

Summary
The CRISPR/Cas9 system has recently been shown to facilitate high levels of precise genome editing using adeno associated viral (AAV) vectors to serve as donor template DNA during homologous recombination (HR). However, the maximum AAV packaging capacity of ~4.5 kilobases limits the donor size. Here we overcome this constraint by showing that two co-transduced AAV vectors can serve as donors during consecutive HR events for integration of large transgenes. Importantly, the method involves a single-step procedure applicable to primary cells with relevance to therapeutic genome editing. We use the methodology in primary human T cells and CD34+ hematopoietic stem and progenitor cells to site-specifically integrate an expression cassette that as a single donor vector would otherwise amount to a total of 6.5 kilobases. This approach now provides an efficient way to integrate large transgene cassettes into the genomes of primary human cells using HR-mediated genome editing with AAV vectors.
eTOC Blurb
Integration of transgenes into specific sites of the genome of primary cells using CRISPR/Cas9 and AAV donor vectors is currently hampered by the limited packaging capacity of AAV. Bak and Porteus now report a method for efficient integration of large transgenes that exceed the capacity of a single AAV.
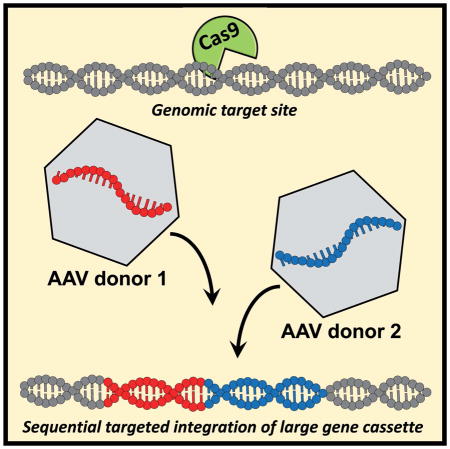
Introduction
Precise genome editing can be accomplished using designer nucleases (e.g. ZFNs and TALENs) or RNA-guided nucleases (e.g. CRISPR/Cas9), which create site-specific double-strand breaks that stimulate homologous recombination (HR) when supplied with a homologous donor DNA template. This method can facilitate targeted integration of transgenes for gene therapy or studies of gene function.
Viral vectors derived from the non-pathogenic, single-stranded DNA virus, adeno-associated virus (AAV), can transduce both dividing and non-dividing cells and have recently been effectively used as donor vectors for HR both in vitro and in vivo (Sather et al., 2015, Wang et al., 2015, DeWitt et al., 2016, Yang et al., 2016, Yin et al., 2016, Dever et al., 2016, De Ravin et al., 2017). However, despite decades of research, the 4.5 kb packaging capacity of recombinant AAV (rAAV) vectors has not been successfully extended, though strategies that enable episomal expression of transgenes that exceed the packaging capacity have been devised (Grieger and Samulski, 2005, Chamberlain et al., 2016, Nakai et al., 2000, Sun et al., 2000, Halbert et al., 2002). The constraint in vector capacity limits applications of genome editing with AAV donor vectors since the homology arms required for efficient HR add a minimum of 2 x 0.4 kilobases (kb) to the vector (Hendel et al., 2014), leaving 3.7 kb for promoter, polyadenylation signal, and transgene. Several genetic diseases involve mutations in genes that exceed this limit, such as Duchenne Muscular Dystrophy (dystrophin: 11 kb;), hemophilia A (Factor VIII: 7 kb), and Cystic Fibrosis (CFTR: 4.4 kb). If required, post-transcriptional regulatory elements would add further to the vector, and depending on gene size the use of multi-cistronic cassettes would be limited.
Here we present a CRISPR/Cas9-based methodology that enables site-specific integration of large transgenes that are split between two AAV donor vectors, and show that this process occurs at high frequencies in the K562 cell line, primary human T cells, and in CD34+ hematopoietic stem and progenitor cells (HSPCs) with long-term repopulation capacity.
Results
Since HR is a seamless process, we envisaged that two parts of a large transgene could be fused together by consecutive HR events using two different AAV donor vectors. Donor A integrates ‘Part A’ of the transgene and a key feature of Donor A is that it contains the same sgRNA target site that mediated its integration so that the site is reconstituted in the genome after integration (Figure 1). 400 bp stuffer DNA after the target site serves as homology arm for Donor B to avoid using the same homology arm as Donor A, which could enable Donor B to be integrated instead of Donor A during the first HR step using only a single homology arm as has previously been reported (Basiri et al., 2017, Lombardo et al., 2007). Homologous recombination of Donor B fuses ‘Part B’ of the transgene to ‘Part A’ using the introduced sgRNA target site.
Figure 1. Sequential homologous recombination of two AAV6 donors with a split gene.

Schematic overview of a two-step HR platform, in which a gene is split between two HR donors (Donor A and B), which undergo sequential HR. Donor A carries a sgRNA target site (red box) immediately after ‘part A’ of the transgene. This allows HR of Donor B using the same sgRNA, which seamlessly fuses ‘part B’ of the transgene to ‘part A’. Stuffer DNA (white box) after the sgRNA target site is used as homology arm for Donor B to avoid re-using the right homology arm from Donor A.
As a first proof of principle of this approach, we designed a donor pair targeting the proposed safe harbor locus CCR5, where GFP was split between two donors designed as described above (Figure 2a and 2b). In addition, the two donors each carried different expression cassettes for other fluorescent proteins (BFP and mCherry, respectively) allowing us to confirm that neither donor alone expressed GFP (Figure S1a and S1b), and that only Donor A could serve as the initial HR donor during CCR5 targeting (Figure S1c). We first tested the system in the K562 cell line by co-electroporation of Cas9 mRNA, CCR5-targeting chemically modified sgRNAs (Hendel et al., 2015), and the two plasmid donors with the split GFP. We observed an average of 0.02 % GFP+ cells when only the two plasmid donors were delivered while 0.45 % of cells stably expressed GFP when the CRISPR components were co-electroporated (Figure S1d). We next tested the system with the two donors delivered as AAV6 vectors immediately following electroporation. In mock-electroporated cells receiving both AAV6 donors, transient and low expression of GFP was observed at day 4 after electroporation and transduction, which was lost by day 16 (Figure 2c and Figure S2a). In contrast, electroporation of the CRISPR components and transduction with both AAV6 donors gave rise to a stable population of GFPhigh-expressing cells observed at both day 4 and day 16 in about 40 % of the cells (Figure 2c and Figure S2a), indicative of chromosomal expression of the GFP expression cassette as previously observed (Dever et al., 2016). As expected, the GFP+ population was highly enriched for BFP/mCherry double-positive cells confirming that integration of both donors is required for reconstitution of the GFP expression cassette (Figure S2b). To confirm that HR did occur through a sequential process that relied on the incorporated sgRNA target site in Donor A, we made a new variant of Donor A with a mutation in the PAM site of the sgRNA. Using this PAM-mutated Donor A with Donor B as before, we observed stable GFP expression in an average of only 2.4 % of cells (compared to 38.4 % if the gRNA site was preserved) confirming that the majority of Donor B integration events relied on CRISPR activity at the sgRNA target site in Donor A (Figure S3c).
Figure 2. Sequential HR targeting a GFP gene split between two AAV donor vectors to the CCR5 locus in K562 cells.

(a) Overview of the donor design for splitting GFP between two AAV donors. The endogenous CCR5 target site is shown with the PAM in red and the 20nt target site in purple. The Cas9 cut site is between nucleotide 17 and 18 of the target sequence. Donor A is designed with 2 x 400 bp homology arms (LHA and RHA) that are split at the CCR5 cut site. The homology arms flank a PGK-BFP expression cassette, part A of the GFP expression cassette (SFFV-GFP (A)), a sgRNA target site for the same CCR5 sgRNA, and stuffer DNA (to serve as homology arm for Donor B to avoid having to re-use the 400bp CCR5 left homology arm). After HR of Donor A, Donor B is designed to seamlessly integrate the rest of GFP using the sgRNA target site present in Donor A. Donor B has a LHA homologous to GFP (begins at amino acid 57 of GFP), a RHA consisting of part of the sgRNA target site and the stuffer DNA, and it carries an EF1a-mCherry expression cassette. Neither donor expresses GFP on its own (Figure S1a and S1b). (b) GFP is split at a PAM site for the CCR5 sgRNA. Codons are depicted above the nucleotides. Donor A carries LHA and RHA which are split directly at the Cas9 cut site in CCR5 as depicted in (a). Donor A carries a truncated GFP sequence that ends after the PAM site identified in the GFP gene. Directly after the PAM, the 20nt target site for the same CCR5 sgRNA is introduced. Note that the last codon (Pro) of the truncated GFP sequence is maintained with the fusion to the sgRNA target sequence. Thus, the LHA of Donor B ends right after this proline codon. The right homology arm begins immediately after the Cas9 cut site (scissors). The two homology arms flank the remaining part of GFP and an mCherry expression cassette, see (a), that upon seamless HR of Donor B will reconstitute a functional GFP open reading frame. (c) K562 cells were mock-electroporated or electroporated with Cas9 mRNA and CCR5 synthetic sgRNAs (CRISPR) followed by transduction with the split GFP AAV6 donor pair. GFP expression was measured either by total percent GFP+ cells after 16 days or percent GFPhigh cells 8 days after transduction (see also Figure S2a). Left panel, representative FACS plots. Right panel, frequencies of cells stably expressing GFP, N = 7, error bars represent SD.
We next tested this split GFP system in activated primary human T cells and CD34+ HSPCs with the CRISPR system delivered by electroporation of precomplexed Cas9 ribonucleoprotein (RNP) and the two AAV donors delivered immediately after. In cells electrorated with Cas9 RNP but not receiving AAV6 donors, >90 % INDEL rates were measured, confirming high activity of the Cas9 RNP system in both cells types (Figure S3a-c). In mock-electroporated cells receiving only the AAV6 donor pair, the frequency of GFP+ cells was less than 1.0 % in both cell types on day 4 after transduction. In contrast, electroporation with Cas9 RNP and transduction with both AAV6 donors gave rise to 8.5 % and 9.5 % GFP+ T cells and HSPCs, respectively (Figure 3a and Figure S3d). In comparison, with a single AAV6 donor vector encoding GFP, average targeting rates were 46 % and 19 % (Figure S3e and S3f). Evaluation of cell death and apoptosis in T cells showed little impact of the treatment on viability of the cells (Figure S4g). We next assessed if the sequential HR process was able to target early progenitor cells in the HSPC population capable of forming colonies in methylcellulose. Sorted GFP+ cells formed erythroid, myeloid, and mixed colonies at ratios comparable to mock-electroporated cells, but an overall lower colony formation frequency indicated a lower frequency of progenitor cells in the GFP+ population than in the mock-electroporated population (Figure 3b upper panel and Figure S3h). However, the extent of this decrease was donor-dependent. In-Out PCRs, in which one primer is located in the targeted genomic locus outside the region of the homology arm and the other primer located in the donor DNA, was used to confirm on-target integration (Figure S3i). On genomic DNA derived from GFP+ colonies, we confirmed on-target integration of both Donor A and Donor B in all colonies analyzed (41 colonies total), and sequencing confirmed seamless HR (Figure 3b lower panel and Figure S3i).
Figure 3. Sequential homologous recombination of two AAV6 donors with a split GFP gene in human T cells and CD34+ hematopoietic stem and progenitor cells.

(a) Primary human T cells and CD34+ hematopoietic stem and progenitor cells (HSPCs) were mock-electroporated or electroporated with Cas9 protein precomplexed with CCR5 chemically modified sgRNAs (CRISPR) followed by transduction with a split GFP AAV6 donor pair (Figure 2a). GFP expression was measured by flow cytometry four days after transduction. Left panel, representative FACS plots from the two cell types. Right panel, frequencies of GFP+ cells for the two cell types, N = 11 (T cells, all from different buffy coat donors), N = 12 (HSPCs, all from different cord blood donors). (b) HSPCs were treated as in (a) and at day 4 post-transduction, GFP+ cells were single-cell sorted into 96-well plates containing methylcellulose and progenitor-derived clones were visualized 14 days after seeding. Top panel, fluorescent microscopy images of formed GFP+ colonies from erythroid (BFU-E), granulocyte/macrophage (CFU-GM), and multi-lineage (CFU-GEMM) progenitors (scale bars: blue=200μm, red=1000μm, green=400μm). Bottom panel, In-Out PCR was performed on colony-derived genomic DNA to confirm targeted integration at the 5’ end (Donor A) and at the 3’ end (Donor B) (Figure S3i). Representative gel image of 6 clones of a total of 41 clones analyzed. Input control is PCR amplification of a part of the HBB gene. (c) HSPCs were treated as in (a) and at day 4 post-transduction, GFP+ cells were isolated by FACS and 40,000 cells were transplanted into each of three male NSG mice sublethally irradiated 24 hrs previously. Engraftment of human cells in the bone marrow was assessed 16 weeks post-transplant by flow cytometric detection of HLA-ABC/hCD45 double-positive cells and GFP expression was analyzed within the human population. FACS plots show a negative control mouse not transplanted with human cells and a representative FASC plot from one of the mice transplanted with GFP+ HSPCs (CRISPR + donors), N = 3.
Only a small fraction of the CD34+ HSPCs are stem cells that are capable of long-term repopulation. To examine if the two-step HR process occurred in long-term repopulating stem cells, we transplanted sorted GFP+ HSPCs into three irradiated immunodeficient NOD scid gamma (NSG) mice. 16 weeks after transplant, all mice showed human chimerism in the bone marrow with an average of 91% GFP+ cells in the human population (Figure 3c). Collectively, these data show that a split rAAV donor system can efficiently undergo sequential HR stimulated by the CRISPR system in the K562 cell line, in primary human T cells, and in HSPCs.
The epidermal growth factor receptor (EGFR) with an open reading frame of 3.6 kb can modulate cell migration and proliferation, and has been shown to play roles in HSPC expansion and G-CSF–induced HSPC mobilization (Takahashi et al., 1998, Ryan et al., 2010). We next applied the methodology to try and integrate EGFR into the CCR5 locus. With the EF1α promoter, the Woodchuck Hepatitis Virus Posttranscriptional Regulatory Element (WPRE), bovine growth hormone (BGH) polyadenylation signal (polyA), and two 400 bp homology arms, such targeting vector would be 6.5 kb, greatly exceeding the packaging capacity of AAV. The EGFR gene was split between two donors as before, but to avoid introducing stuffer DNA as in the split GFP system, the WPRE and BGH polyA was introduced along EGFR part A after the sgRNA target site, so that part of WPRE could serve as homology arm for Donor B (Figure 4a). Using this split AAV6 donor pair in T cells and HSPCs, donor only controls yielded less than 1.0 % EGFR+ cells in both cell types, but with Cas9 RNP electroporation we detected an average of 9.8 % and 9.1 % EGFR+ cells in the two cell types, respectively, with similar rates of EGFR+ cells in the CD4 and CD8 sub-populations (Figure 4b and Figure S4a). Quantification of INDEL rates showed that alleles that had not undergone HR mainly harbored INDELs (Figure S4b). Minimal toxicity was observed in T cells, while modest toxicity was observed in CD34+ HSPCs, which was mainly caused by the high MOI used for AAV6 transduction (Figure S4c and S4d). Colony-forming unit assays on the EGFR+ HSPC population showed formation of erythroid, myeloid, and mixed colonies at comparable ratio to mock-electroporated cells and a small, but non-statistical significant decrease in overall formation frequency (Figure S4e). Finally, In-Out PCRs on colony-derived genomic DNA confirmed on-target integration in all analyzed colonies (20 colonies total), and sequencing confirmed seamless integration by HR (Figure 4c).
Figure 4. Sequential homologous recombination of two AAV6 donors with a split EGFR gene in human T cells and CD34+ hematopoietic stem and progenitor cells.

(a) Schematic overview of a two-step HR platform integrating an EGFR expression cassette into the CCR5 gene. Donor A carries all elements of the expression cassette, but only ‘part A’ of the EGFR coding sequence followed by the same sgRNA target site (red box) used for HR of Donor A. ‘Part B’ is introduced by HR using this sgRNA target site and is fused seamlessly with ‘part A’ thereby constituting a full EGFR open reading frame. (b) Primary human T cells and CD34+ HSPCs were mock-electroporated or electroporated with Cas9 protein precomplexed with CCR5 sgRNA (CRISPR) followed by transduction with the split EGFR AAV6 donor pair. Left panel, representative FASC plots showing EGFR expression four days post-transduction. Right panel, frequencies of EGFR+ cells measured four days post-transduction, N = 14 (T cells, all from different buffy coat donors), N = 9 (HSPCs, all from different cord blood donors). (c) HSPCs were treated as in (b) and at day 4 post-transduction, EGFR+ cells were single-cell sorted into 96-well plates containing methylcellulose and In-Out PCR was performed on genomic DNA from progenitor-derived clones 14 days after seeding. Representative gel image shows targeted integration of Donor A and B, confirmed by the 5’ end and 3’ end PCR, respectively, in 6 out of 20 total colonies. Input control is PCR amplification of part of the HBB gene.
Discussion
Our findings establish that efficient iterative homologous recombination after simultaneous delivery of the genome editing components can occur in human cells, which may enable complex genome engineering through intracellular genomic DNA assembly. Importantly, the system is not only highly efficient in human cancer cell lines, but is also very efficient in primary human blood cells including primary T cells and CD34+ HSPCs. A key aspect of the system is that it does not involve having to serially transfect and transduce cells, but instead can be performed in a single step in which the intracellular homologous recombination machinery naturally iterates the process. This is particularly important when working with stem cells like CD34+ HSPCs that do not tolerate repeated genetic manipulations well and differentiate during extended culturing. While other viral vectors with larger carrying capacity have been used to deliver HR templates, e.g. gutless adenoviral vectors and integration defective lentiviral vectors (IDLV) (Knipping et al., 2017, Hoban et al., 2016, Holkers et al., 2014, Zhang et al., 2014a, Genovese et al., 2014, Zhang et al., 2014b), AAV is currently the vector platform of choice for gene editing in primary T cells and HSPCs since it supports high rates of homologous recombination (Sather et al., 2015). However, in other cell types, different viral vectors may be superior in donor template delivery. Nonetheless, since rates of homologous recombination decrease with increasing insert size, a sequential two-step HR approach may prove to be equivalent to or even more efficient than a single-step integration of a large insert (Perez et al., 2005, Kung et al., 2013). Of note, the principle of sequential HR should be applicable to other viral vectors system as well.
Existing methods for expression of long transgenes split between two AAV vectors rely either on an approach where two overlapping vectors after transduction recombine or anneal before second strand-synthesis to produce the full-length large expression cassette, or on an approach where two vectors are designed with splice donor and acceptor in each vector so that upon intermolecular head-to-tail concatemerization the full-length mRNA transcript is produced. Both these approaches rely on interaction between the two donor vectors and the production of a full-length episomal expression vector. Our approach differs mechanistically from these as it relies on two sequential events of homologous recombination between the donor vectors and the genome, thus reestablishing the full-length expression cassette upon integration into the genome. Interestingly, in the K562 cell line we do observe episomal reconstitution of the GFP expression cassette (Figure S2a). We hypothesize that this episomal expression could be generated by annealing of the left homology arm of Donor B to the complementary sequence in Donor A, which would prime upstream second strand synthesis and regenerate the GFP cassette (see Figure 1). Mutating the PAM of the sgRNA target site in Donor A, we did observe low frequencies of GFP reconstitution above background, and we cannot rule out that episomal DNA forms may be generated that serve as donor template for a single-step targeted integration of the full expression cassette. However, we find it more likely that these events are due to on-target integration of Donor B through nuclease-independent HR (Barzel et al., 2015). The sequential HR platform uses the same sgRNA for both HR events, which simplifies the design and avoids the use of different sgRNAs, which would presumably double the required Cas9 RNP dose and potentially lead to higher rates of off-target activity and translocations. One rate-limiting step of the procedure is that the sgRNA target site may be mutated by non-homologous end-joining (NHEJ). When this happens after the first HR event it can prevent the second HR step from occurring thereby leaving a truncated but functional expression cassette. To avoid production of a truncated protein, Donor A may be designed so that the truncated mRNA transcript does not contain a stop codon in any reading frame downstream of the sgRNA target site so that the transcripts undergo nonstop decay (van Hoof et al., 2002, Frischmeyer et al., 2002), and it may be designed with miRNA binding sites downstream of the sgRNA target site for rapid RNAi-mediated degradation of the transcripts (Brown et al., 2006). Alternatively, a reporter gene may be included for selection of cells that have undergone both HR steps, or the order of the HR steps may be reversed so the promoter is integrated at the second step.
In conclusion, we demonstrate that the homologous recombination machinery in primary human blood cells is robust enough to facilitate sequential HR for integration of gene expression cassettes that exceed the packaging capacity of AAV. This is desirable for therapeutic genome editing that involves integration of large transgenes, in settings where a multi-cistronic cassette is introduced, or in the setting where two or more full transgene cassettes (Promoter-Transgene-polyA signal) need to be integrated. Each of these examples has features that will enable specific therapeutic and research applications in the future.
Experimental Procedures
AAV vector production
The backbone for all AAV vector plasmids were the pAAV-MCS plasmid (Agilent Technologies, Santa Clara, CA), which contains ITRs from AAV serotype 2. All homology arms used were 400bp each. Plasmids were produced using standard molecular cloning techniques. Plasmid pDGM6 plasmid (a kind gift from David Russell, University of Washington, Seattle, WA, USA) was used in the virus production, which contains the AAV6 cap genes, AAV2 rep genes, and adenovirus helper genes. AAV6 vectors were produced by iodixanol gradient purification as described in (Dever et al., 2016). Vectors were titered using quantitative PCR to measure number of vector genomes as described here (Aurnhammer et al., 2012).
Cell isolation and culture
CD34+ HSPCs from cord blood were acquired from donors under informed consent via the Binns Program for Cord Blood Research at Stanford University. CD34+ cells were purified using the CD34+ Microbead Kit Ultrapure (Miltenyi Biotec, San Diego, CA, USA) according to manufacturer’s protocol. All CD34+ HSPCs were used fresh without freezing, and cultured at 37°C, 5% CO2, and 5% O2 in StemSpan SFEM II (Stemcell Technologies, Vancouver, Canada) supplemented with SCF (100 ng/ml), TPO (100 ng/ml), Flt3-Ligand (100 ng/ml), IL-6 (100 ng/ml), StemRegenin1 (0.75 μM), and UM171 (35nM). Primary human CD3+ T cells were isolated from buffy coats obtained from the Stanford School of Medicine Blood Center using a human Pan T Cell Isolation Kit (Miltenyi Biotec, San Diego, CA, USA) according to manufacturer’s instructions. CD3+ cells were cultured at 37°C, 5% CO2, and 20% O2 in X-VIVO 15 (Lonza, Walkersville, MD, USA) supplemented with 5% human serum (Sigma-Aldrich, St. Louis, MO, USA), 100 IU/mL human recombinant IL-2 (Peprotech, Rocky Hill, NJ, USA), and 10 ng/mL human recombinant IL-7 (BD Biosciences, San Jose, CA, USA). Before electroporation, T cells were activated for three days with immobilized anti-CD3 antibodies (clone: OKT3, Tonbo Biosciences, San Diego, CA, USA) and soluble anti-CD28 antibodies (clone: CD28.2, Tonbo Biosciences). K562 cells were purchased from ATCC (Manassas, VA, USA) and cultured at 37°C, 5% CO2, and 20% O2 in RPMI 1640 (HyClone) supplemented with 10% bovine growth serum, 100 mg/ml streptomycin, 100 units/ml penicillin, and 2 mM L-glutamine.
Electroporation and transduction of cells
The CCR5 synthetic sgRNA used were purchased HPLC-purified from TriLink BioTechnologies (San Diego, CA, USA) and contained chemically modified nucleotides (2'O-Methyl 3'phosphorothioate) at the three terminal positions at both the 5' and 3' ends. The sequence of the CCR5 sgRNA with the modified nucleotides underlined: 5'-GCAGCATAGTGAGCCCAGAAGUUUUAGAGCUAGAAAUAGCAAGUUAAAAUAAGGCUAG UCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCUUUU-3'. Cas9 mRNA containing 5-methylcytidines and pseudouridines was purchased from TriLink BioTechnologies. Cas9 protein was purchased from IDT (San Jose, CA, USA). Cas9 protein and sgRNAs were complexed by incubation at a molar ratio of 1:2.5 at 25°C for 10 min immediately prior to electroporation. CD34+ HSPCs were electroporated 2 days after isolation and T cells were electroporated 3 days after stimulation. All electroporations were performed on the Lonza Nucleofector IIb (program U-014 for HSPCs and T cells, and program T-016 for K562 cells). For CD34+ HSPCs and T cells, either the buffer from the Human T Cell Nucleofection Kit (VPA-1002, Lonza) or the 1M electroporation buffer described in (Chicaybam et al., 2013) were used. The following conditions were used for electroporation: 5–10×106 cells/mL, 300 μg/mL Cas9 protein complexed with sgRNA at 1:2.5 molar ratio. For K562 electroporations, an electroporation buffer containing 100 mM KH2PO4, 15 mM NaHCO3, 12 mM MgCl2 x 6H20, 8 mM ATP, 2 mM glucose (pH 7.4) was used with 50 μg/mL Cas9 mRNA and 50 μg/mL sgRNA. For experiments with plasmid donors, 2.5 μg of each plasmid was used. For experiments using AAV6 donors, directly following electroporation, cells were incubated for 15 min at 37°C after which they were added AAV6 at 20% of the final culture volume (multiplicity of infection [MOI] was typically ~2–5×105 vg/cell per AAV donor unless otherwise stated).
Flow cytometry
Expression of fluorescent proteins or cell surface markers was analyzed by flow cytometry on a BD FACSAria II SORP (Franklin Lakes, NJ, USA) or a CytoFLEX Flow Cytometer (Brea, CA, USA). The following antiobodies were used: anti-EGFR (PE or APC, clone: AY13, BioLegend, San Diego, CA, USA), anti-CCR5 (APC, clone: 2D7/CCR5, BD Biosciences), anti-CD3 (BV605, clone: UCHT1, Biolegend), anti-CD4+ (PE-Cy7, clone: RPA-T4, Tonbo Biosciences), anti-CD8 (VF450, clone: RPA-T8, Tonbo Biosciences). The blue or violet LIVE/DEAD Fixable Dead Cell Stain Kit (Life Technologies) or the Ghost Dye Red 780 (Tonbo Biosceiences) was used to discriminate live and dead cells according to manufacturer's instructions. For discrimination of apoptotic cells, PE or APC labeled annexin V (BioLegend) was used following manufacturer’s instructions.
Methylcellulose colony-forming unit (CFU) assay and PCR detection of integration
The CFU assay was performed by FACS sorting of single cells into 96-well plates containing MethoCult Optimum or MethoCult Enriched (Stemcell Technologies) four days after electroporation and transduction. After 12–16 days, colonies were counted and scored based on their morphological appearance in a blinded fashion. Confirmation of on-target integration was performed by PCR on colony-derived genomic DNA that was extracted from colonies by adding PBS to the colonies, mixing followed by pelleting of cells. After washing with PBS, cells were resuspended in 25 μl QuickExtract DNA Extraction Solution (Epicentre, Madison, WI, USA) and incubated at 65°C for 10 min followed by 100°C for 2 min. Integration was detected by PCR using the following primers: GFP integration 5’ end fw: 5’-cccaacagagccaagctctcc-3’; GFP integration 5’ end rv: 5’-ccggtggatgtggaatgtgtgc-3’; GFP integration 3’ end fw: 5’-ggctcgcagccaacgtc-3’; GFP integration 3’ end rv: 5’-catgatggtgaagataagcctcacagc-3’; EGFR integration 5’ end fw: 5’-cccaacagagccaagctctcc-3; EGFR integration 5’ end rv: 5’-gcaccggttcaattgccgacc-3’; EGFR integration 3’ end fw: 5’-ccaaatggcatctttaagggctcc-3; EGFR integration 3’ end rv: 5’-gtgcctcttcttctcatttcgacacc-3’; HBB fw: 5'-ccaactcctaagccagtgccagaagag-3'; HBB rv: 5'-agtcagtgcctatcagaaacccaagag-3'.
Analysis of INDEL rates
Genomic DNA was extracted using QuickExtract DNA (Epicentre, Madison, WI, USA) following manufacturer's instructions, but using 50uL QuickExtract solution per 100,000 cells and extending the last incubation step at 100 °C to 10 min. The targeted region of CCR5 was PCR-amplified with primers spanning the sgRNA target sites: CCR5 (fw): 5'-GCACAGGGTGGAACAAGATGG-3'; CCR5 (rv): 5'-CACCACCCCAAAGGTGACCGT-3'. The iProof High-Fidelity Master Mix was used for PCR-amplification for 35 cycles (Bio-Rad, Hercules, CA, USA), and the purified PCR products were run on a 1% agarose gel, gel-extracted, and then Sanger-sequenced using both PCR primers. Each sequence chromatogram was analyzed using the TIDE software (http://tide.nki.nl). Mock-electroporated samples were used as reference sequence and parameters were set to an INDEL size of 30 nucleotides and the decomposition window to cover the largest possible window with high quality traces. For TOPO cloning, gel-purified PCR amplicons were cloned into the TOPO plasmid using the Zero Blunt TOPO PCR Cloning Kit (Life Technologies) according to manufacturer's protocol. TOPO plasmids were transformed into XL-1 Blue competent E.coli, plated on agar plates with kanamycin, and single colonies were sequenced by McLab (South San Francisco, CA, USA) by rolling circle amplification and sequencing using the forward primer used for PCR amplification.
Transplantation and analysis of human cells in NSG mice
For in vivo studies, 6 to 8 week-old NOD scid gamma (NSG) mice were purchased from the Jackson laboratory (Bar Harbor, ME USA). The experimental protocol was approved by Stanford University’s Administrative Panel on Lab Animal Care. Four days after electroporation and transduction, GFP+ cells were sorted and 40,000 cells were injected intrafemorally into each of three male mice, which had been sublethally irradiated the day before transplantation at 200 cGy. 16 weeks post-transplantation, mice were euthanized, bones (2x femur, 2x tibia, 2x pelvis, sternum, and spine) were collected and crushed using mortar and pestle. Mononuclear cells (MNCs) were isolated using Ficoll gradient centrifugation (Ficoll-Paque Plus, GE Healthcare, Sunnyvale, CA, USA) for 25min at 2,000xg, room temperature. Cells were blocked for nonspecific antibody binding (10% vol/vol, TruStain FcX, BioLegend) and stained (30min, 4°C, dark) with the following antibodies: anti-human CD45 (BV786, clone: HI30, BD Biosciences), anti-HLA-ABC (APC-Cy7, clone: W6/32, BioLegend), anti-mouse CD45.1 (PE-Cy7, clone: A20, eBioScience, San Diego, CA, USA), anti-mouse Ter-119 (BUV395, clone: TER-119, eBioscience). The LIVE/DEAD Fixable Blue Cell Stain Kit (Life Technologies) was used to discriminate live and dead cells according to manufacturer's instructions. Human engraftment was defined by the presence of human CD45+/ HLA-ABC+ double-positive cells.
Supplementary Material
Highlights.
Two AAV donors can be designed to undergo sequential homologous recombination (HR)
A transgene split between two AAV donors can be fused during HR
CRISPR and two AAV donors can mediate integration of large transgene cassettes
Acknowledgments
R.O.B. was supported through an Individual Postdoctoral grant (DFF–1333-00106B) and a Sapere Aude, Research Talent grant (DFF–1331-00735B) both from the Danish Council for Independent Research, Medical Sciences. M.H.P. gratefully acknowledges the support of the Amon Carter Foundation, the Laurie Kraus Lacob Faculty Scholar Award in Pediatric Translational Research, and NIH grant support R01-AI120766. We thank David Russell (University of Washington) for the pDGM6 plasmid, the Binns Program for Cord Blood Research (Stanford University) for cord blood-derived CD34+ HSPCs, and Sruthi Mantri (Stanford University) for isolation of CD34+ HSPCs from cord blood. We also thank Daniel Dever and other members of the Porteus lab for helpful input, comments and discussion. M.H.P. is a consultant and has equity interest in CRISPR Tx, but CRISPR Tx had no input into the design, execution, interpretation, or publication of the results herein.
Footnotes
Author contributions
R.O.B. performed and designed all experiments. M.H.P directed the research, and participated in the design and interpretation of the experiments. R.O.B. wrote the manuscript with help from M.H.P.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- AURNHAMMER C, HAASE M, MUETHER N, HAUSL M, RAUSCHHUBER C, HUBER I, NITSCHKO H, BUSCH U, SING A, EHRHARDT A, BAIKER A. Universal real-time PCR for the detection and quantification of adeno-associated virus serotype 2-derived inverted terminal repeat sequences. Hum Gene Ther Methods. 2012;23:18–28. doi: 10.1089/hgtb.2011.034. [DOI] [PubMed] [Google Scholar]
- BARZEL A, PAULK NK, SHI Y, HUANG Y, CHU K, ZHANG F, VALDMANIS PN, SPECTOR LP, PORTEUS MH, GAENSLER KM, KAY MA. Promoterless gene targeting without nucleases ameliorates haemophilia B in mice. Nature. 2015;517:360–4. doi: 10.1038/nature13864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BASIRI M, BEHMANESH M, TAHAMTANI Y, KHALOOGHI K, MORADMAND A, BAHARVAND H. The Convenience of Single Homology Arm Donor DNA and CRISPR/Cas9-Nickase for Targeted Insertion of Long DNA Fragment. Cell J. 2017;18:532–539. doi: 10.22074/cellj.2016.4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BROWN BD, VENNERI MA, ZINGALE A, SERGI SERGI L, NALDINI L. Endogenous microRNA regulation suppresses transgene expression in hematopoietic lineages and enables stable gene transfer. Nat Med. 2006;12:585–91. doi: 10.1038/nm1398. [DOI] [PubMed] [Google Scholar]
- CHAMBERLAIN K, RIYAD JM, WEBER T. Expressing Transgenes That Exceed the Packaging Capacity of Adeno-Associated Virus Capsids. Hum Gene Ther Methods. 2016;27:1–12. doi: 10.1089/hgtb.2015.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHICAYBAM L, SODRE AL, CURZIO BA, BONAMINO MH. An efficient low cost method for gene transfer to T lymphocytes. PLoS One. 2013;8:e60298. doi: 10.1371/journal.pone.0060298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE RAVIN SS, LI L, WU X, CHOI U, ALLEN C, KOONTZ S, LEE J, THEOBALD-WHITING N, CHU J, GAROFALO M, SWEENEY C, KARDAVA L, MOIR S, VILEY A, NATARAJAN P, SU L, KUHNS D, ZAREMBER KA, PESHWA MV, MALECH HL. CRISPR-Cas9 gene repair of hematopoietic stem cells from patients with X-linked chronic granulomatous disease. Sci Transl Med. 2017:9. doi: 10.1126/scitranslmed.aah3480. [DOI] [PubMed] [Google Scholar]
- DEVER DP, BAK RO, REINISCH A, CAMARENA J, WASHINGTON G, NICOLAS CE, PAVEL-DINU M, SAXENA N, WILKENS AB, MANTRI S, UCHIDA N, HENDEL A, NARLA A, MAJETI R, WEINBERG KI, PORTEUS MH. CRISPR/Cas9 beta-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539:384–389. doi: 10.1038/nature20134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEWITT MA, MAGIS W, BRAY NL, WANG T, BERMAN JR, URBINATI F, HEO SJ, MITROS T, MUNOZ DP, BOFFELLI D, KOHN DB, WALTERS MC, CARROLL D, MARTIN DI, CORN JE. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci Transl Med. 2016;8:360ra134. doi: 10.1126/scitranslmed.aaf9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FRISCHMEYER PA, VAN HOOF A, O'DONNELL K, GUERRERIO AL, PARKER R, DIETZ HC. An mRNA surveillance mechanism that eliminates transcripts lacking termination codons. Science. 2002;295:2258–61. doi: 10.1126/science.1067338. [DOI] [PubMed] [Google Scholar]
- GENOVESE P, SCHIROLI G, ESCOBAR G, DI TOMASO T, FIRRITO C, CALABRIA A, MOI D, MAZZIERI R, BONINI C, HOLMES MC, GREGORY PD, VAN DER BURG M, GENTNER B, MONTINI E, LOMBARDO A, NALDINI L. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014;510:235–40. doi: 10.1038/nature13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRIEGER JC, SAMULSKI RJ. Packaging capacity of adeno-associated virus serotypes: impact of larger genomes on infectivity and postentry steps. J Virol. 2005;79:9933–44. doi: 10.1128/JVI.79.15.9933-9944.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HALBERT CL, ALLEN JM, MILLER AD. Efficient mouse airway transduction following recombination between AAV vectors carrying parts of a larger gene. Nat Biotechnol. 2002;20:697–701. doi: 10.1038/nbt0702-697. [DOI] [PubMed] [Google Scholar]
- HENDEL A, BAK RO, CLARK JT, KENNEDY AB, RYAN DE, ROY S, STEINFELD I, LUNSTAD BD, KAISER RJ, WILKENS AB, BACCHETTA R, TSALENKO A, DELLINGER D, BRUHN L, PORTEUS MH. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat Biotechnol. 2015;33:985–9. doi: 10.1038/nbt.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HENDEL A, KILDEBECK EJ, FINE EJ, CLARK JT, PUNJYA N, SEBASTIANO V, BAO G, PORTEUS MH. Quantifying genome-editing outcomes at endogenous loci with SMRT sequencing. Cell Rep. 2014;7:293–305. doi: 10.1016/j.celrep.2014.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOBAN MD, LUMAQUIN D, KUO CY, ROMERO Z, LONG J, HO M, YOUNG CS, MOJADIDI M, FITZGIBBON S, COOPER AR, LILL GR, URBINATI F, CAMPO-FERNANDEZ B, BJURSTROM CF, PELLEGRINI M, HOLLIS RP, KOHN DB. CRISPR/Cas9-Mediated Correction of the Sickle Mutation in Human CD34+ cells. Mol Ther. 2016;24:1561–9. doi: 10.1038/mt.2016.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOLKERS M, MAGGIO I, HENRIQUES SF, JANSSEN JM, CATHOMEN T, GONCALVES MA. Adenoviral vector DNA for accurate genome editing with engineered nucleases. Nat Methods. 2014;11:1051–7. doi: 10.1038/nmeth.3075. [DOI] [PubMed] [Google Scholar]
- KNIPPING F, OSBORN MJ, PETRI K, TOLAR J, GLIMM H, VON KALLE C, SCHMIDT M, GABRIEL R. Genome-wide Specificity of Highly Efficient TALENs and CRISPR/Cas9 for T Cell Receptor Modification. Mol Ther Methods Clin Dev. 2017;4:213–224. doi: 10.1016/j.omtm.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUNG SH, RETCHLESS AC, KWAN JY, ALMEIDA RP. Effects of DNA size on transformation and recombination efficiencies in Xylella fastidiosa. Appl Environ Microbiol. 2013;79:1712–7. doi: 10.1128/AEM.03525-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOMBARDO A, GENOVESE P, BEAUSEJOUR CM, COLLEONI S, LEE YL, KIM KA, ANDO D, URNOV FD, GALLI C, GREGORY PD, HOLMES MC, NALDINI L. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol. 2007;25:1298–306. doi: 10.1038/nbt1353. [DOI] [PubMed] [Google Scholar]
- NAKAI H, STORM TA, KAY MA. Increasing the size of rAAV-mediated expression cassettes in vivo by intermolecular joining of two complementary vectors. Nat Biotechnol. 2000;18:527–32. doi: 10.1038/75390. [DOI] [PubMed] [Google Scholar]
- PEREZ C, GUYOT V, CABANIOLS JP, GOUBLE A, MICHEAUX B, SMITH J, LEDUC S, PAQUES F, DUCHATEAU P. Factors affecting double-strand break-induced homologous recombination in mammalian cells. Biotechniques. 2005;39:109–15. doi: 10.2144/05391GT01. [DOI] [PubMed] [Google Scholar]
- RYAN MA, NATTAMAI KJ, XING E, SCHLEIMER D, DARIA D, SENGUPTA A, KOHLER A, LIU W, GUNZER M, JANSEN M, RATNER N, LE CRAS TD, WATERSTRAT A, VAN ZANT G, CANCELAS JA, ZHENG Y, GEIGER H. Pharmacological inhibition of EGFR signaling enhances G-CSF-induced hematopoietic stem cell mobilization. Nat Med. 2010;16:1141–6. doi: 10.1038/nm.2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SATHER BD, ROMANO IBARRA GS, SOMMER K, CURINGA G, HALE M, KHAN IF, SINGH S, SONG Y, GWIAZDA K, SAHNI J, JARJOUR J, ASTRAKHAN A, WAGNER TA, SCHARENBERG AM, RAWLINGS DJ. Efficient modification of CCR5 in primary human hematopoietic cells using a megaTAL nuclease and AAV donor template. Sci Transl Med. 2015;7:307ra156. doi: 10.1126/scitranslmed.aac5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUN L, LI J, XIAO X. Overcoming adeno-associated virus vector size limitation through viral DNA heterodimerization. Nat Med. 2000;6:599–602. doi: 10.1038/75087. [DOI] [PubMed] [Google Scholar]
- TAKAHASHI T, YAMADA K, TANAKA T, KUMANO K, KUROKAWA M, HIRANO N, HONDA H, CHIBA S, TSUJI K, YAZAKI Y, NAKAHATA T, HIRAI H. A potential molecular approach to ex vivo hematopoietic expansion with recombinant epidermal growth factor receptor-expressing adenovirus vector. Blood. 1998;91:4509–15. [PubMed] [Google Scholar]
- VAN HOOF A, FRISCHMEYER PA, DIETZ HC, PARKER R. Exosome-mediated recognition and degradation of mRNAs lacking a termination codon. Science. 2002;295:2262–4. doi: 10.1126/science.1067272. [DOI] [PubMed] [Google Scholar]
- WANG J, EXLINE CM, DECLERCQ JJ, LLEWELLYN GN, HAYWARD SB, LI PW, SHIVAK DA, SUROSKY RT, GREGORY PD, HOLMES MC, CANNON PM. Homology-driven genome editing in hematopoietic stem and progenitor cells using ZFN mRNA and AAV6 donors. Nat Biotechnol. 2015;33:1256–1263. doi: 10.1038/nbt.3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG Y, WANG L, BELL P, MCMENAMIN D, HE Z, WHITE J, YU H, XU C, MORIZONO H, MUSUNURU K, BATSHAW ML, WILSON JM. A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat Biotechnol. 2016;34:334–8. doi: 10.1038/nbt.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YIN H, SONG CQ, DORKIN JR, ZHU LJ, LI Y, WU Q, PARK A, YANG J, SURESH S, BIZHANOVA A, GUPTA A, BOLUKBASI MF, WALSH S, BOGORAD RL, GAO G, WENG Z, DONG Y, KOTELIANSKY V, WOLFE SA, LANGER R, XUE W, ANDERSON DG. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat Biotechnol. 2016;34:328–33. doi: 10.1038/nbt.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG W, CHEN H, ZHENG X, WANG D, JI H, XIA H, MAO Q. Targeted genome correction by a single adenoviral vector simultaneously carrying an inducible zinc finger nuclease and a donor template. J Biotechnol. 2014a;188:1–6. doi: 10.1016/j.jbiotec.2014.08.003. [DOI] [PubMed] [Google Scholar]
- ZHANG W, WANG D, LIU S, ZHENG X, JI H, XIA H, MAO Q. Multiple copies of a linear donor fragment released in situ from a vector improve the efficiency of zinc-finger nuclease-mediated genome editing. Gene Ther. 2014b;21:282–8. doi: 10.1038/gt.2013.83. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.