely basic protein 1 (PB1), basic protein 2 (PB2) and acidic protein (PA). These subunits together with the nucleoprotein form the complex which is accountable for viral RNA transcription and replication [14–16]. Genome segment 2 encodes the PB1 subunit, the central component of the polymerase complex with RNA polymerase activity; PB1 interacts directly with PA and PB2 subunits [17–21]. Segment 2 also encodes another protein, PB1-F2, from an alternate reading frame of PB1 mRNA [22, 23]. PB1-F2 is known to increase virulence through the induction of cell death, promoting inflammation and up-regulating viral polymerase activity [24–26]. Genome segment 1 encodes the PB2 subunit, a key player in the initiation of viral transcription through cap-snatching activity in conjunction with endonuclease function of PA subunit [17, 27–29]. Finally, segment 3 encodes the PA subunit, which is responsible for generalized proteolysis of viral and host proteins [30, 31] and plays a role in virus assembly [32]. Segment 3 also encodes a second protein called PA-X [33], which modulates viral pathogenesis [34, 35]. In addition to their basic functions in viral genome expression, the polymerase proteins play a pivotal role in host adaptation and viral pathogenesis [34, 36, 37]. Several mutations in the polymerase subunits of avian and human influenza viruses have been implicated in enhancing virus replication and adaptation to various hosts [38–42]. However, the factors dictating the genetic changes of polymerase genes of EIV H3N8 especially in terms of codon usage bias patterns have not been explored in detail.
The codon usage pattern has a significant role in the evolution of viruses. Several studies have testified the species-specific synonymous codon usage bias [43–45], which shows preference for certain codons encoding for the same amino acids [46]. Such preference for codon usage offers evolutionary force for determining the overall fitness of the virus influencing various cellular processes [47–49]. So far, limited studies have highlighted the factors responsible in shaping the codon usage bias in different influenza viruses. For instance, while mutation pressure plays a key role in shaping the codon usage bias in H1N1 (human influenza virus) [50] and H9N2 (avian influenza virus) [51], natural selection primarily dictates the bias of synonymous codon usage for EIV [52]. One study on the PB1-F2 gene of EIV showed that mutational bias along with selection pressure and gene length influenced the codon usage pattern [53]. Furthermore, the codon usage bias analysis provides a different perspective regarding virus evolution studies in comparison to phylogenetic studies. Nevertheless, comprehensive analysis of codon usage pattern of polymerase of EIVs has not been elucidated so far.
Thus the present study focuses on comprehensive analysis of genetic evolution, synonymous codon usage pattern and factors involved in shaping the codon usage pattern of three polymerase genes across the lineages of EIV H3N8 strains circulating worldwide from 1963 to 2015. Combining the codon usage bias and traditional phylogenetic analyses of polymerase genes of EIV H3N8 will help in understanding the novel perspective of molecular evolution dynamics of EIVs.
Results
Amino acid variations in the phylogenetic clusters of polymerase genes
The deduced amino acid sequences of polymerase genes of EIV H3N8 were aligned and compared against A/equine/Richmond/07 isolate - representative strain of Florida clade 2 sub-lineage of EIV H3N8. The detailed amino acid substitutions observed in PA, PA-X, PB1, PB1-F2 and PB2, proteins encoded by the polymerase genes of EIV H3N8 are presented in additional file (see Additional file 1: Table S1a, S1b, S1c, S1d & S1e).
In the PA protein, four amino acid substitutions (I62V, I270M, I432V & V/F450I) were found in EIVs from 1993 (A/equine/Newmarket/1/93) onwards which persisted in Florida clade 1 & 2 isolates (Table 1 and Additional file 1: Table S1a). Moreover, from 2002 onwards five amino acid substitutions (R213K, A337T, A343E, L345I & K353R) could be observed in all Florida clade 1 and 2 isolates except A/equine/Wisconsin/03, A/equine/Cheshire/06 and A/equine/Linconshire/06. Florida clade 2 isolates from A/equine/Richmond/07 onwards showed six amino acid changes (E64D, M86I, E237K, S/N321 N, A476T & K626R) which are not seen in A/equine/Newmarket/5/03 isolate. The PA sequences of all Asian isolates till 2013 showed almost 100% similarities with A/equine/Richmond/07 isolate from Europe except one amino acid change (K to R/G) at position 158.
Table 1.
Amino acid substitutions in the polymerase genes of EIV H3N8 among different lineages/clades
| Genes | Residue no. | Pre-divergent | Eurasian | Florida clade 1 | Florida clade 2 |
|---|---|---|---|---|---|
| PA | 62 | I | V | V | V |
| 64 | E | E | E | D | |
| 86 | M | M | M | I | |
| 158 | K | K | K | R/G/K | |
| 213 | R | R | K | K | |
| 231 | A | V | V | V | |
| 237 | E | E | E | K | |
| 269 | R | K | K | K | |
| 270 | I | I/M | M | M | |
| 321 | S/N | S | S | N | |
| 337 | A | A | T | T | |
| 343 | A | A | E | E | |
| 345 | L | L | I | I | |
| 353 | K | K | R | R | |
| 354 | T/I | T/D | I | I | |
| 388 | S | N | S | S | |
| 409 | S | S | N/S | S | |
| 432 | I | I | V | V | |
| 450 | V | V/F | I/V | I | |
| 476 | A | A | A | T | |
| 532 | L | F | F | F | |
| 626 | K | K | K | R | |
| PA-X | 213 | G | G | S | S |
| PB1 | 114 | V | V | I | I |
| 119 | V | V | V | M | |
| 154 | D | D | G | G | |
| 198 | K | R | R | R | |
| 221 | A | A | T/A | T | |
| 317 | M | M | I | I | |
| 329 | Q | Q | Q | R | |
| 377 | D | D | D | E | |
| 618 | E | E | E | D | |
| 738 | E | E/D | D | D | |
| PB1-F2 | 4 | G | G | G | E |
| 18 | T | T | I | I | |
| 21 | R | R | K | R | |
| 41 | R | R | H | H | |
| 79 | R | R | R | Q | |
| PB2 | 12 | S | S | L | L |
| 105 | T | T | T | A | |
| 251 | R | R/K | R/K | K | |
| 344 | V | M | M | M | |
| 511 | V | I | I | I | |
| 590 | G | S | S | S |
In PA-X protein, amino acid substitutions at positions 62, 64, 86, & 158 were similar to what was observed in complete PA protein (Additional file 1: Table S1b). However, one specific consensus point mutation (G213S) was observed in Florida clade 1 & 2 isolates from 2002 onwards except A/equine/Cheshire/06 & A/equine/Linconshire/06 isolates. However, a non-sense point mutation was observed at position 210 in A/equine/Richmond/07 isolate. These results in a truncated PA-X protein, which was reported previously [54].
In PB1 protein, lineage - specific amino acid changes were found (Table 1 and Additional file 1: Table S1c). Four point mutations (V119 M, Q329R, D377E & E618D) were observed subsequently in all Florida clade 2 isolates from Europe (A/equine/Richmond/07) and Asia (A/equine/Xinjiang/07). Furthermore, four consensus amino acid changes (V114I, D154G, A221T & M317I) were noticed in EIVs of Florida sublineage clades (1&2) from 1993/2002 onwards.
In the PB1-F2 protein, two point mutations (T18I in A/equine/Newmarket/1/93 & R41H in A/equine/New York/99) appeared during 1993–99 and were subsequently observed in all Florida clade 1 & 2 isolates (Additional file 1: Table S1d). However, a Florida clade 1 specific mutation (R21K) was continuously observed in EIVs from 2003 onwards. Furthermore, two amino acid substitutions (G4E & R79Q), first observed during the 2007 outbreaks in Europe (A/equine/Richmond/07) and Asia (A/equine/Almaty/07 & A/equine/Xinjiang/07) were subsequently found in all Florida clade 2 isolates except A/equine/Perthshire/09.
In PB2 protein, two consensus amino acid changes (T105A & R251K) were observed in Florida clade 2 lineage (Table 1) wherein, R251K substitution started in Athens/03 isolate (data not shown) and other substitution (T105A) found in isolates subsequently from 2007 outbreak in Europe (A/equine/Richmond/07) and Asia (A/equine/Xinjiang/07). Three amino acid substitutions (V344 m, V511I & G590S) were found consistently in EIVs from 1985 onwards. All Indian isolates had two unique amino acid substitutions (V606I & R380K) (Additional file 1: Table S1e).
Phylogenetic analysis
The phylograms of polymerase genes broadly showed that the segment 3 (PA) is more evolutionarily divergent as compared to segment 1 (PB2) & 2 (PB1). The phylogram of PA gene divided EIVs circulating globally into four major groups: Florida clade (1& 2), American, Eurasian and Pre-divergent viruses (Fig. 1) corresponding to those observed in the HA1gene phylogram [5]. The topogram of PA tree also revealed time scale specific grouping of EIVs, wherein A/equine/Newmarket/1/93 was the closest sister lineage of Florida clade-1 & 2 lineages and A/equine/Newmarket/5/03 was the parent isolate of the Florida clade 2 sub-lineage (Fig. 1). Furthermore, in Florida clade 2 lineage, from A/equine/Richmond/07 onwards, EIVs formed a separate group along with 2007 isolates from China (A/equine/Xinjiang/07) and Kazakhstan (A/equine/Almaty/07). The PA-X phylogenetic tree depicted a similar pattern of EIV H3N8 grouping into four major groups as observed for PA gene (Additional file 2). However, two exceptions were found, where A/equine/Shropshire/10 isolate grouped close to A/equine/Newmarket/5/03 in Florida clade 2 lineage and A/equine/Snailwell/98 isolate clustered between A/equine/Newmarket/1/93 and other Florida clade 1 members.
Fig. 1.
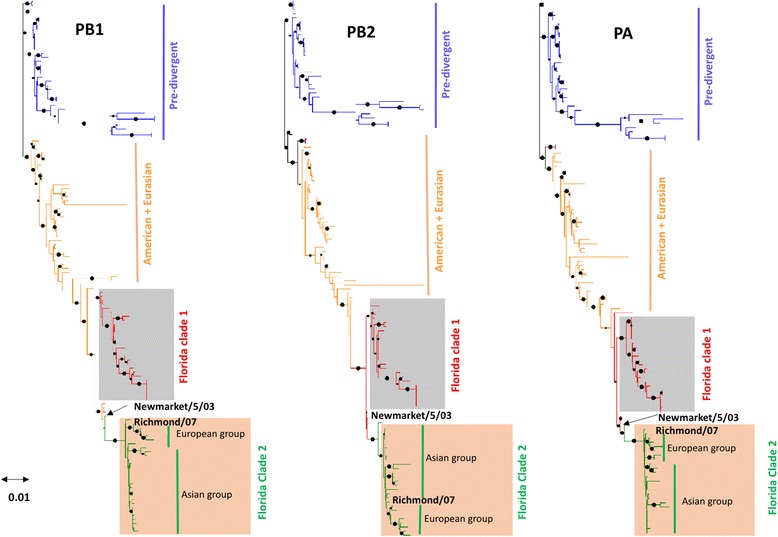
Phylogenetic analyses of polymerase genes of EIVs. The trees were generated by the maximum likelihood model implemented in the software MEGA 5.0. The reliability of the trees was assessed by bootstrap with 1000 replications. The bootstrap values higher than 0.7 are highlighted with solid black dots. The distinct Florida clade 1 and clade 2 sublineages of EIVs have been represented by colored boxes
The phylogram of PB1 gene divided EIV H3N8 into Florida clade 1 & 2, America + Eurasian and Pre-divergent lineages (Fig. 1). The EIVs from A/equine/Richmond/07 onwards were divided into two separate groups – the isolates from Asian countries including China, Kazakhstan & India formed one group and European isolates formed other group. The phylogeny of PB1 also revealed that A/equine/Newmarket/1/93 isolate although clustered separately with A/equine/Cheshire/06, but broadly grouped under Eurasian lineage (Fig. 1). The evolutionary pattern of PB1-F2 gene was different from the PB1 gene, where EIVs grouped into Florida clade 2, Florida clade 1+ clade 2, American, Eurasian and pre-divergent (Additional file 2). The A/equine/Richmond/07 isolate clustered closely with recent European isolates and A/equine/Newmarket/5/03 isolate grouped with Florida clade 1 viruses.
The phylogram of PB2 gene classified EIVs into separate groups - Florida clade 1 & 2, American + Eurasian and Pre-divergent, however A/equine/Newmarket/5/03 clustered in between Florida clade 1 and American lineages (Fig. 1). In Florida clade 2 lineage, A/equine/Richmond/07 isolate clustered separately from all other isolates. Similar to PB1 gene, EIVs in Florida clade 2 lineage again subdivided into two separate sub-groups from A/equine/Richmond/07 onwards. The Asian isolates from China, Kazakhstan & India formed one sub-group and European isolates formed other sub-group within Florida clade 2.
Nucleotide composition affects the codon usage bias
The influence of compositional constraints on the codon usage was assessed by computing the nucleotide compositions of the polymerase genes of EIV and correlating these with the principal axes generated on correspondence analysis (COA) which is used to study the trends in codon usage variations. The mean compositions (%) of nucleotides A (PA = 34.27 ± 0.36, PB1 = 35.10 ± 0.42, PB2 = 34.58 ± 0.48) and G (PB1 = 22.47 ± 0.39, PB2 = 24.50 ± 0.44, U in PA = 24.0 ± 0.31) were highest, and C (%) being the lowest (PA = 18.63 ± 0.44, PB1 = 20.37 ± 0.24, PB2 = 18.94 ± 0.19). The nucleotides at the third positions of synonymous codons (A3, G3, U3, and C3) showed slight variations in the compositional trends. The mean values of A3 (PA = 32.24 ± 0.84, PB1 = 33.09 ± 1.33, PB2 = 36.44 ± 1.35) continued to be highest. Other nucleotides (G3, U3, and C3) in PA and PB2 showed similar trends as their individual nucleotides, while PB1 displayed almost analogous mean values for C3 (22.44 ± 0.67), G3 (22.30 ± 1.21), and U3 (22.17 ± 0.54).
Moreover, correlation of different nucleotide compositions with the two principal axes of COA was performed. In the case of PB1, axis 1 has distinct positive correlation with G3 (r = 0.959, p < 0.0001), GC3s (r = 0.954, p < 0.0001) and negative correlation with A3 (r = −0.956, p < 0.0001). Furthermore, the Enc (Effective number of codons) values were computed to assess the degree of codon usage bias in the polymerase coding sequences of EIV. There is significant positive correlation between ENc and GC3s (r = 0.613, p < 0.0001), while ENc has positive (r = 0.668, p < 0.0001) and negative (r = −0.678, p < 0.0001) correlation with axis 1 and 2, respectively. The case of PB2 is quite different, where axis 1 has distinct positive correlation with A3 (r = 0.852, p < 0.0001) and distinct negative correlation with G3 (r = −0.871, p < 0.0001), C3 (r = −0.821, p < 0.0001), GC3s (r = −0.880, p < 0.0001), and ENc (r = −0.840, p < 0.0001). Similarly, axis 2 also showed distinct correlations with these nucleotide compositions in an opposite polarity pattern (Additional file 3). In the case of PA, axis 1 has distinct positive correlation with A3 (r = 0.910, p < 0.0001), and distinct negative correlation with C3 (r = −0.892, p < 0.0001), GC3s (r = −0.840, p < 0.0001), ENc (r = −0.659, p < 0.0001). These results demonstrate that compositional constraints indeed affect codon usage bias in all the polymerase genes of EIV, but to a different extent.
Relative synonymous Codon usage (RSCU) analysis
The RSCU analysis determines the patterns of synonymous codons usage without the confounding influence of amino acid compositions. We analyzed RSCU values of polymerase genes of EIVs and compared them with their clinical host species (Table 2). The RSCU values of the majority of preferred and non-preferred codons fell between 0.6 and 1.6. Amongst the 18 most frequently used codons in EIVs, fourteen in PA genes (eight A-ended; six U-ended), thirteen in PB1 (nine A-ended; four U-ended) and twelve codons in PB2 (eight A-ended; four U-ended) were A/U-ended, while remaining were G/C-ended codons. We furthermore analyzed the over- and under-represented codons and found that almost all the over-represented codons (RSCU ≥1.6) in PA [CCA (Pro), ACA (Thr), CGA (Arg), GGA (Gly)]; PB1 [CCA (Pro), ACA (Thr), GCA (Ala), CGA/CGG (Arg)] and PB2 [CCA (Pro), ACA (Thr), GCA (Ala), UCA (Ser), UGC (Cys), GGA (Gly)] were A-ended, while nearly all the under-represented codons (RSCU ≤0.6) were G-ended except CCU (Pro), CGU (Arg), UGU (Cys) in PA; CGU (Arg) in PB1 and UUA (Leu), UCU (Ser), UGU (Cys) in PB2 genes. Furthermore, the RSCU values of 59 sense codon from all the polymerase genes of EIV were compared with RSCU values of their host species- horse, donkey and dog. None of the over-represented codon in any of the polymerase genes was common to their host species. Nevertheless, five codons [CCG (Pro), ACG (Thr), GCG (Ala), UCG (Ser) and CGU (Arg)] were under-represented in both polymerase genes and their host species. There were some differences in the RSCU values of polymerase genes but the overall trend is somewhat similar (Additional file 4).
Table 2.
The Relative synonymous codon usage (RSCU) patterns of polymerase genes of EIVs and their host species
| Amino acid | Codon | PB2 | PB1 | PA | Horse | Donkey | Dog | Amino acid | Codon | PB2 | PB1 | PA | Horse | Donkey | Dog |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Phe | UUU | 0.98 | 0.63 | 1.22 | 0.83 | 0.88 | 0.82 | Ser | UCA | 2.2 | 1.58 | 1.18 | 0.80 | 0.83 | 0.81 |
| UUC | 1.02 | 1.38 | 0.77 | 1.17 | 1.12 | 1.16 | UCG | 0.43 | 0.17 | 0.15 | 0.34 | 0.24 | 0.38 | ||
| Leu | UUA | 0.55 | 0.72 | 1.08 | 0.33 | 0.24 | 0.35 | AGU | 0.76 | 0.68 | 1.25 | 0.86 | 1.10 | 0.89 | |
| UUG | 1.45 | 1.29 | 0.92 | 0.72 | 0.82 | 0.68 | AGC | 1.24 | 1.32 | 0.74 | 1.48 | 1.27 | 1.56 | ||
| CUU | 1.16 | 0.65 | 1.35 | 0.73 | 0.80 | 0.67 | Arg | AGA | 1.4 | 1.57 | 1.13 | 1.30 | 1.49 | 1.20 | |
| CUC | 0.88 | 0.79 | 1.05 | 1.32 | 1.63 | 1.25 | AGG | 0.6 | 0.43 | 0.87 | 1.32 | 1.86 | 1.32 | ||
| CUA | 0.95 | 1.11 | 0.66 | 0.34 | 0.18 | 0.37 | CGU | 0.63 | 0.00 | 0.57 | 0.55 | 0.49 | 0.46 | ||
| CUG | 1 | 1.44 | 0.92 | 2.56 | 2.33 | 2.45 | CGC | 0.99 | 0.48 | 1.13 | 1.15 | 0.79 | 1.26 | ||
| Ile | AUU | 1.31 | 0.91 | 1.24 | 0.92 | 0.58 | 0.96 | CGA | 0.98 | 1.91 | 2.1 | 0.61 | 0.74 | 0.67 | |
| AUC | 0.52 | 0.85 | 0.83 | 1.66 | 1.95 | 1.61 | CGG | 1.4 | 1.61 | 0.17 | 1.08 | 0.62 | 1.31 | ||
| AUA | 1.18 | 1.24 | 0.93 | 0.42 | 0.47 | 0.45 | Cys | UGU | 0.01 | 1.19 | 0.59 | 0.89 | 0.62 | 0.85 | |
| Val | GUU | 0.76 | 0.91 | 0.96 | 0.60 | 0.64 | 0.58 | UGC | 1.99 | 0.81 | 1.4 | 1.11 | 1.38 | 1.10 | |
| GUC | 0.66 | 1.12 | 0.63 | 1.08 | 1.42 | 1.1 | His | CAU | 1.4 | 1.11 | 0.92 | 0.81 | 0.89 | 0.78 | |
| GUA | 1.18 | 1.08 | 1.04 | 0.35 | 0.29 | 0.42 | CAC | 0.6 | 0.89 | 1.07 | 1.19 | 1.11 | 1.22 | ||
| GUG | 1.4 | 0.89 | 1.34 | 1.97 | 1.65 | 1.98 | Gln | CAA | 1.25 | 1.24 | 1.25 | 0.52 | 0.84 | 0.50 | |
| Pro | CCU | 1.1 | 1.13 | 0.39 | 1.19 | 0.83 | 1.08 | CAG | 0.75 | 0.76 | 0.74 | 1.48 | 1.16 | 1.46 | |
| CCC | 0.96 | 0.75 | 0.85 | 1.38 | 1.60 | 1.47 | Asn | AAU | 1.2 | 1.15 | 0.97 | 0.84 | 0.66 | 0.87 | |
| CCA | 1.79 | 1.66 | 2.16 | 0.97 | 1.06 | 1.05 | AAC | 0.8 | 0.85 | 1.02 | 1.16 | 1.34 | 1.12 | ||
| CCG | 0.14 | 0.47 | 0.58 | 0.45 | 0.51 | 0.51 | Lys | AAA | 1.37 | 1.32 | 1.33 | 0.79 | 0.79 | 0.79 | |
| Thr | ACU | 0.83 | 1.07 | 0.77 | 0.94 | 0.82 | 0.89 | AAG | 0.63 | 0.89 | 0.67 | 1.21 | 1.21 | 1.13 | |
| ACC | 0.93 | 0.85 | 1.11 | 1.58 | 1.77 | 1.58 | Asp | GAU | 1.15 | 1.27 | 1.25 | 0.83 | 0.84 | 0.86 | |
| ACA | 1.85 | 1.89 | 2 | 0.96 | 0.79 | 1.05 | GAC | 0.85 | 0.73 | 0.74 | 1.17 | 1.16 | 1.09 | ||
| ACG | 0.39 | 0.19 | 0.1 | 0.52 | 0.61 | 0.53 | Glu | GAA | 1.36 | 1.34 | 1.16 | 0.76 | 0.84 | 0.79 | |
| Ala | GCU | 0.94 | 1.03 | 1.26 | 1.05 | 1.20 | 1 | GAG | 0.64 | 0.66 | 0.83 | 1.24 | 1.16 | 1.23 | |
| GCC | 0.93 | 0.94 | 0.81 | 1.72 | 1.74 | 1.78 | Gly | GGU | 0.7 | 0.74 | 0.84 | 0.65 | 0.87 | 0.65 | |
| GCA | 1.79 | 1.75 | 1.44 | 0.77 | 0.77 | 0.81 | GGC | 0.67 | 0.5 | 0.49 | 1.43 | 1.42 | 1.45 | ||
| GCG | 0.35 | 0.29 | 0.48 | 0.45 | 0.30 | 0.47 | GGA | 1.99 | 1.53 | 1.7 | 0.95 | 0.85 | 1.02 | ||
| Tyr | UAU | 0.76 | 0.96 | 1.23 | 0.75 | 0.63 | 0.79 | GGG | 0.64 | 1.23 | 0.96 | 0.97 | 0.86 | 1.05 | |
| UAC | 1.24 | 1.04 | 0.76 | 1.25 | 1.37 | 1.15 | |||||||||
| Ser | UCU | 0.53 | 0.68 | 1.38 | 1.09 | 1.10 | 1.09 | ||||||||
| UCC | 0.84 | 1.57 | 1.27 | 1.43 | 1.46 | 1.52 |
Over- (RSCU ≥1.6) and under-represented (RSCU ≤0.6) codons are displayed in bold and italics, respectively
Codon usage bias among polymerase genes varies and is not clade specific
The ENc values were calculated to estimate the magnitude of codon bias in the polymerase genes of EIV H3N8. The mean ENc values of PB1 (53.04 ± 0.63) was highest, followed by PA (52.12 ± 0.55) and PB2 (49.48 ± 0.88). The ENc values of PB2 were significantly lower compared to PB1 and PA (p < 0.0001). Furthermore, the ENc values were analyzed clade wise in each polymerase i.e. in four well defined clusters of EIV H3N8 (Pre-divergent, Eurasian, Florida clade 1 and Florida clade 2). There was no significant differences among these four clusters in a specific polymerase gene (p > 0.05). However, difference between the ENc values of PB1 and PB2 was more pronounced in clade 1 cluster (p < 0.001) compared to other clusters (p < 0.01). Similarly, we observed comparatively higher difference between the ENc values of PA and PB2 in clade 1&2 clusters (p < 0.01) compared to other clusters (p < 0.05).
Mutation bias plays a minor role in the codon usage bias of polymerase genes of EIV H3N8
An ENc-plot is widely used to find whether mutation bias/mutation pressure influence the codon usage bias or not, whereas the Parity rule 2 (PR2) plot estimates the effects of mutation pressure and natural selection on the codon usage. Hence, to examine whether codon usage patterns in the polymerase genes of EIV have been governed by mutation pressure or not, ENc–GC3s and PR2 plots were constructed. Firstly, a PR2 plot was constructed to examine whether the biased codon choices are restricted in highly biased protein-coding genes. For this, the relationship between purines (A and G) and pyrimidines (C and T) in the four fold degenerate codon families (Ala, Arg, Gly, Leu, Pro, Ser, Thr and Val) were determined. This plot showed that AU bias predominated in the fourfold degenerate codon families in all the polymerase genes coding sequences of EIV (Fig. 3). Furthermore elucidation was derived from the ENc–GC3s plot. In this plot, all the polymerase genes of EIV H3N8 strains clustered below the expected ENc curve (Fig. 3). None of the strains of polymerase genes fell on the expected curve, which would have indicated a major role of mutation pressure in the coding sequences of polymerase genes. These under-curve clustering indicated that factors other than mutation pressure played a major role in the codon usage bias of polymerase genes of EIV H3N8.
Fig. 3.
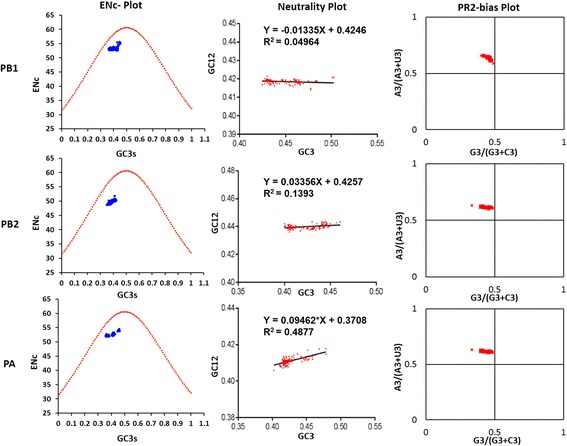
Magnitude of significant determining factors of codon usage bias in polymerase genes of EIVs. ENc-plot: the red dotted line represents the expected curve of positions of strains when the codon usage was only determined by the GC3s composition. Neutrality plot: the red dotted line is the linear regression of GC12 against GC3. PR2 bias plot: AU-bias [A3/(A3 + U3)] at the third codon position of the four-codon amino acids of entire genes were plotted against the GC-bias [G3/(G3 + C3)] and the centre of the plot represents no bias between the influence of the mutation pressure and natural selection
Natural selection predominates in shaping the codon usage patterns of polymerase genes of EIV H3N8
ENc–GC3s plot analysis demonstrated the minor contribution of mutational pressures in shaping the codon usage patterns of the polymerase genes. We next sought to determine the magnitude of natural selection or mutation pressure in generating codon usage bias by constructing the neutrality plots, which determine the mutation-selection equilibrium in shaping the codon usage bias [55]. The distribution range of GC3 was narrow in all the polymerase genes, i.e. PA (40.2 to 47.8%), PB1 (42.5 to 50.3%), and PB2 (40 to 46.1%). There was noticeable correlation between GC1 and GC3 in PA (r = 0.280, p < 0.05) and PB1 (r = 0.610, p < 0.0001), which seemed to indicate the qualitative role of mutation pressure in codon usage bias.
In the neutrality plot analysis, a significant positive correlation was observed between the GC12 and GC3 values of PA (r = 0.61, p < 0.0001) and PB2 (r = 0.29, p = 0.001) genes of the EIV. However, the slopes of the regression line in PA and PB2 were calculated to be 0.0946 and 0.0335, respectively (Fig. 3). This indicates that the influence of direct mutation pressure for codon usage bias in PA and PB2 genes is only 9.46% and 3.35%, respectively. The contribution of natural selection in influencing the codon usage bias was high i.e. 90.54% in PA and 96.65% in PB2 genes. In the PB1 gene of EIV, a negative correlation (−0.332, p < 0.05) was observed, with a slope of −0.0133, and the mutation pressure and natural selection were determined to be 1.33% and 98.67%, respectively, again demonstrating the dominant influence of natural selection. Despite the observed correlation in all the polymerase genes, natural selection emerged as the dominant factor influencing the codon usage bias. Similar results were also obtained when selection pressure was executed on the polymerase genes, with PB1 (dN/dS = 0.082) being under the strongest purifying selection followed by PB2 (dN/dS = 0.129) and PA (dN/dS = 0.147).
Translational selection affects codon usage bias
The influence of translational selection on codon usage bias is usually assessed based on how frequently preferred codons are recognized by the most abundant isoacceptor tRNAs of the host species. The most preferred codons (for each amino acid) were compared with their respective tRNA isotypes in equine cells. This analysis revealed that out of a total of nine, seven two-fold synonymous codon families (Asn, Lys, Asp, Glu, His, Gln, Cys) in PB1 & PB2 and six in PA (Phe, Lys, Asp, Glu, Gln, Tyr) were found to have ‘non-optimal codon–anticodon usage’ (Table 3). The remaining two (Phe and Tyr) had ‘optimal codon–anticodon usage’ in PB1 and PB2. But, these two aromatic amino acid (Phe and Tyr) in PA have their corresponding less frequent tRNA isotypes. We noticed optimal codon–anticodon usage, especially in hydrophobic amino acid (Val, Leu, Ile) of PA which is lacking in PB1 and PB2. Overall findings suggest that PA segment appears to have more adaptability to the tRNA pool of equine cells compared to PB1 and PB2 segments.
Table 3.
Frequency of tRNA genes in equine cells for most preferentially used codons in polymerase genes of EIVs
| AA | Preferentially used codons | tRNA isotypes in equine cells | Total count | ||
|---|---|---|---|---|---|
| PB1 | PB2 | PA | |||
| Ala | GCA | GCA | GCA | AGC (27), GGC (0), CGC (8), TGC (10) | 45 |
| Gly | GGA | GGA | GGA | ACC (0), GCC (10), CCC (8), TCC (5) | 23 |
| Pro | CCA | CCA | CCA | AGG (10), GGG (0), CGG (3), TGG (7) | 20 |
| Thr | ACA | ACA | ACA | AGT (9), GGT (0), CGT (3), TGT (7) | 19 |
| Val | GTC | GTC | GTG | AAC (12), GAC (3), CAC (16), TAC (6) | 37 |
| Ser | TCA | TCA | TCT | AGA (12), GGA (0), CGA (4), TGA (4), ACT (0), GCT (12) | 32 |
| Arg | AGA/CGG | CGA/CGG | CGA | ACG (10), GCG (1), CCG (4), TCG (5), CCT (7), TCT (6) | 33 |
| Leu | TTG | CTG | CTT | AAG (8), GAG (0), CAG (3), TAG (5), CAA (6), TAA (4) | 26 |
| Phe | TTC | TTC | TTT | AAA (0), GAA (13) | 13 |
| Asn | AAT | AAT | AAC | ATT (1), GTT (21) | 22 |
| Lys | AAA | AAA | AAA | CTT (18), TTT (15) | 33 |
| Asp | GAT | GAT | GAT | ATC (1), GTC (10) | 11 |
| Glu | GAA | GAA | GAA | CTC (50), TTC (11) | 61 |
| His | CAT | CAT | CAC | ATG (1), GTG (12) | 13 |
| Gln | CAA | CAA | CAA | CTG (10), TTG (6) | 16 |
| Ile | ATT | ATA | ATT | AAT (20), GAT (0), TAT (4) | 24 |
| Tyr | TAC | TAC | TAT | ATA (1), GTA (14) | 15 |
| Cys | TGC | TGT | TGC | ACA (0), GCA (26) | 26 |
Codons which are likely to be paired with respective anticodon are displayed in bold
Trends in codon usage variations assessed by correspondence analysis
To examine the variations in the synonymous codons usage among the coding sequences of polymerase genes of EIVs, a multivariate statistical method, correspondence analysis (COA) was executed on the RSCU values of complete coding sequences and of codons. The first (ƒ´1) and second (ƒ´2) principal axes accounted for majority of data inertia (PB1: ƒ´1 = 41.4%, ƒ´2 = 15.3%; PB2: ƒ´1 = 40.1%, ƒ´2 = 25.5%; PA: ƒ´1 = 47.7%, ƒ´2 = 19.6%). COA analysis built on RSCU of codons revealed that codons in PB1 and PB2 were frequently distributed along the first (ƒ´1) and second (ƒ´2) principal axes, respectively (Fig. 4), while diffusely distributed in the case of PA. The PB1 and PA gene segments of EIV strains grouped into four defined clusters on both of the principal axes. Moreover, investigation in this type of clustering in PB1 and PB2 revealed that the strains clustered steadily in the format of clusters generated in the phylogenetic analysis, i.e. Pre-divergent, Eurasian, Florida clade 1 and clade 2 clusters (Fig. 4). This was not in the case of PA, where strains did not form these defined clusters and rather frequently distributed along the second (ƒ´2) principal axis.
Fig. 4.
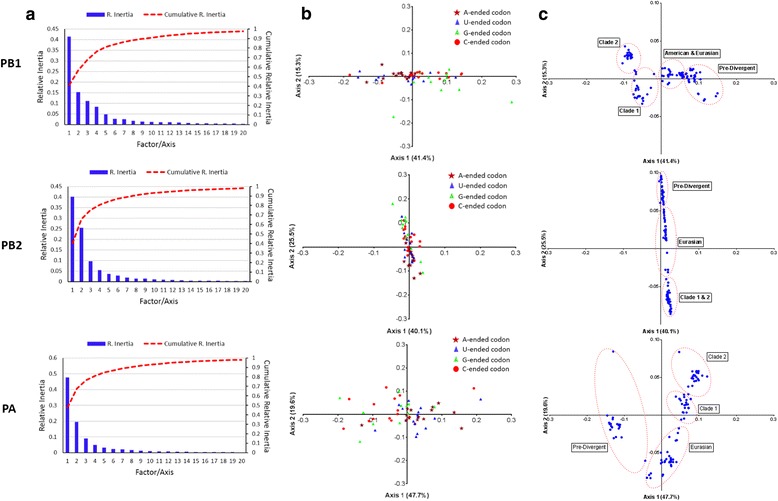
Correspondence analysis (COA) of the synonymous codon usage in EIVs polymerase genes. a COA generated axes contributions. The relative and cumulative inertia of the first 20 factors from a COA of the synonymous codon usage frequencies, (b) COA of the synonymous codon usage towards codons. This analysis was built on the RSCU values of the 59 synonymous codons. The positions of each codon were plotted on the first two-main-dimensional coordinates. Different base ended codons were color labelled. c COA of the synonymous codon usage in coding sequences of polymerase genes of EIV isolates. The positions of each polymerase genes of EIV isolates were plotted on the first two-main-axes
PB2 gene shows highest codon usage deoptimization for Equus caballus
The relative codon deoptimization index (RCDI) compares the similarity in codon usage of a given coding sequence with that of a reference genome. The RCDI values of each polymerase genes of EIV were computed to compare the similarity of the codon usage of these genes and the codon usage of Equus caballus. Mean RCDI values were highest for PB2 (1.43 ± 0.017) followed by PB1 (1.33 ± 0.013) and PA (1.32 ± 0.010) in relation to their clinical host, Equus caballus. Furthermore, the RCDI values were estimated cluster-wise in these polymerase genes to examine whether these variations are due to inherent properties of genes or arose during the course of evolution. We found that Florida clade 1 (PB1 = 1.37 ± 0.015, PB2 = 1.49 ± 0.020) and clade 2 (PB1 = 1.37 ± 0.003, PB2 = 1.47 ± 0.013) isolates displayed higher RCDI values than pre-divergent (PB1 = 1.28 ± 0.015, PB2 = 1.36 ± 0.014) and Eurasian (PB1 = 1.31 ± 0.019, PB2 = 1.38 ± 0.022) isolates in PB1 and PB2 genes (Fig. 5).
Fig. 5.
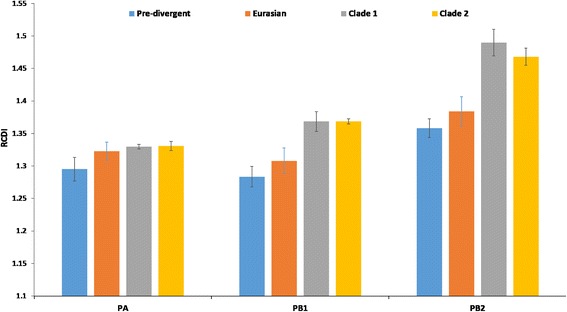
The RCDI analysis of phylogenetic derived clusters of EIVs polymerase genes in relation to their host species, Equus caballus
Equus caballus-induced selection pressure is variable among EIV polymerase genes
A similarity (SiD) analysis was performed to examine whether codon usage patterns of Equus caballus influence the evolution of the codon usage patterns of the polymerase genes coding sequences. We found that Equus caballus exerted more selective pressure on the codon usage patterns of PB1 followed by PB2 and PA (Additional file 5).
Amino acid hydropathicity plays a significant role in codon usage bias rather than aromaticity or genome length
The relationship among the hydropathicity, aromaticity, genome length, ENc and first two principal axes of COA were assessed through Spearman’s rank correlation analysis. Both General average hydropathicity (GRAVY) and aromaticity (AROMO) are indices of amino acid usage, and the variation in amino acid compositions can also influence the results of codon usage analysis. The GRAVY values of PB1 and PB2 had strong negative correlation with ENc (PB1: r = −0.287, p < 0.05; PB2: r = −0.388, p < 0.0001) and GC3s (PB1: r = −0.466, p < 0.0001, PB2: r = −0.393, p < 0.0001) (Additional file 6: Table S6a & S6b). The AROMO values had no significant correlation with ENc or GC3s or principal axes. Furthermore, the GRAVY values of PB1 and PB2 had negative strong correlation with axis 1 (r = −0.429, p < 0.0001) and axis 2 (r = −0.400, p < 0.0001), respectively suggesting the comparatively higher role of hydropathicity in influencing the codon usage bias in PB1. Likewise, in the PA gene segment, both AROMO and GRAVY values had strong significant correlation with ENc (GRAVY: r = 0.492, p < 0.0001, AROMO: r = −0.407, p < 0.0001), GC3s (GRAVY: r = 0.641, p < 0.0001, AROMO: r = −0.753, p < 0.0001) and principal axis 1 (GRAVY: r = −0.697, p < 0.0001, AROMO: r = 0.689, p < 0.0001) (Additional file 6: Table S6c) suggesting that both hydropathicity and aromaticity significantly influences the codon usage bias in PA gene segment.
Discussion
The polymerase genes of EIVs like other gene segments undergo evolutionary changes. We showed that group - specific consensus amino acid substitutions occurred in polymerase genes among EIVs (H3N8) circulating since 1963, which led to divergence of EIVs. Furthermore, we analyzed codon usage pattern of these genes to understand the factors involved in evolution of EIV and their fitness towards host.
Currently EIVs belonging to clade 1 and 2 of Florida sublineage are circulating globally. Analysis of various isolates representing two lineages showed consistent 14 consensus amino acid changes from 2007 onwards. However, unavailability of sequences of polymerase genes between 2003 and 2007 obscures the exact origin of these changes. Availability of partial sequences of a Greek isolate (A/equine/Athens/03) with some of these mutations indicates their origin some time before [9]. Among polymerase proteins, PB2 is more conserved than the other two subunits (PA & PB1) of the RNP complex and interestingly, few stable point mutations in PB2 were found in all Asian isolates and A/equine/Richmond/07 isolate which are located in the cellular mRNA cap - binding site (318–483 amino acid residues) of this protein [28]. Similarly, the D377E change observed in PB1 of all Florida clade 2 EIVs may lead to alteration of stability of the protein as previously described [56]. An amino acid substitution G70E in PB1-F2 protein, which lies in the region essential for virus mediated apoptosis pathway [57–59], was found to be stable in all clade 2 viruses, but variable in European and Florida clade 1 lineages.
The surveillance of internal genes along with HA1 gene may help in better prediction of evolution pattern [60, 61] and severity of infections [62]. The phylogenetic analyses of the polymerase genes of EIVs circulating globally till 2015 have revealed differing evolutionary patterns (Figs. 1, 2, 3, 4 and 5). The isolates such as A/equine/Wisconsin/03 and A/equine/Newmarket/05/03 which represent clade 1 and clade 2 of Florida sublineage according to HA gene analysis did not group in the respective clades. The PA gene showed more evolutionary divergence and classified EIVs into specific lineages which, barring few isolates as mentioned above, followed the evolutionary pattern of HA1 and corroborate with the earlier analysis of equine influenza [63] and other influenza A viruses in various host animals [64, 65]. Our findings suggest that the divergence of Florida clade 2 sublineage of EIVs circulating in Asian region is occurring not only on the basis of stable point mutations in surface encoding HA protein [66], but also in the RNP complex proteins as well as Matrix (M1 and M2) protein [67]. The point mutations in polymerase complex subunits of influenza A viruses may have implication in host adaptation, virus replication and virulence as supported by several previous research findings [38, 68–70].
Fig. 2.
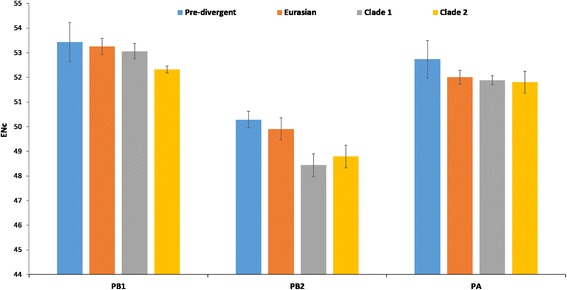
Comparison of phylogenetic derived clusters of EIVs polymerase genes based on their effective number of codon (ENc) values
Various factors shaping the synonymous codon usage bias of the EIV polymerase genes were analyzed systematically. The correspondence analysis was carried out to assess the trends in codon usage variations of polymerase genes. This analysis has previously been used for demonstration of the evolutionary trends and classification of envelope glycoprotein genes or mitochondrial genome [71–74]. This revealed EIV strains to be grouped in well-defined clusters generated in the phylogenetic analysis in respect of PA and PB1 genes, whereas strains did not form defined clusters for PB2 gene.
The overall nucleotide composition affected the codon usage bias especially for codons ending with A/U, which is supported by the fact that over-represented codons (RSCU ≥1.6) were A-ended. Earlier studies on genome based codon usage bias analysis in different influenza viruses specifically H3N8 [52], H5N1 [75], H1N1 and H3N2 [50] also demonstrated a preference towards A- or U-ended codons. Interestingly, none of the over-represented codons in any of the polymerase genes was common to their host species. The ENc values indicated that overall codon usage bias was significantly lower in PB2 (49.48 ± 0.88) as compared to PB1 (53.04 ± 0.63) and PA (52.12 ± 0.55), while the difference was not significant amongst the four clusters of respective polymerase gene. ENc values dropped steadily during the course of evolution from 1963 to current isolates which may reduce competition between the virus and the host for utilizing cellular machinery and thus favour efficient replication of the virus [74, 76]. Similar low codon bias has also been observed in other RNA viruses such as Ebola virus [77], Zika virus [78], H1N1pdm IAV [79]; equine influenza virus H3N8 [52], Foot-and-Mouth Disease Virus [80]; H5N1 influenza virus [75] and equine infectious anaemia virus [81].
However, the low codon bias as revealed by the ENc does not reflect its underlying cause (mutation pressure and/or natural selection). To investigate this ENc–GC3s and PR-2 bias plots were constructed where the under-curve clustering of polymerase genes of EIVs showed that mutation pressure is not the sole factor that influenced the codon usage patterns of the EIV polymerase genes coding sequences. The magnitude of these factors in generating codon usage bias as determined by the neutrality plots, which indicated the predominant role of natural selection in influencing the codon usage bias in all the three polymerase genes (highest in PB1) of EIVs. However, previous studies reported that mutational pressure dominated in structuring the codon usage patterns of H1N1 pdm IAV [79], various RNA [82] and DNA viruses [83]. Contrastingly, PB1-F2, a protein encoded by an alternative open reading frame (ORF) of segment 2 of EIV having role in enhanced pathogenicity was governed by multiple factors such as mutation pressure, natural selection and gene length [53]. Host specific mutation bias was detected while comparing the human and avian influenza viruses [84].
The host imposed translational selection pressure also influenced the codon usage bias of the EIV polymerase genes which was assessed by comparing how most preferred codons are recognized by their respective tRNA isotypes in equine cells. Our previous studies showed that overall codon usage of EIVs does not seem to be well adapted to tRNA pool of equine cells [52]. However, more stringent analysis of polymerase genes in this study suggests that PA segment in comparison to PB1 and PB2 appears to have better adaptability to its clinical host, horse especially by utilizing the high copy number tRNA-anticodon for valine, serine leucine, aaparagine, histidine, isoleucine, cysteine in equine cells. Overall, the polymerase genes by virtue of this could capture the more copy of respective tRNA-anticodon in the equine cells and allow the virus to quickly replicate. However, overall adaptability of polymerase genes to tRNA pool of equine cells was moderate. Other determining parameters of natural selection driven codon bias, GRAVY and AROMO values (indices of amino acid usage) were estimated and correlated with two main axes of COA [85]. Interestingly, aromatic amino acid (Phe and Tyr) had ‘optimal codon–anticodon usage’ in PB1 and PB2, but not in PA. Also, both hydropathicity and aromaticity significantly influence the codon usage bias in PA gene segment, but only hydropathicity in both PB1 and PB2 gene segments.
The high RCDI in PB2 as compared to PB1 and PA genes indicates less adaptation to the codon usage towards highly expressed gene in Equus caballus. The increment in RCDI during the course of evolution in PB1 and PB2 furthermore suggests their less adaptability towards the host. This might lead to low replication rate for successful establishment of virus in its host with alternative codon usage patterns [86]. A similarity (SiD) index revealed that Equus caballus exerted more selective pressure on the codon usage patterns of PB1 as compared to PB2 and PA. Earlier studies also estimated low adaptability in HIV-1 [86] and H1N1pdm IAV in human cells [50]. Destite this, HIV-1 modulate the tRNA pool by selectively enhancement of tRNAs translating A-ending codons (86). This suggests that polymerase genes of EIV having moderate adaptability, may modulate tRNA pool and selectively enhance tRNAs translating A-ending codons for maintaining the efficient replication.
Conclusions
The genetic analysis of the three polymerase genes of EIVs in the present study and matrix gene in our previous study revealed that clade 2 EIVs underwent major changes which led to emergence of a phylogenetically distinct group of EIVs originating from Richmond/1/07 (other sequences being unavailable). These consensus amino acid changes especially in clade 2 EIVs may have repercussions on pathologenicity and virulence of EIVs and needs further studies. The polymerase genes of EIVs exhibit a weak codon usage bias. Multiple factors such as nucleotide composition, mutation pressure, aromaticity and hydropathicity influenced the synonymous codon usage bias in polymerase genes, with natural selection being the major influencing factor.
Methods
Viruses and sequencing
The equine influenza virus (EIV) H3N8 isolates of Florida clade 2 sub-lineage viz. A/equine/Katra-Jammu/06/08, A/equine/Mysore/01/08 and A/equine/Ahmedabad/1/09 isolated by our laboratory from EI epizootic (2008–09) in India [66], were included in the study. The isolates were propagated in embryonated hen’s eggs. Viral RNA was extracted from 200 μl of allantoic fluid using a RNA extraction kit (QIAamp(R) Viral RNA Mini Kit; Qiagen, Valencia, CA), cDNA synthesized using Uni12 primer [87] and polymerase genes were amplified using designed primers (Additional file 7). Amplifications of polymerase genes were carried out using 0.5 μl (3 U) of Hot-Start Taq DNA polymerase (Qiagen, Valencia, CA) in a 50 μl reaction volume with cycling conditions of 95 °C for 5 min followed by 34 cycles of 95 °C for 1 min, 52 °C for 1 min, 72 °C for 3 min and final extension at 72 °C for 10 min. All amplicons were cloned into pTZ57R/T vector (MBI Fermentas, Burlington, Canada) and three recombinant plasmids of each polymerase genes were sequenced commercially using Dye Deoxy Terminator Cycle Sequencing strategy from EuroFins Genomics India Pvt. Ltd. Bangalore, India.
Sequence data
The full length nucleotide sequences of three polymerase genes of EIVs across different lineages reported worldwide between 1963 to 2015 were retrieved from the Influenza Virus Resource at the National Center for Biotechnological Information (http://www.ncbi.nlm.nih.gov/genomes/FLU/FLU.html). Few sequences from GISAID (www.gisaid.org) were also included in the present study in consultation with the Animal Health Trust, UK. The data set comprised 387 nucleotide sequences (124 of PA gene, 134 of PB1 gene & 129 of PB2 gene). The details of the EIV strains and accession numbers of sequences used in this study are provided in Additional file 8.
Sequence and phylogenetic analyses
The consensus nucleotide sequences of 3 clones of each polymerase gene of Indian isolates were generated. Deposited in GenBank, NCBI and accession numbers are provided in Table S2. For comparative studies, the nucleotides as well as deduced amino acid sequences of these genes of EIV H3N8 circulating throughout the globe were aligned using ClustalW programme of MEGA5.0 software [88].
Phylogenetic trees of the polymerase genes were inferred using MEGA5.0 software employing the maximum likelihood model using nucleotide sequences including parameters of transitions and transversions of nucleotide substitutions, equal substitution rates among sites but heterogenous rate among lineages and gaps treated by pairwise-deletion [88]. The bootstrap analyses of the phylograms were carried out with 1000 replicates of dataset to determine the robustness of the individual nodes of the tree.
Selection pressure analysis
Site specific selection pressure was estimated on three gene segments (PB1, PB2, and PA) of EIV using the HyPhy software implemented in the Datamonkey webserver [89]. The similar/duplicate sequences were excluded by this server. The best nucleotide substitution models, PA (012212), PB1 (012210), and PB2 (010210) were chosen based on Akaike Information Criterion (AIC) on a defined Neighbor-Joining (NJ) phylogenetic tree. We also performed GARD (Genetic Algorithm for Recombination Detection) to detect any recombination [90]. We compared four models, Single Likelihood Ancestor Counting (SLAC) model, Fixed Effect Likelihood (FEL) model, Mixed Effects Model of Evolution (MEME), and Fast Unbiased Bayesian Approximation (FUBAR) for estimation of sites under selection pressure [91–93]. The ratio of non-synonymous (dN) to synonymous (dS) substitutions per site (x = dN/dS) was estimated to know the strength of selection pressure. The sites with p values <0.1 for SLAC, FEL and MEME models, and a posterior probability >0.90 for FUBAR were accepted as candidates for selection.
Nucleotide composition analysis
The nucleotide compositional parameters were calculated for polymerase genes of each EIV strain. These includes the frequencies of occurrence of each nucleotide (A %, U %, C %, and G %); each nucleotide at the third position of the synonymous codons (A3%, U3%, C3%, and G3%); G + C at the first (GC1), second (GC2), and third codon positions (GC3); G + C at the first and the second positions (GC12). The codons for Met (AUG), Trp (UGG) and termination codons (UAA, UGA, UAG) were removed from the analysis, as they were not expected to contribute in the codon usage bias.
Relative synonymous codon usage (RSCU)
The RSCU values for all the coding sequences of polymerase genes of EIV were calculated to determine the patterns of synonymous codon usage without the confounding influence of amino acid composition. The RSCU value of a codon is measured as the ratio of its observed frequency to its expected frequency given that all codons for a particular amino acid are used equally [89]. RSCU values are not affected by sequence length and amino acid frequency since these factors are eliminated during the computation. The RSCU values were estimated as per equation given below [94]:
Where gij is the observed number of the ith codon for the jth amino acid which has ni kinds of synonymous codons. Synonymous codons showing the RSCU values of <1.0 represent negative codon usage bias, while of 1.0 and >1.0 represent no bias and positive codon usage bias, respectively.
Effective number of codons (ENC)
The ENc values were computed to assess the degree of codon usage bias in the polymerase coding sequences of EIV using the formula given below:
Where F(i = 2,3,4,6) is the mean of Fi values for the i-fold degenerate amino acid. The Fi values were calculated using the formula given below:
Where n is the total number of occurrences of the codons for that amino acid and nj is the total number of occurrences of the jth codon for that amino acid. The ENc values range from 20 to 61 [95]. The ENc value of 20 states an extreme codon usage bias (only one of the possible synonymous codons is used for the corresponding amino acid), while that of 61 states no bias at all (all possible synonymous codons are used equally for the corresponding amino acid). Consequently, the smaller the ENc value, the greater will be the extent of codon usage bias. Altogether, a gene of ENc ≤ 35 is designated to possess strong codon bias [95, 96].
ENc-plot
An ENc-plot is widely employed to see whether mutation bias/mutation pressure influence the codon usage bias or not. In this plot, the ENc values are the ordinate and the GC3s values (frequency of either a guanine or cytosine at the third codon position of the synonymous codons, excluding Met, Trp, and stop codons) are the abscissa [95]. If predicted ENc values lies on or around the standard curve (functional relation between expected ENc and GC3s), then codon usage is constrained only by G + C mutation bias. Else, other factors such as natural selection play significant role in shaping the codon usage bias. Expected ENc values were calculated as below:
Where ‘s’ is the frequency of G + C at the third codon position of synonymous codons (i.e. GC3s).
Parity rule 2 analysis
The Parity rule 2 (PR2) plot was constructed to estimate the effects of mutation pressure and natural selection on the codon usage of EIVs polymerase genes. In this plot, the AU-bias [A3/(A3 + U3)] at the third codon position of the four-codon amino acid of entire coding sequences is the ordinate and the GC-bias [G3/(G3 + C3)] is the abscissa. The center of the plot, where both coordinates crosses at 0.5, denotes no bias between the influence of the mutation pressure and natural selection [97, 98].
Neutrality plot
A neutrality plot was generated where, GC12 is the ordinate and GC3 is abscissa. A plot regression with a slope of 0 (the points positioned on the parallel lines of the abscissa) indicates no effect of directional mutation pressure, while a slope of 1 (the points positioned on the diagonal line) is indicative of complete neutrality [99].
General average hydropathicity (GRAVY) and aromaticity (AROMO)
The GRAVY values were calculated as a sum of the hydropathy values of all the amino acid in the gene product divided by the number of residues in the polymerase gene sequences [100]. These values range from −2 to 2; where positive and negative values are indicative of hydrophobic and hydrophilic proteins, respectively. AROMO value denotes the frequency of aromatic amino acid (Phe, Tyr, Trp). Both GRAVY and AROMO values are indices of amino acid usage, and the variation in amino acid compositions can also influence the results of codon usage analysis.
Relative codon deoptimization index
The relative codon deoptimization index (RCDI) compares the similarity in codon usage of a given coding sequence with that of a reference genome. The RCDI values for the polymerase genes of EIV were computed using web-based RCDI/eRCDI server (http://genomes.urv.es/CAIcal/RCDI/). This server also calculates expected RCDI values for a set of sequences by generating random sequences with similar G + C content and amino acid compositions. The expected RCDI provides a direct threshold value for discerning whether the differences in the RCDI value are statistically significant and arise from the codon preferences or whether they are merely artifacts that arise from internal biases in the G + C composition and/or amino acid composition of the query sequences. The RCDI value of 1 specifies that the virus trails the host codon usage pattern and shows a host-adapted codon usage pattern. Contrariwise, RCDI values higher than 1 indicate the deoptimization of the codon usage patterns of the virus from that of its host(s) [101, 102].
Similarity index
The similarity (SiD) index provides an insight into the influence of the overall codon usage pattern of the host on the formation of the overall codon usage of the virus, and was calculated as follows:
Where R(A,B) is defined as the cosine value of the angle included between the A and B spatial vectors, and represents the degree of similarity between the virus and host overall codon usage patterns. ai is defined as the RSCU value for a specific codon among the 59 synonymous codons of the virus coding sequence. bi is the RSCU value for the same codon in the host. D(A,B) represents the potential effect of the overall codon usage of the host on that of the virus, and its value range from 0 to 1.0 [103].
Statistical analysis
Correspondence analysis (COA), a multivariate statistical method is widely used to study the trends in codon usage variations. In this analysis, the degrees of freedom were condensed to 40 (from 59 synonymous codons) by eliminating the variations caused by the unequal usage of amino acid while generating a correspondence analysis of RSCU [104]. The major trends within the dataset were estimated based on the measurement of relative inertia, and strains arranged according to their positions along the axes of major inertia. COA was performed on the RSCU values of codons and complete coding sequences of polymerase genes. In addition, Spearman’s rank correlation and linear regression analysis were executed by XLSTAT Version 2016 and GraphPad Prism 7.01 (GraphPad Software, San Diego, California, USA).
Software and databases
Nucleotide compositions, relative synonymous codon usage (RSCU), GRAVY and AROMO, and correspondence analysis were calculated using the program CodonW 1.4.2 (by John Peden and available at http://sourceforge.net/projects/codonw/) [105]. The Effective Number of Codons (ENc) for each polymerase gene of EIV strains were computed using INCA2.1 [106]. Codon usage data of horse (Equus caballus), donkey (Equus asinus), and dog (Canis familiaris) was obtained from the codon usage database (available at: http://www.kazusa.or.jp/codon/) [107]. The RCDI was calculated using the approach of Puigbo et al. [80] (available at: http://genomes.urv.es/CAIcal/RCDI/). The frequencies of tRNAs in equine cells were retrieved from the GtRNAdb database [108].
Additional files
Consensus amino acid changes in the predicted PA protein compared to Richmond/1/07; Table S1b. Consensus amino acid changes in the predicted PA-X protein compared to Richmond/1/07; Table S1c. Consensus amino acid changes in the predicted PB1 protein compared to Richmond/1/07; Table S1d. Consensus amino acid changes in the predicted PB1-F2 protein compared to Richmond/1/07; Table S1e. Consensus amino acid changes in the predicted PB2 protein compared to Richmond/1/07. (DOC 742 kb)
PA-X and PB1-F2 phylograms. (TIFF 2690 kb)
Correlation coefficients among the position of genes along the first two major axes with various indexes of PA polymerase genes’ codon usage. (DOCX 33 kb)
Comparison of Relative Synonymous Codon Usage (RSCU) patterns of EIVs polymerase genes with their host species. (TIFF 1828 kb)
The similarity index analysis of of EIVs polymerase genes in relation to their host species, Equus caballus. (TIFF 358 kb)
Correlation analysis among length (amino acid), GRAVY, AROMO, ENC, GC3s, ENc and the first two principle axes of COA of PB1 gene; Table S6b. Correlation analysis among length (amino acid), GRAVY, AROMO, ENC, GC3s, ENc and the first two principle axes of COA of PB2 gene; Table S6c. Correlation analysis among length (amino acid), GRAVY, AROMO, ENC, GC3s, ENc and the first two principle axes of COA of PA gene. (DOCX 18 kb)
Primers used for amplification of polymerase genes of EIV. (DOCX 12 kb)
Details of polymerase gene sequences of Equine Influenza virus (EIV) strains of equid origin used in the study. (DOC 69 kb)
Acknowledgments
We would like to thank technician, Mukesh Chand for his assistance in propagation of equine influenza virus and young professional, Sunita Kundu for her assistance in molecular work. We sincerely thank the anonymous reviewers for their useful comments and suggestions.
Funding
The present study was funded by Indian Council of Agricultural Research, New Delhi, India from an institutional grant. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Availability of data and materials
The sequence data of polymerase genes of equine influenza virus strains used in the analysis was retrieved from the Influenza Virus Resource at the National Center for Biotechnological Information (NCBI, http://www.ncbi.nlm.nih.gov/genomes/FLU/FLU.html). Few sequences from Global Initiative on Sharing All Influenza Data (GISAID, www.gisaid.org) were also included in the present study in consultation with scientist from the Animal Health Trust, UK. The new sequences reported in this study were deposited in GenBank (NCBI) and assigned accession numbers. The accession numbers of the sequences used in the study have been presented in Additional file 8.
Abbreviations
- A3s
A content on the third synonymous codon position
- AROMO
Aromaticity
- C3s
C content on the third synonymous codon position
- COA
Correspondence analysis
- EIV
Equine influenza virus
- ENc
Effective number of codons
- FEL
Fixed effect likelihood model
- FUBAR
Fast unbiased bayesian approximation
- G3s
G content on the third synonymous codon position
- GARD
Genetic algorithm for recombination detection
- GC1
GC content in the first position of the codons
- GC12
Stands for the average value of GC content in the first and second position of the codons
- GC2
GC content in the second position of the codons
- GC3
GC content in the third position
- GC3s
GC content on the third synonymous codon position
- GRAVY
General average hydropathicity
- MEME
Mixed effects model of evolution
- PR2
Parity rule 2 analysis
- RCDI
Relative codon deoptimization index
- RSCU
Relative synonymous codon usage
- SiD
Similarity Index
- SLAC
Single likelihood ancestor counting
Authors’ contributions
Conceived and designed the experiments: BCB, NV, NK. Performed the experiments: BCB, NK, TA, NV, PS. Analyzed the data: NK, BCB, TA, NV, DE, PS. Manuscript writing: NK, BCB, NV, DE, AR, NR, SB, TA, BNT, RS, PS, RKS. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-4063-1) contains supplementary material, which is available to authorized users.
Contributor Information
Bidhan Ch. Bera, Email: bcbpatent@gmail.com
Nitin Virmani, Email: nvirmani@gmail.com.
Naveen Kumar, Email: navyog.yadav84@gmail.com.
Taruna Anand, Email: tarunandri@gmail.com.
S Pavulraj, Email: pavulraj.vet@gmail.com.
Adam Rash, Email: adam.rash@aht.org.uk.
Debra Elton, Email: debra.elton@aht.org.uk.
Nicola Rash, Email: nicola.rash@aht.org.uk.
Sandeep Bhatia, Email: bhatia1967@gmail.com.
Richa Sood, Email: rsood@hsadl.nic.in.
Raj Kumar Singh, Email: rks_virology@gmail.com.
Bhupendra Nath Tripathi, Email: bntripathi1@yahoo.co.in.
References
- 1.Newton JR, Daly JM, Spencer L, Mumford JA. Description of the outbreak of equine influenza (H3N8) in the united kingdom in 2003, during which recently vaccinated horses in Newmarket developed respiratory disease. Vet Rec. 2006;158:185–192. doi: 10.1136/vr.158.6.185. [DOI] [PubMed] [Google Scholar]
- 2.Barbica L, Madica J, Turka N, Dalyb J. Vaccine failure caused an outbreak of equine influenza in Croatia. Vet Microbiol. 2009;133:164–171. doi: 10.1016/j.vetmic.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 3.Steinbrücka L, Klingena TR, McHardya AC. Computational prediction of vaccine strains for human influenza a (H3N2) viruses. J Virol. 2014;88:12123–12132. doi: 10.1128/JVI.01861-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ping J, Lopes TJS, Nidom CA, Ghedin E, Macken CA, Fitch A, Imai M, Maher EA, Neumann G, Kawaoka Y. Development of high-yield influenza a virus vaccine viruses. Nat Commun. 2015;6:8148. doi: 10.1038/ncomms9148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bryant NA, Rash AS, Russell CA, Ross J, Cooke A, Bowman S, MacRae S, Lewis NS, Paillot R, Zanoni R, Meier H, Griffiths LA, Daly JM, Tiwari A, Chambers TM, Newton JR, Elton DM. Antigenic and genetic variations in European and north American equine influenza virus strains (H3N8) isolated from 2006 to 2007. Vet Microbiol. 2009;138:41–52. doi: 10.1016/j.vetmic.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 6.Damiani AM, Scicluna MT, Ciabatti I, Cardeti G, Sala M, Vulcano G, Cordioli P, Martella V, Amaddeo D, Autorino GL. Genetic characterization of equine influenza viruses isolated in Italy between 1999 and 2005. Virus Res. 2008;131:100–105. doi: 10.1016/j.virusres.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 7.Lai AC, Chambers TM, Holland RE, Jr, Morley PS, Haines DM, Townsend HG, Barrandeguy M. Diverged evolution of recent equine-2 influenza (H3N8) viruses in the western hemisphere. Arch Virol. 2001;146:1063–1074. doi: 10.1007/s007050170106. [DOI] [PubMed] [Google Scholar]
- 8.Daly JM, Lai AC, Binns MM, Chambers TM, Barrandeguy M, Mumford JA. Antigenic and genetic evolution of equine H3N8 influenza a viruses. J Gen Virol. 1996;77:661–671. doi: 10.1099/0022-1317-77-4-661. [DOI] [PubMed] [Google Scholar]
- 9.Bountouri M, Ntafis V, Fragkiadaki E, Kanellos T, Xylouri E. Phylogenetic analysis of the five internal genes and evolutionary pathways of the Greek H3N8 equine influenza virus. Nat Sci. 2012;4:839–847. [Google Scholar]
- 10.Kirkland PD, Finlaison DS, Crispe E, Hurt AC. Influenza virus transmission from horses to dogs, Australia. Emerg Infect Dis. 2010;16:699–702. doi: 10.3201/eid1604.091489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Daly JM, Blunden AS, Macrae S, Miller J, Bowman SJ, Kolodziejek J, Nowotny N, Smith KC. Transmission of equine influenza virus to English foxhounds. Emerg Infect Dis. 2008;14:461–464. doi: 10.3201/eid1403.070643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crawford PC, Dubovi EJ, Castleman WL, Stephenson I, Gibbs EP, Chen L, Smith C, Hill RC, Ferro P, Pompey J, Bright RA, Medina MJ, Johnson CM, Olsen CW, Cox NJ, Klimov AI, Katz JM, Donis RO. Transmission of equine influenza virus to dogs. Science. 2005;310:482–485. doi: 10.1126/science.1117950. [DOI] [PubMed] [Google Scholar]
- 13.Tu J, Zhou H, Jiang T, Li C, Zhang A, Guo X, Zou W, Chen H, Jin M. Isolation and molecular characterization of equine H3N8 influenza viruses from pigs in China. Arch Virol. 2009;154:887–890. doi: 10.1007/s00705-009-0381-1. [DOI] [PubMed] [Google Scholar]
- 14.Fodor E. The RNA polymerase of influenza a virus: mechanisms of viral transcription and replication. Acta Virol. 2013;57(2):113–122. doi: 10.4149/av_2013_02_113. [DOI] [PubMed] [Google Scholar]
- 15.Resa-Infante P, Jorba N, Coloma R, Ortin J. The influenza virus RNA synthesis machine: advances in its structure and function. RNA Biol. 2011;8(2):207–215. doi: 10.4161/rna.8.2.14513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jambrina E, Barcena J, Uez O, Portela A. The three subunits of the polymerase and the nucleoprotein of influenza B virus are the minimum set of viral proteins required for expression of a model RNA template. Virology. 1997;235:209–217. doi: 10.1006/viro.1997.8682. [DOI] [PubMed] [Google Scholar]
- 17.Chu C, Fan S, Li C, Macken C, Kim JH, Hatta M, Neumann G, Kawaoka Y. Functional analysis of conserved motifs in influenza virus PB1 protein. PLoS One. 2012;7(5):e36113. doi: 10.1371/journal.pone.0036113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuan P, Bartlam M, Lou Z, Chen S, Zhou J, He X, Lv Z, Ge R, Li X, Deng T, Fodor E, Rao Z, Liu Y. Crystal structure of an avian influenza polymerase PA(N) reveals an endonuclease active site. Nature. 2009;458(7240):909–913. doi: 10.1038/nature07720. [DOI] [PubMed] [Google Scholar]
- 19.Sugiyama K, Obayashi E, Kawaguchi A, Suzuki Y, Tame JR, Nagata K, Park SY. Structural insight into the essential PB1-PB2 subunit contact of the influenza virus RNA polymerase. EMBO J. 2009;28(12):1803–1811. doi: 10.1038/emboj.2009.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He X, Zhou J, Bartlam M, Zhang R, Ma J, Lou Z, Li X, Li J, Joachimiak A, Zeng Z, Ge R, Rao Z, Liu Y. Crystal structure of the polymerase PA(C)-PB1(N) complex from an avian influenza H5N1 virus. Nature. 2008;454(7208):1123–1126. doi: 10.1038/nature07120. [DOI] [PubMed] [Google Scholar]
- 21.Kobayashi M, Toyoda T, Ishihama A. Influenza virus PB1 protein is the minimal and essential subunit of RNA polymerase. Arch Virol. 1996;141(3–4):525–539. doi: 10.1007/BF01718315. [DOI] [PubMed] [Google Scholar]
- 22.Biswas SK, Nayak DP. Mutational analysis of the conserved motifs of influenza a virus polymerase basic protein 1. J Virol. 1994;68:1819–1826. doi: 10.1128/jvi.68.3.1819-1826.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wise HM, Foeglein A, Sun J, Dalton RM, Patel S, Howard W, Anderson EC, Barclay WS, Digard P. A complicated message: identification of a novel PB1-related protein translated from influenza a virus segment 2 mRNA. J Virol. 2009;83:8021–8031. doi: 10.1128/JVI.00826-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen W, Calvo PA, Malide D, Gibbs J, Schubert U, Bacik I, Basta S, O’Neill R, Schickli J, et al. A novel influenza a virus mitochondrial protein that induces cell death. Nat Med. 2001;7:1306–1312. doi: 10.1038/nm1201-1306. [DOI] [PubMed] [Google Scholar]
- 25.Weeks-Gorospe JN, Hurtig HR, Iverson AR, Schuneman MJ, Webby RJ, McCullers JA, Huberb VC. Naturally occurring swine influenza a virus PB1-F2 phenotypes that contribute to Superinfection with gram-positive RespiratoryPathogens. J Virol. 2012;86(17):9035–9043. doi: 10.1128/JVI.00369-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Varga ZT, Ramos I, Hai R, Schmolke M. Garcı’a-Sastre a, Fernandez-Sesma A, Palese P. The influenza virus protein PB1-F2 inhibits the induction of type I interferon at the level of the MAVS adaptor protein. PLoS Pathog. 2011;7:e1002067. doi: 10.1371/journal.ppat.1002067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dias A, Bouvier D, Crépin T, McCarthy AMINOACID, Hart DJ, Baudin F, Cusack S, Ruigrok RWH. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature. 2009;458:914–918. doi: 10.1038/nature07745. [DOI] [PubMed] [Google Scholar]
- 28.Guilligay D, Tarendeau F, Resa-Infante P, Coloma R, Crepin T, Sehr P, Lewis J, Ruigrok RW, Ortin J, Hart DJ, Cusack S. The structural basis for cap binding by influenza virus polymerase subunit PB2. Nat Struct Mol Biol. 2008;15:500–506. doi: 10.1038/nsmb.1421. [DOI] [PubMed] [Google Scholar]
- 29.Fechter P, Mingay L, Sharps J, Chambers A, Fodor E, Brownlee GG. Two aromatic residues in the PB2 subunit of influenza a RNA polymerase are crucial for cap binding. J Biol Chem. 2003;278:20381–20388. doi: 10.1074/jbc.M300130200. [DOI] [PubMed] [Google Scholar]
- 30.Rodriguez A, Perez-Gonzalez A, Nieto A. Influenza virus infection causes specific degradation of the largest subunit of cellular RNA polymerase II. J Virol. 2007;81:5315–5324. doi: 10.1128/JVI.02129-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hara K, Shiota M, Kido H, Ohtsu Y, Kashiwaqi T, Iwahashi J, Hamada N, Mizoue K, Tsumura N, Kato H, Toyoda T. Influenza virus RNA polymerase PA subunit is a novel serine protease with Ser624 at the active site. Genes Cells. 2001;6:87–97. doi: 10.1046/j.1365-2443.2001.00399.x. [DOI] [PubMed] [Google Scholar]
- 32.Regan JF, Liang Y, Parslow TG. Defective assembly of influenza a virus due to a mutation in the polymerase subunit PA. J Virol. 2006;80:252–261. doi: 10.1128/JVI.80.1.252-261.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jagger BW, Wise HM, Kash JC, Walters KA, Wills NM, Xiao YL, Dunfee RL, Schwartzman LM, Ozinsky A, Bell GL, Dalton RM, Lo A, Efstathiou S, Atkins JF, Firth AE, Taubenberger JK, Digard P. An overlapping protein-coding region in influenza a virus segment 3 modulates the host response. Science. 2012;337:199–204. doi: 10.1126/science.1222213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hayashi T, MacDonald LA, Takimoto T. Influenza a virus protein PA-X contributes to viral growth and suppression of the host antiviral and immune responses. J Virol. 2015;89:6442–6452. doi: 10.1128/JVI.00319-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Desmet EA, Bussey KA, Stone R, Takimoto T. Identification of the N-terminal domain of the influenza virus PA responsible for the suppression of host protein synthesis. J Virol. 2013;87:3108–3118. doi: 10.1128/JVI.02826-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ping J, Dankar SK, Forbes NE, Keleta L, Zhou Y, Tyler S, Brown EG. PB2 and hemagglutinin mutations are major determinants of host range and virulence in mouse-adapted influenza a virus. J Virol. 2010;84(20):10606–10618. doi: 10.1128/JVI.01187-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Naffakh N, Tomoiu A, Rameix-Welti MA, van der Werf S. Host restriction of avian influenza viruses at the level of the ribonucleoproteins. Annu Rev Microbiol. 2008;62:403–424. doi: 10.1146/annurev.micro.62.081307.162746. [DOI] [PubMed] [Google Scholar]
- 38.Rodriguez-Frandsen A, Alfonso R, Nieto A. Influenza virus polymerase: functions on host range, inhibition of cellular response to infection and pathogenecity. Virus Res. 2015;209:23–38. doi: 10.1016/j.virusres.2015.03.017. [DOI] [PubMed] [Google Scholar]
- 39.Zhang H, Li X, Guo J, Li L, Chang C, Li Y, Bian C, Xu K, Chen H, Sun B. The PB2 E627 K mutation contributes to the high polymerase activity and enhanced replication of H7N9 influenza virus. J Gen Virol. 2014;95:779–786. doi: 10.1099/vir.0.061721-0. [DOI] [PubMed] [Google Scholar]
- 40.Linster M, van Boheemen S, de Graaf M, Schrauwen EJ, Lexmond P, Manz B, Bestebroer TM, Baumann J, van Riel D, Rimmelzwaan GF, Osterhaus AD, Matrosovich M, Fouchier RA, Herfst S. Identification, characterization, and natural selection of mutations driving airborne transmission of a/H5N1 virus. Cell. 2014;157(2):329–339. doi: 10.1016/j.cell.2014.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mehle A, Dugan VG, Taubenberger JK, Doudna JA. Reassortment and mutation of the avian influenza virus polymerase PA subunit overcome species barriers. J Virol. 2012;86(3):1750–1757. doi: 10.1128/JVI.06203-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Almond JW. A single gene determines the host range of influenza virus. Nature. 1977;270(5638):617–618. doi: 10.1038/270617a0. [DOI] [PubMed] [Google Scholar]
- 43.Pepin KM, Domsic J, McKenna R. Genomic evolution in a virus under specific selection for host recognition. Infect Genet Evol. 2008;8(6):825–834. doi: 10.1016/j.meegid.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 44.Chen H, Sun S, Norenburg JL, Sundberg P. Mutation and selection cause codon usage and bias in mitochondrial genomes of ribbon worms (Nemertea) PLoS One. 2014;9(1):e85631. doi: 10.1371/journal.pone.0085631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grantham R, Gautier C, Gouy M, Mercier R, Pave A. Codon catalog usage and the genome hypothesis. Nucleic Acids Res. 1980;8:49–62. doi: 10.1093/nar/8.1.197-c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Agashe D, Gomez NCM, Drummond DA, Marx CJ. Good codons, bad transcript: large reductions in gene expression and fitness arising from synonymous mutations in a key enzyme. Mol Biol Evol. 2013;30:549–560. doi: 10.1093/molbev/mss273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Plotkin JB, Kudla G. Synonymous but not the same: the causes and consequences of codon bias. Nat Rev Genet. 2011;12:32–42. doi: 10.1038/nrg2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hershberg R, Petrov DA. Selection on codon bias. Annu Rev Genet. 2008;42:287–299. doi: 10.1146/annurev.genet.42.110807.091442. [DOI] [PubMed] [Google Scholar]
- 49.Parmley JL, Hurst LD. How do synonymous mutations affect fitness? Bioessays. 2007;29(6):515–519. doi: 10.1002/bies.20592. [DOI] [PubMed] [Google Scholar]
- 50.Goñi N, Iriarte A, Comas V, Soñora M, Moreno P, Moratorio G, et al. Pandemic influenza a virus codon usage revisited: biases, adaptation and implications for vaccine strain development. Virol J. 2012;9:263. doi: 10.1186/1743-422X-9-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu X, Wu C, Chen AYH. Codon usage bias and recombination events for neuraminidase and hemagglutinin genes in Chinese isolates of influenza a virus subtype H9N2. Arch Virol. 2010;155:685–693. doi: 10.1007/s00705-010-0631-2. [DOI] [PubMed] [Google Scholar]
- 52.Kumar N, Bera BC, Greenbaum BD, Bhatia S, Sood R, Selvaraj P, Anand T, Tripathi BN, Virmani N. Revelation of influencing factors in overall Codon usage bias of equine influenza viruses. PLoS One. 2016;11(4):e0154376. doi: 10.1371/journal.pone.0154376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lu G, Guo W, Qi T, Ma J, Zhao S, Tian Z, Pan J, Zhu C, Wang X, Xiang W. Genetic analysis of the PB1-F2 gene of equine influenza virus. Virus Genes. 2013. doi:10.1007/s11262-013-0935-x. [DOI] [PubMed]
- 54.Rash A, Woodward A, Bryant N, McCauley J, Elton D. An efficient genome sequencing method for equine influenza [H3N8] virus reveals a new polymorphism in the PA-X protein. Virol J. 2014;11:159. doi: 10.1186/1743-422X-11-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sharp PM, Li WH. An evolutionary perspective on synonymous codon usage in unicellular organisms. J Mol Evol. 1986;24:28–38. doi: 10.1007/BF02099948. [DOI] [PubMed] [Google Scholar]
- 56.Lee DY, Kim KA, Yu YG, Kim KS. Substitution of aspartic acid with glutamic acid increases the unfolding transition temperature of a protein. Biochem Biophys Res Commun. 2004;320(3):900–906. doi: 10.1016/j.bbrc.2004.06.031. [DOI] [PubMed] [Google Scholar]
- 57.Gibbs JS, Malide D, Hornung F, Bennink JR, Yewdell JW. The influenza a virus PB1-F2 protein targets the inner mitochondrial membrane via a predicted basic amphipathic helix that disrupts mitochondrial function. J Virol. 2003;77:7214–7224. doi: 10.1128/JVI.77.13.7214-7224.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yamada H, Chounan R, Higashi Y, Kurihara N, Kido H. Mitochondrial targeting sequence of the influenza a virus PB1-F2 protein and its function in mitochondria. FEBS Lett. 2004;578:331–336. doi: 10.1016/j.febslet.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 59.Krumbholz A, Philipps A, Oehring H, Schwarzer K, Eitner A, Wutzler P, Zell R. Current knowledge on PB1-F2 of influenza a viruses. Med Microbiol Immunol. 2011;200:69–75. doi: 10.1007/s00430-010-0176-8. [DOI] [PubMed] [Google Scholar]
- 60.Blazejewska P, Koscinski L, Viegas N, Anhlan D, Ludwig S, Schughart K. Pathogenicity of different PR8 influenza a virus variants in mice is determined by both viral and host factors. Virology. 2011;412:36–45. doi: 10.1016/j.virol.2010.12.047. [DOI] [PubMed] [Google Scholar]
- 61.Schnitzler SU, Schnitzler P. An update on swine-origin influenza virus a/H1N1: a review. Virus Genes. 2009;39:279–292. doi: 10.1007/s11262-009-0404-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bogs J, Veits J, Gohrbandt S, Hundt J, Stech O, Breithaupt A, Teifke JP, Mettenleiter TC, Stech J. Highly pathogenic H5N1 influenza viruses carry virulence determinants beyond the polybasic hemagglutinin cleavage site. PLoS One. 2010;5:e11826. doi: 10.1371/journal.pone.0011826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Murcia PR, Wood JL, Holmes EC. Genome-scale evolution and Phylodynamics of equine H3N8 influenza a virus. J Virol. 2011;85(11):5312–5322. doi: 10.1128/JVI.02619-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gorman OT, Bean W, Kawaoka Y, Webster R. Evolution of the nucleoprotein gene of influenza a virus. J Virol. 1990;64:1487–1497. doi: 10.1128/jvi.64.4.1487-1497.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gorman OT, Donis R, Kawaoka Y, Webster R. Evolution of influenza a virus PB2 genes: implications for evolution of the ribonucleoprotein complex and origin of human influenza a virus. J Virol. 1990;64:4893–4902. doi: 10.1128/jvi.64.10.4893-4902.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Virmani N, Bera BC, Singh BK, Shanmugasundaram K, Gulati BR, Barua S, Vaid RK, Gupta AK, Singh RK. Equine influenza outbreak in India (2008–09): virus isolation, sero-epidemiology and phylogenetic analysis of HA gene. Vet Microbiol. 2010;143:224–237. doi: 10.1016/j.vetmic.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 67.Virmani N, Bera BC, Shanumugasundaram K, Singh BK, Gulati BR, Singh RK. Genetic analysis of the matrix and non-structural genes of equine influenza virus (H3N8) from epizootic of 2008–09 in India. Vet Microbiol. 2011;152:169–175. doi: 10.1016/j.vetmic.2011.04.011. [DOI] [PubMed] [Google Scholar]
- 68.Fodor E, Crow M, Mingay LJ, Deng T, Sharps J, Fechter P, Brownlee GG. A single amino acid mutation in the PA subunit of the influenza virus RNA polymerase inhibits endonucleolytic cleavage of capped RNAs. J Virol. 2002;76:8989–9001. doi: 10.1128/JVI.76.18.8989-9001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Puthavathana P, Auewarakul P, Charoenying PC, Sangsiriwut K, Pooruk P, Boonnak K. Molecular characterization of the complete genome of human influenza H5N1 virus isolates from Thailand. J Gen Virol. 2005;86:423–433. doi: 10.1099/vir.0.80368-0. [DOI] [PubMed] [Google Scholar]
- 70.Zhu W, Zhu Y, Qin K, Yu Z, Gao R, Yu H, Zhou J, Shu Y. Mutations in polymerase genes enhanced the virulence of 2009 pandemic H1N1 influenza virus in mice. PLoS One. 2012;7(3):e33383. doi: 10.1371/journal.pone.0033383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Smith GJD, Vijaykrishna D, Bahl J, Lycett SJ, Worobey M, Pybus OG, Ma SK, Cheung CL, Raghwani J, Bhatt S, Peiris JSM, Guan Y, Rambaut A. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza a epidemic. Nature. 2009;459(7250):1122–1125. doi: 10.1038/nature08182. [DOI] [PubMed] [Google Scholar]
- 72.Wong E, Smith DK, Rabadan R, Peiris M, Poon L. Codon usage bias and the evolution of influenza a viruses. Codon usage biases of influenza virus. BMC Evol Biol. 2010;10:253. doi: 10.1186/1471-2148-10-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhao Y, Zheng H, Xu A, Yan D, Jiang Z, Qi Q, Sun J. Analysis of codon usage bias of envelope glycoprotein genes in nuclear polyhedrosis virus (NPV) and its relation to evolution. BMC Genomics. 2016;17:677. doi: 10.1186/s12864-016-3021-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wei L, He J, Jia X, Qi Q, Liang Z, Zheng H, Ping Y, Liu S, Sun J. Analysis of codon usage bias of mitochondrial genome in Bombyx Mori and its relation to evolution. BMC Evol Biol. 2014;14:262. doi: 10.1186/s12862-014-0262-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhou T, Gu W, Ma J, Sun X, Lu Z. Analysis of synonymous codon usage in H5N1 virus and other influenza a viruses. Biosystems. 2005;81:77–86. doi: 10.1016/j.biosystems.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 76.Jenkins GM, Holmes EC. The extent of codon usage bias in human RNA viruses and its evolutionary origin. Virus Res. 2003;92:1–7. doi: 10.1016/S0168-1702(02)00309-X. [DOI] [PubMed] [Google Scholar]
- 77.Cristina J, Moreno P, Moratorio G, Musto H. Genome-wide analysis of codon usage bias in Ebolavirus. Virus Res. 2015;196:87–93. doi: 10.1016/j.virusres.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 78.Butt AM, Nasrullah I, Qamar R, Tong Y. Evolution of codon usage in Zika virus genomes is host and vector specific. Emerg Microbes Infect. 2016;5(10):e107. doi: 10.1038/emi.2016.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Anhlan D, Grundmann N, Makalowski W, Ludwig S, Scholtissek C. Origin of the 1918 pandemic H1N1 influenza a virus as studied by codon usage patterns and phylogenetic analysis. RNA. 2011;17:64–73. doi: 10.1261/rna.2395211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhong J, Li Y, Zhao S, Liu S, Zhang Z. Mutation pressures shapes codon usage in the GC-rich genome of foot-and-mouth disease virus. Virus Genes. 2007;35:767–776. doi: 10.1007/s11262-007-0159-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yin X, Lin Y, Cai W, Wei P, Wang X. Comprehensive analysis of the overall codon usage patterns in equine infectious anemia virus. Virol J. 2013;10:356. doi: 10.1186/1743-422X-10-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cheng X, Virk N, Chen W, Ji S, Ji S, et al. CpG usage in RNA viruses: data and hypotheses. PLoS One. 2013;8:e74109. doi: 10.1371/journal.pone.0074109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shackelton LA, Parrish CR, Holmes EC. Evolutionary basis of codon usage and nucleotide composition bias in vertebrate DNA viruses. J Mol Evol. 2006;62:551–563. doi: 10.1007/s00239-005-0221-1. [DOI] [PubMed] [Google Scholar]
- 84.Rabadan R, Levine AJ, Robins H. Comparison of avian and human influenza a viruses reveals a mutational bias on the viral genomes. J Virol. 2006;80:11887–11891. doi: 10.1128/JVI.01414-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tao P, Dai L, Luo M, Tang F, Tien P, Pan Z. Analysis of synonymous codon usage in classical swine fever virus. Virus Genes. 2009;38:104–112. doi: 10.1007/s11262-008-0296-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.van Weringh A, Ragonnet-Cronin M, Pranckeviciene E, Pavon-Eternod M, Kleiman L, Xia X. HIV-1 modulates the tRNA pool to improve translation efficiency. Mol Biol Evol. 2011;28:1827–1834. doi: 10.1093/molbev/msr005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hoffmann E, Stech J, Guan Y, Webster RG, Perez DR. Universal primer set for the full-length amplification of all influenza a viruses. Arch Virol. 2001;146(12):2275–2289. doi: 10.1007/s007050170002. [DOI] [PubMed] [Google Scholar]
- 88.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pond SLK, Frost SDW. Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics. 2005;21(10):2531–2533. doi: 10.1093/bioinformatics/bti320. [DOI] [PubMed] [Google Scholar]
- 90.Pond SLK, Posada D, Gravenor MB, Woelk CH, Frost SDW. GARD: a genetic algorithm for recombination detection. Bioinformatics. 2006;22(24):3096–3098. doi: 10.1093/bioinformatics/btl474. [DOI] [PubMed] [Google Scholar]
- 91.Pond SLK, Frost SDW. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol. 2005;22(5):1208–1222. doi: 10.1093/molbev/msi105. [DOI] [PubMed] [Google Scholar]
- 92.Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Pond SLK. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012;8(7):e1002764. doi: 10.1371/journal.pgen.1002764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Murrell B, Moola S, Mabona A, Weighill T, Sheward D, Pond SLK, Scheffler K. FUBAR: A Fast, Unconstrained Bayesian AppRoximation for inferring selection. Mol Biol Evol. 2013. doi:10.1093/molbev/mst030. [DOI] [PMC free article] [PubMed]
- 94.Sharp PM, Li WH. Codon usage in regulatory genes in Escherichia Coli does not reflect selection for ‘rare’ codons. Nucleic Acids Res. 1986;14:7737–7749. doi: 10.1093/nar/14.19.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wright F. The ‘effective number of codons’ used in a gene. Gene. 1990;87:23–29. doi: 10.1016/0378-1119(90)90491-9. [DOI] [PubMed] [Google Scholar]
- 96.Comeron JM, Aguade M. An evaluation of measures of synonymous codon usage bias. J Mol Evol. 1998;47:268–274. doi: 10.1007/PL00006384. [DOI] [PubMed] [Google Scholar]
- 97.Sueoka N. Intra strand parity rules of DNA base composition and usage biases of synonymous codons. J Mol Evol. 1995;40:318–325. doi: 10.1007/BF00163236. [DOI] [PubMed] [Google Scholar]
- 98.Sueoka N. Translation-coupled violation of parity rule 2 in human genes is not the cause of heterogeneity of the DNA G+C content of third codon position. Gene. 1999;238:53–58. doi: 10.1016/S0378-1119(99)00320-0. [DOI] [PubMed] [Google Scholar]
- 99.Sueoka N. Directional mutation pressure and neutral molecular evolution. Proc Natl Acad Sci U S A. 1988;85:2653–2657. doi: 10.1073/pnas.85.8.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157(1):105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 101.Puigbo P, Aragones L, Garcia-Vallve S. RCDI/eRCDI: a web-server to estimate codon usage deoptimization. BMC Res Notes. 2010;3:87. doi: 10.1186/1756-0500-3-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mueller S, Papamichail D, Coleman JR, Skiena S, Wimmer E. Reduction of the rate of poliovirus protein synthesis through large-scale codon deoptimization causes attenuation of viral virulence by lowering specific infectivity. J Virol. 2006;80:9687–9696. doi: 10.1128/JVI.00738-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhou JH, Zhang J, Sun DJ, et al. The distribution of synonymous codon choice in the translation initiation region of dengue virus. PLoS One. 2013;8:e77239. doi: 10.1371/journal.pone.0077239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Greenacre M. Theory and applications of correspondence analysis. London: Academic; 1984. [Google Scholar]
- 105.Peden JF. Analysis of Codon usage [dissertation] Nottingham University: Department of Genetics; 1999. [Google Scholar]
- 106.Supek F, Vlahovicek K. INCA: synonymous codon usage analysis and clustering by means of self-organizing map. Bioinformatics. 2004;20(14):2329–2330. doi: 10.1093/bioinformatics/bth238. [DOI] [PubMed] [Google Scholar]
- 107.Nakamura Y, Gojobori T, Ikemura T. Codon usage tabulated from international DNA sequence databases: status for the year 2000. Nucleic Acids Res. 2000;28:292. doi: 10.1093/nar/28.1.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chan PP, Lowe TM. GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res. 2009;37:D93–D97. doi: 10.1093/nar/gkn787. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Consensus amino acid changes in the predicted PA protein compared to Richmond/1/07; Table S1b. Consensus amino acid changes in the predicted PA-X protein compared to Richmond/1/07; Table S1c. Consensus amino acid changes in the predicted PB1 protein compared to Richmond/1/07; Table S1d. Consensus amino acid changes in the predicted PB1-F2 protein compared to Richmond/1/07; Table S1e. Consensus amino acid changes in the predicted PB2 protein compared to Richmond/1/07. (DOC 742 kb)
PA-X and PB1-F2 phylograms. (TIFF 2690 kb)
Correlation coefficients among the position of genes along the first two major axes with various indexes of PA polymerase genes’ codon usage. (DOCX 33 kb)
Comparison of Relative Synonymous Codon Usage (RSCU) patterns of EIVs polymerase genes with their host species. (TIFF 1828 kb)
The similarity index analysis of of EIVs polymerase genes in relation to their host species, Equus caballus. (TIFF 358 kb)
Correlation analysis among length (amino acid), GRAVY, AROMO, ENC, GC3s, ENc and the first two principle axes of COA of PB1 gene; Table S6b. Correlation analysis among length (amino acid), GRAVY, AROMO, ENC, GC3s, ENc and the first two principle axes of COA of PB2 gene; Table S6c. Correlation analysis among length (amino acid), GRAVY, AROMO, ENC, GC3s, ENc and the first two principle axes of COA of PA gene. (DOCX 18 kb)
Primers used for amplification of polymerase genes of EIV. (DOCX 12 kb)
Details of polymerase gene sequences of Equine Influenza virus (EIV) strains of equid origin used in the study. (DOC 69 kb)
Data Availability Statement
The sequence data of polymerase genes of equine influenza virus strains used in the analysis was retrieved from the Influenza Virus Resource at the National Center for Biotechnological Information (NCBI, http://www.ncbi.nlm.nih.gov/genomes/FLU/FLU.html). Few sequences from Global Initiative on Sharing All Influenza Data (GISAID, www.gisaid.org) were also included in the present study in consultation with scientist from the Animal Health Trust, UK. The new sequences reported in this study were deposited in GenBank (NCBI) and assigned accession numbers. The accession numbers of the sequences used in the study have been presented in Additional file 8.