

Abstract
It is a challenge to develop a universal single drug that can treat breast cancer at single or multiple-stage complications, yet remains nontoxic to normal cells. The challenge is even greater when breast cancer-specific estrogen-based drugs are being developed which cannot act against multi-staged breast cancer complications owing to cells’ differential ER expression status and their possession of drug-resistant and metastatic phenotypes. We report here the development of a first cationic lipid-conjugated estrogenic derivative (ESC8) that kills breast cancer cells independent of their estrogen receptor (ER) expression status. This ESC8 molecule apparently is nontoxic to normal breast epithelial cells, as well as to other non-cancer cells. ESC8 induces apoptosis through an intrinsic pathway in ER-negative MDA-MB-231 cells. In addition, ESC8-treatment induces autophagy in these cells by interfering with the mTOR activity. This is the first example of an estrogen structure-based molecule that co-induces apoptosis and autophagy in breast cancer cells. Further in vivo study confirms the role of this molecule in tumor regression. Together, our results open new perspective of breast cancer chemotherapy through a single agent, which could provide the therapeutic benefit across all stages of breast cancer.
Keywords: Breast cancer, ESC8, Apoptosis, Autophagy, Mice model
INTRODUCTION
Breast cancer drugs are often based on targeting estrogen receptors (ER) because of ER’s relatively high abundance in most types of breast cancers. The status of this important biomarker provides favorable prognostic features and helps individualize breast cancer therapy (1, 2). Several estrogen-based modulators that rely on mechanisms such as antagonism to estrogen-ER binding, ER-mediated transcription, or estrogen-based hybrid DNA-alkylating agents have been developed over time (1, 3–12). But adjuvant therapy is usually warranted in highly aggressive breast cancer phenotypes that are not responsive to conventional single-agent estrogen. Many other selective ER modulators (SERMs) (e.g., fulvestrant, a lipid-modified analogue of endogenous natural ligand 17 β-estradiol; the non-estrogenic molecules tamoxifen and raloxifen) were largely limited to the treatment of ER-positive breast cancer (13–16). However, developing a universal anti-breast cancer estrogenic molecule that is nontoxic to normal cells, yet acts against both ER-positive and ER-negative breast cancers with drug-resistant and metastatic phenotypes, is challenging.
Most anti-cancer, agent-mediated cancer cell killing occurs through induction of apoptosis. Apoptosis is the type I process involved in programmed cell death (PCD), the other mechanism being autophagy (type II). The ability of tumor cells to evade engagement of apoptosis plays a significant role in their resistance to conventional therapeutic regimens. Of the two major pathways of apoptosis (i.e., intrinsic or extrinsic), activation of the mitochondria-associated intrinsic pathway leads to an increase in caspase-9 and caspase-3, resulting in apoptotic cell death (17).
Autophagy, the self-cannibalism in cells, is often triggered for cyto-protective or survival purposes, including facilitating drug resistance in breast cancer cells and protecting cells against pro-apoptotic insults (18–20). However, further studies also suggest a definitive role for autophagy in cancer and in determining the response of tumor cells (e.g., breast tumor cells) to anti-cancer therapy (21). It has been shown that anti-estrogen treatment of ER-positive breast cancer cells leads to induction of autophagy (22) but to our knowledge there is no evidence of anti-estrogen-mediated involvement of autophagy in ER-negative breast cancer cells. The PI3K-Akt-mTOR signaling pathway, which negatively regulates autophagy, is involved in tumorigenic progression in many cancers and is one of the two mechanisms of induction of autophagy (23).
In this study, we developed a cationic, lipid-conjugated, estrogenic molecule that, in contrast to other estrogenic drugs and SERMs, induced cell death in both ER-positive and ER-negative breast cancer cells. This estrogenic drug-mediated killing of ER-negative breast cancer cells prompted us to conduct an in-depth mechanistic study in ER-negative MDA-MB-231. We found that the apoptosis was induced through up-regulation of the BAX/Bcl-2 ratio, leading to activation of initiator caspase 9 and effecter caspase 3. In this study we also found that ESC8 treatment evoked the upregulation of autophagy via inhibition of the mTOR pathway and that activation of autophagy promoted the cell death of MDA-MB-231 cells. We observed a decrease in phosphorylation of mTOR at ser-2448 and a simultaneous increase in phosphorylation of Akt1/2/3 at ser-473 on ESC8 challenge. We assume that ESC8 treatment inhibited mTOR activity, thereby increasing phosphorylation of Akt in a feedback loop similar to that of rapamycin treatment. The molecule was also tested in vivo to show a significant reduction of tumor aggression. To our knowledge, this is the first example of a concomitant induction of estrogenic drug-mediated autophagy and apoptosis in breast cancer through regulation of PI3K-Akt-mTOR signaling and uncovering of this pathway, which we believe is novel, for promoting tumor cell death may have therapeutic implications in the treatment of breast cancer.
MATERIALS and METHODS
Antibodies
Anti-caspase-3 (#9662), caspase-8 (#9746), caspase-9 (#9502), p-mTOR (#2971), mTOR (#2972), p-p70S6K (#9234), Bcl-2 (# 2876), Atg5 (#2630), Atg12 (#2010), Beclin-1 (#3738), and LC-3B (#2775) antibodies were purchased from Cell Signaling (Danvers, MA), anti-mouse β-Actin antibody was purchased from BD-Pharmingen (San Diego, CA), anti-p-Akt-1/2/3 (Ser-473) (sc-7985), anti-Akt-1 (sc-1618), anti-BAX (sc-6236), anti-Bid (sc-11423), and anti-cytochrome c (sc-13156) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Compound synthesis and characterization
A detailed synthesis of ESC8 is described in the supplementary section.
Cells and cell culture conditions
Detailed cell culture conditions have been provided in the Supplementary “Materials & Methods” section.
Isolation and maintenance of rat hepatocytes
Hepatocytes were isolated from adult male wistar rats (250 g). A detailed procedure is available in the supplemental data.
Isolation of normal human breast epithelium cells
Normal human breast epithelial cells were isolated from human normal breast epithelial tissue of donor breast cancer patient and obtained as a kind gift from Indo-American Cancer Center, Hyderabad, India. A detailed procedure is available in the supplemental data.
Preparation of samples and sample treatment
The compounds were dissolved in cell culture-grade DMSO to get a primary stock. The stock was progressively diluted with DMSO to get secondary stocks. Finally, the working concentrations of the derivatives were obtained by adding the secondary DMSO stocks in 10% fetal bovine serum containing cell culture medium. For controls, cells were treated with only DMSO containing serum-medium and volume of DMSO was equal to the volume of DMSO in which the drugs were dissolved and added to the serum containing medium before the treatment. The amount of DMSO in working solutions never exceeded more than 0.2% with respect to the serum containing culture medium. For the viability studies, 100 µl of cell culture solutions containing respective concentrations of compounds are given to cells pre-washed with phosphate buffer saline. For flow cytometric analysis, 1.5 mL of culture media containing a respective concentration of ESC8 was added to each well of six-well plates.
Cytotoxicity study
Cytotoxicities of the compounds were evaluated by the 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) reduction assay. A detailed procedure is available in supplemental data.
Annexin V staining
The annexin V-FITC-labeled apoptosis detection kit (Sigma Chemical Co., MO). was used to detect and quantify apoptosis by flow cytometry as per manufacturer’s instructions. In brief, cells (1 × 106 cells/well) were seeded in six-well plates and cultured overnight in 10% serum media. The next day, cells were treated with 10 µM ESC8 for 16 hours, harvested in PBS, and collected by centrifugation for 5 minutes at 1,000 rpm. Cells were then resuspended at a density of 1 × 106 cells/mL in 1X binding buffer (HEPES buffer 10 mM & pH 7.4, 150 mM NaCl, 5 mM KCl, 1 mM MgCl2, and 1.8 mM CaCl2) and stained simultaneously with FITC-labeled annexin V (25 ng/mL) and propidium iodide (50 ng/mL) according to the manufacturer’s protocol (Sigma Chemical Co., MO). Cells were analyzed using a flow cytometer (Becton Dickinson, San Jose, CA), and data were analyzed with CellQuest software.
4,6-Diamidino-2-phenylindole (DAPI) staining
MDA-MB-231 cells were grown on the cover slip were first treated with DMSO or ESC8 (5µM) for 16 hours. Cells were then washed with PBS and fixed with 3% paraformaldehyde for 15 minutes at room temperature, and again washed thrice with PBS. Cells were then stained with DAPI (1 µg/ml, Sigma) for 30 minutes. Stained nuclei were visualized using microscope, Axiovert 200M (Carl Zeiss, Inc.) and photographed. Apoptotic cells were morphologically identified by cytoplasmic and nuclear shrinkage.
siRNA knockdown
MDA-MB-231 cells were treated with 100 nM of control/ Atg5 siRNA using Dharmafect-1 (Dharmacon). After 48 hours cells were trypsinized and 5000 cells were plated in 96-well plates and kept overnight. Then cells were treated with either DMSO or ESC8 (5 µM) for 16 hours and cell viability assay was done as described in supplementary.
Autophagic flux Assay
Detection of autophagic flux, the specific processing of autophagy protein LC3 with or without a lysosomal protease inhibitor was determined by Western analysis. MDA-MB-231 cells were treated with or without both pepstatin A and E46d (10 µM) (24) along with ESC8 (1 or 5 µM) for 16 hours and LC3-II level was detected by western blot analysis.
Visualization of monodansylcadaverine (MDC) labeled vacuoles
MDA-MB-231 cells were grown on the cover slip, were first treated with DMSO or ESC8 (5µM) for 16 hours. Autophagic vacuoles were then labeled with MDC by incubating cells with 0.05 mM MDC in PBS for 10 minutes at 37°C. After incubation, cells were washed thrice with PBS and fixed with 3% paraformaldehyde for 15 minutes at room temperature, and again washed thrice with PBS. Cells were then permeabilized with 0.05% Triton-X for 15 minutes at room temperature, washed with PBS and followed by mounting in Vectashield (Vector Labs, CA) without DAPI. Confocal microscopy was performed using a Zeiss LSM 510 confocal laser scan microscope.
Cytosolic Extract Preparation
cytoslic extracts were prepared using MDA-MB-231 cells after ESC8 treatment for 16 hours following a standard protocol, with modifications (25, 26). Cells were washed in cold PBS, suspended in buffer A [10 mm HEPES (pH 7.8), 10 mm KCl, 2 mm MgCl2, 0.1 mm EDTA, 10 µg/ml aprotinin, 3 mm DTT, and 0.1 mm phenylmethylsulfonyl fluoride] and incubated for 15 minutes on ice. Cells were then lysed with 10% Igepal and then centrifuged at 14, 000 rpm for 5 mins. Clear supernatants containing the cytosolic proteins were collected and stored at −70°C. Cytochrome c levels in cysolic extracts were detected by Western blot analysis.
Western Blot Analysis
Western blot analysis was performed to detect the levels of Bcl-2, BAX, Bid, Atg5-Atg12 complex, Beclin-1 and LC-3 expressions, Akt-1/2/3, mTOR, p70S6K phosphorylations, and caspase activation in ESC8-treated MDA-MB-231 cells. Cells were washed with PBS and lysed with RIPA buffer supplemented with a protease inhibitor cocktail after 16 hours of treatment. Supernatant was collected by centrifugation at 13,000 rpm for 10 minutes. Samples were then subjected to SDS-PAGE and then transferred to polyvinyl difluoride membranes and immunoblotted. Antibody-reactive bands were detected by enzyme-linked chemiluminescence (Amersham, Piscataway, NJ) and quantified by laser densitometry. These experiments were repeated at least three times.
Animal xenograft model
5 million MDA-MB-231 cells were orthotopically implanted in the right lower mammary fat pad of each of 6–7 week-old female Balb C SCID mice (NCI-Frederick, MD). When the average tumor sizes were 30–35 cubic mm, three groups of five mice each were segregated into the following treatment groups: a) Untreated group injected with 5% glucose solution intraperitoneally, b) ESC8 (10 mg/Kg/mice)- injected group, c) ESC8 (10 mg/kg/mice)- injected group in which, injections were started when tumor size reached ~130–135 mm. Tumor sizes were measured twice a week. Only two alternate-day injections were given to groups a and b, whereas four consecutive-day injections were given to group c. All the experiments were done under the animal protocol approved by the Mayo Clinic’s Institutional Animal Care and Use Committee.
Histological study
MDA-MB-231 tumors were removed and fixed in neutral buffered 10% formalin at room temperature for 24 hours prior to embedding in paraffin and sectioning. Sections were deparaffinized and then subjected to TUNEL staining for apoptosis according to the manufacturer’s instructions (DeadEnd™ Colorimetric TUNEL system, Promega). The TUNEL-positive nuclei were counted in the entire section. Thereafter, ten fields of vision were photographed, the total number of nuclei was counted, and the average number of nuclei/unit area was calculated. Photographs of the entire cross-section were digitized using an Olympus camera (DP70). The apoptosis index was calculated as the number of positive nuclei divided by the total number of nuclei.
Statistical Analysis
Data were expressed as mean ±S.D. and statistically analyzed by the two-tailed, unpaired, student's t-test using Microsoft Excel (Seattle). Data were considered significant when p<0.05 for animal studies and p<0.01 for cell culture-based studies.
RESULTS
Synthesis and anti-cancer effect of ESC8
We synthesized ESC8 by inserting an eight-carbon (C8), twin-chain, cationic lipid at the 17β position of estradiol (ES) (Figure 1a). Our previous study indicated that any lipid-based substitution at the cyclopentyl-ring (ring d), especially at the 17β-position, largely retains its selective ability to target ER-expressing cancer cells when associated with a liposomal delivery system (27). The detailed synthesis of ESC8 is provided in the supplementary data. Cationic lipids containing twin carbon chains show natural affinity towards negatively-charged cellular membrane making them excellent agents to deliver bioactive molecules such as DNA inside the cells. We hypothesized that they may enhance the uptake of small molecule ligands also. In accordance with our hypothesis, co-treatment of cationic lipid moiety and estrogen induced increased toxicity in comparison to individual treatments (Figure 1b). We also presumed that a chemical conjugation would be more effective than co-treatment due to proximity and better co-operative effect. This presumption indeed seems to be true as the conjugated moiety showed additional level of anticancer effect (Figure 1b). In an attempt to justify our hypothesis, we tested the anti-cancer effect of ESC8, an estrogenic molecule, in different kinds of breast cancer cells. The anti-cancer effect of ESC8 in ER-positive, MCF-7 and ER-negative, MDA-MB-231 cells were compared with the anti-cancer effects of the estrogenic drug 2-methoxyestradiol (2OMe-ES) and non-estrogenic anti–breast-cancer agents such as 4-hydroxytamoxifen (4OH-Tam), tamoxifen (Tam) and epirubicin (Epi). As indicated in Figure 1c–d, ESC8 was clearly the most efficient killer of both ER-positive and ER-negative cells. We found lower IC50 for ESC8 (MCF-7 3.0 µM and MDA-MB-231 3.2 µM) compared with 4OH-Tam (4.3 µM and 7.7 µM) and 2OMe-ES (10.5 µM and 18.0 µM) under the experimental conditions. Figure 1e shows the effect of ESC8 on other breast cancer and non-cancer cells. Five breast cancer cells and four non-cancer cells were tested. The other tested breast cancer cells were T47D (ductal, ER+), ZR-75-1 (ductal, ER+), and MDA-MB-435S (ductal, ER-). The non-cancer cells were COS-1, CHO, freshly isolated rat hepatocytes, and normal breast epithelia (NBE, isolated from human breast cancer patients). Figure 1e clearly shows that ESC8 induced killing of all breast cancer cells but was unable to induce toxicity to any of the normal or healthy cells at the treated concentration. Moreover, use of ER-antagonist, ICI182780, was able to abrogate the anti-cancer effect of ESC8 in ER-positive MCF-7 cells but had no effect in ER-negative MDA-MB-231 (Figure 1f). Therefore, ESC8-mediated toxicity via ER-antagonism can be ruled out for MDA-MB-231. In conclusion, ESC8 was clearly the most potent anti-cancer agent in comparison to other breast cancer-specific estrogenic, non-estrogenic, and ER-antagonizing molecules and – most importantly – ESC8 induced killing of these cells irrespective of ER-expression status.
Figure 1.
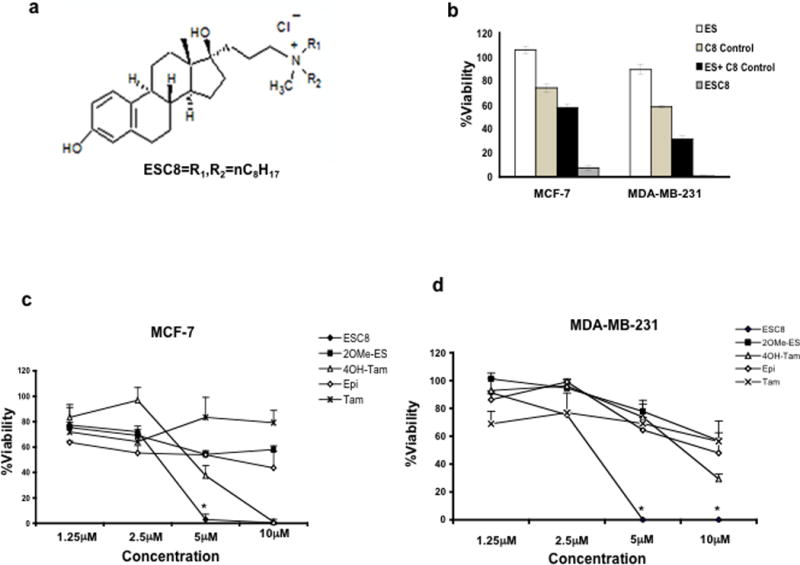
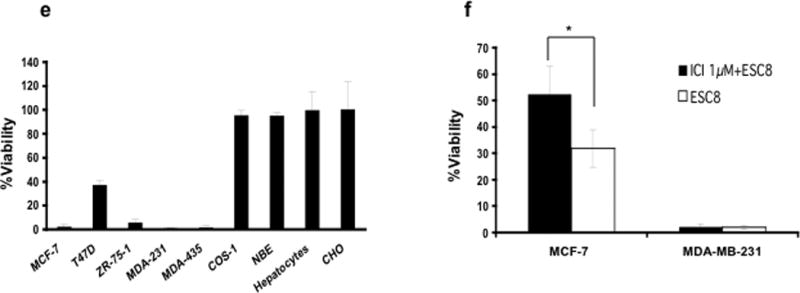
a. Structure of ESC8. Anti-cancer effect of ESC8. b. MCF-7 and MDA-MB-231 cells were treated with ES (white), C8 control (light grey), ES + C8 control (dark grey) or ESC8 (black) for 48 hours at a concentration 10 µM (of each component) followed by determination of viabilities of cells using MTT assay. c–d. Viability studies in MCF-7, and in MDA-MB-231 cells in presence of following compounds, ESC8 (●), 2OMe-ES (■), 4OH-Tam (∆), Epi (◊), Tam (X) at the indicated concentrations for 48 hours. 5µM ESC8 treatment for MCF-7 and 5µM and 10 µM ESC8 treatments for MDA-MB-231 * p values are < 0.005 compared to other drugs treatments. At 10µM dose in MCF-7, * p<0.005 between ESC8 treatment and other drugs except 4OH-Tam. e. Representation of viabilities of ESC8 (10 µM, 48 hours)-treated breast cancer cells (MCF-7, T47D, ZR-75-1, MDA-MB-231 and MDA-MB-435S) and non-cancer cells (COS-1, normal-breast epithelial (NBE), hepatocytes and CHO). f. Comparison of the viabilities of ESC8-treated (10 µM, 4 hours, left for 44 hours) cells in presence and absence of the pretreatment of ICI (ICI182780, an ER antagonist, 1 µM, 2 hours) [*p< 0.001 for ICI182780 treated vs. untreated MCF7 cells]. Figures are representative of minimum of four separate experiments with similar results. For normal breast epithelial cells (NBE) cells the experiments were done only thrice.
ESC8 induces apoptosis in breast cancer cells
To ascertain the induction of apoptosis in cancer cells following ESC8 treatment, we first studied the Annexin V/ Propidium iodide (PI) binding profile of ESC8-treated MCF-7 and MDA-MB-231 cells using flow cytometry analysis. As shown in Figure 2a, the ESC8 treatment (10 µM, 16 hours) led to the accumulation of 70% of Annexin and PI positive MCF-7 cells. In MDA-MB-231, 40% apoptotic (Annexin and PI positive) cells were accumulated. In normal cells, COS-1 and isolated rat hepatocytes, ESC8 could not induce any significant apoptosis. Clearly, ESC8 induced apoptosis in both ER-positive/negative breast cancer cells. MDA-MB-231 cells were also stained with DAPI after 5 µM ESC8 treatment to show the cytoplasmic and nuclear shrinkage due to induction of apoptosis (Figure S1a). Thus it became a point of interest to ascertain the actual pathway through which apoptosis-induction was triggered, especially in ER-negative cells. ESC8, an estrogen-like molecule, was naturally expected to have a anti-cancer effect on ER-positive cells, at least, via possible ER-antagonism but similar reason could not be expected in ER-negative cells.
Figure 2. ESC8 induces apoptosis in breast cancer cells.
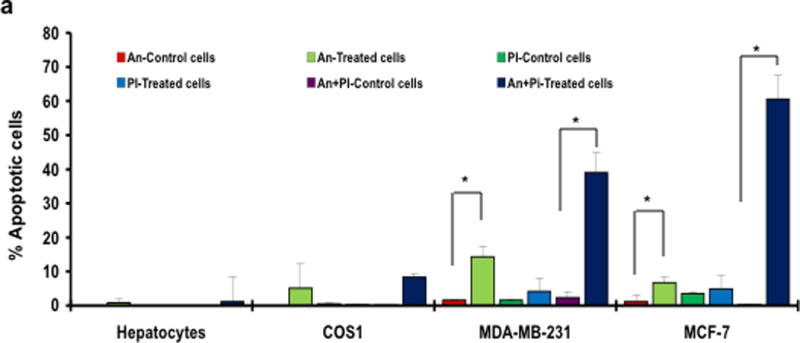
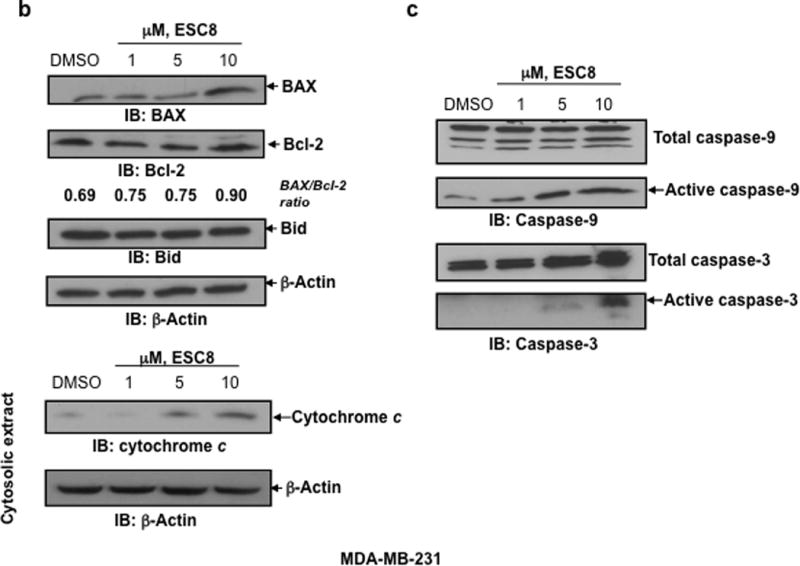
a. Annexin V binding and propidium iodide (PI) uptake in cells treated with ESC8 were measured using flow cytometry. MCF-7, MDA-MB-231, COS-1, hepatocyte cells were treated with ESC8 (10 µM) for 16 hours or kept untreated and stained with annexin V and PI for further flow cytometric analysis. At a 10 µM concentration significantly higher levels of Annexin/PI staining were recorded in ESC8-treated MCF-7 and MDA-MB-231 cells (* p <0.05 for both An & An+PI positive, MDA-MB-231 & MCF7 cells with ESC8 treatment vs. control and * p values <0.001 between An+PI positive cancer cells with that of non cancer cells). The data shown are representative of three independent experiments with similar findings. b. The 10 µM dose of estradiol derivative ESC8 increased the BAX (an apoptosis promoter) to Bcl-2 (an apoptosis inhibitor) protein ratio in MDA-MB-231 cells after 16 hours of treatment. ESC8-treatment also up-regulated the cysolic level of cytochrome c. Bid expression did not change with ESC8 treatments. c. The 1 µM and 5 µM doses of ESC8 were sufficient for the induction of activated caspase-9 and -3 activities, respectively, in MDA-MB-231 cells after 16 hours of treatment, consistent with the increase in Annexin V/PI staining. β-Actin was used as a loading control. The relative fold expressions of protein levels have been indicated as required. Figures represent three separate experiments with similar results.
Effect of ESC8 on MDA-MB-231 apoptosis: initiation of intrinsic pathway and increased caspase activity
Apoptosis provides a conceptual framework to link cancer genetics with cancer therapy. The intrinsic apoptotic pathway hinges on the balance of activity between pro- and anti-apoptotic members of the Bcl-2 family, which act to regulate the permeability of mitochondrial membranes and subsequently induce apoptosis. The cells were treated with increasing doses of ESC8. The 10 µM dose of estradiol derivative ESC8 increased the BAX (an apoptosis promoter) to Bcl-2 (an apoptosis inhibitor) protein ratio in MDA-MB-231 cells after 16 hours of treatment (Figure 2b). ESC8 treatment caused significant up-regulation of the level of cytochrome c in cytosolic extracts of ESC8 treated cells (Figure 2b). We also found that a 1 µM and 5 µM dose of ESC8 were sufficient for the induction of activated caspase-9 and -3 activities (Figure 2c), respectively, in MDA-MB-231 cells after 16 hours of treatment, consistent with the increase in Annexin V/ PI staining (Figure 2a). However, no change in the levels of activated caspase-8 (data not shown) and Bid (Figure 2b) were detected in MDA-MB-231 cells after ESC8 treatment. The data clearly indicate that the induction of ESC8-mediated apoptosis in MDA-MB-231 was due to activation of caspases such as initiator caspase-9 and effecter caspase-3.
ESC8 induces autophagy
he role of autophagy in cancer is a topic of intense debate. Many anti-cancer agents have been reported to induce autophagy, suggesting tumor cell death by these agents (21). As a matter of interest, we chose to see if ESC8 also falls in the above-mentioned category of agents that induce autophagy as a mediator of cancer cell death. As part of our investigation, we checked the status of autophagy induction in MDA-MB-231 cells. MDA-MB-231 cells were treated with three different doses of ESC8 for 16 hours. A cytosolic form of LC3 (LC3-I) associates to phosphatidylethanolamine to form a LC3-phosphatidylethanolamine conjugate (LC3-II). This is recruited to autophagosomal membranes, and the total turnover of LC3-II reflects the autophagic turnover in the cell (21, 28). Figure 3a clearly shows the significant up-regulation of LC3B-II, starting with a 1 µM dose of ESC8, indicating a substantial increase in autophagy. Interestingly, no significant changes in the expression levels of autophagy-related proteins, such as Atg5-Atg12 complex with anti-Atg5 or anti-Atg12 antibody and Beclin-1 (Figure 3a), were detected. Additionally, we have shown that upon ESC8 treatment MDC (a specific marker for autolysosomes) (29) labeled autophagic vesicles were induced in MDA-MB-231 cells (Figure S1b).
Figure 3. ESC8 induces autophagy in MDA-MB-231 cells.
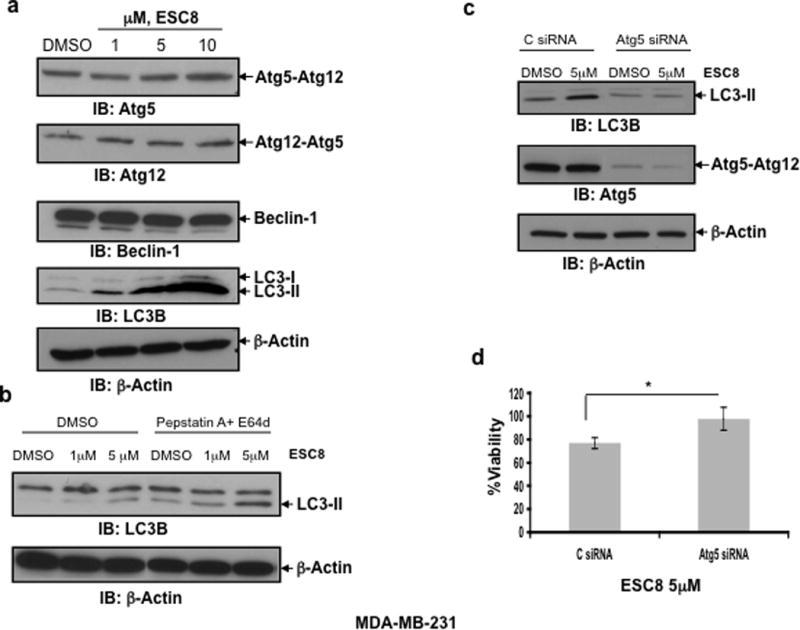
a. The significant up-regulation of LC3B-II, starting with a 1 µM dose of ESC8, was detected, indicating a substantial increase in autophagy in MDA-MB-231 cells. The levels of Atg5-Atg12 complex and Beclin-1 did not change significantly upon ESC8 treatment. b. MDA-MB-231 cells were treated with ESC8 for 16 hours in presence or absence of pepstatin A and E64d. Processing of LC3-II was determined by Western blot analysis. c. LC3-II levels in MDA-MB-231 cells upon ESC8 treatment (5 µM) after Atg5 knockdown. d. ESC8-mediated killing was significantly reduced (* p< 0.01) after Atg5 knockdown in MDA-MB-231 cells. Figures represent three separate experiments with similar results.
At the late stage of autophagy, LC3-II itself, is degraded by lysosome. Therefore, autophagic flux assay is recently being performed to measure the amount of LC3-II delivered to lysosomes by comparing LC3-II levels with or without the treatment of lysosomal protease inhibitors (30). ESC8 treatment in combination with pepstatin A and E64d showed an increase in LC3-II level compared to ESC8 treatment alone in MDA-MB-231 cells, indicating the accumulation of LC3-II due to the blockade of LC3-II degradation in lysosomes (Fig. 3b), which further suggests an autophagic response caused by ESC8.
Next, we were interested in evaluating whether autophagy is responsible for cell death caused by the exposure to ESC8. MDA-MB-231 cells were pretreated with Atg5 siRNA to block the induction of autophagy and then treated with 5 µM of ESC8. After 16 hours of ESC8 treatment, cell survival was measured by MTT assay. After Atg5 knockdown, ESC8 could not induce LC3-II formation (Figure 3c) and survival of MDA-MB-231 cells was significantly increased (p< 0.01) (Figure 3d) as evidenced from the MTT assay result. Atg5 knockdown has no effect on the Annexin V/PI staining of the MDA-MB-231 cells compared to control siRNA treated cells upon ESC8 treatment (data not shown).
Taken together, our data (Figure 2–3) indicated that ESC8 led to simultaneous induction of apoptosis and autophagy to kill the ER-negative breast cancer cells. Finally, we wanted to understand which pathway might be involved in this regulation.
Effect of ESC8 on Akt and mTOR phosphorylation
The P13K/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR)-signaling pathways has been implicated in autophagy and apoptosis regulation (31–33). First we examined whether PI3K-Akt pathway inhibition could have any effect on ESC8-mediated killing. Pre-treatment of MDA-MB-231 cells with PI3K-Akt pathway inhibitor LY294002 (LY) followed by treatment with ESC8 clearly and considerably inhibited the ESC8-mediated toxicity in the cells (Figure 4a). Further western-blot results show that a 1 µM ESC8 treatment blocked the phosphorylation of mTOR at ser-2448 in MDA-MB-231 cells (Figure 4b). Inhibition of mTOR phosphorylation resulted in decreased phosphorylation of p70S6 kinase at Thr-389 (Figure 4b), the downstream effecter of mTOR and indicting the inhibition of mTOR kinase activity upon ESC8 treatment. In contrast, a 10 µM dose of ESC8 in MDA-MB-231 cells induced phosphorylation of Akt1/2/3 at ser-473 (Figure 4b). To replicate these anti-cancer effects in a preclinical set-up, we further developed an orthotopic breast cancer model with MDA-MB-231 cells.
Figure 4. ESC8 kills ER- breast cancer cells by manipulating PI3K-Akt pathway.
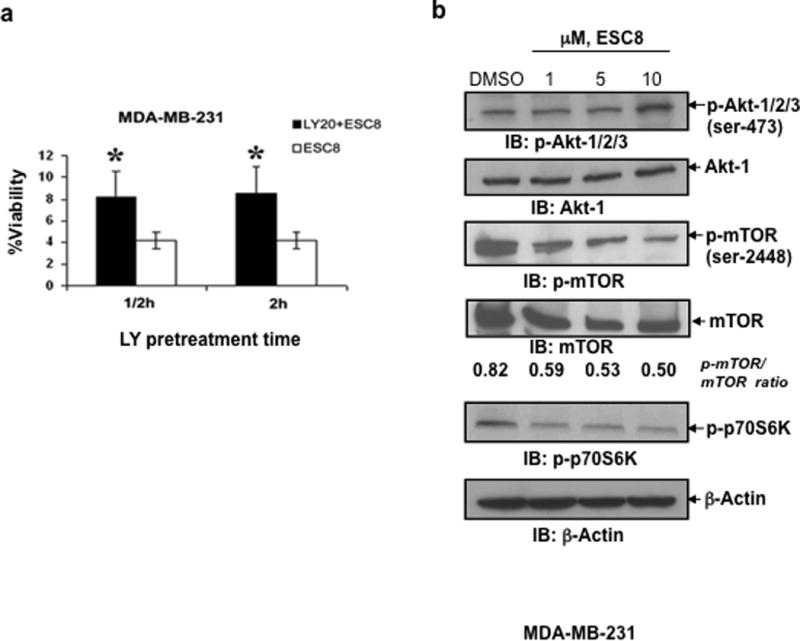
a. MDA-MB-231 cells were pre-treated with 20µM LY294002 (LY20) (black bar) or untreated cells (white bar) treated with ESC8 and viability of cells were assayed (* p< 0.005), (n=6). b. In MDA-MB-231 cells, a 1 µM ESC8 treatment blocked mTOR phosphorylation at ser-2448 and its kinase activity, as determined by Western blot with antibodies against p-mTOR and p-p70S6 kinase. In contrast, a 10 µM dose of ESC8 in MDA-MB-231 cells induced phosphorylation of Akt-1/2/3 at ser-473. β-Actin was used as a loading control. The relative fold expressions of protein levels have been indicated as required. Figures represent three separate experiments with similar results.
Effect of ESC8 on tumor regression and tumor cell apoptosis
The naked molecule ESC8 was injected intraperitoneally into mice containing a mammary pad tumor xenograft of MDA-MB-231 cells. The 10-mg/kg ESC8 injection began when average tumor volumes were about 30 to 40 cubic mm. In another group of mice, 10 mg/kg of ESC8 was injected intraperitoneally when the average tumor volume reached 130 cubic mm. Figure 5a clearly indicates the tumor-inhibitory effect of ESC8, even at a limited number of injections. The significant inhibitory effect of ESC8 was pronounced even when the tumor size was bigger. To further confirm the mechanism of the observed tumor-suppressive activities, we examined the effect of ESC8 on MDA-MB-231 tumor cell apoptosis in vivo with the TUNEL assay (Figure 5b–c). The average number of TUNEL-positive cells in ten randomly selected microscopic fields in both control and ESC8-treated groups were calculated. A significant increase in the number of apoptotic cells was observed in the group treated with ESC8 (p < 0.001, compared to the control group).
Figure 5. Effect of ESC8 on tumor formation, aggressiveness and apoptosis induction in vivo.
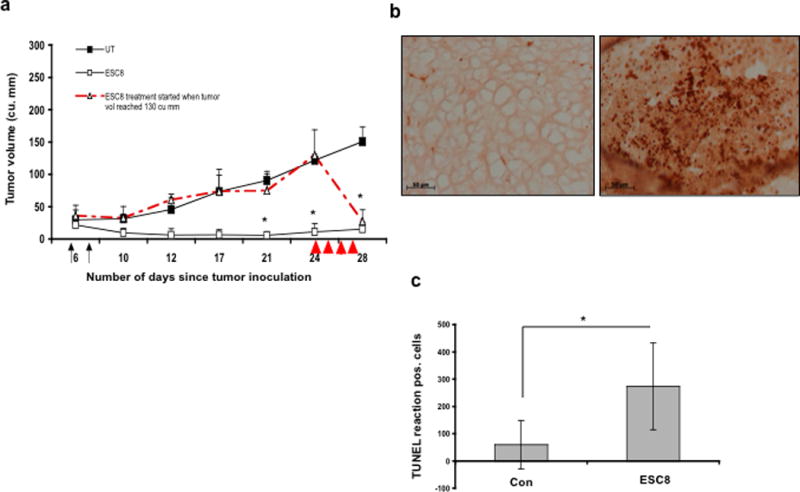
a. When the average tumor became 30 mm,3 two injections (black arrow) of ESC8 (10 mg/Kg, open square) and 5% glucose (UT, black square) were given intraperitoneally to respective groups of mice. In other group (open triangle) ESC8 treatment started when tumor volume in mice reached 130 mm3. In this group mice received 4 injections of ESC8 (10 mg/Kg, red arrows head) intraperitoneally for four consecutive days [n=5, * p < 0.01]. b. To detect apoptosis, a TUNEL assay was performed on two sets of tumors obtained from mice either ESC8-treated or from control groups which received 5% glucose. TUNEL-positive nuclei in control and treatment groups are shown. Control group (left) received only 5% glucose and treatment group (right) received 10 mg/Kg ESC8 only. c. The average number of TUNEL-positive cells was scored in 10 randomly selected microscopic fields. ESC8 as a single therapeutic agent caused significant tumor cell apoptosis. *, p < 0.01(treated group vs. control group).
DISCUSSION
Breast cancer, the leading cause of death in women worldwide, is associated with ER and its functional activity in 60% to 70% of cancer cases. Conventional treatments of early-stage or ER-responsive advanced-stage breast cancer rely on either hormone ablation or antagonizing ER through the use of small molecule drugs. The use of conventional therapeutics for advanced breast cancer becomes wasteful, non-effective and, in some cases, exacerbates the situation through the induction of thrombosis and endometrial carcinoma — especially for tamoxifen-treated cases (34, 35).
The complexity in the advanced or post–ER-reliance stage of cancer, which includes the failure to activate apoptotic signals, heralds the transformation of primary cancer cells into a drug-resistant, metastatic phenotype. In a recent study, we could selectively manipulate glucocorticoid receptor (GR), a nuclear hormone receptor (NHR), in cancer cells using a cationic liposomal formulation associated with GR-ligand dexamethasone for selective access to the GR response element in cancer cells’ nuclei (36). In trying to replicate a similar observation with breast cancer cell-associated NHR, ER using its endogenous ligand, ES, we found to our surprise that ES in mere association with an eight-carbon, twin-chain, quaternary ammonium cationic lipid could efficiently kill both ER-positive and ER-negative breast cancer cells.
On the basis of the above observations, we hypothesized that instead of mere association, if we chemically conjugate this eight-carbon cationic lipid to an estrogen molecule, we might get enhanced anti-breast cancer activity. We found that ESC8 could initiate ER expression-status nonspecific, anti–breast-cancer activity, but with minimal toxicity to the non-cancer cells. The observation is in contrast to previous evidence in the literature in which anti-cancer molecules with ES-moiety or ES-targeting antagonists, such as tamoxifen or Faslodex, show selective activity against ER-positive breast cancer cells. Our data clearly indicate that ESC8 is like any other common estrogenic drug-type molecule that is prone to induce cell death in ER-positive cells. At least in ER-positive breast cancer cells, the cationic molecule in the ER-bound state would have proximity to the estrogen responsive promoter region of genomic DNA, thereby possibly damaging the DNA through irreversible electrostatic interaction. In ER-negative breast cancer cells, ESC8 exerts its effect possibly by manipulating PI3K/Akt/mTOR pathway.
Our subsequent studies revealed ESC8-mediated prominent induction of apoptosis in both ER-positive and ER-negative cancer cells. On investigating the specific reason for the activation of apoptosis in ER-negative MDA-MB-231 cells, we found significant apoptosis induction through the mitochondria-mediated intrinsic pathway in which caspase 3 formation was triggered by an elevated level of caspase 9 on ESC8 treatment. We also noticed that autophagy was highly induced but with no significant change in the level of autophagy-related proteins such as Atg5-Atg12 and Beclin-1. Because one of the autophagy regulators is the PI3K-Akt-mTOR pathway, we investigated the effect of ESC8 on the protein levels of p-Akt-1/2/3 (ser-473), p-mTOR (Ser-2448), and p-p70S6K (Thr-389). ESC8 treatment inhibited mTOR and p70S6K phosphorylations, however we observed an increase in phosphorylation of Akt at ser-473 with the 10 µM dose of ESC8. Although increased Akt phosphoryation is pro-survival, this finding resembles the rapamycin-mediated mTOR inhibition, which accompanies elevation of the phospho-Akt level in a negative feedback loop. Previous literatures have shown that rapamycin-induced Akt phosphorylation is dependent on PI3K and IGF signaling pathway (37, 38). Therefore, mTOR is a possible target of ESC8 and we think that ESC8 in combination with Akt Inhibitor would be better option for breast cancer therapy. Additional in-depth studies are currently being done to further look into the novel mechanism.
Our observation of simultaneous induction of mitochondria-assisted apoptotic signal and autophagy induction followed by inhibition of mTOR activity contradicts the previous literature, which showed that inhibition of mTOR fortifies cells against pro-apoptotic stimuli with the concomitant induction of autophagy (18, 19). Here in ESC8-mediated killing of breast cancer cells, autophagy accompanies apoptosis, and our data also clearly prove that under these circumstances, the induction of autophagy is not cytoprotective in nature.
In conclusion, we report the development of a new class of highly efficient anti-breast cancer agents that contain a novel combination of estradiol and cationic lipid moieties. This estrogenic molecule, in contrast to other available estrogenic drugs, exhibits highly efficient anti-cancer activity in all ER-positive and ER-negative primary and advanced breast cancer cells. Hence, this anti-cancer agent is the first estrogenic drug that, with this special chemico-structural trait, shows unusual co-induction of autophagy and apoptosis in that it kills even ER-negative breast cancer cells. This unique structural trait provides the potential scope to treat even multiple-staged breast cancer using a single drug-based therapy.
Supplementary Material
Acknowledgments
B.S.R. thanks U.G.C, S.R., K.P. and K.R.R. thanks CSIR, Govt. of India, for their respective doctoral fellowships. This work is supported by NIH grant CA78383, a Mayo Clinic Breast Cancer SPORE Development grant, and a generous gift from Bruce and Martha Atwater (DM). DM is a Scholar of the American Cancer Society. RB acknowledges IUSSTF for a visiting fellowship to Mayo Clinic, USA, and Department of Science & Technology (Govt. of India) for a research grant. We also acknowledge Santanu Bhattacharya and Jim Tarara, Mayo Clinic for helping with MDC staining and confocal microscopy, respectively.
References
- 1.Cleator SJ, Ahamed E, Coombes RC, Palmieri C. A 2009 update on the treatment of patients with hormone receptor-positive breast cancer. Clin Breast Cancer. 2009;9(Suppl 1):S6–S17. doi: 10.3816/CBC.2009.s.001. [DOI] [PubMed] [Google Scholar]
- 2.Osborne CK. Steroid hormone receptors in breast cancer management. Breast Cancer Res Treat. 1998;51:227–38. doi: 10.1023/a:1006132427948. [DOI] [PubMed] [Google Scholar]
- 3.Boivin RP, Luu-The V, Lachance R, Labrie F, Poirier D. Structure-activity relationships of 17alpha-derivatives of estradiol as inhibitors of steroid sulfatase. J Med Chem. 2000;43:4465–78. doi: 10.1021/jm0001166. [DOI] [PubMed] [Google Scholar]
- 4.Brueggemeier RW, Bhat AS, Lovely CJ, et al. 2-Methoxymethylestradiol: a new 2-methoxy estrogen analog that exhibits antiproliferative activity and alters tubulin dynamics. J Steroid Biochem Mol Biol. 2001;78:145–56. doi: 10.1016/s0960-0760(01)00090-5. [DOI] [PubMed] [Google Scholar]
- 5.Cushman M, He HM, Katzenellenbogen JA, Lin CM, Hamel E. Synthesis, antitubulin and antimitotic activity, and cytotoxicity of analogs of 2-methoxyestradiol, an endogenous mammalian metabolite of estradiol that inhibits tubulin polymerization by binding to the colchicine binding site. J Med Chem. 1995;38:2041–9. doi: 10.1021/jm00012a003. [DOI] [PubMed] [Google Scholar]
- 6.Jones GB, Hynd G, Wright JM, et al. Target-directed enediynes: designed estramycins. J Org Chem. 2001;66:3688–95. doi: 10.1021/jo0055842. [DOI] [PubMed] [Google Scholar]
- 7.Jordan VC. Tamoxifen: a most unlikely pioneering medicine. Nat Rev Drug Discov. 2003;2:205–13. doi: 10.1038/nrd1031. [DOI] [PubMed] [Google Scholar]
- 8.Kasiotis KM, Mendorou C, Haroutounian SA, Alexis MN. High affinity 17alpha-substituted estradiol derivatives: synthesis and evaluation of estrogen receptor agonist activity. Steroids. 2006;71:249–55. doi: 10.1016/j.steroids.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 9.Lerner LJ, Jordan VC. Development of antiestrogens and their use in breast cancer: eighth Cain memorial award lecture. Cancer Res. 1990;50:4177–89. [PubMed] [Google Scholar]
- 10.Mitra K, Marquis JC, Hillier SM, et al. A rationally designed genotoxin that selectively destroys estrogen receptor-positive breast cancer cells. J Am Chem Soc. 2002;124:1862–3. doi: 10.1021/ja017344p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Purohit A, Hejaz HA, Walden L, et al. The effect of 2-methoxyoestrone-3-O-sulphamate on the growth of breast cancer cells and induced mammary tumours. Int J Cancer. 2000;85:584–9. [PubMed] [Google Scholar]
- 12.Rao PN, Cessac JW, Tinley TL, Mooberry SL. Synthesis and antimitotic activity of novel 2-methoxyestradiol analogs. Steroids. 2002;67:1079–89. doi: 10.1016/s0039-128x(02)00066-1. [DOI] [PubMed] [Google Scholar]
- 13.Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;365:1687–717. doi: 10.1016/S0140-6736(05)66544-0. [DOI] [PubMed] [Google Scholar]
- 14.Chia S, Gradishar W. Fulvestrant: expanding the endocrine treatment options for patients with hormone receptor-positive advanced breast cancer. Breast. 2008;17(Suppl 3):S16–21. doi: 10.1016/j.breast.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 15.Peng J, Sengupta S, Jordan VC. Potential of selective estrogen receptor modulators as treatments and preventives of breast cancer. Anticancer Agents Med Chem. 2009;9:481–99. doi: 10.2174/187152009788451833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shelly W, Draper MW, Krishnan V, Wong M, Jaffe RB. Selective estrogen receptor modulators: an update on recent clinical findings. Obstet Gynecol Surv. 2008;63:163–81. doi: 10.1097/OGX.0b013e31816400d7. [DOI] [PubMed] [Google Scholar]
- 17.Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–6. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 18.Ravikumar B, Berger Z, Vacher C, O'Kane CJ, Rubinsztein DC. Rapamycin pre-treatment protects against apoptosis. Hum Mol Genet. 2006;15:1209–16. doi: 10.1093/hmg/ddl036. [DOI] [PubMed] [Google Scholar]
- 19.Ravikumar B, Vacher C, Berger Z, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–95. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 20.Schoenlein PV, Periyasamy-Thandavan S, Samaddar JS, Jackson WH, Barrett JT. Autophagy facilitates the progression of ERalpha-positive breast cancer cells to antiestrogen resistance. Autophagy. 2009;5:400–3. doi: 10.4161/auto.5.3.7784. [DOI] [PubMed] [Google Scholar]
- 21.Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5:726–34. doi: 10.1038/nrc1692. [DOI] [PubMed] [Google Scholar]
- 22.Bursch W, Ellinger A, Kienzl H, et al. Active cell death induced by the anti-estrogens tamoxifen and ICI 164 384 in human mammary carcinoma cells (MCF-7) in culture: the role of autophagy. Carcinogenesis. 1996;17:1595–607. doi: 10.1093/carcin/17.8.1595. [DOI] [PubMed] [Google Scholar]
- 23.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–77. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 24.Sato K, Tsuchihara K, Fujii S, et al. Autophagy is activated in colorectal cancer cells and contributes to the tolerance to nutrient deprivation. Cancer Res. 2007;67:9677–84. doi: 10.1158/0008-5472.CAN-07-1462. [DOI] [PubMed] [Google Scholar]
- 25.Mukhopadhyay D, Knebelmann B, Cohen HT, Ananth S, Sukhatme VP. The von Hippel-Lindau tumor suppressor gene product interacts with Sp1 to repress vascular endothelial growth factor promoter activity. Mol Cell Biol. 1997;17:5629–39. doi: 10.1128/mcb.17.9.5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pal S, Datta K, Khosravi-Far R, Mukhopadhyay D. Role of protein kinase Czeta in Ras-mediated transcriptional activation of vascular permeability factor/vascular endothelial growth factor expression. J Biol Chem. 2001;276:2395–403. doi: 10.1074/jbc.M007818200. [DOI] [PubMed] [Google Scholar]
- 27.Reddy BS, Banerjee R. 17Beta-estradiol-associated stealth-liposomal delivery of anticancer gene to breast cancer cells. Angew Chem Int Ed Engl. 2005;44:6723–7. doi: 10.1002/anie.200501793. [DOI] [PubMed] [Google Scholar]
- 28.Tanida I, Ueno T, Kominami E. LC3 and Autophagy. Methods Mol Biol. 2008;445:77–88. doi: 10.1007/978-1-59745-157-4_4. [DOI] [PubMed] [Google Scholar]
- 29.Munafo DB, Colombo MI. A novel assay to study autophagy: regulation of autophagosome vacuole size by amino acid deprivation. J Cell Sci. 2001;114:3619–29. doi: 10.1242/jcs.114.20.3619. [DOI] [PubMed] [Google Scholar]
- 30.Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–5. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 31.Castedo M, Ferri KF, Kroemer G. Mammalian target of rapamycin (mTOR): pro- and anti-apoptotic. Cell Death Differ. 2002;9:99–100. doi: 10.1038/sj.cdd.4400978. [DOI] [PubMed] [Google Scholar]
- 32.Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C. PI3K/Akt and apoptosis: size matters. Oncogene. 2003;22:8983–98. doi: 10.1038/sj.onc.1207115. [DOI] [PubMed] [Google Scholar]
- 33.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Killackey MA, Hakes TB, Pierce VK. Endometrial adenocarcinoma in breast cancer patients receiving antiestrogens. Cancer Treat Rep. 1985;69:237–8. [PubMed] [Google Scholar]
- 35.Lipton A, Harvey HA, Hamilton RW. Venous thrombosis as a side effect of tamoxifen treatment. Cancer Treat Rep. 1984;68:887–9. [PubMed] [Google Scholar]
- 36.Mukherjee A, Narayan KP, Pal K, et al. Selective cancer targeting via aberrant behavior of cancer cell-associated glucocorticoid receptor. Mol Ther. 2009;17:623–31. doi: 10.1038/mt.2009.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun SY, Rosenberg LM, Wang X, et al. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005;65:7052–8. doi: 10.1158/0008-5472.CAN-05-0917. [DOI] [PubMed] [Google Scholar]
- 38.Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene. 2007;26:1932–40. doi: 10.1038/sj.onc.1209990. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.