

Abstract
An efficient process for the purification of synthetic phosphorothioate and native DNA sequences is presented. The process is based on the use of an aminopropylated silica gel support functionalized with aminooxyalkyl functions to enable capture of DNA sequences through an oximation reaction with the keto function of a linker conjugated to the 5′-terminus of DNA sequences. Deoxyribonucleoside phosphoramidites carrying this linker, as a 5′-hydroxyl protecting group, have been synthesized for incorporation into DNA sequences during the last coupling step of a standard solid-phase synthesis protocol executed on a controlled pore glass (CPG) support. Solid-phase capture of the nucleobase- and phosphate-deprotected DNA sequences released from the CPG support is demonstrated to proceed near quantitatively. Shorter than full-length DNA sequences are first washed away from the capture support; the solid-phase purified DNA sequences are then released from this support upon reaction with tetra-n-butylammonium fluoride in dry dimethylsulfoxide (DMSO) and precipitated in tetrahydrofuran (THF). The purity of solid-phase-purified DNA sequences exceeds 98%. The simulated high-throughput and scalability features of the solid-phase purification process are demonstrated without sacrificing purity of the DNA sequences.
Keywords: Synthetic DNA sequences, Solid-phase purification, High-throughput capability, Large scale purification, Cost-effective process
INTRODUCTION
The use of synthetic DNA sequences and their analogues for recognition and binding to mRNAs encoding disease-causing proteins has led to the development of nucleic acid-based drugs capable of inhibiting the expression of those proteins (Crooke, 2004). This approach to control the expression of disease-causing proteins requires the production of synthetic nucleic acid sequences in sufficient quantities and of high purity to support preclinical and clinical studies. In contrast, total gene synthesis for synthetic biology applications demands minute amounts of numerous highly pure synthetic DNA sequences. Although the phosphoramidite approach to the chemical synthesis of nucleic acid sequences (Beaucage and Caruthers, 1981; Sinha et al., 1984; UNIT 3.3) is efficient and amenable to pharmaceutical scale production, the purification of those sequences is a daunting challenge. Despite the fact that the coupling efficiency of phosphoramidite monomers is near quantitative, when performed on a controlled pore glass support, the full-length nucleic acid sequences are mixed with impurities consisting of shorter sequences, resulting from incomplete phosphoramidite coupling at each cycle of the nucleic acid sequence assembly. Other process-related impurities include deletion sequences caused by failure to quantitatively prevent the growth of shorter than full-length sequences or to completely remove the 5′-hydroxyl protecting group at each step of the nucleic acid sequence synthesis protocol. It should also be noted that longer than full-length nucleic acid sequences can occur when the activation of phosphoramidite monomers by a weak acid causes, to a limited extent, the premature cleavage of the acid-labile group protecting the 5′-hydroxyl function of the newly extended nucleic acid sequence (Eleuteri et al., 2000; Wei, 2013). Although longer than full-length nucleic acid sequences are produced in minute amounts, the physicochemical similarity of these impurities to the desired nucleic acid sequence makes their removal very challenging. In the context of large-scale production of nucleic acid-based drugs, HPLC-based methods (UNIT 10.5) including reversed-phase and/or anion-exchange HPLC are currently the preferred techniques for purifying nucleic acid sequences. These techniques require high capacity instruments and accessories (e.g., preparative columns) in addition to large volumes of consumables (e.g., buffered aqueous and organic elution solvents). The HPLC-based purification of nucleic acid sequences is also time-consuming given that, depending on the nature of individual nucleic acid sequence, more than one purification run may be required to achieve the level of purity needed for pharmaceutical applications. Furthermore, a major technical drawback associated with any HPLC purification process is the tedious removal of relatively large volumes of aqueous solvents that are generated during the course of the purification procedure. Thus, HPLC-based methods for the purification of nucleic acid sequences are neither cost-effective nor amenable to high-througput purification processes; only a single nucleic acid sequence can be purified per run unless numerous instruments are dedicated for this purpose.
This unit describes an innovative solid phase method for the purification of synthetic unmodified and modified (i.e., phosphorothioate) DNA sequences. Specifically, the preparation of a capture support that is critically needed for the proposed purification process is detailed in Basic Protocol 1. The chemical synthesis of linkers for solid-phase capture of DNA sequences and for determining the surface density of aminooxy functions covalently bound to the capture support is delineated in Basic Protocol 2. The preparation of 5′-functionalized deoxyribonucleosides and deoxyribonucleoside phosphoramidites for solid-phase capture of DNA sequences is described in Basic Protocol 3. The solid-phase synthesis of 5′-functionalized DNA sequences for solid-phase purification is reported in Support Protocol 1. Capture of crude 5′-functionalized DNA sequences by the capture solid support and release of the purified sequences from the support is outlined in Basic Protocol 4. A scale up process for the solid-phase purification of phosphorothioate DNA sequences is detailed in Alternate Protocol 1.
BASIC PROTOCOL 1. GENERAL PROCEDURE FOR THE PREPARATION OF A CAPTURE SUPPORT
As illustrated in Fig. 1, this protocol provides a general method for the preparation of capture support 3 from commercial 3-aminopropylated silica gel, 7-oxooctanoic acid (1) and O,O′-1,3-propanediylbishydroxylamine dihydrochloride.
Figure 1.

Preparation of the capture support 3. CDI, 1,1′-carbonyldiimidazole; Si, 3-aminopropyl silica gel.
Materials
7-Oxooctanoic acid (1, Aldrich)
O,O′-1,3-Propanediylbishydroxylamine dihydrochloride (Aldrich)
1,1′-Carbonyldiimidazole (CDI, Aldrich)
3-Aminopropyl silica gel (~1 mmol NH2, Aldrich)
Tetrahydrofuran, dry (THF, Acros)
Acetonitrile (MeCN, Aldrich)
Triethylamine (Et3N, Aldrich)
Cap Mix A (Glen research)
Cap Mix B (Glen Research)
10-mL screw cap glass vials with caps
25- and 50-mL round-bottom flasks
100-mL Erlenmeyer flasks
50-mL glass cylinder
Rubber septa for 14/20-glass joints
Magnetic stir bars
Magnetic stirrer
8-mesh Drierite with indicator (Aldrich)
Drierite guard tube
Reflux condenser (LabGlass)
Vacuum line (ChemGlass)
High-vacuum oil pump (Edwards)
Temperature controlled orbital shaker (Thomas Scientific)
15-mL, 30-mL fritted glass vacuum filtration Buchner funnel of coarse porosity (ChemGlass)
5- and 10-mL Luer-tipped glass syringes
21-G stainless steel syringe needles
Prepare the solid support 2
-
1
Place 7-oxooctanoic acid (1, 0.58 g, 3.0 mmol) and a magnetic stir bar into a flame-dried 25-mL round bottom flask. Add 1,1′-carbonyldiimidazole (CDI, 0.49 g, 3.0 mmol). Seal the flask with a reflux condenser and a Drierite drying tube.
-
2
Remove the drying tube. Add 4-mL dry THF through the reflux condenser using a 5-mL glass syringe. Connect the drying tube to the reflux condenser. Stir the solution for 2 hr at ~25 °C using a magnetic stirrer.
-
3
Place commercial 3-aminopropyl silica gel (1.0 g, ~1 mmol NH2) into a 30-mL fritted glass vacuum filtration Buchner funnel of coarse porosity connected to a 100-mL Erlenmeyer. Add 20-mL of 1:5 (v/v) Et3N/MeCN to the funnel using a 50-mL glass cylinder. Allow the filtrate to drip into the Erlenmeyer. Connect the Buchner funnel to an in-house vacuum line. Apply vacuum to air-dry the support.
-
4
Remove the reflux condenser/drying tube set from the flask of step 2. Add the dry support of step 3 to the solution. Securely seal the flask with a rubber septum. Agitate the suspension at 65 °C for 24 hr using an orbital shaker.
-
5
Transfer the suspension to a 30-mL fritted glass vacuum filtration Buchner funnel of coarse porosity connected to a 100-mL Erlenmeyer. Connect the Buchner funnel to an in-house vacuum line. Apply vacuum and wash the support successively with 20-mL THF and 20-mL MeCN using a 50-mL glass cylinder. Air-dry the support.
-
6
Place the support in a flame dried 50-mL round bottom flask. Seal the flask with a rubber septum. Add 20-mL of a commercial solution of acetic anhydride, 1-methylimidazole and pyridine in THF [Cap Mix A:Cap Mix B (1:1 v/v)] through the septum using a 10-mL glass syringe and a 21-G syringe needle as a vent. Remove the needle. Mechanically agitate the solution for 30 min at ~25 °C using an orbital shaker.
This step is required to inactivate amino functions left unreacted on solid support 2. -
7
Repeat step 5 with the following exception: Apply vacuum and wash the support twice with 20-mL MeCN using a 50-mL glass cylinder. Air dry the support. Transfer the support to a 25-mL round bottom flask connected to a high vacuum line/oil vacuum pump system. Apply vacuum for 1 hr at ~25 °C. Store the solid support 2 (981 mg) at −20°C until needed.
Loss of solid support 2 occurred when transferring the support from the fritted glass vacuum filtration Buchner funnel to a round-bottom flask (i.e., steps 3 to 4, 5 to 6 and step 7.
Prepare the capture support 3
-
8
Place the solid support 2 (1.0 g) into a 10-mL screw cap glass vial. Add a solution of O,O′-1,3-propanediylbishydroxylamine dihydrochloride (537 mg, 3.00 mmol) in 4-mL H2O using a 5-mL glass syringe. Seal the glass vial and mechanically agitate the suspension for 6 hr at 65 °C using an orbital shaker.
-
9
Repeat step 5 with the following exception: Apply vacuum and wash the support successively with 20-mL DMF and 20-mL MeCN using a 50-mL glass cylinder. Transfer the support to a 25-mL round bottom flask connected to a high vacuum line/oil vacuum pump system. Apply vacuum over a period of 1 hr at ~25 °C. Store the solid support 3 (995 mg) at −20°C until needed.
BASIC PROTOCOL 2. PREPARATION OF CHEMICAL LINKERS FOR THE SOLID-PHASE CAPTURE OF DNA SEQUENCES AND FOR DETERMINING THE SURFACE DENSITY OF AMINOOXY FUNCTIONS COVALENTLY BOUND TO SUPPORT 3
Functionalization of the terminal 5′-hydroxyl of DNA sequences with carbonyl-derived linkers is necessary for capture by the solid support 3 upon reaction with its highly reactive aminooxy groups. Fig. 2 depicts the reaction of N,N′-dimethylethylenediamine with -γ-butyrolactone (4a) or 5,5-dimethyldihydrofuran-2-one (4b) to provide the aminated amido alcohol 5a or 5b. The reaction of CDI-activated 7-oxooctanoic acid 1 with 5a or 5b gives the diamidated keto alcohol 6a or 6b; monomethoxytritylation of 6a affords the linker 6c for measuring the surface density of aminooxy functions covalently attached to the capture support 3.
Figure 2.

Preparation of the diamidated keto alcohol linkers 6a–c. Conditions: (i) MMTr-Cl, dry pyridine, 25 °C, 4 hr. CDI, 1,1′-carbonyldiimidazole; MMTr, 4-monomethoxytrityl.
Materials
N,N′-Dimethylethylenediamine (Aldrich)
γ-Butyrolactone (Aldrich)
5,5-Dimethyldihydrofuran-2-one (ACS Scientific)
7-Oxooctanoic acid (1, Aldrich)
1,1′-Carbonyldiimidazole (CDI, Aldrich)
4-Methoxytrityl chloride (Aldrich)
Chloroform (CHCl3, Fisher Scientific)
Dichloromethane (CH2Cl2, Fisher Scientific)
Methanol (MeOH, FisherScientific)
Acetonitrile (MeCN, Aldrich)
Tetrahydrofuran, dry (THF; Acros)
Pyridine, dry (Acros)
Toluene (Fisher Scientific)
Triethylamine (Et3N, Aldrich)
Dimethyl sulfoxide (DMSO, Aldrich)
Deuterated dimethylsulfoxide (DMSO-d6, Aldrich)
Phosphomolybdic acid (Aldrich)
3% Trichloroacetic acid in dichloromethane (Deblock solution, Glen Research)
Anhydrous sodium sulfate (Aldrich)
8-mesh Drierite with indicator (Aldrich)
Drierite guard tube
4- and 25-mL screw cap glass vials with caps
25- 50- and 100-mL round-bottom flasks (Kontes)
Rubber septa for 14/20 glass joints (Aldrich)
100-mL Separatory funnel
50- and 100-mL Erlenmeyer flasks
Reflux condenser (LabGlass)
50-mL glass cylinder
Pipettor (Corning)
1-mL pipette tips (Fisher Scientific)
5- and 10-mL Luer-tipped glass syringes
21-G stainless steel syringe needles
10-mL volumetric flask (Thomas Scientific)
13×100-mm disposable glass tubes
5 × 20-cm glass chromatography columns (LabGlass)
Silica gel (60-Å, 230 to 400 mesh; EMD)
2.5 × 7.5-cm TLC plates precoated with a 250-μm layer of silica gel 60 F254 (EMD)
Pasteur pipettes (VWR)
Vacuum line (LabGlass)
15-mL, 30-mL fritted glass vacuum filtration Buchner funnel of coarse porosity (ChemGlass)
Magnetic stir bars
Magnetic stirrer
Magnetic hot-plate stirrer (Corning)
Dry heat block (VWR)
High-vacuum oil pump (Edwards)
Temperature controlled orbital shaker (Thomas Scientific)
Rotary evaporator connected to a vacuum pump (Büchi)
Hand-held UV254 lamp (UVP)
UV/vis spectrophotometer (Agilent Technologies)
NMR spectrometer (Bruker)
Additional reagents and equipment for column chromatography (APPENDIX 3E) and TLC (APPENDIX 3D)
Synthesize 4-hydroxy-N-methyl-N-(2-(methylamino)ethyl)butanamide (5a)
-
1
Place N,N′-Dimethylethylenediamine (3.25 g, 40.0 mmol) and commercial γ-butyrolactone (4a, 1.72 g, 20.0 mmol) in a 25-mL screw-cap glass vial. Seal the vial and heat it at 90 °C for 16 hr using a dry heat block.
-
2
Pour the liquid into a 25-mL round-bottom flask. Rinse the vial with 5-mL MeCN and pour the liquid into the round bottom flask. Concentrate to an oil using a rotary evaporator connected to a high vacuum oil pump.
Purify and isolate 5a
-
3
Use a Pasteur pipette to spread the oily material on top of a 5 × 20-cm chromatography glass column packed with silica gel (~40 g) that has been equilibrated in 9:1 (v/v) CHCl3/MeOH.
-
4
Elute the column using a gradient of methanol (10% → 20%) in chloroform and collect 6-mL fractions in 13 × 100 mm disposable glass tubes.
-
5
Analyze fractions by TLC (APPENDIX 3D) on a 2.5 × 7.5-cm EMD silica gel 60 F254 TLC plate using 9:1 (v/v) CHCl3/MeOH as the eluent. Pool appropriate fractions in an appropriately sized round-bottom flask and rotoevaporate under reduced pressure (~20 mmHg) to afford the linker 5a (2.32 g, 14.4 mmol) as an oil in a yield of 72%.
The product is visualized on TLC [Rf (9:1 (v/v) CHCl3/MeOH)= 0.2] by soaking the silica gel glass plate in a commercial phosphomolybdic acid solution. The product appears as a dark blue spot upon heating the glass plate on a magnetic hot plate stirrer.4-Hydroxy-N-methyl-N-(2-(methylamino)ethyl)butanamide (5a).1HNMR (300 MHz, DMSO-d6): δ 3.39 (dt, J = 6.6, 2.0 Hz, 2H), 3.32 (dt, J = 6.6, 2.0 Hz, 2H), 2.95 (s, 1.5H), 2.79 (s, 1.5H), 2.59 (t, J = 6.6 Hz, 1H), 2.53 (t, J = 6.6 Hz, 1H), 2.34 (t, J = 7.4 Hz, 1H), 2.29 (t, J = 7.4 Hz, 1H), 2.28 (s, 1.5H), 2.25 (s, 1.5H), 1.63 (m, 2H). 13C NMR (75 MHz, DMSO-d6): δ 171.92, 171.91, 60.3, 49.7, 49.0, 48.9, 46.6, 36.2, 36.0, 35.4, 33.1, 29.3, 28.6, 28.3, 28.0. +ESI-TOF-HRMS calcd for C8H18N2O2 [M + H]+ 175.1400, found 175.1405.
Synthesize 4-hydroxy-N,4-dimethyl-N-(2-(methylamino)ethyl)-pentanamide (5b)
-
6
Repeat step 1 with the exception of replacing 4a with commercial 5,5-dimethyldihydrofuran-2-one (4b, 2.28 g, 20.0 mmol).
-
7
Repeat step 2.
Purify and isolate 5b
-
8
Repeat steps 3 through 5.
The linker 5b is isolated as an oil (2.99 g, 14.8 mmol) in a yield of 74%. The product is visualized on TLC [Rf (9:1 (v/v) CHCl3/MeOH)= 0.2] as reported in the annotation of step 5.4-Hydroxy-N,4-dimethyl-N-(2-(methylamino)ethyl)-pentanamide (5b). 1H NMR (300 MHz, DMSO-d6): δ 4.23 (br s, 1H), 3.33 (q, J = 6.6 Hz, 2H), 2.97 (s, 1.5H), 2.79 (s, 1.5H), 2.61 (t, J = 6.6 Hz, 1H), 2.53 (t, J = 6.6 Hz, 1.5H), 2.37-2.27 (m, 3H), 2.28 (s, 1.5H), 2.25 (s, 1.5H), 1.57 (m, 2H), 1.08 (s, 3H), 1.07 (s, 3H). 13C NMR (75 MHz, DMSO-d6): δ 172.5, 172.4, 79.1, 49.7, 49.0, 48.9, 46.6, 38.7, 38.3, 36.2, 35.9, 33.1, 28.9, 28.1, 27.4. +ESI-TOF-HRMS calcd for C10H22N2O2 [M + H]+ 203.1754, found 203.1763.
Synthesize N-(2-(4-hydroxy-N-methylbutanamido)ethyl)-N-methyl-7-oxooctanamide (6a)
-
9
Add 1,1′-carbonyldiimidazole (2.52 g, 15.5 mmol) and a magnetic stir bar to a solution of 7-oxooctanoic acid (1, 2.45 g,15.5 mmol) in 15-mL dry THF in a 50-mL round-bottom flask. Seal the flask with a reflux condenser and a Drierite drying tube. Stir the solution using a magnetic stirrer for 2 hr at ~25 °C.
-
10
Remove the reflux consenser/drying tube set. Add 5a (2.32 g, 14.4 mmol) to the solution using a pipettor. Seal the flask with a rubber septum. Place the sealed flask on a magnetic hot plate stirrer. Stir the reaction at 65 °C over a period of 24 hr.
-
11
Remove the flask from the magnetic hot plate stirrer. Remove the rubber septum. Rotoevaporate the liquid to a thick oil under reduced pressure (~20 mmHg) using a vacuum pump.
-
12
Add 40-mL CHCl3 using a 50-mL glass cylinder to dissolve the oily material. Pour the solution into a 100-mL separatory funnel.
-
13
Add 20-mL water using a 50-mL glass cylinder. Seal the separatory funnel and shake vigorously. Allow phase separation. Collect the organic phase into a 100-mL round-bottom flask and rotoevaporate to an oil under reduced pressure (~20 mmHg) using a vacuum pump.
Purify and isolate 6a
-
14
Use a Pasteur pipette to add 4-mL CHCl3 and spread the solution on top of a 5 × 20-cm chromatography glass column packed with silica gel (~40 g) that has been equilibrated in CHCl3.
-
15
Elute the product off the column using a gradient of methanol (0→4%) in chloroform and collect 6-mL fractions in 13 × 100-mm disposable glass tubes.
-
16
Analyze fractions by TLC (APPENDIX 3D) on a 2.5 × 7.5–cm EMD silica gel 60 F254 TLC plate using 9:1 (v/v) CHCl3/MeOH as the eluent. Pool appropriate fractions in an appropriately sized round-bottom flask and rotoevaporate under reduced pressure (~20 mmHg) to afford the linker 6a (3.57g, 11.3 mmol) as an oil in a yield of 78%.
The linker 6a is visualized on TLC [Rf (9:1 (v/v) CHCl3/MeOH)= 0.6; 5a: Rf (9:1 (v/v) CHCl3/MeOH)= 0.2] as reported in the annotation of step 5.N-(2-(4-Hydroxy-N-methylbutanamido)ethyl)-N-methyl-7-oxooctanamide (6a). 1H NMR (300 MHz, DMSO-d6): δ 4.22 (m, 1H), 3.47 (s, 0.4H), 3.39 (m, 3H), 3.33 (s, 0.4H), 2.98 (s, 0.6H), 2.96 (s, 0.6H), 2.94 (s, 1H), 2.91 (s, 1H), 2.82 (d, J = 2.1 Hz, 0.6H), 2.80 (s, 1H), 2.40 (t, J = 7.3 Hz, 2H), 2.33-2.21 (m, 3H), 2.17 (t, J = 7.3 Hz, 0.8H), 2.06 (s, 3H), 1.56-1.52 (m, 1.8H), 1.50–1.38 (m, 4.2H), 1.22 (m, 2H), 1.08 (s, 1.4H), 1.06 (s, 3.8H); 13C NMR (75 MHz, DMSO-d6): δ 208.4, 172.9, 172.7, 172.5, 172.4, 172.2, 172.0, 171.8, 171.7, 79.1, 68.25, 68.21, 47.3, 47.2, 46.6, 46.4, 45.8, 45.6, 44.1, 44.0,42.6, 38.7, 38.2, 36.0, 35.8, 35.1, 35.0, 33.40, 33.37, 33.05, 33.01, 32.4, 32.3, 31.4, 29.6, 29.2, 28.4, 28.3, 28.2, 28.14, 28.06, 27.2, 24.7, 24.6, 24.3, 24.2, 23.1. +ESI-TOF-HRMS calcd for C18H34N2O4 [M + H]+ 343.2591, found 343.2623.
Synthesize N-(2-(4-hydroxy-N,4-dimethylpentanamido)ethyl)-N-methyl-7-oxooctanamide (6b)
-
17
Repeat step 9.
-
18
Repeat step 10 with the exception of replacing 5a with 5b (2.91 g, 14.4 mmol).
-
19
Repeat step 11 through 16.
The linker 6b is isolated as an oil (3.97 g, 11.6 mmol) in a yield of 80%. The product is visualized on TLC [Rf (9:1 (v/v) CHCl3/MeOH)= 0.6; 5b: Rf (9:1 (v/v) CHCl3/MeOH)= 0.2] as reported in the annotation of step 5.N-(2-(4-Hydroxy-N,4-dimethylpentanamido)ethyl)-N-methyl-7-oxooctanamide (6b). 1HNMR (300 MHz, DMSO-d6): δ 4.22 (m, 1H), 3.47 (s, 0.4H), 3.39 (m, 3H), 3.33 (s, 0.4H), 2.98 (s, 0.6H), 2.96 (s, 0.6H), 2.94 (s, 1H), 2.91 (s, 1H), 2.82 (d, J = 2.1 Hz, 0.6H), 2.80 (s, 1H), 2.40 (t, J = 7.3 Hz, 2H), 2.33-2.21 (m, 3H), 2.17 (t, J = 7.3 Hz, 0.8H), 2.06 (s, 3H), 1.56-1.52 (m, 1.8H), 1.50-1.38 (m, 4.2H), 1.22 (m, 2H), 1.08 (s, 1.4H), 1.06 (s, 3.8H). 13C NMR (75 MHz, DMSO-d6): δ 208.4, 172.9, 172.7, 172.5, 172.4, 172.2, 172.0, 171.8, 171.7, 79.1, 68.25, 68.21, 47.3, 47.2, 46.6, 46.4, 45.8, 45.6, 44.1, 44.0, 42.6, 38.7, 38.2, 36.0, 35.8, 35.1, 35.0, 33.40, 33.37, 33.05, 33.01, 32.4, 32.3, 31.4, 29.6, 29.2, 28.4, 28.3, 28.2, 28.14, 28.06, 27.2, 24.7, 24.6, 24.3, 24.2, 23.1. +ESI-TOF-HRMS calcd for C18H34N2O4 [M + H]+ 343.2591, found 343.2623.
Synthesize N-(2-(4-((4-methoxyphenyl)diphenylmethoxy)-N-methylbutanamido)ethyl)-N-methyl-7-oxooctanamide (6c)
-
20
Place linker 6a (628 mg, 2.00 mmol) and a magnetic stir bar in a 100-mL round bottom flask. Seal the flask with a rubber septum.
-
21
Add sequentially 10-mL dry pyridine and a solution of 4-methoxytrityl chloride (927 mg, 3.00 mmol) in 10-mL dry pyridine through the rubber septum using a 10-mL glass syringe and a 21-G syringe needle as a vent. Remove the syringe needle vent and stir the reaction mixture at ~25 °C over a period of 4 h using a magnetic stirrer.
-
22
Remove the rubber septum. Quench the reaction by adding 20-mL water using a 50-mL glass cylinder.
Extract the crude product
-
23
Add 30-mL CHCl3 using a 50-mL glass cylinder. Pour the liquid into a 100-mL separatory funnel. Seal the funnel and shake vigorously. Allow phase separation.
-
24
Collect the organic phase and mix with 5 g anhydrous sodium sulfate. Filter the suspension through a 30-mL fritted glass vacuum filtration Buchner funnel of coarse porosity connected to a 100-mL round bottom flask. Connect the Buchner funnel to an in-house vacuum line and apply vacuum. Remove the funnel and rotoevaporate the solvent to an oil under reduced pressure (~20 mmHg) using a vacuum pump.
Purify and isolate the crude linker 6c
-
25
Use a Pasteur pipette to add 4-mL CHCl3 and spread the solution on top of a 5 × 20-cm chromatography glass column packed with silica gel (~40 g) that has been equilibrated in a 99.5:0.5 (v/v) CHCl3/pyridine.
-
26
Elute the product from the column using a gradient of 0 → 2% methanol in a 99.5:0.5 (v/v) CHCl3/pyridine. Collect 6-mL fractions in 13 × 100-mm disposable glass tubes.
-
27
Analyze fractions by TLC (APPENDIX 3D) on a 2.5 × 7.5–cm EMD silica gel 60 F254 TLC plate using 9:1 (v/v) CHCl3/MeOH as the eluent and UV254 for detection. Pool appropriate fractions [6c: Rf = 0.8; 6a: Rf (9:1 (v/v) CHCl3/MeOH)= 0.6] in an appropriately sized round-bottom flask and rotoevaporate under reduced pressure (~20 mmHg) using a vacuum pump.
The linker 6c is needed to determine the concentration or surface density of aminooxy functions covalently bound to the capture support 3. -
28
Dissolve the purified material in 5-mL toluene. Rotoevaporate the solvent to dryness using an oil vacuum pump. Repeat this step twice.
Pure linker 6c (1.01 g, 1.7 mmol) is isolated as an oil in a yield of 85%.N-(2-(4-((4-Methoxyphenyl)diphenylmethoxy)-N-methylbutanamido)ethyl)-N-methyl-7-oxooctanamide (6c). 1H NMR (300 MHz, DMSO-d6): δ 7.39-2.29 (m, 8H), 7.25-7.20 (m, 4H), 6.89 (d, J = 8.8 Hz, 2H), 3.74 (s, 3H), 3.42 (s, 0.5H), 3.36 (s, 2H), 2.98 (m, 2H), 2.93 (s, 0.7H), 2.90 (s, 0.8H), 2.88 (s, 1H), 2.87 (s, 1H), 2.79 (m, 2H), 2.35 (m, 3H), 2.26-2.12 (m, 3H), 2.04 (s, 1H), 2.03 (s, 1H), 2.02 (s, 1H), 1.77 (m, 2H), 1.42 (m, 4H), 1.19 (m, 2H). 13C NMR (75 MHz, DMSO-d6): δ 208.3, 172.1, 172.0, 171.9, 171.7, 171.6, 171.5, 171.4, 158.1, 144.59, 144.57, 135.4, 129.8, 128.8, 128.1, 127.9, 127.8, 126.7, 125.2, 113.1, 85.5, 62.6, 62.5, 62.4, 55.0, 47.3, 47.2, 46.4, 46.3, 45.8, 45.6, 44.13, 44.10, 42.6, 35.9, 35.8, 35.1, 35.0, 33.4, 33.3, 33.0, 32.34, 32.28, 31.4, 29.58, 29.56, 29.3, 29.2, 28.38, 28.35, 28.3, 28.2, 25.4, 25.1, 24.7, 24.5, 24.3, 24.2, 23.1, 21.0. +ESI-TOF-HRMS calcd for C36H46N2O5Cs [M + Cs]+ 719.2456, found 719.2458.
Determine the surface density of aminooxy functions covalently bound to capture support 3
-
29
Place 150 mg of solid support 3 into a 15-mL fritted glass vacuum filtration Buchner funnel of coarse porosity connected to a 50-mL Erlenmeyer. Pour 5-mL of 3:7 (v/v) Et3N/MeCN into the funnel. Allow the filtrate to drip into the Erlenmeyer.
-
30
Pour 10-mL MeCN into the funnel. Connect the Erlenmeyer to an in-house vacuum line. Apply vacuum and air-dry the support. Transfer 20 mg of the support to a 4-mL screw cap glass vial.
Functionalize the capture support 3 with the linker 6c
-
31
Add a solution of linker 6c (60 mg, 100 μmol) in 300-μL 5:1 (v/v) DMSO/H2O. Seal the vial and mechanically agitate it at ~25 °C using an orbital shaker over a period of 24 hr
-
32
Pour the suspension into a 15-mL fritted glass vacuum filtration Buchner funnel of coarse porosity connected to a 100-mL Erlenmeyer. Connect the funnel to an in-house vacuum line and apply vacuum.
-
33
Pipet 10 mL MeCN into the glass funnel. Apply vacuum. Repeat this step twice and air dry the support. Remove the funnel from the Erlenmeyer.
Measure the surface density of the covalently attached aminooxy functions
-
34
Pipet 1-mL 3% trichloroacetic acid in dichloromethane in the glass funnel. Allow the yellow-colored liquid to drip into a 10-mL volumetric flask. Repeat this step nine times. Add dichloromethane as needed to bring the volume to the mark of the 10-mLvolumetric flask.
-
35
Measure the absorbance of the solution at 478 nm (ε = 56 mL.cm−1.μmol−1) against a blank solution of 3% trichloroacetic acid in dichloromethane using a UV/vis spectrophotometer.
The surface density of aminooxy functions is 146 ± 7 μmol per gram of capture support 3. This measurement serves as a reference standard to ensure consistency/reproducibity in the preparation of support 3. The importance of this measurement relates to the determination of optimal amount of support 3 needed to ensure complete capture of the DNA sequences to be purified.
BASIC PROTOCOL 3. PREPARATION OF 5′-FUNCTIONALIZED DEOXYRIBONUCLEOSIDES AND DEOXYRIBONUCLEOSIDE PHOSPHORAMIDITES FOR SOLID-PHASE CAPTURE OF DNA SEQUENCES
The conjugation of the linker 6b to the 5′-hydroxy function of 2′-deoxythymidine and N-protected 2′-deoxyribonucleosides (7a–d) upon reaction with dichlorodiisopropylsilane in the presence of imidazole is detailed in this protocol. As schematically shown in Figure 3, conversion of the 5′-functionalized deoxyribonucleosides 8a–d to their corresponding deoxyribonucleoside phosphoramidite derivatives 9a–d is also described.
Figure 3.

Synthesis of 5′-functionalized deoxyribonucleosides (8a–d) and deoxyribonucleoside phosphoramidites (9a–d) for solid-phase capture of DNA sequences. DIEA, N,N-diisopropylethylamine; BP, (a) N6-benzoyladenin-9-yl, (b) N4-benzoylcytosin-1-yl, (c) N2-isobutyrylguanin-9-yl, or (d) thymin-1-yl; i-Pr, isopropyl; DMF, N,N-dimethylformamide.
Materials
N-(2-(4-hydroxy-N,4-dimethylpentanamido)ethyl)-N-methyl-7-oxooctanamide (6b, Basic Protocol 2)
1H-Imidazole (Aldrich)
N,N-Dimethylformamide, dry (DMF, Acros)
N,N-Diisopropylethylamine (Aldrich)
Dichlorodiisopropylsilane (Aldrich)
N6-Benzoyl-2′-deoxyadenosine (7a, ChemGenes)
N4-Benzoyl-2′-deoxycytidine (7b, ChemGenes)
N2-Isobutyryl-2′-deoxyguanosine (7c, ChemGenes)
2′-Deoxythymidine (7d, ChemGenes)
Acetone (Fisher Scientific)
Benzene (C6H6, Aldrich)
Deuterated benzene (C6D6, Aldrich)
Deuterated dimethyl sulfoxide (DMSO-d6, Aldrich)
Triethylamine (Et3N, Aldrich)
Ethyl acetate (EtOAc, VWR)
Dichloromethane, dry (CH2Cl2, Acros)
Chloroform (CHCl3, Fisher Scientific)
Methanol (MeOH, Fisher Scientific)
Isopropyl ether (Aldrich)
2-Cyanoethyl N,N-diisopropylchlorophosphoramidite (Aldrich)
Sodium bicarbonate (NaHCO3, Fisher Scientific)
Anhydrous sodium sulfate (Fisher Scientific)
Argon or nitrogen sources
30-mL fritted glass vacuum filtration Buchner funnel of coarse porosity (ChemGlass)
50- and 100-mL round-bottom flasks (Kontes)
100-mL Erlenmeyer flask (Kontes)
Rubber septa for 14/20 glass joints (Aldrich)
50-mL glass cylinder
100-mL separatory funnel
Chromatography glass column (LabGlass)
Silica gel (60-Å, 230 to 400 mesh; EMD)
13×100-mm disposable glass tubes
Pasteur pipettes (VWR)
1- and 10-mL glass syringes
21-G stainless steel syringe needles
Magnetic stir bars
Magnetic stirrer
Pipettor (Corning)
1-mL pipet tips (Fisher Scientific)
Dry ice-isopropyl ether bath
Dry ice-acetone bath
Wet ice bath
Rotary evaporator connected to a vacuum pump (Büchi)
Hand-held UV254 lamp (UVP)
UV/vis spectrophotometer (Agilent Technologies)
NMR spectrometer (Bruker)
Lyophilizer
Synthesize the 5′-functionalized deoxyribonucleosides 8a-d
-
1
Place the linker 6b (780 mg, 2.28 mmol), imidazole (184 mg, 2.70 mmol) and a magnetic stir bar in a flame-dried 50-mL round bottom flask.
-
2
Add, under an argon atmosphere, 5-mL dry DMF using a 10-mL syringe and pipet 2.35 mL N,N-diisopropylethylamine (13.5 mmol) using a pipettor. Seal the flask with a rubber septum and cool the solution to ~0 °C using a crushed wet ice bath.
-
3
Add 730-μL dichlorodiisopropylsilane (4.50 mmol) through the rubber septum using a 1-mL glass syringe. Stir the cold solution for 1 hr at ~0 °C using a magnetic stirrer.
-
4
Allow the reaction mixture to warm up to room temperature while stirring over a period of 4 hr. Cool the reaction mixture to ~ −60 °C in a dry ice-isopropyl ether bath.
-
5
Add dropwise, over a period of 1 min, a 5-mL solution of 2′-deoxythymidine (7a, 1.92 g, 5.40 mmol) and imidazole (368 mg, 5.40 mmol) in dry DMF, through the rubber septum using a 10-mL glass syringe. Stir the solution for 1 hr at ~ −60 °C. Allow the reaction mixture to warm up to ~0 °C in a crushed wet ice bath while keeping stirring it at this temperature for 3 h.
Quench the reaction
-
6
Remove the rubber septum and pour 35-mL of a cold (~0 °C) 5% aqueous solution NaHCO3.
Extract the product
-
7
Pour the aqueous solution into a 100-mL separatory funnel. Add 40-mL EtOAc into the separatory funnel using a glass cylinder. Seal the funnel and shake it vigorously. Allow complete phase separation.
-
8
Collect the organic layer into a 100-mL round bottom flask and rotoevaporate to an oil under reduced pressure (~ 20 mmHg) using a vacuum pump.
Purify and isolate the 5′-functionalized deoxyribonucleosides 8a
-
9
Use a Pasteur pipette to add 4-mL CHCl3 and spread the solution on top of a 5 × 20-cm chromatography glass column packed with silica gel (~40 g) that has been equilibrated in CHCl3.
-
10
Elute the product from the column using a gradient of 0 → 6% methanol in chloroform. Collect 6-mL fractions in 13 × 100-mm disposable glass tubes.
-
11
Analyze the fractions by TLC (APPENDIX 3D) on a 2.5 × 7.5–cm EMD silica gel 60 F254 TLC plate using 9:1 (v/v) CHCl3/MeOH as the eluent and UV254 for detection. Pool appropriate fractions [Rf (9:1 (v/v) CHCl3/MeOH) = 0.7] in an appropriately sized round-bottom flask and rotoevaporate the volatiles under reduced pressure (~20 mmHg) using a vacuum pump.
The purified 5′-functionalized deoxyribonucleoside 8a (1.32 g, 1.59 mmol) is isolated as a solid in a yield of 70%.8a. 1H NMR (300 MHz, DMSO-d6): δ 11.23 (s, 1H), 8.74 (s, 1H), 8.62 (s, 0.7H), 8.59 (s, 0.3H), 8.32 (s, 1H), 8.07 (d, J = 7.8 Hz, 2H), 7.65 (dd, J = 7.6, 7.4 Hz, 1H), 7.56 (dd, J = 7.6, 7.4 Hz, 2H), 6.51 (t, J = 6.8 Hz, 1H), 5.45 (m, 1H), 4.54 (br s, 1H), 3.98 (m, 2H), 3.85 (m, 1H), 3.38 (m, 4H), 2.92 (m, 4H), 2.79 (m, 2H), 2.38 (t, J = 7.3 Hz, 2H), 2.31-2.13 (m, 4H), 2.05 (s, 3H), 1.66 (m, 2H), 1.43 (m, 4H), 1.21 (m, 7H), 0.97 (m, 16H). 13C NMR (75 MHz, DMSO-d6): δ 208.3, 172.4, 172.2, 172.1, 172.0, 171.9, 171.8, 171.71, 171.66, 165.6, 151.9, 151.4, 150.3, 143.0, 142.8, 133.4, 132.4, 128.5, 128.4, 125.9, 125.8, 87.2, 83.74, 83.66, 79.2, 73.2, 73.1, 70.1, 70.4, 68.3, 68.2, 62.9, 62.7, 47.2, 46.5, 46.4, 45.8, 45.7, 44.1, 44.0, 42.6, 38.5, 35.9, 35.8, 35.0, 33.5, 33.3, 33.2, 33.0, 32.4, 32.3, 31.5, 29.6, 29.3, 29.2, 28.4, 28.3, 28.2, 28.1, 28.0, 27.3, 27.1, 24.7, 24.6, 24.3, 24.2, 23.1, 17.60, 17.52, 17.47, 17.43, 17.3, 17.21, 17.18, 17.11, 17.0, 12.9, 12.7, 12.6, 12.32, 12.29. +ESI-TOF-HRMS calcd for C41H63N7O8SiNa [M + Na]+ 832.4399, found 832.4411. -
12
Repeat steps 1 through 11 with the following exception:
Replace the deoxyribonucleoside 7a in step 5 with 7b (1.79 g, 5.40 mmol), 7c (1.82 g, 5.40 mmol) or 7d (1.31g, 5.40 mmol). The purified 5′-functionalized deoxyribonucleoside 8b (1.16 g, 1.48 mmol), 8c (890 mg, 1.13 mmol) or 8d (1.08 g, 1.54 mmol) is isolated as a solid in a yield of 65%, 50%, and 68% respectively.8b. 1H NMR (300 MHz, DMSO-d6): δ 11.25 (s, 1H), 8.27 (d, J = 7.3 Hz, 1H), 8.01 (d, J = 7.3 Hz, 2H), 7.63 (m, 1H), 7.52 (m, 2H), 7.37 (d, J = 7.3 Hz, 1H), 6.16 (t, J = 6.2 Hz, 1H), 5.37 (d, J = 4.4 Hz, 1H), 4.28 (m, 1H), 4.01-3.89 (m, 3H), 3.47 (s, 0.4H), 3.38 (m, 3H), 2.98 (s, 0.7H), 2.95 (s, 0.7H), 2.94 (s, 1.1H), 2.90 (s, 1.1H), 2.81 (m, 2H), 2.38 (m, 5H), 2.23 (t, J = 7.3 Hz, 2H), 2.15 (m, 2H), 2.05 (s, 3H), 1.70 (m, 2H), 1.43 (m, 4H), 1.27 (s, 6H), 1.20 (m, 2H), 1.02 (m, 14H). 13C NMR (75 MHz, DMSO-d6): δ 208.29, 208.27, 172.4, 172.2, 172.0, 171.9, 171.8, 171.70, 171.66, 167.3, 163.0, 154.3, 144.3, 133.1, 132.7, 128.41, 128.38, 95.8, 87.1, 86.0, 73.34, 73.30, 69.3, 62.3, 47.3, 47.2, 46.5, 46.4, 45.8, 45.7, 44.1, 44.0, 42.6, 40.8, 35.9, 35.8, 35.1, 35.0, 33.5, 33.3, 33.2, 33.0, 32.4, 32.3, 31.5, 29.6, 29.39, 29.37, 28.4, 28.3, 28.2, 28.13, 28.07, 27.3, 27.1, 24.7, 24.6, 24.31, 24.25, 23.1, 17.61, 17.59, 17.51, 17.48, 12.8, 12.6. +ESI-TOF-HRMS calcd for C40H63N5O9SiNa [M + Na]+ 808.4287, found 808.4298.8c. 1HNMR (300 MHz, DMSO-d6): δ 12.07 (s, 1H), 11.68 (s, 1H), 8.16 (s, 1H), 6.22 (t, J = 6.6 Hz, 1H), 5.37 (d, J = 4.2 Hz, 1H), 4.42 (m, 1H), 3.88 (d, J = 7.8 Hz, 1H), 3.83 (m, 2H), 3.44-3.34 (m, 4H), 2.95 (s, 0.8H), 2.94 (s, 0.8H), 2.91 (s, 1.2H), 2.89 (s, 1.2H), 2.77 (m, 3H), 2.63 (m, 2H), 2.38 (t, J = 7.3 Hz, 2H), 2.33-2.13 (m, 5H), 2.05 (s, 3H), 1.66 (m, 2H), 1.43 (m, 4H), 1.21 (m, 7H), 1.12 (d, J = 6.8 Hz, 6H), 0.95 (m, 14H). 13C NMR (75 MHz, DMSO-d6): δ 208.3, 180.1, 172.4, 172.2, 172.1, 172.0, 171.9, 171.8, 171.69, 171.66, 154.8, 148.3, 148.0, 137.2, 128.3, 120.3, 87.1, 82.8, 73.2, 73.1, 70.0, 62.9, 47.3, 47.2, 46.5, 46.3, 45.7, 45.6, 44.1, 44.0, 42.6, 35.9, 35.8, 35.05, 35.02, 34.7, 33.5, 33.3, 33.2, 33.0, 32.4, 32.3, 31.4, 29.6, 29.3, 28.4, 28.3, 28.2, 28.1, 28.0, 27.2, 27.1, 24.7, 24.5, 24.3, 24.2, 23.1, 18.83, 18.78, 17.55, 17.52, 17.48, 17.43, 17.1, 12.7, 12.6. +ESI-TOF-HRMS calcd for C38H65N7O9SiNa [M + Na]+ 814.4505, found 814.4518.8d. 1H NMR (300 MHz, DMSO-d6): δ 11.33 (s, 1H), 7.42 (s, 1H), 6.17 (t, J = 6.8 Hz, 1H), 5.31 (d, J = 4.5 Hz, 1H), 4.25 (m, 1H), 3.85 (m, 3H), 3.46 (s, 0.4H), 3.39 (s, 2 H), 3.36 (m, 1.1H), 2.97 (s, 0.8H), 2.95 (s, 0.7H), 2.92 (s, 1H), 2.90 (s, 1H), 2.80 (m, 2H), 2.39 (t, J = 7.3 Hz, 2H), 2.13 (m, 1H), 2.22 (m, 2H), 2.12 (m, 3H), 2.06 (s, 3H), 1.76 (s, 3H), 1.67 (m, 2H), 1.44 (m, 4H), 1.21 (m, 8H), 1.00 (m, 13H), 0.95 (s, 1H), 0.93 (s, 1H). 13C NMR (75 MHz, DMSO-d6): δ 208.3, 172.4, 172.2, 172.0, 171.9, 171.8, 171.71, 171.67, 163.1, 150.3, 135.6, 109.4, 86.5, 83.6, 73.3, 73.2, 70.0, 62.8, 47.3, 47.2, 46.5, 46.4, 45.8, 45.7, 44.1, 44.0, 42.6, 35.9, 35.8, 35.1, 35.0, 33.5, 33.3, 33.1, 33.0, 32.4, 32.3, 31.5, 29.6, 29.4, 28.4, 28.3, 28.2, 28.1, 28.0, 27.3, 27.1, 24.7, 24.6, 24.3, 24.2, 23.1, 17.60, 17.56, 17.50, 17.48, 17.3, 12.8, 12.63, 12.59, 12.7. +ESI-TOF-HRMS calcd for C34H60N4O9SiNa [M + Na]+ 719.4022, found 719.4031.
Synthesize the 5′-functionalized deoxyribonucleoside phosphoramidites 9a-d
-
13
Place a magnetic stir bar into a flame-dried 100-mL round-bottom flask. Seal the vessel with a rubber septum.
-
14
Add a solution of the 5′-functionalized 2′-deoxythymidine (8a, 810 mg, 1.00 mmol) in 10-mL anhydrous CH2Cl2 through the rubber septum using a 10-mL glass syringe and a 21-G syringe needle as a vent.
-
15
Add 335-μL 2-cyanoethyl N,N-diisopropylchlorophosphoramidite (1.50 mmol) using a 1-mL glass syringe. Stir the reaction mixture at ~25°C for 3 hr using a magnetic stirrer.
Quench the reaction
-
16
Add 10-mL water through the rubber septum using a 10-mL glass syringe and a 21-G syringe needle as a vent. Remove the rubber septum and add 25-mL CH2Cl2 in the flask using a 50-mL glass cylinder.
Extract the product
-
17
Pour the liquid into a 100-mL separatory funnel. Seal the funnel and shake vigorously. Allow phase separation.
-
18
Collect the organic layer in a 100-mL Erlenmeyer and mix with 5 g anhydrous sodium sulfate. Filter the suspension through a 30-mL fritted glass vacuum filtration Buchner funnel of coarse porosity connected to a 100-mL round bottom flask. Connect the funnel to an in-house vacuum line. Apply vacuum.
-
19
Remove the funnel and rotoevaporate the solvent to an oil under reduced pressure (~20 mmHg) using a vacuum pump.
Purify and isolate the 5′-functionalized deoxyribonucleoside phosphoramidites 9a
-
20
Use a Pasteur pipette to add 4-mL 9:1 (v/v) C6H6/Et3N and disperse the solution on top of a 5 × 20-cm chromatography glass column packed with ~25 g silica gel that has been equilibrated in 9:1 (v/v) C6H6/Et3N.
-
21
Elute the product from the column using 9:1 (v/v) C6H6/Et3N as the eluent. Collect 6-mL fractions in 13 × 100-mm disposable glass tubes.
-
22
Analyze the fractions by TLC (APPENDIX 3D) on a 2.5 × 7.5–cm EMD silica gel 60 F254 TLC plate using 9:1 (v/v) C6H6/Et3N as the eluent and UV254 for detection. Pool appropriate fractions [Rf (9:1 (v/v) C6H6/Et3N) = 0.7] into a 100-mL round bottom flask and rotoevaporate the solvents to a colorless oil under reduced pressure (~20 mmHg) using a vacuum pump.
Lyophilize the 5′-functionalized dexoyribonucleoside phosphoramidite 9a
-
23
Dissolve the purified phosphoramidite 9a in 10-mL dry benzene. Freeze the solution by manually swirling it in a dry-ice/acetone bath. Lyophilize the frozen solution under high vacuum using an oil vacuum pump connected to a glass trap cooled in liquid nitrogen-filled Dewar flask.
Triethylamine-free 9a (837 mg, 830 μmol) is isolated as a colorless oil in a yield of 83%.9a. 31P NMR (121 MHz, C6D6): δ 149.0, 148.8, 148.76, 148.72. +ESI-TOF-HRMS calcd for C50H80N9O9PSi [M + H]+ 1010.5659, found1010.5675. -
24
Repeat steps 13 through 23 with the following exception:
Replace the 5′-functionalized deoxyribonucleoside 8a in step 14 with 8b (785 mg, 1.00 mmol), 8c (791 mg, 1.00 mmol) or 8d (696 mg, 1.00 mmol).The purified triethylamine-free 5′-functionalized deoxyribonucleoside phosphoramite 9b (788 mg, 800 μmol), 9c (743 mg, 795 μmol) or 9d (717 mg, 800 μmol) is isolated as a colorless oil in a yield of 80%, 75%, and 80% respectively.9b. 31P NMR (121 MHz, C6D6): δ 148.3, 148.25, 148.22, 148.1, 147.91, 147.89. +ESI-TOF-HRMS calcd for C49H80N7O10PSi [M+ H]+ 986.5546, found 986.5548.9c. 31P NMR (121 MHz, C6D6): δ 148.77, 148.73, 148.0, 147.92, 147.88. +ESI-TOF-HRMS calcd for C47H82N9O10PSi [M + H]+ 992.5764, found 992.5773.9d. 31P NMR (121 MHz, C6D6) δ 148.7, 148.6, 148.49, 148.46, 148.37. +ESI-TOF-HRMS calcd for C43H77N6O10PSiCs [M + Cs]+ 1029.4257, found 1029.4266.
SUPPORT PROTOCOL 1. SOLID-PHASE SYNTHESIS OF 5′-FUNCTIONALIZED PHOSPHOROTHIOATE AND NATIVE DNA SEQUENCES
This protocol delineates the synthesis and deprotection of 5′-functionalized phosphorothioate and native DNA sequences (Fig. 4).
Figure 4.

5′-Functionalized DNA sequences for solid-phase capture and purification.
Additional Materials
Long-chain alkylamine controlled-pore glass (500 Å LCAA-CPG or 2000 Å LCAA-CPG) support functionalized with 1-μmol 5′-O-(4,4′-dimethoxytrityl)-2′-deoxythymidine as the leader nucleoside (Glen Research).
5′-O-(4,4′-dimethoxytrityl)-dABz, -dGiBu, -dCBz and -dT phosphoramidite monomers (ChemGenes)
5′-Functionalized deoxyribonucleoside phosphoramidites 9a–d (Basic Protocol 3)
Anhydrous acetonitrile diluent (MeCN, Glen Research)
-
Reagents for oligonucleotide synthesis (all available from Glen Research):
0.45 M 1H-tetrazole or 0.25 M 5-ethythio-1H-tetrazole in MeCN
Cap A solution: acetic anhydride in tetrahydrofuran (THF)/pyridine
Cap B solution: 1-methylimidazole in THF
Oxidation solution: 0.05 M 3H-1,2-benzodithiol-3-one 1,1-dioxide in MeCN or 0.02 M iodine in THF/pyridine/water
Deblocking solution: 3% trichloroacetic acid in CH2Cl2
Concentrated aqueous ammonia (NH3, Aldrich)
4-mL screw cap glass vial with caps (Fisher Scientific)
1-mL glass syringe
1-mL plastic syringe
21-G stainless steel syringe needles
Heat block (VWR)
DNA/RNA synthesizer (394 DNA/RNA synthesizer, Applied Biosystems)
Synthesize the DNA Sequences
-
1
Perform a 1-μmol scale solid-phase synthesis of the phosphorothioate DNA sequence 10a–d (Fig. 4) using an Applied Biosystems 394 DNA/RNA synthesizer in trityl-on mode according to the manufacturer’s instructions (also see APPENDIX 3C).
Each synthesis is performed using a long-chain alkylamine controlled-pore glass (500 Å LCAA-CPG or 2000 Å LCAA-CPG for the native DNA sequence 10f) support functionalized with 5′-O-(4,4′-dimethoxytrityl)-2′-deoxythymidine as the leader nucleoside. The syntheses are carried out using a DNA/RNA synthesizer and commercial appropriately protected deoxyribonucleoside phosphoramidites, which are each dissolved in dry MeCN to give a 0.1 M solution. Each 5′-functionalized deoxyribonucleoside phosphoramidite 9a–d is dissolved in dry MeCN to provide a 0.15 M solution. Each solution is placed in a distinct vial connected to the DNA/RNA synthesizer through an additional delivery port. Commercial 1H-tetrazole solution was used for phosphoramidite activation in the solid-phase synthesis of DNA sequences 10a–f. The reaction times for the coupling, capping, oxidation and 5′-deblocking steps in the synthesis of native and phosphorothioate DNA sequences are 120, 60, 60 s and 60 s, respectively. However, the capping step in the synthesis of phosphorothioate DNA sequences (10a–d) is performed after the oxidative sulfuration step, which is effected using 0.05 M 3H-1,2-benzodithiol-3-one 1,1-dioxide in MeCN (Iyer et al., 1990); the standard 0.02 M iodine solution in THF/pyridine/water is employed in the oxidation step of native DNA sequences. The last coupling reaction of each synthesis is performed using any of the activated 5′-functionalized deoxyribonucleoside phosphoramidites 9a–d over a period of 180 s.
Deprotect the 5′-functionalized DNA sequences
-
2
Transfer the LCAA-CPG-linked 5′-functionalized DNA sequence 10a from the synthesis column to a 4-mL screw cap glass vial. Add 1-mL of concentrated aqueous ammonia using a 1-mL glass syringe. Tightly seal the vial and place it in a heat block kept at ~65 °C over a period of 16 hr.
-
3
Allow the vial to cool to ~25 °C. Carefully open the vial and transfer the ammoniacal solution to and empty 4-mL screw cap glass vial using a 1-mL plastic syringe. Evaporate the solution to approximately half its original volume using a stream of air.
The deprotected DNA sequence 10a can be used immediately for solid-phase purification or kept frozen at −20 °C in the sealed glass vial for subsequent solid-phase purification. -
4
Repeat steps 2 and 3 for deprotection of each crude DNA sequence 10b–f.
BASIC PROTOCOL 4. GENERAL PROCEDURE FOR THE SOLID-PHASE CAPTURE AND RELEASE OF PHOSPHOROTHIOATE AND NATIVE DNA SEQUENCES
The details of the capture of crude 5′-functionalized DNA sequences 10a–f by the solid support 3 are provided in this protocol. As shown in Fig 5, capture of the DNA sequences occurs from the reaction of the aminooxy groups of support 3 with the keto function of each 5′-functionalized DNA sequences 10a–f and formation of stable oxime conjugates on the surface of solid support 11a–f. Fig. 5 also shows the release of the captured DNA sequences from support 11a–f upon treatment with tetra-n-butylammonium fluoride to provide the highly pure DNA sequences 12a–f (Fig. 6).
Figure 5.

Capture of crude 5′-functionalized DNA sequences (10a–f) by the solid support 3 and release of these sequences from the solid supports 11a–f as purified sequences 12a–f. Si, 3-aminopropylated silica gel; B, adenin-9-yl, cytosin-1-yl, guanin-9-yl, or thymin-1-yl.
Figure 6.

Solid-phase purified DNA sequences released from the supports 11a–f.
Materials
Capture solid support 3 (Basic Protocol 1)
5′-Functionalized DNA sequences 10a–f (Basic Protocol 3)
Acetonitrile (MeCN, Aldrich)
Dimethyl sulfoxide (DMSO, Aldrich)
Peroxide-free tetrahydrofuran (THF, Aldrich)
Triethylamine (Aldrich)
Methoxytrimethylsilane
2 M Triethylammonium acetate solution (Applied Biosystems)
Tetra-n-butylammonium fluoride hydrate (Aldrich)
Tetra-n-butyl ammonium chloride (Aldrich)
Concentrate aqueous ammonia (NH3, Aldrich)
4-mL screw cap glass vial with caps (Fisher Scientific)
1.5-mL microcentrifuge tubes (Fisher Scientific)
15-mL fritted glass vacuum filtration Buchner funnel of coarse porosity (ChemGlass)
100-mL Erlenmeyer flask (Fisher Scientific)
25-mL round-bottom flask
1- and 10-mL plastic syringes (Fisher Scientific)
1- and 10-mL glass syringes (Fisher Scientific)
21-G stainless steel syringe needles
Pasteur pipettes (VWR)
Pipettor (Corning)
1-mL pipet tips (Fisher Scientific)
Spatula
Temperature-controlled orbital shaker (Thomas Scientific)
Heath block (VWR)
RP-HPLC (Agilent Technologies)
5 μm Supelcosil 25 cm × 4.6 mm LC-18S column (Supelco)
Eppendorf microcentrifuge
Rotary evaporator connected to a vacuum pump (Büchi)
High vacuum oil pump
Capture the crude 5′-functionalized phosphorothioate or native DNA sequences
-
1
Capture support 3 (150 mg) is added to a 15-mL fritted glass vacuum filtration Buchner funnel of coarse porosity connected to a 100-mL Erlenmeyer. Add a 10-mL 1:5 (v/v) Et3N/MeCN using a 10-mL plastic syringe. Allow the filtrate to drip into the Erlenmeyer. Connect the funnel to an in-house vacuum line and apply vacuum to air-dry the capture support.
-
2
Transfer the dry support to a 4-mL screw cap glass vial. Add successively solid tetra-n-butylammonium chloride (14 mg, 50 μmol) using a spatula and the solution (~ 500 μL in water) of crude 5′-functionalized DNA sequence 10a (170 OD260) obtained from step 3 of Support Protocol 1 using a 1-mL plastic syringe. Seal the flask and agitate the suspension at 65 °C over a period of 3 hr using an orbital shaker.
-
3
Stop the shaker. Allow the suspension to settle. Verify the near completeness of the capture reaction by RP-HPLC analysis of a 1-μL aliquot of the supernatant.
The completeness of the capture reaction is made based on a side-by-side comparison of the RP-HPLC chromatographic profile of crude 5′-functionalized DNA sequence 10a, prior to capture, with the RP-HPLC profile of the 1-μL supernatant aliquot recorded after capture of 10a. RP-HPLC analyses have been performed using a 5 μm Supelcosil LC-18S column (25 cm × 4.6 mm) under the following conditions: starting from 0.1 M (pH 7.0), a linear gradient of 2.5% MeCN/min is pumped at a flow rate of 1 mL/min for 40 min. -
4
Repeat step 1 through 3 for solid-phase capture of each 5′-functionalized DNA sequence 10b–f.
-
5
Transfer the suspended solid support 11a (Fig. 5) to a 15-mL fritted glass vacuum filtration Buchner funnel of coarse porosity connected to a 100-mL Erlenmeyer. Add 10-mL of 1:1 (v/v) CH3CN/H2O. Connect the funnel to an in-house vacuum line. Apply vacuum to air-dry the support.
-
6
Transfer the support to a 4-mL screw cap glass vial. Add a 1-mL of 1:1 (v/v) concentrated aqueous NH3/CH3CN using a 1-mL glass syringe. Seal the vial and place it in a heat block for 30 min at 65 °C. Allow the flask to cool to ~25 °C.
-
7
Transfer the suspended solid support 11a to a 15-mL fritted glass vacuum filtration Buchner funnel of coarse porosity connected to a 100-mL Erlenmeyer. Connect the funnel to an in-house vacuum line and apply vacuum to air-dry the support.
-
8
Repeat steps 6 and 7 once.
-
9
Add 1-mL DMSO to the Buchner funnel using a 1-mL glass syringe. Apply vacuum. Repeat step 9 four more times and air-dry the support 11a.
The washing steps 5 through 9 are necessary to wash away the shorter than full length DNA sequences that might have adsorbed to the support 11a.
Release of the DNA sequence 12a from the Support 11a
-
10
Transfer the air-dried solid support to a 4-mL screw cap glass vial. Add 0.5-mL of a 1.0 M tetra-n-butylammonium fluoride in dry DMSO using a 1-mL glass syringe. Seal the flask and place it in a heat block over a period of 3 hr at 65 °C. Allow the glass vial to cool to ~25 °C.
1.0 M tetra-n-butylammonium fluoride in dry DMSO is prepared by mixing solid tetra-n-butylammonium fluoride hydrate (262 mg) in 1.0-mL dry DMSO. -
11
Open the glass vial. Add 200 μL methoxytrimethylsilane and 200 μL MeCN to the suspension. Seal the vial and keep it at ~25 °C for 30 min.
Methoxytrimethylsilane is used to consume excess fluoride ions. -
12
Pour the suspension into a 15-mL fritted glass vacuum filtration Buchner funnel of coarse porosity connected to a 25-mL round-bottom flask. Add a 10-mL of 1:5 (v/v) Et3N/MeCN using a 10-mL glass syringe. Allow the filtrate to drip into the flask. Connect the funnel to an in-house vacuum line. Apply vacuum to air-dry the support.
-
13
Repeat step 6.
-
14
Transfer the suspended solid support 11a to a 15-mL fritted glass vacuum filtration Buchner funnel of coarse porosity connected to the 25-mL round bottom flask used in step 12. Allow the filtrate to drip into the flask. Apply vacuum to air-dry the support.
-
15
Repeat successively step 6 and 14. Remove the Buchner funnel. Concentrate the filtrates collected in the 25-mL round bottom flask under reduced pressure to a volume of ~100 μL using a high vacuum oil pump.
The washing steps 12 through 15 are necessary to ensure that the full length DNA sequence 12a is completely washed away from the support 11a.
Precipitate the DNA sequence 12a
-
16
Add 1-mL peroxide-free THF to the concentrated material. Swirl the flask to yield a uniform precipitate of the solid-phase purified DNA sequence 12a.
-
17
Pipet the precipitate into a 1.5-mL microcentrifuge tube using a Pasteur pipette. Centrifuge at 16,000g for 15 min at 25 °C.
-
18
Carefully remove the supernatant using a pipettor. Resuspend the DNA pellet in 1-mL peroxide-free THF. Centrifuge at 16,000g for 15 min at 25 °C.
-
19
Repeat step 18 two more times.
-
20
Carefully remove the supernatant using a pipettor. Dry the DNA pellet under reduced pressure using a SpeedVac system. Store the dried DNA pellet at −20 °C until further use.
-
21
Repeat steps 5 through 20 with each of the solid support 11b–f to provide the solid-phase purified DNA sequences 12b–f.
The solid-phase purification of DNA sequence 10a demonstrates that the sequences 10b–f can each be, technically, solid-phase purified in a parallel or high-throughput manner. The yields of solid-phase purified DNA sequences depend of several factors including: (i) composition (purine or pyrimidine rich; modified or unmodified) and length of the DNA sequence; (ii) solubility of the sequence in the capture solution, (iii) quality of the phosphoramidite 9a–d and (iv) performance of the DNA synthesizer. Under optimal conditions, the yield and purity of solid-phase purified DNA sequences are comparable or better than that obtained by RP-HPLC, as discussed in the background section of this report.
ALTERNATE PROTOCOL 1. SCALABLE SOLID-PHASE PURIFICATION OF A RELEVANT DNA SEQUENCE
This protocol provides evidence that the solid-phase purification of synthetic DNA sequences can be qualified as a scalable process. This is demonstrated through the solid-phase synthesis of ten identical 5′-functionalized phosphorothioate DNA sequences, each on a 1 μmol scale. These individual sequences will be purified together using a10-fold increase of the amount of capture support 3 typically needed for a 1 μmol scale solid-phase purification process.
Materials
5′-Functionalized phosphorothioate DNA sequences (10a, Support Protocol 1)
Capture support 3 (Basic Protocol 1)
Acetonitrile (MeCN, Aldrich)
Dimethyl sulfoxide (DMSO, Aldrich)
Dimethyl sulfoxide, dry (Acros)
Tetra-n-butylammonium chloride (Aldrich)
Tetra-n-butylammonium fluoride hydrate (Aldrich)
Methoxytrimethylsilane (Aldrich)
Peroxide-free tetrahydrofuran (THF, Aldrich)
Triethylamine (Et3N, Aldrich)
25- and 100-mL round-bottom flasks
100-mL Erlenmeyer flask
20-mL screw cap glass vial with caps (Kimble)
10-mL glass cylinder
15-mL fritted glass vacuum filtration Buchner funnel of coarse porosity (ChemGlass)
5-mL plastic syringe
5- and 10-mL glass syringes
21-G stainless steel syringe needles (Fisher Scientific)
Pipettor (Corning)
1-mL pipette tips (Fisher Scientific)
Heat block (VWR)
DNA/RNA synthesizer (394 DNA/RNA synthesizer, Applied Biosystems)
Rotary evaporator connected to a vacuum pump (Büchi)
Temperature-controlled orbital shaker (Thomas Scientific)
Vortexer (VWR)
Synthesize the DNA Sequence 10a
-
1
Repeat step 1 of Support Protocol 1.
The solid-phase synthesis of each of the ten phosphorothioate DNA sequences (10a) has been achieved on a 1-μmole scale using a DNA/RNA synthesizer capable of assembling four DNA sequences in a single run. Thus, two synthesis runs of four identical sequences and a single run of two identical sequences have been performed to provide 10a from ten individual sequences.
Deprotect the 5′-functionalized DNA sequence 10a
-
2
Repeat step 2 of Support Protocol 1 for each of the ten individual sequences.
-
3
Allow each of the ten vials to cool to ~25 °C. Carefully open each vial and pool each ammoniacal solution together into a 25-mL round-bottom flask using a 5-mL plastic syringe. Rotoevaporate to half (~5 mL) of the original volume (~10 mL) under reduced pressure (~ 20 mmHg).
-
4
Add 5.0-mL of a 0.1 M solution of tetra-n-butylammonium chloride in 1:1 (v/v) DMSO/H2O to the crude DNA solution of step 3 using a 5-mL glass syringe.
Capture the crude 5′-functionalized phosphorothioate DNA sequence 10a
-
5
Repeat step 1 of Basic Protocol 4 using 1.50 g of capture support 3.
The amount of capture support 3 is a 10-fold increase of the amount of support used (150 mg) for the solid-phase purification of 10a that was synthesized on a 1- μmol scale (see step1 of Support Protocol 1) -
6
Place the capture support 3 of step 5 into a 20-mL screw cap glass vial. Pour the crude DNA solution of step 4 into the vial. Tightly seal the vial and agitate the suspension over 3 hr at 65 °C using an orbital shaker.
-
7
Repeat step 3 of Basic Protocol 4 using 0.5 μL aliquot of the supernatant.
-
8
Transfer the suspended solid support 11a to a 15-mL fritted glass vacuum filtration Buchner funnel of coarse porosity connected to a 100-mL Erlenmeyer. Connect the funnel to an in-house vacuum line. Apply vacuum to air-dry the support.
-
9
Transfer the support to a 20-mL screw cap glass vial. Add a 10-mL of 10% Et3N in 1:1 (v/v) CH3CN/H2O using a 10-mL glass syringe. Seal the vial and place it in a heat block for 30 min at 65 °C. Allow the flask to cool at ~25 °C.
-
10
Repeat successively steps 8 and 9 two more times.
-
11
Repeat step 8.
-
12
Add 5-mL DMSO to the Buchner funnel using a 5-mL glass syringe. Apply vacuum. Repeat step 12 four more times and air-dry the support 11a.
Release of the DNA sequence 12a from the Support 11a
-
13
Transfer the air-dried solid support to a 20-mL screw cap glass vial. Add 5-mL of a 1.0 M tetra-n-butylammonium fluoride in dry DMSO using a 5-mL glass syringe. Seal the flask and place it in a heat block over a period of 3 hr at 65 °C. Allow the glass vial to cool to ~25 °C.
-
14
Open the glass vial. Add 2 mL methoxytrimethylsilane and 2 mL MeCN to the suspension using a 5-mL glass syringe. Seal the vial and keep it at ~25 °C for 30 min.
-
15
Pour the suspension into a 15-mL fritted glass vacuum filtration Buchner funnel of coarse porosity connected to a 100-mL round bottom flask. Allow the filtrate to drip into the flask. Connect the funnel to an in-house vacuum line. Apply vacuum to air-dry the support.
-
16
Transfer the air-dried solid support to a 20-mL screw cap glass vial. Add 10-mL of 10% Et3N in1:1 (v/v) MeCN/H2O. Seal the flask and place it in a heat block over a period of 30 min at 65 °C. Allow the glass vial to cool to ~25 °C.
-
17
Repeat step 15.
-
18
Repeat successively steps 16 and 17. Remove the funnel from the 100-mL round-bottom flask containing all filtrates collected in step 15, 17 and 18. Concentrate the filtrates to ~ 1-mL by rotoevaporation under reduced pressure (~20 mmHg) using a vacuum pump.
Precipitate the DNA sequence 12a
-
19
Add 19-mL peroxide-free THF to the concentrated material. Seal the vial. Manually swirl the vial to yield a uniform precipitate of the solid-phase-purified DNA sequence 12a. Allow the product to settle.
-
20
Open the vial and carefully pipet most of the clear supernatant using a pipettor. Add 10-mL peroxide-free THF to the vial using a 10-mL glass cylinder. Seal the vial and vortex it for 30 sec, using a vortexer. Allow the product to settle.
-
21
Repeat step 20 three times.
-
22
Open the vial. Pipet approximately half of the clear supernatant using a pipettor. Swirl the vial and pour the suspension into a 25-mL-round bottom flask. Rinse the vial by adding 5-mL peroxide-free THF using a 10-mL glass cylinder. Swirl and pour the wash into the 25-mL round-bottom flask.
-
23
Rotoevaporate the suspension to dryness under reduced pressure (~20 mmHg). Store indefinitely the purified nucleic acid sequence 12a at −20 °C.
COMMENTARY
Background Information
The ability to design and synthesize DNA and RNA sequences has had a massive impact on the rapidly expanding fields of synthetic biology and nucleic acid-based drug development. Typically, DNA/RNA sequences and their analogues have been used for recognition and binding to mRNAs encoding disease-causing proteins and have led to the production of nucleic acid-based drugs capable of inhibiting the expression of these proteins through either an antisense (Crook, 2004) or an RNA interference (Dorsett and Tuschl, 2004) pathway. Both approaches to silencing gene expression require the manufacture of synthetic nucleic acid sequences in large quantities (e.g., millimoles) and high purity for preclinical and clinical investigations. Conversely, total gene synthesis for synthetic biology applications requires small amounts (e.g., nanomoles) of a large number of highly pure synthetic DNA sequences. Although the automated chemical synthesis of nucleic acid sequences is efficient and can be scaled up for pharmaceutical production, the purification of these sequences is challenging; the full-length nucleic acid sequences are mixed with shorter sequences, due to incomplete coupling of activated deoxyribonucleoside phosphoramidites at each chain extension step of the nucleic acid sequence assembly. Even though these shorter than full-length sequences are produced in small amounts, the physicochemical similarity of these sequences to the desired nucleic acid sequence makes them very difficult to chromatographically separate them. HPLC-based methods including reversed-phase HPLC and anion-exchange HPLC are currently the preferred techniques for either small or large-scale purification of nucleic acid sequences. However, these methods require expensive instruments and accessories; considerable volumes of buffered aqueous and organic solvents are also needed for large-scale purification of DNA sequences, thereby contributing to high operational cost. In addition to not being cost-effective, HPLC-based purification methods are not amenable to parallel purification processes; only a single nucleic acid sequence can be purified per run and depending on the nature of each nucleic acid sequence, more than one purification run may be required to achieve the level of sequence purity required for pharmaceutical or synthetic biology applications. One important limitation of any large- or small-scale HPLC purification of DNA sequences is the burdensome removal of relatively large volumes of aqueous solvents generated during the purification process; this operation requires pricey equipment as well. HPLC-based purification methods are time-consuming and often do not completely resolve shorter than full-length sequences from the desired DNA sequence. Alternatively, polyacrylamide gel electrophoresis (PAGE, see UNIT 10.4) can efficiently separate shorter nucleic acid sequences from full-length DNA sequences; however the high-throughput or large scale purification of DNA sequences by PAGE, has to the best of our knowledge not been reported in the scientific literature. Several methods orthogonal to reversed-phase HPLC or electrophoretic techniques have, however, been proposed for the purification of nucleic acid sequences. These methods are based on ion-pair (Swiderski et al., 1994), hydrophobic (Sproat et al., 1999) or affinity (Fang and Bergstrom, 2003a, 2003b, 2003c, 2004; Pearson et al., 2005; Beller and Bannwarth, 2005; Dandapani, 2006; Mishra et al., 2006) chromatography. All of these purification techniques are either not amenable to either highly parallel or large-scale purification of nucleic acid sequences. A conceptually different approach to the purification of synthetic DNA sequences has been proposed (Fang et al., 2011; Pokharel et al., 2014). The method consists of catching shorter than full-length sequences through a polymerization process; the full-length sequence is then extracted from the polymer and isolated by precipitation in n-butanol. This process has been claimed to be potentially useful for large-scale and high-throughput purification of DNA sequences (Pokharel et al., 2014). In this context, an innovative solid-phase-based purification method for the purification of synthetic unmodified and modified (e.g., phosphorothioate) DNA sequences has recently been reported (Grajkowski et. al., 2016). The main objective of this method is to provide an efficient solid-phase capture of synthetic nucleic acid sequences through an oximation reaction and subsequent fluoride-assisted release of the highly purified sequences from the capture support. This solid-phase purification process has been applied to the purification of an unmodified synthetic DNA sequence of moderate length (60-mer) and shorter phosphorothioate DNA sequences (20-mer). The parallel purification of a phosphorothioate DNA sequence and the scalability of the solid-phase purification process in terms of yield and purity of the sequence have been investigated. Specifically, the solid-phase purification process consists of functionalizing a commercial aminopropylated silica gel-based support using 1,1′-carbonyldiimidazole-activated 7-oxooctanoic acid to provide the ketoalkyl amidoalkylated support 2 (Fig. 1). Treatment of the support 2 with O,O′-1,3-propanediylbishydroxylamine dihydrochloride yields the aminooxy-functionalized capture support 3. The selection of aminooxy groups for the functionalization of support 3 is justified based on the high reactivity (Fina and Edwards, 1973) these groups with aldehydes or ketones to form stable aldoximes or ketoximes. Although the formation of stable oxime ethers with nucleosides and nucleic acid sequences (Morvan et al. 1996; Trevisiol et al., 1997; Kawasaki et al., 1999; Salo et al., 1999; Forget et al., 2001a, 2001b, 2001c; Defrancq and Lhomme, 2001; Katajisto et al., 2004; Cieślak et al., 2012, 2013) has been reported for various purposes, oximation reactions have not been used for solid-phase purification of nucleic acid sequences. With the intent of using the aminooxy functions of support 3 to capture full-length synthetic DNA sequences, the 5′-terminus of these sequences should strategically be functionalized with a linker carrying a keto function to enable conjugation of the DNA sequences through stable oxime linkages. Such a linker has been prepared from the reaction of an aqueous solution of N,N′-dimethylethylenediamine with 4,4-dimethyl-γ-butyrolactone (4b) to produce the aminated amido alcohol 5b (Fig. 2). The reaction of CDI-activated 7-oxooctanoic acid 1 with 5b at gives the keto diamidated alcohol 6b in a post-purification yield of 80%. Conjugation of 6b to the 5′-hydroxy function of 2′-deoxythymidine and N-protected 2′-deoxyribonucleosides (7a–d) is achieved upon reaction with dichlorodiisopropylsilane (Fang and Bergstrom, 2003c; Fang and Fueangfung, 2010; Trader and Carlson, 2011) and imidazole to provide the 5′-O-diisopropylsiloxyl ether derivatives (Fang and Bergstrom, 2003a, 2003b, 2004) 8a–d (Fig. 4.xx.3) with yields in the range of 50–70%. With the aim of incorporating the 5′-functionalized deoxyribonucleosides 8a–d into DNA sequences, they have been converted to their corresponding phosphoramidite derivatives 9a–d (Fig. 3) using commercial 2-cyanoethyl N,N-diisopropylchlorophosphoramidite in the presence of N,N-diisopropylethylamine in anhydrous methylene chloride (Beaucage, 1993; Ellington and Pollard, 2000; Wilk et al., 2001).
The 5′-functionalized deoxyribonucleoside phosphosphoramites 9a–d have been purified on silica gel and isolated as triethylamine-free viscous oils after lyophilization from dry benzene in yields ranging from 75–83%. These phosphoramidites have been satisfactorily characterized by 31P NMR spectroscopy and high resolution mass spectrometry.
The solid-phase synthesis of 5′-functionalized phosphorothioate and native DNA sequences (10a–d and 10e–f, respectively) has been conducted using a commercial long-chain alkylamine controlled-pore glass support (LCAA-CPG) and standard deoxyribonucleoside phosphoramidites according to standard protocols. The 5′-functionalized phosphoramidites 9a–d are used in the last coupling cycle of each DNA sequence assembly. It is critically important that phosphoramidite coupling efficiency and the capping of unreacted 5′-hydroxy functions be optimal for solid-phase purification of DNA sequences; less than optimal phosphoramidite coupling and capping reactions will result in poorer recovery of solid-phase-purified DNA sequences. In this regard, the coupling time of 5′-functionalized phosphoramidites 9a–d has been extended to 180 s to ensure the highest coupling efficiency of these 5′-sterically-demanding phosphoramidite monomers. Post-synthesis deprotection and release of the DNA sequences 10a–f from LCAA-CPG have been performed under basic conditions according to standard protocols (Ellington and Pollard, 2000). Each aqueous ammonia solution containing the crude DNA sequence 10a–f (Figs. 4 and 5) and shorter than full-length sequences is evaporated to one-half of its original volume. Solid tetra-n-butylammonium chloride (TBACl) and the capture support 3 are then sequentially added to the aqueous solution of DNA sequences; the suspension is kept at 65 °C over a period of 3 hr. TBACl is used to exchange the ammonium counterions of thiophosphate or phosphate diester functions with tetrabutylammonium ions to enhance the solubility of phosphorothioate DNA sequences in aqueous solutions. RP-HPLC analysis of the pre- and post-capture solutions of each DNA sequence synthesized using the 5′-functionalized deoxyribonucleoside phosphoramidites 9a–d indicates that the oximation reaction resulting in the capture of each 5′-functionalized DNA sequence 10a–f is in all cases near complete (Grajkowski et al. 2016). The solid supports 11a–f (Fig. 5) are then treated twice with a warm (55 °C) solution of aqueous ammonia in acetonitrile to wash off unbound shorter than full-length DNA sequences by filtration; a final wash with anhydrous DMSO is necessary to ensure efficient release of the DNA sequences 12a–f from their respective supports (11a–f) upon treatment with 1.0 M tetra-n-butylammonium fluoride (TBAF) in dry DMSO for 3 hr at 65 °C. The solid-phase purified DNA sequences 12a–f (Fig. 6) are isolated by precipitation in dry THF and characterized by ESI-TOF-MS. The purity of these sequences has been evaluated by RP-HPLC and by PAGE under denaturing conditions (Fig. 7, Fig. 8, Fig. 9, Fig. 10 and Fig. 11).
Figure 7.

Solid-phase purification of the phosphorothioate DNA sequence 10a. RP-HPLC analysis of solid-phase-purified 12a that was released from the support 11a. Inset: Purity analysis of the solid-phase-purified 12a by PAGE. Chromatographic conditions are reported in the annotation of step 3 of Basic Protocol 4; electrophoretic conditions are reported in Grajkowski et al. 2016. Abbreviation: BP, bromophenol blue dye.
Figure 8.

Solid-phase purification of the phosphorothioate DNA sequence 10b. RP-HPLC analysis of solid-phase-purified 12b that was released from the support 11b. Inset: Purity analysis of the solid-phase-purified 12b by PAGE. Abbreviation: BP, bromophenol blue dye.
Figure 9.

Solid-phase purification of the phosphorothioate DNA sequence 10c. RP-HPLC analysis of solid-phase-purified 12c that was released from the support 11c. Inset: Purity analysis of the solid-phase-purified 12c by PAGE. Abbreviation: BP, bromophenol blue dye.
Figure 10.

Solid-phase purification of the phosphorothioate DNA sequence 10d. RP-HPLC analysis of solid-phase-purified 12d that was released from the support 11d. Inset: Purity analysis of the solid-phase-purified 12d by PAGE. Abbreviation: BP, bromophenol blue dye.
Figure 11.

Solid-phase purification of the native DNA sequence 10f. RP-HPLC analysis of solid-phase-purified 12f that was released from the support 11f. Inset: Purity analysis of the solid-phase-purified 12f by PAGE. Abbreviation: BP, bromophenol blue dye.
Although the RP-HPLC chromatograms show the purity of solid-phase-purified DNA sequences is essentially 100%, PAGE analyses of the same sequences show minute amounts of shorter than full-length DNA sequences, indicating that the coupling efficiency of commercial phosphoramidites and that of 9a–d is less than 100%. The presence of tiny amounts of shorter than full-length sequences also underscores the affinity of those sequences for silica gel, which makes it difficult to quantitatively wash them off the supports 11a–f prior to release.
With the objective of demonstrating that the solid-phase purification process can be qualified as highly parallel and scalable, the solid-phase synthesis of ten (10) identical phosphorothioate DNA sequences (10a) is carried out, each on a scale of 1 μmol using the conditions described in Support Protocol 1 and Alternate Protocol 1. Upon completion of the syntheses, deprotection, and release of each DNA sequence from the CPG support, the ammoniacal solution of each sequence are pooled together and concentrated under reduced pressure. The amounts of the capture support 3, reagents, and solvent required for the capture of 10a are increased by 10-fold while keeping the final concentration of the reagents the same as that reported for individual syntheses. The capture reaction is performed under conditions identical to those described in Basic Protocol 4 for individual syntheses in terms of reaction time and temperature. As anticipated, the capture reaction resulted in the near complete (>98%) disappearance of the DNA sequence 10a (Figs. 12A–B). Release of the DNA sequence 12a from the support 11a (Fig. 12C) has been done using a 10-fold increase of 1.0 M TBAF/DMSO solution that had been required for a 1 μmol scale reaction, while keeping reaction time and temperature conditions the same. A 10-fold increase in the volume of THF is necessary to precipitate 12a (Fig. 12D). The yield of the solid-phase-purified DNA sequence has been measured by UV spectroscopy at 260 nm and found to be 995 ODs; this yield is nearly proportional to that of solid-phase-purified 12a (105 ODs) isolated from a 1 μmol process scale. These results conclusively demonstrate that the solid-phase purification of nucleic acid sequences can be achieved in a highly parallel and scalable process. Thus, the proposed solid-phase purification process provides a cost-effective high-throughput alternative to conventional chromatographic methods for the purification and production of phosphorothioate or native DNA sequences with a comparable, if not better, level of purity.
Figure 12.



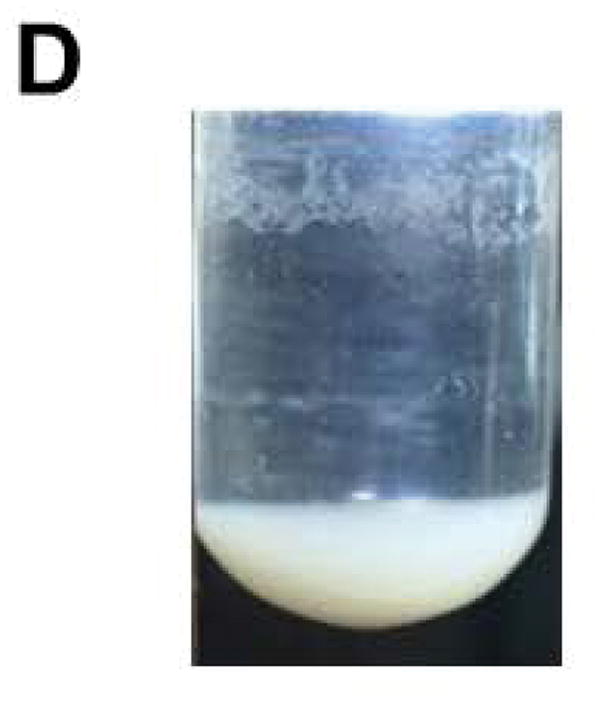
10-Fold scale up solid-phase purification of the phosphorothioate DNA sequence 12a. (A) RP-HPLC profile of unpurified 10a. (B) RP-HPLC profile of unpurified 10a after capture by the support 3. (C) RP-HPLC analysis of solid-phase purified 12a that has been released from the support 11a. (D) Photograph of precipitated 12a accounting for 995 OD260 of the DNA sequence. Inset: Purity analysis of the solid-phase purified 12a by PAGE. Abbreviation: BP, bromophenol blue dye.
Critical Parameters and Troubleshooting
When preparing the capture support 3, it is critically important to adequately execute step 6 of Basic Protocol 1 because any free amine functions, left unreacted on the solid-support 2 and/or 3, has the potential of forming relatively unstable imino-conjugates with 5′-functionalized DNA sequences 10a–f during the capture process. This can lead to premature hydrolysis of the imino-conjugates and ultimately result in poorer recovery of the solid-phase-purified DNA sequences 12a–f.
The 5′-functionalized deoxyribonucleoside phosphoramidites 9a–d (see Basic Protocol 3) must be prepared under strictly anhydrous conditions and must not be exposed to any acidic reagents or conditions, which would make these compounds prone to hydrolysis. With the purpose of preventing this unfavorable event, readers are referred to the Critical Parameters and Troubleshooting sections of UNITS 2.7 & 3.17, which address those critical issues associated with the preparation and use of nucleoside phosphoramidites. Precipitation of the 5′-functionalized deoxyribonucleoside phosphoramidites 9a–d from cold hexane after silica-gel purification is helpful in rendering those phosphoramidites free of H-phosphonate impurities and residual triethylamine; if not completely removed, triethylamine can neutralize 1H-tetrazole or other acidic activators and reduce the coupling efficiency of any phosphoramidites during automated solid-phase DNA synthesis. The presence of moisture can also compromise the coupling efficiency of phosphoramidites and should be taken into consideration; this concern has also been addressed in UNIT 2.7.
The capture support 3 is acidic. It is therefore crucial to neutralize this support by exposing it to a solution of triethylamine in acetonitrile (see step 1 of Basic Protocol 4) prior to capture of the 5′-functionalized DNA sequences 10a–f to avoid depurination of these sequences.
The purity of solid-phase-purified DNA sequences 12a–f is highly dependent on the efficiency of eluting off shorter than full-length DNA sequences from the silica-based support 11a–f. The affinity of shorter than full-length DNA sequences for silica-based supports increases with the size and polarity of these sequences. Steps 5 through 9 of Basic Protocol 4 have been optimized to wash off most of those shorter than full length DNA sequences. The same rationale applies when releasing the full-length DNA sequences (12a–f) from the supports 11a–f; steps 12 through 15 of Basic Protocol 4 have also been optimized to isolate the DNA sequences with the highest recovery yields and purity (ca. 98+%).
Anticipated Results
The two-step functionalization of commercial 3-aminopropylated silica gel with CDI-activated 7-oxooctanoic acid and O,O′-1,3-propanediylbishydroxylamine dihydrochloride should provide the capture support 3 with a surface density of ~ 150 μmol of aminooxy functions per gram of support. The reaction of N,N′-dimethylethylenediamine with 4,4-dimethyl-γ-butyrolactone affords the aminated amido alcohol 5b in a yield of 74%; amidation of 5b with CDI-activated 7-oxooctanoic acid gives the ketodiamidated alcohol 6b in a yield of 80%. Conjugation of the linker 6b to the 5′-hydroxyl of each deoxribonucleoside 7a–d, to provide 5′-functionalized deoxyribonucleosides (8a–d) with yields in the range of 50–70%. Phosphitylation of 8a–d under standard conditions leads to the corresponding phosphoramidite derivatives 9a–d in yields of 75–83%. The coupling efficiency of the 5′-functionalized phosphoramidites 9a–d, used in the last solid-phase synthesis coupling cycle of each DNA sequences listed in Figure 4, should be ~99%. Capture of the 5′-functionalized DNA sequences 10a–f by the solid support 3 is near complete, as shown in Figs. 12A–B. The high-throughput synthesis and 10-fold scale-up solid-phase purification process of the 5′-functionalized DNA sequence 10a is performed under conditions highly similar to those used for the 1-μmole scale process; release of the captured 10a from support 11a yields 995 OD260 of solid-phase purified DNA sequence 12a after precipitation in dry THF (Figs. 12C–D). This yield should be nearly proportional to that measured from the solid-phase-purified 12a (105 OD260) isolated from a 1 μmol-scale purification process.
Time Considerations
Preparation of the intermediate solid support 2 will take ~26 hr to complete. The preparation of capture solid support 3 from support 2 can be achieved within 17 hr. Preparation and purification of each of the amidated alcohols 5a and 5b is accomplished within 24 hr. Synthesis and purification of the ketodiamidated alcohols 6a and 6b are each achieved within 35 hr. Synthesis and purification of the ketodiamidated alcohol 6c needed for measuring the surface density of aminooxy functions conjugated to the capture support 3 will take 10 hr to complete. Measurement of the surface density of aminooxy functions per gram of support 3 is completed ~26 hr. Preparation and purification of each 5′-functionalized deoxyribonucleoside 8a–d is achieved within 16 hr. Phosphitylation of 8a–d to the corresponding phosphoramidite derivatives 9a–d and purification of the monomeric phosphoramidites including lyophilization can each be done within 25 hr. Solid phase synthesis and deprotection of each DNA sequence 10a–f are completed within 24 hr. Solid-phase capture of each DNA sequence including washing off shorter than full-length DNA sequences from the support 11a–f is done within 5 hr. Release of solid-phase purified DNA sequences 12a–f from the support 11a–f including precipitation, centrifugation and washes are each done with 6 hr. A 10-fold scale-up solid-phase purification of any DNA sequences can be achieved essentially within the time required for a small scale purification process. Many of the above tasks can be performed in a parallel manner.
Acknowledgments
This work was supported through FDA intramural funds.
Literature Cited
- Beaucage SL, Caruthers MH. Deoxynucleoside phosphoramidites -- A new class of key intermediates for deoxypolynucleotide synthesis. Tetrahedron Lett. 1981;22:1859–1862. [Google Scholar]
- Beaucage SL. Oligodeoxyribonucleotides synthesis -- Phosphoramidite Approach. In: Agrawal S, editor. Methods in Molecular Biology, Vol.20: Protocols for Oligonucleotides and Analogs. Humana Press, Inc; Totowa: 1993. pp. 33–61. [DOI] [PubMed] [Google Scholar]
- Beller C, Bannwarth W. Noncovalent attachment of nucleotides by fluorous-fluorous interactions: Application to a simple purification principle for synthetic DNA fragments. Helv Chim Acta. 2005;88:171–179. [Google Scholar]
- Cieślak J, Grajkowski A, Ausín C, Gapeev A, Beaucage SL. Permanent or reversible conjugation of 2′-O- or 5′-O-aminooxymethylated nucleosides with functional groups as a convenient and efficient approach to the modification of RNA and DNA sequences. Nucleic Acids Res. 2012;40:2312–2329. doi: 10.1093/nar/gkr896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieślak J, Ausín C, Grajkowski A, Beaucage SL. The 2-cyano-2,2-dimethylethanimine-N-oxymethyl group for the 2′-hydroxyl protection of ribonucleosides in the solid-phase synthesis of RNA sequences. Chem Eur J. 2013;19:4623–4632. doi: 10.1002/chem.201204235. [DOI] [PubMed] [Google Scholar]
- Cooke ST. Antisense strategies. Curr Mol Med. 2004;4:465–487. doi: 10.2174/1566524043360375. [DOI] [PubMed] [Google Scholar]
- Dandapani S. Recent applications of fluorous separation methods in organic and bioorganic chemistry. QSAR Comb Sci. 2006;25:681–688. [Google Scholar]
- Defrancq E, Lhomme J. Use of an aminooxy linker for the functionalization of oligodeoxyribonucleotides. Bioorg Med Chem Lett. 2001;11:931–933. doi: 10.1016/s0960-894x(01)00108-1. [DOI] [PubMed] [Google Scholar]
- Dorsett Y, Tuschl T. siRNAs: applications in fuctional genomics and potential as therapeutics. Nat Rev Drug Discovery. 2004;3:318–329. doi: 10.1038/nrd1345. [DOI] [PubMed] [Google Scholar]
- Eleuteri A, Capaldi DC, Krotz AH, Cole DL, Ravikumar VT. Pyridinium trifluoroacetate/N-methylimidazole as an efficient activator for oligonucleotide synthesis via the phosphoramidite method. Org Process Res Develop. 2000;4:182–189. [Google Scholar]
- Ellington A, Pollard JD., Jr . Introduction to the synthesis and purification of oligonucleotides. In: Beaucage SL, Bergstrom DE, Glick GD, Jones RA, editors. Current Protocols in Nucleic Acid Chemistry. I. John Wiley & Sons, Inc; Hoboken: 2000. pp. A.3C.1–A.3C.22. [DOI] [PubMed] [Google Scholar]
- Fang S, Bergstrom DE. Reversible biotinylation phosphoramidite for 5′-end-labeling, phosphorylation, and affinity purification of synthetic oligonucleotides. Bioconjug Chem. 2003a;14:80–85. doi: 10.1021/bc025626o. [DOI] [PubMed] [Google Scholar]
- Fang S, Bergstrom DE. Fluoride cleavable biotinylation phosphoramidite for 5′-end-labeling and affinity purification of synthetic oligonucleotides. Nucleic Acids Res. 2003b;31:708–715. doi: 10.1093/nar/gkg130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang S, Bergstrom DE. Reversible Biotinylation of the 5′-Terminus of Oligodeoxyribonucleotides and its Application in Affinity Purification. In: Beaucage SL, Bergstrom DE, Glick GD, Jones RA, editors. Current Protocols in Nucleic Acid Chemistry. I. John Wiley & Sons, Inc; Hoboken: 2003c. pp. 4.20.1–4.20.17. [DOI] [PubMed] [Google Scholar]
- Fang S, Bergstrom DE. Reversible 5′-end biotinylation and affinity purification of synthetic RNA. Tetrahedron Lett. 2004;45:7987–7990. [Google Scholar]
- Fang S, Fueangfung S. Scalable synthetic oligodeoxynucleotide purification with use of a catching by polymerization, washing, and releasing approach. Org Lett. 2010;12:3720–3723. doi: 10.1021/ol101316g. [DOI] [PubMed] [Google Scholar]
- Fang S, Fueangfung S, Lin X, Zhang X, Mai W, Bi L, Green SA. Synthetic oligodeoxynucleotide purification by polymerization of failure sequences. Chem Commun. 2011;47:1345–1347. doi: 10.1039/c0cc04374e. [DOI] [PubMed] [Google Scholar]
- Fina NJ, Edwards JO. The alpha effect. A review. Int J Chem Kinet. 1973;5:1–26. [Google Scholar]
- Forget D, Boturyn D, Defrancq E, Lhomme J, Dumy P. Highly efficient synthesis of peptide-oligonucleotide conjugates: Chemoselective oxime and thiazolidine formation. Chem Eur J. 2001a;7:3976–3984. doi: 10.1002/1521-3765(20010917)7:18<3976::aid-chem3976>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Forget D, Renaudet O, Defrancq E, Dumy P. Efficient preparation of carbohydrate-oligonucleotide conjugates (COCs) using oxime bond formation. Tetrahedron Lett. 2001b;42:7829–7832. [Google Scholar]
- Forget D, Renaudet O, Boturyn D, Defrancq E, Dumy P. 3′-Oligonucleotides conjugation via chemoselective oxime bond formation. Tetrahedron Lett. 2001c;42:9171–9174. [Google Scholar]
- Grajkowski A, Cieslak J, Beaucage SL. Solid-phase purification of synthetic DNA sequences. J Org Chem. 2016;8:6165–6175. doi: 10.1021/acs.joc.6b01020. [DOI] [PubMed] [Google Scholar]
- Iyer RP, Phillips LR, Egan W, Regan JB, Beaucage SL. The automated synthesis of sulfur-containing oligodeoxyribonucleotides using 3H-1,2-benzodithiol-3-one 1,1-dioxide as a sulfur-transfer reagent. J Org Chem. 1990;55:4693–4698. [Google Scholar]
- Katajisto J, Virta P, Lönnberg H. Solid-phase synthesis of multiantennary oligonucleotide glycoconjugates utilizing on-support oximation. Bioconjug Chem. 2004;15:890–896. doi: 10.1021/bc049955n. [DOI] [PubMed] [Google Scholar]
- Kawasaki AM, Casper MD, Prakash TP, Manalili S, Sasmor H, Manoharan M, Cook PD. Synthesis, hybridization, and nuclease resistance properties of 2′-O-aminooxyethyl (2′-O-AOE) modified oligonucleotides. Tetrahedron Lett. 1999;40:661–664. [Google Scholar]
- McBride LJ, McCollum C, Davidson S, Efcavitch JW, Andrus A, Lombardi SJ. A new, reliable cartridge for the rapid purification of synthetic DNA. BioTechniques. 1988;6:362–367. [PubMed] [Google Scholar]
- Mishra R, Mishra S, Misra K. Synthesis and application of fluorous-tagged oligonucleotides. Chem Lett. 2006;(10):1184–1185. [Google Scholar]
- Morvan F, Sanghvi YS, Perbost M, Vasseur JJ, Bellon L. Oligonucleotide mimics for antisense therapeutics: Solution phase and automated solid-support synthesis of MMI linked oligomers. J Am Chem Soc. 1996;118:255–256. [Google Scholar]
- Pearson WH, Berry DA, Stoy P, Jung K-Y, Sercel AD. Fluorous affinity purification of oligonucleotides. J Org Chem. 2005;70:7114–7122. doi: 10.1021/jo050795y. [DOI] [PubMed] [Google Scholar]
- Pokharel D, Yuan Y, Fueangfung S, Fang S. Synthetic oligodeoxynucleotide purification by capping failure sequences with a methacrylamide phosphoramidite followed by polymerization. RSC Adv. 2014;4:8746–8757. [Google Scholar]
- Salo H, Virta P, Hakala H, Prakash TP, Kawasaki AM, Manoharan M, Lönnberg H. Aminooxy functionalized oligonucleotides: Preparation, on-support derivatization, and postsynthetic attachment to polymer support. Bioconjug Chem. 1999;10:815–823. doi: 10.1021/bc990021m. [DOI] [PubMed] [Google Scholar]
- Sinha ND, Biernat J, McManus J, Köster H. Polymer support oligonucleotide synthesis XVIII: Use of β-cyanoethyl-N,N-dialkylamino-/N-morpholino phosphoramidite of deoxynucleosides for the synthesis of DNA fragments simplifying deprotection and isolation of the final product. Nucl Acids Res. 1984;12:4539–4557. doi: 10.1093/nar/12.11.4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sproat BS, Rupp T, Menhardt N, Keane D, Beijer B. Fast and simple purification of chemically modified hammerhead ribozymes using a lipophilic capture tag. Nucleic Acids Res. 1999;27:1950–1955. doi: 10.1093/nar/27.8.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiderski PM, Bertrand EL, Kaplan BE. Polystyrene reverse-phase ion-pair chromatography of chimeric ribosymes. Anal Biochem. 1994;216:83–88. doi: 10.1006/abio.1994.1011. [DOI] [PubMed] [Google Scholar]
- Trader DJ, Carlson EE. Siloxyl ether functionalized resins for chemoselective enrichment of carboxylic acids. Org Lett. 2011;13:5652–5655. doi: 10.1021/ol202376m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevisiol E, Renard A, Defrancq E, Lhomme J. The oxyamino-aldehyde coupling reaction: An efficient method for the derivatization of oligonucleotides. Tetrahedron Lett. 1997;38:8687–8690. [Google Scholar]
- Urdea MS, Horn T. Solid-supported synthesis, deprotection and enzymatic purification of oligodeoxyribonucleotides. Tetrahedron Lett. 1986;27:2933–2936. [Google Scholar]
- Wei X. Coupling activators for the oligonucleotide synthesis via phosphoramidite approach. Tetrahedron. 2013;69:3615–3637. [Google Scholar]
- Wilk A, Grajkowski A, Chmielewski MK, Phillips LR, Beaucage SL. Deoxyribonucleoside Phosphoramidites. In: Beaucage SL, Bergstrom DE, Glick GD, Jones RA, editors. Current Protocols in Nucleic Acid Chemistry. I. John Wiley & Sons, Inc; Hoboken: 2001. pp. 2.7.1–2.7.12. [DOI] [PubMed] [Google Scholar]