

Abstract
Background
Gene expression analyses demand appropriate reference genes (RGs) for normalization, in order to obtain reliable assessments. Ideally, RG expression levels should remain constant in all cells, tissues or experimental conditions under study. Housekeeping genes traditionally fulfilled this requirement, but they have been reported to be less invariant than expected; therefore, RGs should be tested and validated for every particular situation. Microarray data have been used to propose new RGs, but only a limited set of model species and conditions are available; on the contrary, RNA-seq experiments are more and more frequent and constitute a new source of candidate RGs.
Results
An automated workflow based on mapped NGS reads has been constructed to obtain highly and invariantly expressed RGs based on a normalized expression in reads per mapped million and the coefficient of variation. This workflow has been tested with Roche/454 reads from reproductive tissues of olive tree (Olea europaea L.), as well as with Illumina paired-end reads from two different accessions of Arabidopsis thaliana and three different human cancers (prostate, small-cell cancer lung and lung adenocarcinoma). Candidate RGs have been proposed for each species and many of them have been previously reported as RGs in literature. Experimental validation of significant RGs in olive tree is provided to support the algorithm.
Conclusion
Regardless sequencing technology, number of replicates, and library sizes, when RNA-seq experiments are designed and performed, the same datasets can be analyzed with our workflow to extract suitable RGs for subsequent PCR validation. Moreover, different subset of experimental conditions can provide different suitable RGs.
Electronic supplementary material
The online version of this article (doi:10.1186/s12938-017-0356-5) contains supplementary material, which is available to authorized users.
Keywords: Reference genes, Normalization, Real-time PCR, Quantitative PCR, Olive (Olea europaea L.), Cancer
Background
Traditionally, gene expression studies have been carried out by non-quantitative or semi-quantitative RNA gel blotting and later by reverse transcription-polymerase chain reaction (RT-PCR) analyses. Development of real-time, quantitative PCR (qPCR) [1] took the place of these techniques due to its higher specificity, sensitivity and broad quantification range. The use of an appropriate reference gene (RG) to avoid false results and for proper interpretation of gene expression data soon emerged as a significant concern in these experiments, mainly due to the increased sensitivity of qPCR with respect to Northern blotting and RT-PCR. The first RGs were brought from Northerns and usually encoded proteins involved in structural functions and basic cell metabolism due to their theoretical expression invariability in most tissues. This initial election was revealed inappropriate [2–4] and the quest of more reliable RGs has been pursued in the literature [5–8].
Conclusions of any qPCR experiment are depending on RGs, but also on the selection of an appropriate normalization method. Relative quantification is the most widely used method for normalization, where gene expression level is normalized by an internal RG that should remain constant in all experimental conditions under study. BestKeeper [9], geNorm [10] and NormFinder [11] are the most popular methods for normalization and confirming RGs. Based on the raw, relative quantities, geNorm calculates the minimal number of RGs for each experiment and NormFinder also provides a stability value for each gene. BestKeeper employs a pair-wise correlation analysis based on a geometric mean to determine the optimal RGs. But all of them are based on qPCR data, which produce a recursive problem since a qPCR is required to decide if a RG is appropriate for qPCR. Reports describing that several of the most commonly used housekeeping genes exhibit substantial variability in microarray data sets or under different experimental conditions are becoming more and more frequent [12, 13]. Consequently, the choice of the best RGs should be based on preliminary experimental evidence when comparing different developmental stages, tissues, cell types or environmental conditions, as well as in careful testing and validation [14]. Hence, the selection of both RGs and normalization method are critical for obtaining reliable quantitative gene expression assessments to correct for non-specific variation, such as differences in RNA quantity and quality.
In the search for appropriate, stable RGs, a data mining strategy based on the use of publicly available microarray data repositories was envisaged. It was available only for some species, usually model organisms [15], and provided useful RGs [7]. When microarray data are unavailable (i.e. for non-model organisms or unusual experimental conditions), other strategies must be regarded to establish suitable RGs. Such is the case of the olive tree (Olea europaea L.), one of the most important oil-producing plant species all over the world. Although a first draft genome of this plant has just been published [16] and some gene expression analyses have been reported [17, 18], further and longer studies will be required to select reliable RGs. Several attempts for the identification of putative RGs in this species have been carried out, by evaluating olive genes orthologs to the best-ranked RGs from other crops. They were selected according to their stability in olive tissues, as it occurs in other plants, and throughout different experimental conditions: different developmental stages of the olive mesocarp tissue across different cultivars [19], and several fruit developmental/ripening stages and leaves subjected to wounding [20, 21]. The peculiarity of plant reproductive tissues makes the search of these RGs particularly tricky, as some well known housekeeping genes display differential expression in pistil, pollen and other floral organs [15]. Nevertheless, other analyses indicate that a large proportion of constitutive transcripts are shared by most somatic, reproductive, and haploid tissues [22]. Consequently, a reasonable thought is that RGs can be more easily detected in model organism such as Arabidopsis, mice or humans, where more microarray data are available. The only problem with these species relies on the experimental conditions for which new RGs are required. It can be concluded that ideally RGs for qPCR validation could be inferred from the experimental data to be analyzed.
The falling cost of NGS (next-generation sequencing) technologies has made their use more and more frequent. This has resulted in an explosive growth of data that are gathered into the Sequence Read Archive (SRA) [23]. This public repository allows for new discoveries by comparing the archived data sets. Since any RNA-seq study requires further, experimental validation, and qPCR has become the de facto standard, we thought that NGS data can also be analyzed as a source of RGs. With this aim, an automatic workflow has been constructed to obtain highly, but invariantly, expressed RGs for particular experimental conditions based on the coefficient of variation (CV) of normalized expression values by RPMM (reads per mapped million) and managing both Roche/454 and Illumina reads. Candidate RGs have been proposed for reproductive tissues of olive tree, Arabidopsis thaliana flowers, and three different human cancers. Experimental validation of olive tree RGs is also included.
Methods
Sequence reads and reference transcriptomes
Olea europaea (olive tree) reads (SRA BioProject PRJNA287107) correspond to a Roche GS-FLX Titanium + sequencing experiment for different developmental stages of pollen and pistil, as described in [17]. Reads (mean length 385 nt) were mapped only against transcripts coding for a complete protein (9157 transcripts) in the reproductive transcriptome described in ReprOlive (http://reprolive.eez.csic.es) [17].
Arabidopsis thaliana reads were obtained from SRA BioProject PRJEB9470. A late flowering strain (Columbia) and the reference Killean were compared to determine genes involved in early flowering [24]. Three biological replicates from ten day-old seedlings were paired-end sequenced (100 bp) on a HiSeq 1000. The Arabidopsis reference transcriptome (35,386 transcripts) was downloaded from Phytozome (https://phytozome.jgi.doe.gov) and refers to TAIR10 [25].
Sequencing reads from matched normal and malignant tissues from the same patient were considered for the study in humans. Matched normal and malignant prostate tissues from 14 Chinese [26] were obtained from SRA BioProject PRJEB2449 (HiSeq 2000, 90 nt paired-end reads). Sequencing of matched samples of normal lung and small-cell lung cancer of 17 patients [27] were available under permission at EGA under accession EGAS00001000334 (HiSeq 2000, 75 nt, paired-end reads). Matched samples of normal lung and lung adenocarcinoma from 50 patients [28] were downloaded from ENA under accession number ERP001058 (HiSeq 2000, 100 nt, paired-end reads). The three data sets of human reads were mapped onto the cDNA data set of 176,241 transcripts (downloaded from the ENSEMBL repository) deduced from the human GRCh38 genome.
Read count table
A scheme of the automatic workflow executed in this work for obtaining the matrix of read counts of transcripts vs. experimental conditions is shown in Fig. 1a for the Arabidopsis datasets. It has been constructed using AutoFlow [29], a workflow manager developed in Ruby in our laboratory. The workflow receives as input files the raw reads and the transcriptome, both in Fasta format. Each file of raw reads is considered an experimental condition. Raw reads are then pre-processed using SeqTrimNext [30] to remove noisy sequences and retain only reliable reads. Useful reads are then mapped to the corresponding transcriptome using Bowtie2 [31] with default parameters and the −a option to allow each read to map in all possible transcripts. Mapped reads are then counted with Bio-samtools from BioRuby [32] with the −f2 option to count only reads where both ends are mapping on the same transcript (only for Illumina reads). The output is the tab-delimited, read count table where columns are experimental conditions, rows are transcripts, and the values are the number of counts of each transcript at each condition. Other pre-processing and mapping approaches can also be used provided that a tab-delimited read count table of transcripts vs. experimental conditions is obtained.
Fig. 1.

Flow diagram as provided by AutoFlow for the detection of RGs using the SRA datasets of PRJEB9470 from Arabidopsis. a The first workflow that prepares the reads, maps them on the transcriptome and provides the read count table. b The findRGs workflow for detecting candidate RGs. In this example, several filtering parameters were tested: 10 and 20% for the maximum CV, and 10,000, 30,000 and 100,000 reads for the minimum counted reads per transcript and condition. One Venn diagram by each CV cut-off is obtained, as shown in Figs. 2, 4, 5 and 6
Detection of reference genes
A second workflow (called findRGs, Fig. 1b) will look for the candidate RGs and is the main contribution of this work. It is also based on AutoFlow, although the basic functions could be also implemented in a spreadsheet. The input required are the previous read count table and, optionally, a tab-delimited annotation table with at least two columns: the first column containing the ID of each transcript, and the second column containing a reference ID of an orthologous gene. For convenience, it is recommended to add a third column containing the description of the ortholog. As shown in Fig. 1b, counts in the table are then normalized as the number of reads mapped on a transcript divided by the number of transcriptome-mapped reads of the corresponding sample (RPMM: reads per mapped million). Coefficient of variation (CV, ratio of the standard deviation to the mean, expressed as a percentage) of RPMM along all conditions is obtained per transcript. This normalized table containing the RPMMs and the CVs is analyzed by two customizable parameters: (1) CV (10% maximum, although the range can move from 0 up to 20) to select for genes whose expression is as invariant as possible, and (2) counted reads per transcript and condition (minimum of 10; we propose to start with a minimum value resulting from the multiplication of 0.00003 by the lowest library size) to select for RG with the highest level of expression to warrant the correct amplification by PCR. The workflow allows combining different values of these two parameters in a single execution. When the reference transcriptome used for mapping overestimates the number of transcripts, many of them will refer to the same gene. In this case, the optional annotation table serves to detect transcripts sharing the same ortholog or ID, and filter them to retain only the one with the highest RPMM value. Finally, a Venn diagram showing the number of specific and common orthologs between the different combinations of tested parameters is generated, in order to visualize the results and check the suitability of such parameters.
To install findRGs, first install Ruby 1.9.3 or higher and R 3.0.2 or higher, and then install AutoFlow, the workflow manager, as a gem with the command gem install autoflow. Other dependiencies have to be downloaded with the command git clone ‘https://github.com/seoanezonjic/general_scripts.git’, and then placed in a ‘custom’ directory. This custom directory containing AutoFlow scripts must be included in the $PATH environment variable of your computer as export PATH = "path_to_custom_directory:$PATH" in the.baschrc file. Finally download findRGs to the AutoFlow environment with the commad AutoFlow –get_template_repository ‘https://github.com/rosariocarmona/autoflow_templates.git’. The workflow can be executed as AutoFlow −w findRGs −V ‘$input_file = read_count_table,$min_reads = [10;50;100],$cv_filter = [10, 20],$annot = annotation_table, where −w indicates that findRGs is the workflow template to be executed and −V sets the default parameters.
Experimental validation of candidates to RG in olive tree
Preliminary validation of some olive tree candidates was performed by semi-quantitative, real time PCR analysis according to Alché et al. [33] using 25 cycles to ensure an exponential amplification rate. Primers used are listed in Table 1. A total of 5 μl of the PCR reaction were loaded per lane and separated on 2% agarose gels in Tris–borate-EDTA (TBE). Equal loading of the RT mixture used for PCR was ensured by using Bioanalyzer (Agilent Technologies) accurate quantitation.
Table 1.
Primers used for PCR amplification
| Gene | Direction | Sequence |
|---|---|---|
| 18S | Forward | 5′-TTT GAT GGT ACC TGC TAC TCG GAT AAC C |
| Reverse | 5′-CTC TCC GGA ATC GAA CCC TAA TTC TCC | |
| Ubiquitin monomer to pentamer | Forward | 5′-ATGCAGAT(C/T)TTTGTGAAGAC |
| Reverse | 5′-ACCACCACG(G/A)AGACGGAG | |
| Actin | Forward | 5′-TTG CTC TCG ACT ATG AAC AGG |
| Reverse | 5′-CTC TCG GCC CCA ATA GTA ATA | |
| Mitogen-activated protein kinase | Forward | 5′-CCAGGCGAGATTTCAGAGAC |
| Reverse | 5′-TCGGTTTAAGGTCTCGATGG | |
| Proline transporter | Forward | 5′-TTGTAGTGAGGGGCGGTTAC |
| Reverse | 5′-CATGCAACCAAAGAAGCAGA | |
| l-Ascorbate oxidase homolog | Forward | 5′-ACAAAAGGCATTGCTTGGTC |
| Reverse | 5′-GGCCAAAACGAAGTTTACCA | |
| Gliceraldehyde-3-phosphate dehydrogenase | Forward | 5′-GGGCAAGATCAAGATTGGAA |
| Reverse | 5′-GTCTTCTCGCCGAACAAAAG | |
| Salicylic acid-binding protein | Forward | 5′-GCATTGACCCGAAAATCCTA |
| Reverse | 5′-AGGATGGCGGATTTGTAGTG | |
| S-adenosylmethionine decarboxylase proenzyme | Forward | 5′-AGCTTCTGGCATCAGGAAAA |
| Reverse | 5′-AGCCAGTACCCTCTCAAGCA |
Results and discussion
Pre-processed reads
NGS reads cannot be used for mapping as obtained from the corresponding sequencing platform [30]. Therefore, they were pre-processed with SeqTrimNext and then mapped with Bowtie2. Dataset sizes collected for this study were intentionally very heterogeneous (Additional file 1) to test the workflow in different settings. The percent of useful reads seems to be homogeneous within Illumina reads (>64%, mean 85%, depending on datasets), as well as in 454/Roche reads (>50%, mean 54%). The percent of mapped reads with respect to useful reads is also homogeneous within technologies (last column on Additional file 1), suggesting the appropriateness of the pre-processing. Dataset size heterogeneity is regular within three samples: in olive tree, raw reads range from 217,163 to 262,749 (1.2 times) and useful reads from 111,760 to 150,185 (1.3 times); in Arabidopsis, raw reads range from 9,107,610 to 13,076,233 (1.4 times) and useful reads from 8,859,088 to 12,678,437 (1.4 times); and in prostate cancer, data are also very homogeneous, with a range of 1.2 times for both raw and useful reads (Additional file 1). But more extreme situations can be found on lung cancer samples: in small-cell lung cancer, raw reads range from 68.5 to 19.3 million raw reads (3.5 times) and useful reads from 16.8 to 62.6 million reads (3.72 times); and on adenocarcinoma, raw reads range from 10.9 to 109.3 million reads (5.5 times) and useful reads from 8.7 to 93.4 million reads (10.7 times). With such an heterogeneity, and in contrast to previously published [34], the transcript RNA abundance must be normalized within samples in order to remove the bias due to the sequencing depth of a sample.
Since no comparison between transcripts is performed, normalization by the length or the RNA species is not required. A widely used method of count normalization is RPKM (reads per kilobase per million reads) for single-end reads and the FPKM (single fragment per kilobase and million reads) [35], even though they have been revealed to be inconsistent for comparisons within the same sample [36]. However, in this work, gene expression is compared for one gene along all samples, making unnecessary that normalization by length, as each transcript count along samples will be divided by the same constant (transcript length). Moreover, in some non-model organisms (such as olive tree), the transcript length is not well known since only a fragment of the transcript has been reconstructed, or the transcript is divided in several independent contigs. That is why we have introduced a simplification of RPKM as the RPMM (reads per mapped million) based on the counts per million.
Workflow execution times
Execution times for the different groups of tasks of the complete workflow (pre-processing, mapping and analysis with findRGs; Fig. 1) using three datasets from different species, different sequencing technology and increasing number of reads was assessed (Table 2). Using the same number of CPUs, the pre-processing task is by far the longest stage in olive tree and Arabidopsis (98% of total workflow time in olive tree and almost 70% in Arabidopsis), being much more lasting in olive tree pistil than in Arabidopsis, mainly due to the longer Roche/454 read length. In contrast, mapping is the longest task using human prostate reads (around 85% of total workflow time), due to the larger size of the transcriptome on which reads try to align. Analysis with findRGs is quite fast (below 1 min per 100,000 reads) in the three cases, regardless of read type or length, the species and the number of experimental conditions. Therefore, the analysing workflow is considerably fast and can be offered as a web tool, even though long reads or large transcriptomes might decrease its performance.
Table 2.
Workflow execution times estimated for three datasets
| Species/tissue | No. raw reads | Mean length (nt) | No. transcripts | Pre-processing | Mapping | FindRGs | Total |
|---|---|---|---|---|---|---|---|
| Olive tree pistil | 767,963 | 525 | 9157 | 24 min 45 s | 26 s | 5 s | 25 min 16 s |
| 8 nodes, 192 cpus | 3 nodes, 72 cpus | 1 node, 9 cpus | |||||
| Arabidopsis | 23,821,198 (x2) | 100 (x2) | 35,386 | 43 s | 19 s | 0.2 s | 1 min 2 s |
| 8 nodes, 192 cpus | 2 nodes, 48 cpus | 1 node, 9 cpus | |||||
| Human prostate | 969,884,666 (x2) | 90 (x2) | 176,241 | 28 s | 2 min 37 s | 0.03 s | 3 min 5 s |
| 96 nodes, 2304 cpus | 24 nodes, 576 cpus | 1 node, 9 cpus |
All time values are referred to 100,000 reads when executed on SUSE® Linux Enterprise Server v12 using Opteron processors with 4 GB/core of RAM
Candidate RGs in reproductive tissues of olive tree
Some transcripts are better suited RGs for the analysis of gene expression within a given tissue. Moreover, some of these RGs can even be considered appropriate for gene expression analyses involving several tissues [37]. For this reason, three different executions of the workflow were made in olive: pollen, pistil and both together. Less variant transcripts were retained with two CV cut-off values: 10% (default) and 20% (non-stringent). Taking into account that reads come from Roche/454 platform, the minimum number of mapped reads per gene was set to 10, 50 or 100. A comparative summary of results is shown on Fig. 2. While a significant number of candidates are obtained for pollen and pistil, respectively, with the most stringent conditions (>100 reads and CV < 10%), no candidate is obtained for both pollen and pistil and just one with less rigorous parameters (>50 reads, CV < 20%). This suggests that comparative expression analyses of reproductive tissues require a careful selection of RGs.
Fig. 2.

Venn diagrams summarizing the number of RGs obtained for reproductive tissues of olive tree. Two cut-off values were used for CV and three for counted reads. Reproductive RGs were obtained after combining both pollen and pistil reads
A detailed analysis of RGs obtained for these experimental conditions in olive trees have already been suggested and/or used as RGs in others species. In the pistil of the olive tree (Fig. 2a), one of the best RG is ubiquitin (with three different sequences: rp11_olive_006479, rp11_olive_031243, rp11_olive_045557; Additional file 2). Ubiquitin is a traditional and extensively used RG in plants, for example, in banana [38], peach [39] and rice [40], among others. Polyubiquitin 10 (rp11_olive_006473; Additional file 2) also appears as RG and its highly stable expression has been also proven in Arabidopsis [15]. It has been validated as an RG in blueberry [41], cotton [42] and poplar [14], and used for normalizing in a work regarding olive fruit development and ripening [43]. Elongation factor 1−α (two different sequences: rp11_olive_008243 and rp11_olive_009319; Additional file 2) also emerges as a candidate RG in the olive pistil. It was evaluated as candidate for RG in potato (Solanum tuberosum), resulting in the most stable among the group tested during biotic and abiotic stresses [44]. It is therefore suggested that pistil is another organ where this gene is stable. It has also been validated as a good RG in many species. Another candidate is glyceraldehyde-3-phosphate dehydrogenase (rp11_olive_003751; Additional file 2), which was identified as one of the best RGs for olive fruit development and ripening [20] and used as normalizer for the analysis of cDNAs associated with alternate bearing in olive [45]. Other candidates obtained in olive pistil have never been used before as RGs, but they show outstanding RPMM values and very low CV. This is the case, among others, of salicylic acid-binding protein 2 (rp11_olive_003751) and methylesterase 1 (rp11_olive_015883). Their use should be carefully considered and evaluated in the near future.
A larger number of candidate RGs are suggested for olive pollen (Fig. 2b; Additional file 3). No evidence is present in literature about their use as RGs for most of them, in spite of their apparently low variation in their expression. This is the case of MOB kinase activator (rp11_olive_000239; Additional file 3) or cytochrome P450 (rp11_olive_006957; Additional file 3). Their effectiveness as RGs merits the testing. The only gene proposed by the workflow as suitable RG for olive pollen that has been previously used is cysteine proteinase (rp11_olive_005653; Additional file 3). This gene has been validated as stable and as a suitable RG in Coffea arabica [46]. It was also evaluated as RG in olive fruit, but it was not among the most stable tested genes [21], at least in this tissue.
For both pollen and pistil tissues (“Reproductive” in Fig. 2c), two recognized RGs have been proposed in our analysis (Table 3). One of them, S-adenosylmethionine decarboxylase (rp11_olive_005197_split_1), was previously pointed out as one of the most abundant sequence in expressed sequence tag (ESTs) libraries of potato (Solanum tuberosum) [47]. In fact, this gene also emerged as candidate RG in pollen (Additional file 3) and pistil (Additional file 2). On the other hand, actin 7 (rp11_olive_005099; Table 3) has been extensively employed as RG in many species, such as chicory [48], berry [49] and pea [50]. Likewise, actin 7 also appears as a RG in the pollen analysis (Additional file 3) and it can also be observed in pistil under slightly less restrictive conditions (>50 reads, CV < 10%; results non shown). Once again, unknown candidate RGs are obtained. Shaggy-related protein kinase eta (rp11_olive_006695; Table 3), for instance, would be another interesting gene to test. We can conclude that the approach followed here for reproductive tissues, alone or in combination, yields a set of RGs which is widely supported by previous results described in the literature.
Table 3.
Best RGs in reproductive tissues (combination of pollen and pistil) of olive tree according to Fig. 2c and ranked by CV
| Transcript_id | RPMM | CV (%) | Mean RPMM | Best hit | Description | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| PM | PG1 | PG5 | S2 | S3 | S4 | |||||
| rp11_olive_006695 | 205 | 209 | 206 | 208 | 177 | 154 | 10.72 | 193.2 | Q39011 | Shaggy-related protein kinase eta Arabidopsis thaliana |
| rp11_olive_000781 | 105 | 140 | 112 | 104 | 108 | 127 | 11.36 | 116 | Q94A41 | Alpha-amylase 3, chloroplastic Arabidopsis thaliana |
| rp11_olive_006061 | 327 | 272 | 238 | 343 | 285 | 253 | 13.16 | 286.3 | Q8VZ80 | Polyol transporter 5 Arabidopsis thaliana |
| rp11_olive_006091 | 283 | 221 | 211 | 208 | 177 | 190 | 15.65 | 215 | A0A022R151 | Uncharacterized protein Erythranthe guttata |
| rp11_olive_010107 | 228 | 213 | 184 | 250 | 295 | 199 | 15.98 | 228.2 | O23254 | Serine hydroxymethyltransferase 4 Arabidopsis thaliana |
| rp11_olive_005197_split_1 | 366 | 430 | 381 | 343 | 423 | 552 | 16.37 | 415.8 | Q42679 | S-adenosylmethionine decarboxylase proenzyme Catharanthus roseus |
| rp11_olive_003279 | 122 | 179 | 153 | 114 | 118 | 145 | 16.64 | 138.5 | Q9LV37 | Mitogen-activated protein kinase 9 Arabidopsis thaliana |
| rp11_olive_000623 | 94 | 94 | 58 | 104 | 108 | 100 | 17.68 | 93 | A0A068V6W8 | Coffea canephora DH200 = 94 genomic scaffold, scaffold_132 Coffea canephora |
| rp11_olive_007981 | 144 | 149 | 108 | 156 | 147 | 91 | 18.2 | 132.5 | Q93Y40 | Oxysterol-binding protein-related protein 3C Arabidopsis thaliana |
| rp11_olive_005099 | 888 | 728 | 678 | 530 | 550 | 579 | 18.84 | 658.8 | P53492 | Actin-7 Arabidopsis thaliana |
| rp11_olive_005815 | 311 | 272 | 256 | 322 | 364 | 444 | 19.03 | 328.2 | P17598 | Catalase isozyme 1 Gossypium hirsutum |
| rp11_olive_000209_split_1 | 161 | 115 | 117 | 166 | 118 | 100 | 19.16 | 129.5 | Q67YI9-2 | 2 of Clathrin interactor EPSIN 2 Arabidopsis thaliana |
| rp11_olive_001245 | 161 | 175 | 108 | 166 | 187 | 118 | 19.16 | 152.5 | A5A7I7 | Calcium-dependent protein kinase 4 Solanum tuberosum |
| rp11_olive_008079 | 239 | 204 | 197 | 343 | 275 | 263 | 19.34 | 253.5 | M1AVD3 | Uncharacterized protein Solanum tuberosum |
| rp11_olive_008883 | 128 | 119 | 144 | 187 | 108 | 118 | 19.51 | 134 | Q9LZI2 | UDP-glucuronic acid decarboxylase 2 Arabidopsis thaliana |
| rp11_olive_035033 | 178 | 166 | 224 | 177 | 285 | 199 | 19.76 | 204.8 | P62201 | Calmodulin Lilium longiflorum |
| rp11_olive_029725 | 211 | 128 | 184 | 177 | 157 | 118 | 19.82 | 162.5 | O04834 | GTP-binding protein SAR1A Arabidopsis thaliana |
They were obtained for different stages of pollen and pistil with CV < 20% and minimum counted reads of 10. Transcript_id: transcript identifiers in the ReprOlive transcriptome
Since RG candidates for reproductive tissues were only obtained when less stringent parameters than in pollen or pistil separately were used (Fig. 2), special care should be taken with such reproductive candidates to RGs. Moreover, these differences may reflect substantial differences in the differentiation of both tissues, in such a way that nearly none gene has the same expression level in both tissues.
Experimental validation of RGs in olive tree
It can be thought that the number of reads in a 454/Roche sequencing experiment is not enough to obtain a reliable prediction of RGs. Therefore, an experimental validation was envisaged to further support the predicted RGs obtained with findRGs. Polyubiquitin and actin were validated by RT-PCR in different olive tissues in comparison to 18S, a widely used RG (Fig. 3). Both genes show thick and similar expression levels in reproductive tissues (mature pollen and pistil). However, while ubiquitin seems to be a good RG in pollen and pistil, as well as in inflorescences, leafs and seeds, actin was not a good RG for seed. The other commented RGs in previous section resulted in the following outcomes: Two of the transcripts with lower variation in mature pollen (rp11_olive_002359: Mitogen-activated protein kinase, and rp11_olive_009589: Proline transporter 2) showed a good level of expression in both the mature pollen and the whole olive inflorescence, however, they presented lower/null expression in the pistil, seed and leaf. As expected, RT-PCR amplification of rp11_olive_004773, l-ascorbate oxidase homolog (one of the most expressed transcripts in olive pollen), also displayed a similar pattern, with bands of very high intensity corresponding to both the mature pollen and the inflorescence. Regarding transcripts proposed as RGs for the olive pistil due to their low variation, testing of the transcript rp11_olive_003751, glyceraldehyde-3-phosphate dehydrogenase, by RT-PCR resulted in high expression in the pistil, and lower expression in the remaining tissues, including vegetative tissues as those of the leaf. The highly expressed transcript in the pistil rp11_olive_019507, salicylic acid-binding protein 2, generated an intense amplification band in the pistil, the mature pollen and the whole inflorescence, with no amplification in vegetative/seed tissues. Finally, a similar pattern of expression was detected when the proposed RG for reproductive (pollen + pistil) tissues was validated by RT-PCR. In this case, bands of identical intensity were present in both the pollen and the pistil, and with lower intensity, in the whole inflorescence. Overall, RT-PCR validations showed a high degree of consistency with the results obtained by bioinformatics methods, even though a limited number of long reads were obtained by 454/Roche platform. This finding suggests that our bioinformatic approach should be widely used before any RT-PCR or qPCR experiment is carried out. In conclusion, the preliminary RT-PCR validation of the predicted RGs provides reliability to the findRGs workflow approach.
Fig. 3.
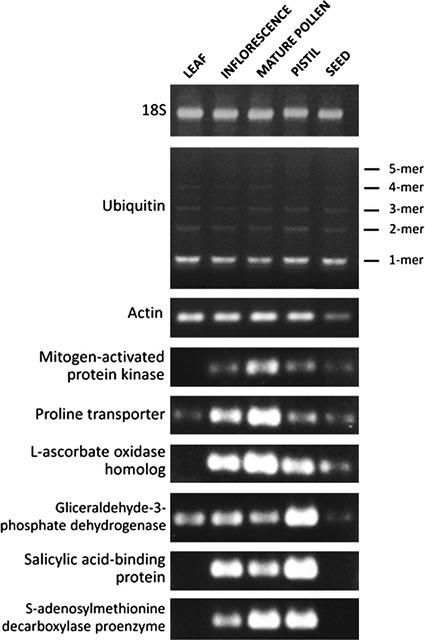
Preliminary RT-PCR validation of RGs predicted in this work in olive tissues in comparison to 18S
Candidate RGs in Arabidopsis thaliana
The successful use of findRGs in olive tree drove us to extend it use to more complex datasets. Using publicly available SRA data in PRJEB9470 from Arabidopsis, three executions (per replicate) of the workflows in Fig. 1 were carried out (Fig. 4). As before, less variant transcripts were retained with two CV cut-off values: 10% (default) and 20% (non-stringent), but, since these reads were obtained with the Illumina platform, a greater minimum value of counted reads per gene is required. This minimum was set to 10,000, 30,000, and 100,000 for comparative reasons. The lower number of candidates shown in Fig. 4 with respect to Fig. 2 can be explained by the different count threshold due to the different sequencing technology. The number of candidate RGs with the most stringent conditions (>100,000 reads, CV < 10%) is very homogeneous for the three replicates (Fig. 4a–c) and they refer almost exclusively to the same gene, ribulose-1,5-bisphosphate carboxylase/oxygenase (rubisco) (AT1G67090, AT5G38410, AT5G38420, AT5G38430; Table 4), being by far the best candidate in all the cases since their RPMM is much greater than other candidate RGs emerging in less stringent conditions (Additional file 4). Rubisco has been previously used as RG in tea leaf tissues [51]. However, it is not a good candidate for non-green (non-photosynthetic) tissues like those of the anther, neither for pollen [52].
Fig. 4.
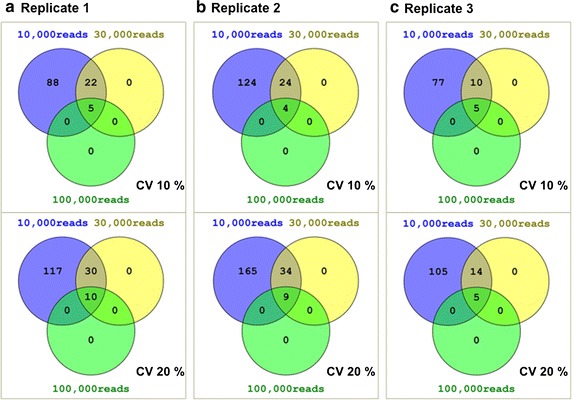
Venn diagrams summarizing the number of RGs obtained for Arabidopsis thaliana. Two cut-off values were used for CV and three for counted reads
Table 4.
Best RGs in Arabidopsis thaliana according to Fig. 4 and ranked by CV
| Arabidopsis | transcript_id | RPMM | CV (%) | Mean RPMM | Description | |
|---|---|---|---|---|---|---|
| Col_0 | Kil_0 | |||||
| Replicate 1 | AT1G67090.1 | 9476 | 9191 | 1.53 | 9333.5 | Ribulose bisphosphate carboxylase small chain 1A |
| AT5G38410.1 | 7760 | 8135 | 2.36 | 7947.5 | Ribulose bisphosphate carboxylase (small chain) family protein | |
| AT5G38430.1 | 7054 | 7548 | 3.38 | 7301 | Ribulose bisphosphate carboxylase (small chain) family protein | |
| AT2G39730.1 | 4051 | 4343 | 3.48 | 4197 | Rubisco activase | |
| AT5G38420.1 | 7329 | 7889 | 3.68 | 7609 | Ribulose bisphosphate carboxylase (small chain) family protein | |
| Replicate 2 | AT2G39730.1 | 3906 | 4323 | 5.07 | 4114.5 | Rubisco activase |
| AT1G67090.1 | 8523 | 9636 | 6.13 | 9079.5 | ribulose bisphosphate carboxylase small chain 1A | |
| AT1G21310.1 | 7013 | 7976 | 6.42 | 7494.5 | Extensin 3 | |
| AT5G38410.1 | 7047 | 8438 | 8.98 | 7742.5 | Ribulose bisphosphate carboxylase (small chain) family protein | |
| Replicate 3 | AT5G38420.1 | 8708 | 8526 | 1.06 | 8617 | Ribulose bisphosphate carboxylase (small chain) family protein |
| AT5G38430.1 | 8424 | 8169 | 1.54 | 8296.5 | Ribulose bisphosphate carboxylase (small chain) family protein | |
| AT2G39730.1 | 4365 | 4524 | 1.79 | 4444.5 | Rubisco activase | |
| AT5G38410.1 | 9172 | 8822 | 1.95 | 8997 | Ribulose bisphosphate carboxylase (small chain) family protein | |
| AT1G67090.1 | 11,051 | 9694 | 6.54 | 10,372.5 | Ribulose bisphosphate carboxylase small chain 1A | |
They were obtained for the three replicates with CV < 10% and minimum counted reads of 100,000. Transcript_id: transcript identifiers in TAIR database
Making a comparison between candidate RGs obtained with less rigorous conditions (>10,000 reads and CV < 10%), 77 candidates are shared by the three replicates (Additional file 4). Several of these genes have been previously commented in this paper as widespread used RGs, such as glyceraldehyde-3-phosphate dehydrogenase (AT3G26650, AT1G42970, AT3G04120 and AT1G13440; Additional file 4), and S-adenosylmethionine decarboxylase (AT3G02470; Additional file 4). Additionally, other RGs commonly used in literature emerged, such as phosphoglycerate kinase 1 (AT3G12780; Additional file 4), typically used as control. It shows constant expression levels in leaves, fruit and flowers in tomato [37] and it has also been described as one of the best RGs in Chrysanthemum species subjected to different kind of stresses [53]. Several ribosomal proteins are within the candidates (AT1G43170, AT1G02780, AT3G25520, AT5G39740, AT1G56070 and AT5G20290; Additional file 4). They are listed as housekeeping genes and have been suggested as RGs based on analysis of microarray data [54]. However, since all these genes have significant expression variation across tissues, their suitability should be tested in every particular situation. α-Tubulin (AT1G50010 and AT1G04820; Additional file 4) also emerged as candidate. Although it has been extensively used as RG, controversial data have been reported on its reliability, being considered the best in certain species and the worst one in others [20]. There are many others well positioned candidate RGs emerging in all executions that would deserve experimental testing, for instance fructose-bisphosphate aldolase (AT2G21330 and AT4G38970; Additional file 4) or GTP binding elongation factor Tu (AT1G07920, AT1G07930, AT1G07940 and AT5G60390; Additional file 4).
The fact that the same gene appears as candidate RG in the three replicates separately indicates that the possible variability between replicates appears not to be affecting the estimation. The calculation of RGs combining all replicates extracted the same candidate RGs (results not shown), suggesting that an average number of reads per replicate of ~11,000,000 (Additional file 1) could be enough for the aim of the workflow. In conclusion, the list of candidate RGs obtained by means of our workflow offers a first and reliable estimation of the most appropriate RGs for expression studies between these two Arabidopsis strains in these particular experimental conditions. It also suggests that mapping with less than 11 millions of reads could be enough to obtain a reliable prediction.
RGs and human cancers
Many studies on cancer perform multiple comparisons (between tumors and normal tissues, different stages, response to treatments…). RGs needed for these comparisons should have consistent expression level in the conditions to be analyzed. The search of appropriate RGs in such cases becomes particularly tricky and challenging, since cancer is associated with changes in gene expression involving many pathways, and it is demonstrated a huge heterogeneity within and among cancers [55]. Even traditional housekeeping genes are likely to change their expression level during the course of the disease [56], since they might not only be implicated in the basal cell metabolism but also in other cell functions [57]. Therefore, it is crucial to perform preliminary evaluations for identifying the most stably expressed genes in each situation. Moreover, it is not unusual that cancer experiments have many tens of replicates [28]. Therefore, this is a good situation to test if findRGs can cope with large amount of samples with sample size heterogeneity in a high throughput experiment. Taking into account the higher number of samples and the possible increase of variability, less stringent filtering parameters values of maximum CV and minimum counted reads were tested and adapted in each particular situation.
Candidate RGs for prostate cancer
Figure 5a shows the number of candidate RGs using different cut-off values for CV and counted reads in prostate samples. No candidate RG is obtained in the most stringent conditions (>100,000 and CV < 15%) and only one with a slightly more permissive maximum CV value of 20%. More RGs were obtained using less stringent conditions; those obtained with >30,000 and CV < 20% are presented in Table 5. Many of them have been used as RGs in cancer studies. For example, the tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein zeta polypeptide (ENST00000395957.6; Table 5) has been repeatedly studied as RG candidate in prostate cancer, although it has not been between the most stable genes [58, 59]. Nevertheless, it is one of the best ranked as stable genes for the comparison between cancer stem cells and native cells [60]. Nascent-polypeptide-associated complex alpha polypeptide (ENST00000356769.7; Table 5) is a human housekeeping gene evaluated and sometimes proposed as RG in several types of cancer, such as breast cancer [61] or colon cancer [11]. Phosphoglycerate kinase 1 (ENST00000373316.4; Table 5), another human housekeeping gene, was typically used as RG, as in plants, and has been demonstrated to be affected between normal and malignant tissues in certain malignancies, but not in others [60, 62]. Genes encoding the different subunits of ATP synthase mitochondrial are considered human housekeeping genes [63], some of them considered RGs for some tumors [11], whereas no in all [64]. Following our results, ENST00000398752.10 and ENST00000495596.5 (Table 5) should merit experimental consideration. Several ribosomal proteins are also proposed as RGs (ENST00000314138.10, ENST00000519807.5, ENST00000338970.10 and ENST00000456530.6; Table 5). Despite their widely spread use as RGs, expression variations of these genes have been detected not only between tumors and healthy tissues [65], but also across normal tissues [54].
Fig. 5.

Venn diagrams summarizing the number of RGs obtained for matched samples of normal and malignant tissues of three different human cancers: prostate, small-cell lung cancer and lung adenocarcinoma. Two cut-off values per cancer were used for CV and different counted reads depending on the tissue
Table 5.
Best candidate RGs for normal and malignant prostate tissues according to Fig. 5a and ranked by CV
| Transcript_id | CV (%) | Mean RPMM | Gene | Description |
|---|---|---|---|---|
| ENST00000510199.5 | 8.95 | 99.8 | GNB2L1 | Guanine nucleotide binding protein (G protein), beta polypeptide 2-like 1 |
| ENST00000425566.1 | 9.91 | 127.3 | RPL23AP87 | Ribosomal protein L23a pseudogene 87 |
| ENST00000314138.10 | 10.52 | 134.9 | RPL27A | Ribosomal protein L27a |
| ENST00000412331.6 | 11.18 | 108.1 | EIF3L | Eukaryotic translation initiation factor 3 subunit L |
| ENST00000494591.1 | 11.49 | 78.4 | RPSAP36 | Ribosomal protein SA pseudogene 36 |
| ENST00000519807.5 | 11.5 | 168.1 | RPS20 | Ribosomal protein S20 |
| ENST00000356769.7 | 11.58 | 92.8 | NACA | Nascent polypeptide-associated complex alpha subunit |
| ENST00000496593.5 | 12.28 | 253.5 | RPLP0P2 | Ribosomal protein, large, P0 pseudogene 2 |
| ENST00000338970.10 | 12.63 | 176.9 | RPL14 | Ribosomal protein L14 |
| ENST00000610672.4 | 12.74 | 244 | MED22 | Mediator complex subunit 22 |
| ENST00000395957.6 | 12.97 | 95.5 | YWHAZ | Tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, zeta |
| ENST00000234831.9 | 13.69 | 108.1 | TMEM59 | Transmembrane protein 59 |
| ENST00000353047.10 | 14.01 | 156.1 | CTSB | Cathepsin B |
| ENST00000556083.1 | 14.36 | 137.5 | ACTN1 | Actinin, alpha 1 |
| ENST00000558264.5 | 14.59 | 129.8 | TPM1 | Tropomyosin 1 (alpha) |
| ENST00000394621.6 | 14.87 | 189.2 | STEAP2 | STEAP2 metalloreductase |
| ENST00000335508.10 | 15.15 | 116 | SF3B1 | Splicing factor 3b subunit 1 |
| ENST00000341423.9 | 15.26 | 134.4 | HMGB1 | High mobility group box 1 |
| ENST00000564521.6 | 15.84 | 167 | ALDOA | Aldolase, fructose-bisphosphate A |
| ENST00000398752.10 | 16.45 | 200.6 | ATP5A1 | ATP synthase, H + transporting, mitochondrial F1 complex, alpha subunit 1, cardiac muscle |
| ENST00000264657.9 | 16.84 | 137.7 | STAT3 | Signal transducer and activator of transcription 3 (acute-phase response factor) |
| ENST00000357214.5 | 17.4 | 105.5 | SFPQ | Splicing factor proline/glutamine-rich |
| ENST00000456530.6 | 17.44 | 118.5 | RPL15 | Ribosomal protein L15 |
| ENST00000495596.5 | 17.64 | 164.2 | ATP5G2 | ATP synthase, H + transporting, mitochondrial Fo complex subunit C2 (subunit 9) |
| ENST00000391959.5 | 17.67 | 125.6 | PPP1R12B | protein phosphatase 1 regulatory subunit 12B |
| ENST00000369936.2 | 17.97 | 235 | KIAA1324 | KIAA1324 |
| ENST00000300619.11 | 19.01 | 104.5 | ZNF91 | Zinc finger protein 91 |
| ENST00000401722.7 | 19.21 | 156.4 | SLC25A3 | Solute carrier family 25 (mitochondrial carrier; phosphate carrier), member 3 |
| ENST00000618621.4 | 19.43 | 405.9 | LPP | LIM domain containing preferred translocation partner in lipoma |
| ENST00000249822.8 | 19.53 | 107.3 | ARPP19 | cAMP regulated phosphoprotein 19 kDa |
| ENST00000353411.10 | 19.68 | 122.8 | SKP1 | S-phase kinase-associated protein 1 |
| ENST00000375856.4 | 19.76 | 151.8 | IRS2 | Insulin receptor substrate 2 |
| ENST00000373316.4 | 19.79 | 118.1 | PGK1 | Phosphoglycerate kinase 1 |
| ENST00000306085.10 | 19.9 | 159.2 | TRIM56 | Tripartite motif containing 56 |
| ENST00000357308.8 | 20 | 105 | GFPT1 | Glutamine–fructose-6-phosphate transaminase 1 |
They were obtained with CV < 20% and minimum counted reads of 30,000. Transcript_id: human transcript identifiers in ENSEMBL database
Candidate RGs for small-cell lung cancer
No candidates were obtained with the same cut-offs of prostate cancer. Then, RG selection was carried out with minimum number of mapped reads set to 10,000 and 20,000 and CV cut-off values 15 and 20% (Fig. 5b). Even so, the number of candidates with the most stringent CV cut-off (<15%) was very low, indicating variability between samples. The candidate RGs obtained with the less stringent combination of filtering parameters (>10,000 and CV < 20%) are given in Table 6. Ribosomal proteins (ENST00000456530.6, ENST00000422514.6 and ENST00000338970.10; Table 6) and a gene encoding a subunit of ATP synthase mitochondrial (ENST00000495596.5; Table 6) are present, as in prostate. Ubiquitin A-52 residue ribosomal protein fusion product 1 (ENST00000442744.6; Table 6) is also suggested and has been identified as RG in breast cancer [61], as well as in bladder or testis through microarray meta-analysis of human clinical samples [66]. Therefore, these less stringent cut-offs are providing reliable RGs.
Table 6.
Best candidate RGs for normal lung and small-cell lung cancer according to Fig. 5b and ranked by CV
| Transcript_id | CV (%) | Mean RPMM | Gene | Description |
|---|---|---|---|---|
| ENST00000425566.1 | 12.68 | 76.2 | RPL23AP87 | Ribosomal protein L23a pseudogene 87 |
| ENST00000338970.10 | 12.96 | 103.3 | RPL14 | Ribosomal protein L14 |
| ENST00000442744.6 | 13.28 | 69.4 | UBA52 | Ubiquitin A-52 residue ribosomal protein fusion product 1 |
| ENST00000456530.6 | 16.02 | 76.7 | RPL15 | Ribosomal protein L15 |
| ENST00000553521.5 | 16.21 | 50.2 | SRSF5 | Serine/arginine-rich splicing factor 5 |
| ENST00000373242.6 | 16.8 | 73 | SAR1A | Secretion associated, Ras related GTPase 1A |
| ENST00000261890.6 | 16.88 | 55.3 | RAB11A | RAB11A, member RAS oncogene family |
| ENST00000510199.5 | 17.11 | 66 | GNB2L1 | Guanine nucleotide binding protein (G protein), beta polypeptide 2-like 1 |
| ENST00000234115.10 | 17.59 | 63.6 | PLEKHB2 | Pleckstrin homology domain containing B2 |
| ENST00000401722.7 | 17.69 | 83.6 | SLC25A3 | Solute carrier family 25 (mitochondrial carrier; phosphate carrier), member 3 |
| ENST00000412331.6 | 17.76 | 54.6 | EIF3L | Eukaryotic translation initiation factor 3 subunit L |
| ENST00000422514.6 | 18.83 | 80.3 | RPL23A | Ribosomal protein L23a |
| ENST00000342374.4 | 19.13 | 45.2 | SERINC3 | Serine incorporator 3 |
| ENST00000483316.1 | 19.26 | 77.6 | BAZ2B | Bromodomain adjacent to zinc finger domain 2B |
| ENST00000335508.10 | 19.41 | 72.4 | SF3B1 | Splicing factor 3b subunit 1 |
| ENST00000471227.3 | 19.62 | 66.4 | RPL23AP2 | Ribosomal protein L23a pseudogene 2 |
| ENST00000334256.8 | 19.77 | 46.9 | KPNA4 | Karyopherin alpha 4 (importin alpha 3) |
| ENST00000332361.5 | 19.79 | 64.5 | RPL23AP57 | Ribosomal protein L23a pseudogene 57 |
| ENST00000416139.1 | 19.81 | 64.5 | RPL23AP18 | Ribosomal protein L23a pseudogene 18 |
| ENST00000495596.5 | 19.84 | 71.5 | ATP5G2 | ATP synthase, H + transporting, mitochondrial Fo complex subunit C2 (subunit 9) |
| ENST00000446445.1 | 19.87 | 64.1 | RPL23AP43 | Ribosomal protein L23a pseudogene 43 |
They were obtained with CV < 20% and minimum counted reads of 10,000. Transcript_id: human transcript identifiers in ENSEMBL database
Candidate RGs for lung adenocarcinoma
Figure 5c shows that samples of normal lung and lung adenocarcinoma are the more variant instances analyzed in this work since only 7 RGs are obtained using >10,000 mapped reads and CV < 15%. Therefore the list of candidate RGs was obtained with a minimum counted reads of 30,000 and a CV cut-off of 20% (Table 7). Some of the RGs (ENST00000270460.10, ENST00000323443.6, ENST00000367975.6, ENST00000528973.1, ENST00000262160.10, ENST00000398004.3, ENST00000396444.7, ENST00000258711.7, ENST00000329627.11 and ENST00000238831.8; Table 7) have been described as human housekeeping genes [63], but there are no evidence about their use as RGs. Several zinc finger proteins (ENST00000328654.9, ENST00000307635.3 and ENST00000253115.6; Table 7) have already been suggested as RGs in cancerous kidney and lymph node tissues [66], but are not suitable RGs for normal and colorectal cancer tissues [64]; however, according our results, they seem to be appropriate for studies in normal and cancerous lung. Some of the candidate RGs previously commented for prostate or small-cell lung cancer, such as ribosomal proteins (Tables 4, 5), are also retained for lung adenocarcinoma with the less stringent conditions (>10,000 reads and CV < 20%; non shown results). This prompted us to think that those transcripts could finally be suitable for studies involving several types of normal and cancerous cells.
Table 7.
Best candidate RGs for normal normal lung and lung adenocarcinoma according to Fig. 5c and ranked by CV
| Transcript_id | CV (%) | Mean RPMM | Gene | Description |
|---|---|---|---|---|
| ENST00000411857.2 | 16.34 | 224.7 | HNRNPA1P54 | Heterogeneous nuclear ribonucleoprotein A1 pseudogene 54 |
| ENST00000270460.10 | 18.06 | 204.1 | EPN1 | Epsin 1 |
| ENST00000373191.8 | 18.17 | 195.4 | AGO3 | Argonaute 3, RISC catalytic component |
| ENST00000323443.6 | 18.2 | 218.4 | LRRC57 | Leucine rich repeat containing 57 |
| ENST00000367975.6 | 18.35 | 204.8 | SDHC | Succinate dehydrogenase complex subunit C |
| ENST00000528973.1 | 18.42 | 211 | PCSK7 | Proprotein convertase subtilisin/kexin type 7 |
| ENST00000262160.10 | 18.7 | 214 | SMAD2 | SMAD family member 2 |
| ENST00000607772.5 | 18.73 | 200.3 | CNKSR3 | CNKSR family member 3 |
| ENST00000261854.9 | 18.85 | 198.2 | SPPL2A | Signal peptide peptidase like 2A |
| ENST00000398004.3 | 19.12 | 316.1 | SLC35E3 | Solute carrier family 35 member E3 |
| ENST00000396444.7 | 19.21 | 294 | USP8 | Ubiquitin specific peptidase 8 |
| ENST00000304177.9 | 19.28 | 212.4 | C15orf40 | Chromosome 15 open reading frame 40 |
| ENST00000328654.9 | 19.31 | 241.8 | ZNF26 | Zinc finger protein 26 |
| ENST00000307635.3 | 19.34 | 218.1 | ZNF556 | Zinc finger protein 556 |
| ENST00000258711.7 | 19.38 | 323.7 | CHST12 | Carbohydrate (chondroitin 4) sulfotransferase 12 |
| ENST00000329627.11 | 19.41 | 318.1 | PEX26 | Peroxisomal biogenesis factor 26 |
| ENST00000322122.7 | 19.49 | 192.7 | TRIM72 | Tripartite motif containing 72, E3 ubiquitin protein ligase |
| ENST00000238831.8 | 19.5 | 291.1 | YIPF4 | Yip1 domain family member 4 |
| ENST00000258149.9 | 19.71 | 222.8 | MDM2 | MDM2 proto-oncogene, E3 ubiquitin protein ligase |
| ENST00000253115.6 | 19.72 | 227.3 | ZNF426 | Zinc finger protein 426 |
| ENST00000614987.4 | 19.74 | 346.8 | RPS6KA5 | Ribosomal protein S6 kinase, 90 kDa, polypeptide 5 |
They were obtained with CV < 20% and minimum counted reads of 30,000. Transcript_id: human transcript identifiers in ENSEMBL database
Candidate RGs for different combination of malignant and normal tissues
Although every cancer is an independent scenario that should be analyzed separately in the search of RGs, different combinations or tissues and/or states were tested. When combining normal and malignant lung samples, filtering parameters were still less stringent: CV is maintained in 15 and 20% and the minimum counted reads was set to 7500 and 10,000 (Fig. 6). The analysis was performed with only normal lung and combining normal and malignant samples. The resulting RG candidates for normal lung are listed in Additional file 5. The resulting RGs for combined normal and malignant lung samples are listed in Additional file 6.
Fig. 6.
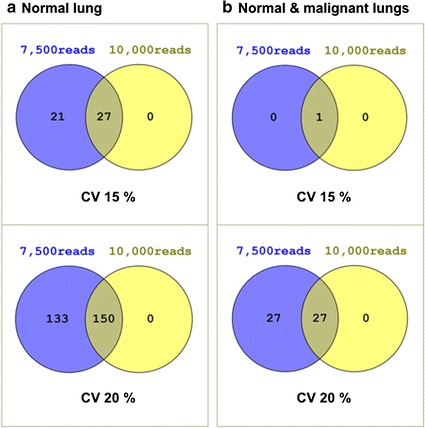
Venn diagrams summarizing the number of RGs obtained for different combinations of lung samples: samples from only normal lung (a) or normal and malignant lung (b) were analyzed with two CV cut-off values per combination and two different counted reads
Candidate RGs for normal lung tissues revealed some known RGs, such as nascent-polypeptide-associated complex alpha polypeptide (ENST00000356769.7; Additional file 5) [11, 61], ornithine decarboxylase antizyme 1 (ENST00000586054.2; Additional file 5) [66], although with some up-regulation in cancer [67], and small ubiquitin-like modifier 2 gene [66]. For the whole set of lung samples (Fig. 6b; Additional file 6), the ribosomal protein L14 (ENST00000338970.10; Additional files 5, 6), already described in normal and cancerous kidney tissues [66], was found. Interestingly, ATP synthase subunit alpha mitochondrial (ENST00000398752.10; Additional files 5, 6) has not been described as RG in literature and deserves a careful testing.
The analysis combining the whole normal and malignant human cancers samples described here (prostate cancer, small-cell cancer lung and lung adenocarcinoma) did not provide any RG, revealing that the addition of a completely different tissue (prostate) supposes a new source of variability between samples.
Conclusion
The automatic workflow presented in this work takes advantage of new and publicly available NGS data to predict the suitable RGs for every experimental situation. Those candidate RGs can be useful for qPCR validation in further expression analyses. The analysis is particularly suitable in non-model species, for which few or no RGs have been identified, or in new experimental conditions where no previous data are available. The workflow seems to be independent of the sequencing technology that generates the reads, the number of reads, as well as the read length, since it seems to work equally well with many short reads (Illumina from Arabidopsis and human) than with a few long reads (Roche/454 from olive tree). It supports massive data analyses with low (Arabidopsis) and high (adenocarcinoma) number of samples. Time executions for the different tasks of the workflow are reasonably short, since the time consuming parts (pre-processing and mapping) are required for any NGS analysis and are performed only once. Our workflow is customizable and adaptable to the requirements of each experimental case. The algorithm in findRGs has been acceptably tested for three species (olive, Arabidopsis and human) in comparison studies focused in very different biological aspects, so as different developmental, physiological or pathological stages (reproductive tissues, flower and cancer). Lists of candidate RGs have been generated in every case, some of which have even been already described in the literature and others have been preliminarily validated here (Fig. 3); both findings are supporting this experimental approach. More interestingly, new and more suitable RGs can be discovered with findRGs. Even though the expression level and stability of these new RGs may require some experimental validation prior to their utilization for normalization, we encourage the utilization of findRGs where possible, since it can be quite helpful as a preliminary approximation about the best RG candidates, prior to each single expression experiment.
Additional files
Additional file 1. Statistics related to pre-processing with SeqTrimNext and mapping with Bowtie2 for the different datasets: olive tree libraries, Arabidopsis libraries, normal/malignant prostate samples, normal/small-cell cancer lung samples and normal/adenocarcinoma lung samples.
Additional file 2. Best RGs in olive tree pistil according to Fig. 2a, ranked by CV. They were obtained for different stages of pistil development with CV < 10% and minimum counted reads of 100. Transcript_id: transcript identifiers in the ReprOlive transcriptome.
Additional file 3. Best RGs in olive tree pollen according to Fig. 2b, ranked by CV. They were obtained for different stages of pollen development, with CV < 10% and minimum counted reads of 100. Transcript_id: transcript identifiers in the ReprOlive transcriptome.
Additional file 4. Best candidate RGs in Arabidopsis thaliana for replicates 1, 2 and 3 with their respective RPMM values for each replicate. They were obtained with CV < 10% and a minimum counted reads of 10,000. Transcript_id: transcript identifiers in TAIR database.
Additional file 5. Best candidate RGs for normal lung tissues according to Fig. 6a and ranked by CV. They were obtained with CV < 15% and minimum counted reads of 10,000. Transcript_id: human transcript identifiers in ENSEMBL database.
Additional file 6. Best candidate RGs for normal and malignant lung samples according to Fig. 6b, ranked by CV. They were obtained with CV < 20% and minimum counted reads of 10,000. Transcript_id: human transcript identifiers in ENSEMBL database.
Declarations
Authors’ contributions
RC, JDA and MGC conceived and designed this study. RC, PS and RL have developed and executed the workflows. RC analyzed and interpreted the results of olive and Arabidopsis data. MA analyzed and interpreted the results of cancer data. MJJ-Q and AZ performed the biological assays. RC, JDA and MGC wrote the manuscript. MGC coordinated the study. All authors contributed, corrected the final manuscript. All authors read and approved the final manuscript.
Acknowledgements
The authors also thankfully acknowledge the computer resources and the technical support provided by the Plataforma Andaluza de Bioinformática of the University of Malaga.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All references to data analysed during this study are included in this published article.
Funding
This research was supported by co-funding from the European Union through the ERDF 2014–2020 “Programa Operativo de Crecimiento Inteligente” to the projects RTA2013-00068-C03 and RTA2013-00023-C02 of the Spanish INIA; BFU2011-22779 and RECUPERA2020-3.1.4 from the Spanish MINECO, P11-CVI-7487 from the regional PAI and NEUMOSUR grant 12/2015 entitled “Expresión de retrotransposones en pacientes con adenocarcinoma intervenido. Comparación entre tejjido sano y tumoral”. Publication costs were funded by the mentioned grants.
About this supplement
This article has been published as part of BioMedical Engineering OnLine Volume 16 Supplement 1, 2017: Selected articles from the 4th International Work-Conference on Bioinformatics and Biomedical Engineering-IWBBIO 2016. The full contents of the supplement are available online at https://biomedical-engineering-online.biomedcentral.com/articles/supplements/volume-16-supplement-1.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12938-017-0356-5) contains supplementary material, which is available to authorized users.
Contributor Information
Rosario Carmona, Email: rosario.carmona@eez.csic.es.
Macarena Arroyo, Email: macarroyo@gmail.com.
María José Jiménez-Quesada, Email: mariajose.jimenez@eez.csic.es.
Pedro Seoane, Email: seoanezonjic@uma.es.
Adoración Zafra, Email: dori.zafra@eez.csic.es.
Rafael Larrosa, Email: rlarrosa@uma.es.
Juan de Dios Alché, Email: juandedios.alche@eez.csic.es.
M. Gonzalo Claros, Email: claros@uma.es.
References
- 1.Freeman WM, Walker SJ, Vrana KE. Quantitative RT-PCR: pitfalls and potential. Biotechniques. 1999;26:112–125. doi: 10.2144/99261rv01. [DOI] [PubMed] [Google Scholar]
- 2.Rocha-Martins M, Njaine B, Silveira MS. Avoiding pitfalls of internal controls: validation of reference genes for analysis by qRT-PCR and Western blot throughout rat retinal development. PLoS ONE. 2012;7:e43028. doi: 10.1371/journal.pone.0043028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lanoix D, Lacasse A-A, St-Pierre J, Taylor SC, Ethier-Chiasson M, Lafond J, Vaillancourt C. Quantitative PCR pitfalls: the case of the human placenta. Mol Biotechnol. 2012;52:234–243. doi: 10.1007/s12033-012-9539-2. [DOI] [PubMed] [Google Scholar]
- 4.Wang Y, Chen Y, Ding L, Zhang J, Wei J, Wang H. Validation of reference genes for gene expression by quantitative real-time RT-PCR in stem segments spanning primary to secondary growth in Populus tomentosa. PLoS ONE. 2016;11:e0157370. doi: 10.1371/journal.pone.0157370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu G, Qiu X, Cao L, Zhang Y, Zhan Z, Han R. Evaluation of reference genes for reverse transcription quantitative PCR studies of physiological responses in the ghost moth, Thitarodes armoricanus (Lepidoptera, Hepialidae) PLoS ONE. 2016;11:e0159060. doi: 10.1371/journal.pone.0159060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu J, Zhang H, Liu L, Li W, Wei Y, Shi S. Validation of reference genes for RT-qPCR studies of gene expression in preharvest and postharvest longan fruits under different experimental conditions. Front Plant Sci. 2016;7:780. doi: 10.3389/fpls.2016.00780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peng X, McCormick DL. Identification of reliable reference genes for quantitative gene expression studies in oral squamous cell carcinomas compared to adjacent normal tissues in the F344 rat model. Oncol Rep. 2016;36:1076–1084. doi: 10.3892/or.2016.4883. [DOI] [PubMed] [Google Scholar]
- 8.Cassol D, Cruz FP, Espindola K, Mangeon A, Müller C, Loureiro ME, Corrêa RL, Sachetto-Martins G. Identification of reference genes for quantitative RT-PCR analysis of microRNAs and mRNAs in castor bean (Ricinus communis L.) under drought stress. Plant Physiol Biochem. 2016;106:101–107. doi: 10.1016/j.plaphy.2016.02.024. [DOI] [PubMed] [Google Scholar]
- 9.Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper–Excel-based tool using pair-wise correlations. Biotechnol Lett. 2004;26:509–515. doi: 10.1023/B:BILE.0000019559.84305.47. [DOI] [PubMed] [Google Scholar]
- 10.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:RESEARCH0034. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andersen CL, Jensen JL, Ørntoft TF. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004;64:5245–5250. doi: 10.1158/0008-5472.CAN-04-0496. [DOI] [PubMed] [Google Scholar]
- 12.Lee PD, Sladek R, Greenwood CMT, Hudson TJ. Control genes and variability: absence of ubiquitous reference transcripts in diverse mammalian expression studies. Genome Res. 2002;12:292–297. doi: 10.1101/gr.217802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suzuki T, Higgins PJ, Crawford DR. Control selection for RNA quantitation. Biotechniques. 2000;29:332–337. doi: 10.2144/00292rv02. [DOI] [PubMed] [Google Scholar]
- 14.Brunner AM, Yakovlev IA, Strauss SH. Validating internal controls for quantitative plant gene expression studies. BMC Plant Biol. 2004;4:14. doi: 10.1186/1471-2229-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Czechowski T, Stitt M, Altmann T, Udvardi MK. Genome-wide identification and testing of superior reference genes for transcript normalization. Society. 2005;139:5–17. doi: 10.1104/pp.105.063743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cruz F, Julca I, Gómez-Garrido J, Loska D, Marcet-Houben M, Cano E, Galán B, Frias L, Ribeca P, Derdak S, Gut M, Sánchez-Fernández M, García JL, Gut IG, Vargas P, Alioto TS, Gabaldón T, Birol I, Raymond A, Jackman S, Pleasance S, Coope R, Taylor G, Yuen M, Keeling C, Brand D, Vandervalk B, Kirk H, Pandoh P, Moore R, et al. Genome sequence of the olive tree, Olea europaea. Gigascience. 2016;5:29. doi: 10.1186/s13742-016-0134-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carmona R, Zafra A, Seoane P, Castro AJ, Guerrero-Fernández D, Castillo-Castillo T, Medina-García A, Cánovas FM, Aldana-Montes JF, Navas-Delgado I, de Dios Alché J, de Claros MG. ReprOlive: a database with linked data for the olive tree (Olea europaea L.) reproductive transcriptome. Front Plant Sci. 2016;6:625. doi: 10.3389/fpls.2015.00625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galla G, Barcaccia G, Ramina A, Collani S, Alagna F, Baldoni L, Cultrera NGM, Martinelli F, Sebastiani L, Tonutti P. Computational annotation of genes differentially expressed along olive fruit development. BMC Plant Biol. 2009;9:128. doi: 10.1186/1471-2229-9-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ray DL, Johnson JC. Validation of reference genes for gene expression analysis in olive (Olea europaea) mesocarp tissue by quantitative real-time RT-PCR. BMC Res Notes. 2014;7:304. doi: 10.1186/1756-0500-7-304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nonis A, Vezzaro A, Ruperti B. Evaluation of RNA extraction methods and identification of putative reference genes for real-time quantitative polymerase chain reaction expression studies on olive (Olea europaea L.) fruits. J Agric Food Chem. 2012;60:6855–6865. doi: 10.1021/jf300419w. [DOI] [PubMed] [Google Scholar]
- 21.Resetic T, Stajner N, Bandelj D, Javornik B, Jakse J. Validation of candidate reference genes in RT-qPCR studies of developing olive fruit and expression analysis of four genes involved in fatty acids metabolism. Mol Breed. 2013;32:211–222. doi: 10.1007/s11032-013-9863-7. [DOI] [Google Scholar]
- 22.Ma J, Skibbe DS, Fernandes J, Walbot V. Male reproductive development: gene expression profiling of maize anther and pollen ontogeny. Genome Biol. 2008;9:R181. doi: 10.1186/gb-2008-9-12-r181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kodama Y, Shumway M, Leinonen R. The sequence read archive: explosive growth of sequencing data. Nucleic Acids Res. 2012;40(Database issue):54–56. doi: 10.1093/nar/gkr854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lutz U, Posé D, Pfeifer M, Gundlach H, Hagmann J, Wang C, Weigel D, Mayer KFX, Schmid M, Schwechheimer C. Modulation of ambient temperature-dependent flowering in Arabidopsis thaliana by Natural Variation of FLOWERING LOCUS M. PLoS Genet. 2015;11:e1005588. doi: 10.1371/journal.pgen.1005588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lamesch P, Berardini TZ, Li D, Swarbreck D, Wilks C, Sasidharan R, Muller R, Dreher K, Alexander DL, Garcia-Hernandez M, Karthikeyan AS, Lee CH, Nelson WD, Ploetz L, Singh S, Wensel A, Huala E. The Arabidopsis information resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res. 2012;40:1202–1210. doi: 10.1093/nar/gkr1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ren S, Peng Z, Mao J-H, Yu Y, Yin C, Gao X, Cui Z, Zhang J, Yi K, Xu W, Chen C, Wang F, Guo X, Lu J, Yang J, Wei M, Tian Z, Guan Y, Tang L, Xu C, Wang L, Gao X, Tian W, Wang J, Yang H, Wang J, Sun Y. RNA-seq analysis of prostate cancer in the Chinese population identifies recurrent gene fusions, cancer-associated long noncoding RNAs and aberrant alternative splicings. Cell Res. 2012;22:806–821. doi: 10.1038/cr.2012.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, Bergbower EA, Guan Y, Shin J, Guillory J, Rivers CS, Foo CK, Bhatt D, Stinson J, Gnad F, Haverty PM, Gentleman R, Chaudhuri S, Janakiraman V, Jaiswal BS, Parikh C, Yuan W, Zhang Z, Koeppen H, Wu TD, Stern HM, Yauch RL, Huffman KE, Paskulin DD, Illei PB, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet. 2012;44:1111–1116. doi: 10.1038/ng.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seo JS, Ju YS, Lee WC, Shin JY, Lee JK, Bleazard T, Lee J, Jung YJ, Kim JO, Shin JY, Yu SB, Kim J, Lee ER, Kang CH, Park IK, Rhee H, Lee SH, Kim JI, Kang JH, Kim YT. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res. 2012;22:2109–2119. doi: 10.1101/gr.145144.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seoane P, Ocana S, Carmona R, Bautista R, Madrid E, Torres AM, Claros MG. AutoFlow, a versatile workflow engine illustrated by assembling an optimised de novo transcriptome for a non-model species, such as faba bean (Vicia faba) Curr Bioinform. 2016;11:440–450. doi: 10.2174/1574893611666160212235117. [DOI] [Google Scholar]
- 30.Falgueras J, Lara AJ, Fernández-Pozo N, Cantón FR, Pérez-Trabado G, Claros MG. SeqTrim: a high-throughput pipeline for pre-processing any type of sequence read. BMC Bioinformatics. 2010;11:1–12. doi: 10.1186/1471-2105-11-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goto N, Prins P, Nakao M, Bonnal R, Aerts J, Katayama T. BioRuby: bioinformatics software for the Ruby programming language. Bioinformatics. 2010;26:2617–2619. doi: 10.1093/bioinformatics/btq475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Dios Alché J, Castro AJ, Olmedilla A, Fernández MC, Rodríguez R, Villalba M, Rodríguez-García MI. The major olive pollen allergen (Ole e I) shows both gametophytic and sporophytic expression during anther development, and its synthesis and storage takes place in the RER. J Cell Sci. 1999;112(Pt 1):2501–2509. doi: 10.1242/jcs.112.15.2501. [DOI] [PubMed] [Google Scholar]
- 34.Carmona R, Seoane P, Zafra A, Jiménez-Quesada MJ, de Dios Alché J, Claros MG: Automatic workflow for the identification of constitutively-expressed genes based on mapped NGS reads. In: Bioinformatics and biomedical engineering, vol 9656; 2016 (Lecture Notes in Computer Science). p. 403–14.
- 35.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 36.Wagner GP, Kin K, Lynch VJ. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012;131:281–285. doi: 10.1007/s12064-012-0162-3. [DOI] [PubMed] [Google Scholar]
- 37.Coker JS, Davies E. Selection of candidate housekeeping controls in tomato plants using EST data. Biotechniques. 2003;35:740–748. doi: 10.2144/03354st04. [DOI] [PubMed] [Google Scholar]
- 38.Chen L, Zhong H, Kuang J, Li J, Lu W, Chen J. Validation of reference genes for RT-qPCR studies of gene expression in banana fruit under different experimental conditions. Planta. 2011;234:377–390. doi: 10.1007/s00425-011-1410-3. [DOI] [PubMed] [Google Scholar]
- 39.Tong Z, Gao Z, Wang F, Zhou J, Zhang Z. Selection of reliable reference genes for gene expression studies in peach using real-time PCR. BMC Mol Biol. 2009;10:71. doi: 10.1186/1471-2199-10-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jain M, Nijhawan A, Tyagi AK, Khurana JP. Validation of housekeeping genes as internal control for studying gene expression in rice by quantitative real-time PCR. Biochem Biophys Res Commun. 2006;345:646–651. doi: 10.1016/j.bbrc.2006.04.140. [DOI] [PubMed] [Google Scholar]
- 41.Vashisth T, Johnson LK, Malladi A. An efficient RNA isolation procedure and identification of reference genes for normalization of gene expression in blueberry. Plant Cell Rep. 2011;30:2167–2176. doi: 10.1007/s00299-011-1121-z. [DOI] [PubMed] [Google Scholar]
- 42.Artico S, Nardeli SM, Brilhante O, Grossi-de-Sa MF, Alves-Ferreira M. Identification and evaluation of new reference genes in Gossypium hirsutum for accurate normalization of real-time quantitative RT-PCR data. BMC Plant Biol. 2010;10:49. doi: 10.1186/1471-2229-10-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vezzaro A, Krause ST, Nonis A, Ramina A, Degenhardt J, Ruperti B. Isolation and characterization of terpene synthases potentially involved in flavor development of ripening olive (Olea europaea) fruits. J Plant Physiol. 2012;169:908–914. doi: 10.1016/j.jplph.2012.01.021. [DOI] [PubMed] [Google Scholar]
- 44.Nicot N. Housekeeping gene selection for real-time RT-PCR normalization in potato during biotic and abiotic stress. J Exp Bot. 2005;56:2907–2914. doi: 10.1093/jxb/eri285. [DOI] [PubMed] [Google Scholar]
- 45.Yanik H, Turktas M, Dundar E, Hernandez P, Dorado G, Unver T. Genome-wide identification of alternate bearing-associated microRNAs (miRNAs) in olive (Olea europaea L.) BMC Plant Biol. 2013;13:10. doi: 10.1186/1471-2229-13-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barsalobres-Cavallari CF, Severino FE, Maluf MP, Maia IG. Identification of suitable internal control genes for expression studies in Coffea arabica under different experimental conditions. BMC Mol Biol. 2009;10:1. doi: 10.1186/1471-2199-10-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ronning CM, Stegalkina SS, Ascenzi RA, Bougri O, Hart AL, Utterbach TR, Vanaken SE, Riedmuller SB, White JA, Cho J, Pertea GM, Lee Y, Karamycheva S, Sultana R, Tsai J, Quackenbush J, Griffiths HM, Restrepo S, Smart CD, Fry WE, Van Der Hoeven R, Tanksley S, Zhang P, Jin H, Yamamoto ML, Baker BJ, Buell CR. Comparative analyses of potato expressed sequence tag libraries. Plant Physiol. 2003;131:419–429. doi: 10.1104/pp.013581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maroufi A, Van Bockstaele E, De Loose M. Validation of reference genes for gene expression analysis in chicory (Cichorium intybus) using quantitative real-time PCR. BMC Mol Biol. 2010;11:15. doi: 10.1186/1471-2199-11-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reid KE, Olsson N, Schlosser J, Peng F, Lund ST. An optimized grapevine RNA isolation procedure and statistical determination of reference genes for real-time RT-PCR during berry development. BMC Plant Biol. 2006;6:27. doi: 10.1186/1471-2229-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Die JV, Román B, Nadal S, González-Verdejo CI. Evaluation of candidate reference genes for expression studies in Pisum sativum under different experimental conditions. Planta. 2010;232:145–153. doi: 10.1007/s00425-010-1158-1. [DOI] [PubMed] [Google Scholar]
- 51.Gohain Bornali. Rubisco-bis-phosphate oxygenase (RuBP)—a potential housekeeping gene forqPCR assays in tea. Afr J Biotechnol. 2012;11:11193–11199. [Google Scholar]
- 52.Hoedemaekers K, Derksen J, Hoogstrate SW, Wolters-Arts M, Oh S-A, Twell D, Mariani C, Rieu I. BURSTING POLLEN is required to organize the pollen germination plaque and pollen tube tip in Arabidopsis thaliana. New Phytol. 2015;206:255–267. doi: 10.1111/nph.13200. [DOI] [PubMed] [Google Scholar]
- 53.Gu C, Chen S, Liu Z, Shan H, Luo H, Guan Z, Chen F. Reference gene selection for quantitative real-time PCR in Chrysanthemum subjected to biotic and abiotic stress. Mol Biotechnol. 2011;49:192–197. doi: 10.1007/s12033-011-9394-6. [DOI] [PubMed] [Google Scholar]
- 54.Thorrez L, Van Deun K, Tranchevent LC, Van Lommel L, Engelen K, Marchal K, Moreau Y, Van Mechelen I, Schuit F. Using ribosomal protein genes as reference: a tale of caution. PLoS ONE. 2008;3:e1854. doi: 10.1371/journal.pone.0001854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, Kiezun A, Hammerman PS, McKenna A, Drier Y, Zou L, Ramos AH, Pugh TJ, Stransky N, Helman E, Kim J, Sougnez C, Ambrogio L, Nickerson E, Shefler E, Cortés ML, Auclair D, Saksena G, Voet D, Noble M, DiCara D, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Waxman S, Wurmbach E. De-regulation of common housekeeping genes in hepatocellular carcinoma. BMC Genom. 2007;8:243. doi: 10.1186/1471-2164-8-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thellin O, Zorzi W, Lakaye B, De Borman B, Coumans B, Hennen G, Grisar T, Igout A, Heinen E. Housekeeping genes as internal standards: use and limits. J Biotechnol. 1999;75:291–295. doi: 10.1016/S0168-1656(99)00163-7. [DOI] [PubMed] [Google Scholar]
- 58.Ohl F, Jung M, Xu C, Stephan C, Rabien A, Burkhardt M, Nitsche A, Kristiansen G, Loening SA, Radonic A, Jung K. Gene expression studies in prostate cancer tissue: which reference gene should be selected for normalization? J Mol Med. 2005;83:1014–1024. doi: 10.1007/s00109-005-0703-z. [DOI] [PubMed] [Google Scholar]
- 59.Souza AFD, Brum IS, Neto BS, Berger M, Branchini G. Reference gene for primary culture of prostate cancer cells. Mol Biol Rep. 2013;40:2955–2962. doi: 10.1007/s11033-012-2366-5. [DOI] [PubMed] [Google Scholar]
- 60.Lemma S, Avnet S, Salerno M, Chano T, Baldini N. Identification and validation of housekeeping genes for gene expression analysis of cancer stem cells. PLoS ONE. 2016;11:e0149481. doi: 10.1371/journal.pone.0149481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Popovici V, Goldstein DR, Antonov J, Jaggi R, Delorenzi M, Wirapati P. Selecting control genes for RT-QPCR using public microarray data. BMC Bioinform. 2009;10:42. doi: 10.1186/1471-2105-10-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rubie C, Kempf K, Hans J, Su T, Tilton B, Georg T, Brittner B, Ludwig B, Schilling M. Housekeeping gene variability in normal and cancerous colorectal, pancreatic, esophageal, gastric and hepatic tissues. Mol Cell Probes. 2005;19:101–109. doi: 10.1016/j.mcp.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 63.Eisenberg E, Levanon EY. Human housekeeping genes, revisited. Trends Genet. 2013;29:569–574. doi: 10.1016/j.tig.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 64.Bianchini M, Levy E, Zucchini C, Pinski V, Macagno C, De Sanctis P, Valvassori L, Carinci P, Mordoh J. Comparative study of gene expression by cDNA microarray in human colorectal cancer tissues and normal mucosa. Int J Oncol. 2006;29:83–94. [PubMed] [Google Scholar]
- 65.Dupasquier S, Delmarcelle A-S, Marbaix E, Cosyns J-P, Courtoy PJ, Pierreux CE. Validation of housekeeping gene and impact on normalized gene expression in clear cell renal cell carcinoma: critical reassessment of YBX3/ZONAB/CSDA expression. BMC Mol Biol. 2014;15:9. doi: 10.1186/1471-2199-15-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cheng WC, Chang CW, Chen CR, Tsai ML, Shu WY, Li CY, Hsu IC. Identification of reference genes across physiological states for qRT-PCR through microarray meta-analysis. PLoS ONE. 2011;6:e17347. doi: 10.1371/journal.pone.0017347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Martin JL. Validation of reference genes for oral cancer detection panels in a prospective blinded cohort. PLoS ONE. 2016;11:e0158462. doi: 10.1371/journal.pone.0158462. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Statistics related to pre-processing with SeqTrimNext and mapping with Bowtie2 for the different datasets: olive tree libraries, Arabidopsis libraries, normal/malignant prostate samples, normal/small-cell cancer lung samples and normal/adenocarcinoma lung samples.
Additional file 2. Best RGs in olive tree pistil according to Fig. 2a, ranked by CV. They were obtained for different stages of pistil development with CV < 10% and minimum counted reads of 100. Transcript_id: transcript identifiers in the ReprOlive transcriptome.
Additional file 3. Best RGs in olive tree pollen according to Fig. 2b, ranked by CV. They were obtained for different stages of pollen development, with CV < 10% and minimum counted reads of 100. Transcript_id: transcript identifiers in the ReprOlive transcriptome.
Additional file 4. Best candidate RGs in Arabidopsis thaliana for replicates 1, 2 and 3 with their respective RPMM values for each replicate. They were obtained with CV < 10% and a minimum counted reads of 10,000. Transcript_id: transcript identifiers in TAIR database.
Additional file 5. Best candidate RGs for normal lung tissues according to Fig. 6a and ranked by CV. They were obtained with CV < 15% and minimum counted reads of 10,000. Transcript_id: human transcript identifiers in ENSEMBL database.
Additional file 6. Best candidate RGs for normal and malignant lung samples according to Fig. 6b, ranked by CV. They were obtained with CV < 20% and minimum counted reads of 10,000. Transcript_id: human transcript identifiers in ENSEMBL database.
Data Availability Statement
All references to data analysed during this study are included in this published article.