

Abstract
Introduction
‘Multi-omics’ datasets obtained from an organism of interest reared under different environmental treatments are increasingly common. Identifying the links among metabolites and transcripts can help to elucidate our understanding of the impact of environment at different levels within the organism. However, many methods for characterizing physiological connections cannot address unidentified metabolites.
Objectives
Here, we use Eigenvector Metabolite Analysis (EvMA) to examine links between metabolomic, transcriptomic, and phenotypic variation data and to assess the impact of environmental factors on these associations. Unlike other methods, EvMA can be used to analyze datasets that include unidentified metabolites and unannotated transcripts.
Methods
To demonstrate the utility of EvMA, we analyzed metabolomic, transcriptomic, and phenotypic datasets produced from 20 Drosophila melanogaster genotypes reared on four dietary treatments. We used a hierarchical distance-based method to cluster the metabolites. The links between metabolite clusters, gene expression, and overt phenotypes were characterized using the eigenmetabolite (first principal component) of each cluster.
Results
EvMA recovered chemically related groups of metabolites within the clusters. Using the eigenmetabolite, we identified genes and phenotypes that significantly correlated with each cluster. EvMA identifies new connections between the phenotypes, metabolites, and gene transcripts. Conclusion EvMA provides a simple method to identify correlations between metabolites, gene expression, and phenotypes, which can allow us to partition multivariate datasets into meaningful biological modules and identify under-studied metabolites and unannotated gene transcripts that may be central to important biological processes. This can be used to inform our understanding of the effect of environmental mechanisms underlying physiological states of interest.
Keywords: Eigenvector metabolite analysis, Linkage analyses, Environment, Enrichment analyses
1 Introduction
With growing use of high throughput techniques, the availability of ‘omics’ datasets obtained from an organism of interest under the same experimental conditions is increasing (see Culibrk et al. (2016); Gehlenborg et al. (2010); Rebollar et al. (2016); Zhang et al. (2010) for reviews of using ‘multi-omics’ datasets in different systems). In particular, studies utilizing a ‘multiomics’ approach that combine metabolomic and transcriptomic datasets can address a variety of questions that include: species identification and taxonomy (Raupach et al. 2016), personalized medicine and natural product development (Katz and Baltz 2016; Gligorijević et al. 2016), and the impact of different environments on phenotypes and/or traits of interest (Reed et al. 2014; Williams et al. 2015; Jia et al. 2016; Fei et al. 2016). Furthermore, identifying linkages between these datasets using network (Gustafsson et al. 2014; Valcàrcel et al. 2014) and correlation (Redestig and Costa 2011) analyses and can help to elucidate a systems level understanding of the impact of environment on an organism of interest or the mechanisms underlying human diseases.
Many current methods for integrating data on metabolites, transcripts, and phenotypes are network or pathway based (Gehlenborg et al. 2010). Programs are available that allow the user to analyze and visualize interactions between metabolomic and transcriptomic data [e.g., 3Omics (Kuo et al. 2013) and MassTRIX (Wägele et al. 2012)]. Other programs use this type of data to conduct pathway analyses [e.g., MarVis-Pathway (Kaever et al. 2015)] or examine regulatory networks [e.g., FlexFlux (Marmiesse et al. 2015)]. In some cases, a method is focused on identifying a single pathway of interest [e.g., methionine salvage; Cho et al. (2014)]. These methods focus on identifying the effect of the environment (or other external variables) on the linkages between endophenotypes (e.g., metabolite and transcript expression patterns) (Lakshmanan et al. 2015; Osorio et al. 2012; Serra et al. 2015; Trikka et al. 2015), rather than identifying how these endophenotypes relate to an organism’s visible external phenotype. Furthermore, these methods require that metabolites be identified or the transcripts be annotated with a molecular function. However, untargeted metabolomic analyses identify a large number of metabolites, but not all of them will be identified (Dunn et al. 2012), and transcriptomic analyses frequently recover gene products with no available annotation (Alvarez et al. 2015; Pavey et al. 2012). Thus, these pathway-based methods exclude portions of the data that could prove informative to a study.
In the enclosed paper, we use a newer approach to—omics integration, Eigenvector Metabolite Analysis (EvMA), for examining the impact of environmental factors on linkages between metabolomic, transcriptomic, and phenotypic variation data. EvMA can identify linkages in datasets that include metabolites and transcripts that are not fully annotated. Similar approaches have successfully identified linkages across other—omics data and have shown promise in a range of metabolomics datasets (McHardy et al. 2013; Peng et al. 2014). The resulting linkages can be used to inform functional predictions of genes with previously unknown functions and categorize unidentified metabolites into broad chemical and biological categories. We demonstrate the utility of EvMA by reexamining the datasets from Reed et al. (2014). This data was obtained from a population of wild-derived isofemale lines of Drosophila melanogaster reared on four different diets. The impact of environment on the linkages is assessed for each diet set individually and a dataset combined across diets. We also show that EvMA identifies patterns not found by the other analysis methods used on this dataset (Reed et al. 2014; Williams et al. 2015) and can be used to improve functional categorization of unknown transcripts and metabolites.
2 Eigenvector metabolite analysis
Eigenvector metabolite analysis (EvMA; Fig. 1) can be used to examine metabolomic, transcriptomic, and phenotypic data, as well as assess the effect of environment on both metabolite clustering patterns and linkages between metabolites, gene expression, and phenotypes of interest. Unlike gene-set pathway-based methods that require annotated metabolites and transcripts, it is possible to quantify linkages between unidentified metabolites and functionally uncharacterized transcripts using EvMA. To identify the connections between these datasets, this method incorporates: (1) distance-based hierarchical clustering analysis (HCA) of metabolic features, (2) principal component analysis (PCA) of metabolic clusters to identify an eigenmetabolite for the cluster, (3) Pearson correlation analysis between eigenmetabolite, gene transcripts, and phenotypes, and (4) enrichment analyses of significantly correlated genes. The distance-based hierarchical clustering analysis examines the effect of different environments on variation in metabolite concentration patterns. The threshold for defining clusters in the analysis should be chosen to maximize the fraction of the variance explained by the first few principal components of each cluster, while keeping the size of the clusters manageable and interpretable, and thus are somewhat subjective. However, as the field of clustering analysis of biological data matures, ways to set the clustering threshold in a biologically meaningful way entirely algorithmically could be developed. After identifying the clustering patterns of the metabolic features, PCA is used to describe the major axis of variance in each cluster, the eigenmetabolite. A common concern of using PCA is the effect of noise in the dataset on the clustering patterns (Halouska and Powers 2006). Through using PCA to describe the variance of discrete modules (e.g., clusters) instead of the complete dataset, EvMA reduces the impact of noise blurring the underlying biological signal. By completing a Pearson correlation analysis using the eigenvector of the first principal component of each cluster (e.g., eigenmetabolite), gene transcripts and phenotypes that are significantly correlated with each metabolite cluster can be identified. A gene-set enrichment analysis is then used to identify overrepresented biological annotations associated with the list of significantly correlated genes for each cluster to aid in biological interpretation of the quantitative summaries. The resulting output from these analyses can help to quantify the impact of the environment on metabolites, gene transcripts, and phenotypes, and can place unknown metabolites and transcripts in a biological context.
Fig. 1.

Workflow of eigenvector metabolite analysis. A distance based hierarchical clustering analysis is used to group the metabolic features obtained under a specific treatment. The makeup of each cluster is classified based on the chemical classes of the metabolites. To describe the variance of a cluster, a PCA is completed, and the eigenvector of the first principal component of the cluster (e.g., the eigenmetabolite) is then used for the further analyses. To identify significantly correlated gene transcripts and overt phenotypes, a Pearson correlation analysis is completed using the eigenmetabolite. For significantly correlated gene transcripts, overexpressed biological annotations are identified using an enrichment analysis
3 Materials and methods
To demonstrate the utility of EvMA, this method was applied to the metabolomic, transcriptomic, and phenotypic data of Reed et al. (2014) that was collected from D. melanogaster reared on four environmental (dietary) treatments. We applied EvMA to the data from the dietary treatments, both individually and in combination, and compared the linkages identified using EvMA to the findings from each of these previous studies.
3.1 Experimental dataset
To represent a diverse natural population, 20 experimental D. melanogaster lines were chosen that exhibited a diversity of phenotypic reaction norms (e.g., pupal body weight and larval triglyceride storage) to different dietary treatments (Reed et al. 2010). The larvae of each line were reared on four environmental (dietary) treatments that varied in the sugar and fat composition. The examined treatments included a normal diet (e.g., used to maintain the flies in the lab; 4 % sucrose and <0.2 % fat), a low calorie diet (0.75 % glucose and <0.2 % fat), a high glucose diet (4 % glucose and <0.2 % fat), and a high fat diet (0.75 % glucose and 3 % coconut oil (saturated fat)). See Reed et al. (2010) and Reed et al. (2014) for a detailed description and rationale of the diets used.
Samples consisted of three food vials per experimental line per diet per time replicate. Each vial was seeded with 50 early first instar larvae. Randomized blocks of four synchronized lines were run on all dietary treatments, and three independent time replicates were completed for each combination of line and diet. Late third instar larvae in the food vials were pooled, fasted, and samples were then drawn for metabolomics, gene expression, and the overt larval phenotypes of larval triglyceride storage (lipid) and trehalose level of larvae (sugar). Additional vials were allowed to develop through the pupal stage to allow for the measurement the overt pupal phenotypes of body weight (weight), percentage of larvae that survive to pupation (larval survival), percentage of pupae that successfully mature (pupal survival), and time from first instar larvae to mature pupae (development time). Reed et al. (2010); Reed et al. (2014) provide a complete description of the methods used to measure the phenotypes.
A Nimblegen 12-plex Drosophila expression array was used to obtain whole genome expression data following the manufacturers’ protocols (Nuwaysir et al. 2002). Of 15,595 possible genes, 11,650 genes were expressed at detectable levels. This data is available in the GEO database (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE50745.
Metabolomic profiling was conducted on samples of six larvae pooled from three replicate vials of a genotype and diet. Gas chromatography-mass spectrometry (GC–MS) was used to analyze the pooled samples (see Reed et al. (2014) for a complete description of the metabolomic analyses). In the metabolic profiles, 189 reliably detectable metabolic features were identified (Reed et al. 2014). This data was pre-processed, and the tolerant cut-off value for metabolites missing proportion was set at 25 %, leading to the removal of two features from further analyses. The chemical category of the remaining 187 features was determined by searching profiles against the National Institute of Standards and Technology (NIST) database (Linstrom and Mallard 2016; http://webbook.nist.gov; retrieved July 18, 2012). Standards for candidate compounds were run on the GC–MS, and the identity of 58 of the metabolites was confirmed (Table 1). Of the remaining metabolic features, 126 were annotated with confidence to a chemical class (e.g., saturated fatty acid or amino acid). The identities of the three remaining features are uncertain.
Table 1.
Primary metabolites identified to Level 1 of the Metabolite Standards Initiative and number used to represent the peak in Figs. 2 and 3
| Metabolite class | Metabolite | ID# |
|---|---|---|
| Amino acid/amino acid-like | Alanine | 3 |
| Arginine monohydrochloride | 23 | |
| Asparagine | 31 | |
| Aspartic acid | 26 | |
| Glutamic acid | 28 | |
| Glycineb | 4, 7, 16 | |
| Isoleucinec | 12, 13 | |
| Leucine | 10 | |
| Methionine | 27 | |
| Norleucine | 17 | |
| Phenylalanine | 29 | |
| Proline | 15 | |
| Serineg | 8, 20 | |
| Threonineh | 14, 21 | |
| Tyrosinej | 41, 42 | |
| Urea or alanine | 2 | |
| Valine | 6 | |
| Fatty acid/fatty acid-like | Arachidate | 52 |
| Caprate | 22 | |
| Elaidate or linolenate | 50 | |
| Laurate | 30 | |
| Margarate | 48 | |
| Hexadecanoic acid, palmitate | 46 | |
| Linoleate or olenic acid or petroselinate | 49 | |
| Myristic acid, myristate | 37 | |
| Stearate | 51 | |
| Palmitoleate | 45 | |
| Sugar/sugar-like | Fructosea | 34, 35, 38, 40 |
| Glucose | 36 | |
| Glucose or fructose | 39 | |
| Glucose or myo-inositol | 44 | |
| Glycerol | 9 | |
| Malic acid | 24 | |
| Maltosed | 53, 55 | |
| Mannose or fructosee | 32, 33 | |
| Meso-erythritol/erythritol | 25 | |
| Myo-inositol | 43 | |
| Propanoic acid | 1 | |
| Succinic acid | 18 | |
| Trehalosei | 57, 58 | |
| Other | Arachidonoyl dopamine | 56 |
| l-Dopa | 47 | |
| Adenosine | 54 | |
| Uracil | 19 | |
| Phosphoric acidf | 5, 11 |
Multiple peaks identified as same compound
3.2 Eigenvector metabolite analysis
3.2.1 Hierarchical clustering analysis
A hierarchical clustering analysis based on distance methods was applied to each dietary treatment and the combined dataset. This analysis was completed using the hclust.r package in the R programming language (RCoreTeam 2013). The Euclidean distance method was applied to all clustering analyses. For each dietary treatment and the combined dataset, a range of cluster numbers (e.g., 5–8 clusters) was examined for collapsing data into eigenmetabolites. The number of clusters (Figs. 2, 3a–d) used in the downstream analysis was chosen to maximize the variance explained by the first and second principal component of each cluster and to disrupt large clusters. This selection process combines an algorithmic analysis (e.g., comparison of the results of hierarchical clustering analysis over a range of numbers) with the judgment of the investigators as to what was the optimal cluster number, which maximized variance but resulted in few large clusters. This is often referred to as a “Elbow method” of cluster selection. The Elbow method looks at the percentage of variation explained by the first eigenvector of the metabolites in each cluster as a function of the number of clusters. The results were visually inspected, and the number of clusters was chosen such that that adding another cluster doesn’t give much better modeling of the data. More precisely, if one plots the percentage of variance explained by the clusters against the number of clusters, the first clusters will add much information (explain a lot of variance), but at some point the marginal gain will drop, giving an angle in the graph. The number of clusters is chosen at this point, hence the “elbow criterion” (Thorndike 1953). The elbow approach was performed independently for each diet, thus resulting meaningful groups of metabolites with manageable (and interpretable) cluster sizes.
Fig. 2.
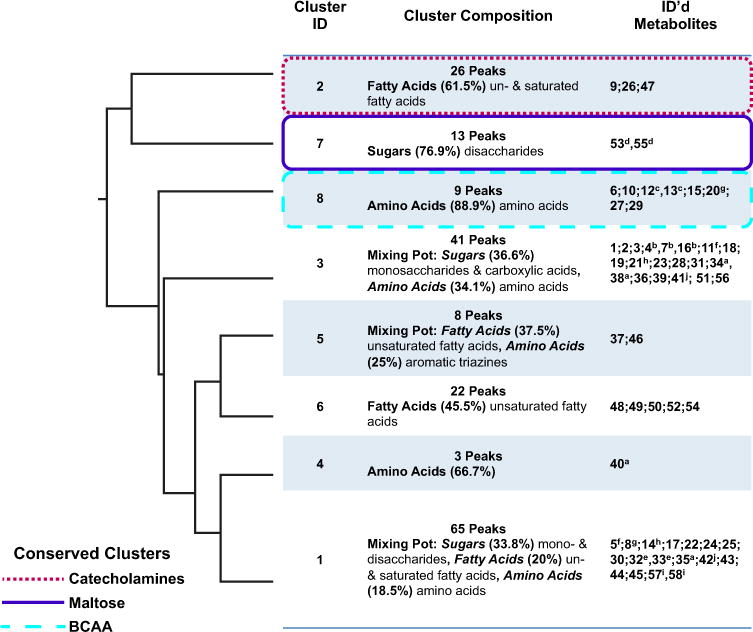
Hierarchical clustering of combined dataset. The topology recovered among the eight metabolite clusters in the combined dataset of all diets. For each cluster, the assigned identification number (cluster ID), the number of features (peaks) found in the cluster, the chemical classification of the cluster (cluster composition), and the identification number based on Table 1 of metabolites identified using standards are provided (ID’ed metabolites). Metabolite clusters that are conserved across and within the four dietary treatments (Catecholamines, Maltose, and BCAA) are identified using different colored boxes: dotted red (Catecholamines), solid purple (Maltose), and broken blue (BCAA)
Fig. 3.
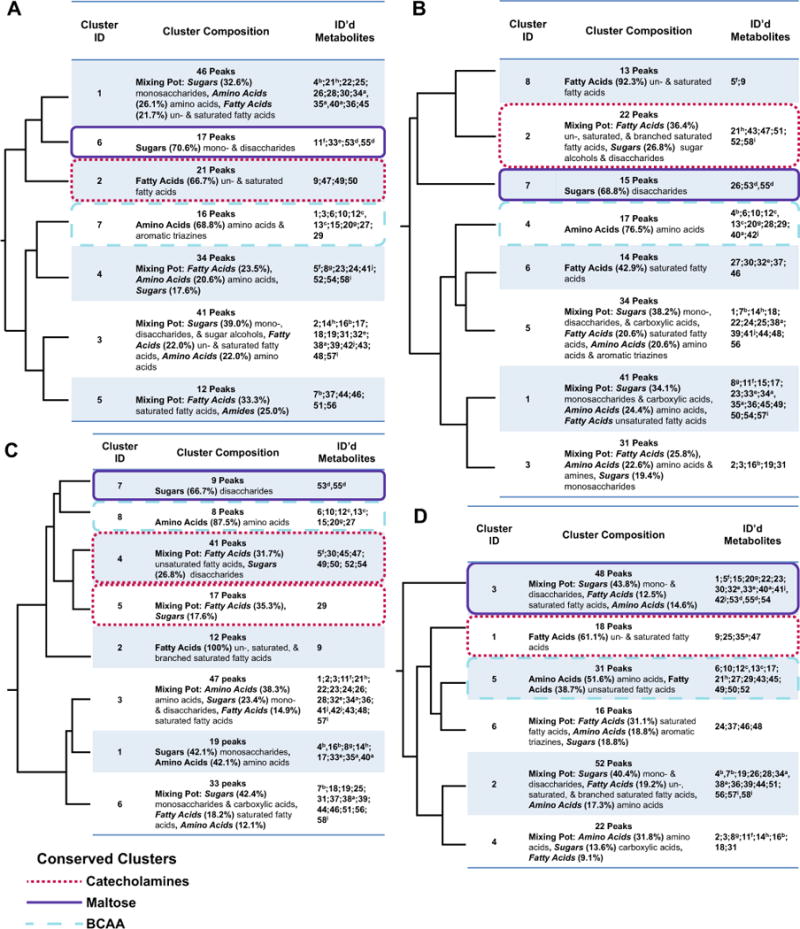
Hierarchical clustering of individual diets. The topology recovered among metabolite clusters in individual dietary treatments: Normal (a), Low Calorie (b), High Glucose (c), and High Fat (d). Labeled as in Fig. 2
To aid in interpretation, the makeup of each cluster was classified based on the chemical classes of the metabolic features that were found in the cluster. Clusters that did not contain a majority of a single chemical class (e.g., fatty acid/fatty acid-like) were defined as “Mixing Pots”, and the chemical classes that primarily composed the clusters were described. See Supplemental Data 1 for a complete list of the metabolites, retention times, identifications, and cluster placement for each dataset.
3.2.2 Principal component analysis
After the final cluster number was determined, a principal component analysis (PCA) was conducted for the metabolites within each cluster. Screeplots and R2 values for each principal component were examined to determine which principal component(s) should be used to characterize the metabolite cluster. Based on these analyses, the eigenmetabolite (e.g., eigenvector of the first principal component of a cluster) was found to well represent the variation of the clusters. See Supplemental Data 2 for the R2 of the eigenmetabolite of each cluster. The empirical results lent further confidence in the interpretability of the eigenmetabolites, as clusters that were composed of metabolites from similar chemical classes had eigenvector(s) that explained large amounts of variation, and clusters with metabolites from across different chemical classes had lower R2 values. A previous study has found similar results (Halouska and Powers 2006). This is a benefit of the multistage approach, because removing the noisy metabolites from the more interpretable, similar clusters makes it easier to find the biological signal in a complex dataset (improving the signal to noise ratio).
3.2.3 Pearson correlation analysis
To identify gene transcripts and overt phenotypes that significantly correlated with each metabolite cluster, the eigenmetabolite of each cluster recovered for the combined dataset and each dietary treatment was used in a Pearson correlation analysis. The Pearson correlation analyses were completed using the cor() function in R, and tested using a two-sided t test for correlations. Correlation analysis was used to assess the associations between each of the 11,650 individual, detected transcripts and the metabolite clusters represented by the eigenmetabolite. To control for issues with multiple comparisons, P-values were adjusted using both the Bonferroni correction method and the False Discovery Rate as calculated according to Storey (2002) (Supplemental Data 2). Transcripts found to significantly correlate with a cluster (q-value < 0.05) were retained for further analysis. The correlation analysis was also completed individually for each of the six measured overt phenotypes and the eigenmetabolite of a cluster, and considered significant at p-value < 0.05.
3.2.4 Enrichment analysis
The significantly correlated gene transcripts from the Nimblegen 12-plex microarray were identified with Fly-Base (http://flybase.org/; dos Santos et al. 2015) transcript IDs from the fourth release (Dmel Release 4.3, 2006 March). We used the FlyBase Upload/Convert IDs tools to update the identification of all significantly correlated transcripts to the sixth release (FB2015_2). In some cases, it was not possible to update a transcript ID (e.g., transcript was associated with a pseudogene). Genes that could not be updated were excluded from further analysis (see Supplemental Data 2 for exact numbers). The FlyBase Batch Downloader Tool was used to obtain the associated name or gene ID for each updated transcript (see Supplemental Data 3 for a complete list of the updated significantly correlated genes). The lists of genes were then assessed for overrepresented biological annotations using the functional annotation tool in the DAVID Bioinformatics Resource v6.7 [http://david.abcc.ncifcrf.gov/; (Huang et al. 2009a, 2009b)]. The functional annotation chart was obtained for each list (Supplemental Data 4), and the five most significantly enriched Gene Ontology (GO) Terms and KEGG pathways (p-value < 0.05) are presented in Tables 2 and 3.
Table 2.
Summary of Pearson correlation and enrichment analysis for combined dataset§
| Cluster ID and classification(s) | Number of correlated genes | Named genes | Enriched GO terms | Enriched KEGG pathways | Overt phenotypes |
|---|---|---|---|---|---|
| 2 Fatty acids (Catecholamines) | 0 | – | – | – | Weight Larval survival |
| 7 Sugars (Maltose) | 845 | Stress induced DNase Vacuolar Peduncle Start1 Congested-like trachea Heterochromatin Protein 1D3 chromoshadow domain |
GO:0055114 ~ oxidation reduction GO:0045333 ~ cellular respiration GO:0015980 ~ energy derivation by oxidation of organic compounds GO:0022904 ~ respiratory electron transport chain GO:0022900 ~ electron transport chain |
dme00260:Glycine, serine and threonine metabolism dme00903:Limonene and pinene degradation dme00350:Tyrosine metabolism |
Sugar Weight Pupal survival |
| 8 Amino acids (BCAA) | 688 | Adenosine deaminase-related growth factor A* Astray* Prolyl-4-hydroxylase-alpha EFB* Utx histone demethylase Amyrel |
GO:0055114 ~ oxidation reduction GO:0009408 ~ response to heat GO:0009266 ~ response to temperature stimulus GO:0001666 ~ response to hypoxia‡ GO:0070482 ~ response to oxygen levels‡ |
dme04144:Endocytosis dme02010:ABC transporters |
Weight |
| 3 Mixing pot | 3134 | Cuticular protein 31A Fumarylacetoacetase Cyp49a1 LambdaTry Enhancer of split mα, Bearded family member |
GO:0055114 ~ oxidation reduction GO:0044429 ~ mitochondrial part GO:0005739 ~ mitochondrion GO:0006091 ~ generation of precursor metabolites and energy GO:0045333 ~ cellular respiration |
dme00190:Oxidative phosphorylation dme03050:Proteasome dme00650:Butanoate metabolism dme00280:Valine, leucine and isoleucine degradation dme00020:Citrate cycle (TCA cycle) |
Sugar Lipid Weight |
| 5 Mixing pot | 9 | Dawdle Adipokinetic hormone receptor Acetyl-CoA carboxylase |
– | – | Larval survival Pupal survival Development time |
| 6 Fatty acids | 3013 | Heat shock gene 67Ba Maltase A2 Prolyl-4-hydroxylase-alpha EFB Heat shock gene 67Bc* Esterase P* |
GO:0044429 ~ mitochondrial part GO:0005739 ~ mitochondrion GO:0003735 ~ structural constituent of ribosome GO:0033279 ~ ribosomal subunit GO:0005840 ~ ribosome |
dme00190:Oxidative phosphorylation dme03010:Ribosome dme00020:Citrate cycle dme04330:Notch signaling pathway dme00650:Butanoate metabolism |
Weight Development time |
| 1 Mixing pot | 227 | Keren Kinesin-like protein at 68-D Dead box protein 73D Acetyl-CoA carboxylase Astray* Lethal (2) 34Fd* |
GO:0042254 ~ ribosome biogenesis GO:0022613 ~ ribonucleoprotein complex biogenesis GO:0006364 ~ rRNA processing GO:0016072 ~ rRNA metabolic process GO:0034660 ~ ncRNA metabolic process |
– | Larval survival Pupal survival Development time |
Includes the classification(s) of a cluster, the total number of genes correlated with a cluster, and the five most significantly correlated named genes, enriched GO Terms, KEGG Pathways, and Overt Phenotypes
Equal q-values
Equal p-values
Only metabolite clusters correlated with genes or overt phenotypes are included
Table 3.
Summary of Pearson correlation and enrichment analysis for Individual Dietary Treatments§
| Diet | Cluster ID and classification (s) | Number of correlated genes | Named genes | Enriched GO terms | Enriched KEGG pathways | Overt phenotypes |
|---|---|---|---|---|---|---|
| Normal |
6 Sugars (Maltose) |
33 | GDAP1 Ortholog* Senescence marker protein-30* Serpent* Nicastrin* LambdaTry* |
– | – | Sugar Weight |
|
2 Fatty acids (Catecholamines) |
0 | – | – | – | Weight | |
|
7 Amino acids (BCAA) |
0 | – | – | – | Weight | |
|
4 Mixing pot |
3 | Phosphoribosylamidotransferase 2 | – | – | – | |
|
3 Mixing pot |
0 | – | – | – | Weight | |
|
5 Mixing pot |
0 | – | – | – | Sugar Weight |
|
| Low calorie |
8 Fatty acids |
440 | Pasilla Next 184 significantly correlated named genes have same q-value. |
G0:0007444 ~ imaginal disc development G0:0035220 ~ wing disc development G0:0009791 ~ post-embryonic development G0:0048569 ~ post-embryonic organ development G0:0002165 ~ instar, larval, or pupal development |
dme04320:Dorso-ventral axis formation dme04013:MAPK signaling pathway dme04330:Notch signaling pathway |
Weight |
|
2 Mixing pot (Catecholamines) |
0 | – | – | – | Weight | |
|
4 Amino acids (BCAA) |
0 | – | – | – | Development time | |
|
6 Fatty acids |
303 | Cuticular protein 62Bc* Fatty Acid Synthase 2* Obstructor-E* Cuticular protein 31A* LambdaTry* |
GO:0055114 ~ oxidation reduction GO:0044429 ~ mitochondrial part GO:0005739 ~ mitochondrion GO:0005740 ~ mitochondrial envelope G0:0031966 ~ mitochondrial membrane |
– | Pupal survival Development time |
|
|
3 Mixing pot |
92 | Succinyl-CoA: 3-ketoacid CoA transferase* Holes in muscle* Esterase 6* Cuticular protein 31A* Next 5 significantly correlated named genes have same q-value |
GO:0055114 ~ oxidation reduction GO:0009072 ~ aromatic amino acid family metabolic process GO:0005759 ~ mitochondrial matrix‡ GO:0031980 ~ mitochondrial lumen‡ GO:0006816 ~ calcium ion transport |
– | Weight Development Time |
|
| High glucose |
7 Sugars (Maltose) |
0 | – | – | – | Development time |
|
8 Amino acids (BCAA) |
4 | Astray | GO:0006911 ~ phagocytosis, engulfment GO:0006909 ~ phagocytosis |
– | – | |
|
4 Mixing pot (Catecholamines) |
0 | – | – | – | Weight | |
|
5 Mixing pot (Catecholamines) |
2575 | Kallmann syndrome 1 ortholog Esterase P* Tachykinin* CAP-D2 condensin subunit* Matrix metalloproteinase 2* |
GO:0044429 ~ mitochondrial part GO:0005739 ~ mitochondrion GO:0005761 ~ mitochondrial ribosome‡ GO:0000313 ~ organellar ribosome‡ GO:0005740 ~ mitochondrial envelope |
dme00190:0xidative phosphorylation dme00650:Butanoate metabolism dme04330:Notch signaling pathway dme00770:Pantothenate and CoA biosynthesis |
Sugar Weight | |
|
2 Fatty acids |
0 | – | – | – | Larval survival Pupal survival |
|
|
3 Mixing pot |
0 | – | – | – | Weight | |
|
6 Mixing pot |
276 | Peroxidase Nicotinic Acetylcholine Receptor betal Peroxin 7 Esterase P Utx histone demethylase |
GO:0042302 ~ structural constituent of cuticle GO:0005198 ~ structural molecule activity GO:0005184 ~ neuropeptide hormone activity GO:0005214 ~ structural constituent of chitin-based cuticle GO:0005576 ~ extracellular region |
– | Lipid Weight |
|
| High fat |
1 Fatty acids (Catecholamies) |
0 | – | – | – | Weight Development time |
|
2 Mixing pot |
434 | Scheggia Fumarylacetoacetase Cuticular protein 67Fal* Cyp4adl* Sclp* Fatty acid synthase 2* |
GO:0055114 ~ oxidation reduction GO:0048037 ~ cofactor binding GO:0044429 ~ mitochondrial part GO:0005759 ~ mitochondrial matrix‡ GO:0031980 ~ mitochondrial lumen‡ |
dme00280:Valine, leucine and isoleucine degradation | Weight | |
|
4 Mixing pot |
0 | – | – | – | Lipid |
Includes the classification(s) of a cluster, the total number of genes correlated with a cluster, and the five most significantly correlated named genes, enriched GO Terms, KEGG Pathways, and Overt Phenotypes
Equal q-values
Equal p-values
Only metabolite clusters correlated with genes or overt phenotypes are included
4 Results and discussion
Below we discuss the results from our EvMA of the metabolite, transcript, and phenotype dataset of Reed et al. (2014) and compare them with the results from previous analyses of the dataset (Reed et al. 2014; Williams et al. 2015) to identify findings unique to EvMA.
4.1 Environmental effect on metabolite clustering
To assess the effect of environment on metabolite clustering with EvMA, we used a distance based HCA of the combined dataset and each of the four dietary treatments (Figs. 2, 3a–d). The metabolomic dataset consisted of 187 features, which were present in more than 75 % of the samples. These features were predominantly composed of three general chemical classes: sugars/sugar-like compounds (53 features), fatty acids/fatty acid-like compounds (46 features), and amino acids/amino acid-like compounds (42 features). The specific identity of 58 of the metabolic features was empirically confirmed using known chemical standards (Table 1), while the chemical class of the remaining metabolites was determined through comparison to the NIST database (Linstrom and Mallard 2016).
One of the most compelling overall findings of our EvMA is that the hierarchical clustering recovered groups of metabolites of similar chemical character based on correlation alone. Often a cluster contained only a few empirically confirmed compounds, but the other metabolites in the cluster showed generally similar characteristics that allowed us to categorize them to a general chemical class such as amino acid-like compounds. Since the correlation and clustering data recovered groups of metabolites that fall within the same general chemical category, we have confidence that the chemical category assignment is appropriate. This assignment allows us to learn something of the biological significance of these unknown metabolites even though we do not have definitive chemical identities for them. The categorical information we can assign to unknowns will improve our ability to identify them definitively in the future. Given that a lack of chemical identification is a major challenge in metabolomics, EvMA provides one method to begin to sort and prioritize the unknowns.
In a previous analysis of this dataset, Reed et al. (2014) determined that environmental factors (e.g., diet) were responsible for a significant portion of the variation (9 %) in the metabolome. Williams et al. (2015) also found that within these metabolite profiles the effects of different diets were well detected. Similarly, EvMA demonstrated that diet did affect the clustering patterns of some specific metabolites. However, EvMA also highlighted that the correlation patterns of other metabolites were conserved across the combined dataset and in individual dietary treatments. For example, the three branched chain amino acids (BCAA) were found in a single cluster composed primarily of amino acids in each of the treatments, but the other metabolites that occurred in the BCAA cluster varied between diets and include aromatic amino acids and other amino acids. Other metabolites whose correlation patterns were conserved include the neurotransmitter L-DOPA and two additional unidentified features that are putatively characterized in the catecholamine chemical class (TARGET_526 AND TARGET_559; Supplemental Data 1) as determined from their best matches to the NIST database. These three metabolic features were always found in the same cluster (Figs. 2, 3a, b, d) or adjacent clusters [e.g., high glucose diet (Fig. 3c)]. In addition, the clusters that contained these three metabolic features were primarily composed of fatty acids or were classified as “Mixing Pots” that contained a substantial number of fatty acids. Thus, EvMA analysis demonstrates that environment influences the clustering pattern of some but not all metabolic features.
In addition to identifying metabolites whose clustering patterns were not affected by diet, EvMA identified environmental factors that were especially perturbing to the metabolite correlation patterns. For example, in all of the examined datasets, except that of the high glucose diet, we consistently found a cluster composed predominantly of sugars and sugar-like compounds that included maltose and an additional 11 disaccharides. In the HCA analysis of the high glucose diet (Fig. 3c) these disaccharides were divided between two non-adjacent clusters (Cluster 7 and Cluster 4); thus, the high glucose diet was highly perturbing to the metabolic processes that usually regulate disaccharide levels. These results demonstrate that EvMA can detect distinct impacts on correlation patterns that result from different environments.
4.2 Function of correlated genes
Using EvMA, we identified transcripts whose expression significantly correlated (q-value < 0.05) with the variation observed in a metabolite cluster, the eigenmetabolite. Some clusters were associated with a large number of genes, while others were correlated with fewer than 10 genes. This suggests that the linkage between gene expression and metabolome varies dramatically across clusters. This is consistent with the findings of Reed et al. (2014) that showed genetic variation contributed more greatly to gene expression variation, while diet played a more significant role in metabolome variation. An important conclusion then is that gene expression based regulation does not influence all metabolites equally. For example, we found that the conserved BCAA cluster was only significantly correlated with genes in the combined dataset (688 genes; Cluster 8; Table 2) and the high glucose diet (4 genes; Cluster 8; Table 3), but did not show a significant association with any transcripts on the other treatments. See Supplemental Data 3 for lists of all significantly correlated genes for each cluster and treatment.
4.2.1 Individual named genes
EvMA identifies genes involved in the metabolism of metabolites in the metabolite clusters, which can be used as a tool to hypothesize function for genes that otherwise have no known function. We demonstrate this principle through examining the function of the named genes associated with clusters. We found several instances where individual correlated genes are active in physiological processes that utilize the metabolites (or their byproducts) found in the correlated metabolite cluster. For example, the BCAA cluster in the combined dataset and the high glucose diet contained serine and was significantly correlated with Astray, a phosphoserine phosphatase gene that is active in the metabolism and biosynthesis of serine by modifying phosphoserine, a precursor metabolite (Tables 2, 3). The combined dataset BCAA cluster also contained proline and was correlated with prolyl-4-hydroxylase-alpha EFB, which acts to hydroxylate peptidyl-proline. In addition, several clusters from individual diets or the combined dataset that were primarily composed of fatty acids (Cluster 6; Fig. 3b; Table 3) or contained a substantial number of fatty acids (Cluster 1 and 5; Fig. 2; Table 2) were correlated with genes that act in fatty acid biosynthesis (e.g., Fatty acid synthase 2 and acetyl-CoA carboxylase) and lipid catabolism (e.g., adipokinetic hormone receptor). A similar pattern of correlation with genes that function in pathways that utilize associated metabolites was found in the cluster of the combined dataset that contains the aromatic amino acid tyrosine (Cluster 3). This cluster was correlated with two genes that function in the aromatic amino acid metabolic process (e.g., fumarylacetoacetase and Cyp49a1). Thus, we provide proof of concept that using EvMA, it is possible to predict the function of individual genes associated with a given cluster based on its composition, which can in turn be leveraged to improve gene annotations.
4.2.2 Biological annotations of gene lists
A major finding in our EvMA is that by using an enrichment analysis of all genes associated with a given cluster, we often found cases where the overrepresented biological annotations (e.g., GO terms, KEGG pathways) included physiological processes that make use of correlated metabolites or their byproducts. Since we demonstrated these logical connections based on correlations alone, we can leverage the correlations between eigenmetabolites and transcripts to assign biological function to genes and metabolites that have not been functionally characterized previously. A limitation to this analysis is that short gene lists are unlikely to demonstrate enrichment. With the exception of the four genes correlated with Cluster 8 in the high glucose diet (Table 3), no list that included 30 or fewer genes was enriched for either GO terms or KEGG pathways. Furthermore, only lists with >400 genes had significantly enriched biological annotations that included KEGG pathways.
Combined Dataset Transcript-Eigenmetabolite Associations
An example of a logical association between the members of a metabolite cluster and the correlated gene transcripts occurred in the maltose cluster of the combined dataset (Cluster 7; Fig. 2; Table 2), which was primarily composed of sugars and correlates with 845 genes. These genes were enriched for cellular respiration (GO:0045333) and energy derivation by oxidation of organic compounds (GO:0015980). Similarly, Cluster 6 of the combined dataset was primarily composed of fatty acids and the 3,013 genes associated with Cluster 6 were enriched for the citrate cycle (00020:KEGG pathway), which requires the metabolite acetyl coenzyme A. While acetyl coenzyme A was not detected in our analysis, this metabolite is a product of fatty acid metabolism and is also active in the synthesis of fatty acids (Berg et al. 2002), making the link between the metabolites and transcripts for this cluster a logical one. A logical association was also present between the 3,134 genes correlated with the eigenmetabolite of Cluster 3 of the combined dataset and the metabolites found in the cluster (Table 2). The correlated genes were enriched for butanoate metabolism (00650:KEGG pathway) and oxidative phosphorylation (00190:KEGG pathway). Cluster 3 contained succinic acid, which participates directly in both of these metabolic pathways, and the amino acids tyrosine and arginine that can be degraded to provide metabolites necessary for butanoate metabolism. The cluster also contained the neuropeptide arachidonoyl dopamine. Previously, Williams et al. (2015) found that the genes correlated with this metabolite were enriched for enzymes in tyrosine metabolism, a pathway that gives rise to precursors of arachidonoyl dopamine. Our enrichment analysis also identified tyrosine metabolism as an enriched biological annotation of the gene list in the combined dataset. However, in the individual dietary treatments, the gene lists correlated with the metabolite cluster containing arachidonoyl dopamine were not significantly enriched for tyrosine metabolism. Thus, analysis of the data across environments in combination can assist in the detection of patterns not detectable in environment specific analyses.
Individual Diet Transcript-Eigenmetabolite Associations
Our analysis of the gene enrichment in the individual dietary treatments also recovered overrepresented biological annotations that are part of physiological pathways that utilize the metabolites in a cluster. For example, Cluster 6 of the high glucose diet contained the neuropeptide and catecholamine arachidonoyl dopamine described above and correlated with 276 genes, which are enriched for neuropeptide hormone activity (GO:0005184) and functions involved in the synthesis of cuticle (Fig. 3c; Table 3). Catecholamines play a role in behavior and learning in Drosophila (Martínez-Ramírez et al. 1992; Burke et al. 2012; Van Swinderen and Andretic 2011) and also act as crosslinking agents in insect cuticle (Evans 1980). Intriguingly, of the 15 transcripts that bear no functional classification in the 100 genes most significantly correlated with the eigenmetabolite for Cluster 6, six (CG14374, CG13059, CG12239, CG10311, CG15021, CG13230) are expressed at high levels in the adult head, carcass, or both, consistent with the enriched functions of neuropeptide hormone activity and cuticle synthesis found in the functionally characterized genes. Thus, these genes with unknown function may be good candidates for involvement in neuropeptide metabolism contributing to diet-specific overt phenotype variation. A logical association between a metabolite cluster and the biological functions of the correlated genes was also observed in Cluster 3 of the low calorie diet dataset (Fig. 3b; Table 3), which contained tyrosine (aromatic amino acid). The 90 genes correlated with the cluster were enriched for aromatic amino acid family metabolic process (GO:0009072).
Using EvMA, we demonstrate that collectively there are many examples of logical associations between the metabolites in the clusters and the genes correlated with the eigenmetabolite of that cluster. Since the function of significantly correlated genes (and overrepresented biological functions associated with correlated genes) can be predicted in part by their associated metabolites, we provide the proof of concept that correlations between known and unknown genes or metabolites found by EvMA can facilitate the generation of hypotheses about the function of the unknowns. For example, of the 100 most highly correlated gene transcripts with the eigenmetabolite for Cluster 7 of the combined dataset, 10 have no functional classification at all (Supplemental Data 3). Given that Cluster 7 is composed primarily of disaccharides we could hypothesize that the function of these uncharacterized genes may contribute to metabolism of disaccharides and may share functional similarity to genes in the overrepresented GO category of energy derivation by oxidation of organic compounds. Thus, they would be good candidates for further scrutiny by researchers interested in generating complete pathways for disaccharide metabolism.
Another example of an important novel finding of EvMA is the gene CG14005, which was the nineteenth most significantly correlated gene transcript (q = 0.0001) for the eigenmetabolite of Cluster 8 in the combined dataset that contains the BCAAs. This same transcript was also highly significantly correlated with Clusters 3 and 6 (q = 0.0005 and q = 0.003 respectively), and the eigenmetabolites of all three of these clusters were correlated with the overt phenotype of weight. CG14005 has no annotated function, but is characterized as having a Myb/SANT-like DNA-binding domain, which also has no known function. Given that this gene transcript is important for three metabolite clusters associated with weight, it is a prime candidate for functional testing to determine why it appears to have a central role in metabolism and thereby, perhaps also elucidating the biological function of a Myb/SANT-like DNA-binding domain.
4.3 Mechanistic links between phenotypes and environment
Using the EvMA, we identified which of six specific phenotypes significantly correlated with metabolic clusters. Previously, Reed et al. (2014) examined the impact of diet, genotype, and genotype-by-diet interactions on each of the phenotypes and found that certain metabolites could be used to predict phenotypes. Expanding upon this finding, Williams et al. (2015) assessed the modularity of the trehalose level of the hemolymph (sugar), triglyceride storage (lipid), and weight phenotypes and noted that the metabolites correlated with different phenotypes varied significantly between diets. The added findings in the results of our EvMA of the linkages between metabolic clusters and each of the six phenotypes highlight phenotypes that either correlate with unique metabolite clusters only in the combined dataset, correlate with metabolite clusters only in a diet specific manner, or correlate with specific metabolite clusters under all experimental conditions.
4.3.1 Combined dataset phenotype-eigenmetabolite associations
Using the EvMA to identify correlated phenotypes, we found some metabolites were only correlated with a phenotype of interest in the combined dataset. For example, the pupal survival phenotype was correlated with three metabolite clusters in the combined dataset (Clusters 1, 5, and 7; 65, 8, and 13 features; Table 2). Clusters 1 and 5 of the combined dataset were Mixing Pots. Cluster 7 contained mostly sugars (including maltose), whose correlation pattern was conserved across all treatments. However, the maltose cluster in the individual dietary treatments was not correlated significantly with pupal survival. Pupal survival was also correlated with a single metabolic cluster in the low calorie (Cluster 6; 14 features Table 3) and high glucose diets (Cluster 2; 12 features; Table 3). Both of these individual diet clusters were composed primarily of fatty acids, but there was little overlap between the metabolites found in these clusters and Clusters 1 and 5 of the combined dataset. These findings indicate that the manner in which metabolites mediate pupal survival differs across diets. The larval survival phenotype also demonstrated unique correlation patterns in the combined dataset. Larval survival was correlated with three clusters in the combined dataset (Clusters 1, 2, and 5) and Cluster 2 of the high glucose diet. There was no overlap between the metabolites found in Clusters 1 and 5 of the combined dataset, which were classified as Mixing Pots, and the high glucose cluster. Thus, this analysis provides proof of concept that examination of the combined dataset allows for the identification of distinct associations between metabolic features and a phenotype of interest that may not be detected when only analyzing correlations between metabolic clusters and phenotypes on a specific diet.
4.3.2 Individual diet phenotype-eigenmetabolite associations
In addition to identifying phenotypes with distinct metabolic correlations in the combined dataset, the results of the EvMA demonstrated that some phenotypes correlate with metabolic clusters in a diet specific manner. For example, the lipid phenotype was significantly correlated with a single cluster in the high glucose diet (Cluster 6; 33 features; Table 3) and the high fat diet (Cluster 4; 22 features; Table 3). These individual clusters shared only seven metabolic features, including succinic acid and asparagine. Another phenotype that demonstrated a diet specific correlation pattern was the sugar phenotype that was associated with a single cluster in the high glucose diet (Cluster 5; 17 features; Table 3) and two metabolite clusters in the normal diet (Cluster 5; 12 features; Cluster 6; 17 features; Table 3). The larval survival phenotype also showed a diet specific correlation where it was only correlated with a metabolite cluster in the high glucose diet, which was composed of only fatty acids. Collectively, these findings suggest that EvMA can be used to identify context (environment) dependent mechanistic links between some phenotypes and metabolites, which can inform our understanding of how the metabolites correlated with phenotypes associated with complex diseases can vary depending on the environment.
4.3.3 Diet independent phenotype-eigenmetabolite associations
Although some of the phenotypes only correlated with metabolites under specific dietary conditions or when the combined dataset was examined, the EvMA analysis identified correlations between phenotypes and metabolic clusters that are conserved under all of the treatment conditions. For example, the body weight phenotype was always correlated with the conserved metabolite cluster(s) that contains L-DOPA and the two unidentified catecholamines. These clusters were composed primarily of fatty acids or contained a substantial number of fatty acids. Similarly, Williams et al. (2015) found that the catecholamine L-DOPA was correlated with weight and sugar phenotypes across multiple diets, but did not find a correlation between fatty acids and the weight phenotype. They also found that arachidonoyl dopamine correlated with weight and lipid phenotypes on multiple diets. Using the EvMA analysis, arachidonoyl dopamine was only significantly correlated with both the weight and lipid phenotypes in the combined dataset (Cluster 3; 41 features; Table 2) and the high sugar dataset (Cluster 6; 33 features; Table 3). Each of these clusters was classified as a Mixing Pot, and they shared 19 metabolites (including arachidonoyl dopamine) that were primarily sugars (52.6 %). The clusters containing arachidonoyl dopamine in the normal diet (Cluster 5; 12 features; Table 3) and the high fat diet (Cluster 2; 52 features; Table 3) were significantly correlated with weight, but not the lipid phenotype. Thus, the EvMA recovers some similar findings to previous studies of the test dataset for the weight and lipid phenotypes, but also expands upon these findings by highlighting novel linkages between the overt phenotypes and metabolites and metabolite classes that had not been recovered previously. Some of these novel linkages include: (1) the correlation of the weight phenotype to clusters containing a substantial number of fatty acids as well as L-DOPA, (2) the correlation of both the weight and lipid phenotypes to the cluster containing arachidonoyl dopamine only in the combined and high sugar datasets, and (3) the additional 18 metabolic features that are primarily sugars shared between these two clusters containing arachidonoyl dopamine (Cluster 3 and 6 respectively).
4.3.4 Clusters correlated with multiple phenotypes
In addition to examining overt phenotypes individually, the EvMA identified metabolite clusters that correlated with multiple phenotypes under a given environmental condition. For example, metabolite clusters in the combined dataset (Cluster 6; Table 2), low calorie diet (Cluster 3; Table 3), and high fat diet (Cluster 1; Table 3) were correlated with both weight and development time phenotypes. The combined dataset and high fat diet clusters were composed primarily of fatty acids, and the low calorie diet cluster was classified as a “Mixing Pot” that contained mostly fatty acids. Although the clusters all contained fatty acids, there was limited overlap in the metabolic features that were found in each cluster. Thus, under certain conditions weight and development time phenotypes are linked by fatty acid metabolites, but the fatty acids vary based on the environment. Other phenotypes that correlated with the same cluster in a treatment were the larval and pupal survival phenotype. Both of these phenotypes correlated with Cluster 2 of the high glucose diet (Table 3), and along with the development time correlated with Clusters 1 and 5 of the combined dataset (Table 2). The high glucose diet cluster contained fatty acids and has no overlap with either of the combined dataset clusters that were “Mixing Pots”. Collectively, these findings provide proof of concept that EvMA can be used to identify metabolites that link multiple overt phenotypes under different conditions, which can help to inform our understanding of the manner in which certain phenotypes are correlated with the same metabolites under a given condition.
4.4 Associations between genes and phenotypes
The EvMA examined the correlation patterns between eigenmetabolites, gene transcripts, and phenotypes. While examining these correlations, we identified instances where eigenmetabolite of the clusters was correlated with genes enriched for a biological annotation that is known to contribute to a phenotype of interest. For example, in the combined dataset and the high glucose diet, we identified an eigenmetabolite (Cluster 3) correlated with the sugar phenotype, and the correlated gene list was enriched for oxidative phosphorylation (00190:KEGG pathway). Defects in oxidative phosphorylation have been found to play a role in insulin resistance (Boirie 2003; Buchner et al. 2011; Cree-Green et al. 2015). Thus, using EvMA, it is possible to identify metabolites that are useful in predicting gene expression or phenotype under different environmental conditions.
5 Concluding remarks
Different ‘omics’ datasets obtained from an organism of interest under the same experimental conditions are becoming more common. In this work, we use EvMA, a novel method for identifying linkages between metabolomic, transcriptome, and phenotypic data and assessing the impact of environment on these correlations. Environment can be responsible for a significant amount of variance in the metabolome (e.g., Reed et al. (2014)). As such, a PCA analysis would be expected to recover clusters. However, a common concern associated with PCA is the effect of noise on the resulting clusters (Halouska and Powers 2006). One goal of EvMA is to lessen the impact of noise in the dataset by completing the PCA on discrete modules (e.g., clusters) instead of the whole dataset. Based on correlation alone, EvMA recovers clusters of metabolites with similar chemical characters, which can be used to inform identifications of unknown metabolites. Furthermore, the EvMA identifies the effects of different environmental factors on metabolite correlation patterns, and highlights correlations that are maintained across different environments. When clusters are not primarily composed of metabolites of similar chemical classes, the resulting eigenmetabolite (eigenvector of metabolite clusters) is lower. However, the impact of this noise is limited to the individual module as opposed to impacting the entire analysis. Thus, when examining linkages between the eigenmetabolites and gene transcripts, EvMA demonstrates that some metabolite clusters are significantly correlated with individual genes and groups of genes enriched for physiological processes that utilize these metabolites. Thus, providing proof of concept that when using EvMA, it is possible to predict the type of genes whose expression patterns are associated with metabolites of interest. In turn, these findings can be used to generate hypotheses of the biological function of unannotated genes. In addition, the EvMA found specific phenotypes associate with specific metabolite clusters in an environment specific manner, indicating a context dependent mechanistic link. Furthermore, EvMA identified clusters that correlated with multiple phenotypes. Thus, highlighting metabolites that link phenotypes under specific environmental conditions. When identifying linkages between metabolite clusters, transcripts, and phenotypes, some correlations were only identified in the EvMA of the combined dataset. Thus, we suggest including a combined analysis to identify distinct, underlying associations that may not be detectable in datasets from individual environments. Finally, EvMA identified clusters correlated with specific phenotypes and genes enriched for functions that influence the associated phenotype. Thus, this method may be useful in identifying metabolites that can predict gene expression or phenotypes in a context dependent manner. These results demonstrate that EvMA provides a simple method to identify correlations between metabolites, gene expression, and phenotypes, which can allow us to partition multivariate datasets into biologically meaningful modules and identify under-studied metabolites and gene transcripts that may presently have unknown function but are central to important biological processes. This can, in turn, be used to inform our understanding of the effect of environment mechanisms underlying physiological states of interest, including human disease.
Supplementary Material
Acknowledgments
Funding for this study was provided by the National Institute of Health (NIH)-R01 GM098856 to LR, NIH-NRSA Fellowship to LR, NIH-R01 GM61600 to Greg Gibson, and Australian Research Council (ARC) DP0880204 to Greg Gibson. We thank Vishal Oza, Pablo Chialvo, and members of the Reed Lab for helpful suggestions and critical comments on this manuscript.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s11306-016-1117-3) contains supplementary material, which is available to authorized users.
Compliance with ethical standards
Conflicts of interest The authors declare they have no potential conflicts of interest.
Research involving human participants and/or animals This article does not include any studies involving humans or regulated animal models.
Informed consent The studies presented in this article did not include human subjects.
References
- Alvarez M, Schrey AW, Richards CL. Ten years of transcriptomics in wild populations: what have we learned about their ecology and evolution? Molecular Ecology. 2015;24(4):710–725. doi: 10.1111/mec.13055. [DOI] [PubMed] [Google Scholar]
- Berg JM, Tymoczko JL, Stryer L. Biochemistry. 5th. New York: W. H. Freeman; 2002. Section 22.5, Acetyl Coenzyme A carboxylase plays a key role in controlling fatty acid metabolism. Available from: http://www.ncbi.nlm.nih.gov/books/NBK22381/ [Google Scholar]
- Boirie Y. Insulin regulation of mitochondrial proteins and oxidative phosphorylation in human muscle. Trends in Endocrinology and Metabolism. 2003;14(9):393–394. doi: 10.1016/j.tem.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Buchner DA, Yazbek SN, Solinas P, Burrage LC, Morgan MG, Hoppel CL, et al. Increased mitochondrial oxidative phosphorylation in the liver is associated with obesity and insulin resistance. Obesity (Silver Spring) 2011;19(5):917–924. doi: 10.1038/oby.2010.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke CJ, Huetteroth W, Owald D, Perisse E, Krashes MJ, Das G, et al. Layered reward signalling through octopamine and dopamine. Drosophila Nature. 2012;492(7429):433–437. doi: 10.1038/nature11614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho K, Evans BS, Wood BM, Kumar R, Erb TJ, Warlick BP, et al. Integration of untargeted metabolomics with transcriptomics reveals active metabolic pathways. Metabolomics. 2014 doi: 10.1007/s11306-014-0713-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cree-Green M, Newcomer BR, Coe G, Newnes L, Baumgartner A, Brown MS, et al. Peripheral insulin resistance in obese girls with hyperandrogenism is related to oxidative phophorylation and elevated serum free fatty acids. American Journal of Physiology-Endocrinology and Metabolism. 2015;308:E726–E733. doi: 10.1152/ajpendo.00619.2014.-Hyperandrogenic. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culibrk L, Croft CA, Tebbutt SJ. Systems biology approaches for host-fungal interactions: An expanding multiomics frontier. OMICS: A Journal of Integrative Biology. 2016;20(3):127–138. doi: 10.1089/omi.2015.0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Santos G, Schroeder AJ, Goodman JL, Strelets VB, Crosby MA, Thurmond J, et al. FlyBase: Introduction of the Drosophila melanogaster release 6 reference genome assembly and large-scale migration of genome annotations. Nucleic Acids Research. 2015;43:D690–697. doi: 10.1093/nar/gku1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn WB, Erban A, Weber RJM, Creek DJ, Brown M, Breitling R, et al. Mass appeal: Metabolite identification in mass spectrometry-focused untargeted metabolomics. Metabolomics. 2012;9(S1):44–66. doi: 10.1007/s11306-012-0434-4. [DOI] [Google Scholar]
- Evans PD. Biogenic amines in the insect nervous system. Advances in Insect Physiology. 1980;15:317–473. [Google Scholar]
- Fei F, Mendonca ML, McCarry BE, Bowdish DME, Surette MG. Metabolic and transcriptomic profiling of Streptococcus intermedius during aerobic and anaerobic growth. Metabolomics. 2016;12(3):1–13. doi: 10.1007/s11306-016-0966-0. [DOI] [Google Scholar]
- Gehlenborg N, O’Donoghue SI, Baliga NS, Goesmann A, Hibbs MA, Kitano H, et al. Visualization of omics data for systems biology. Nature Methods. 2010;7(3S):S56–S68. doi: 10.1038/nmeth.1436. [DOI] [PubMed] [Google Scholar]
- Gligorijevic V, Malod-Dognin N, Przulj N. Integrative methods for analyzing big data in precision medicine. Proteomics. 2016;16(5):741–758. doi: 10.1002/pmic.201500396. [DOI] [PubMed] [Google Scholar]
- Gustafsson M, Nestor CE, Zhang H, Barabási AL, Baranzini S, Brunak S, et al. Modules, networks and systems medicine for understanding disease and aiding diagnosis. Genome Medicine. 2014;6:82. doi: 10.1186/s13073-014-0082-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halouska S, Powers R. Negative impact of noise on the principal component analysis of NMR data. Journal of Magnetic Resonance. 2006;178:88–95. doi: 10.1016/j.jmr.2005.08.016. [DOI] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Research. 2009a;37(1):1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols. 2009b;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Jia X, Sun C, Zuo Y, Li G, Li G, Ren L, et al. Integrating transcriptomics and metabolomics to characterise the response of Astragalus membranaceus Bge. var. mongolicus (Bge.) to progressive drought stress. BMC Genomics. 2016;17(1):188. doi: 10.1186/s12864-016-2554-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaever A, Landesfeind M, Feussner K, Mosblech A, Heilmann I, Morgenstern B, et al. MarVis-pathway: Integrative and exploratory pathway analysis of non-targeted metabolomics data. Metabolomics. 2015;11(3):764–777. doi: 10.1007/s11306-014-0734-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz L, Baltz RH. Natural product discovery: Past, present, and future. Journal of Industrial Microbiology and Biotechnology. 2016;43(2–3):155–176. doi: 10.1007/s10295-015-1723-5. [DOI] [PubMed] [Google Scholar]
- Kuo TC, Tian TF, Tseng YJ. 3Omics: A web-based systems biology tool for analysis, integration and visualization of human, transcriptomic, proteomic and metabolomic data. BMC Systems Biology. 2013;7:64. doi: 10.1186/1752-0509-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshmanan M, Lim SH, Mohanty B, Kim JK, Ha SH, Lee DY. Unraveling the light-specific metabolic and regulatory signatures of rice through combined in silico modeling and multiomics analysis. Plant Physiology. 2015;169(4):3002–3020. doi: 10.1104/pp.15.01379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linstrom PJ, Mallard WG. NIST chemistry webbook, NIST standard reference database number. Vol. 69. Gaithersburg, MD 20899: National Institute of Standards and Technology; 2016. (Vol Retrieved July 18, 2012) [Google Scholar]
- Marmiesse L, Peyraud R, Cottret L. FlexFlux: Combining metabolic flux and regulatory network analyses. BMC Systems Biology. 2015;9:93. doi: 10.1186/s12918-015-0238-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Ramírez AC, Ferré J, Silva FJ. Catecholamines in Drosophila melanogaster: Dopa and dopamine accumulation during development. Insect Biochemistry and Molecular Biology. 1992;22(5):491–494. [Google Scholar]
- McHardy IH, Goudarzi M, Tong M, Ruegger PM, Schwager E, Weger JR, et al. Integrative analysis of the microbiome and metabolome of the human intestinal mucosal surface reveals exquisite inter-relationships. Microbiome. 2013;1:17. doi: 10.1186/2049-2618-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuwaysir EF, Huang W, Albert TJ, Singh J, Nuwaysir K, Pitas A, et al. Gene expression analysis using oligonucleotide arrays produced by maskless photolithography. Genome Research. 2002;12:1749–1755. doi: 10.1101/gr.362402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osorio S, Alba R, Nikoloski Z, Kochevenko A, Fernie AR, Giovannoni JJ. Integrative comparative analyses of transcript and metabolite profiles from pepper and tomato ripening and development stages uncovers species-specific patterns of network regulatory behavior. Plant Physiology. 2012;159(4):1713–1729. doi: 10.1104/pp.112.199711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavey SA, Bernatchez L, Aubin-Horth N, Landry CR. What is needed for next-generation ecological and evolutionary genomics? Trends in Ecology & Evolution. 2012;27(12):673–678. doi: 10.1016/j.tree.2012.07.014. [DOI] [PubMed] [Google Scholar]
- Peng J, Zeng J, Cai B, Yang H, Cohen MJ, Chen W, et al. Establishment of quantitative severity evalution model for spinal cord injury by metabolomic fingerprinting. PLoS One. 2014;9(4):e93736. doi: 10.1371/journal.pone.0093736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raupach MJ, Amann R, Wheeler QD, Roos C. The application of “-omics” technologies for the classification and identification of animals. Organisms Diversity & Evolution. 2016;16(1):1–12. doi: 10.1007/s13127-015-0234-6. [DOI] [Google Scholar]
- RCoreTeam. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2013. URL http://www.R-project.org. [Google Scholar]
- Rebollar EA, Antwis RE, Becker MH, Belden LK, Bletz MC, Brucker RM, et al. Using “Omics” and integrated multi-omics approaches to guide probiotic selection to mitigate chytridiomycosis and other emerging infectious diseases. Front Microbiol. 2016;7:68. doi: 10.3389/fmicb.2016.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redestig H, Costa IG. Detection and interpretation of metabolite-transcript coresponses using combined profiling data. Bioinformatics. 2011;27(13):i357–365. doi: 10.1093/bioinformatics/btr231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed LK, Lee K, Zhang Z, Rashid L, Poe A, Hsieh B, et al. Systems genomics of metabolic phenotypes in wild-type Drosophila melanogaster. Genetics. 2014;197:781–793. doi: 10.1534/genetics.114.163857/-/DC1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed LK, Williams S, Springston M, Brown J, Freeman K, DesRoches CE, et al. Genotype-by-diet interactions drive metabolic phenotype variation in Drosophila melanogaster. Genetics. 2010;185(3):1009–1019. doi: 10.1534/genetics.109.113571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra AA, Couee I, Heijnen D, Michon-Coudouel S, Sulmon C, Gouesbet G. Genome-wide transcriptional profiling and metabolic analysis uncover multiple molecular responses of the grass species lolium perenne under low-intensity xenobiotic stress. Front Plant Sci. 2015;6:1124. doi: 10.3389/fpls.2015.01124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD. A direct approach to false discovery rates. Journal of the Royal Statistical Society. Series B, Methodological. 2002;64(3):479–498. [Google Scholar]
- Thorndike RL. Who belongs in the family? Psychometrika. 1953;18(4):267–276. [Google Scholar]
- Trikka FA, Nikolaidis A, Ignea C, Tsaballa A, Tziveleka LA, Ioannou E, et al. Combined metabolome and transcriptome profiling provides new insights into diterpene biosynthesis in S. pomifera glandular trichomes. BMC Genomics. 2015;16(1):935. doi: 10.1186/s12864-015-2147-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valcàrcel B, Ebbels TM, Kangas AJ, Soininen P, Elliot P, Ala-Korpela M, et al. Genome metabolome integrated network analysis to uncover connections between genetic variants and complex traits: An application to obesity. Journal of the Royal Society, Interface. 2014;11(94) doi: 10.1098/rsif.2013.0908. 20130908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Swinderen B, Andretic R. Dopamine in Drosophila: Setting arousal thresholds in a miniature brain. Proc Biol Sci. 2011;278(1707):906–913. doi: 10.1098/rspb.2010.2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wägele B, Witting M, Schmitt-Kopplin P, Suhre K. MassTRIX reloaded: Combined analysis and visualization of transcriptome and metabolome data. PLoS One. 2012;7(7):e39860. doi: 10.1371/journal.pone.0039860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams S, Dew-Budd K, Davis KC, Anderson J, Bishop R, Freeman K, et al. Metabolomic and gene expresion profiles exhibit modular genetic and dietary structure linking metabolic syndrome phenotypes Drosophila. G3. 2015;(5):2817–2829. doi: 10.1534/g3.115.023564/-/DC1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Li F, Nie L. Integrating multiple ‘omics’ analysis for microbial biology: application and methodologies. Microbiology. 2010;156(Pt 2):287–301. doi: 10.1099/mic.0.034793-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.