

Abstract
Traditional structure and ligand based virtual screening approaches rely on the availability of structural and ligand binding information. To overcome this limitation, hybrid approaches were developed that relied on extraction of ligand binding information from proteins sharing similar folds and hence, evolutionarily relationship. However, they cannot target a chosen pocket in a protein. To address this, a pocket centric virtual ligand screening approach is required. Here, we employ a new, iterative implementation of a pocket and ligand-similarity based approach to virtual ligand screening to predict small molecule binders for the olfactomedin domain of human myocilin implicated in glaucoma. Small-molecule binders of the protein might prevent the aggregation of the protein, commonly seen during glaucoma. First round experimental assessment of the predictions using differential scanning fluorimetry with myoc-OLF yielded 7 hits with a success rate of 12.7%; the best hit had an apparent dissociation constant of 99 nM. By matching to the key functional groups of the best ligand that were likely involved in binding, the affinity of the best hit was improved by almost 10,000 fold from the high nanomolar to the low picomolar range. Thus, this study provides preliminary validation of the methodology on a medically important glaucoma associated protein.
Keywords: Binding, Myocilin, Glaucoma, drug discovery, virtual ligand screening, thermal shift assay
Graphical abstract
The art of drug discovery relies on identifying small molecules that bind to and modulate the behavior of a target protein. Attempts have been made to achieve this objective using both experimental high throughput screening (HTS) and computational virtual ligand screening (VLS) approaches 1. Though identifying small-molecules employing experimental HTS approaches would be ideal, its cost and time-consuming nature can be greatly reduced by efficient VLS approaches that narrow down the set of candidate small molecule ligands that will be experimentally screened. Traditional VLS exclusively relies on structure-based or ligand-based approaches with their associated advantages and disadvantages as delineated in 2–8. Structure-based VLS require no a priori knowledge of binding ligands and could target a specific pocket of interest 9, but most variants require a high-resolution structure and even then, their success rate for a novel protein target is quite low 3–5, 7, 8, 10. In contrast, ligand-based VLS approaches require at least one known bioactive molecule that serves as a seed to fish out database molecules with similar chemotypes and thus cannot explore new regions of chemical space. In practice, many high value therapeutically important protein targets lack either structural and/or significant ligand binding information. For these targets, ligand copying from evolutionarily related homologous proteins is a quite effective alternative which has higher success rates than traditional approaches even when the crystal structure is unknown 3–5, 7, 8, 10. However, these methods lack the ability to target a specific protein pocket and were limited to the identification of ligands similar to that in the template protein. To address the former issue, we previously demonstrated that since the number of distinct small molecule ligand binding pockets in proteins is small, copying ligands from pockets similar to the target pocket performs fairly well 11. Here, we apply a generalization that enables both pocket selection and exploration of a broader range of ligand space to a medically relevant protein myocilin for which only very poor quality binders were previously known 12 (Fig 1).
Fig 1.

Schematic representation of pocket-based VLS approach. Given a target protein (red), seed ligands (orange and magenta) from the holo proteins are copied based on either global (blue template) or local (green template) pocket similarities reported by TM-align and APoc respectively. Next, the ligand library is screened against the seeds using shape or fragment similarities. The results are then validated experimentally using thermal shift assays. Finally, the “hits” are used as new seeds for additional iterative rounds of VLS.
In a previous study, we implemented a pocket based VLS approach called PoLi to determine small-molecule binders for Escherichia coli dihydrofolate reductase, DHFR, with an 18.4% success rate 11. Here, success is defined as identifying small molecules with μM or better binding affinities. DHFR is a well-studied protein with a lot of structural and small-molecule binding information available making it a relatively easy case. However, this methodology has not been implemented on less well-documented proteins that lack extensive structural or small-molecule binding information, nor does it allow for the exploration of a broader range of ligand space. In this study, we address these issues and demonstrate the successful application of our improved pocket based VLS approach to myocilin-OLF. Olfactomedin or oligosaccharide-lipid flippase (OLF) domains have been implicated in several disorders in humans ranging from inflammatory bowel disorders, cancers to glaucoma 13. Mutant variants of human myocilin are implicated in causing primary open-angle glaucoma (POAG) 12. There has been a paucity of information on the small-molecules that show binding to myoc-OLF as a possible ameliorative tool to prevent its aggregation. A recent study reported 14 compounds that bind to the protein, with two, GW5074 and apigenin, capable of inhibiting myoc-OLF amyloid formation in vitro 14. However, the binding affinities of GW5074 and apigenin for myoc-OLF, as determined by SPR, were 33.5 ± 20.9 and 41.6 ± 39.6 μM. Of greater concern, apigenin gave a somewhat incomprehensible negative ΔTm of −0.3 °C, possibly indicative of very weak binding, while GW5074 gave a minuscule ΔTm of 0.2 °C. Thus, these molecules are quite weak hits. Here, we use a hybrid pocket- and ligand-based virtual screening algorithm to predict novel ligands that can bind to myoc-OLF, see Fig 1. Furthermore, we employ a newly developed iterative ligand seeding approach, instead of traditional quantitative structure-activity relationship studies (QSAR), to predict small molecules with tighter binding potential (Fig 1). This search based on iterative geometric and chemical similarity represents a promising approach towards identifying the active pharmacophore and is an alternative to traditional QSAR.
Following the protocol schematically outlined in Fig. 1, pocket detection was carried out for the olfactomedin domain of myocilin as follows: The experimentally solved crystal structure of Myocilin Olfactomecin Domain (PDB_ID: 4WXQ 15) was employed to detect potential small molecule binding pockets. In the first step, Concavity with default parameters 16 identified cavities on Myocilin’s surface and the associated pocket residues were identified. Subsequently, we used APoc 17 to calculate the pocket-pocket similarity between myocilin and the holo pockets of the holo-protein database, PDB-holo (having more than 50,000 entries), used previously 18) in order to copy ligands using pocket similarity. A ligand is copied only if the pocket similarity score has a statistically significant p-value (less than 0.01). Two different methods were utilized to find similar ligands to the seed ligand (the ligand copied from the template pocket). A) If a small and localized fragment of a seed ligand was responsible for enthalpic interactions (e.g. hydrogen bonds) given by PoseView 19, and the region of interaction was aligned to the pocket of Myocilin-OLF, the fragment was used as a template for pharmacophore search. Given a pharmacophore of interest, the OpenBabel 20 cheminformatics tool was used to generate the SMARTS patterns. Next, the pattern was used to screen against the ligand library (NCI diversity set). B) If multiple non-localized regions (separated by more than one rotatable bond) of the seed ligand participate in ab interaction with the template’s pocket, then LIGSIFT21 was used to calculate the 3D shape and chemical similarities of the seed molecule with the ligand library.
Myocilin’s structure was scanned for potential ligand binding pockets and similar holo-structures as described above. This resulted in a total of two potential binding pockets, PKT1 (green) and PKT2 (blue) shown in Fig 2. Each pocket was then used to scan PDB-holo for similar pockets. Initially, we found 40 small molecules with their pocket’s having significant similarities to either PKT1 or PKT2. This list was then assessed manually and reduced to 26 molecules that had “drug-like” properties22 (Table S1). Finally, three seed molecules from PKT1 and two from PKT2 were chosen to scan against the NCI diversity set (Table 1).
Fig 2.

Structure of Myocilin Olfactomecin Domain (white). The binding sites (PKT1: green, PKT2: blue, PKT3: yellow, PKT4: magenta) and their corresponding seed molecules are highlighted and color-coded. Dashed lines around fragments of a ligand indicate the pharmacophore region used for finding similar molecules. LIGSIFT was used to find similar small molecules to the seed ligands marked with asterisk. The identification of each pocket’s amino acids is found in Table S4.
Table 1.
List of seed molecules and their template copied using global or local similarities.
| PDB ID | Ligand ID | Npkt | Naligned | TM-Score | P-value (Pocket) | Npred | Nhit | |
|---|---|---|---|---|---|---|---|---|
| PKT1 | 3V5L | 0G1 | 15 | 15 | 0.27 | 0.001 | 5 | 1 |
| 4BDK | RQQ | 12 | 12 | 0.25 | 0.008 | 10 | 3 | |
| 1QCA | FUA | 12 | 11 | 0.33 | 0.003 | 8 | 0 | |
| PKT2 | 4YKA | TYC | 12 | 11 | 0.36 | 0.002 | 11 | 0 |
| 4J93 | 0OE | 10 | 10 | 0.30 | 0.0003 | 10 | 2 | |
| PKT3 | 2XZG | VH1 | 13 | 12 | 0.61 | N/A | 5 | 0 |
| 4G55 | VH2 | 12 | 11 | 0.61 | N/A | 3 | 0 | |
| PKT4 | 4MSL | 2ET | 15 | 11 | 0.50 | N/A | 4 | 1 |
Npkt: Number of the template PDB amino acids in contact with its ligand.
Naligned: Number of the amino acids that are aligned to Myocilin’s structure.
Npred: Number of predictions from the NCI diversity set.
Nhit: Number of cases showing a positive thermal shift.
Global structure similarity to holo-proteins also provided alternative binding pockets (Figure 2) of PKT3 (yellow) and PKT4 (magenta). Ligands were also copied based on the global similarity of the proteins using TM-align (a widely used protein structural alignment algorithm) 23 to identify holo-proteins with high structural similarity (TM-score > 0.5) whose ligands will then be copied to the myocilin pocket of interest. The structure of myocilin was compared to PDB-homo. A ligand is copied from a holo template protein if more than two thirds of its holo pocket residues are aligned (distance < 3 Å) to those in myocilin. Since the templates of these two pockets provided us with seed ligands (Table 1), we simply used them to scan the NCI set and didn’t use PKT3 and PKT4 to search for similar pockets. Scanning the NCI diversity set using the set of 8 seed molecules from PKT1–PKT4 gave 55 molecules (Table S2).
To experimentally assess the validity of the predictions, high-throughput thermal shift assays were carried out following established guidelines employing differential scanning fluorimetry (DSF) 24, 25. Briefly, samples in 96-well PCR plates were heated from 25 °C to 74 °C using a 1 °C/min heating ramp in a RealPlex quantitative PCR instrument with 5 X Sypro orange as the reporter fluorescent dye. A single data point was acquired for each degree increment. The reaction was carried out in a volume of 20 μl, with 100 mM HEPES pH 7.3, 150 mM NaCl and 5 μM of the respective protein 8, 11, 26, 27. The experiments were performed with biological replicates and the mean value was considered for further analysis. Curves showing a single sigmoidal thermal transition were selected and normalized for further analysis. The curves were fit to Boltzmann’s equation (Eq. 1) to obtain the melting temperature, Tm, from the observed fluorescence intensity, I by:
| (1) |
where Imin and Imax are the minimum and maximum intensities; a denotes the slope of the curve at the unfolding transition midpoint temperature, Tm. To estimate the approximate ligand-binding affinity at Tm, Eq. (2) was used; ΔCp is ignored.
| (2) |
KL(Tm) is the ligand association constant and [L] is the free ligand concentration at Tm ([LTm] ~ [L]total, when [L]total > > the total concentration of protein. KD is the inverse of KL(Tm). ΔH is the enthalpy change and R is the gas constant. ΔH was estimated employing both van’t Hoff and Gibbs-Helmholtz analyses. This methodology has been described previously for apparent KD computation in 8.
A total of 55 small molecule predictions belonging to approximately the top 1 % of the ligand library were selected to be assessed with DSF. As stated above, these hits were derived from 4 pockets and 8 seed molecules [0G1, 0OE, FUA, RQQ, TYC, 2ET, VH1, VH2]. All 55 molecules yielded interpretable curves, and seven (Fig 3A) showed positive shifts with the magnitude of the shifts indicated in Fig 3B and Table 2. This indicates a 12.7% success rate in the screening of the top 55 molecules, reflecting a 50% success in seed selection. Fig 3B shows the resulting experimental curves. The best hit was NSC37433, with a thermal shift (ΔTm) of 16.4 °C with respect to the protein alone control. It should be noted that the reported ΔTm for NSC37433 is an approximate estimate given that the shape of the curve deviates substantially from a typical two-state unfolding curve and has a small plateau making the fit have broad confidence intervals. This ΔTm corresponds to an approximate dissociation constant of 99 nM. Note that the pocket similarity of the template’s small-molecule (ligand ID: RQQ from PKT1) binding site with the myoc-OLF domain’s NSC37433 binding site has a P-value of 0.008. This site has two amino acids (Ile477 and Asn428) that partly overlap with Ca2+ binding site of myoc-OLF comprised of five amino acids (Asp380, Asn428, Ala429, Asn428 and Ile477). This indicates the likely functional significance of the predicted and experimentally assessed binding of NSC37433. In this initial round of screening, there were 6 additional hits that showed high μM or better affinities for myoc-OLF domain (Table 2).
Fig 3.

Initial round of screening with Myoc-OLF (A) Structure of small molecules showing binding to Myoc-OLF as assessed by thermal shift assay methodology. The SDF files for the small molecules were downloaded from Pubchem (http://pubchem.ncbi.nlm.nih.gov) and the figure was generated using ChemBioDraw 15.1. (B) Thermal unfolding curves for Myoc-OLF in the presence of the various small-molecules that were screened from PoLi VLS predictions and yielded positive shifts.
Table 2.
Summary of virtual ligand screening, thermal shift assay and binding parameters for the initial hits obtained on myoc-OLF.
| Molecules | Seed | Tm (°C) | ΔTm | KD (approx.)(M) | |
|---|---|---|---|---|---|
| Myoc-OLF | NA | NA | 49.83 | NA | NA |
| NSC37433 | RQQ | PKT1 | 66.27 | 16.4 | 9.9 × 10−8 |
| NSC643148 | 0G1 | PKT1 | 51.85 | 2.02 | 1.7 × 10−4 |
| NSC408734 | 0OE | PKT2 | 51.61 | 1.78 | 1.9 × 10−4 |
| NSC93033 | 0OE | PKT2 | 51.49 | 1.96 | 2.0 × 10−4 |
| NSC79688 | 2ET | PKT4 | 51.43 | 1.60 | 2.1 × 10−4 |
| NSC11437 | RQQ | PKT1 | 51.33 | 1.50 | 2.2 × 10−4 |
| NSC16722 | RQQ | PKT1 | 50.74 | 0.91 | 3.0 × 10−4 |
NA, not applicable
Next, we took the strongest binder (NSC37433), which was predicted to bind to PKT1 based on pocket similarity and used it as a seed for the second round of screening with the hope of increasing binding affinity. For this purpose, we used NSC37433’s scaffold (two benzene rings with hydrogen bond donor or acceptors groups, connected by three or four linear linkers) to search for similar molecules in the NCI diversity set. The second round of screening provided us with 35 (Table S3) new predictions.
Very few VLS studies have explored the power of iterative screening whereby predictions are experimentally assessed and hits from the first round are used to identify additional and better ligands using ligand-similarity metrics 28–30. Here, in contrast, a total of 35 small molecule predictions were tested with DSF. Analysis indicated that 7 curves were uninterpretable and 25 curves showed either no shift or negative shifts vis-à-vis the protein alone control indicating that there was no potential binding. However, we identified an additional set of 3 new ligands as potential binders of Myoc-OLF. Three small molecules (Fig 4A) showed positive shifts with the magnitude of the shifts as indicated in Fig 4B and Table 3. However, it would have to be once again pointed out that the reported ΔTm for the three top hits are gross approximations given that the shape of the curves deviates substantially from a typical two-state unfolding curve with NSC80735 and NSC201634 showing biphasic transitions possibly indicative of multiple unfolding intermediates. Further, most of the curves have minimal plateau regions with the resultant fits having broad confidence intervals. Nevertheless, despite these concerns, the results indicate an 11% (3/28) success rate. While two of the three hits did not yield better apparent binding affinities than the parent NSC37433, all three hits from this round of screening had affinities less than ~10 μM with the best hit showing an approximate pM affinity for the protein (Table 3). However, these affinities have to be treated with caution given the biphasic nature of the curves and the lack of a proper plateau that results in an increased error. The best hit in this seeded round of screening was NSC201634, with an approximate thermal shift (ΔTm) of 32°C with respect to the protein alone control. This indicates an approximate dissociation constant of 8 pM. Furthermore, this represents a ~10,000-fold increase in the affinity of the best hit compared to the best hit from the initial round of screening i.e. NSC37433.
Fig 4.

Second round of screening with Myoc-OLF predictions done using seeding and ligand similarity algorithm LIGSIFT (A) Structure of small molecules showing binding to Myoc-OLF as assessed by thermal shift assay methodology. The SDF files for the small molecules were downloaded from Pubchem (http://pubchem.ncbi.nlm.nih.gov) and the figure was generated using ChemBioDraw 15.1. (B) Thermal unfolding curves for Myoc-OLF in the presence of the various small-molecules that were screened from PoLi VLS predictions after seeding and that yielded positive shifts.
Table 3.
Summary of virtual ligand screening, thermal shift assay and binding parameters for the hits obtained after initial seeding with known binders of myoc-OLF from the previous round of virtual screening.
| Molecules | Tm (°C) | ΔTm | KD (approx.)(M) |
|---|---|---|---|
| NSC201634 | 83.79 | ~32* | 8.0 × 10−12 |
| NSC80735 | 62.86 | 11.2 | 6.5 × 10−7 |
| NSC46492 | 57.96 | 6.3 | 1.1 × 10−5 |
S. No., serial number; NA, not applicable,
The confidence interval of the curve was too wide and hence the estimate represents an approximate fit.
The olfactomedin domain of human myocilin has been implicated in POAG. Small-molecule probes binding to this protein target might have potential applications in alleviating conditions caused by aberrant form of this protein. Mutations on Myocilin inhibit the proteolytic processing of the full length protein which occurs in the linker (Arg226-Ile227) between amino-terminal domain and the C-terminal OLF domain 31. Thus, compounds stabilizing the protein’s native conformation could potentially restore this proteolytic cleavage, thus reducing the secretion of the aggregation prone protein. With this goal, we employed our pocket based VLS in combination with high-throughput experimental screening to find novel small molecule binders of this protein. As shown in Fig 5, the application of iterative pocket-based VLS on myoc-OLF demonstrates that it is not only able to predict novel small molecules with better binding affinities for a target of interest than were previously known, but on iteration it can yield better binding ligands. On the other hand, ligand identification based on global fold similarity alone was much less successful, with only a single 2μM hit, NSC79688, found. Thus, this study paves the way for future efforts employing this approach to predict better binding ligands for hitherto intractable but therapeutically important targets.
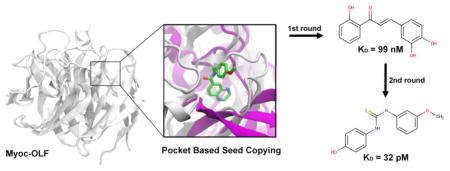
Fig 5.

Application of iterative pocket-based VLS to myoc-OLF. Ligand RQQ was copied based on a statistically significant pocket similarity (P-value = 0.008) of a pocket in template protein 4BDK (purple) to myoc-OLF (white). The first round of screening provided a nM binder (NSC37433) and the second round increased the affinity into pM (NSC201634) regime.
Supplementary Material
Acknowledgments
This project was funded by R35GM118039 of the Division of General Medical Sciences of the NIH. The authors wish to thank Prof. Raquel Lieberman, Georgia Institute of Technology, for providing purified human myocilin olfactomedin domain. We would also like to thank the Developmental Therapeutics Program of the National Cancer Institute and the Medicines for Malaria Venture (MMV) for providing the small molecules used in this study.
Abbreviations
- OLF
eukaryotic oligosaccharide-lipid flippase superfamily
- POAG
primary open-angle glaucoma
- MYOC
myocilin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hung CL, Chen CC. Computational approaches for drug discovery. Drug Dev Res. 2014;75(6):412–418. doi: 10.1002/ddr.21222. [DOI] [PubMed] [Google Scholar]
- 2.Klebe G. Virtual ligand screening: strategies, perspectives and limitations. Drug Discov Today. 2006;11(13–14):580–594. doi: 10.1016/j.drudis.2006.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brylinski M, Skolnick J. A threading-based method (FINDSITE) for ligand-binding site prediction and functional annotation. Proc Natl Acad Sci U S A. 2008;105(1):129–134. doi: 10.1073/pnas.0707684105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brylinski M, Skolnick J. FINDSITE: a threading-based approach to ligand homology modeling. PLoS Comput Biol. 2009;5(6):e1000405. doi: 10.1371/journal.pcbi.1000405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Skolnick J, Brylinski M. FINDSITE: a combined evolution/structure-based approach to protein function prediction. Brief Bioinform. 2009;10(4):378–391. doi: 10.1093/bib/bbp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Geppert H, Vogt M, Bajorath J. Current trends in ligand-based virtual screening: molecular representations, data mining methods, new application areas, and performance evaluation. J Chem Inf Model. 2010;50(2):205–216. doi: 10.1021/ci900419k. [DOI] [PubMed] [Google Scholar]
- 7.Zhou H, Skolnick J. FINDSITE(comb): a threading/structure-based, proteomic-scale virtual ligand screening approach. J Chem Inf Model. 2013;53(1):230–240. doi: 10.1021/ci300510n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Srinivasan B, Zhou H, Kubanek J, Skolnick J. Experimental validation of FINDSITE(comb) virtual ligand screening results for eight proteins yields novel nanomolar and micromolar binders. J Cheminform. 2014;6:16. doi: 10.1186/1758-2946-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lyne PD. Structure-based virtual screening: an overview. Drug Discov Today. 2002;7(20):1047–1055. doi: 10.1016/s1359-6446(02)02483-2. [DOI] [PubMed] [Google Scholar]
- 10.Zhou H, Skolnick J. FINDSITE(X): a structure-based, small molecule virtual screening approach with application to all identified human GPCRs. Mol Pharm. 2012;9(6):1775–1784. doi: 10.1021/mp3000716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roy A, Srinivasan B, Skolnick J. PoLi: A Virtual Screening Pipeline Based on Template Pocket and Ligand Similarity. J Chem Inf Model. 2015;55(8):1757–1770. doi: 10.1021/acs.jcim.5b00232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Donegan RK, Lieberman RL. Discovery of Molecular Therapeutics for Glaucoma: Challenges, Successes, and Promising Directions. J Med Chem. 2016;59(3):788–809. doi: 10.1021/acs.jmedchem.5b00828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hill SE, Donegan RK, Nguyen E, Desai TM, Lieberman RL. Molecular Details of Olfactomedin Domains Provide Pathway to Structure-Function Studies. PLoS One. 2015;10(6):e0130888. doi: 10.1371/journal.pone.0130888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Orwig SD, Chi PV, Du Y, et al. Ligands for glaucoma-associated myocilin discovered by a generic binding assay. ACS Chem Biol. 2014;9(2):517–525. doi: 10.1021/cb4007776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Donegan RK, Hill SE, Freeman DM, et al. Structural basis for misfolding in myocilin-associated glaucoma. Hum Mol Genet. 2015;24(8):2111–2124. doi: 10.1093/hmg/ddu730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Capra JA, Laskowski RA, Thornton JM, Singh M, Funkhouser TA. Predicting protein ligand binding sites by combining evolutionary sequence conservation and 3D structure. PLoS Comput Biol. 2009;5(12):e1000585. doi: 10.1371/journal.pcbi.1000585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao M, Skolnick J. APoc: large-scale identification of similar protein pockets. Bioinformatics. 2013;29(5):597–604. doi: 10.1093/bioinformatics/btt024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tonddast-Navaei S, Srinivasan B, Skolnick J. On the importance of composite protein multiple ligand interactions in protein pockets. Journal of computational chemistry. 2016 doi: 10.1002/jcc.24523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stierand K, Rarey M. Drawing the PDB: Protein-Ligand Complexes in Two Dimensions. ACS Med Chem Lett. 2010;1(9):540–545. doi: 10.1021/ml100164p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: An open chemical toolbox. J Cheminform. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roy A, Skolnick J. LIGSIFT: an open-source tool for ligand structural alignment and virtual screening. Bioinformatics. 2015;31(4):539–544. doi: 10.1093/bioinformatics/btu692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walters WP, Ajay, Murcko MA. Recognizing molecules with drug-like properties. Curr Opin Chem Biol. 1999;3(4):384–387. doi: 10.1016/s1367-5931(99)80058-1. [DOI] [PubMed] [Google Scholar]
- 23.Pandit SB, Skolnick J. Fr-TM-align: a new protein structural alignment method based on fragment alignments and the TM-score. BMC bioinformatics. 2008;9:531. doi: 10.1186/1471-2105-9-531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc. 2007;2(9):2212–2221. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- 25.Crowther GJ, He P, Rodenbough PP, et al. Use of thermal melt curves to assess the quality of enzyme preparations. Anal Biochem. 2010;399(2):268–275. doi: 10.1016/j.ab.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Srinivasan B, Tonddast-Navaei S, Skolnick J. Ligand binding studies, preliminary structure-activity relationship and detailed mechanistic characterization of 1-phenyl-6,6-dimethyl-1,3,5-triazine-2,4-diamine derivatives as inhibitors of Escherichia coli dihydrofolate reductase. Eur J Med Chem. 2015;103:600–614. doi: 10.1016/j.ejmech.2015.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Srinivasan B, Skolnick J. Insights into the slow-onset tight-binding inhibition of Escherichia coli dihydrofolate reductase: detailed mechanistic characterization of pyrrolo [3,2-f] quinazoline-1,3-diamine and its derivatives as novel tight-binding inhibitors. FEBS J. 2015;282(10):1922–1938. doi: 10.1111/febs.13244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shiryaev SA, Cheltsov AV, Gawlik K, Ratnikov BI, Strongin AY. Virtual ligand screening of the National Cancer Institute (NCI) compound library leads to the allosteric inhibitory scaffolds of the West Nile Virus NS3 proteinase. Assay Drug Dev Technol. 2011;9(1):69–78. doi: 10.1089/adt.2010.0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perez-Pineiro R, Burgos A, Jones DC, et al. Development of a novel virtual screening cascade protocol to identify potential trypanothione reductase inhibitors. J Med Chem. 2009;52(6):1670–1680. doi: 10.1021/jm801306g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ai N, Welsh WJ, Santhanam U, Hu H, Lyga J. Novel virtual screening approach for the discovery of human tyrosinase inhibitors. PLoS One. 2014;9(11):e112788. doi: 10.1371/journal.pone.0112788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aroca-Aguilar JD, Sanchez-Sanchez F, Ghosh S, Coca-Prados M, Escribano J. Myocilin mutations causing glaucoma inhibit the intracellular endoproteolytic cleavage of myocilin between amino acids Arg226 and Ile227. J Biol Chem. 2005;280(22):21043–21051. doi: 10.1074/jbc.M501340200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.