

SUMMARY
Horizontally transferred genetic elements such as viruses and conjugative plasmids move DNA between organisms, increasing genetic diversity but destabilizing engineered biological systems. Here we used a genomically recoded Escherichia coli strain lacking UAG stop codons and its recognition protein release factor 1 to study how an alternate genetic code influences horizontally transferred genetic element propagation. The alternate genetic code conferred resistance to multiple viruses (λ, M13, P1, MS2) at titers up to 1011 PFU/mL and impaired conjugative plasmids (F and RK2) up to 105 -fold. By recoding UAG codons to UAA in viruses and plasmids, we restored viral infectivity and conjugative function. Propagating viruses on a mixed community of cells with standard and alternate genetic codes reduced viral titer, and over time viruses adapted to the alternate genetic code. This work demonstrates that altering the genetic code broadly obstructs the propagation of horizontally transferred genetic elements and supports the use of genomic recoding as a strategy to stabilize engineered biological systems.
eTOC
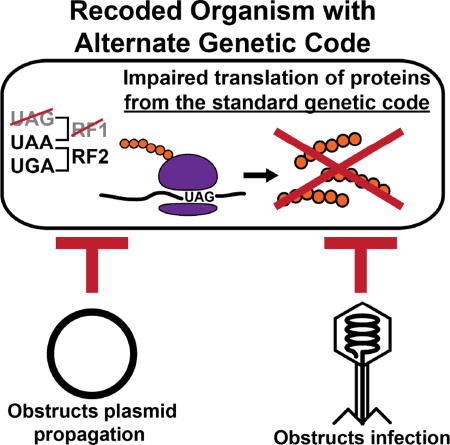
The conservation of the genetic code enables horizontal gene transfer, increasing genetic diversity but destabilizing engineered biological systems. Altering the genetic code obstructs the propagation of multiple viruses and conjugative plasmids, and motivates the use of genomic recoding as a strategy to genetically isolate and stabilize engineered organisms and microbial communities.
INTRODUCTION
Horizontal gene transfer (HGT), distinct from vertical transmission of genetic material from parents to offspring, is the intra-generational movement of genetic material between organisms and across species. It occurs through transformation, transduction, and conjugation, and often requires the presence of horizontally transferred genetic elements in the form of viruses and plasmids (Thomas and Nielsen, 2005). While HGT contributes to genetic diversity in nature (Ochman et al., 2000), it can introduce phenotypic changes that destabilize engineered biological systems through gene-gene interactions or global changes in cellular fitness (Baltrus, 2013), resulting in impaired growth or compromised functions. For example, biological-based production suffers losses from viral infection (Calendar, 2006), such as the 2009 infection of Genzyme’s bioreactors that caused ~$300 million in losses of high-value therapeutics and alleged loss of human life (Bethencourt, 2009). Genes from horizontally transferred genetic elements can also compromise mechanisms designed to isolate and contain genetically modified organisms (GMOs) from natural species (Kong et al., 2008; Ronchel and Ramos, 2001). As biotechnology’s societal and economic impact grows (Carlson, 2016), new approaches to minimize HGT and ensure the stability and safety of engineered biological systems are needed (Moe-Behrens et al., 2013; Schmidt and de Lorenzo, 2012).
Horizontally transferred genetic elements require expression of their genes to propagate, relying on the standard genetic code shared by most organisms to ensure accurate translation of their proteins. A genomically recoded Escherichia coli strain (C321.ΔA) containing an alternate genetic code exhibited increased tolerance to infection with the bacteriophage T7 (Lajoie et al., 2013b), suggesting that an alternate genetic code could confer resistance to viral infection and more broadly, all types of horizontal gene transfer. While these preliminary results demonstrated a 25% attenuation of infection for phage T7, the earlier study did not examine broad resistance to viruses or explore other mechanisms of HGT such as conjugation. How the alternate genetic code could cause resistance and the role of recoded organisms in HGT resistance within microbial communities also remains unknown. The alternate genetic code of C321.ΔA was achieved by mutating all instances of the UAG stop codon to the synonymous UAA stop codon and deleting the gene encoding the release factor 1 (RF1) protein, which terminates translation at UAG codons (Isaacs et al., 2011; Lajoie et al., 2013b). We hypothesize that this alternate genetic code would obstruct HGT by compromising translation of UAG-ending genes in horizontally transferred genetic elements, impairing their propagation.
Here we assessed the ability of horizontally transferred genetic elements to propagate on the genomically recoded E. coli strain and find impaired infection by multiple viruses, plasmid conjugation, and plasmid propagation compared with control strains containing the standard genetic code. We then conducted rescue experiments with horizontally transferred genetic elements that had UAG codons mutated to UAA to determine which genes must be accurately translated to ensure propagation and establish a causal link between the alternate genetic code of C321.ΔA and resistance to HGT. To extend our investigations beyond the level of an individual host and to the level of microbial communities, we investigated viral fitness and capacity for adaptation on mixed populations of bacteria comprising alternate and standard genetic codes. Our findings demonstrate that an alternate genetic code confers broad resistance to horizontally transferred genetic elements and motivates the use of genomic recoding as a strategy to genetically isolate and stabilize engineered organisms and microbial communities for biological-based production and deployment of safe GMOs into open systems (e.g., clinical medicine, environmental bioremediation).
RESULTS
Organisms with alternate genetic codes exhibit multi-viral resistance
We assayed a diverse panel of viruses that vary in proportion and type of UAG-ending genes (Table 1) for their ability to infect three strains of E. coli harboring standard or alternate genetic codes: (i) wild-type strain containing both UAG codons and RF1 (+UAG+RF1), (ii) strain lacking UAG codons but retaining RF1 (ΔUAG+RF1), and (iii) the C321.ΔA strain with an alternate genetic code lacking UAG codons and RF1 (ΔUAGΔRF1) (Figure 1A). Using serially-diluted phage to quantify plaque forming units per mL (PFU/mL), we found that viruses showed PFU/mL within 10-fold of each other on +UAG+RF1 and ΔUAG+RF1 hosts, but five (λ cI857, P1vir, P1 C1-100, M13, and MS2) of seven viruses produced no detectable plaques on ΔUAGΔRF1 (Figure 1B, S1A, S1B, p < 0.001). The two phages able to infect ΔUAGΔRF1 were Mu, which lacks UAG codons (Morgan et al., 2002), and T5, in which 12 of 13 UAG-ending genes are putative (Wang et al., 2005). Phage λ cI857, a temperature-sensitive variant of the lysogenic λ phage, produced clear plates on ΔUAGΔRF1 at high titers (109 PFU/mL, n = 3) (Figure S1C), but no visible plaques at up to 107 PFU/mL (p = 1.9e-006). P1vir and P1 c1-100, obligately lytic and temperature-inducible variants of the transducing phage P1 (Lobocka et al., 2004; Yarmolinsky, 1988), produced no plaques on ΔUAGΔRF1 at any concentration, including undiluted phage titer at 109 PFU/mL (p = 2.5e-005 for P1vir, p = 1.7e-006 for P1 c1-100). The pF-dependent phages MS2 and M13 (Loeb and Zinder, 1961; van Wezenbeek et al., 1980) also yielded no plaques on ΔUAGΔRF1 at any concentration up to titers of 1011 (MS2, p = 3.8e-005) and 1012 (M13, p = 3e-4) PFU/mL, respectively.
Table 1.
Horizontally transferred genetic elements used in this study
| Name | Type | Genome Type |
Size (kb) | # UAG-ending/Total genes |
Adhesion Site |
|---|---|---|---|---|---|
| Mu | Virus | dsDNA | 36.7 | 0/55 | Lipopolysaacharide component |
| λ cI857 | Virus | dsDNA | 48.5 | 4/92 | Maltoporin (lamB) |
| T5 | Virus | dsDNA | 121.8 | 13/195 | Ferrichrome receptor (fhuA) |
| M13 | Virus | ssDNA | 6.4 | 1/10 | pF conjugative pilus |
| P1vir | Virus | dsDNA | 94.0 | 12/116 | Lipopolysaacharide component |
| P1 c1-100 | Virus | dsDNA | 94.5 | 13/117 | Lipopolysaacharide component |
| MS2 | Virus | ssRNA | 3.6 | 2/4 | pF conjugative pilus |
| pF | Conjugative Plasmid | dsDNA | 100.3 | 10/106 | N/A |
| pRK2 | Conjugative Plasmid | dsDNA | 60.1 | 9/74 | N/A |
Figure 1. The alternate genetic code obstructs viral infection.

(A) Schematic depicting viral infection of cells with standard genetic codes or alternate genetic codes with no assigned meaning for the UAG codon. (B) Relative titers of viruses on strains +UAG+RF1, ΔUAG+RF1, and ΔUAGΔRF1. (C) Mutation analysis of 94 λ plaques isolated after recoding using MAGE. Colors represent number of mutations and the bar pattern represents proportion of mutants with UAG-to-UAA recoding in egrN or lgrQ. (D) Relative titers of λ phages with varying recoded loci (x-axis). (E) Relative titers of wild-type and recoded M13 phages infected on hosts with wild-type or partially recoded (Fpr) pF. For all relative titers, data are mean with standard deviation, n=3. “0” indicates zero plaque forming units (PFU)/mL. P-values are as follows: * is P ≤ 0.05, ** is P ≤ 0.01, *** is P ≤ 0.001, and **** is P ≤ 0.0001.
While λ and P1 c1-100 were capable of adhering to ΔUAGΔRF1, they produced less than one progeny per cell infection (Figure S2), suggesting that the barrier to infection occurred after cell adhesion. To determine if restoring accurate translation of viral gene products can rescue infectivity, we used MAGE (Gallagher et al., 2014; Wang et al., 2009) to recode the four UAG-ending genes (ea31, egrN, lgrQ, Rz) in phage λ (Table S1, Figure S3A). This differs from gene deletion or mutagenesis of the coding region, since the coding region is unchanged and can produce the wild-type protein if placed in a host containing RF1. Phage λ variants propagated on ΔUAGΔRF1 showed enrichment for UAG-to-UAA mutations, and 100% of phages propagated on ΔUAGΔRF1 contained at least one UAG-to-UAA mutation in either egrN or lgrQ (Fig. 1C). To confirm the recovery of infection and determine which UAG-to-UAA reassignments were causative in restoring viral fitness, we infected all λ variants on ΔUAG ΔRF1 and found that while recoding of the UAG codon terminating egrN to UAA partially restored viral titer (p = 1.2e-4), recoding of the UAG codon terminating lgrQ to UAA fully restored viral titer (p = 2.9e-6) on ΔUAGΔRF1 to levels observed when infecting +UAG+RF1 (Figure 1D, S3B, S3C). Both egrN and lgrQ encode proteins that promote host transcription of viral genes by preventing rho-dependent transcription termination (Grayhack et al., 1985; Schauer et al., 1987). These data suggest that recoding of the lgrQ stop codon from UAG-to-UAA could be necessary and sufficient to fully restore λ infection on ΔUAGΔRF1.
We next recoded M13 to create M13rec via PCR, mutating the UAG codon to UAA at the end of Gene IV, the sole UAG-ending gene that encodes the phage assembly protein (Table S1). We found that despite recoding, M13rec was unable to infect ΔUAGΔRF1 (Figure 1E, S6, p = 3.0e-005). In viral adhesion and progeny per cell assays (Figure S3D, S3E), M13 bound all host strains similarly but showed one progeny per cell on ΔUAGΔRF1 hosts, suggesting a failure to enter the cell. Additionally, phage MS2, which binds to the same surface receptor as M13, showed impaired binding on ΔUAGΔRF1 hosts (p = 3.2e-4). This indicated a defect in the binding receptor for these phages, which is a conjugative pilus encoded by the plasmid pF, another horizontally transferred genetic element (Table 1) (Deng and Perham, 2002; Ou, 1973; Paranchych et al., 1971). Analysis of the pF sequence revealed ten UAG-ending genes (Table S1). Of these ten genes, one gene (repE) is involved in plasmid maintenance, two genes (traY and traL) are implicated in plasmid transfer, and one gene (ybhA) is a duplicate of an E. coli gene that forms a dimer, potentially producing dominant negative effects (Frost et al., 1994; Nelson et al., 1995; Watson et al., 1982). Using MAGE, we recoded the UAG codons terminating repE, traY, traL, and ybhA to UAA to produce a partially recoded pF (pFpr) and attempted infection with M13. We found that M13 was unable to infect ΔUAGΔRF1 carrying pFpr (p = 0.0013), but when M13rec was infected on ΔUAGΔRF1 carrying pFpr, the phage recovered its ability to infect (Figure 1E, S3D, S3E, p = 1.8e-005).
The alternate genetic code obstructs the transfer and replication of conjugative plasmids
We next investigated the conjugation efficiency of pF in ΔUAGΔRF1 to assess whether the alternate genetic code could obstruct forms of HGT beyond transduction. We quantified conjugative transfer efficiency of pF and pFpr from +UAG+RF1, ΔUAG+RF1, and ΔUAGΔRF1 donors to +UAG+RF1, ΔUAG+RF1, and ΔUAGΔRF1 recipients. Transfer of pF from ΔUAGΔRF1 donors was below the 1% limit of detection for our assay (Figure 2A, p = 1.8e-6) and conjugation events were 100,000-fold less frequent than from +UAG+RF1 cells (Figure S4A, p = 1.0e-4), while transfer of pFpr from the ΔUAGΔRF1 donor exhibited a 2% transfer efficiency (Figure 2A, p = 3.7e-5). Recoding all 10 UAG codons to UAA (pFrec) resulted in a ~12-fold increase in transfer efficiency from ΔUAGΔRF1 donors to 25% (Figure 2A, p = 4.8e-3), indicating UAG codons in pF impaired conjugative transfer. To more broadly investigate the effect of an alternate genetic code on conjugative plasmids, we also tested the ability of the broad host-range pRK2 (Thomas and Smith, 1987) to transfer and replicate in ΔUAGΔRF1 strains. We found that ΔUAGΔRF1 experienced 25–50% reduction in conjugative transfer as a recipient and exhibited a 27% increase in doubling time (Figure 2B, S4B, Table S2, p < 0.01), which were recovered with UAG-to-UAA recoding in the plasmid, suggesting UAG codons in RK2 impaired both plasmid transfer and replication.
Figure 2. The alternate genetic code obstructs conjugation.

Conjugation efficiency from donors with standard and alternate genetic codes (x-axis) to recipients with standard and alternate genetic codes (bars) for wild-type and recoded (A) pF and (B) pRK2 conjugative plasmids. “0” indicates transfer efficiency was below limit of detection of 1%. Data are mean with standard deviation, n=3. P-values are as follows: * is P ≤ 0.05, ** is P ≤ 0.01, *** is P ≤ 0.001, and **** is P ≤ 0.0001. (C) Mutation analysis of 96 pF variants isolated after recoding using MAGE and conjugation from +UAG+RF1 and ΔUAGΔRF1. (D) Mutation analysis of 96 pRK2 variants isolated after recoding using MAGE and conjugation to +UAG+RF1 or ΔUAGΔRF1. For mutation analysis, colors represent number of mutations and pattern represents mutants with UAG-to-UAA recoding in indicated genes.
To determine if restoring accurate translation of conjugative plasmid gene products can rescue plasmid transfer and replication, we used MAGE to recode UAG-ending pF and pRK2 genes to UAA and assayed propagation between and within cells by measuring conjugation rates and doubling times. After creating a library of pF variants with diverse UAG-to-UAA recoding in +UAG+RF1 and ΔUAGΔRF1 donors, we mated donors to a +UAG+RF1 recipient to select for recovered conjugative function. All pF variants screened from ΔUAGΔRF1 donors contained UAG-to-UAA mutations in traY, traL, or repE, of which 97.8% clones contained UAG-to-UAA mutations in traY or traL (Figure 2C) compared to only 55.3% of pF variants from +UAG+RF1 donors. Assays of conjugative function confirmed that UAG-to-UAA mutations in traY and traL recover conjugative efficiency 10,000-fold (Figure S4A, p = 2.0e-4). In contrast, the MAGE-derived pRK2 library conjugated into both +UAG+RF1 and ΔUAGΔRF1 recipients identified a single gene, trfA, which restored both conjugative function and host fitness. Gene trfA encodes proteins that initiate vegetative replication in pRK2 (Pansegrau et al., 1994). In ΔUAGΔRF1 recipients, 100% of pRK2 variants contained a UAG-to-UAA mutation in trfA, compared to 23.4% from +UAG+RF1 recipients (Figure 2D). Recoding trfA restored both conjugative efficiency and propagation of pRK2 in ΔUAGΔRF1 to levels seen in +UAG+RF1 (Figure 2B, S4B, Table S2, p = < 1.0e-4). These results demonstrate that an alternate genetic code can impair replication and conjugation of plasmid-based horizontally transferred genetic elements, impeding their propagation within and between hosts.
Recoded organisms reduce viral titer in mixed microbial communities and prompt adaptation to the alternate genetic code
We examined how the presence of recoded organisms in a microbial community affects viral fitness by propagating phage λ on communities comprising varying ratios of two types of cells: permissive hosts with standard genetic codes and non-permissive hosts with alternate genetic codes (Figure 3A). As we increased the proportion of non-permissive hosts to permissive hosts, we expected viral titer to decrease as the virus is increasingly likely to bind and infect non-permissive hosts. We found that in the presence of 90 to 100% ΔUAGΔRF1, viral populations that start at 109 PFU/mL decrease continuously and are extinct by days five and three, respectively (Figure 3B, p < 0.001). In contrast, viral populations propagated on communities comprising 0%, 10%, or 50% ΔUAGΔRF1 all showed increases in viral titer to 1010 on day one and then diverged. The populations of viruses propagated on 50% ΔUAGΔRF1 declined on days two and three, stabilizing at a PFU/mL of 5- to 10-fold less than viral populations propagated on 10% or no ΔUAGΔRF1 (p < 0.05).
Figure 3. Recoded organisms reduce viral population fitness in microbial communities and select for viral mutations that eliminate UAG codon use.

(A) Schematic of microbial community assays. Phages are infected on a co-culture containing varying ratios of ΔUAG+RF1 and ΔUAGΔRF1, extracted the next day, and propagated on a co-culture with the same cell ratio. Viral populations of λ were quantified by infection on ΔUAG+RF1, and ability of phage MS2 to infect ΔUAGΔRF1 was assayed by plating on ΔUAGΔRF1 containing pFpr. (B) Titers of phage λ viral populations propagated on microbial communities containing cells with standard and alternate genetic codes. Lines are mean of 3 biological replicates for each population. (C) Location of mutations eliminating UAG codon usage in the MS2 genome (Calendar, 2006; Fiers et al., 1976). (D) Relative titers of wild-type and recoded MS2 (MS2rec2) phages infected on ΔUAGΔRF1 with pF or pFpr, which is required for phage infection. Data are mean with standard deviation, n=3. P-values are as follows: * is P ≤ 0.05, ** is P ≤ 0.01, *** is P ≤ 0.001, and **** is P ≤ 0.0001.
To determine whether horizontally transferred genetic elements could adapt to the alternate genetic code, we subjected the highly mutagenic ssRNA phage MS2 (1.5×10−3 substitutions per bp per replication) (Drake, 1993) to selective pressure by propagating it on a microbial community containing changing ratios of strains ΔUAG+RF1 and ΔUAGΔRF1 carrying pFpr (Figure 3A). After five days of propagation in soft agar, the MS2 population produced opaque plaques when infected on only ΔUAGΔRF1 carrying pFpr. Sequencing of eight plaques revealed that phages contained the following two mutations (designated as MS2rec): (1) a UAG-to-UAA mutation in the last codon of the rep gene, and (2) a nonsense AGA-to-UGA mutation in the penultimate codon of the mat gene to create a premature stop codon (Figure 3C) (Fiers et al., 1976). These mutations eliminate UAG codon use and provide direct experimental evidence that viruses adapt their genetic code to achieve compatibility with the alternate genetic code of the ΔUAGΔRF1 host. To improve plaque clarity, we further evolved MS2rec on decreasing ratios of ΔUAG+RF1 to ΔUAGΔRF1 until phage produced clearer plaques when infected on ΔUAGΔRF1. We sequenced three clones from this population and found two clones contained an A->C transversion in the overlapping lysis and coat protein genes and designated this phage MS2rec2. MS2rec2 demonstrated a 109 increase over MS2 in ability to infect hosts with an alternate genetic code in the presence of pFpr (Figure 3D, p = 0.026), recovering infection to within 10-fold of MS2 on hosts with standard genetic codes and demonstrating viral adaptation to the alternate genetic code.
DISCUSSION
Horizontally transferred genetic elements have driven both the evolution of natural mechanisms to restrict HGT such as restriction enzymes and CRISPR-Cas (Thomas and Nielsen, 2005), but these mechanisms are often sequence-specific. Additionally, anthropogenic strategies to reduce HGT exist (Getino et al., 2015), but these require continuous input of a small molecule to prevent HGT from occurring. In this study, we demonstrate that altering the genetic code of a cell can reduce HGT by broadly obstructing propagation of horizontally transferred genetic elements in a host and within microbial communities. Despite the rarity of UAG codons (Table 1), an alternate genetic code conferred resistance to various viruses with different life cycles (lytic, lysogenic, and non-lytic) and conjugative plasmids containing selectively advantageous genes (e.g., antibiotic resistance). Additionally, in the F-dependent phages M13 and MS2, restoring viral infection required recoding UAG codons in both the viral genome and pF, suggesting a two-layer model of immunity against F-dependent viruses that may extend to other viruses dependent on conjugative plasmids for infection.
Recoding UAG codons to UAA in phages and conjugative plasmids identified key causative genes and established that UAG codons caused impaired propagation. We found that a single UAG codon could serve as a barrier to propagation of both viruses (λ, M13) and conjugative plasmids (RK2). Given that 93% of all Enterobacteria and Escherichia phages on NCBI’s Viral Genomes Resource have at least one UAG-ending gene (Table S3) (Brister et al., 2015), our data suggest that the alternate genetic code lacking UAG assignment may confer broad multiviral resistance. Though the molecular mechanism of this impairment has not been experimentally investigated in this study, we hypothesize that the lack of RF1 causes ribosomal stalling at UAG and results in two possible outcomes that would cause impaired propagation: (1) tmRNA-mediated rescue that results in degradation of the translated peptide (Keiler, 2015), or (2) near-cognate or amber suppression (Eggertsson and Soll, 1988), creating a C-terminal tail that disrupts protein function.
Our study also demonstrates that alternate genetic codes impair viral population fitness in microbial communities, reducing viral titer and prompting emergence of viral variants that have adapted to the alternate genetic code. In microbial communities, the presence of organisms with alternate genetic codes reduced λ viral titer when they comprised at least half of the host population, and we interpret this as a reduction in viral population fitness. Previous work demonstrated that non-permissive host cells overexpressing viral surface receptors could reduce viral titer when mixed with permissive host cells (Dennehy et al., 2007), as viral binding kinetics favor attachment and attempted infection of non-permissive hosts. However, we did not modify surface receptor expression in our ΔUAGΔRF1, suggesting that alternate genetic codes can reduce viral titer and viral population fitness in a microbial community more broadly than surface receptor-based strategies. Furthermore, over time the ssRNA phage MS2 mutated away from UAG codon use when propagated in the presence of the alternate genetic code, indicating that viruses experience a selective pressure to reduce the presence of UAG codons in their genomes.
From an applied perspective, the ability to impair propagation of horizontally transferred genetic elements provides a useful strategy for genetic isolation in diverse biomanufacturing and biocontainment applications. Utilizing organisms with alternate genetic codes in industrial settings could increase stability and reduce the risk and cost of biological production (Calendar, 2006). In GMOs that function as engineered probiotics (Steidler, 2003) or in bioremediation applications (Pieper and Reineke, 2000), alternate genetic codes can act as barriers to gene transfers that could compromise biocontainment mechanisms such as natural or synthetic auxotrophies (Mandell et al., 2015; Rovner et al., 2015; Steidler et al., 2003) and toxin kill switches (Cai et al., 2015; Chan et al., 2016; Gallagher et al., 2015). While a virus adapted to the alternate genetic code with few mutations (Figure 3C), we anticipate that greater barriers achieved by further recoding within E. coli and additional organisms could prove insurmountable to horizontally transferred genetic elements. Research into sense codon reassignment is already underway in E. coli and other bacterial species (Krishnakumar et al., 2013; Lajoie et al., 2013a), paving the way for alternate genetic codes with multiple codon reassignments. By expanding recoding efforts to multiple species, we envision the development of synthetic microbial communities with alternate genetic codes that are genetically isolated and robust to perturbation by HGT. In assaying the ability of horizontally transferred genetic elements to utilize a host with an alternate genetic code, we step into an ancient evolutionary arms race between selfish genetic elements and the hosts they exploit. This interplay has generated a panoply of adaptations in the history of life (Syvanen, 2012) and driven the evolution of microbial defense systems against horizontally transferred genetic elements (Bickle and Kruger, 1993; Makarova et al., 2011). Since metagenomic studies have revealed that alternate genetic codes are more prevalent than previously thought (Ivanova et al., 2014) and reassignments of the same codons have occurred in multiple lineages (Knight et al., 2001), some have suggested that alternate genetic codes could serve as defense systems against horizontally transferred genetic elements (Shackelton and Holmes, 2008). While previous research into codon usage showed that synthesis of viral genes utilizing underrepresented codon pairs reduced the virulence of both poliovirus and influenza virus (Coleman et al., 2008; Nogales et al., 2014), our work demonstrates that an alternate genetic code can impede horizontally transferred genetic element propagation and create barriers to HGT. These results support the hypothesis that alternate genetic codes act as defense against exploitation by horizontally transferred genetic elements, suggesting an evolutionary interplay between the genetic code and horizontal gene transfer that may have driven the evolution of alternate genetic codes.
Experimental Procedures
Viral Relative Titers
To quantify relative titers, we mixed 100-fold dilutions of phage with 300 µL of mid-log (OD600=0.5) cells in 3 mL of TK soft agar and poured onto TK solid agar plates. We found that MS2 produced clearer plaques if propagated on late log (OD600 = 1.0) cells and used this OD instead. We plated each dilution in triplicate, incubated plates overnight, and counted plaques the next day.
Recoding of Viral Genomes
To recode lambda phage UAG codons to UAA, we lysogenized λ cI857 in an MG1655 strain with a mutS deletion and a constitutively expressed lambda beta protein on a plasmid. At mid-log, we rinsed cells, re-suspended them in oligo, and electroporated using the methods described previously (Gallagher et al., 2014). We then allowed cells to return to mid-log density and repeated for a total of 5 MAGE cycles. The oligonucleotide sequences we used to convert UAG codons to UAA are available in the Oligonucleotides Excel spreadsheet. After MAGE, we induced lytic cycle by shifting an aliquot of cells to 42 °C for 20 minutes, and then plated 100-fold serial dilutions of this in the same manner as viral relative titers on +UAG+RF1 or ΔUAGΔRF1. We then assayed 47 plaques from each host for UAG-to-UAA mutations via sequencing.
We utilized PCR with a UAG-to-UAA mutation in primer overhangs to recode the M13 genome. To minimize error in replication, we used High-fidelity PCR Mix from Kapa Biosystems (KK2602). After PCR, we circularized linear amplified phage genome by mixing 100–300 ng of DNA in Gibson Assembly Master Mix from NEB and incubating for 1 hour at 50°C. We then drop-dialyzed the assembly for 1 hour to remove salts and transformed phage genome into +UAG+RF1 with pF to extract virus. We sequenced isolated virus to verify the UAG-to-UAA mutation and to detect any accessory mutations.
Microbial community assays
Microbial community assays were performed using three phage populations as biological replicates propagated on mixed populations of ΔUAG+RF1 (viable host) and ΔUAGΔRF1 (nonviable host) cells at varying ratios. ΔUAG+RF1 was chosen instead of +UAG+RF1 because its doubling time is similar to ΔUAGΔRF1, simplifying cell ratio calculations. Each day, we extracted phage as described above, diluted viral population 104, and re-infected on fresh mixed populations of cells with the same ratio. We titered phage using methods described above.
We evolved MS2 via serial propagation on mixed populations of ΔUAG+RF1 (viable host) and ΔUAGΔRF1 (nonviable host) cells. ΔUAG+RF1 was chosen instead of +UAG+RF1 because its doubling time is similar to ΔUAG ΔRF1, simplifying cell ratio calculations. Each day, we extracted phage as described above, diluted viral population to get 104 to 105 plaques, and re-infected on mixture of strains. We propagated MS2 on agar overlays with 1:1 ratio of ΔUAG+RF1 to ΔUAGΔRF1 cells for 5 days, when we plated viral populations on only ΔUAGΔRF1 cells and sequenced 8 resulting plaques. We further evolved MS2rec using the following propagation: 2 days 1:1 ratio ΔUAG+RF1 to ΔUAGΔRF1, 4 days 1:3 ratio ΔUAG+RF1 to ΔUAGΔRF1, 2 days 1:9 ratio ΔUAG+RF1 to ΔUAGΔRF1, 1 day on ΔUAGΔRF1 only. We picked 3 plaques from this population for sequencing.
Recoding of conjugative plasmids and quantifying selective pressure on UAG->UAA mutations
To recode conjugative plasmids, we mated plasmids into cells carrying the lambda red cassette and performed MAGE as described previously (Gallagher et al., 2014) with UAG-to-UAA mutation oligonucleotides whose sequences are available in the Oligonucleotides Excel spreadsheet. For pF, we performed 8 cycles of MAGE in ΔUAGΔRF1 and +UAG+RF1 backgrounds and then conjugated to +UAG+RF1 strains to identify mutations that recover conjugative ability. For pRK2, we performed 10 cycles of MAGE in a +UAG+RF1 background and then conjugated to +UAG+RF1 or ΔUAGΔRF1 strains to identify mutations that recover plasmid maintenance and conjugative efficiency. In both cases, the clone with the most UAG-to-UAA mutations was then chosen for subsequent cycles of MAGE until all UAG codons were converted to UAA.
Quantifying Conjugation Efficiency
We used conjugation conditions described previously (Ma et al., 2014). Briefly, we grew cultures of donor and recipient cells to late log in antibiotics selecting for plasmid or recipient and then rinsed and re-suspended in media to remove antibiotics. We then concentrated cells and normalized to OD600=20 by doing 100-fold dilution and normalizing to OD600=0.2, then mixed donors and recipients in 1:1 ratio and spotted onto pre-warmed LB Lennox agar plates in 2× 20uL and 6× 10uL pattern. We incubated plates at 37°C for 1 hour (pRK2) or 2 hours (pF), then rinsed cells from the plate, diluted serially 10-fold, and plated on plates containing antibiotic selecting for recipient strain and incubated overnight at 37°C. We then picked 86 colonies from plates selecting for the recipient strain and patched them onto plates selecting for recipient strain with the conjugative plasmid, incubated plates overnight at 37°C, and counted the number of patched colonies that grew.
Statistical Analysis
P values were calculated using unpaired T-tests in GraphPad Prism 6 with no assumption of consistent standard deviation and a false discovery rate threshold of 0.01.
Supplementary Material
Highlights.
An alternate genetic code obstructs the propagation of viruses and conjugative plasmids
Recoding viruses and plasmids to match the altered genetic code restores propagation
Recoded organisms reduce viral population fitness within microbial communities
Viruses adapt to match the alternate genetic code and infect recoded organisms
Acknowledgments
We gratefully acknowledge Paul Turner (Yale University) for his experimental advice and manuscript feedback. NJM gratefully acknowledges support from the NIH (T32GM007499, T32GM007223) and the Gruber Foundation. FJI gratefully acknowledges support from DARPA (N66001-12-C-4211, HR0011-15-C-0091), DOE (152339.5055249.100), NIH (1R01GM117230-01), Gen9 Inc., DuPont Inc., and the Arnold and Mabel Beckman Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
N.J.M. and F.J.I. conceived the study, designed experiments and wrote the paper. N.J.M. conducted experiments and F.J.I. supervised the study.
References
- Baltrus DA. Exploring the costs of horizontal gene transfer. Trends Ecol Evol. 2013;28:489–495. doi: 10.1016/j.tree.2013.04.002. [DOI] [PubMed] [Google Scholar]
- Bethencourt V. Virus stalls Genzyme plant. Nat Biotech. 2009;27:681–681. [Google Scholar]
- Bickle TA, Kruger DH. Biology of DNA restriction. Microbiol Rev. 1993;57:434–450. doi: 10.1128/mr.57.2.434-450.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calendar R. The bacteriophages. 2. Oxford ; New York: Oxford University Press; 2006. [Google Scholar]
- Carlson R. Estimating the biotech sector's contribution to the US economy. Nature biotechnology. 2016;34:247–255. doi: 10.1038/nbt.3491. [DOI] [PubMed] [Google Scholar]
- Coleman JR, Papamichail D, Skiena S, Futcher B, Wimmer E, Mueller S. Virus attenuation by genome-scale changes in codon pair bias. Science. 2008;320:1784–1787. doi: 10.1126/science.1155761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng LW, Perham RN. Delineating the site of interaction on the pIII protein of filamentous bacteriophage fd with the F-pilus of Escherichia coli. J Mol Biol. 2002;319:603–614. doi: 10.1016/S0022-2836(02)00260-7. [DOI] [PubMed] [Google Scholar]
- Drake JW. Rates of spontaneous mutation among RNA viruses. Proc Natl Acad Sci U S A. 1993;90:4171–4175. doi: 10.1073/pnas.90.9.4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggertsson G, Soll D. Transfer ribonucleic acid-mediated suppression of termination codons in Escherichia coli. Microbiol Rev. 1988;52:354–374. doi: 10.1128/mr.52.3.354-374.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiers W, Contreras R, Duerinck F, Haegeman G, Iserentant D, Merregaert J, Min Jou W, Molemans F, Raeymaekers A, Van den Berghe A, et al. Complete nucleotide sequence of bacteriophage MS2 RNA: primary and secondary structure of the replicase gene. Nature. 1976;260:500–507. doi: 10.1038/260500a0. [DOI] [PubMed] [Google Scholar]
- Frost LS, Ippen-Ihler K, Skurray RA. Analysis of the sequence and gene products of the transfer region of the F sex factor. Microbiol Rev. 1994;58:162–210. doi: 10.1128/mr.58.2.162-210.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher RR, Li Z, Lewis AO, Isaacs FJ. Rapid editing and evolution of bacterial genomes using libraries of synthetic DNA. Nat Protoc. 2014;9:2301–2316. doi: 10.1038/nprot.2014.082. [DOI] [PubMed] [Google Scholar]
- Getino M, Sanabria-Rios DJ, Fernandez-Lopez R, Campos-Gomez J, Sanchez-Lopez JM, Fernandez A, Carballeira NM, de la Cruz F. Synthetic Fatty Acids Prevent Plasmid-Mediated Horizontal Gene Transfer. MBio. 2015;6:e01032–01015. doi: 10.1128/mBio.01032-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayhack EJ, Yang XJ, Lau LF, Roberts JW. Phage lambda gene Q antiterminator recognizes RNA polymerase near the promoter and accelerates it through a pause site. Cell. 1985;42:259–269. doi: 10.1016/s0092-8674(85)80121-5. [DOI] [PubMed] [Google Scholar]
- Isaacs FJ, Carr PA, Wang HH, Lajoie MJ, Sterling B, Kraal L, Tolonen AC, Gianoulis TA, Goodman DB, Reppas NB, et al. Precise manipulation of chromosomes in vivo enables genome-wide codon replacement. Science. 2011;333:348–353. doi: 10.1126/science.1205822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova NN, Schwientek P, Tripp HJ, Rinke C, Pati A, Huntemann M, Visel A, Woyke T, Kyrpides NC, Rubin EM. Stop codon reassignments in the wild. Science. 2014;344:909–913. doi: 10.1126/science.1250691. [DOI] [PubMed] [Google Scholar]
- Keiler KC. Mechanisms of ribosome rescue in bacteria. Nat Rev Microbiol. 2015;13:285–297. doi: 10.1038/nrmicro3438. [DOI] [PubMed] [Google Scholar]
- Knight RD, Freeland SJ, Landweber LF. Rewiring the keyboard: evolvability of the genetic code. Nat Rev Genet. 2001;2:49–58. doi: 10.1038/35047500. [DOI] [PubMed] [Google Scholar]
- Kong W, Wanda SY, Zhang X, Bollen W, Tinge SA, Roland KL, Curtiss R., 3rd Regulated programmed lysis of recombinant Salmonella in host tissues to release protective antigens and confer biological containment. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:9361–9366. doi: 10.1073/pnas.0803801105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnakumar R, Prat L, Aerni HR, Ling J, Merryman C, Glass JI, Rinehart J, Soll D. Transfer RNA misidentification scrambles sense codon recoding. Chembiochem. 2013;14:1967–1972. doi: 10.1002/cbic.201300444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lajoie MJ, Kosuri S, Mosberg JA, Gregg CJ, Zhang D, Church GM. Probing the limits of genetic recoding in essential genes. Science. 2013a;342:361–363. doi: 10.1126/science.1241460. [DOI] [PubMed] [Google Scholar]
- Lajoie MJ, Rovner AJ, Goodman DB, Aerni HR, Haimovich AD, Kuznetsov G, Mercer JA, Wang HH, Carr PA, Mosberg JA, et al. Genomically recoded organisms expand biological functions. Science. 2013b;342:357–360. doi: 10.1126/science.1241459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobocka MB, Rose DJ, Plunkett G, 3rd, Rusin M, Samojedny A, Lehnherr H, Yarmolinsky MB, Blattner FR. Genome of bacteriophage P1. Journal of bacteriology. 2004;186:7032–7068. doi: 10.1128/JB.186.21.7032-7068.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb T, Zinder ND. A bacteriophage containing RNA. Proceedings of the National Academy of Sciences of the United States of America. 1961;47:282–289. doi: 10.1073/pnas.47.3.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma NJ, Moonan DW, Isaacs FJ. Precise manipulation of bacterial chromosomes by conjugative assembly genome engineering. Nat Protoc. 2014;9:2285–2300. doi: 10.1038/nprot.2014.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, Moineau S, Mojica FJ, Wolf YI, Yakunin AF, et al. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol. 2011;9:467–477. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moe-Behrens GH, Davis R, Haynes KA. Preparing synthetic biology for the world. Front Microbiol. 2013;4:5. doi: 10.3389/fmicb.2013.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan GJ, Hatfull GF, Casjens S, Hendrix RW. Bacteriophage Mu genome sequence: analysis and comparison with Mu-like prophages in Haemophilus, Neisseria and Deinococcus. J Mol Biol. 2002;317:337–359. doi: 10.1006/jmbi.2002.5437. [DOI] [PubMed] [Google Scholar]
- Nelson WC, Howard MT, Sherman JA, Matson SW. The traY gene product and integration host factor stimulate Escherichia coli DNA helicase I-catalyzed nicking at the F plasmid oriT. J Biol Chem. 1995;270:28374–28380. [PubMed] [Google Scholar]
- Nogales A, Baker SF, Ortiz-Riano E, Dewhurst S, Topham DJ, Martinez-Sobrido L. Influenza A virus attenuation by codon deoptimization of the NS gene for vaccine development. J Virol. 2014;88:10525–10540. doi: 10.1128/JVI.01565-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H, Lawrence JG, Groisman EA. Lateral gene transfer and the nature of bacterial innovation. Nature. 2000;405:299–304. doi: 10.1038/35012500. [DOI] [PubMed] [Google Scholar]
- Ou JT. Inhibition of formation of Escherichia coli mating pairs by f1 and MS2 bacteriophages as determined with a Coulter counter. Journal of bacteriology. 1973;114:1108–1115. doi: 10.1128/jb.114.3.1108-1115.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pansegrau W, Lanka E, Barth PT, Figurski DH, Guiney DG, Haas D, Helinski DR, Schwab H, Stanisich VA, Thomas CM. Complete nucleotide sequence of Birmingham IncP alpha plasmids. Compilation and comparative analysis. J Mol Biol. 1994;239:623–663. doi: 10.1006/jmbi.1994.1404. [DOI] [PubMed] [Google Scholar]
- Paranchych W, Ainsworth SK, Dick AJ, Krahn PM. Stages in phage R17 infection. V. Phage eclipse and the role of F pili. Virology. 1971;45:615–628. doi: 10.1016/0042-6822(71)90176-0. [DOI] [PubMed] [Google Scholar]
- Ronchel MC, Ramos JL. Dual system to reinforce biological containment of recombinant bacteria designed for rhizoremediation. Appl Environ Microbiol. 2001;67:2649–2656. doi: 10.1128/AEM.67.6.2649-2656.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauer AT, Carver DL, Bigelow B, Baron LS, Friedman DI. lambda N antitermination system: functional analysis of phage interactions with the host NusA protein. J Mol Biol. 1987;194:679–690. doi: 10.1016/0022-2836(87)90245-2. [DOI] [PubMed] [Google Scholar]
- Schmidt M, de Lorenzo V. Synthetic constructs in/for the environment: managing the interplay between natural and engineered Biology. FEBS Lett. 2012;586:2199–2206. doi: 10.1016/j.febslet.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackelton LA, Holmes EC. The role of alternative genetic codes in viral evolution and emergence. J Theor Biol. 2008;254:128–134. doi: 10.1016/j.jtbi.2008.05.024. [DOI] [PubMed] [Google Scholar]
- Syvanen M. Evolutionary implications of horizontal gene transfer. Annu Rev Genet. 2012;46:341–358. doi: 10.1146/annurev-genet-110711-155529. [DOI] [PubMed] [Google Scholar]
- Thomas CM, Nielsen KM. Mechanisms of, and barriers to, horizontal gene transfer between bacteria. Nat Rev Microbiol. 2005;3:711–721. doi: 10.1038/nrmicro1234. [DOI] [PubMed] [Google Scholar]
- Thomas CM, Smith CA. Incompatibility group P plasmids: genetics, evolution, and use in genetic manipulation. Annu Rev Microbiol. 1987;41:77–101. doi: 10.1146/annurev.mi.41.100187.000453. [DOI] [PubMed] [Google Scholar]
- van Wezenbeek PM, Hulsebos TJ, Schoenmakers JG. Nucleotide sequence of the filamentous bacteriophage M13 DNA genome: comparison with phage fd. Gene. 1980;11:129–148. doi: 10.1016/0378-1119(80)90093-1. [DOI] [PubMed] [Google Scholar]
- Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, Church GM. Programming cells by multiplex genome engineering and accelerated evolution. Nature. 2009;460:894–898. doi: 10.1038/nature08187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Jiang Y, Vincent M, Sun Y, Yu H, Wang J, Bao Q, Kong H, Hu S. Complete genome sequence of bacteriophage T5. Virology. 2005;332:45–65. doi: 10.1016/j.virol.2004.10.049. [DOI] [PubMed] [Google Scholar]
- Watson LA, Phua SH, Bergquist PL, Lane HE. An Mr 29000 protein is essential for mini-F maintenance in E. coli. Gene. 1982;19:173–178. doi: 10.1016/0378-1119(82)90003-8. [DOI] [PubMed] [Google Scholar]
- Yarmolinsky MB, Sternberg N. Bacteriophage P1. In: Calendar R, editor. The Bacteriophages. New York, N.Y: Plenum Publishing Corporation; 1988. pp. 291–438. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.