

Abstract
B-lineage and myeloid leukemia cells are often transformed by the same oncogenes, but have different biological and clinical characteristics. While B-lineage acute lymphoblastic leukemia (ALL) cells are characterized by a state of chronic energy deficit, myeloid leukemia cells show abundant energy reserve. Interestingly, fasting has been demonstrated to selectively inhibit the development of B-lineage ALL, but not myeloid leukemia, further suggesting that lineage identity may be linked to divergent metabolic states in hematopoietic malignancies. B-lymphoid transcription factors IKZF1, EBF1 and PAX5 are essential for early B cell development and commitment to B cell identity. However, in >80% of human pre-B ALL cases, the leukemic clones harbor genetic lesions of these transcription factors. The significance of these defects has only recently been investigated. Here we discuss the unexpected function of a B-lymphoid transcriptional program as a metabolic barrier against malignant transformation of B cell precursor cells. The metabolic gatekeeper function of B-lymphoid transcription factors may force silent pre-leukemic clones carrying potentially oncogenic lesions to remain in a latent state. In addition, this program sets the threshold for responses to glucocorticoids in pre-B ALL. Finally, the link between tumor suppressor and metabolic functions of B-lymphoid transcription factors is matched by observations in clinical trials: Obesity and hyperglycemia are associated with poor clinical outcome in patients with pre-B ALL.
A mouse model to demonstrate potential link between obesity, LEPR and B-lineage ALL
Several lines of evidence suggest that glucose and energy supply represents a rate-limiting factor in oncogenic transformation of B cells. For instance, obese children and children with type 2 diabetes mellitus (T2DM) with higher glucose and insulin serum levels are more likely to relapse with B cell lineage acute lymphoblastic leukemia (ALL). They have significantly inferior outcomes compared to children with normal glucose/insulin levels [1–3]. In addition, pre-B ALL patients with high glucose levels at the time of induction chemotherapy were less likely to achieve durable remissions of disease, and had worse overall outcomes compared to those with normal glucose levels [4]. Likewise, previous studies demonstrated that obesity was associated with increased risk for patients with mature B cell lymphoma, including non-Hodgkin lymphoma (B-NHL), diffuse large B-cell lymphoma (DLBCL), follicular lymphoma (FL) and also in multiple myeloma [5–7]. Obesity is associated with leptin resistance characterized by attenuated leptin receptor (LEPR) signaling [8]. Interestingly, positive associations were reported between incidence of B-NHL and polymorphisms in genes encoding molecules that regulate energy homeostasis, including leptin, LEPR and adiponectin [6,9]. Collectively, these findings suggest that high abundance of glucose and energy supply in the context of obesity and T2DM represents a risk factor for patients with B cell malignancies.
Studying a mouse model of N-Myc-driven B-cell lineage ALL, a link between obesity, Lepr signaling and development of ALL has recently been identified [10]. Notably, fasting selectively inhibited the development of B-lineage ALL, but not myeloid leukemia, through induction of Lepr expression. Furthermore, fasting arrested growth of B-lineage ALL in a human xenograft model, and expression levels of LEPR were found to be lower in human ALL samples in comparison to normal bone marrow samples [10]. Thus, downregulation of LEPR signaling contributes to development and maintenance of ALL, and the impacts of fasting are cell-type dependent, raising the question whether lineage identities (myeloid vs. B-lineage) are coupled with distinct metabolic states and control in hematopoietic malignancies.
Divergent clinical characteristics of B-lineage and myeloid malignancies
B-lineage and myeloid malignancies arise from lesions affecting oncogenes (e.g. BCR-ABL1, RAS, MYC, MLL) in multipotent progenitor cells. While often transformed by the same oncogenes, B-lineage and myeloid leukemias are markedly different in clinical characteristics [11–13]. For instance, while > 95% of chronic myeloid leukemia (CML) patients achieve long-term disease-free survival under treatment with tyrosine kinase inhibitors [12], patients with Philadelphia chromosome-positive (Ph+) ALL invariably relapse within months after initial remission [13]. In addition, glucocorticoids (GCs) are highly active in the treatment of pre-B ALL and B cell lymphoma [14], while patients with myeloid leukemias do not benefit from GC treatment [15]. Furthermore, a recent study demonstrated that GCs favor survival of hematopoietic stem and myeloid progenitor cells [16], which is in striking contrast to induction of cell death in B cells [17].
Divergent responses to GCs in B-cell vs. myeloid malignancies represent an empirically established standard in clinical medicine; however, the underlying reason for this difference has only recently been investigated. Expression levels of the glucocorticoid receptor (NR3C1) depend on a B-lymphoid transcriptional program. The B-lymphoid transcription factors PAX5 and IKZF1 set the threshold for GC responses in patient-derived pre-B ALL cells [18]. It was previously reported that phosphorylation of NR3C1 by AKT1 inhibits nuclear translocation of the glucocorticoid receptor upon GC treatment, and that pharmacological inhibition of AKT1 reverses GC resistance in ALL [19]. In addition to positively regulating NR3C1 expression, inducible reconstitution of PAX5 or IKZF1 in patient-derived haploinsufficient pre-B ALL cells reduced phosphorylation of AKT-S473 [18], suggesting that PAX5 and IKZF1 may also modulate sensitivity to GCs through suppression of AKT activity.
B cell precursors are defined by a state of chronic energy depletion
With respect to their biological characteristics, B-lineage and myeloid leukemia cells differ in their proliferation kinetics, cell size, cytoplasmic volume as well as mitochondrial number and volume (TABLE 1). Compared to myeloid leukemia, pre-B ALL cells divide faster (5- to 24-fold shorter doubling-times), are significantly smaller (5-fold smaller cytoplasmic volume) and carry half the number of mitochondria with about one-third of the mitochondrial volume observed in myeloid leukemia cells (TABLE 1). Similarly, patient-derived myeloid leukemia cells have abundant ATP reserves (low AMP:ATP ratios), while AMP:ATP ratios in patient-derived pre-B ALL cells are very high and inverted compared to myeloid leukemia cells [18; Table 1]. High baseline AMP:ATP ratios indicate a state of chronic energy deficit which triggers constitutive activation of the energy-stress sensor LKB1-AMPK pathway [20]. Low baseline levels of ATP as well as restriction of mitochondrial volume and number suggest that B cell precursors are defined by a state of chronic energy depletion [18].
Table 1.
Comparison of biological characteristics of myeloid and B-lineage leukemia cells
| Characteristic | Myeloid | B-lineage |
|---|---|---|
| Transforming oncogenes | RAS (AML) | RAS |
| BCR-ABL1 (CML) | BCR-ABL1 (Ph+ ALL) | |
|
| ||
| Lesions in transcription factors | CEBPA (5–14%; AML)60, GATA2 (11%; CML-blast crisis)61, PU.1 (7%; AML)62 | IKZF1 (84% of Ph+ ALL cases), EBF1 (14%), PAX5 (51%)63 |
|
| ||
| AMP:ATP ratio | 0.06 ± 0.03 (CML)18 | 4.5 ± 1.2 (Ph+ ALL) |
|
| ||
| Doubling time | 11 – 14 days (AML)64 | 2 days66 |
| 48 days (CML)65 | ||
|
| ||
| Cytoplasmic volume (μm3) | 73967 | 12168 |
|
| ||
| Number of mitochondrial per cell | 15.9 ± 3.8 (AML)69 | 7.8 ± 3.769 |
|
| ||
| Mitochondrial volume (μm3) | 42.9 ± 6.767 | 15.0 ± 5.968 |
Genetic lesions in B-lymphoid transcription factors represent near-obligate defects in human ALL
Myeloid (CEBPA, PU.1, GATA2) and B-lymphoid (e.g. IKZF1, EBF1, PAX5) transcription factors determine myeloid vs. B-lineage identity, respectively. B-lymphoid transcription factors antagonize myeloid differentiation as an alternative lineage fate by repressing the myeloid master regulator CEBPα and vice versa [21–25]. The transcription factors IKZF1, EBF1 and PAX5 are critical for B cell development at the level of B-lineage commitment, V(D)J recombination and pre-B cell receptor signaling [26]. IKZF1 primes lymphoid gene expression in multipotent progenitor (MPP) cells, allowing differentiation into common lymphoid progenitors [CLPs; 27], and it is also required for pre-BCR signaling [28]. EBF1 is essential for the formation of pro-B cells and restricts lineage fate option. EBF1 antagonizes expression of myeloid transcription factors CEBPα and PU.1, while positively regulating expression of PAX5 [24]. PAX5 is essential for the commitment to the B cell lineage and maintenance of B-cell identity [21].
While myeloid leukemia cells frequently acquire genetic lesions in myeloid transcription factors [29], pre-B ALL clones often carry genetic lesions that results in reduced activity or inactivation of B-lymphoid transcription factors [18,30,31]. Such genetic lesions may reduce the stringency of B-cell or myeloid lineage commitment; however, the significance of genetic inactivation of B-lymphoid transcription factors has only been recently elucidated. Studying patient samples from clinical trials for pre-B ALL in children and adults, inactivating lesions in B-lymphoid transcription factors PAX5, IKZF1, EBF1 and TCF3 were identified in the vast majority of ALL cases studied [18]. These lesions resulted in decreased protein levels of PAX5 as well as expression of truncated dominant-negative IKZF1 proteins (IK6) owing to intragenic deletions of IKZF1 [18]. Therefore, defects in B-lymphoid transcription factors represent near-obligate lesions in human pre-B ALL. Interestingly, CEBPA not only promotes myeloid development at the expense of B-lymphoid differentiation, but also functions as a mediator of glucose uptake in adipocytes [32]. Conversely, Ebf1−/− mice exhibit increased glucose transport and glucose transporter type 4 (GLUT4) expression in adipose tissues [33], suggesting that EBF1 may function as a negative regulator of glucose transport.
PI3K-AKT signaling couples B cell fate with metabolic state through regulation of transcription factors FOXO1 and IRF4
Previous studies demonstrated that hematopoietic stem and progenitor cells differ in their dependency on glycolysis [34]. More recently, asymmetric transmission of nutrient-sensing PI3K/AKT/mTOR signaling has been shown to dictate B cell fate through regulation of transcription factors FOXO1and IRF4 [35]. The transcription factor FOXO1 is known to induce PAX5 expression and promote B cell lineage commitment [36]. AKT-mediated phosphorylation causes export of FOXO1 from the nucleus to the cytoplasm, inhibiting the transcriptional activity of FOXO1 [37]. While expression of PAX5 is downregulated upon B cell differentiation into plasma cells [38], IRF4 mediates plasma cell differentiation [39]. In asymmetric cell divisions of B cells, high PI3K-AKT signaling characterized by FOXO1 inactivation, IRF4 induction and repression of PAX5 drives plasma cell differentiation. In contrast, the sibling cells with lower PI3K-AKT signaling undergo self-renewal and exhibit features of a memory or germinal center B cell fate [35].
Both IRF4 and FOXO1 were previously implicated in regulation of glucose and energy metabolism. For instance, IRF4 was shown to activate expression of glycolytic genes, including Glut1/3, Hk2, Pfkfb3, Pfkm, Pfkp, Pgk1, Pgm2, Pgm2l1, Eno1 and Ldh1, in activated B cells [35]. On the contrary, FOXO1 plays a key role in modulating expression of gluconeogenic genes during fasting [40]. Furthermore, it has been shown that FOXO1 impairs both glycolysis and mitochondrial respiration by antagonizing MYC signaling [41]. These findings raise the question whether B cell identity is linked to a specific metabolic state. Using pharmacological as well as genetic approaches, Adams et al. demonstrated that plasma cell differentiation is coupled with glycolysis, and that self-renewal of B cells is maintained by oxidative phosphorylation [42]. For instance, inhibiting glycolysis using 2-Deoxy-D-glucose (2-DG) stimulated self-renewal, while using oligomycin to inhibit oxidative phosphorylation shifted cell fate toward plasma cell differentiation [42]. Dynamin-related protein 1 (Drp1) is essential for mitochondrial dynamics by forming helices around mitochondria to modulate mitochondrial division [43]. Interestingly, overexpression of the dominant negative mutant (Drp1K38A) or treatment with a pharmacological inhibitor of Drp1 (mDivi−1) resulted in increased plasma cell differentiation [42], suggesting that mitochondrial fission and its impact on energy metabolism may play a role in determining B cell fate.
A B-lymphoid transcriptional program that negatively regulates glucose uptake and energy metabolism
More recently, our group studied whether B-lymphoid transcription factors can regulate glucose and energy supply in B-cell precursor cells, and whether this has implications for transformation and leukemogenesis [18]. Combining gene expression and ChIP-seq analyses, a novel B-lymphoid program for transcriptional repression of glucose uptake/utilization (INSR, GLUT 1/3/6, HK2/3 and G6PD) and activation of inhibitors of glucose transport, including NR3C1 [glucocorticoid receptor; 44], TXNIP [glucose feedback sensor; 45] and CNR2 [cannabinoid receptor; 46] has been identified [18]. Consistent with changes in gene expression observed, inducible reconstitution of PAX5 or IKZF1 in patient-derived pre-B ALL cells lacking functional PAX5 or IKZF1 resulted in a state of chronic energy deficit. Reconstitution of PAX5 or IKZF1 resulted in decreased glucose uptake and ATP levels, leading to activation of the energy stress sensor LKB1-AMPK [18]. B→ myeloid reprogramming mediated by the myeloid transcription factor CEBPa subverts B-lineage commitment by repression of PAX5 [18,22]. Notably, gene expression analysis revealed transcriptional activation of multiple mediators of glucose uptake/metabolism and energy supply (Insr, Glut1, Glut6, Hk3, Pygl and G6pd) upon B→ myeloid reprogramming [22]. Likewise, reversal of B-lymphoid lineage commitment relieves restriction of glucose and energy supply. B→myeloid reprogramming was shown to salvage chronic energy deprivation and restore a state of energy abundance characterized by increased glucose uptake and cellular ATP levels [18].
Mechanistic contribution of PAX5-targets to negative regulation of glucose metabolism
CRISPR/Cas9-based genetic screens identified NR3C1, TXNIP and CNR2 as central effectors of the tumor suppressive function of PAX5 [18]. Importantly, loss of Nr3c1 [47], Txnip [48] or Cnr2 [49] function increased both glucose uptake and cellular ATP levels in murine pre-B ALL cells, validating specific aspects of regulation of glucose uptake and energy metabolism by these molecules [18]. Furthermore, agonists of TXNIP and CNR2 synergized with GCs to induce cell death of patient-derived pre-B ALL cells, exacerbating ATP deficit in pre-B cells [18]. Taken together, these findings suggest that B-lymphoid transcription factors (PAX5 and IKZF1) restrict glucose uptake, which is upstream of both glycolysis and oxidative phosphorylation, limiting glucose and energy supply to levels not sufficient for transformation of pre-B cells. Therefore, B-lymphoid transcription factors PAX5 and IKZF1 function as metabolic gatekeepers.
Transport-independent pyruvate- and TCA cycle metabolites can bypass the gatekeeper function of PAX5 and IKZF1
During early B cell development, pre-B cells undergo intense selective pressure and sustain DNA damage during multiple rounds of V(D)J recombination of immunoglobulin heavy and light chain genes [50]. Acquisition of genetic lesions during the V(D)J recombination process at the pre-B cell stage represents a major driver of clonal evolution [51,52], which is compounded by the high rate of proliferation of pre-B cells. Given the inherent risk of malignant transformation during early B cell development, the B-lymphoid transcriptional program for the restriction of glucose and energy supply may function as an additional safeguard against malignant transformation. Indeed, transport-independent lipophilic methyl-conjugates of pyruvate and TCA cycle intermediates (oxaloacetate and dimethyl succinate) can bypass the tumor suppressor function of PAX5 and IKZF1, jumpstarting oncogenic signaling and enabling leukemic transformation of pre-B cells [18; FIGURE 1].
Figure 1. Metabolic gatekeeper function of B-lymphoid transcription factors.
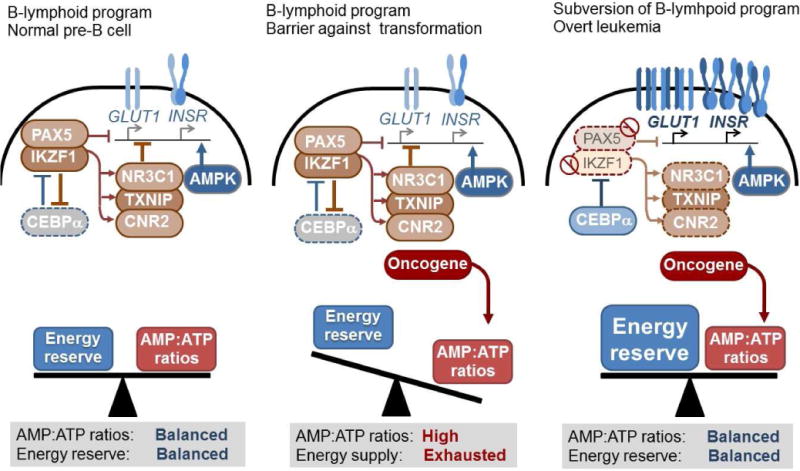
B-lymphoid transcription factors (e.g. PAX5 and IKZF1) are essential for early B cell development, and they oppose myeloid differentiation as an alternative lineage fate through repression of the myeloid transcription factor CEBPα and vice versa. While often transformed by the same oncogenes, B-lineage and myeloid leukemias are distinct with respect to their biological and clinical characteristics. In contrast to myeloid leukemia cells, pre-B ALL cells have high AMP:ATP ratios indicative of a state of chronic energy deficit, resulting in the activation of the energy-stress sensor AMPK. Transforming oncogenes (e.g. BCR-ABL1, RAS) impose significant metabolic requirements on ATP supply. In addition to their function in B cell development, B-lymphoid transcription factors function as a metabolic barrier against malignant transformation. Genetic lesions in B-lymphoid transcription factors represent near-obligate defects in human pre-B ALL. B-lymphoid transcriptional program represses glucose uptake as well as utilization (e.g. GLUT1, INSR), while activating inhibitors of glucose transport (NR3C1, TXNIP and CNR2). B-lymphoid lineage commitment limits the amount of glucose and energy supply to levels that are not sufficient for leukemic transformation. Genetic deletion of NR3C1, TXNIP and CNR2 revealed these inhibitors of glucose transport as key effects of PAX5-mediated tumor suppression.
Latent persistence of pre-leukemic B cell clones during early childhood and in healthy adults
Under this scenario, one would predict that healthy individuals frequently carry pre-malignant B cells that harbor an oncogenic lesion in the presence of fully functional PAX5 and IKZF1. Indeed, pre-leukemic B cell clones carrying multiple oncogenic lesions BCR-ABL1 [31], MLL-AF4 [53], ETV6-RUNX1 [54] gene rearrangements and hyperdiploidy [55] are frequently found in cord blood samples from healthy neonates. Furthermore, silent oncogenes including BCR-ABL1 [56,57] and mutant RAS [58] are often found in small fractions of normal B cells in healthy adults. In the bone marrow of asymptomatic CML patients in remission, ~10% of pre-B cells carry a silent BCR-ABL1 oncogene [59]. In patients with Noonan syndrome and RAS-associated lymphoproliferative disorder (RALD), RAS pathway mutations cause pre-B cell expansions but give rise to leukemia only in very few cases [58]. These findings collectively suggest that pre-leukemic B cell clones frequently occur in healthy individuals, during early childhood and in healthy adults. The high frequency of genetic lesions observed in B-lymphoid transcription factors in pre-B ALL takes on new significance from the finding that B-lymphoid transcriptional program limits glucose and energy supply, and thus functions as a metabolic barrier against oncogenic transformation [18].
Highlights.
Glucocorticoid activity in leukemia depends on B-lymphoid transcription factors.
B cell identity is marked by chronic energy deficit.
PAX5 and IKZF1 restrict glucose uptake and energy metabolism.
PAX5 and IKZF1 safeguard against leukemic transformation of B cell precursors.
Acknowledgments
Research in the Müschen laboratory is funded by the NIH/NCI through Outstanding Investigator Award R35CA197628 (to M.M.), R01CA137060, R01CA157644 and R01CA172558 (to M.M.), a Wellcome Trust Senior Investigator Award and a Leukemia and Lymphoma Scholar award (to M.M.), the Howard Hughes Medical Institute HHMI-55108547 (to M.M.), the Alex’s Lemonade Stand Foundation for Childhood Cancer (to M.M.), the William Lawrence & Blanche Hughes Foundation for childhood cancer (to M.M.), the Norman and Sadie Lee Foundation (for Pediatric Cancer, to M.M.), and the Falk Trust through a Falk Medical Research Trust Catalyst Award (to M.M.), Cancer Research Institute (CRI) through a Clinic and Laboratory Integration Program (CLIP) grant (to M.M.). M.M. is a Howard Hughes Medical Institute (HHMI) Faculty Scholar.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest disclosure
The authors declare no competing financial interests.
References
- 1.Butturini AM, Dorey FJ, Lange BJ, et al. Obesity and outcome in pediatric acute lymphoblastic leukemia. J Clin Oncol. 2007;25:2063–2069. doi: 10.1200/JCO.2006.07.7792. [DOI] [PubMed] [Google Scholar]
- 2.Gelelete CB, Pereira SH, Azevedo AM, et al. Overweight as a prognostic factor in children with acute lymphoblastic leukemia. Obesity (Silver Spring, Md) 2011;19:1908–1911. doi: 10.1038/oby.2011.195. [DOI] [PubMed] [Google Scholar]
- 3.Orgel E, Tucci J, Alhushki W, et al. Obesity is associated with residual leukemia following induction therapy for childhood B-precursor acute lymphoblastic leukemia. Blood. 2014;124:3932–3938. doi: 10.1182/blood-2014-08-595389. [DOI] [PubMed] [Google Scholar]
- 4.Weiser MA, Cabanillas ME, Konopleva M, et al. Relation between the duration of remission and hyperglycemia during induction chemotherapy for acute lymphocytic leukemia with a hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone/methotrexate-cytarabine regimen. Cancer. 2004;100:1179–1185. doi: 10.1002/cncr.20071. [DOI] [PubMed] [Google Scholar]
- 5.Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348:1625–1638. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- 6.Skibola CF, Holly EA, Forrest MS, et al. Body mass index, leptin and leptin receptor polymorphisms, and non-Hodgkin lymphoma. Cancer Epidemiol Biomarkers Prev. 2004;13:779–786. [PubMed] [Google Scholar]
- 7.Castillo JJ, Ingham RR, Reagan JL, et al. Obesity is associated with increased relative risk of diffuse large B-cell lymphoma: a meta-analysis of observational studies. Clinical Lymphoma, Myeloma & Leukemia. 2014;14:122–130. doi: 10.1016/j.clml.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 8.Myers MG, Jr, Leibel RL, Seeley RJ, Schwartz MW. Obesity and leptin resistance: distinguishing cause from effect. Trends Endocrinol Metab. 2010;21:643–651. doi: 10.1016/j.tem.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Willett EV, Skibola CF, Adamson P, et al. Non-Hodgkin’s lymphoma, obesity and energy homeostasis polymorphisms. Br J Cancer. 2005;93:811–816. doi: 10.1038/sj.bjc.6602762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu Z, Xie J, Wu G, et al. Fasting selectively blocks development of acute lymphoblastic leukemia via leptin-receptor upregulation. Nat Med. 2017;23:79–90. doi: 10.1038/nm.4252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Futreal PA, Coin L, Marshall M, et al. A census of human cancer genes. Nat Rev Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Druker BJ, Guilhot F, O’Brien SG, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408–2417. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- 13.Druker BJ, Sawyers CL, Kantarjian H, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344:1038–1042. doi: 10.1056/NEJM200104053441402. [DOI] [PubMed] [Google Scholar]
- 14.Lin KT, Wang LH. New dimension of glucocorticoids in cancer treatment. Steroids. 2016;111:84–88. doi: 10.1016/j.steroids.2016.02.019. [DOI] [PubMed] [Google Scholar]
- 15.Kaspers GJ, Kardos G, Pieters R, et al. Different cellular drug resistance profiles in childhood lymphoblastic and non-lymphoblastic leukemia: a preliminary report. Leukemia. 1994;8:1224–1229. [PubMed] [Google Scholar]
- 16.Guo B, Huang X, Cooper S, Broxmeyer HE. Glucocorticoid hormone-induced chromatin remodeling enhances human hematopoietic stem cell homing and engraftment. Nat Med. 2017;23:424–428. doi: 10.1038/nm.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gruver-Yates AL, Quinn MA, Cidlowski JA. Analysis of glucocorticoid receptors and their apoptotic response to dexamethasone in male murine B cells during development. Endocrinology. 2014;155:463–474. doi: 10.1210/en.2013-1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chan LN, Chen Z, Braas D, et al. Metabolic gatekeeper function of B-lymphoid transcription factors. Nature. 2017;542:479–483. doi: 10.1038/nature21076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Piovan E, Yu J, Tosello V, et al. Direct reversal of glucocorticoid resistance by AKT inhibition in acute lymphoblastic leukemia. Cancer Cell. 2013;24:766–776. doi: 10.1016/j.ccr.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shaw RJ, Kosmatka M, Bardeesy N, et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci USA. 2004;101:3329–3335. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nutt SL, Heavey B, Rolink AG, Busslinger M. Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. Nature. 1999;401:556–562. doi: 10.1038/44076. [DOI] [PubMed] [Google Scholar]
- 22.Xie H, Ye M, Feng R, Graf T. Stepwise reprogramming of B cells into macrophages. Cell. 2004;117:663–676. doi: 10.1016/s0092-8674(04)00419-2. [DOI] [PubMed] [Google Scholar]
- 23.Hsu CL, King-Fleischman AG, Lai AY, Matsumoto Y, Weissman IL, Kondo M. Antagonistic effect of CCAAT enhancer-binding protein-alpha and Pax5 in myeloid or lymphoid lineage choice in common lymphoid progenitors. Proc Natl Acad Sci USA. 2006;103:672–677. doi: 10.1073/pnas.0510304103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pongubala JM, Northrup DL, Lancki DW, et al. Transcription factor EBF restricts alternative lineage options and promotes B cell fate commitment independently of Pax5. Nat Immunol. 2008;9:203–215. doi: 10.1038/ni1555. [DOI] [PubMed] [Google Scholar]
- 25.Reynaud D, Demarco IA, Reddy KL, et al. Regulation of B cell fate commitment and immunoglobulin heavy-chain gene rearrangements by Ikaros. Nat Immunol. 2008;9:927–936. doi: 10.1038/ni.1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Medina KL, Pongubala JM, Reddy KL, et al. Assembling a gene regulatory network for specification of the B cell fate. Dev Cell. 2004;7:607–617. doi: 10.1016/j.devcel.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 27.Ng SY-M, Yoshida T, Zhang J, Georgopoulos K. Genome-wide lineage-specific transcriptional networks underscore Ikaros-dependent lymphoid priming in hematopoietic stem cells. Immunity. 2009;30:493–507. doi: 10.1016/j.immuni.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schwickert TA, Tagoh H, Gültekin S, et al. Stage-specific control of early B cell development by the transcription factor Ikaros. Nat Immunol. 2014;15:283–293. doi: 10.1038/ni.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tenen DG. Disruption of differentiation in human cancer: AML shows the way. Nat Rev Cancer. 2003;3:89–101. doi: 10.1038/nrc989. [DOI] [PubMed] [Google Scholar]
- 30.Fazio G, Palmi C, Rolink A, Biondi A, Cazzaniga G. PAX5/TEL acts as a transcriptional repressor causing down-modulation of CD19, enhances migration to CXCL12, and confers survival advantage in pre-BI cells. Cancer Res. 2008;68:181–189. doi: 10.1158/0008-5472.CAN-07-2778. [DOI] [PubMed] [Google Scholar]
- 31.Cazzaniga G, van Delft FW, Lo Nigro L, et al. Developmental origins and impact of BCR-ABL1 fusion and IKZF1 deletions in monozygotic twins with Ph+ acute lymphoblastic leukemia. Blood. 2011;118:5559–5564. doi: 10.1182/blood-2011-07-366542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu N, Rosen ED, Brun R, et al. Cross-regulation of C/EBP alpha and PPAR gamma controls the transcriptional pathway of adipogenesis and insulin sensitivity. Mol Cell. 1999;3:151–158. doi: 10.1016/s1097-2765(00)80306-8. [DOI] [PubMed] [Google Scholar]
- 33.Fretz JA, Nelson T, Xi Y, Adams DJ, Rosen CJ, Horowitz MC. Altered metabolism and lipodystrophy in the early B-cell factor 1-deficient mouse. Endocrinology. 2010;151:1611–1621. doi: 10.1210/en.2009-0987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang YH, Israelsen WJ, Lee D, et al. Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell. 2014;158:1309–1323. doi: 10.1016/j.cell.2014.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin W-HW, Adams WC, Nish SA, et al. Asymmetric PI3K signaling driving developmental and regenerative cell fate bifurcation. Cell Reports. 2015;13:2203–2218. doi: 10.1016/j.celrep.2015.10.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin YC, Jhunjhunwala S, Benner C, et al. A global network of transcription factors, involving E2A, EBF1 and Foxo1, that orchestrates B cell fate. Nat Immunol. 2010;11:635–643. doi: 10.1038/ni.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eijkelenboom A, Burgering BM. FOXOs: signaling integrators for homeostasis maintenance. Nature Rev Mol Cell Biol. 2013;14:83–97. doi: 10.1038/nrm3507. [DOI] [PubMed] [Google Scholar]
- 38.Shi W, Liao Y, Willis SN, et al. Transcriptional profiling of mouse B cell terminal differentiation defines a signature for antibody-secreting plasma cells. Nat Immunol. 2015;16:663–673. doi: 10.1038/ni.3154. [DOI] [PubMed] [Google Scholar]
- 39.Klein U, Casola S, Cattoretti G, et al. Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nat Immunol. 2006;7:773–782. doi: 10.1038/ni1357. [DOI] [PubMed] [Google Scholar]
- 40.Liu Y, Dentin R, Chen D, et al. A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange. Nature. 2008;456:269–273. doi: 10.1038/nature07349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilhelm K, Happel K, Eelen G, et al. FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature. 2016;529:216–220. doi: 10.1038/nature16498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adams WC, Chen Y-H, Kratchmarov R, et al. Anabolism-associated mitochondrial stasis driving lymphocyte differentiation over self-renewal. Cell Reports. 2016;17:3142–3152. doi: 10.1016/j.celrep.2016.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mishra P, Chan DC. Metabolic regulation of mitochondrial dynamics. J Cell Biol. 2016;212:379–387. doi: 10.1083/jcb.201511036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brennan-Speranza TC, Henneicke H, Gasparini SJ, et al. Osteoblasts mediate the adverse effects of glucocorticoids on fuel metabolism. J Clin Invest. 2012;122:4172–4189. doi: 10.1172/JCI63377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu N, Zheng B, Shaywitz A, et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol Cell. 2013;49:1167–1175. doi: 10.1016/j.molcel.2013.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nogueiras R, Diaz-Arteaga A, Lockie SH, et al. The endocannabinoid system: role in glucose and energy metabolism. Pharmacol Res. 2009;60:93–98. doi: 10.1016/j.phrs.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 47.Mittelstadt PR, Monteiro JP, Ashwell JD. Thymocyte responsiveness to endogenous glucocorticoids is required for immunological fitness. J Clin Invest. 2012;122:2384–2394. doi: 10.1172/JCI63067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yoshioka J, Imahashi K, Gabel SA, et al. Targeted deletion of thioredoxin-interacting protein regulates cardiac dysfunction in response to pressure overload. Circ Res. 2007;101:1328–1338. doi: 10.1161/CIRCRESAHA.106.160515. [DOI] [PubMed] [Google Scholar]
- 49.Pereira JP, An J, Xu Y, Huang Y, Cyster JS. Cannabinoid receptor 2 mediates the retention of immature B cells in bone marrow sinusoids. Nat Immunol. 2009;10:403–411. doi: 10.1038/ni.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Buchner M, Swaminathan S, Chen Z, Müschen M. Mechanisms of pre-B-cell receptor checkpoint control and its oncogenic subversion in acute lymphoblastic leukemia. Immunol Rev. 2015;263:192–209. doi: 10.1111/imr.12235. [DOI] [PubMed] [Google Scholar]
- 51.Papaemmanuil E, Rapado I, Li Y, et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat Genet. 2014;46:116–125. doi: 10.1038/ng.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Swaminathan S, Klemm L, Park E, et al. Mechanisms of clonal evolution in childhood acute lymphoblastic leukemia. Nat Immunol. 2015;16:766–774. doi: 10.1038/ni.3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gale KB, Ford AM, Repp R, et al. Backtracking leukemia to birth: identification of clonotypic gene fusion sequences in neonatal blood spots. Proc Natl Acad Sci USA. 1997;94:13950–13954. doi: 10.1073/pnas.94.25.13950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wiemels JL, Cazzaniga G, Daniotti M, et al. Prenatal origin of acute lymphoblastic leukaemia in children. Lancet. 1999;354:1499–1503. doi: 10.1016/s0140-6736(99)09403-9. [DOI] [PubMed] [Google Scholar]
- 55.Maia AT, Tussiwand R, Cazzaniga G, et al. Identification of preleukemic precursors of hyperdiploid acute lymphoblastic leukemia in cord blood. Genes Chromosomes Cancer. 2004;40:38–43. doi: 10.1002/gcc.20010. [DOI] [PubMed] [Google Scholar]
- 56.Biernaux C, Loos M, Sels A, Huez G, Stryckmans P. Detection of major bcr-abl gene expression at a very low level in blood cells of some healthy individuals. Blood. 1995;86:3118–3122. [PubMed] [Google Scholar]
- 57.Bose S, Deininger M, Gora-Tybor J, Goldman JM, Melo JV. The presence of typical and atypical BCR-ABL fusion genes in leukocytes of normal individuals: biologic significance and implications for the assessment of minimal residual disease. Blood. 1998;92:3362–3367. [PubMed] [Google Scholar]
- 58.Takagi M, Shinoda K, Piao J, et al. Autoimmune lymphoproliferative syndrome-like disease with somatic KRAS mutation. Blood. 2011;117:2887–2890. doi: 10.1182/blood-2010-08-301515. [DOI] [PubMed] [Google Scholar]
- 59.Reinhold U, Hennig E, Leiblein S, Niederwieser D, Deininger MW. FISH for BCR-ABL on interphases of peripheral blood neutrophils but not of unselected white cells correlates with bone marrow cytogenetics in CML patients treated with imatinib. Leukemia. 2003;17:1925–1929. doi: 10.1038/sj.leu.2403077. [DOI] [PubMed] [Google Scholar]
- 60.Wouters BJ, Löwenberg B, Erpelinck-Verschueren CAJ, van Putten WLJ, Valk PJM, Delwel R. Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood. 2009;113:3088–3091. doi: 10.1182/blood-2008-09-179895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang S-J, Ma L-Y, Huang Q-H, et al. Gain-of-function mutation of GATA-2 in acute myeloid transformation of chronic myeloid leukemia. Proc Natl Acad Sci USA. 2008;105:2076–2081. doi: 10.1073/pnas.0711824105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mueller BU, Pabst T, Osato M, et al. Heterozygous PU.1 mutations are associated with acute myeloid leukemia. Blood. 2002;100:998–1007. doi: 10.1182/blood.v100.3.998. [DOI] [PubMed] [Google Scholar]
- 63.Mullighan CG, Miller CB, Radtke I, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. 2008;453:110114. doi: 10.1038/nature06866. [DOI] [PubMed] [Google Scholar]
- 64.Ommen HB, Schnittger S, Jovanovic JB, et al. Strikingly different molecular relapse kinetics in NPM1c, PML-RARA, RUNX1-RUNX1T1, and CBFB-MYH11 acute myeloid leukemias. Blood. 2010;115:198–205. doi: 10.1182/blood-2009-04-212530. [DOI] [PubMed] [Google Scholar]
- 65.Branford S, Yeung DT, Prime JA, et al. BCR-ABL1 doubling times more reliably assess the dynamics of CML relapse compared with the BCR-ABL1 fold rise: implications for monitoring and management. Blood. 2012;119:4264–4271. doi: 10.1182/blood-2011-11-393041. [DOI] [PubMed] [Google Scholar]
- 66.Naumovski L, Morgan R, Hecht F, Link MP, Glader BE, Smith SD. Philadelphia chromosome-positive acute lymphoblastic leukemia cell lines without classical breakpoint cluster region rearrangement. Cancer Res. 1988;48:2876–2879. [PubMed] [Google Scholar]
- 67.Sokol RJ, Hudson G, James NT, Frost IJ, Wales J. Human macrophage development: a morphometric study. J Anat. 1987;151:27–35. [PMC free article] [PubMed] [Google Scholar]
- 68.Boesen AM. Stereologic analysis of the ultrastructure in isolated human T and non-T lymphoid cells. II. Data on blasts in ALL; correlation with immunologic studies and FAB-morphology. Virchows Arch B Cell Pathol Incl Mol Pathol. 1983;42:303–314. doi: 10.1007/BF02890392. [DOI] [PubMed] [Google Scholar]
- 69.Iwama Y, Eguchi M. Quantitative evaluation of leukemic mitochondria with a computer-controlled image analyzer. Virchows Arch B Cell Pathol Incl Mol Pathol. 1986;51:375–384. doi: 10.1007/BF02899046. [DOI] [PubMed] [Google Scholar]