

Summary
Clearance of pathogens or tumor cells by antibodies traditionally requires both Fab and Fc domains of IgG. Here we show the Fc domain of IgG alone mediates recognition and clearance of herpes simplex virus (HSV1) infected cells. The human natural killer (NK) cell surface is naturally coated with IgG bound by its Fc domain to the Fcγ receptor CD16a. NK cells utilize the Fc domain of bound IgG to recognize gE, an HSV1-encoded glycoprotein that also binds the Fc domain of IgG but at a site distinct from CD16a. The bridge formed by Fc domain between the HSV1-infected cell and the NK cell results in NK cell activation and lysis of the HSV1-infected cell in the absence of HSV1-specific antibody in vitro, and prevents fatal HSV1 infection in vivo. This mechanism also explains how bacterial IgG-binding proteins regulate NK cell function, and may be broadly applicable to Fcγ receptor-bearing cells.
Keywords: NK cells, herpes virus, HSV1, ADCC, CD16a, gE, protein A, protein G
Introduction
Natural killer (NK) cells are innate lymphoid cells capable of directly recognizing cancer cells and virally infected cells without prior antigen exposure (Orr and Lanier, 2010). The functional status of NK cells is regulated by signal inputs from cytokines and a wide variety of NK cell activating or inhibitory receptors, for which many viral or bacterial proteins are natural ligands (Orr and Lanier, 2010). Almost all circulating NK cells express the low affinity Fcγ receptor (FcγR) IIIA/CD16a (CD16a hereafter), which is required for antibody dependent cell-mediated cytoxicity (ADCC) (Nimmerjahn and Ravetch, 2008). CD16a is a type I transmembrane protein whose extracellular domain binds the Fc domain of IgG (IgGFc) at the hinge region (Sondermann et al., 2000) and its transmembrane helix and intracellular domain couple with the signal transducer CD3ζ (Anderson et al., 1990; Lanier et al., 1989). In classical ADCC, polyvalent antigen-antibody complexes bind CD16a and consequently cluster CD3ζ, whose subsequent phosphorylation results in the activation of NK cells and lysis of antibody-coated target cells (O’Shea et al., 1991). Monomeric binding of CD16a by IgGFc, however, is not able to activate NK cells (Metzger, 1992). Upon activation, human NK cells undergo dramatic phenotypic and functional changes, including expression of early activation marker CD69 (Testi et al., 1994) and degranulation marker CD107a (Alter et al., 2004), loss of CD16a and adhesion molecule CD62L (Romee et al., 2013), secreting cytokine IFN-γ and lysing target cells by releasing pre-stored perforin and granzyme B (Orr and Lanier, 2010)
Human NK cells constitute the first line of defense against infection with herpes simplex virus (HSV1). While HSV1 infection in a heathy population is usually self-limited and shows no or very mild symptoms (Gilden et al., 2007), patients with NK cell deficiencies can suffer severe, recurrent, and sometimes fatal HSV1 infection (Biron et al., 1989), suggesting a critical role for NK cells in control of HSV1 infection. The HSV1 genome contains 84 open reading frames, encoding 74 unique viral proteins (Szpara et al., 2010); only few genes have been studied for their roles in modulating the function of NK cells with some controversial results (Bishop et al., 1983; Chisholm et al., 2007; Fitzgerald-Bocarsly et al., 1991; Huard and Fruh, 2000; Imai et al., 2013; Lopez-Guerrero et al., 1988). How NK cells mechanistically recognize and clear primary HSV1 infection (i.e., before antigen specific immunity is generated) is unclear. Given the increasing importance of HSV1 in the immunotherapy of cancer (Andtbacka et al., 2015) and the lack of approved vaccines for most human pathogens in the herpesviridae family (Gilden et al., 2007), we set out to systematically dissect the contribution of individual HSV1 genes to NK cell activation.
We developed a cytotoxicity assay, Differential Cytolysis Mediated by Ectopic Gene Expression (DC-MEGE), and screened HSV1 genes for their ability to modulate human NK cell function when individually expressed in human glioma (brain tumor) cells. We found that HSV1 glycoprotein E (gE), which can bind IgGFc, activated human NK cells through direct interaction. We found that IgGFc can bridge gE and CD16a to form the gE-IgGFc-CD16a ternary complex, which conveyed the activation signal for NK cells. In vivo infusion of human IgG1Fc fragments alone protected mice from lethal HSV1 infection in a manner dependent on NK cells and gE, as did other human IgG1 therapeutic antibodies not targeting any HSV1 antigens. Furthermore, we found bacterial IgGFc binding proteins also activate NK cells through the IgGFc-mediated bridging. Overall, we provide evidence for a basic mechanism by which immune cells expressing FcγR recognize certain primary viral and bacterial infections expressing Fc-binding proteins via bridging mediated by IgGFc in the absence of antigen-specific antibodies or prior sensitization.
Result
HSV1 gE is a human NK cell activator
We chose glioma as the target cells for HSV1 infection and NK cell activation for three reasons: (1) HSV1 is neurotropic virus and has been exploited for treating glioma; (2) primary NK cells are relatively inert to native glioma cells, (3) HSV1-infected glioma causes a rapid migration of NK cells and strong NK activation (Alvarez-Breckenridge et al., 2012). We developed DC-MEGE to measure how NK cells respond to glioma cells expressing a single HSV1 gene (Figure 1A, and S1A-S1D). Each HSV1 gene was cloned upstream of the “self-cleaving” T2a sequence and green fluorescence protein (GFP) in the lentiviral expression vector called pCDH. Therefore, GFP reports the expression of viral proteins (Figure S1A) (Szymczak et al., 2004). Following transfection, glioma cells were divided into two equal portions, one cultured alone and the other cultured with primary NK cells (Figure S1B). The percentage of GFP+ living glioma cells were recorded 5 hours later in parallel as GFP(+NK)% when NK cells are present, or GFP(−NK)% when glioma cells are cultured alone. As shown in the sample analysis result (Figure S1C and S1D), DC-MEGE is an unbiased assay because: (1) following transfection GFP+ and GFP− cells are treated under the same condition and (2) cytotoxicity of DC-MEGE is measured by relative change of percent GFP and not affected by initial transfection efficacy. Applying the DC-MEGE assay, we screened 65 HSV1 genes and found that glioma cells expressing UL12, UL30, Us3, Us8 and Us12 were more susceptible to NK cell cytolysis, while expression of UL48, Us5, or Us6 made glioma cells resistant to NK cell cytolysis (Figure 1B, S1C and S1D).
Figure 1. DC-MEGE identifies HSV1 gE as an NK cell activating molecule.
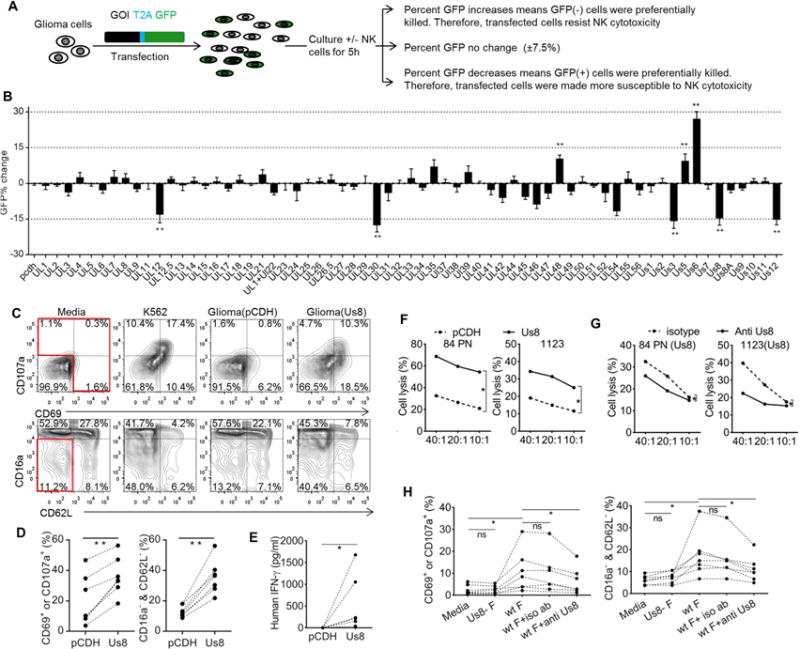
(A) Flow diagram of the DC-MEGE assay. (B) DC-MEGE results for all 65 HSV1 genes (mean ± sem, n≥4). (C) Phenotype of primary human NK cells from a representative normal donor after a 7 hour culture in media, with K562 cells (positive control), or transfected glioma cells. This was repeated with 7 individual normal donors, and a summary of the percentages of NK cells gaining the expression of CD69 or CD107a, or NK cells losing both CD16a and CD62L is provided in (D). (E) Human primary NK cells were treated as in c for 20 hours and IFNγ production was measure at 20 hours of culture by ELSIA (n=5, mean of triplicates). (F) Cytotoxicity of primary human NK cells against transfected human glioma cell lines at the specified effector: target ratio (x-axis). (G) Cytotoxicity of primary human NK cells against glioma cells expressing Us8 in the presence of isotype or mouse anti-Us8 specific antibody. (H) Summary of phenotypical changes of primary human NK cells after culturing for 7 hours in plates precoated with inactivated pure viruses. In some cases, isotype or mouse Us8-specific antibody was added into plates to block Us8 (n=5–7). Each dotted line in d, e and h links data acquired from the same donor. * p<0.05, ** p<0.01. Please see also Figure S1.
HSV1 Us8 encodes gE, which alone is a low affinity human IgGFc-interacting protein, binding human IgG1, IgG2 and IgG4 at the CH2-CH3 interface (Sprague et al., 2006). Previous studies proposed that gE can compete with human Fcγ receptors (including CD16a) to bind IgGFc, thereby inhibiting NK cell activation via antibody dependent cellular cytotoxicity (ADCC) (Corrales-Aguilar et al., 2014; Dubin et al., 1991; Frank and Friedman, 1989). However, our results showed that expression of gE activated NK cells and stimulated lysis of glioma, we thus focused on HSV1 Us8 to resolve how gE modulates NK cells mechanistically.
K562 cells are leukemia cells negative for MHC I molecule and widely used as a positive control for NK cell activation (Lozzio and Lozzio, 1979). A characteristic activating phenotype, including the increase of CD69 and CD107a and the loss of CD62L and CD16a, is shown in human NK cells cultured with K562 or glioma cells expressing Us8 (referred as glioma Us8 hereafter) (Figure 1C). Because the expression of CD69 and CD107a in activated NK cells were highly correlated (Figure S1E and S1F), we chose to combine CD69 and CD107a when summarizing staining results (Figure 1D). This activating phenotype was barely shown on NK cells cultured with glioma cell transfected with an empty pCDH vector (glioma pCDH hereafter) (Figure 1C and 1D). Culture with glioma Us8 also induced NK cells to produce IFN-γ (Figure 1E). Consistent with the DC-MEGE result, the transfection of Us8 and consequent expression of gE also sensitized both a proneural (84PN) and a mesenchymal 1123 (Mao et al., 2013) human glioma cell line to NK cytotoxicity (Figure 1F).
To answer whether gE activated NK cells directly or indirectly, we treated glioma Us8 with isotype or gE-specific mouse monoclonal antibody to block gE, and found that NK cytotoxicity towards glioma Us8 was attenuated by gE-specific antibody (Figure 1G). Furthermore, we purified wild type (wt) HSV1 F strain virus, of which gE is a major protein component, and coated plates with detergent-inactivated WT virus. NK cells cultured in plates coated with WT virus also showed the activating phenotype. This appeared to be dependent on gE because a mutant HSV1 F strain with targeted deletion of Us8 (Us8−) (Suenaga et al., 2014) was not able to activate NK cells in the same experimental setting. Further, blocking WT virus with gE-specific antibody also aborted NK cell activation (Figure 1H and S1G). Taken together, these results demonstrated HSV1 gE activated human NK cells through direct interaction.
Human IgG links gE and NK cell activation
HSV1 gE can heterdimerize with glycoprotein I (gI), and the resultant gE-gI complex binds human IgG with much higher affinity than gE does alone (Johnson et al., 1988; Sprague et al., 2006). The HSV1 gI is encoded by the Us7 gene and gI alone does not bind human IgG (Johnson et al., 1988). Human NK cells cultured with glioma cells expressing Us7 (glioma Us7, hereafter) did not undergo activation (Figure 1B, S1C and S1D). Because the IgG binding property of gE has been linked with an immune evasion role by several studies (Corrales-Aguilar et al., 2014; Dubin et al., 1991; Frank and Friedman, 1989), our first thoughts were that NK cell activation and IgG binding were two independent functions and fulfilled by different structural domains of gE. If this was true, glioma expressing both Us7 and Us8 (glioma Us7+Us8, hereafter), with formation of the high affinity IgG-binding gE-gI complex, should cause no stronger NK cell activation than glioma Us8 alone, assuming both have equal gE expression. We found, however, that glioma Us7+Us8 activated NK cells much more potently than glioma Us8 although both express similar amount of gE (Figure 2A, 2B, and S1H), suggesting NK cell activation by gE involves the IgG binding function of gE.
Figure 2. IgG links gE and NK cell activation.
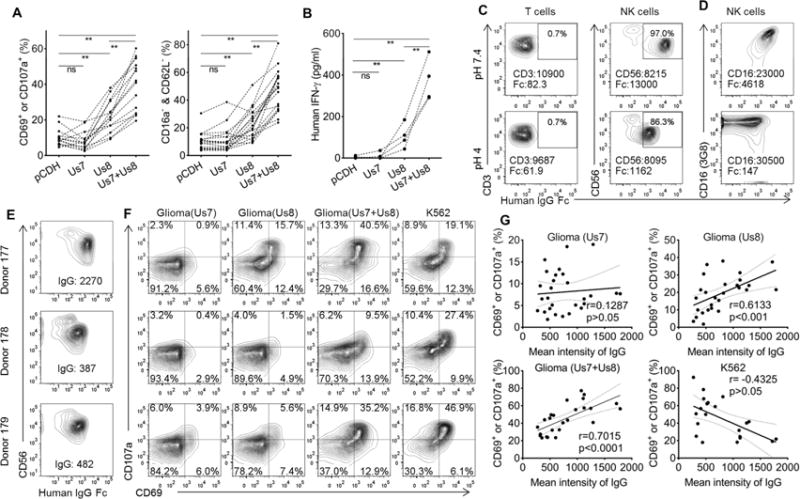
(A) Summary for the phenotypical changes of primary human NK cells after culturing with transfected glioma cells for 7 hours (n=14). (B) Primary human NK cells were treated as in (A), supernatants were collected at 20 hours and measured for human IFNγ (mean of triplicates, n=4). (C and D) Human PBMC were washed with pH7.4 or pH4 media, and subsequently stained with lineage markers and mouse anti human IgGFc antibody. Mean intensity of cell markers and Fc are shown in figures. One representative of at least five donors is shown. (E-G) NK cells from healthy human donors were stained for surface IgG immediately after isolation (E), and stained for CD69 and CD107a after coculturing with specified cells with for 7 hours (F) Percentage of NK cells positive for CD69 or CD107a after coculturing with specified cells were plotted against surface IgG intensity of corresponding human donors. Correlation of surface IgG and NK cell activation were calculated using linear regression (G) (n≥20) Each dotted line in a-c links data acquired from the same donor. * p<0.05, ** p<0.01. Please see also Figure S1.
Human IgG was not added to the glioma-NK cell co-culture thus far, however, we found that human IgG molecules were naturally present on the surface of primary human NK cells (Figure 2C). Washing NK cells briefly with acidic media decreased surface IgG (Figure 2C) and increased binding of an anti-human CD16a antibody (3G8) that competes with human IgG for the same binding site on CD16a (Perussia et al., 1984) (Figure 2D), demonstrating that IgG molecules are anchored on human NK cells via CD16a. Primary NK cells from a variety of healthy human donors had very different amount of surface IgG (Figure 2E) and their response to stimuli varied (Figure 2F). We found that the response of human NK cells towards glioma Us8 (measured by the percentage of CD69+ or CD107a+ NK cells) directly correlated with the surface density of IgG; and the correlation became even stronger when NK cells were cultured with glioma Us7+Us8 (Figure 2G). In contrast, the response of NK cells to K562 cells showed no correlation with the surface density of IgG (Figure 2G). Taken together, surface IgG on human NK cells links gE and NK cell activation.
CD16a, IgG1Fc and HSV1 gE form a ternary complex
Previous studies assumed that IgG bound by gE could no longer bind human Fcγ receptors due to competition or steric hindrance, and thus contributing to immune evasion (Corrales-Aguilar et al., 2014; Dubin et al., 1991; Frank and Friedman, 1989). However, CD16a binding sites on IgG1Fc are located far away from the CH2-CH3 interface where gE binds IgG1Fc, and structure modeling using the known gE-IgG1Fc (Sprague et al., 2006) and CD16a-IgG1Fc (Sondermann et al., 2000) crystal structures supported that gE and CD16a could bind the same IgG1Fc molecule without interfering with each other (Figure 3A).
Figure 3. Structural basis for the gE-IgG-CD16a interaction.
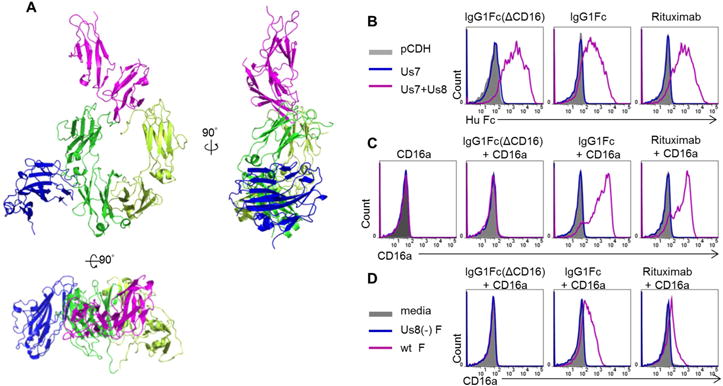
(A) Model structure of gE-IgG1Fc-CD16a ternary complex. Front view, side view and top view are shown respectively. CD16a is shown as magenta, gE is shown as blue, two monomers of IgGFc dimer are shown as green and lime. (B and C) Flowcytometry-based method to detect the binding of different human IgG1Fc products to transfected glioma cells (B), and the binding of CD16a ectodomain to transfected glioma cells in the absence or presence of different human IgG1Fc products (C). Below each histogram is corresponding graphic illustration and binding result. (D), Binding of CD16a ectodomain to infected glioma cells in the presence of different human IgG1Fc products. Data in (B–D) are the representative of at least 4 repetitions. Please see also Figure S2.
To validate the existence of such a CD16a-IgG1Fc-gE complex, we tested the binding between gE, IgG1Fc and CD16a using a flow cytometry-based method (Figure 3B and 3C). IgG1Fc(ΔCD16) is a recombinant human IgG1Fc fragment without the CD16a binding sites; while IgG1Fc has intact CD16a binding sites and rituximab is a humanized IgG1 monoclonal antibody against human CD20 that also has intact CD16a binding sites (Edwards et al., 2004). All three forms of IgG1Fc could bind glioma Us7+Us8 but none bound glioma Us7, demonstrating that Fc specifically binds gE and that the CD16a binding sites on IgG1Fc are not involved in binding gE (Figure 3B). The CD16a ectodomain did not directly bind either glioma Us7 or glioma Us7+Us8, however the CD16a ectodomain bound glioma Us7+Us8 only when IgG1Fc or rituximab was present but not when IgG1Fc (ΔCD16) was present (Figure 3C). We also performed similar CD16a binding tests using glioma cells infected with Us8− or WT HSV1, and showed gE was required for capturing IgG1Fc and CD16a (Figure 3D). Taken together, these results demonstrate the CD16a-IgG1-gE complex exists.
Staphylococcus aureus (S.A) and group G streptococcus also produce human IgG-binding proteins (protein A and protein G, respectively). Like HSV1 gE, protein A and protein G bind IgGFc mainly at the CH2-CH3 interface (Deis et al., 2015; Sauer-Eriksson et al., 1995), and structural modeling supported that IgG occupied by CD16a could still bind protein A and protein G (Figure S2A and S2B). In support of the structure modeling, we found that protein A and protein G could bind primary human NK cells. A prior brief acidic wash of NK cells, which removes surface IgG bound to CD16a (Figure 2D), reduced protein A and protein G binding (Figure S2C and S2D). These results further supported that, contrary to conventional assumptions (Deis et al., 2015), surface IgG molecules coated on NK cells via CD16a have a freely accessible CH2-CH3 interface to bind gE, protein A and protein G.
The CD16a-IgG-gE complex is essential for NK cell activation by gE
To test whether the formation of CD16a-IgG-gE complex is responsible for NK cell activation by gE, we incubated primary human NK cells with an excess of soluble protein A or protein G to occupy all CH2-CH3 binding sites prior to culturing the NK cells with different stimuli. Preincubation of NK cells with either soluble protein A or protein G did not change human NK cell activation when co-cultured with K562 cells, or cytokines (IL12+IL18) that activate NK cells through STAT4, STAT3 and MAPK pathways (Kalina et al., 2000; Watford et al., 2004) (Figure S3A–3C). However, blocking with soluble protein A or G completely inhibited phenotypic changes, cytotoxicity and IFN-γ production of NK cell induced by co-culture with glioma Us8 and glioma Us7+Us8 (Figure 4A–4D). Additionally, plates coated with pure and inactivated wt HSV1 could no longer activate NK cells that were pre-incubated with soluble protein A or protein G (Figure S3D and S3E).
Figure 4. Human IgG bridging is essential for NK cell activation by gE.

(A and B) Phenotypes of primary human NK cells, which were first incubated with media, or media supplemented with protein A or protein G for 30 minutes, followed by culturing with different stimuli for another 7 hours. One representative donor (A) and statistical summary for 7–8 donors (B) are provided. (C) Cytotoxicity of primary human NK cells, which were pre-incubated with media, or media supplemented with protein A or protein G for 30 minutes, against transfected glioma cells. (D) NK cells were treated as in (A), IFNγ secretion was accessed at 20 hours of culture (mean of triplicates, n=5). (E and F) Summary for the phenotypical changes of primary human NK cells after 7 hours culture with infected glioma cells (E) or transfected glioma cells (F) in the absence or presence of human IgG1Fc (n=7–9). (G and H) Cytotoxicity of primary human NK cells against infected glioma cells (G) or transfected glioma cells (H) in the absence or presence of human IgG1Fc or other antibodies. (I, J) Phosphorylation of CD3ζ in NK cells following 1 hour stimulation. Representative result (I) and statistical summary (n=7) (J). Each dotted line in (B, D, E, F and J) links data acquired from the same donor. (C, G and H) were repeated at least 3 times with different donors. * p<0.05. * * p<0.01. Please see also Figure S3
Experiments described above were all conducted without adding extra human IgG, and NK cell activation by gE was mediated by surface IgG naturally present on primary NK cells (Figure 2C). During primary HSV1 infection in vivo, infected cells are likely to be coated with human IgG due to the expression of gE and the abundance of IgG in the human body. To test whether the non-immune IgG coating on HSV1 infected glioma or glioma Us8 or glioma Us7+Us8 could provide additional anchoring and activating sites for NK cells, we treated each with IgG1Fc before culture with NK cells. The presence of IgG1Fc fragments indeed further enhanced activation of NK cells by infected or transfected glioma in a gE-dependent manner (Figure 4E, 4F, S3F and S3G).
Anti-HSV1 IgG increased NK cytotoxicity to glioma cells infected with HSV1 WT F strain or Us8− F strain (classical ADCC), but human IgG1Fc and an antibody targeting an irrelevant antigen (i.e., rituximab) increased killing of glioma cells infected with WT F strain but not Us8− F strain (Figure 4G). Similarly, NK cytotoxicity against glioma Us8 and glioma Us7+Us8 cells was also increased when IgG1Fc was present (Figure 4H). Although CD3ζ phosphorylation is a very transient event, we were able to capture specific CD3ζ phosphorylation induced by glioma Us7+Us8 and positive control H2O2 stimulation (Figure 4I and 4J). CD3ζ phosphorylation was not induced by either glioma Us7 or cytokines IL12+IL18, demonstrating the specificity of this staining (Figure 4I and 4J). Taken together, CD16a-IgG-gE is a functional complex fully responsible for the NK cell activation by target cells expressing HSV1 gE.
IgG bridging promotes the clearance of HSV1 infection in vivo
HSV1 gE does not bind mouse IgG (Chapman et al., 1999), however, mouse FcγR binds human IgG with high affinity as reported by others (Ober et al., 2001). We also found injecting mice with human IgG1Fc or whole humanized antibody rituximab, while not affecting the phenotype of mouse NK cells, resulted the coating of human IgG on mouse NK cells (Figure S4A). Therefore, we propose that provision of human IgG or human IgGFc should be able to bridge mouse NK cells and HSV1 infected cells, and promote clearance of HSV1 infection both in vitro and in vivo. Consistent with this hypothesis, we found NK cells isolated from C57BL/6 and BALB/c mice displayed enhanced cytotoxicity towards glioma Us7+Us8 when human IgG1Fc was present (Figure 5A). Using a HSV1 F strain expressing luciferase (Zerboni et al., 2013) we demonstrated that administration of rituximab promoted clearance of HSV1 infection in vivo (Figure 5B). In a lethal HSV1 infection model, rituximab alleviated the symptoms of infection (Figure 5C and S4B), and provided mice complete protection from lethal HSV1 infection (Figure 5D). Importantly, this protection is not dependent on Fab of rituximab because human IgG1Fc and daratumumab (a human IgG1 antibody directed against human CD38) demonstrated a similar protection against HSV1 infection (Figure 5C, 5D and S4B).
Figure 5. IgG bridging mediated clearance of HSV1 infection in vivo.
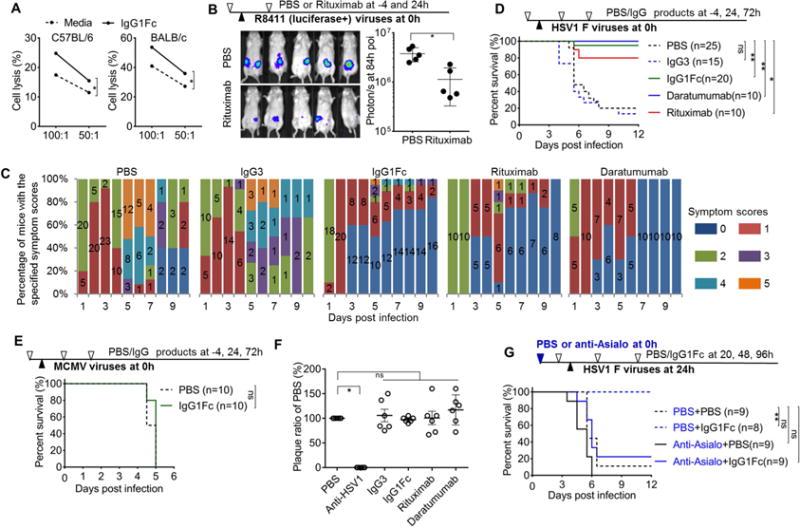
(A) Cytotoxicity of primary NK cells from two different mouse species against glioma Us7+Us8 in the presence or absence of human IgG1Fc. Each dot is mean of triplicates, and data represent two independent experiments. (B) HSV1 virus load at 84 hours post infection was measured by bioluminescence (n=5). (C and D) Symptom (C) and survival curve (D) of mice receiving the specified injection regimens composed of HSV1 and different human IgG products. Clinical scores of mice are represented by colored columns, in which is the number of mice with specified symptom score (see methods for detail). (E) Survival curve of mice receiving lethal MCMV infection and human IgG1Fc. (F) HSV1 F strain viruses were incubated with PBS plus different human IgG products for 30min at room temperature, and their infectivity was tested using plaque assay on BHK cells. All numbers are normalized to PBS control. (G) NK cell depletion by anti-asialo aborted the protection of lethal HSV1 infection by human IgG1Fc. * p<0.05, ** p<0.001. Please see also Figure S4
HSV1 gE is required for the cell-to-cell spread of HSV1 in vivo (Polcicova et al., 2005), and Us8− HSV1 F strain did not cause productive infection in vivo (data not shown), therefore we could not use the Us8− F strain in vivo to directly confirm the dependence on gE for this type of protection. However, human IgG3, which does not bind HSV1 gE (Sprague et al., 2006), failed to provide any protection against HSV1 infection (Figure 5C, 5D and S4B). Furthermore, human IgG1Fc was not able to protect mice from lethal infection of mouse cytomegalovirus (MCMV) (Figure 5E), another member of the herpesviridae family, which caused a similar progressively fatal infection as seen with HSV1 (Figure 5D) but did not produce a protein able to bind human IgG (Figure S4C). These results support the notion that the IgG1Fc-mediated protection of HSV1 infection is dependent on the interaction of IgG1Fc and gE.
Although human IgG1Fc, daratumumab and rituximab can all bind HSV1 through gE, this binding had no direct effect on the infectivity of HSV1 in vitro (Figure 5F), indicating the protective role of these reagents against HSV1 infection in vivo must be executed by immune cells. When NK cells were depleted by anti-asialo GM1 antibody (Kasai et al., 1981) during the infection (Figure S4D), human IgG1Fc failed to protect mice from lethal HSV1 infection (Figure 5G). These results demonstrate that human IgG1Fc functions as a bridge, engaging viral-infected cells and NK cells, which provide robust protection against primary HSV1 infection before the adaptive immune system generates a pathogen-specific antibody. We focused only on NK cells in this study, however, human monocytes and macrophages express high affinity FcγRI/CD64, which binds IgG in the same fashion as CD16a (Kiyoshi et al., 2015). We therefore do not exclude the possibility of their involvement in clearance of HSV1 infection through IgGFc-mediated bridging.
Taken together, we have described a previously unappreciated mechanism by which NK cells recognize HSV1 infected cells utilizing IgGFc to form a bridge between CD16a and the Fc-binding protein gE, resulting in NK cell activation and viral clearance (Figure S4E-S4H). It differs from the classical IgG function of ADCC by not requiring any antigen-specific antibody, and limits virus infection before the establishment of adaptive immunity. We propose to call this type of NK cell recognition, activation and cytotoxicity Fc bridged cell-mediated cytotoxicity (FcBCC).
Bacterial IgG binding proteins activate NK cells through crosslinking surface IgG
In the 1970s and 1980s, Protein A was extensively studied for its ability to induce “natural killing activity” of peripheral blood mononuclear cells (PBMC). The consensus from these earlier studies was that the natural killing activity of PBMC could only be increased when PBMC were exposed to soluble protein A for at least 20 hours (Catalona et al., 1981; Kasahara et al., 1981; Kasahara et al., 1982; Kay et al., 1977; Olinescu et al., 1983; Patel et al., 1981; Ratliff et al., 1981). However, whether the enhanced “natural killing activity” is contributed by NK cells, or whether protein A activates NK cells directly or indirectly through other mononuclear cells remained unknown.
Given our demonstration that soluble protein A and protein G bind to IgG molecules presented on the surface of NK cells (Figure S2C and S2D) we asked if this IgG bridge might also be responsible for the activation of NK cells by bacterial IgG binding proteins. However, we did not observe activation of purified human NK cells by soluble protein A or protein G even after 20 hours of culture (Figure 6A–6C). NK cell activation through CD16a minimally requires two CD16a binding sites physically close enough so as to cluster CD3ζ and initiate phosphorylation (Metzger, 1992). HSV1 gE expressed on cells or coated in plates are de facto multimers, able to cluster many CD16a molecules and initiate activation (Figure 2A and 2B). We therefore assessed the ability of multimeric forms of protein A or protein G, as would exist in bacteria or as adhered to a plate, to activate NK cells.
Figure 6. Bacterial IgG binding proteins activate human NK cells through crosslinking IgG bridge.
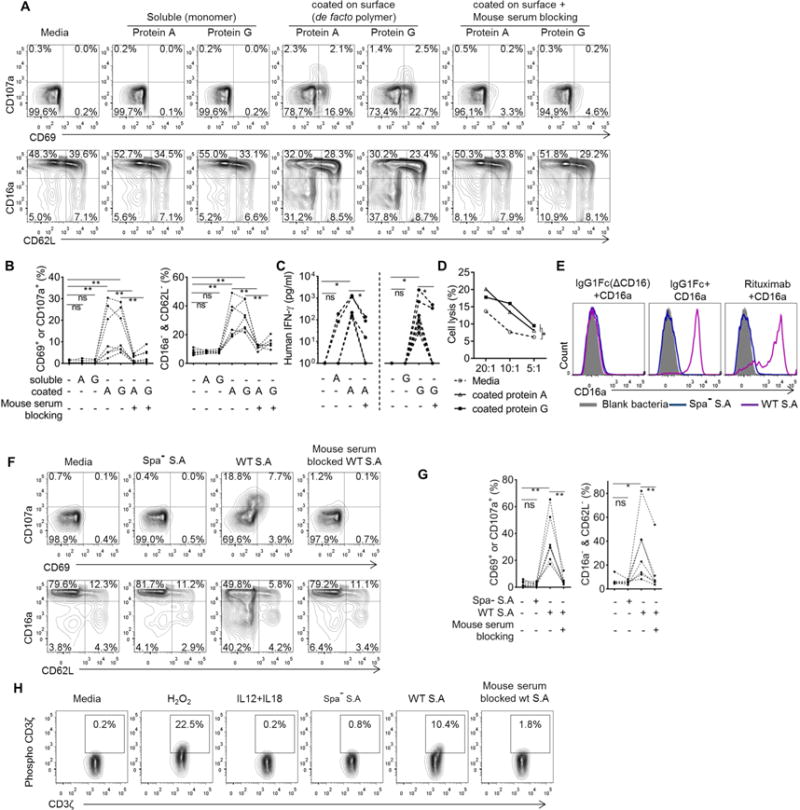
(A and B) Phenotypes (A) and statistical summary (B) of primary human NK cells after 7 hours culture with different forms of bacterial IgG binding proteins (n=7). (C) Human NK cells were treated as in (A), and IFNγ concentration was measured by ELISA at 20th hour of culture (mean of triplicates, n=6). Each dotted line in (B) and (C) links data acquired from one donor. (D) Human NK cells were cultured in plates coated with media, protein A or protein G for 30min and cytotoxicity was assessed against glioma cells. Numbers are mean of triplicates and data represent two independent experiments. (E) S.A bacteria were incubated first with different human IgG products, and subsequently with biotinylated CD16a. The binding of CD16a to bacteria was detected with fluorescent streptavidin. (F and G) Phenotypes (F) and statistical summary (G) of primary human NK cells after culturing with bacteria for 7 hours (n=7). (H), Phosphorylation of CD3ζ after exposing primary human NK cells to stimuli for 1h. (E) and (H) were repeated minimally on 4 donors. * p<0.05, ** p<0.01. Please see also Figure S5
Purified primary human NK cells were indeed activated after culturing in plates precoated with protein A or G (Figure 6A and 6B), producing IFN-γ (Figure 6C) and showing enhanced NK cytotoxicity (Figure 6D). These NK cell functional enhancements were abrogated when protein A- or G-coated plates were blocked with mouse serum (Figure 6A–6C), which prevented all potential interactions between protein A or protein G and human IgG on the surface of NK cells. Furthermore, preincubating NK cells with either soluble protein A or soluble protein G inhibited the activation of NK cells cultured in plate coated with protein A or G, demonstrating the essentiality of multimeric binding for NK cell activation and a common mechanism shared by protein A and protein G to activate NK cells (Figure S5A and S5B).
We next tested if S.A bacteria could activate NK cells by culturing human primary NK cells with WT S.A Newman strain and a protein A deficient Newman strain (Spa−) (Patel et al., 1987) (Figure S5C). We tested the binding of CD16a to S.A bacteria in the presence of human IgG (rituximab), IgG1Fc, or IgG1Fc (ΔCD16), and confirmed that a CD16a-IgGFc-protein A complex formed (Figure 6E). We found that WT S.A induced activation of NK cells, which was dependent on protein A because NK cell activation was not induced by Spa− strain or by WT S.A blocked with mouse serum (Figure 6F and 6G). NK cell activation by wt S.A was also blocked by pretreatment with soluble protein A or protein G (Figure S5D). CD3ζ phosphorylation of primary human NK cells cultured under these conditions completely mirrored the phenotypic results (Figure 6H). Taken together, WT S.A, polymerized protein A and protein G use the same IgG-bridging mechanism as HSV1 to activate NK cells.
Although protein A has been shown to bind tumor-necrosis factor-α receptor 1 (TNFR1) and induce inflammatory signals via the NF-κB pathway (Gomez et al., 2004), TNFR1 should not be involved in NK cell activation by protein A, because (1) human NK cells barely express TNFR1 as previously described (Liu and Tang, 2014) and validated here (Figure S5E), (2) soluble protein A does not activate NK cells, and (3) protein G does not bind TNFR1 (Gomez et al., 2004), and (4) we showed here polymerized protein G and polymerized protein A used the same mechanism to activate NK cells. However, TNFR1 was expressed on monocytes and granulocytes (Figure S5E), which therefore can be activated by soluble protein A, and in turn activate bystander NK cells, potentially explaining why prolonged (20 h) protein A treatment was required to increase “natural killing activity” of PBMC (Catalona et al., 1981; Kasahara et al., 1981; Kasahara et al., 1982; Kay et al., 1977; Olinescu et al., 1983; Patel et al., 1981; Ratliff et al., 1981). However, we did not infect mice with WT or Spa− S.A to study NK cell activation by polymerized protein A in vivo. Lipid, protein and nucleic acid components of S.A can all elicit strong innate immunity signals from monocytes, neutrophils, endothelial cells, epithelial cells et al (Fournier and Philpott, 2005). Protein A deficiency alone would not be enough to nullify S.A’s ability to activate NK cells, because bystander activation of NK cells cannot be avoided in the in vivo settings. Taken together, our study demonstrates that bacterial IgG binding proteins can directly activate NK cells through IgGFc-mediated bridging.
Discussion
Here we have reported a previously unappreciated mechanism of innate immune cell recognition of and response to a primary viral infection mediated only by the Fc domain of IgG. These findings were supported by crystal structure as well as in vitro and in vivo functional validation. We demonstrated that this mechanism of recognition and activation is identical among three different pathogens and their genetically distinct Fc-binding proteins. Our work suggests that during primary HSV1 infection when anti-HSV1 antibody is not yet generated, NK cells can utilize what we have now termed as FcBCC to recognize and then clear HSV1-infected cells. This result is consistent with the observation that most primary HSV1 infections in man are clinically asymptomatic and/or self-limited. Protein A has long been proposed as a virulent factor for S.A, and Spa− S.A causes a milder symptoms in mice than WT S.A (Palmqvist et al., 2002). Our findings that plate-coated protein A and wt S.A activated NK cells, and Spa− S.A did not activate NK cells, provide a mechanistic explanation for this phenotype.
It is also highly likely that FcBCC is at least in part responsible for the rapid NK cell clearance of oncolytic HSV1 in the setting of malignant glioma (Alvarez-Breckenridge et al., 2012), and clearance of infection by many other members of the herpesviridae family encoding similar or identical Fc-binding proteins (Corrales-Aguilar et al., 2014; De Miranda-Santos and Campos-Neto, 1981; Litwin et al., 1992; Loukas et al., 2001; Olson et al., 1997; Sprague et al., 2008). Given the presence of IgG binding proteins in many different pathogens, FcBCC may have broad implications in controlling infectious diseases. In addition, protein A and protein G are widely used in the industry for antibody purification; our findings may provide insights for other uses for these two proteins.
HSV1 gE complexes have been shown to participate in “antibody bipolar bridging”, whereby a single anti-HSV1 specific IgG antibody simultaneously binds to a HSV1-antigen using its Fab domain and to gE via its Fc domain (Frank and Friedman, 1989). It has been proposed that such antibody bipolar bridging could block access of the Fc portion of the anti-HSV1 specific IgG antibody to FcγR expressed on innate immune effector cells, and thereby reduce classical ADCC and presumably provide a mechanism for immune evasion following HSV1 infection (Corrales-Aguilar et al., 2014; Dubin et al., 1991). However, previous studies suggesting an inhibition of NK cell activation by HSV1 gE have been conducted in the presence of anti-HSV1 specific antibody (Corrales-Aguilar et al., 2014; Dubin et al., 1991; Frank and Friedman, 1989). These studies all relate to classical ADCC, one of the strongest NK cell activation signals. Considering that the configuration of gE-IgGFc may not be as optimal as that of the antigen-antibody complex to be recognized by CD16a, it is not surprising that an immunosuppressive role has been assigned to gE under such conditions. The function of HSV1 gE binding non-immune IgG under the condition of primary viral infection, however, was not pursued in these studies.
Our knowledge regarding other functions of gE other than IgG-binding is limited, although few studies showed that gE was required for the spread of HSV1 between cells in vivo (Collins and Johnson, 2003). Why does HSV1 encode gE to limit the infection of human host by itself? Viruses are completely dependent on host cells for replication and transmission. Weakening the host can also weaken the virus, and to the extreme, extinction of host leads to the extinction of the virus. From the standpoint of evolution, viruses can not only cause disease and do harm without benefiting the host in some way. A recent study lends support to a less popular notion, suggesting that viral infection can compensate the host with a stronger immune response (Furman et al., 2015). In the current study, we found gE limited the uncontrolled replication and spread of HSV1 during its lytic stage of infection. Under this circumstance, the human host is not overwhelmed and HSV1 infection can proceed to its latent stage of infection. Consequently, gE may play a beneficial role during HSV1 infection in that it allows the virus to maintain a constant existence in the human population.
Our study also establishes a functional role for surface IgG on the NK cell and other immune cells. Each monomeric IgG has only one FcγR binding site and thus prevents surface coated IgG from binding another FcγR thereby causing autoimmune disease (Sondermann et al., 2000). CD16a binds IgG with low affinity, so there is an equilibrium between binding and disassociation, and only a certain percentage of CD16a on NK cells are occupied by IgG. As such, other CD16a molecules unoccupied by IgG and thus can be stained by the 3G8 mAb. The same phenomenon happens to host cells expressing gE following HSV1 infection. Therefore, FcBCC can be triggered bidirectionally: gE binds the IgG-CD16a complex, and unoccupied CD16a binds the IgG-gE complex.
It is noteworthy that gE, protein A and protein G share no sequence or structure similarity, yet each of these IgG-binding proteins can be recognized by the Fc domain of IgG that is bound to the surface of NK cells. Human cells may also express such IgG-binding proteins. Due to the importance of Fc-binding in mediating every facet of immune responses (Nimmerjahn and Ravetch, 2008), screening human proteins that are able to form a ternary complex with IgGFc and human FcγRs or complement may identify auto-antigens, define autoimmune diseases and the cause of Infusion reactions to therapeutic monoclonal antibodies, leading to the development of therapeutic interventions and improved antibody-based therapy.
STAR METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| BV421 anti-human CD3 (clone HIT3a) | BD | Cat# 740073 |
| APC-H7 anti-human CD14 (M5E2) | BD | Cat# 561384 |
| PE anti-human CD19 (clone HIB19) | BD | Cat# 555413 |
| PC7 anti-human CD56 (clone N901) | Beckman Coulter | Cat# A51078 |
| FITC anti-human CD69 (clone FN50) | BD | Cat# 555530 |
| PE anti-human CD62L (clone DREG-56) | BD | Cat# 555544 |
| V421anti-human CD107a (clone H4a3) | Biolegend | Cat# 328626 |
| APC/Cy7 anti-human CD16 (clone 3G8) | Biolegend | Cat# 302018 |
| FITC CD64 (clone 10.1) | BD | Cat# 555527 |
| PE Ms IgG1, κ (clone MOPC-21) | BD | Cat# 555749 |
| APC anti-human IgG (polyclonal) | Jackson ImmunoResearch | Cat# 209605098 |
| APC anti-human IFNgamma (clone B27) | BD | Cat# 554702 |
| Anti-HSV1 gE (clone 9H3) | abcam | Cat# Ab6510 |
| FITC anti-human CD3ζ (clone 6B10.2) | Biolegend | Cat# 644104 |
| APC anti-human CD3zeta (pY142) (clone K25-407.69) | BD | Cat# 558489 |
| Anti asialo gm1 | Biolegend | Cat# 146002 |
| PE anti-human CD120a (TNF-RI) (clone REA252) | Miltenyi Biotech | Cat# 130-106-360 |
| IgG1, Kappa from human myeloma plasma | Sigma | Cat# I5154-1MG |
| IgG3 Kappa | Sigma | Cat# I5654 |
| Immune globulin, GamaStan S/D | GRIFOLS | N/A |
| Rituximab | Genentech | N/A |
| Obinutuzumab | Janssen | N/A |
| Immunoglobulin G-Fc Fragment; Purified | Scripps Laboratories | Cat# I2724 |
| Bacterial and Virus Strains | ||
| HSV1 F strain | ATCC | Cat# VR-733 |
| HSV1 Us8- F strain | Suenaga et al., 2014 | N/A |
| R8411 (HSV1 F strain expresses luciferase) | Zerboni et al., 2013 | N/A |
| MCMV | Orr MT et al., 2010 | N/A |
| Staphylococcus newman strain (wt) | ATCC | Cat# 25904 |
| Staphylococcus newman strain (spa-) | Patel et al., 1987 | N/A |
| Biological Samples | ||
| Umbilical cord blood | American Red Cross, Columbus, OH, USA | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| FCGR3A (CD16a), 176 Phe/Val Recombinant Human Protein, His. AVI Tag, Biotinylated | Sinobiological | Cat# 10389-H27H1-B-25 |
| Recombinant Protein G | Thermo | Cat# 10-1200 |
| Protein G, Alexa Fluor® 488 conjugate | Thermo | Cat# P-11065 |
| Recombinant Protein A | Thermo | Cat# 21184 |
| Protein A, Alexa Fluor® 647 conjugate | Thermo | Cat# P-21462 |
| Human EGF | ThermoFisher | Cat# PHG0311 |
| Human basic FGF | ThermoFisher | Cat# PHG0021 |
| Human IL12 | Genetics Institute | N/A |
| Human IL-18 | R&D system | Cat# 9124-IL-050 |
| EasySep™ Human NK Cell Enrichment Kit | STEMCELL Technologies | Cat# 19055 |
| CD56 MicroBeads, human | Miltenyi Biotech | Cat# 130-050-401 |
| NK Cell Isolation Kit II, mouse | Miltenyi Biotech | Cat# 130-096-892 |
| LS Columns | Miltenyi Biotech | Cat# 130-042-401 |
| LD Columns | Miltenyi Biotech | Cat# 130-042-901 |
| Human IFN-γ ELISA Kit () | R&D Systems | Cat# SIF50 |
| Dead Cell Removal Kit | Miltenyi Biotech | Cat# 130-090-101 |
| PhosSTOP | sigma | Cat# 4906845001 |
| Phosflow™ Fix Buffer I, | BD | Cat# 557870 |
| Phosflow™ Perm Buffer III | BD | Cat# 558050 |
| Q5® High-Fidelity DNA Polymerase | NEB | Cat# M0491S |
| GeneJET Gel Extraction Kit | ThermoFisher | Cat# K0692 |
| DMEM | ThermoFisher | Cat# 11965118 |
| RPMI 1640 | ThermoFisher | Cat# 11875119 |
| DMEM/F12 | ThermoFisher | Cat# 10565-042 |
| heparin | sigma | Cat# H3393 |
| MycoAlert | Lonza | Cat# LT07-703 |
| Tryptic Soy Broth | Sigma | Cat# 22092-500G |
| D-Luciferin, Potassium Salt | GOLD BIOTECHNOLOGY | Cat# LUCK-1G |
| Basic Glial Cells Nucleofector™ Kit | LONZA | Cat# VPI-1006 |
| B-27® Supplement (50X), serum free | ThermoFisher | Cat# 17504044 |
| Protein A Magnetic Beads | NEB | Cat# S1425S |
| Critical Commercial Assays | ||
| Human IFN-γ ELISA Kit | R&D Systems | Cat# SIF50 |
| Experimental Models: Cell Lines | ||
| BHK cells | ATCC | Cat# CCL-10 |
| Vero cells | ATCC | Cat# CCL-181 |
| K562 | ATCC | Cat# CCL-243 |
| #83 | Mao et al. 2013 | N/A |
| #84 | Mao et al. 2013 | N/A |
| #1123 | Mao et al. 2013 | N/A |
| Experimental Models: Organisms/Strains | ||
| BALB/cJ | Jackson lab | Cat# 000651 |
| C57BL/6J | Jackson lab | Cat# 000664 |
| Oligonucleotides | ||
| HSV1 UL1 forward: GTCTACACTAGTATGGGGATTTTGGGTTGGGTCGGG |
this study | N/A |
| HSV1 UL1 reverse: GTCTACTTAATTAAGATGCGCCGGGAGTGGGGTCGTC |
this study | N/A |
| HSV1 UL2 forward: GTCTACACTAGTATGAAGCGGGCCTGCAGCCGAAG |
this study | N/A |
| HSV1 UL2 reverse: GTCTACTTAATTAAAACCGACCAGTCGATGGGTG |
this study | N/A |
| HSV1 UL3 forward: GTCTACACTAGTATGGTTAAACCTCTGGTCTCATAC |
this study | N/A |
| HSV1 UL3 reverse: GTCTACTTAATTAACTCGGCCCCCGAGGCCAGCATG |
this study | N/A |
| HSV1 UL4 forward: GTCTACACTAGTATGTCCAATCCACAGACGACCATC |
this study | N/A |
| HSV1 UL4 reverse: GTCTACTTAATTAAGGACCCCAAAAGTTTGTCTGCG |
this study | N/A |
| HSV1 UL5 forward: GTCTACACTAGTATGGCGGCGGCCGGCGGGGAG |
this study | N/A |
| HSV1 UL5 reverse: GTCTACTTAATTAAATATACAATGACCACGTTCGGATCG |
this study | N/A |
| HSV1 UL6 forward: GTCTACACTAGTATGACCGCACCACGCTCGCGG |
this study | N/A |
| HSV1 UL6 reverse: GTCTACTTAATTAATCGTCGGCCGTCGCGGCGGCCATCC |
this study | N/A |
| HSV1 UL7 forward: GTCTACACTAGTATGGCCGCCGCGACGGCCGAC |
this study | N/A |
| HSV1 UL7 reverse: GTCTACTTAATTAAACAAAACTGATAAAACAGCGACG |
this study | N/A |
| HSV1 UL8 forward: GTCTACACTAGTATGGACACCGCAGATATCGTGTGG |
this study | N/A |
| HSV1 UL8 reverse: GTCTACTTAATTAAGGCAAACAGAAACGACATCTTG |
this study | N/A |
| HSV1 UL9 forward: GTCTACACTAGTATGCCTTTCGTGGGGGGCGCGGAG |
this study | N/A |
| HSV1 UL9 reverse: GTCTACTTAATTAATAGGGTGCTAAAGTTCACCG |
this study | N/A |
| HSV1 UL10 forward: GTCTACACTAGTATGGGACGCCCGGCCCCCAG |
this study | N/A |
| HSV1 UL10 reverse: GTCTACTTAATTAACCAACGGCGGACGGTGCTGTAC |
this study | N/A |
| HSV1 UL11 forward: GTCTACACTAGTATGGGCCTCTCGTTCTCCGGGGC |
this study | N/A |
| HSV1 UL11 reverse: GTCTACTTAATTAATTCGCTATCGGACATGGGGGGTG |
this study | N/A |
| HSV1 UL12 forward: GTCTACACTAGTATGGAGTCCACGGTAGGCCC |
this study | N/A |
| HSV1 UL12 reverse: GTCTACTTAATTAAGCGAGACGACCTCCCCGTCG |
this study | N/A |
| HSV1 UL13 forward: GTCTACACTAGTATGGATGAGTCCCGCAGACAGCG |
this study | N/A |
| HSV1 UL13 reverse: GTCTACTTAATTAACGACAGCGCGTGCCGCGCGCAC |
this study | N/A |
| HSV1 UL14 forward: GTCTACACTAGTATGGACCGAGATGCCGCCCACG |
this study | N/A |
| HSV1 UL14 reverse: GTCTACTTAATTAATTCGCCATCGGGATAGTCCCG |
this study | N/A |
| HSV1 UL15 forward: GTCTACACTAGTATGTTTGGTCAGCAGCTGGCGTC |
this study | N/A |
| HSV1 UL15 reverse: GTCTACTTAATTAACGAAACGCGTGTGATGGGAGCG |
this study | N/A |
| HSV1 UL16 forward: GTCTACACTAGTATGGCGCAGCTGGGACCCCGGCG |
this study | N/A |
| HSV1 UL16 reverse: GTCTACTTAATTAATTCGGGATCGCTTGAGGAGGCCCG |
this study | N/A |
| HSV1 UL17 forward: GTCTACACTAGTATGAACGCGCACTTGGCCAACGAGGTC |
this study | N/A |
| HSV1 UL17 reverse: GTCTACTTAATTAAGCGAGAACGGCCGTTCCCGGA |
this study | N/A |
| HSV1 UL18 forward: GTCTACACTAGTATGCTGGCGGACGGCTTTGAAAC |
this study | N/A |
| HSV1 UL18 reverse: GTCTACTTAATTAAGGGATAGCGTATAACGGG |
this study | N/A |
| HSV1 UL19 forward: GTCTACACTAGTATGGCCGCTCCCAACCGCGACCC |
this study | N/A |
| HSV1 UL19 reverse: GTCTACTTAATTAACAGAGCCAGTCCCTTGAGCGGGGATG |
this study | N/A |
| HSV1 UL20 forward: GTCTACACTAGTATGACCATGCGGGATGACCTTCCTC |
this study | N/A |
| HSV1 UL20 reverse: GTCTACTTAATTAAGAACGCGACGGGTGCATTCAAG |
this study | N/A |
| HSV1 UL21 forward: GTCTACACTAGTATGGAGCTTAGCTACGCCACC |
this study | N/A |
| HSV1 UL21 reverse: GTCTACTTAATTAACACAGACTGTCCGTGTTTGG |
this study | N/A |
| HSV1 UL22 forward: GTCTACACTAGTATGGGGAATGGTTTATGGTTCG |
this study | N/A |
| HSV1 UL22 reverse: GTCTACTTAATTAATTCGCGTCTCCAAAAAAACGGGACAC |
this study | N/A |
| HSV1 UL23 forward: GTCTACACTAGTATGGCTTCGTACCCCTGCCATC |
this study | N/A |
| HSV1 UL23 reverse: GTCTACTTAATTAAGTTAGCCTCCCCCATCTCCCGGGCAAACG |
this study | N/A |
| HSV1 UL24 forward: GTCTACACTAGTATGGCCGCGAGAACGCGCAGC |
this study | N/A |
| HSV1 UL24 reverse: GTCTACTTAATTAATTCGGAGGCGGCTCGGGGTTTG |
this study | N/A |
| HSV1 UL25 forward: GTCTACACTAGTATGGACCCGTACTGCCCATTTG |
this study | N/A |
| HSV1 UL25 reverse: GTCTACTTAATTAAAACCGCCGACAGGTACTGTGG |
this study | N/A |
| HSV1 UL26 forward: GTCTACACTAGTATGGCAGCCGATGCCCCGGGAG |
this study | N/A |
| HSV1 UL26 reverse: GTCTACTTAATTAAGCGGGCCCCCATCATCTGAGAG |
this study | N/A |
| HSV1 UL26.5 forward: GTCTACACTAGTATGAACCCCGTTCCGGCATCGGGC |
this study | N/A |
| HSV1 UL26.5 reverse: GTCTACTTAATTAAGCGGGCCCCCATCATCTGAGAG |
this study | N/A |
| HSV1 UL27 forward: GTCTACACTAGTATGCGCCAGGGCGCCCCCGC |
this study | N/A |
| HSV1 UL27 reverse: GTCTACTTAATTAACAGGTCGTCCTCGTCGGCGTC |
this study | N/A |
| HSV1 UL28 forward: GTCTACACTAGTATGGCCGCCCCGGTGTCCGAGCCC |
this study | N/A |
| HSV1 UL28 reverse: GTCTACTTAATTAACGGGGGCCCGTCGTGCCCCC |
this study | N/A |
| HSV1 UL29 forward: GTCTACACTAGTATGGAGACAAAGCCCAAGACGGC |
this study | N/A |
| HSV1 UL29 reverse: GTCTACTTAATTAACAGCATATCCAACGTCAGGTCTC |
this study | N/A |
| HSV1 UL30 forward: GTCTACACTAGTATGTTTTCCGGTGGCGGCGG |
this study | N/A |
| HSV1 UL30 reverse: GTCTACTTAATTAATGCTAGAGTATCAAAGGCTC |
this study | N/A |
| HSV1 UL31 forward: GTCTACACTAGTATGTATGACACCGACCCCCATC |
this study | N/A |
| HSV1 UL31 reverse: GTCTACTTAATTAACGGCGGAGGAAACTCGTCGAATG |
this study | N/A |
| HSV1 UL32 forward: GTCTACACTAGTGCCCAGCCATGGCAACTTCG |
this study | N/A |
| HSV1 UL32 reverse: GTCTACTTAATTAATACATAGGTACACAGGGTGTGC |
this study | N/A |
| HSV1 UL33 forward: GTCTACACTAGTGAAGTTGCCATGGCTGGGC |
this study | N/A |
| HSV1 UL33 reverse: GTCTACTTAATTAAGCCCCGCAGAATCTGGTGCAGGTC |
this study | N/A |
| HSV1 UL34 forward: GTCTACACTAGTATGGCGGGACTGGGCAAGCCC |
this study | N/A |
| HSV1 UL34 reverse: GTCTACTTAATTAATAGGCGCGCGCCAGCACCAAC |
this study | N/A |
| HSV1 UL35 forward: GTCTACACTAGTATGGCCGTCCCGCAATTTCAC |
this study | N/A |
| HSV1 UL35 reverse: GTCTACTTAATTAACGGGGTCCCGGGCGTCGAAGG |
this study | N/A |
| HSV1 UL36 forward: GTCTACACTAGTATGATCGCGGGCACCCCACCGCAC |
this study | N/A |
| HSV1 UL36 reverse: GTCTACTTAATTAAGCCCAGTAACATGCGCACGTGATG |
this study | N/A |
| HSV1 UL37 forward: GTCTACACTAGTATGGCAGACCGCGGTCTCCCGTCCG |
this study | N/A |
| HSV1 UL37 reverse: GTCTACTTAATTAATTGGTAACTCGTTAACGGCAAGTC |
this study | N/A |
| HSV1 UL38 forward: GTCTACACTAGTATGAAGACCAATCCGCTACCCG |
this study | N/A |
| HSV1 UL38 reverse: GTCTACTTAATTAACGCGCATGCCCGCCACTCGCC |
this study | N/A |
| HSV1 UL39 forward: GTCTACACTAGTATGGCCAGCCGCCCAGCCGC |
this study | N/A |
| HSV1 UL39 reverse: GTCTACTTAATTAACAGCGCGCAGCTCGTGCAGAC |
this study | N/A |
| HSV1 UL40 forward: GTCTACACTAGTATGGATTCCGCGGCCCCAG |
this study | N/A |
| HSV1 UL40 reverse: GTCTACTTAATTAACAGATCGTTGACGACCGC |
this study | N/A |
| HSV1 UL41 forward: GTCTACACTAGTATGGGTTTGTTCGGGATGATGAAG |
this study | N/A |
| HSV1 UL41 reverse: GTCTACTTAATTAACTCGTCCCAGAATTTGGCCAG |
this study | N/A |
| HSV1 UL42 forward: GTCTACACTAGTATGACGGATTCCCCTGGCGGTG |
this study | N/A |
| HSV1 UL42 reverse: GTCTACTTAATTAAGGGGAATCCAAAACCAGAC |
this study | N/A |
| HSV1 UL43 forward: GTCTACACTAGTATGCTCCGCAACGACAGCCACC |
this study | N/A |
| HSV1 UL43 reverse: GTCTACTTAATTAAATCGCCCGACCGCCCGCCCGTTG |
this study | N/A |
| HSV1 UL44 forward: GTCTACACTAGTGCTTTGCCGGGAACGCTAGC |
this study | N/A |
| HSV1 UL44 reverse: GTCTACTTAATTAACCGCCGATGACGCTGCCGCGAC |
this study | N/A |
| HSV1 UL45 forward: GTCTACACTAGTATGCCTCTGCGGGCATCGGAAC |
this study | N/A |
| HSV1 UL45 reverse: GTCTACTTAATTAACGGCAGCCCCAGCGCGTTGC |
this study | N/A |
| HSV1 UL46 forward: GTCTACACTAGTCTGGACGCGGCATAACTCCGAC |
this study | N/A |
| HSV1 UL46 reverse: GTCTACTTAATTAACCGGCTCCGGCGTCCTTCGCGTTTAAG |
this study | N/A |
| HSV1 UL47 forward: GTCTACACTAGTATGTCGGCTCGCGAACCCGC |
this study | N/A |
| HSV1 UL47 reverse: GTCTACTTAATTAATGGGCGTGGCGGGCCTCCCAG |
this study | N/A |
| HSV1 UL48 forward: GTCTACACTAGTATGGACCTCTTGGTCGACGAGCTG |
this study | N/A |
| HSV1 UL48 reverse: GTCTACTTAATTAACCCACCGTACTCGTCAATTCCAAG |
this study | N/A |
| HSV1 UL49 forward: GTCTACACTAGTATGACCTCTCGCCGCTCCGTGAAG |
this study | N/A |
| HSV1 UL49 reverse: GTCTACTTAATTAACTCGACGGGCCGTCTGG |
this study | N/A |
| HSV1 UL49A forward: GTCTACACTAGTCTCATCTTCCTGTTAGGGACGATG |
this study | N/A |
| HSV1 UL49A reverse: GTCTACTTAATTAAGGCGTGCCCGGCAGCCAGTAG |
this study | N/A |
| HSV1 UL50 forward: GTCTACACTAGTGTCCCTAACAGGAAGATGAGTCAG |
this study | N/A |
| HSV1 UL50 reverse: GTCTACTTAATTAAAATACCGGTAGAGCCAAAACC |
this study | N/A |
| HSV1 UL51 forward: GTCTACACTAGTATGGCTTCTCTTCTCGGGGC |
this study | N/A |
| HSV1 UL51 reverse: GTCTACTTAATTAATTGACCCAAAACACACGGAGCTGC |
this study | N/A |
| HSV1 UL52 forward: GTCTACACTAGTATGGGGCAGGAAGACGGGAAC |
this study | N/A |
| HSV1 UL52 reverse: GTCTACTTAATTAAAGACGACGGTTGAGAGGTGCTGC |
this study | N/A |
| HSV1 UL53 forward: GTCTACACTAGTATGCTCGCCGTCCGTTCCCTGCAG |
this study | N/A |
| HSV1 UL53 reverse: GTCTACTTAATTAATACATCAAACAGGCGCCTCTGGATC |
this study | N/A |
| HSV1 UL54 forward: GTCTACACTAGTATGGCGACTGACATTGATATGCTAATTG |
this study | N/A |
| HSV1 UL54 reverse: GTCTACTTAATTAAAAACAGGGAGTTGCAATAAAAATATTTGC |
this study | N/A |
| HSV1 UL55 forward: GTCTACACTAGTCTTTTGCACTATGACAGCGACC |
this study | N/A |
| HSV1 UL55 reverse: GTCTACTTAATTAACGCCTTAATTTTAATCTTGAC |
this study | N/A |
| HSV1 UL56 forward: GTCTACACTAGTCATCCATGGCTTCGGAGGCGGCGC |
this study | N/A |
| HSV1 UL56 reverse: GTCTACTTAATTAACCGCCACAGGAATACCAGAATAATG |
this study | N/A |
| HSV1 RS1 forward: GTCTACACTAGTATGGCGTCGGAGAACAAGCAGCG |
this study | N/A |
| HSV1 RS1 reverse: GTCTACTTAATTAACAGCACCCCGTCCCCCTCGAACGCG |
this study | N/A |
| HSV1 US1 forward: GTCTACACTAGTATGGCCGACATTTCCCCAGG |
this study | N/A |
| HSV1 US1 reverse: GTCTACTTAATTAACGGCCGGAGAAACGTGTCGCTG |
this study | N/A |
| HSV1 US2 forward: GTCTACACTAGTATGGGCGTTGTTGTCGTCAACG |
this study | N/A |
| HSV1 US2 reverse: GTCTACTTAATTAACAGGGTGGTAACCGGATAGCAGATG |
this study | N/A |
| HSV1 US3 forward: GTCTACACTAGTATGGCCTGTCGTAAGTTTTGTCG |
this study | N/A |
| HSV1 US3 reverse: GTCTACTTAATTAATTTCTGTTGAAACAGCGGCAAAC |
this study | N/A |
| HSV1 US4 forward: GTCTACACTAGTCATCATGTCGCCGGGCGCCATG |
this study | N/A |
| HSV1 US4 reverse: GTCTACTTAATTAACCCGCGTTCGGACGGCAGGCAC |
this study | N/A |
| HSV1 US5 forward: GTCTACACTAGTATGTCTCTGCGCGCAGTCTG |
this study | N/A |
| HSV1 US5 reverse: GTCTACTTAATTAATACGACAACTGGGTCCATGTAGG |
this study | N/A |
| HSV1 US6 forward: GTCTACACTAGTGTGTGGTGCGTTCCGGTATG |
this study | N/A |
| HSV1 US6 reverse: GTCTACTTAATTAAGTAAAACAAGGGCTGGTGCGAG |
this study | N/A |
| HSV1 US7 forward: GTCTACACTAGTGTCCCGTTCCGGGATGCCGTG |
this study | N/A |
| HSV1 US7 reverse: GTCTACTTAATTAATACCAACAGGGGAGGCGTTG |
this study | N/A |
| HSV1 US8 forward: GTCTACACTAGTGACATGGATCGCGGGGCGGTG |
this study | N/A |
| HSV1 US8 reverse: GTCTACTTAATTAACCAGAAGACGGACGAATCGG |
this study | N/A |
| HSV1 US8A forward: GTCTACACTAGTATGGATCCGGCTTTGAGATC |
this study | N/A |
| HSV1 US8A reverse: GTCTACTTAATTAATGCGCCTCGGGCAATTGACGTC |
this study | N/A |
| HSV1 US9 forward: GTCTACACTAGTATGACGTCCCGGCTCTCCG |
this study | N/A |
| HSV1 US9 reverse: GTCTACTTAATTAAGCGGAGCAGCCACATCAGGAG |
this study | N/A |
| HSV1 US10 forward: GTCTACACTAGTGTGATAATGATCAAGCGGCGG |
this study | N/A |
| HSV1 US10 reverse: GTCTACTTAATTAAGCACAGGGGTGGGGTTAGG |
this study | N/A |
| HSV1 US11 forward: GTCTACACTAGTGTGGCTCTCGAGATGAGCCAG |
this study | N/A |
| HSV1 US11 reverse: GTCTACTTAATTAATACAGACCCGCGAGCCGTACGTG |
this study | N/A |
| HSV1 US12 forward: GTCTACACTAGTATGTCGTGGGCCCTGGAAATGGC |
this study | N/A |
| HSV1 US12 reverse: GTCTACTTAATTAAACGGGTTACCGGATTACGGGGAC |
this study | N/A |
| Recombinant DNA | ||
| pfuse-hg1fc1 | InvivoGen | Cat# pfuse-hg1fc1 |
| pCDH vector | System Bioscience | Cat# CD510B |
| Software and Algorithms | ||
| FlowJo v7 | TreeStar Inc | N/A |
| GraphPad Prism 7 | GraphPad | N/A |
| ZDOCK | Pierce et al., 2014 | http://zdock.umassmed.edu/ |
| PyMol | The PyMOL Molecular Graphics System | https://www.pymol.org/ |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and request for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michael Caligiuri (michael.caligiuri@osumc.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All animal experiments were approved by The Ohio State University Institutional Animal Care and Use Committee. Blinding and randomization were not adopted for designing animal experiments. 8-to-12 weeks old female C57BL/6 and BALB/c mice (Jackson Lab) were used for all the studies.
METHODS DETAILS
Cloning of HSV1 genes
Genomic DNA of HSV1 F strain (sequence accession number: GU734771) was used as the template to amplify individual HSV1 genes, which were cloned into pCDH vector (System Bioscience, CD510B) following standard protocol. Gene specific primers are flanked with SpeI site at the 5′ and PacI site at the 3′ end, respectively. Primer sequences are available upon request.
Culture and transfection of human glioma spheres
All experiments involving human tissue were performed with the approval of The Ohio State University Institutional Review Board and informed consent from patients. Glioma cells were derived from primary human brain tumors and grown in DMEM/F12 supplemented with B27, heparin (5 ug/mL), basic FGF (20 ng/mL), and EGF (20 ng/mL) as described previously (Mao et al., 2013). Except otherwise noted, #83 glioma cells were used throughout this study (Mao et al., 2013). Cells were tested with MycoAlert to exclude from mycoplasma contamination. For one single transfection, 10 million glioma cells were washed once with DMEM/F12 media, and resuspended in 100 μl of basic nucleofector solution. Subsequently, cell suspension was mixed with 6 μg of plasmid and nucleofected using Nucleofector (Amaxa GmBH) with program A33. Following nucleofection, cells were immediately mixed with 1 ml media and transferred into one well of 6-well plates containing 4 ml DMEM/F12 with all supplements.
Human NK cell Isolation and stimulation
All NK cells in this study were freshly enriched from peripheral blood leukopacks of healthy donors using RosetteSep cocktail and CD56 microbeads. Fresh NK cells were cultured at the density of 5 million NK cells /ml RPMI1640 media supplemented with 10% heat inactivated FBS and antibiotic-antimycotic. 5*105 human NK cells were cultured with 1*105 infected or transfected glioma cells, K562 cells or with IL12 (10 ng/mL)+ IL-18 (10 ng/mL), 1*108 cfu of 1*108 cfu of paraformaldehyde-fixed wt or Spa-S.A bacteria. To saturate the CH2-CH3 interface of IgG molecules anchored on human NK cells, 5ug/ml protein A or protein G was added into NK cells 30min prior to coculture/stimulation. In some cases, human NK cells were cultured in protein- or virus-coated 96 well plates, which were pre-incuabated with 50ul protein A (0.1ug/ul), protein G (0.1ug/ul), wt or Us8− HSV1 F (0.1ug/ul) overnight at 4 °C. Flowcytometry staining were conducted following standard procedures at 7th or 20th hour of coculture/stimulation, and all flowcytometry data were collected with LSRII (BD bioscience) and analyzed with FlowJo. For all the CD107a staining, CD107a antibody was added at the beginning of coculture. Cell supernatants were collected after 20 hours’ culture, and quantification of human IFN-γ was performed using ELISA Kit.
DCMEGE assay
24h after transfection, glioma cells were resuspended and centrifuged down at 50g for 5 min to remove cell debris and dead cells. Subsequently, 1*104 glioma cells were resuspended with 100 μl DMEM/F12 media and seeded into each well of a U bottom 96-well plate. Purified human NK cells were resuspended in complete RPMI1640 media to the final concentration of 5*106/ml, and 100 μl of NK cells were added to culture with transfected glioma cells. In a parallel control experiment, 100 μl complete RPMI1640 media, instead of human NK cells, were added to the seeded glioma cells. Culture samples were collected using LSRII at 5th hour of culture. Living glioma cells were gated in based on their FSC and SSC and measured for the percentage of GFP+ cells (Figure S1B). Differential cytotoxicity contributed by the expression of individual viral gene is calculated with the formula: .
HSV1 virus production, purification and inactivation
Vero cells were seeded at a density of 7*106 cells per 100 mm dish and inoculated with 2.5 pfu per cell of HSV-1 F strain or Us8- mutants (Suenaga et al., 2014). At 24 hours post-inoculation, culture media and cell debris were collected. After three freeze-thaw cycle to release viruses, cell debris were removed by low speed centrifugation (2,000×g for 5 minutes), supernatants were loaded on a 5 ml 35% sucrose gradient and centrifuged in a Beckman SW27 rotor at 25,000 rpm for 1 hour. Virus pallet was collected, washed and concentrated in PBS. Pure viruses were inactivated with 0.2% trition-100 for 30min at room temperature, and subsequently diluted to 0.1ug protein/ml PBS for coating plates.
Plaque assay for HSV1
Sequentially diluted viruses were loaded on single layer of vero cells and incubated at 37°c; pooled human IgG (final concentration 0.1%) was added 1h later to restrain viruses spread. Plaques were counted after 48 hours culture. To determine the effect of human IgG3, human IgGFc, Rituximab, daratumumab, and human IgG (GamaStan) on the infectivity of HSV1, these reagents were added into sequentially diluted viruses at 1ug/ml and incubated at room temperature for 30min prior to plaque assay. Treated viruses were tittered with standard plaque assay and all results were normalized to PBS as the control.
Chromium release cytotoxicity assay
Target glioma cells were either transfected with HSV1 genes at 24 hours, or infected with HSV1 at MOI=1 at 12 hours before radiolabelling. Target cells were labeled by incubating 5*105 cells in 50 μCi 51Cr for 90min at 37°C. Radiolabeled cells were washe d 3 times and resuspended in complete RPMI 1640 media, and seeded in U bottom 96-well plates at a concentration of 5*103 cells/100ul media/well. In some cases, antibodies or IgG products (5ug/ml) were added into radiolabeled target cells and incubated on ice for 30 minutes for binding or blocking certain interactions. Human or mouse NK cells were added in at specified effector-to-target ratios (E:T, x-axis on figures) and incubated at 37°C for 4–6 hours. Each E:T ratio were run in t riplicates and data represent two independent experiments. After incubation, plates were centrifuged to pellet the cells to the bottom of the plate, and supernatants were collected from each of the wells and quantitated using a gamma counter. Radiolabelled cells were treated with 1% SDS as a measure of maximal cytolysis, or incubated without NK cells were used to measure spontaneous release. Cytolysis was calculated with the formula: . Cytolysis data were presented as mean of triplicates at each E:T ratio and each experiment was repeated with NK cells from at least two different donors and only one representative was shown.
Structure Modeling
All docking predictions were conducted on the ZDOCK server (Pierce et al., 2014), and crystal structure of CD16a-Fc (PDB ID: 1E4K) was used as the host. For modeling gE-Fc-CD16a complex, gE subunit of gE-Fc (PDB ID: 2GJ7) was uploaded to the ZDOCK server to dock 1E4K. HSV1 gE residues 225, 245–247, 249–250, 256, 258, 311, 316, 318–322, 324, and 338–342, and IgG1Fc residues 252–258, 307, 309–311, 314–315, 382, 428, and 433–436 were specified as contact residues (Sprague et al., 2006). For modeling protein A-Fc-CD16a complex, protein A domain C of protein A-Fc complex (PDB ID: 4WWI) was used to dock 1E4K. Protein A domain C residues Q9, Q10, N11, F13, Y14, and L17, and IgGFc residues S254, Q311, L432, and N434, were defined as contacting residues (Deis et al., 2015). For modeling protein G-Fc-CD16a complex, protein G C2 fragment of crystal structure of protein G-Fc complex (PDB ID: 1FCC) was used to dock 1E4K. Protein G C2 residues 27, 28, 31, 32, 35, 40, 42, 43 and IgGFc residues 253, 254, 380, 382, 433–436, were defined as contacting residues(Sauer-Eriksson et al., 1995). The top-scoring complexes were manually examined for consistency with original crystal structures. All structure images were generated using PyMol.
Interaction of human CD16a, IgG and IgG-binding proteins from pathogens
Transfected glioma cells, infected glioma cells, or bacteria were first incubated for 30min on ice with PBS with or without IgG1Fc (ΔCD16), IgG1Fc, rituximab, respectively. After one wash with FACS buffer, samples were incubated with Alex647 labeled mouse anti-human IgGFc (209–605–098, Jackson ImmunoResearch) in the presence of excess unlabeled mouse serum to detect the binding between Fc with gE or protein A. Otherwise, cells or bacteria were further incubated with biotinylated CD16a and apc-streptavidin (554067, BD) on ice for 20min to detect binding of CD16a to IgG. Stained cells or bacteria and relevant staining controls were checked on LSRII (BD).
CD3ζ phosphorylation staining
Half million NK cells were rested at 37°C for 1h an d stimulated with H2O2 (11mM), IL12 (10ng/ml)+IL18 (10ng/ml), 2*105 transfected glioma cells, or 1*108 cfu of bacteria for 1h. NK cells were fixed with Phosflow Fix Buffer I, permeabilized with Phosflow™ Perm Buffer III, blocked with normal mouse immunoglobulin, and then stained with anti-CD 3ζ (pY142) and anti-CD3ζ. Phosphatase inhibitor (Roche) was supplemented in all the staining steps.
Mouse experiments
For bioluminescence imaging to track virus load, BALB/c mice were i.p injected with 1.2*105 pfu of R8411 viruses (Zerboni et al., 2013). BALB/c mice were i.p injected with 200ug rituximab at 4 hours prior to virus challenge, and at 24 hours after virus challenge. Each mouse was given 3mg luciferin potassium 10 minutes prior to isoflurane anesthesia to ensure consistent photon flux. Images were taken at OSU Small Animal Imaging Facility using an IVIS Spectrum (Perkin Elmer) at 84h post infection. Each group was recorded for 4 sections of 2 minute exposure. Bioluminescence values were measured from the whole mouse and calculated as photon flux (photons/s/) using Living Image 4.0 (Perkin Elmer).
For survival study, BALB/c mice were i.p injected with 3*106 pfu HSV1 F strain viruses or 5*105 pfu MCMV. PBS, 200ug human IgG3, 200ug human IgGFc, 200ug rituximab or 200ug daratumumab were given via i.p injection at 4 hours prior to virus challenge, and at 24 hours and 72 hours after virus challenge. Mice were weighed every 24h beginning on the day of infection, and monitored for neurological symptoms (scoring from 0 to 5: 0 = no symptoms; 1 = ruffled fur, hunched posture, but can easily be made to move around; 2 = hunched and slow to move; 3 = hunched, some movement, and labored breathing; 4 = hunched, labored breathing, and little or no movement; 5 = moribund or dead, as described in (Karaba et al., 2012)).
To study the involvement of NK cells in mediating the protection of HSV1 infection by human IgGFc, mice were given i.p injection of 20ul anti-Asialo GM1 antibody or equal volume of PBS at 24 hours before virus challenge. For the in vitro mouse NK cell stimulation and cytotoxicity, NK cells were enriched from spleens of 8 to 12 weeks old C57BL/6 and BALB/c mice using NK cell isolation kit (Miltenyi Biotec) following manufacturer’s instruction. Purified mouse NK cells were cultured the same as human NK cells described above to 51Cr release cytotoxicity assay.
QUANTIFICATION AND STATISTICAL ANALYSIS
Two-sample t test was used to compare two independent groups and paired t test was used to compare two paired groups. Data transformation was performed if the original distribution is non-normal. Linear mixed models were used to account for the covariance structure due to repeated measures from the same donor when three or more groups were compared. Kaplan-Meier method was used to estimate survival functions and log-rank test was used to compare survival curves of different groups. P values were adjusted for multiple comparisons by Holm’s procedure. A p value of < 0.05 was considered significant. Experiments were repeated at least three or more times. Data are displayed as mean ± SEM.
Supplementary Material
Highlights.
DC-MEGE Identified HSV1 viral proteins regulating NK cell cytotoxicity
HSV1 gE activates NK cells through an IgGFc bridge with CD16a on NK cells
IgGFc alone protects mice from lethal HSV1 infection; NK cells are required
Bacterial IgG binding proteins activate NK cells through the IgGFc bridge
Acknowledgments
We would like thank Roselyn J. Eisenberg, Gary Cohen, Harvey Friedman and Timothy Frost, Bernard Roizman, and Lewis Lanier for sharing reagents. Thanks to Clark Anderson, Susheela Tridandapani, Jon Butchar, Ansel Natlin for critically reading the manuscript. Thanks to Kathleen McConnell and Victoria Sellers for help with mouse experiments. HD is a Pelotonia fellow. This research has been supported by NIH grant P01CA163205 awarded to EAC, NI, TC, BK, JY, and MAC, P01CA095426 awarded to JY and MAC, and NCI grant CA155521 awarded to JY.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors have declared that no conflict of interest exists.
SUPPLEMENTAL INFORMATION
Supplemental Information includes nine figures and can be found with this article online at:
Author contributions
HSD designed and conducted all experiments with help from NG, CB, JWZ and CM; HSD and JYZ did statistical analysis. TC, TS, HA and NI provided reagents; EAC, BK, and JY provided intellectual input into the conduct and analysis of the experiments; HSD and MAC conceived of the project; analyzed and interpreted the data, and wrote the manuscript.
References
- Alter G, Malenfant JM, Altfeld M. CD107a as a functional marker for the identification of natural killer cell activity. Journal of Immunological Methods. 2004;294:15–22. doi: 10.1016/j.jim.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Alvarez-Breckenridge CA, Yu J, Price R, Wojton J, Pradarelli J, Mao H, Wei M, Wang Y, He S, Hardcastle J, et al. NK cells impede glioblastoma virotherapy through NKp30 and NKp46 natural cytotoxicity receptors. Nat Med. 2012;18:1827–1834. doi: 10.1038/nm.3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P, Caligiuri M, O’Brien C, Manley T, Ritz J, Schlossman SF. Fc gamma receptor type III (CD16) is included in the zeta NK receptor complex expressed by human natural killer cells. Proc Natl Acad Sci U S A. 1990;87:2274–2278. doi: 10.1073/pnas.87.6.2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, Delman KA, Spitler LE, Puzanov I, Agarwala SS, et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J Clin Oncol. 2015 doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- Biron CA, Byron KS, Sullivan JL. Severe herpesvirus infections in an adolescent without natural killer cells. N Engl J Med. 1989;320:1731–1735. doi: 10.1056/NEJM198906293202605. [DOI] [PubMed] [Google Scholar]
- Bishop GA, Glorioso JC, Schwartz SA. Relationship between expression of herpes simplex virus glycoproteins and susceptibility of target cells to human natural killer activity. J Exp Med. 1983;157:1544–1561. doi: 10.1084/jem.157.5.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalona WJ, Ratliff TL, McCool RE. gamma-Interferon induced by S. aureus protein A augments natural killing and ADCC. Nature. 1981;291:77–79. doi: 10.1038/291077a0. [DOI] [PubMed] [Google Scholar]
- Chapman TL, You I, Joseph IM, Bjorkman PJ, Morrison SL, Raghavan M. Characterization of the interaction between the herpes simplex virus type I Fc receptor and immunoglobulin G. J Biol Chem. 1999;274:6911–6919. doi: 10.1074/jbc.274.11.6911. [DOI] [PubMed] [Google Scholar]
- Chisholm Susan E, Howard K, Gómez Mar V, Reyburn Hugh T. Expression of ICP0 Is Sufficient to Trigger Natural Killer Cell Recognition of Herpes Simplex Virus–Infected Cells by Natural Cytotoxicity Receptors. The Journal of Infectious Diseases. 2007;195:1160–1168. doi: 10.1086/512862. [DOI] [PubMed] [Google Scholar]
- Collins WJ, Johnson DC. Herpes simplex virus gE/gI expressed in epithelial cells interferes with cell-to-cell spread. J Virol. 2003;77:2686–2695. doi: 10.1128/JVI.77.4.2686-2695.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrales-Aguilar E, Trilling M, Hunold K, Fiedler M, Le VT, Reinhard H, Ehrhardt K, Merce-Maldonado E, Aliyev E, Zimmermann A, et al. Human cytomegalovirus Fcgamma binding proteins gp34 and gp68 antagonize Fcgamma receptors I, II and III. PLoS Pathog. 2014;10:e1004131. doi: 10.1371/journal.ppat.1004131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Miranda-Santos IK, Campos-Neto A. Receptor for immunoglobulin Fc on pathogenic but not on nonpathogenic protozoa of the Trypanosomatidae. J Exp Med. 1981;154:1732–1742. doi: 10.1084/jem.154.6.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deis LN, Wu Q, Wang Y, Qi Y, Daniels KG, Zhou P, Oas TG. Suppression of conformational heterogeneity at a protein-protein interface. Proc Natl Acad Sci U S A. 2015;112:9028–9033. doi: 10.1073/pnas.1424724112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubin G, Socolof E, Frank I, Friedman HM. Herpes simplex virus type 1 Fc receptor protects infected cells from antibody-dependent cellular cytotoxicity. J Virol. 1991;65:7046–7050. doi: 10.1128/jvi.65.12.7046-7050.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards JC, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, Stevens RM, Shaw T. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350:2572–2581. doi: 10.1056/NEJMoa032534. [DOI] [PubMed] [Google Scholar]
- Fitzgerald-Bocarsly P, Howell DM, Pettera L, Tehrani S, Lopez C. Immediate-early gene expression is sufficient for induction of natural killer cell-mediated lysis of herpes simplex virus type 1-infected fibroblasts. J Virol. 1991;65:3151–3160. doi: 10.1128/jvi.65.6.3151-3160.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier B, Philpott DJ. Recognition of Staphylococcus aureus by the innate immune system. Clin Microbiol Rev. 2005;18:521–540. doi: 10.1128/CMR.18.3.521-540.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank I, Friedman HM. A novel function of the herpes simplex virus type 1 Fc receptor: participation in bipolar bridging of antiviral immunoglobulin G. Journal of virology. 1989;63:4479–4488. doi: 10.1128/jvi.63.11.4479-4488.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman D, Jojic V, Sharma S, Shen-Orr SS, Angel CJ, Onengut-Gumuscu S, Kidd BA, Maecker HT, Concannon P, Dekker CL, et al. Cytomegalovirus infection enhances the immune response to influenza. Sci Transl Med. 2015;7:281ra243. doi: 10.1126/scitranslmed.aaa2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilden DH, Mahalingam R, Cohrs RJ, Tyler KL. Herpesvirus infections of the nervous system. Nat Clin Pract Neurol. 2007;3:82–94. doi: 10.1038/ncpneuro0401. [DOI] [PubMed] [Google Scholar]
- Gomez MI, Lee A, Reddy B, Muir A, Soong G, Pitt A, Cheung A, Prince A. Staphylococcus aureus protein A induces airway epithelial inflammatory responses by activating TNFR1. Nat Med. 2004;10:842–848. doi: 10.1038/nm1079. [DOI] [PubMed] [Google Scholar]
- Huard B, Fruh K. A role for MHC class I down-regulation in NK cell lysis of herpes virus-infected cells. Eur J Immunol. 2000;30:509–515. doi: 10.1002/1521-4141(200002)30:2<509::AID-IMMU509>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Imai T, Koyanagi N, Ogawa R, Shindo K, Suenaga T, Sato A, Arii J, Kato A, Kiyono H, Arase H, et al. Us3 kinase encoded by herpes simplex virus 1 mediates downregulation of cell surface major histocompatibility complex class I and evasion of CD8+ T cells. PLoS One. 2013;8:e72050. doi: 10.1371/journal.pone.0072050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DC, Frame MC, Ligas MW, Cross AM, Stow ND. Herpes simplex virus immunoglobulin G Fc receptor activity depends on a complex of two viral glycoproteins, gE and gI. J Virol. 1988;62:1347–1354. doi: 10.1128/jvi.62.4.1347-1354.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalina U, Kauschat D, Koyama N, Nuernberger H, Ballas K, Koschmieder S, Bug G, Hofmann WK, Hoelzer D, Ottmann OG. IL-18 activates STAT3 in the natural killer cell line 92, augments cytotoxic activity, and mediates IFN-gamma production by the stress kinase p38 and by the extracellular regulated kinases p44erk-1 and p42erk-21. J Immunol. 2000;165:1307–1313. doi: 10.4049/jimmunol.165.3.1307. [DOI] [PubMed] [Google Scholar]
- Kasahara T, Harada H, Shioiri-Nakano K, Imai M, Sano T. Potentiation of natural killer activity of human lymphocytes by Staphylococcus aureus bacteria and its protein A. Immunology. 1981;42:175–183. [PMC free article] [PubMed] [Google Scholar]
- Kasahara T, Harada H, Shioiri-Nakano K, Wakasugi H, Imai M, Mayumi M, Sano T, Sugiura A. Potentiation of natural killer cell activity of human lymphocytes in vitro: the participation of interferon in stimulation with Staphylococcus aureus Cowan I bacteria but not with protein A. Immunology. 1982;45:687–695. [PMC free article] [PubMed] [Google Scholar]
- Kasai M, Yoneda T, Habu S, Maruyama Y, Okumura K, Tokunaga T. In vivo effect of anti-asialo GM1 antibody on natural killer activity. Nature. 1981;291:334–335. doi: 10.1038/291334a0. [DOI] [PubMed] [Google Scholar]
- Kay HD, Bonnard GD, West WH, Herberman RB. A functional comparison of human Fc-receptor-bearing lymphocytes active in natural cytotoxicity and antibody-dependent cellular cytotoxicity. J Immunol. 1977;118:2058–2066. [PubMed] [Google Scholar]
- Kiyoshi M, Caaveiro JM, Kawai T, Tashiro S, Ide T, Asaoka Y, Hatayama K, Tsumoto K. Structural basis for binding of human IgG1 to its high-affinity human receptor FcgammaRI. Nat Commun. 2015;6:6866. doi: 10.1038/ncomms7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier LL, Yu G, Phillips JH. Co-association of CD3 zeta with a receptor (CD16) for IgG Fc on human natural killer cells. Nature. 1989;342:803–805. doi: 10.1038/342803a0. [DOI] [PubMed] [Google Scholar]
- Litwin V, Jackson W, Grose C. Receptor properties of two varicella-zoster virus glycoproteins, gpI and gpIV, homologous to herpes simplex virus gE and gI. J Virol. 1992;66:3643–3651. doi: 10.1128/jvi.66.6.3643-3651.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Tang J. Expression levels of tumor necrosis factor-alpha and the corresponding receptors are correlated with trauma severity. Oncol Lett. 2014;8:2747–2751. doi: 10.3892/ol.2014.2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Guerrero JA, Alarcon B, Fresno M. Mechanism of recognition of herpes simplex virus type 1-infected cells by natural killer cells. J Gen Virol. 1988;69(Pt 11):2859–2868. doi: 10.1099/0022-1317-69-11-2859. [DOI] [PubMed] [Google Scholar]
- Loukas A, Jones MK, King LT, Brindley PJ, McManus DP. Receptor for Fc on the surfaces of schistosomes. Infect Immun. 2001;69:3646–3651. doi: 10.1128/IAI.69.6.3646-3651.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozzio BB, Lozzio CB. Properties and usefulness of the original K-562 human myelogenous leukemia cell line. Leuk Res. 1979;3:363–370. doi: 10.1016/0145-2126(79)90033-x. [DOI] [PubMed] [Google Scholar]
- Mao P, Joshi K, Li J, Kim SH, Li P, Santana-Santos L, Luthra S, Chandran UR, Benos PV, Smith L, et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc Natl Acad Sci U S A. 2013;110:8644–8649. doi: 10.1073/pnas.1221478110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger H. Transmembrane signaling: the joy of aggregation. J Immunol. 1992;149:1477–1487. [PubMed] [Google Scholar]
- Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. 2008;8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- O’Shea JJ, Weissman AM, Kennedy IC, Ortaldo JR. Engagement of the natural killer cell IgG Fc receptor results in tyrosine phosphorylation of the zeta chain. Proc Natl Acad Sci U S A. 1991;88:350–354. doi: 10.1073/pnas.88.2.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ober RJ, Radu CG, Ghetie V, Ward ES. Differences in promiscuity for antibody-FcRn interactions across species: implications for therapeutic antibodies. Int Immunol. 2001;13:1551–1559. doi: 10.1093/intimm/13.12.1551. [DOI] [PubMed] [Google Scholar]
- Olinescu A, Hristescu S, Sjoquist J, Ghetie V. Correlation between the triggering of proliferation and the potentiation of NK activity induced by protein A in human lymphocytes. Immunol Lett. 1983;6:231–237. doi: 10.1016/0165-2478(83)90010-x. [DOI] [PubMed] [Google Scholar]
- Olson JK, Bishop GA, Grose C. Varicella-zoster virus Fc receptor gE glycoprotein: serine/threonine and tyrosine phosphorylation of monomeric and dimeric forms. J Virol. 1997;71:110–119. doi: 10.1128/jvi.71.1.110-119.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr MT, Lanier LL. Natural Killer Cell Education and Tolerance. Cell. 2010;142:847–856. doi: 10.1016/j.cell.2010.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmqvist N, Foster T, Tarkowski A, Josefsson E. Protein A is a virulence factor in Staphylococcus aureus arthritis and septic death. Microb Pathog. 2002;33:239–249. doi: 10.1006/mpat.2002.0533. [DOI] [PubMed] [Google Scholar]
- Patel AH, Nowlan P, Weavers ED, Foster T. Virulence of protein A-deficient and alpha-toxin-deficient mutants of Staphylococcus aureus isolated by allele replacement. Infect Immun. 1987;55:3103–3110. doi: 10.1128/iai.55.12.3103-3110.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel PC, Stefanescu-Soare I, Menezes J. Staphylococcal protein A enhances natural killing activity against lymphoid tumor cell lines. Int J Cancer. 1981;28:277–284. doi: 10.1002/ijc.2910280305. [DOI] [PubMed] [Google Scholar]
- Perussia B, Trinchieri G, Jackson A, Warner NL, Faust J, Rumpold H, Kraft D, Lanier LL. The Fc receptor for IgG on human natural killer cells: phenotypic, functional, and comparative studies with monoclonal antibodies. J Immunol. 1984;133:180–189. [PubMed] [Google Scholar]
- Polcicova K, Goldsmith K, Rainish BL, Wisner TW, Johnson DC. The extracellular domain of herpes simplex virus gE is indispensable for efficient cell-to-cell spread: evidence for gE/gI receptors. J Virol. 2005;79:11990–12001. doi: 10.1128/JVI.79.18.11990-12001.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratliff TL, McCool RE, Catalona WJ. Interferon induction and augmentation of natural-killer activity by Staphylococcus protein A. Cell Immunol. 1981;57:1–12. doi: 10.1016/0008-8749(81)90114-3. [DOI] [PubMed] [Google Scholar]
- Romee R, Foley B, Lenvik T, Wang Y, Zhang B, Ankarlo D, Luo X, Cooley S, Verneris M, Walcheck B, et al. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17) Blood. 2013;121:3599–3608. doi: 10.1182/blood-2012-04-425397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer-Eriksson AE, Kleywegt GJ, Uhlen M, Jones TA. Crystal structure of the C2 fragment of streptococcal protein G in complex with the Fc domain of human IgG. Structure. 1995;3:265–278. doi: 10.1016/s0969-2126(01)00157-5. [DOI] [PubMed] [Google Scholar]
- Sondermann P, Huber R, Oosthuizen V, Jacob U. The 3.2-A crystal structure of the human IgG1 Fc fragment-Fc gammaRIII complex. Nature. 2000;406:267–273. doi: 10.1038/35018508. [DOI] [PubMed] [Google Scholar]
- Sprague ER, Reinhard H, Cheung EJ, Farley AH, Trujillo RD, Hengel H, Bjorkman PJ. The Human Cytomegalovirus Fc Receptor gp68 Binds the Fc CH2-CH3 Interface of Immunoglobulin G. Journal of Virology. 2008;82:3490–3499. doi: 10.1128/JVI.01476-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprague ER, Wang C, Baker D, Bjorkman PJ. Crystal structure of the HSV-1 Fc receptor bound to Fc reveals a mechanism for antibody bipolar bridging. PLoS Biol. 2006;4:e148. doi: 10.1371/journal.pbio.0040148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suenaga T, Kohyama M, Hirayasu K, Arase H. Engineering large viral DNA genomes using the CRISPR-Cas9 system. Microbiology and Immunology. 2014;58:513–522. doi: 10.1111/1348-0421.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szpara ML, Parsons L, Enquist LW. Sequence variability in clinical and laboratory isolates of herpes simplex virus 1 reveals new mutations. J Virol. 2010;84:5303–5313. doi: 10.1128/JVI.00312-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymczak AL, Workman CJ, Wang Y, Vignali KM, Dilioglou S, Vanin EF, Vignali DA. Correction of multi-gene deficiency in vivo using a single ‘self-cleaving’ 2A peptide-based retroviral vector. Nat Biotechnol. 2004;22:589–594. doi: 10.1038/nbt957. [DOI] [PubMed] [Google Scholar]
- Testi R, D’Ambrosio D, De Maria R, Santoni A. The CD69 receptor: a multipurpose cell-surface trigger for hematopoietic cells. Immunol Today. 1994;15:479–483. doi: 10.1016/0167-5699(94)90193-7. [DOI] [PubMed] [Google Scholar]
- Watford WT, Hissong BD, Bream JH, Kanno Y, Muul L, O’Shea JJ. Signaling by IL-12 and IL-23 and the immunoregulatory roles of STAT4. Immunol Rev. 2004;202:139–156. doi: 10.1111/j.0105-2896.2004.00211.x. [DOI] [PubMed] [Google Scholar]
- Zerboni L, Che X, Reichelt M, Qiao Y, Gu H, Arvin A. Herpes simplex virus 1 tropism for human sensory ganglion neurons in the severe combined immunodeficiency mouse model of neuropathogenesis. J Virol. 2013;87:2791–2802. doi: 10.1128/JVI.01375-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.