

Abstract
BACKGROUND
Mast cells are present in the airways of patients who have severe asthma despite glucocorticoid treatment; these cells are associated with disease characteristics including poor quality of life and inadequate asthma control. Stem cell factor and its receptor, KIT, are central to mast-cell homeostasis. We conducted a proof-of-principle trial to evaluate the effect of imatinib, a KIT inhibitor, on airway hyper-responsiveness, a physiological marker of severe asthma, as well as on airway mast-cell numbers and activation in patients with severe asthma.
METHODS
We conducted a randomized, double-blind, placebo-controlled, 24-week trial of imatinib in patients with poorly controlled severe asthma who had airway hyperresponsiveness despite receiving maximal medical therapy. The primary end point was the change in airway hyperresponsiveness, measured as the concentration of methacholine required to decrease the forced expiratory volume in 1 second by 20% (PC20). Patients also underwent bronchoscopy.
RESULTS
Among the 62 patients who underwent randomization, imatinib treatment reduced airway hyperresponsiveness to a greater extent than did placebo. At 6 months, the methacholine PC20 increased by a mean (±SD) of 1.73±0.60 doubling doses in the imatinib group, as compared with 1.07±0.60 doubling doses in the placebo group (P = 0.048). Imatinib also reduced levels of serum tryptase, a marker of mast-cell activation, to a greater extent than did placebo (decrease of 2.02±2.32 vs. 0.56±1.39 ng per milliliter, P = 0.02). Airway mast-cell counts declined in both groups. Muscle cramps and hypophosphatemia were more common in the imatinib group than in the placebo group.
CONCLUSIONS
In patients with severe asthma, imatinib decreased airway hyperresponsiveness, mast-cell counts, and tryptase release. These results suggest that KIT-dependent processes and mast cells contribute to the pathobiologic basis of severe asthma. (Funded by the National Institutes of Health and others; ClinicalTrials.gov number, NCT01097694.)
MANY PATIENTS WITH SEVERE ASTHMA do not have adequate disease control despite the use of high-dose inhaled or systemic glucocorticoids.1 Severe asthma is associated with airway hyperresponsiveness — that is, an exaggerated response to a bronchoconstrictor stimulus — and airway inflammation, both of which persist despite high-dose glucocorticoid therapy.2,3 Increased airway hyper-responsiveness is associated with a progressive loss of lung function,4 and, among patients with moderate-to-severe asthma, those with airway hyperresponsiveness have a poorer quality of life than those without this trait.5 In addition, studies have shown that treatment targeting airway hyperresponsiveness leads to more effective control of asthma6 and reductions in airway remodeling.7
Mast cells are long-lived, tissue-dwelling, hematopoietic effector cells that are implicated in the pathobiologic basis of asthma8 and can persist in the face of glucocorticoid therapy.9 Their presence correlates with airway hyper-responsiveness and asthma disease severity.9,10
Stem cell factor and its receptor, the KIT proto-oncogene receptor tyrosine kinase (hereafter referred to as KIT), are essential for normal mast-cell development and survival in the tissues.11 Levels of soluble stem cell factor are increased in the serum of patients with asthma and correlate with asthma severity.12 Imatinib inhibits the tyrosine kinase activity of KIT13,14 and, as a consequence, markedly reduces bone marrow mast-cell numbers and serum tryptase levels in patients with chronic myeloid leukemia15 and reduces serum tryptase levels in patients with pulmonary hypertension.16 Tryptase, a mast-cell granule–associated protease, is a marker of mast-cell burden and activation when detected in extracellular fluids. Tryptase levels in the bronchoalveolar lavage (BAL) fluid of patients with difficult-to-control asthma exceed those in patients with well-controlled asthma.17 Therefore, we conducted a proof-of-principle trial, involving patients who have severe asthma and airway hyperresponsiveness despite treatment with maximal conventional therapy, to test the hypothesis that KIT inhibition, achieved with the use of imatinib, would improve airway hyper-responsiveness and decrease airway mast-cell counts and activation.
METHODS
TRIAL DESIGN AND OVERSIGHT
We conducted a randomized, double-blind, placebo-controlled, proof-of-principle trial in seven academic centers in the United States from November 2010 through July 2015. The screening and run-in period lasted for a minimum of 4 weeks, after which the patients were randomly assigned to a 24-week intervention period. Randomization was performed centrally by the Brigham and Women’s Hospital investigational drug pharmacy. Novartis provided imatinib tablets in bulk free of charge, and the imatinib and placebo were prepared and packaged by the Brigham and Women’s Hospital investigational drug pharmacy. Novartis reviewed the protocol and the manuscript before initial submission but had no other role in the trial. The authors vouch for the accuracy and completeness of the data and the fidelity of the trial to the protocol, which is available with the full text of this article at NEJM.org.
The patients were stratified according to their use of omalizumab and then were randomly assigned in a 1:1 ratio to the imatinib group or the placebo group. The details of the trial design, screening, and randomization are shown in Figure 1. This trial was approved by the institutional review board at each study site. All the patients provided written informed consent.
Figure 1. Trial Design, Randomization, and Follow-up.
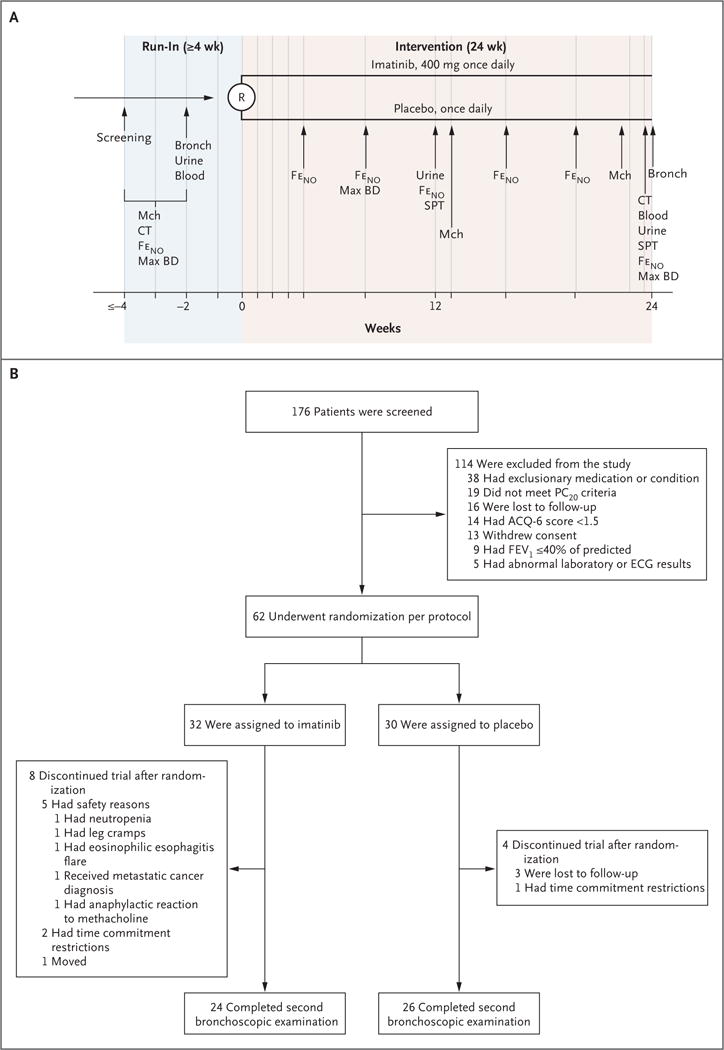
Panel A shows an overview of the trial design and procedures. Blue vertical lines denote study visits. The period between screening and initial bronchoscopy, urine testing, and blood testing was a minimum of 2 weeks in duration; during this period, other tests (computed tomography [CT], fraction of exhaled nitric oxide [FeNO], maximum bronchodilator response [Max BD], and methacholine challenge [Mch]) were performed at intervals that differed among the patients. Spirometry was performed at each study visit. Blood denotes blood testing (assessments of serum tryptase and peripheral-blood eosinophil counts), Bronch bronchoscopy, R randomization, and SPT skin-prick testing. Panel B summarizes the numbers of patients involved in screening, randomization, and trial completion. In the imatinib group, 24 patients completed the trial procedures through the second bronchoscopy and biopsy. In the placebo group, 26 patients completed the trial procedures through the second bronchoscopy and biopsy. Scores on the six-item Asthma Control Questionnaire (ACQ-6) range from 0 to 6, with lower values denoting better asthma control. The minimally important difference is 0.5. ECG denotes electrocardiographic, FEV1 forced expiratory volume in 1 second, and PC20 the concentration of methacholine required to decrease the FEV1 by 20%.
PATIENTS
We enrolled patients 18 to 65 years of age who had severe, refractory asthma that was not controlled despite continuous treatment with inhaled beclomethasone at a dose higher than 960 μg per day or equivalent and at least one additional controller medication. To be eligible for participation, patients were required to have score on the six-item Asthma Control Questionnaire (ACQ-6) of at least 1.5 (scores range from 0 to 6, with lower values denoting better control; minimally important difference, 0.5)18,19 and a provocative concentration of methacholine causing a decrease in forced expiratory volume in 1 second (FEV1) of 20% (PC20) that was 10 mg per milliliter or lower. Additional inclusion criteria were an FEV1 that was at least 40% of the predicted value and adherence of at least 80% to peak flow and diary recordings during the screening period. The full inclusion and exclusion criteria are provided in the protocol.
INTERVENTIONS
The patients were randomly assigned to receive imatinib or placebo once daily. Imatinib treatment was initiated at an oral dose of 200 mg per day for 2 weeks, after which the dose was increased to 400 mg per day, which is the dose that has been shown to inhibit the KIT receptor.20 During the trial period, the patients underwent prerandomization bronchoscopy with airway biopsy, which was repeated at week 24 of the intervention period (Table S1 in the Supplementary Appendix, available at NEJM.org).
OUTCOMES
The primary outcome was the change in airway hyperresponsiveness, assessed as PC20, from baseline to 3 and 6 months of therapy in the imatinib group as compared with the corresponding changes in the control group. We also assessed other physiological and patient-reported measures of asthma control as secondary outcomes, as detailed in the protocol, including FEV1, peak expiratory flow, the number and localization of mast cells in the endobronchial-biopsy samples, tryptase levels in serum and BAL fluid, and asthma-related quality of life. For exploratory prespecified analyses in the imatinib group, we examined, in a correlative manner, the relationship of changes in airway hyperresponsiveness and FEV1 to neutrophil and eosinophil counts in BAL fluid; tryptase-positive mast-cell counts in total airway and smooth-muscle endobronchial-biopsy samples; eosinophil counts in blood; levels of the mast-cell–related mediators histamine, prostaglandin D2, and cysteinyl leukotrienes in BAL fluid; and levels of cysteinyl leukotriene E4 and prostaglandin D2 metabolite in urine.
MEASUREMENTS
Allergy skin testing was performed with the use of the Multi-Test II device (Lincoln Diagnostics). The fraction of exhaled nitric oxide (FeNO) (NIOX MINO, Aerocrine) and patient-reported outcomes (the ACQ-6, the Asthma Quality-of-Life Questionnaire [AQLQ; scores range from 1 to 7, with higher values denoting better quality of life; minimally important difference, 0.5], and the Asthma Symptom Utility Index [ASUI, an assessment of asthma symptoms and treatment side effects weighted according to patient preferences; scores range from 0 to 1.0, with higher scores indicating better control of symptoms and side effects; minimally important difference, 0.09]) were assessed with the use of validated instruments and measures.18,19,21–23 Asthma exacerbations were defined as an increase in asthma symptoms leading to an emergency department visit or hospitalization, initiation of oral glucocorticoid treatment in the patients who were not regularly taking oral glucocorticoids, or a doubling or an increase of 10 mg per day in the dose of glucocorticoids in the patients who were regularly taking glucocorticoids.
Biologic fluids were assessed for the mast-cell activation markers tryptase, histamine, cysteinyl leukotrienes, and prostaglandin D2.24–26 Computed tomographic (CT) assessment of airway-wall thickness was performed with the use of three-dimensional multidetector CT methods in conjunction with advanced image-processing tools. Airway-wall area and thickness percentages were calculated as previously reported.27 Details of the above methods are provided in the Supplementary Appendix.
STATISTICAL ANALYSIS
The primary outcome was the between-group difference in the change in airway hyperresponsiveness to methacholine. We compared the change in log-transformed methacholine reactivity (PC20) in the imatinib group versus the change in the placebo group from baseline at months 3 and 6. We used a mixed-model repeated-measures analysis for comparisons between the groups. The primary analysis was performed with the use of a modified per-protocol population, which included all randomly assigned patients for whom there was at least one postbaseline observation. The target enrollment was 60 patients, under the assumption of a 10% dropout rate, which would result in 54 patients completing the trial; this provided 80% power to detect a doubling-dose change in PC20. We also used a linear model to assess, as secondary outcomes, differences in the mean change from baseline between the groups (with adjustment for baseline values) in physiological and patient-reported outcomes. We compared FEV1 with a mixed model to assess the between-group difference in the change from baseline over weeks 8 through 24. Values are reported as means and standard deviations unless otherwise noted. The Spearman’s correlation coefficient was used to assess the association between specified variables. SAS software, version 9.4 (SAS Institute), was used to perform these analyses.
RESULTS
PATIENTS
A total of 176 potential participants were screened, 62 of whom underwent randomization per protocol (32 in the imatinib group and 30 in the placebo group) and 50 of whom completed the trial (Fig. 1). The baseline characteristics of the patients who underwent randomization are summarized in Table 1. There were no significant differences in baseline characteristics between the groups. A total of 12 patients discontinued participation in the trial, 5 of whom stopped because of adverse events (Fig. 1B).
Table 1.
Baseline Characteristics of the Patients in the Intention-to-Treat Population.*
| Characteristic | Imatinib (N = 32) |
Placebo (N = 30) |
|---|---|---|
| Age — yr | 42.0±10.2 | 37.7±11.8 |
| Female sex — no. (%) | 19 (59) | 18 (60) |
| White race — no. (%)† | 19 (59) | 15 (50) |
| Non-Hispanic ethnic background — no. (%)† | 27 (84) | 25 (83) |
| Age at onset of asthma — yr | 16.1±12.9 | 11.8±13.1 |
| Log2 methacholine PC20‡ | 1.21±1.29 | 1.16±1.66 |
| FEV1 — liters | 2.27±0.64 | 2.13±0.79 |
| Percent of predicted | 71.9±14.6 | 65.5±17.6 |
| Maximum post-bronchodilation FEV1 — liters | 2.54±0.66 | 2.56±0.77 |
| FVC — liters | 3.34±0.85 | 3.31±0.95 |
| Percent of predicted | 83.7±17.8 | 84.0±17.1 |
| PEF — liters/min | ||
| Morning | 349±96 | 331±112 |
| Evening | 358±94 | 338±106 |
| FeNO — PPb | 35.6±26.3 | 35.2±46.6 |
| Peripheral eosinophil count per cubic millimeter | 452±59 | 303±36 |
| Median total IgE level (IQR) — lU/ml | 185 (45–559) | 150 (58–317) |
| No. of positive skin tests | 2.7±2.2 | 2.7±2.2 |
| ACQ-6 score§ | 2.47±0.87 | 2.61±0.94 |
| AQLQ score¶ | 4.56±1.19 | 4.14±1.32 |
| ASUI score‖ | 0.62±0.18 | 0.64±0.18 |
| Other medications used — no. of patients | ||
| Beclomethasone or equivalent inhaled glucocorticoid** | 32 | 30 |
| Oral glucocorticoid | 5 | 2 |
| Omalizumab | 2 | 3 |
| Long-acting beta-agonist | 31 | 30 |
| Long-acting muscarinic antagonist | 2 | 3 |
| Leukotriene modifier | 8 | 17 |
Plus–minus values are means ±SD. There were no significant differences between the groups. To convert the values for IgE to micrograms per liter, multiply by 2.40. BD denotes bronchodilator, FeNO fraction of exhaled nitric oxide, FEV1 forced expiratory volume in 1 second, FVC forced vital capacity, PC20 provocative concentration of methacholine causing a 20% decrease in FEV1, and PEF peak expiratory flow.
Race and ethnic background were reported by the patient.
Data were available for 24 patients in the imatinib group and 27 patients in the placebo group.
Scores on the six-item Asthma Control Questionnaire (ACQ-6) range from 0 to 6, with lower values denoting better asthma control. The minimally important difference is 0.5.
The Asthma Quality-of-Life Questionnaire (AQLQ) is a 32-item scale; scores range from 1 to 7, with higher values denoting better quality of life. The minimally important difference is 0.5.
The Asthma Symptom Utility Index (ASUI) is 10-item scale for the assessment of asthma symptoms and treatment side effects weighted according to patient preferences; scores range from 0 to 1.0, with higher values indicating better control of symptoms and side effects. The minimally important difference is 0.09.
This category includes inhaled beclomethasone (with hydrofluoroalkane propellant) at a dose higher than 960 μg per day (or equivalent).
PRIMARY OUTCOME
Imatinib reduced airway reactivity to a greater extent than did placebo. Imatinib increased the methacholine PC20 by a mean (±SD) of 1.20±0.52 doubling doses from baseline to month 3 (P = 0.03) and by 1.73±0.60 doubling doses from baseline to month 6 (P = 0.008), as compared with increases of 0.03±0.42 (P = 0.94) and 1.07±0.60 (P = 0.08) doubling doses, respectively, in the placebo group (P = 0.048 for the difference between imatinib and placebo over the course of the trial) (Fig. 2).
Figure 2. Change in Airway Methacholine Reactivity.
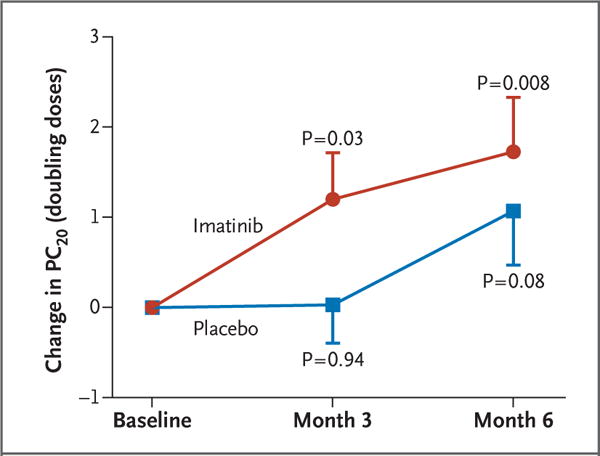
Shown is the mean change (±SE) in the concentration of methacholine required to cause a 20% decrease in FEV1 from baseline (PC20) at month 3 and month 6 relative to the baseline methacholine PC20 obtained before the administration of imatinib or placebo. The P values shown are for the paired t-test evaluating the difference between the indicated time point and baseline. The P value for the between-group difference in the change in values, determined by a mixed-model analysis, is 0.048.
SECONDARY OUTCOMES
Imatinib reduced the evidence of mast-cell activity. At baseline, the mean tryptase level in serum was 4.75±2.59 ng per milliliter in the imatinib group and 4.86±2.13 ng per milliliter in the placebo group. The total tryptase level in serum decreased by 2.02±2.32 ng per milliliter (42.7±31.6%) in the imatinib group, as compared with 0.56±1.39 ng per milliliter (11.5±31.0%) in the placebo group (P = 0.02 for the between-group difference in the change from baseline) (Fig. 3). Tryptase levels in BAL fluid tended to decrease with imatinib, whereas they increased with placebo (a decrease from 1.34±2.19 to 0.60±0.86 ng per milliliter in the imatinib group vs. an increase from 0.96±0.91 to 1.39±2.2 ng per milliliter in the placebo group, P = 0.12 for the between-group difference in the change from baseline) (Table 2). Total and smooth muscle–associated mast-cell counts in endobronchial-biopsy samples had decreased in both groups, with a non-significant trend toward a greater reduction in the imatinib group (Table 2).
Figure 3. Total Tryptase Levels in Serum.
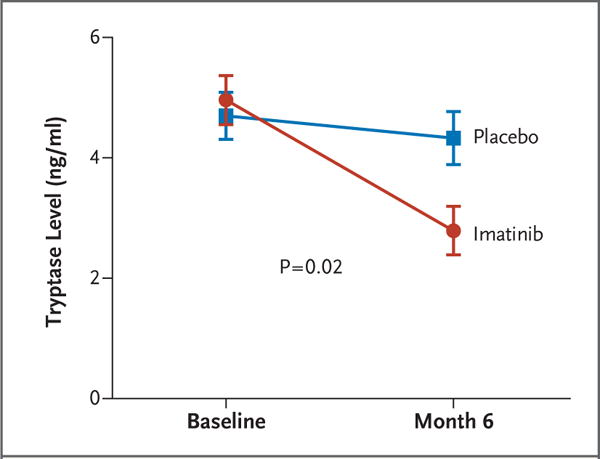
Shown are the mean total tryptase levels (±SE) in serum, as measured by immunoassay (UniCAP, Pharmacia). The P value is for the between-group difference in the change in values from baseline. A 42.7±31.6% decrease in the serum tryptase level was observed in the imatinib group, as compared with an 11.5±31.0% decrease in the placebo group.
Table 2.
Change from Baseline to 6 Months in Clinical and Inflammatory Measures with Imatinib and Placebo.*
| Measure | Change from Baseline | P Value | |
|---|---|---|---|
| Imatinib | Placebo | ||
| PEF (liters/min) | |||
| Morning | 7.3±46.1 | −6.4±39.3 | 0.38 |
| Evening | 8.3±53.6 | −8.2±37.2 | 0.31 |
|
| |||
| FeNO (ppb) | 7.89±33.0 | −5.92±33.2 | 0.11 |
|
| |||
| Maximum post-bronchodilation FEV1 (liters) | 0.01±0.2 | −0.08±0.26 | 0.10 |
|
| |||
| ACQ-6 score | −0.62±0.96 | −0.49±0.89 | 0.31 |
|
| |||
| AQLQ score | 0.55±1.0 | 0.25±0.80 | 0.11 |
|
| |||
| ASUI score | 0.07±0.20 | 0.05±0.18 | 0.62 |
|
| |||
| Airway-wall thickness (%)† | −0.0040± 0.03 | −0.0027±0.02 | 0.13 |
|
| |||
| Airway-wall area (%)† | 0.0002±0.02 | 0.0002±0.02 | 0.25 |
|
| |||
| Peripheral-blood eosinophil count per cubic millimeter | −10.22±50.6 | −2.59±25.6 | 0.94 |
|
| |||
| BAL eosinophils (%) | 2.55±8.8 | −2.63±10.4 | 0.15 |
|
| |||
| Tryptase-positive mast-cell count per square millimeter | |||
| Total airway | −54.2±96.5 | −32.3±79.8 | 0.11 |
| Airway smooth muscle | −102.7±167.9 | −79.2±157.3 | 0.07 |
|
| |||
| No. of positive skin-prick tests | −0.71±1.62 | −0.61±2.4 | 0.87 |
|
| |||
| BAL tryptase (ng/ml) | −0.74±2.35 | 0.43±2.19 | 0.12 |
|
| |||
| BAL prostaglandin D2 (pg/ml) | 12.2±57.2 | −4.2±54.4 | 0.33 |
|
| |||
| BAL cysteinyl leukotrienes (pg/ml) | 3.0±37.3 | 6.5±32.9 | 0.56 |
|
| |||
| BAL histamine (nmol/liter) | 2.1±6.3 | −1.1±13.4 | 0.54 |
|
| |||
| Urinary prostaglandin D2 metabolite (ng/mg creatinine) | −0.30±1.5 | 0.39±1.73 | 0.18 |
|
| |||
| Urinary leukotriene E4 (ng/mg creatinine) | 0.07±0.65 | 0.01±0.18 | 0.47 |
Group comparisons were adjusted for baseline values and assessed with the use of the linear model. All patients who underwent randomization were included. Baseline measures are provided in Table S4 in the Supplementary Appendix. On the basis of a Poisson regression model, from baseline to 6 months, a total of 16 asthma exacerbations occurred in the imatinib group and 20 exacerbations occurred in the placebo group (P = 0.36). BAL denotes bronchoalveolar lavage.
Measurement was made with the use of three-dimensional multidetector computed tomography.
Although results of airway reversibility tests were not among the criteria used when patients were screened for the trial, at week 24 there was a difference of 46 ml (95% confidence interval, 36 to 56) in the change from baseline in mean FEV1 between the imatinib group and the placebo group (P = 0.04) (Fig. S1 in the Supplementary Appendix). The patients who were treated with imatinib had numerically fewer asthma exacerbations, greater reductions in airway-wall thickness, higher morning and evening peak expiratory flows, and greater improvements in patient-reported outcomes as assessed by the ACQ-6,18 AQLQ,19 and ASUI22 than did the patients who received placebo, but none of the differences were significant (Table 2). There were no significant differences between imatinib and placebo with regard to effects on eosinophil counts in peripheral blood and BAL fluid, FeNO, urinary leukotriene E4 or prostaglandin D2 metabolite levels, or cysteinyl leukotriene, prostaglandin D2, or histamine levels in BAL fluid (Table 2).
A total of 27 secondary outcome measures were prespecified in the protocol; we report data for 21 of these outcomes in this article and in the Supplementary Appendix. The data and status for all primary and secondary outcomes are available at ClinicalTrials.gov (https://clinicaltrials.gov/ct2/show/results/NCT01097694).
TERTIARY ANALYSES
In the imatinib group, decreases in airway hyper-responsiveness were inversely correlated with baseline peripheral-blood eosinophil counts (r2 = 0.218, P = 0.03). We found a nonsignificant trend toward correlation between reductions in total airway mast-cell counts and increases in FEV1 (r2 = 0.163, P = 0.06) (Fig. S2 in the Supplementary Appendix). Increases in FEV1 were positively correlated with baseline BAL neutrophil counts (r2 = 0.441, P = 0.003). We found no correlation between changes in airway hyperresponsiveness or FEV1 and baseline eosinophil counts in BAL fluid, tryptase-positive mast-cell counts in endobronchial-biopsy samples of total airway and smooth muscle, or levels of the mast cell–related mediators histamine, prostaglandin D2, and cysteinyl leukotrienes.
SAFETY
The number of total and severe adverse events did not differ significantly between the groups (Table S2 in the Supplementary Appendix). Imatinib was associated with a higher likelihood of muscle cramps and metabolic abnormalities, specifically hypophosphatemia. Two patients in the imatinib group discontinued participation in the trial because of adverse events (neutropenia in one patient and leg cramps in the other) that were thought by investigators, who were unaware of the study-group assignments, to be related to the study agent (Fig. 1B). Overall, 7 severe adverse events were reported in three patients during imatinib therapy, and 10 severe adverse events were reported in five patients who received placebo. When asthma exacerbations were not counted as severe adverse events, there were 5 events in the imatinib group and 3 in the placebo group (Table S3 in the Supplementary Appendix).
DISCUSSION
Patients with severe asthma often have airway hyperresponsiveness and poor disease control despite the use of high-dose inhaled glucocorticoids and additional controller medications. They have a high airway mast-cell burden — a feature linked to airway hyperresponsiveness — as compared with patients with milder asthma,3,28 which suggests a possible pathobiologic role for mast cells in these patients. In this proof-of-principle trial, we sought to determine whether inhibition of KIT could reduce the mast-cell burden and thereby reduce the airway hyperresponsiveness and secondary outcomes associated with clinical asthma. We used the KIT inhibitor imatinib to treat patients who had severe, poorly controlled asthma and methacholine reactivity despite receiving maximum guideline-directed therapy.29
We found that imatinib reduced total serum tryptase levels to a greater extent than did placebo, a finding consistent with decreases in mast-cell numbers and possibly with reductions in mast-cell activation. Imatinib also tended to reduce the tryptase levels in BAL fluid, whereas the levels increased slightly in the placebo group. At the same time, imatinib decreased airway hyper-responsiveness and produced a small but significant increase in FEV1.
The effect of imatinib on airway hyperresponsiveness was apparent at 3 months, at which point the increase in methacholine PC20 was more than 1 doubling dose greater in the imatinib group than in the placebo group; 1 doubling dose is considered a clinically important difference for an individual patient.6 Airway hyperresponsiveness continued to decrease during the next 3 months, such that at the end of the trial, the PC20 in the imatinib group had increased to 330% of the baseline value. Airway hyperresponsiveness also began to decrease in the placebo group after 3 months. This improvement is consistent with the delayed long-term improvement in airway hyperresponsiveness in response to treatment with inhaled glucocorticoids that has been noted by others30 and may be due to persistent adherence in both the imatinib group and the placebo group as a result of the patients’ participation in the trial.
We noted that increases in FEV1 in the imatinib group tended to correlate with reductions in endobronchial mast-cell counts, a trend that was not observed in the placebo group. Although the increase in FEV1 may not seem substantial, it suggests that mast-cell–dependent processes contribute to airway obstruction in these patients despite high-dose antiinflammatory glucocorticoid therapy. The near-50-ml difference in the change from baseline in FEV1 between the imatinib and placebo groups is small, but it is likely to be important in light of the population we studied. The patients in our trial population had less than half the baseline bronchodilator response of patients who are enrolled in most trials of new biologic agents,31 and 50 ml was proportionately half the response shown in those trials.31
It is important to highlight the fact that mast cells can survive for months to years in tissue and that their numbers are increased in states of inflammation. Our data (tryptase levels and endobronchial-biopsy mast-cell counts) show that 6 months of imatinib therapy reduced but did not completely eliminate mast-cell counts in the airways of patients with severe asthma. Data from studies of other diseases have shown that treatment with imatinib for more than 1 year reduces numbers of resident mast cells more than do shorter durations of treatment.15 Thus, larger and perhaps longer clinical trials will be required in order to understand whether specific mast-cell–targeted therapies will be clinically effective.
In addition to effecting classical IgE-mediated activation, mast cells can also serve as effectors of innate and type 1 immune mechanisms and can amplify airway hyperresponsiveness, airway remodeling, and neutrophilic airway inflammation in animal models.32 In exploratory analyses, the decrease in airway hyperresponsiveness associated with imatinib was negatively correlated with baseline blood eosinophil counts, and baseline numbers of neutrophils in BAL fluid were strongly correlated with increases in FEV1 (r2 = 0.441, P = 0.003). Together, these findings support a role for mast cells in noneosinophilic asth ma. Since almost half of the patients with severe asthma have neutrophilic airway inflammation,33 we speculate that KIT inhibition might represent an important approach to treatment for this group.
Several caveats need to be considered. It is possible that some of the positive effects seen in this trial are not related to mast-cell inhibition. KIT is expressed by both group 2 and group 3 innate lymphoid cells, which can produce cytokines that drive eosinophilic and neutrophilic inflammation, respectively.34–36 Imatinib also inhibits platelet-derived growth factor receptor signal transduction, which can mediate airway smooth-muscle proliferation and contractility.37 It is also possible that the effect we found with regard to airway hyperresponsiveness will not translate into a clinical benefit in larger studies.
Imatinib was associated with adverse effects that resolved with discontinuation of treatment. The side-effect profile of this particular KIT inhibitor needs to be taken into account in the design of larger and longer trials of this therapeutic strategy in patients with severe asthma.
In conclusion, this proof-of-principle trial showed that antagonism of KIT and decreases in mast-cell counts were associated with reductions in airway hyperresponsiveness and small increases in FEV1 in a group of patients with severe asthma who were already taking maximal therapy. These data are not clinically directive, but they set the stage for follow-up studies targeting mast cells in patients with severe asthma.
Supplementary Material
Acknowledgments
Supported by grants from the National Heart, Lung, and Blood Institute and the National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIH U01 HL102225, RO1 HL117945, RO1 AI078908, R37 AI052353, T32 AI007306-23, and K23 AI118804), and by contributions from the Vinik family and the Kaye family. Novartis provided imatinib free of charge.
We thank Sumit Narula, Bryce Robinson, Vanessa Curtis, Sylvia Johnson, Necole Harris, Michele Wolff, Holly Eversoll, Gina Crisafi, and Jacqueline Sharp for project management assistance; Stephanie Dutile for assistance in preparing the grant proposal and preparation and execution of the trial protocol; Loren Denlinger for clinical support; Geneline Sajol and Anya Cutler for technical assistance; Usha Govindarajula for statistical assistance; and the members of the data and safety monitoring board: Reynold A. Panettieri, Jr., David Schoenfeld, Mark Liu, Robert Wood, and Carol Freund Taylor.
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
References
- 1.Wenzel S. Severe asthma in adults. Am J Respir Crit Care Med. 2005;172:149–60. doi: 10.1164/rccm.200409-1181PP. [DOI] [PubMed] [Google Scholar]
- 2.Chanez P, Wenzel SE, Anderson GP, et al. Severe asthma in adults: what are the important questions? J Allergy Clin Immunol. 2007;119:1337–48. doi: 10.1016/j.jaci.2006.11.702. [DOI] [PubMed] [Google Scholar]
- 3.Busse WW, Banks-Schlegel S, Wenzel SE. Pathophysiology of severe asthma. J Allergy Clin Immunol. 2000;106:1033–42. doi: 10.1067/mai.2000.111307. [DOI] [PubMed] [Google Scholar]
- 4.Limb SL, Brown KC, Wood RA, et al. Irreversible lung function deficits in young adults with a history of childhood asthma. J Allergy Clin Immunol. 2005;116:1213–9. doi: 10.1016/j.jaci.2005.09.024. [DOI] [PubMed] [Google Scholar]
- 5.Porsbjerg C, Rasmussen L, Nolte H, Backer V. Association of airway hyperresponsiveness with reduced quality of life in patients with moderate to severe asthma. Ann Allergy Asthma Immunol. 2007;98:44–50. doi: 10.1016/S1081-1206(10)60858-7. [DOI] [PubMed] [Google Scholar]
- 6.Sont JK, Willems LN, Bel EH, van Krieken JH, Vandenbroucke JP, Sterk PJ. Clinical control and histopathologic outcome of asthma when using airway hyperresponsiveness as an additional guide to long-term treatment. Am J Respir Crit Care Med. 1999;159:1043–51. doi: 10.1164/ajrccm.159.4.9806052. [DOI] [PubMed] [Google Scholar]
- 7.Ward C, Johns DP, Bish R, et al. Reduced airway distensibility, fixed airflow limitation, and airway wall remodeling in asthma. Am J Respir Crit Care Med. 2001;164:1718–21. doi: 10.1164/ajrccm.164.9.2102039. [DOI] [PubMed] [Google Scholar]
- 8.Bradding P, Walls AF, Holgate ST. The role of the mast cell in the pathophysiology of asthma. J Allergy Clin Immunol. 2006;117:1277–84. doi: 10.1016/j.jaci.2006.02.039. [DOI] [PubMed] [Google Scholar]
- 9.Siddiqui S, Mistry V, Doe C, et al. Airway hyperresponsiveness is dissociated from airway wall structural remodeling. J Allergy Clin Immunol. 2008;122:335–41. doi: 10.1016/j.jaci.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brightling CE, Bradding P, Symon FA, Holgate ST, Wardlaw AJ, Pavord ID. Mast-cell infiltration of airway smooth muscle in asthma. N Engl J Med. 2002;346:1699–705. doi: 10.1056/NEJMoa012705. [DOI] [PubMed] [Google Scholar]
- 11.Da Silva CA, Reber L, Frossard N. Stem cell factor expression, mast cells and inflammation in asthma. Fundam Clin Pharmacol. 2006;20:21–39. doi: 10.1111/j.1472-8206.2005.00390.x. [DOI] [PubMed] [Google Scholar]
- 12.Makowska JS, Cieslak M, Kowalski ML. Stem cell factor and its soluble receptor (c-kit) in serum of asthmatic patients-correlation with disease severity. BMC Pulm Med. 2009;9:27. doi: 10.1186/1471-2466-9-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carroll M, Ohno-Jones S, Tamura S, et al. CGP 57148, a tyrosine kinase inhibitor, inhibits the growth of cells expressing BCR-ABL, TEL-ABL, and TEL-PDGFR fusion proteins. Blood. 1997;90:4947–52. [PubMed] [Google Scholar]
- 14.Heinrich MC, Griffith DJ, Druker BJ, Wait CL, Ott KA, Zigler AJ. Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood. 2000;96:925–32. [PubMed] [Google Scholar]
- 15.Cerny-Reiterer S, Rabenhorst A, Stefanzl G, et al. Long-term treatment with imatinib results in profound mast cell de ficiency in Ph+ chronic myeloid leukemia. Oncotarget. 2015;6:3071–84. doi: 10.18632/oncotarget.3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farha S, Dweik R, Rahaghi F, et al. Imatinib in pulmonary arterial hypertension: c-Kit inhibition. Pulm Circ. 2014;4:452–5. doi: 10.1086/677359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kraft M, Martin RJ, Lazarus SC, et al. Airway tissue mast cells in persistent asthma: predictor of treatment failure when patients discontinue inhaled corticosteroids. Chest. 2003;124:42–50. doi: 10.1378/chest.124.1.42. [DOI] [PubMed] [Google Scholar]
- 18.Juniper EF, O’Byrne PM, Guyatt GH, Ferrie PJ, King DR. Development and validation of a questionnaire to measure asthma control. Eur Respir J. 1999;14:902–7. doi: 10.1034/j.1399-3003.1999.14d29.x. [DOI] [PubMed] [Google Scholar]
- 19.Juniper EF, Guyatt GH, Willan A, Griffith LE. Determining a minimal important change in a disease-specific Quality of Life Questionnaire. J Clin Epidemiol. 1994;47:81–7. doi: 10.1016/0895-4356(94)90036-1. [DOI] [PubMed] [Google Scholar]
- 20.Prescribing information: Gleevec. Novartis. 2016 Sep; https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/gleevec_tabs.pdf.
- 21.Juniper EF, Guyatt GH, Ferrie PJ, Griffith LE. Measuring quality of life in asthma. Am Rev Respir Dis. 1993;147:832–8. doi: 10.1164/ajrccm/147.4.832. [DOI] [PubMed] [Google Scholar]
- 22.Revicki DA, Leidy NK, Brennan-Diemer F, Sorensen S, Togias A. Integrating patient preferences into health outcomes assessment: the multiattribute Asthma Symptom Utility Index. Chest. 1998;114:998–1007. doi: 10.1378/chest.114.4.998. [DOI] [PubMed] [Google Scholar]
- 23.Juniper EF, Guyatt GH, Epstein RS, Ferrie PJ, Jaeschke R, Hiller TK. Evaluation of impairment of health related quality of life in asthma: development of a questionnaire for use in clinical trials. Thorax. 1992;47:76–83. doi: 10.1136/thx.47.2.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morrow JD, Guzzo C, Lazarus G, Oates JA, Roberts LJ., II Improved diagnosis of mastocytosis by measurement of the major urinary metabolite of prostaglandin D2. J Invest Dermatol. 1995;104:937–40. doi: 10.1111/1523-1747.ep12606209. [DOI] [PubMed] [Google Scholar]
- 25.Morrow JD, Minton TA. Improved assay for the quantification of 11-dehydrothromboxane B2 by gas chromatography-mass spectrometry. J Chromatogr. 1993;612:179–85. doi: 10.1016/0378-4347(93)80161-v. [DOI] [PubMed] [Google Scholar]
- 26.Kim DC, Hsu FI, Barrett NA, et al. Cysteinyl leukotrienes regulate Th2 cell-dependent pulmonary inflammation. J Immunol. 2006;176:4440–8. doi: 10.4049/jimmunol.176.7.4440. [DOI] [PubMed] [Google Scholar]
- 27.Aysola RS, Hoffman EA, Gierada D, et al. Airway remodeling measured by multi-detector CT is increased in severe asthma and correlates with pathology. Chest. 2008;134:1183–91. doi: 10.1378/chest.07-2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gibson PG, Saltos N, Borgas T. Airway mast cells and eosinophils correlate with clinical severity and airway hyperresponsiveness in corticosteroid-treated asthma. J Allergy Clin Immunol. 2000;105:752–9. doi: 10.1067/mai.2000.105319. [DOI] [PubMed] [Google Scholar]
- 29.National Asthma Education and Prevention Program. Expert Panel Report 3 (EPR-3): Guidelines for the Diagnosis and Management of Asthma — summary report 2007. J Allergy Clin Immunol. 2007;120(Suppl):S94–138. doi: 10.1016/j.jaci.2007.09.043. [DOI] [PubMed] [Google Scholar]
- 30.Juniper EF, Kline PA, Vanzieleghem MA, Ramsdale EH, O’Byrne PM, Harg-reave FE. Effect of long-term treatment with an inhaled corticosteroid (budesonide) on airway hyperresponsiveness and clinical asthma in nonsteroid-dependent asthmatics. Am Rev Respir Dis. 1990;142:832–6. doi: 10.1164/ajrccm/142.4.832. [DOI] [PubMed] [Google Scholar]
- 31.Ortega HG, Liu MC, Pavord ID, et al. Mepolizumab treatment in patients with severe eosinophilic asthma. N Engl J Med. 2014;371:1198–207. doi: 10.1056/NEJMoa1403290. [DOI] [PubMed] [Google Scholar]
- 32.Yu M, Eckart MR, Morgan AA, et al. Identification of an IFN-γ/mast cell axis in a mouse model of chronic asthma. J Clin Invest. 2011;121:3133–43. doi: 10.1172/JCI43598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wenzel SE, Schwartz LB, Langmack EL, et al. Evidence that severe asthma can be divided pathologically into two inflammatory subtypes with distinct physiologic and clinical characteristics. Am J Respir Crit Care Med. 1999;160:1001–8. doi: 10.1164/ajrccm.160.3.9812110. [DOI] [PubMed] [Google Scholar]
- 34.Boyd A, Ribeiro JM, Nutman TB. Human CD117 (cKit)+ innate lymphoid cells have a discrete transcriptional profile at homeostasis and are expanded during filarial infection. PLoS One. 2014;9(9):e108649. doi: 10.1371/journal.pone.0108649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klose CS, Artis D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat Immunol. 2016;17:765–74. doi: 10.1038/ni.3489. [DOI] [PubMed] [Google Scholar]
- 36.McKenzie AN, Spits H, Eberl G. Innate lymphoid cells in inflammation and immunity. Immunity. 2014;41:366–74. doi: 10.1016/j.immuni.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 37.Ingram JL, Bonner JC. EGF and PDGF receptor tyrosine kinases as therapeutic targets for chronic lung diseases. Curr Mol Med. 2006;6:409–21. doi: 10.2174/156652406777435426. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.