

Abstract
Aggregatibacter actinomycetemcomitans is a Gram negative oral bacterium associated with localized aggressive periodontitis (LAP). Detection of A. actinomycetemcomitans in clinical samples is routinely done by PCR. Our aim was to develop a rapid and reliable PCR method that can be used as a chair-side tool to detect A. actinomycetemcomitans in clinical samples. Sensitivity and specificity assessment was performed on buccal and plaque samples obtained from 40 adolescents enrolled in an ongoing LAP study by comparing 20 A. actinomycetemcomitans-positive subjects and 20 who were negative. In a second study, A. actinomycetemcomitans presence was tested in oral samples from eighty-six primates that included rhesus monkeys, chimpanzees, marmosets, tamarins and baboons. All samples were processed for detection of A. actinomycetemcomitans by means of culture, conventional PCR (cPCR) and rapid PCR (rPCR) using a Super Convection based AmpXpress thermal cycler (AlphaHelix, Sweden). For human samples, culture, cPCR and rPCR showed perfect agreement. Using this method A. actinomycetemcomitans was detected in 27 of 32 rhesus monkeys, 4 of 8 chimpanzees and 1 of 34 marmosets. Rapidity of AmpXpress thermal cycler, combined with Ready-To-Go PCR beads (GE Life sciences), a quick DNA extraction kit (Epicentre Biotechnologies, Madison, Wisconsin, USA) and a bufferless fast agarose gel system, made it possible to obtain results on A. actinomycetemcomitans detection within 35 min. We conclude that AmpXpress fast PCR can be conveniently used as a chair-side tool for rapid detection of A. actinomycetemcomitans in clinical samples.
Keywords: A. actinomycetemcomitans, Localized aggressive periodontitis, rapid PCR, Super convection
1. Introduction
Aggregatibacter actinomycetemcomitans is a Gram negative coccobacillus implicated in localized aggressive periodontitis (Zambon, 1985; Christersson, 1993). A. actinomycetemcomitans colonizes the oral cavities of humans (Slots, 1976; Socransky and Haffajee, 1992) and non-human primates (Eke et al., 1993) and belongs to the HACEK group of organisms believed to be associated with a number of systemic diseases including infective endocarditis (Das et al., 1997; Ellner et al., 1979; Paturel et al., 2004).
In addition to conventional culture based methods used to identify A. actinomycetemcomitans, PCR is now a well-established and widely used technique (Flemmig et al., 1995; Tran and Rudney, 1999). A large number of reports exist in the literature describing PCR based identification of A. actinomycetemcomitans (Flemmig et al., 1995; Goncharoff et al., 1993; Kim et al., 2005). While several studies have aimed at identifying only A. actinomycetemcomitans in the specimen, others have identified additional oral bacteria apart from A. actinomycetemcomitans e.g., using multiplex PCR (Tran and Rudney, 1999). Furthermore, 16S rDNA has been used as the target in PCR in many studies, but other genes such as lktA (Flemmig et al., 1995; Goncharoff et al., 1993; Tonjum and Haas, 1993) have also been used to identify A. actinomycetemcomitans. PCR based detection of bacteria in clinical specimens is sensitive and specific (Ficarra and Eversole, 1992; Olive, 1989). Conventional PCR, however, is time-consuming more often than not. This is mainly due to poor heat transfer on conventional PCR machines, resulting in longer time required to complete the reaction. In a clinical study setting during field screening of patients, generally samples are collected and brought to the laboratory where samples are processed and PCR performed for A. actinomycetemcomitans identification. The overall time for conventional PCR can vary from 2 to 4 h to overnight (Kramer and Coen, 2001). It is advantageous when conducting a screening examination to identify subjects who harbor A. actinomycetemcomitans at chair side within a short time period so that they can be informed that they are carriers of this potentially pathogenic organism. Extended time periods required for conventional PCR are inconvenient and can result in loss of subject interest and participation in ongoing studies. Therefore, rapid attainment of data at chair side during screening examinations could provide a great advantage and should improve recruitment of subjects. In our approach to develop a rapid PCR method for the detection of A. actinomycetemcomitans, we utilized samples from A. actinomycetemcomitans-positive and negative subjects who were involved in a longitudinal study of the relationship of A. actinomycetemcomitans to the initiation of localized aggressive periodontitis. In addition, oral samples from several primate species were used to compare culture to conventional PCR and to a new Super Convection rapid PCR technique. In this report we demonstrate that the new ultrafast PCR technique can be conveniently used as a chair-side tool for rapid A. actinomycetemcomitans detection.
2. Materials and methods
2.1. Bacterial culture
Buccal and plaque samples were suspended in A. actinomycetemcomitans Growth Medium (AAGM) broth [trypticase soy broth with 0.8% glucose (8 g/l), 0.6% yeast extract (6 g/l) and 0.4% sodium bicarbonate (4 g/l), 75 μg/ml bacitracin and 5 μg/ml vancomycin] and brought to the laboratory for processing. In some cases, plaque and buccal samples were collected using cytology brushes, and then the brushes were stabbed in half-strength AAGM agar in small glass vials before being sent to our laboratory. Once samples reached the laboratory, serial 10-fold dilutions were made and spread on AAGM plates. After 3 days of incubation at 37 °C and 10% CO2, A. actinomycetemcomitans colonies were preliminarily identified by colony morphology and catalase positivity. Presumptive A. actinomycetemcomitans colonies were subcultured from each sample. Human A. actinomycetemcomitans isolate IDH781 and Aggregatibacter aphrophilus ATCC® 33389™, a phylogenetic relative of A. actinomycetemcomitans, were also grown on AAGM as above.
2.2. Purification of DNA
DNA from the buccal/plaque samples from humans and primates, and genomic DNA from A. actinomycetemcomitans isolates was purified using DNeasy® blood and tissue kit from Qiagen (QIAGEN Sciences, Germantown, Maryland, USA). Briefly, the samples were treated with a lysis buffer and proteinase-K overnight at 56 °C, followed by extraction and purification of DNA using Qiaquick spin columns (Qiagen). DNA from buccal samples from subjects with or without LAP were extracted using QuickExtract™ DNA extraction kit from Epicentre Biotechnologies (Madison, Wisconsin, USA). The swab samples were suspended first in a quick DNA extract solution and heated at 65 °C for 6 min and then the tubes were transferred to 98 °C and incubated for 2 min. The extracted DNA was stored at —20 °C.
2.3. Human sampling and analysis
Buccal samples from 40 subjects (29 females and 11 males, mean age = 14 year) enrolled in an ongoing LAP study (20 A. actinomycetemcomitans-positive subjects and 20 A. actinomycetemcomitans-negative) were used for detecting A. actinomycetemcomitans by both cPCR and rPCR. All volunteers gave consent, using a form that was reviewed and approved by the Institutional Review Board (IRB) of the University of Medicine & Dentistry of New Jersey (UMDNJ).
2.4. Primate sampling
Monkey samples were collected from the North East Regional Primate Research Center (NEPRC) at Harvard University, Southwest National Primate Research Center (SNPRC), Yerkes Regional Primate Research Center at Emory University and Laboratory Animal Services facility at Rutgers University. All monkeys (Table 2) had an intact dentition and were housed in separate cages. Prior to sampling all primates were anesthetized using ketamine hydrochloride (15 mg/kg) and a supplement of isoflurane. Buccal mucosa of the monkeys was sampled with sterile wooden tongue depressors. Plaque samples were collected using autoclaved dental scalers. The samples were suspended in AAGM broth and processed for bacterial culture as described above. Sample collection from primates was approved by the Institutional Animal Care and Use Committees of the UMDNJ, Harvard University and Rutgers University.
Table 2.
Specificity and sensitivity of cPCR and rPCR in detecting A. actinomycetemcomitans from buccal samples from subjects.
| Culture results | Conventional PCR | Rapid PCR | |
|---|---|---|---|
| Aaa positive | 20 | 16 | 16 |
| Aa negative | 20 | 17 | 17 |
| Culture vs PCR | cPCR vs rPCR | ||
|
| |||
| Sensitivity | 80% | 100% | |
| Specificity | 85% | 100% | |
Aa: A. actinomycetemcomitans.
2.5. Super convection rapid PCR
Table 1 shows primers and amplicon sizes. For all PCR reactions, Ready-To-Go beads (GE HealthCare Biosciences, Buckinghamshire, UK) were used. When each bead was reconstituted to 25 μl final volume, the concentration of each dNTP was 200 μM in 10 mM Tris–HCl (pH 9), 50 mM KCl and 1.5 mM MgCl2. Primers were used at a concentration of 0.5 μM and the amount of template DNA was 50–100 ng per reaction. PCR reactions were performed on the rapid PCR machine AmpXpress (Alpha Helix Molecular Diagnostics AB, Sweden). Rapid PCR is facilitated by a centrifugation based convection technology used in the instrument (Martensson et al., 2006). A typical thermal profile consisted of an initial denaturation of 94 °C for 1 min followed by 30 cycles of 94 °C for 0 s, 55 °C for 6 s and 72 °C for 7 s. No final elongation was required.
Table 1.
PCR primers.
| Primer | Sequence (5′–3′) | Amplicon | Reference |
|---|---|---|---|
| lktA | GGAATTCCTAGGTATTGCGAAACAATTTGATC | 262 | Goncharoff et al. (1993) |
| GGAATTCCTGAAATTAAGCTGGTAATC | |||
| Aa 16S rDNA | TAGCCCTGGTGCCCGAAGC | 428 | Kim et al. (2005) |
| CATCGCTGGTTGGTTACCCTCTG | |||
| Serotype b | TCTCCACCATTTTTGAGTGG | 333 | Kaplan et al. (2001) |
| Serotype c | GAAACCACTTCTATTTCTCC | 268 | Kaplan et al. (2001) |
| Serotype f | CCTTTATCAATCCAGACAGC | 232 | Kaplan et al. (2001) |
| Universal Fwd for sero b, c and f | ARAAYTTYTCWTCGGGAATG | ||
| Serotype a | TGGGTCATGGGAGGTACTCC | 293 | Kaplan et al. (2001) |
| GCTAGGAACAAAGCAGCATC | |||
| Serotype d | GGAACGGGTATGGGAACGG | 411 | Kaplan et al. (2001) |
| GGATGCTCATCTAGCCATGC | |||
| Serotype e | ATTCCAGCCTTTTGGTTCTC | 311 | Kaplan et al. (2001) |
| TGGTCTGCGTTGTAGGTTGG |
2.6. Conventional PCR
A Techne TC-412 PCR machine (Techne Inc. Burlington, NJ, USA) was used. All PCR reagents were the same as described above for rapid PCR. Except for variable annealing temperatures for different primer pairs, the temperature profile was as follows: Initial denaturation 94 °C for 10 min followed by 30 cycles of 94 °C for 1 min, 55 °C for 1 min and 72 °C for 1 min.
2.7. Agarose gel electrophoresis
PCR products were visualized by electrophoresis through a 2% agarose gel after adding the dye EZ-Vision™ Three (Amresco, Ohio, USA) to the entire PCR product, i.e., 25 μl. In rPCR experiments, a rapid and bufferless agarose gel system that completes in 6 min at 250 V (Febe bufferless agarose gel; Biokeystone Co, California, USA) was used. The gels were then exposed to UV light on a trans illuminator and pictures were taken by the attached Kodak DC290 camera.
3. Results
3.1. Validation of the efficacy of rPCR
In order to test the reliability of the Super Convection rPCR, we performed PCR for lktA of A. actinomycetemcomitans strains in parallel both on the AmpXpress machine as well as the conventional PCR machine. Fig. 1A shows that all A. actinomycetemcomitans strains tested produced an expected 262-bp band of similar intensity from both PCR machines. Sensitivity of the super convection rPCR was also compared with that of the conventional PCR. Genomic DNA from a serial 10-fold dilutions of A. actinomycetemcomitans IDH781 was used in the same PCR reaction as above using A. actinomycetemcomitans-specific lktA primers. It was possible to amplify the fragment at a bacterial concentration as low as 103/ml using rPCR, similar to cPCR (Fig. 1B). Comparison of rPCR with cPCR in terms of time requirements is schematically shown in Fig. 2. The time requirement for each step of either PCR method was established after running at least 3 experiments. Regardless of using a quick DNA extraction method and a fast agarose gel electrophoresis system, the cPCR takes approximately 2 h, while rPCR requires as little as 35 min to obtain results (Fig. 2).
Fig. 1.
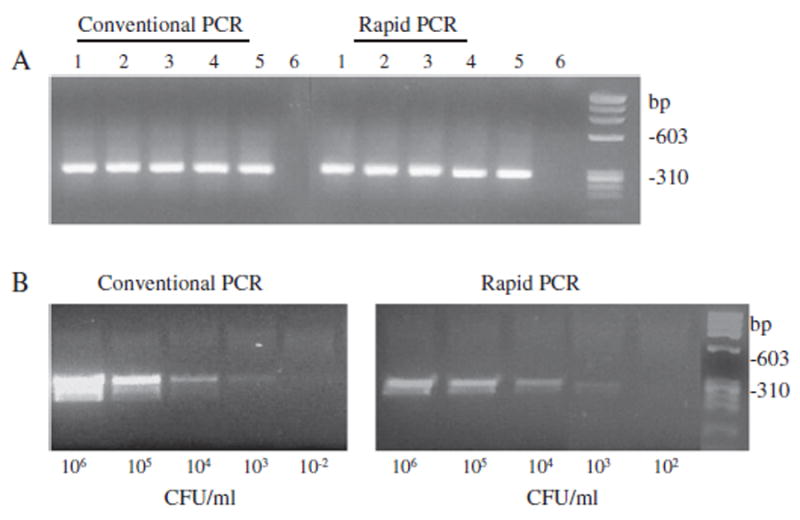
Validation of the efficacy of the AmpXpress rPCR. Panel A: Efficacy of AmpXpress rapid PCR machine was compared to that of a regular PCR machine. A. actinomycetemcomitans-specific lktA primers (Goncharoff et al., 1993), were used to amplify a 262-bp fragment of the lktA gene. Purified genomic DNA from A. actinomycetemcomitans strains was used for PCR at 50 ng per reaction. Lanes: A. actinomycetemcomitans strains IDH781 (lane 1), HK1651 (lane 2), JP2 (lane 3), CU1000 (lane 4), NJ4500 (lane 5) and negative control (lane 6). Panel B: Detection limit of cPCR and rPCR. Purified genomic DNA from a serial 10-fold dilution of A. actinomycetemcomitans IDH781 was used. Equal volume of DNA sample from each dilution was used in PCR reactions.
Fig. 2.
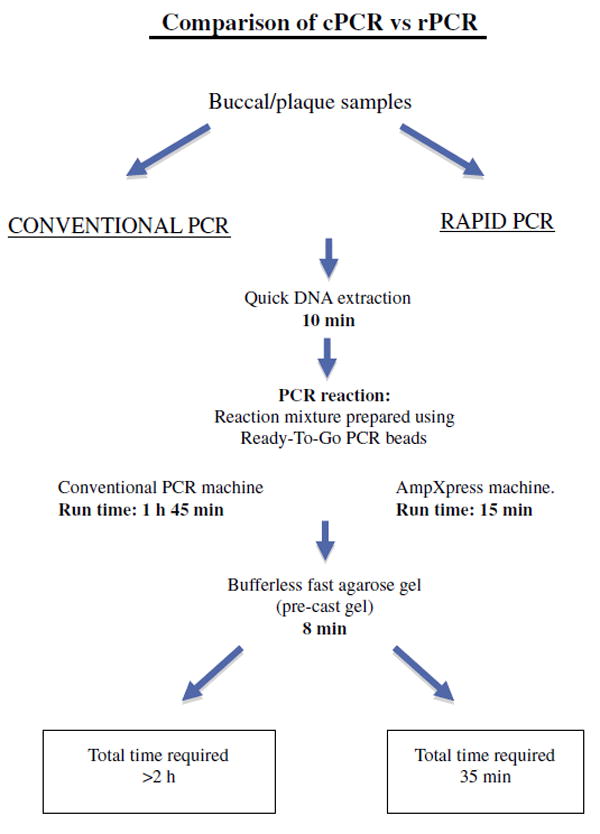
Time requirements by cPCR and rPCR—from sample collection to data acquisition. Schematic presentation of time required for each step in the detection of A. actinomycetemcomitans by rPCR as compared to cPCR. In the case of rPCR, it is necessary that all equipments and reagents be ready before beginning in order to complete quickly in 35 min. Notwithstanding the use of a quick DNA extraction method and a fast agarose gel electrophoresis system, cPCR takes approximately 2 h, which is still outside a “chairside” timeframe.
3.2. Efficacy of super convection AmpXpress PCR with respect to human clinical samples
Buccal samples from 40 subjects (20 each from A. actinomycetemcomitans-positive and -negative subjects) were subjected to A. actinomycetemcomitans detection by using both cPCR and rPCR. Data for 10 A. actinomycetemcomitans-positive samples are shown in Fig. 3. The results showed that 16 of 20 A. actinomycetemcomitans culture-positive were A. actinomycetemcomitans positive by PCR (sensitivity=80%) while 3 of 20 A. actinomycetemcomitans culture-negative were PCR positive for A. actinomycetemcomitans (specificity=85%) (Table 2).
Fig. 3.
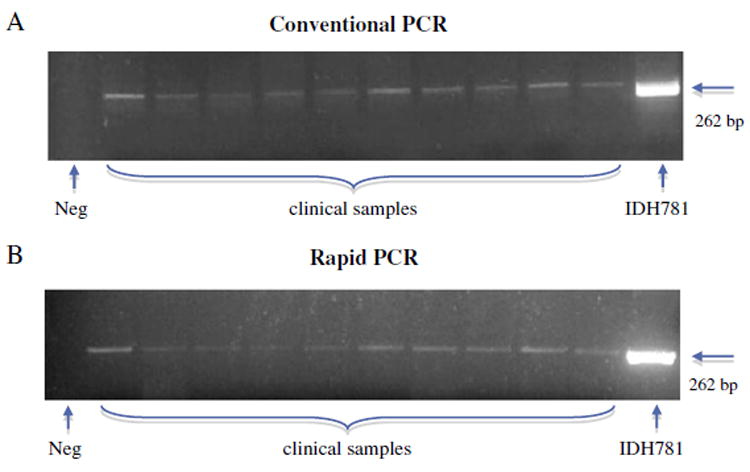
Detection of A. actinomycetemcomitans from human clinical samples by cPCR and rPCR. DNA purified from buccal samples from 40 subjects (20 each from A. actinomycetemcomitans-positive and A. actinomycetemcomitans-negative subjects) were used for PCR detection of A. actinomycetemcomitans. Panel A: cPCR; DNA was purified using the Qiagen kit, PCR performed on a regular PCR machine and PCR products were run on a conventional agarose gel. Panel B: rPCR; DNA was purified using QuickExtract™ solution, PCR performed on the AmpXpress rapid PCR machine, and PCR products were run on a 2% agarose gel in a bufferless agarose gel system for 6 min. Lanes: Data from 10 random A. actinomycetemcomitans-positive samples is shown. Lanes 1–10: LAP samples; lane 11: A. actinomycetemcomitans IDH781, used as positive control. Neg = negative control.
3.3. Identification of A. actinomycetemcomitans isolates from monkeys by rPCR
After establishing the efficacy of rPCR for rapid identification of A. actinomycetemcomitans from clinical samples, we then utilized the method to study the prevalence of A. actinomycetemcomitans among Old World and New World non-human primates. Primers specific for A. actinomycetemcomitans lktA sequence (Goncharoff et al., 1993) were used for the identification of A. actinomycetemcomitans. Data for 11 strains are shown in Fig. 4 panel A. Twenty seven of 32 rhesus monkeys, 4 of 8 chimpanzees and 1 of 34 marmosets harbored A. actinomycetemcomitans as revealed by amplification of lktA fragment (Table 3). No A. actinomycetemcomitans was detected from cynomolgus, baboon or tamarin group of monkeys.
Fig. 4.
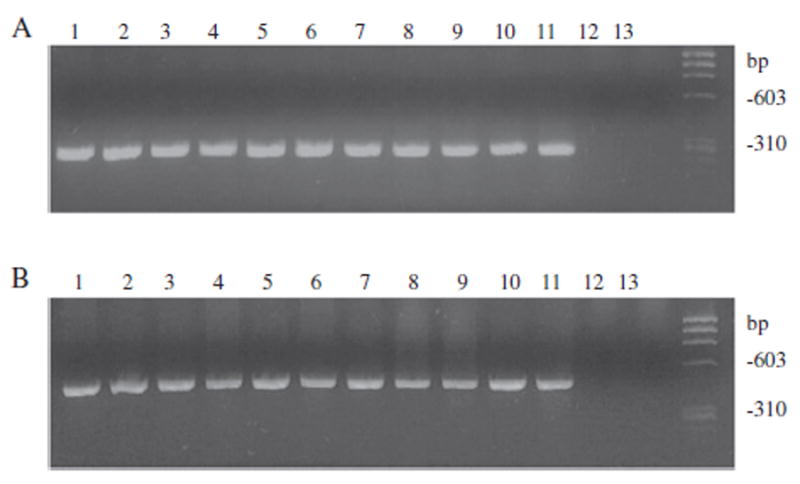
Identification of A. actinomycetemcomitans isolates from monkeys by rPCR. Panel A: A. actinomycetemcomitans-specific lktA primers were used for the identification of A. actinomycetemcomitans by rPCR. Panel B: A. actinomycetemcomitans-specific 16S rDNA primers were used to confirm the results from lktA PCR. Lanes: Eleven randomly selected monkey A. actinomycetemcomitans isolates (lanes 1–11), A. aphrophilus ATCC 33389 (lane 12) and negative control (lane 13).
Table 3.
Monkeys used in this study.
| Monkey group | Age (years) | Sex | A. actinomycetemcomitans-positive | Serotype |
|---|---|---|---|---|
| Mean (range) | ||||
| Rhesus (n=32) | 8.5 (2–15) | Male=16 | 27 (84%) | d=12 |
| Female=16 | b+c+d+e=4 | |||
| c+f=1 | ||||
| f=10 | ||||
| Chimpanzee (n=8) | 28.2 (15–33) | Male=2 | 4 (50%) | b=3 |
| Female=6 | c=1 | |||
| f=4 | ||||
| Cynomolgus (n=4) | 5.7 (5–6) | Male=4 | 0 | |
| Female=0 | ||||
| Baboon (n=4) | 15.7 (14–19) | Male=1 | 0 | |
| Female=3 | ||||
| Tamarin (n=4) | 2 (2–2) | Male=2 | 0 | |
| Female=2 | ||||
| Marmoset (n=34) | 5 (2–8) | Male=20 | 1 (2.9%) | f=1 |
| Female=14 |
Identity of A. actinomycetemcomitans was further confirmed by A actinomycetemcomitans-specific 16S rRNA PCR (Kim et al., 2005). An expected band of 468 bp size was seen in all strains lktA-positive for A actinomycetemcomitans. A. aphrophilus ATCC 33389, a phylogenetic relative of A. actinomycetemcomitans was used as a negative control and did not show any band (Fig. 4, panel B).
3.4. Serotyping of monkey A. actinomycetemcomitans isolates
Serotypes of monkey A. actinomycetemcomitans were determined by PCR using serotype-specific primers (Table 1) (Suzuki et al., 2001). Among 19 A. actinomycetemcomitans-positive rhesus monkeys at NEPRC, 12 had serotype d and four had mixtures of serotypes b, c, d and e but d occurred in 16 of them, while 3 strains were of serotype f (Table 3). In the case of Rutgers rhesus monkeys all four isolates were of serotype f, but two were additionally positive for serotype b or c. From the monkey colony at SNPRC, all three isolates from rhesus monkeys and a single isolate from a marmoset were all serotype f.
4. Discussion
Although microbiological and biochemical tools are an essential part of A. actinomycetemcomitans identification, PCR is used as a routine and common technique. In clinical studies involving subjects, rapid identification of A. actinomycetemcomitans might be of great advantage since chair-side identification could inform patients of the presence of this pathogenic bacterium. In this study, we demonstrated that using a Super Convection rapid PCR technique, A. actinomycetemcomitans could be quickly detected in buccal or plaque samples from humans in 35 min (Fig. 2).
Conventional PCR machines take a longer time for a reaction to complete since heating and cooling are based on diffusion (deMello, 2003; Jia et al., 2007; Kramer and Coen, 2001). In contrast, rapid PCR on AmpXpress is facilitated by the convection heating mechanism, where high velocities of the reaction mixture in rotating tubes impart homogeneous temperature and excellent mixing. This eventually results in less time needed for each cycle (Gidlof et al., 2009; Martensson et al., 2006). Two previous reports have utilized this technology for RT-PCR quantification of non-oral viruses (Gidlof et al., 2009; Martensson et al., 2006). In those reports, although the technology was the same, thermal cycling and duration of reaction were longer because the experimental setup was RT-PCR. Furthermore, several other rapid PCR systems have been reported in the literature, but the limitations of those systems are either commercial unavailability or non-portability of the equipment (Muddu et al., 2011; Oda et al., 1998; Wheeler et al., 2011). Therefore, this is the first report showing successful utilization of AmpXpress, a portable Super Convection PCR machine for rapid bacterial identification. We first validated the efficacy of the rapid PCR instrument by comparing it against a conventional PCR machine. Amplification of an lktA band of similar intensity from both machines suggests that AmpXpress is as efficient as the conventional PCR machine. Furthermore, sensitivity of PCR reaction was also the same on both instruments, i.e., lowest detection limit of 103 cfu/ml. This is in agreement with several earlier studies using conventional PCR machines for A. actinomycetemcomitans identification (Flemmig et al., 1995; Poulsen et al., 2003).
For identification of A. actinomycetemcomitans by PCR, different primers specific for A. actinomycetemcomitans 16S rDNA have been used (Kim et al., 2005; Tran and Rudney, 1999). In this study, we utilized primers that are specific for the lktA fragment of A. actinomycetemcomitans (Goncharoff et al., 1993). These primers are highly specific for A. actinomycetemcomitans and do not cross react with any of the 13 other common oral bacteria (Goncharoff et al., 1993). Additionally, we performed PCR for A. actinomycetemcomitans identification using species-specific 16S rDNA primers (Kim et al., 2005). That A. aphrophilus, a close phylogenetic relative of A. actinomycetemcomitans, did not show any band substantiates the specificity of these primers.
Our initial goal was to determine the feasibility of using the AmpXpress method for chair-side identification in clinical samples so that we could inform subjects who had consented to participate in a longitudinal study that they harbored this potentially dangerous microorganism. As a result of both the sensitivity and specificity of the results obtained in this pilot study we decided to include plaque and buccal samples from several primate species to expand the sample size and also to investigate the carriage of A. actinomycetemcomitans in different primate species. This allowed us to analyze an additional 86 oral primate samples for A. actinomycetemcomitans carriage.
Of interest was the finding that A. actinomycetemcomitans was detected in 84% (27 of 32) of rhesus monkeys obtained from three different primate research centers, and only one of 34 marmosets. Four of eight chimpanzees harbored A. actinomycetemcomitans; while baboons and tamarins did not show any A. actinomycetemcomitans. Admittedly the sample size for these species needs to be expanded in the future. Nevertheless, our preliminary survey of primates shows a significantly higher association of A. actinomycetemcomitans with Old World monkeys as opposed to New World primates which is in line with previous studies demonstrating A. actinomycetemcomitans bound specifically to buccal epithelial cells from Old World monkeys (Yue et al., 2007). Occurrence of periodontitis in Old World and New World monkeys is ambiguous in the literature. Some studies have reported spontaneous development of the disease in monkeys in wild and in captivity (Dreizen and Levy, 1977; Page et al., 1972). It is possible however, that the form of periodontitis in those animals was chronic periodontitis in older monkeys and may not be aggressive periodontitis. To gain a greater insight into A. actinomycetemcomitans carriage in primates and the effect of A. actinomycetemcomitans on disease initiation, we are currently in the process of whole genome sequencing of A. actinomycetemcomitans isolates; comparative genomic analysis of A. actinomycetemcomitans isolates from monkeys versus humans is also in progress in our laboratory. In conclusion, the rPCR method consisting of Ready-To-Go PCR beads, a quick DNA extraction kit (Epicentre Biotechnologies) and a bufferless fast agarose gel system makes it possible to detect A. actinomycetemcomitans in no more than 35 min and thus provides a rapid and accurate method of detecting A. actinomycetemcomitans at chair side within a reasonable time period.
Acknowledgments
This study was supported by NIDCR grants R21DE021172 and R01DE017968 to D. H. Fine.
References
- Christersson LA. Actinobacillus actinomycetemcomitans and localized juvenile periodontitis. Clinical, microbiologic and histologic studies. Swed Dent J Suppl. 1993;90:1–46. [PubMed] [Google Scholar]
- Das M, Badley AD, Cockerill FR, Steckelberg JM, Wilson WR. Infective endocarditis caused by HACEK microorganisms. Annu Rev Med. 1997;48:25–33. doi: 10.1146/annurev.med.48.1.25. [DOI] [PubMed] [Google Scholar]
- deMello AJ. Microfluidics: DNA amplification moves on. Nature. 2003;422:28–29. doi: 10.1038/422028a. [DOI] [PubMed] [Google Scholar]
- Dreizen S, Levy BM. Monkey models in dental research. J Med Primatol. 1977;6:133–144. doi: 10.1159/000459735. [DOI] [PubMed] [Google Scholar]
- Eke PI, Braswell L, Arnold R, Fritz M. Sub-gingival microflora in Macaca mulatta species of rhesus monkey. J Periodontal Res. 1993;28:72–80. doi: 10.1111/j.1600-0765.1993.tb01053.x. [DOI] [PubMed] [Google Scholar]
- Ellner JJ, Rosenthal MS, Lerner PI, McHenry MC. Infective endocarditis caused by slow-growing, fastidious, Gram-negative bacteria. Medicine. 1979;58:145–158. doi: 10.1097/00005792-197903000-00003. [DOI] [PubMed] [Google Scholar]
- Ficarra G, Eversole LR. Polymerase chain reaction: relevance for oral pathology. Minerva Stomatol. 1992;41:425–429. [PubMed] [Google Scholar]
- Flemmig TF, Rudiger S, Hofmann U, Schmidt H, Plaschke B, Stratz A, Klaiber B, Karch H. Identification of Actinobacillus actinomycetemcomitans in subgingival plaque by PCR. J Clin Microbiol. 1995;33:3102–3105. doi: 10.1128/jcm.33.12.3102-3105.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gidlof O, Burvall S, Edvinsson L, Montelius M, Allen M, Molin M. Complete discrimination of six individuals based on high-resolution melting of hypervariable regions I and II of the mitochondrial genome. Biotechniques. 2009;47:671–672 674, 676, passim. doi: 10.2144/000113197. [DOI] [PubMed] [Google Scholar]
- Goncharoff P, Figurski DH, Stevens RH, Fine DH. Identification of Actinobacillus actinomycetemcomitans: polymerase chain reaction amplification of lktA-specific sequences. Oral Microbiol Immunol. 1993;8:105–110. doi: 10.1111/j.1399-302x.1993.tb00554.x. [DOI] [PubMed] [Google Scholar]
- Jia G, Siegrist J, Deng C, Zoval JV, Stewart G, Peytavi R, Huletsky A, Bergeron MG, Madou MJ. A low-cost, disposable card for rapid polymerase chain reaction. Colloids Surf B Biointerfaces. 2007;58:52–60. doi: 10.1016/j.colsurfb.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Kaplan JB, Perry MB, MacLean LL, Furgang D, Wilson ME, Fine DH. Structural and genetic analyses of O polysaccharide from Actinobacillus actinomycetemcomitans serotype f. Infect Immun. 2001;69:5375–5384. doi: 10.1128/IAI.69.9.5375-5384.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SG, Kim SH, Kim MK, Kim HS, Kook JK. Identification of Actinobacillus actinomycetemcomitans using species-specific 16S rDNA primers. J Microbiol. 2005;43:209–212. [PubMed] [Google Scholar]
- Kramer MF, Coen DM. Enzymatic amplification of DNA by PCR: standard procedures and optimization. In: Ausubel Frederick M, et al., editors. Current protocols in molecular biology. Unit 15. Chapter 15. 2001. p. 11. [DOI] [PubMed] [Google Scholar]
- Martensson G, Skote M, Malmqvist M, Falk M, Asp A, Svanvik N, Johansson A. Rapid PCR amplification of DNA utilizing Coriolis effects. Eur Biophys J. 2006;35:453–458. doi: 10.1007/s00249-006-0052-z. [DOI] [PubMed] [Google Scholar]
- Muddu R, Hassan YA, Ugaz VM. Rapid PCR thermocycling using microscale thermal convection. J Vis Exp. 2011;5:49. doi: 10.3791/2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda RP, Strausbauch MA, Huhmer AF, Borson N, Jurrens SR, Craighead J, Wettstein PJ, Eckloff B, Kline B, Landers JP. Infrared-mediated thermocycling for ultrafast polymerase chain reaction amplification of DNA. Anal Chem. 1998;70:4361–4368. doi: 10.1021/ac980452i. [DOI] [PubMed] [Google Scholar]
- Olive DM. Detection of enterotoxigenic Escherichia coli after polymerase chain reaction amplification with a thermostable DNA polymerase. J Clin Microbiol. 1989;27:261–265. doi: 10.1128/jcm.27.2.261-265.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page RC, Simpson DM, Ammons WF, Schectman LR. Host response in chronic periodontal disease. J Periodontal Res. 1972;7:283–296. doi: 10.1111/j.1600-0765.1972.tb01717.x. [DOI] [PubMed] [Google Scholar]
- Paturel L, Casalta JP, Habib G, Nezri M, Raoult D. Actinobacillus actinomycetemcomitans endocarditis. Clin Microbiol Infect. 2004;10:98–118. doi: 10.1111/j.1469-0691.2004.00794.x. [DOI] [PubMed] [Google Scholar]
- Poulsen K, Ennibi OK, Haubek D. Improved PCR for detection of the highly leukotoxic JP2 clone of Actinobacillus actinomycetemcomitans in subgingival plaque samples. J Clin Microbiol. 2003;41:4829–4832. doi: 10.1128/JCM.41.10.4829-4832.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slots J. The predominant cultivable organisms in juvenile periodontitis. Scand J Dent Res. 1976;84:1–10. doi: 10.1111/j.1600-0722.1976.tb00454.x. [DOI] [PubMed] [Google Scholar]
- Socransky SS, Haffajee AD. The bacterial etiology of destructive periodontal disease: current concepts. J Periodontol. 1992;63:322–331. doi: 10.1902/jop.1992.63.4s.322. [DOI] [PubMed] [Google Scholar]
- Suzuki N, Nakano Y, Yoshida Y, Ikeda D, Koga T. Identification of Actinobacillus actinomycetemcomitans serotypes by multiplex PCR. J Clin Microbiol. 2001;39:2002–2005. doi: 10.1128/JCM.39.5.2002-2005.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonjum T, Haas R. Identification of Actinobacillus actinomycetemcomitans by leukotoxin gene-specific hybridization and polymerase chain reaction assays. J Clin Microbiol. 1993;31:1856–1859. doi: 10.1128/jcm.31.7.1856-1859.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran SD, Rudney JD. Improved multiplex PCR using conserved and species-specific 16S rRNA gene primers for simultaneous detection of Actinobacillus actinomycetemcomitans, Bacteroides forsythus, and Porphyromonas gingivalis. J Clin Microbiol. 1999;37:3504–3508. doi: 10.1128/jcm.37.11.3504-3508.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler EK, Hara CA, Frank J, Deotte J, Hall SB, Benett W, Spadaccini C, Beer NR. Under-three minute PCR: probing the limits of fast amplification. Analyst. 2011;136:3707–3712. doi: 10.1039/c1an15365j. [DOI] [PubMed] [Google Scholar]
- Yue G, Kaplan JB, Furgang D, Mansfield KG, Fine DH. A second Aggregatibacter actinomycetemcomitans autotransporter adhesin exhibits specificity for buccal epithelial cells in humans and Old World primates. Infect Immun. 2007;75:4440–4448. doi: 10.1128/IAI.02020-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambon JJ. Actinobacillus actinomycetemcomitans in human periodontal disease. J Clin Periodontol. 1985;12:1–20. doi: 10.1111/j.1600-051x.1985.tb01348.x. [DOI] [PubMed] [Google Scholar]