

Summary
The separation of germline from somatic lineages is fundamental to reproduction and species preservation. Here, we show that Drosophila Germ Cell-Less (GCL) is a critical component in this process by acting as a switch that turns off a somatic lineage pathway. GCL, a conserved BTB (Broad-complex, Tramtrack, and Bric-a-brac) protein, is a substrate-specific adaptor for Cullin3-RING ubiquitin ligase complex (CRL3GCL). We show that CRL3GCL promotes PGC fate by mediating degradation of Torso, a receptor tyrosine kinase (RTK) and major determinant of somatic cell fate. This mode of RTK degradation does not depend upon receptor activation but is prompted by release of GCL from the nuclear lamina during mitosis. The cell cycle-dependent change in GCL localization provides spatiotemporal specificity for RTK degradation and sequesters CRL3GCL to prevent it from participating in excessive activities. This precisely orchestrated mechanism of CRL3GCL function and regulation defines cell fate at the single cell level.
eTOC blurb
Primordial Germ Cells (PGCs) ensure continuity of life through generations. Combining genetic and biochemical analysis, Pae et al. show that GCL and CUL3 promote PGC formation by targeting the Torso RTK for ubiquitylation and degradation. Cell-cycle dependent regulation of GCL subcellular localization confers spatiotemporal control of the Torso pathway.
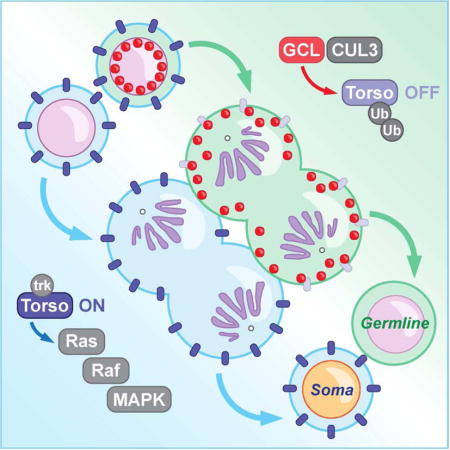
Introduction
Germ cells are specialized cells capable of producing an entirely new organism. Hence, the establishment of the germline precursors, primordial germ cells (PGCs), separately from all somatic cells is one of the first key decisions made in early embryonic development (Cinalli et al., 2008, Nakamura and Seydoux, 2008, Seydoux and Braun, 2006). This germline-soma dichotomy is readily tractable in Drosophila melanogaster. In Drosophila, PGCs are the first cells to form in the embryo, and their formation requires maternally provided “germ plasm,” a specialized protein- and mRNA-rich cytoplasm located at the posterior pole (Gao and Arkov, 2013). Among many germ plasm components, GCL has emerged as a key regulator of PGC formation (Cinalli and Lehmann, 2013, Jongens et al., 1992). Embryos that lack maternally inherited gcl products (gclΔ/Δ embryos, hereafter) completely lack or form a significantly reduced number of PGCs. GCL acts as a rate-limiting factor that controls a spindle-independent cleavage event, which physically separates the future germ cell lineage from the rest of the embryo that will develop into the soma (Cinalli and Lehmann, 2013). Despite its critical function, little is known about the molecular mechanism by which GCL promotes PGC formation and prevents acquisition of somatic fate adopted by neighboring cells.
The evolutionarily conserved BTB domain in GCL provided a first mechanistic insight (Fig. 1A). To date, BTB domain proteins have been implicated in two major biological activities: (1) transcriptional regulation and (2) protein ubiquitylation as subunits of CRL3s, a major class of E3 ubiquitin ligases that are required for fundamental cellular and developmental processes, such as cell cycle progression, cell death, and transcription (Genschik et al., 2013, Pintard et al., 2004). CRL3s are composed of the central scaffold protein Cullin3 (CUL3), a BTB-domain substrate-specific adaptor protein, and the catalytic RBX1 RING-domain protein. While it was previously suggested that GCL could affect transcriptional onset of a subset of somatic genes (Leatherman et al., 2002), other experiments indicated that the major function of GCL is likely independent of transcriptional regulation (Cinalli, 2012, Cinalli and Lehmann, 2013). In support of GCL acting as a substrate-specific adaptor of CRL3, GCL was predicted to contain a specialized BTB fold that can accommodate the interaction with CUL3 (Zhuang et al., 2009).
Figure 1. Assembly of CRL3GCL is required for proper PGC formation in Drosophila.
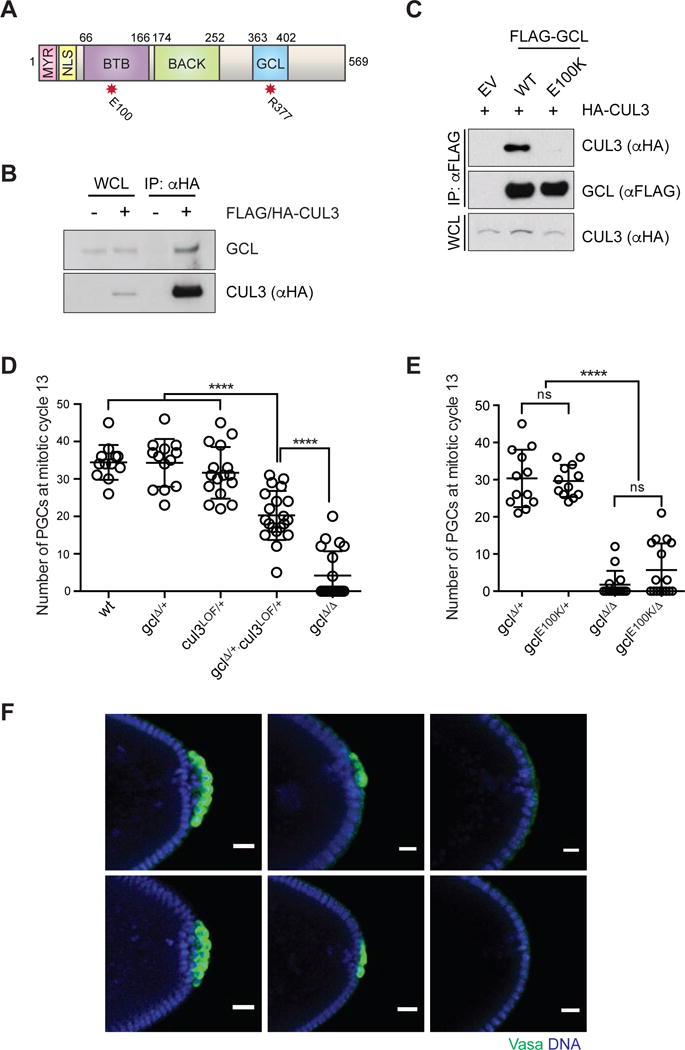
(A) Domain architecture of GCL protein. MYR: myristoylation signal, NLS: nuclear localization signal, BTB: Broad-Complex, Tramtrack and Bric a brac domain, BACK: BTB and C-terminal Kelch domain) and the conserved GCL domain. The red asterisks highlight the position of functional mutants used in this study.
(B) Ovary lysates were prepared from females expressing FLAG-HA-tagged CUL3 (driven by UAS promoter using the germline-specific driver nanos-Gal4) and immunoprecipiated with anti-HA resin. Immuno-complexes were probed with an antibody recognizing endogenous GCL. IP, immunoprecipitation; WCL, whole cell lysate.
(C) Drosophila Schneider 2 (S2) cells were co-transfected with the indicated constructs or empty vector (EV), as indicated. Twenty-four hours after transfection, cells were harvested and lysed. cell lysates were immunoprecipitated (IP) with anti-FLAG resin, and immunocomplexes were probed with indicated antibodies.
(D, E) Number of PGCs in embryos of indicated maternal genotype. Each circle represents the number of PGCs counted in a single embryo. Bars represent the mean ± standard deviation. (n > 12, ****P < 0.0001, ns = not significant, Mann-Whitney test)
(F) Immunostaining of embryos from females of indicated genotype for expression of PGC marker Vasa (green). DAPI for DNA (blue). Posterior poles of representative embryos are shown. Scale bar = 20μm.
See also Figure S1.
In this study, we demonstrate that GCL indeed functions as a CRL3 substrate adaptor to promote proper PGC development. We identify the Receptor Tyrosine Kinase Torso as a novel interactor and substrate of CRL3GCL. Torso was originally identified in a genetic screen for maternal factors required for Drosophila embryo patterning (Klingler et al., 1988) and was later shown to specify somatic cell fates at the anterior and posterior ends of the embryo through activation of the Ras/Raf/MAPK signaling pathway (Duffy and Perrimon, 1994, Li, 2005). Torso and its ligand, Trunk, are expressed ubiquitously, but local presence of the ligand modifier, Torso-like (Tsl), restricts Torso activity to anterior and posterior poles of the early embryo (Casanova and Struhl, 1989, Savant-Bhonsale and Montell, 1993). It was previously shown that overexpression of Torso reduces the number of PGCs, similar to gclΔ/Δ mutants (Martinho et al., 2004). Torso and its downstream signaling pathway are excluded from PGCs (Gabay et al., 1997, Martinho et al.,, 2004), highlighting the importance of controlling the somatic signaling pathway that opposes germline development.
We also investigate the critical regulatory elements that are required for GCL to function properly. GCL lacks any of the substrate recognition domains, such as Kelch and MATH domains, identified in other CRL adaptors. Instead of these canonical substrate recognition domains, GCL contains a unique, previously uncharacterized peptide sequence (the GCL domain) that is highly conserved among all homologs (Fig. S3A). Our studies provide evidence that the GCL domain and cell cycle-dependent localization cues are responsible for substrate recognition and the spatiotemporal regulation of GCL, respectively. These features define the GCL proteins throughout evolution. Moreover, mutations in the mammalian homolog of GCL, Gmcl1, are associated with male fertility disorders such as azoospermia in mice and humans (Kimura et al., 2003, Kleiman et al., 2003), suggesting that the regulatory mechanism of GCL at the crossroad between cell lineages may be broadly applicable across species.
Results
CRL3GCL is required for proper germ cell lineage development in Drosophila
To determine whether GCL acts as a CRL3 substrate adaptor, we tested for physical and genetic interactions between GCL and CUL3, the only Cullin3 homolog in Drosophila. We found that transgenically expressed CUL3 co-immunoprecipitated with endogenous GCL in oocyte lysate extracts (Fig. 1B). We confirmed this interaction using co-expressed tagged CUL3 and GCL in Drosophila S2 cells (Fig. 1C). Structural studies have previously shown that substrate-specific adaptors interact with CUL3 through the conserved ϕ-x-E motif within the BTB domain (Fig. S1A) (Canning et al., 2013). As expected from the structural prediction, substitution of the glutamic acid within the ϕ-x-E motif of GCL with lysine (E100K) disrupted the interaction between GCL and CUL3 (Fig. 1C), without affecting the ability of GCL to homodimerize through its BTB domain (Fig. S1B, C). These data suggest GCL forms protein complexes with CUL3.
CUL3 is essential for Drosophila oogenesis (Hudson and Cooley, 2010), precluding us from directly addressing its role in PGC formation. Therefore, we tested for dosage-dependent genetic interactions between GCL and CUL3. Introduction of a single cul3 loss-of-function (LOF) allele into a gcl heterozygous background (gclΔ/+, cul3LOF/+) led to a significant reduction in PGCs compared to each heterozygous control (Fig. 1D), suggesting an essential role for CUL3 in GCL-dependent PGC formation. In addition, a mutagenesis-induced gclE100K allele (Fig. S2D) failed to complement a gcl deletion allele (gclΔ) (Fig. 1E, F), despite normal levels and localization pattern of gclE100K mRNA transcripts (Fig. S1E, F). These results demonstrate that GCL interacts with CUL3 through the BTB domain to assemble CRL3GCL and that CRL3GCL promotes PGC formation in Drosophila embryos.
CRL3GCL targets the RTK Torso for ubiquitylation and degradation
In a CUL3-based ubiquitin ligase complex, the BTB-domain protein GCL is expected to confer substrate specificity. To identify targets of CRL3GCL, we used FLAG-HA-tagged transgenic constructs to purify GCL-interacting protein complexes from Drosophila embryo lysates and analyzed them by mass spectrometry (Fig. S2A–C, Table S1). We employed two mutant forms of GCL as controls (Fig. S2A): (1) CUL3-binding mutant GCLE100K is expected to act as a “substrate trap,” increasing the chance of co-purification with the substrates and (2) a C-terminal truncation that is expected to lack substrate-binding would serve as a negative control. This unbiased proteomics analysis of the GCL complexes identified peptides derived from CUL3, confirming that GCL and CUL3 form protein complexes (Fig. S2D). Additionally, multiple unique peptides corresponding to the RTK Torso and the Torso interacting protein Closca were specifically found in GCLWT and GCLE100K immunoprecipiates (Fig. S2D).
Given previous genetic studies suggesting a role for Torso in antagonizing PGC formation, we focused on the interaction between Torso and GCL. We confirmed the specificity of the immunopurification result by transiently expressing GCL and Torso, each C-terminally tagged with FLAG or HA, respectively, in HEK293T cells. We found that GCL co-immunoprecipitated with Torso in this heterologous setting (Fig. 2A), where Torso and its ligand are not conserved, suggesting a direct interaction between GCL and Torso. This interaction was more prominent with co-expression of dominant-negative CUL3 (DN CUL3), a truncated form that is defective in binding RBX1 (Fig. 2A), suggesting that Torso could be a CRL3 substrate. Moreover, overexpression of wild-type GCL, but not the CUL3-binding mutant GCLE100K, led to a substantial decrease in Torso that was blocked with a chemical inhibition of Cullin-RING ligase activity using MLN4924 (Fig. 2B). These results indicate that GCL mediates the downregulation of Torso in a CUL3-dependent manner. We then examined whether GCL promotes the ubiquitylation of Torso in cells co-transfected with HA-tagged Torso and MYC-tagged ubiquitin. We found that GCL induced a ladder of several high molecular-weight bands that correspond to ubiquitylated forms of Torso detected by an anti-MYC antibody in Torso immunoprecipitates that were purified under denaturing conditions (Fig. 2C). Importantly, we did not detect these slow migrating bands when the mutant adaptor, GCLE100K, was expressed or MLN4924 was added (Fig. 2C). Altogether, these results demonstrate that CRL3GCL targets Torso for poly-ubiquitylation and degradation.
Figure 2. The Torso RTK is degraded in a CRL3GCL-dependent manner.
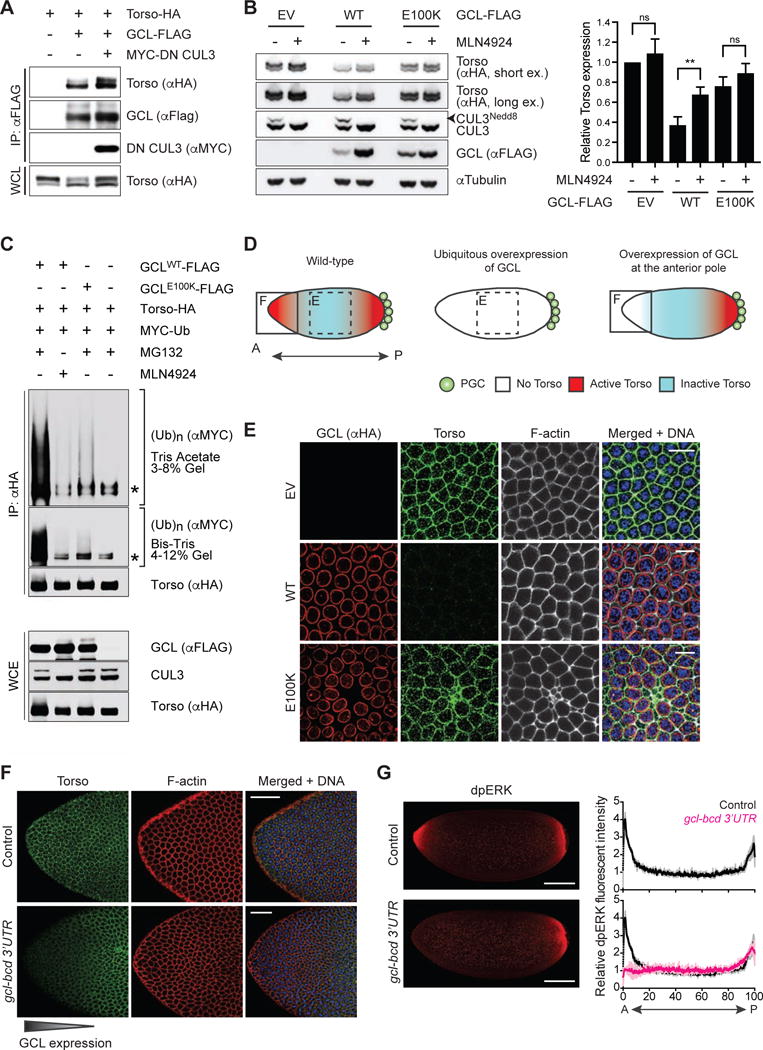
(A) HEK293T cells were transfected with the indicated constructs. Twenty-four hours after transfection, cells were harvested and lysed. Cell lysates were immunoprecipitated (IP) with anti-FLAG resin, and immunocomplexes were probed with indicated antibodies. Dominant negative CUL3 (DN CUL3) is a truncated form of CUL3 that is defective in binding RBX1, thereby stabilizing the GCL-Torso interaction. See also Figure S2.
(B) (Left) HEK293T cells transiently expressing HA-tagged Torso were transfected with empty vector (EV), FLAG-tagged GCLWT, or FLAG-tagged GCLE100K for 24 hours. Where indicated, MLN4924 (a Cullin-RING ligase inhibitor) was added for 4 hours prior to collection, reflected by the absence of neddylated CUL3 (CUL3Nedd8). Total cell lysates were immunoblotted as indicated. (Right) Densitometric scanning quantification of Torso expression levels relative to the corresponding αTubulin (loading control) levels. The averaged relative Torso expression levels from triplicate experiments are plotted with respect to the control (EV without MLN4924 treatment). Error bars represent standard deviation.
(C) HEK293T cells were transfected with MYC-tagged wild-type Ubiquitin (Ub), HA-tagged Torso, and either FLAG-tagged GCLWT or FLAG-tagged GCLE100K. Twenty-four hours post-transfection, cells were treated with either MG132 (a proteasome inhibitor) or MLN4924 (a CRL inhibitor) for 3 hours before collection for immunoprecipitation (IP) under denaturing condition (to dissociate any protein interacting with Torso that may also be ubiquitylated) and immunoblotted as indicated. The bracket on the right indicates a ladder of bands with a relative molecular mass of >150 kDa corresponding to poly-ubiquitylated Torso. WCE, whole-cell extract. The asterisk indicates a non-specific band.
(D) A graphical illustration of Torso protein expression (blue) and catalytic activity (red) in an embryo relative to the site of PGC formation (posterior pole, P). In wild-type embryos, ubiquitously-expressed Torso is activated only at the anterior and posterior termini. Dashed box highlights the central region of a syncytial blastoderm embryo shown in panel E. Solid box highlights the anterior pole (A) shown in panel F.
(E) Embryos ubiquitously expressing either empty vector (EV) or FLAG-HA-tagged GCL (WT or E100K). GCL transgenes were generated with UAS promoter and the k10 3′UTR regulatory sequence and were expressed using the germline-specific driver nanos-GAL4∷VP16. Fixed embryos at interphase of nuclear cycle 12–13 were immunostained with anti-HA (red) and anti-Torso (green). DNA (blue), F-actin (gray). Scale bar = 10μm.
(F) Embryos ectopically expressing GCL at the anterior pole (gcl-bcd 3′UTR) were fixed and stained with an antibody recognizing Torso (green). DAPI (blue) counterstains DNA, and phalloidin (red) labels F-actin. Embryos from wild-type (w−1118) females are used as controls. Scale bar = 50μm.
(G) (Left) Embryos ectopically expressing GCL at the anterior pole (gcl-bcd 3′UTR) were fixed and stained with an antibody recognizing dpERK as a readout of Torso catalytic activity (red). Embryos from wild-type (w−1118) females are used as controls (top). Scale bar = 100μm. (Right) dpERK intensity across the embryo (0 = Anterior, A; 100 = Posterior, P) relative to the center of the embryo. Black line represents averaged relative dpERK intensity of 3 wild-type embryos during nuclear cycle 12–13, magenta line represents averaged relative dpERK intensity of 3 gcl-bcd 3′UTR embryos at the same stage. Grey and pink lines represent standard deviation for wild-type and gcl-bcd 3′UTR embryos, respectively.
To investigate whether CRL3GCL regulates Torso protein levels in vivo, we ectopically expressed GCL across the entire embryo and probed for changes in Torso expression. In wild-type embryos, Torso expression is undetectable in PGCs and ubiquitously expressed at the membrane of all somatic blastoderm cells (Casanova and Struhl, 1989). In contrast, ubiquitous GCL overexpression markedly reduced Torso protein levels throughout the entire embryo, as detected by immunofluorescence and immunoblotting (Fig. 2D, E, 3A, B). These effects were sensitive to GCL dosage, as an ectopic GCL gradient created by using an anterior-specific RNA localization signal (Jongens et al., 1994) caused a concomitant decrease in the Torso expression level (Fig. 2D, F). Ectopic GCL expression also inhibited Torso signaling, as shown by a reduction in doubly phosphorylated ERK (dpERK), which is a readout of Torso activity and is required for activation of the somatic program (Fig. 2D, G) (Gabay et al.,, 1997). To determine whether this effect on Torso protein involved the CUL3-dependent function of GCL, GCLE100K was expressed transgenically. Overexpression of GCLE100K caused no detectable change in Torso protein expression pattern or levels (Fig. 2E, 3A, B). These results show that CRL3GCL can promote a reduction in Torso protein levels and activity in vivo. Notably, both active Torso, at the anterior and posterior poles, and inactive Torso protein, present everywhere else in the embryo, can be depleted in a CRL3GCL-dependent manner.
Figure 3. The conserved GCL domain is essential for substrate recognition.
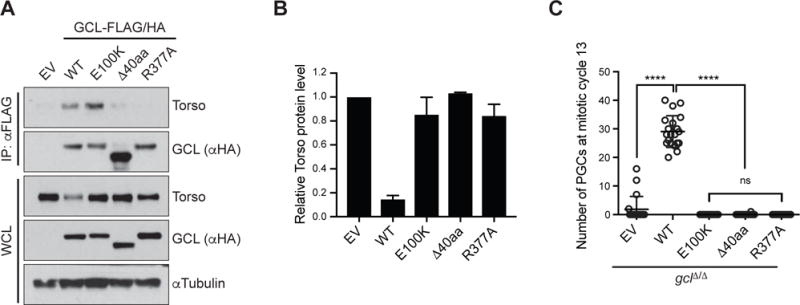
(A) Lysates prepared from 0–2 hours AEL (after egg laying) embryos expressing either empty vector (EV) or a FLAG-HA-tagged GCL variant (WT, E100K, Δ40aa, or R377A) were immunoprecipitated (IP) with anti-FLAG resin. Immunocomplexes were probed with antibodies specific to the indicated endogenous proteins. GCL transgenes, generated with UAS promoter and the k10 3′UTR regulatory sequence, were expressed using the germline-specific driver nanos-GAL4∷VP16.
(B) Densitometric scanning quantification of the Torso expression levels in the embryo lysates relative to the corresponding αTubulin (loading control) levels. The averaged relative Torso expression levels from triplicate experiments are plotted with respect to the EV control. Error bars represent standard deviation.
(C) EV or a FLAG-HA-tagged GCL variant (WT, E100K, Δ40aa, R377A) was expressed in gclΔ/Δ embryos using the germline-specific driver nanos-GAL4∷VP16. Number of PGCs in each embryo was counted and plotted. Bars represent the mean ± standard deviation. (n = 20 for each genotype, ****P < 0.0001, ns = not significant, Mann-Whitney test).
See also Figure S3.
Essential role of conserved GCL domain in substrate recognition
We confirmed the specific interaction between GCL and endogenous Torso by immunoprecipitation and immunoblot analysis using embryo lysates (Fig. 3A, B). GCLE100K, which is unable to interact with CUL3, showed a more robust interaction with Torso (Fig. 3A), suggesting that GCLE100K-bound Torso is unable to be targeted for degradation. Upon examining the role of the conserved GCL domain (Fig. S3A), we found that both a deletion of the entire GCL domain (Δ40aa), as well as an alanine substitution of a highly conserved arginine within this domain (R377A), prevented GCL from interacting with endogenous Torso (Fig. 3A, S3B). Consistent with this finding, neither nor GCLR377A transgene expression was able to downregulate Torso expression in vivo or rescue the PGC formation defect in gclΔ/Δ embryos (Fig. 3A–C). These results show that the biological function of GCL requires interactions with both CUL3 and Torso and that the conserved GCL domain is essential for substrate recognition by CRL3GCL.
CRL3GCL restricts the Torso signaling pathway at the posterior pole
If Torso degradation were the major function of CRL3GCL, we would expect that PGC formation would be sensitive to Torso dosage. Supporting this hypothesis, we found a significant increase in the number of PGCs in embryos carrying LOF mutations in the Torso ligand-modifier, Tsl (tsl−/−), which is required for Torso receptor activation (Johnson et al., 2015, Stevens et al., 1990) (Fig. 4A). To directly test the hypothesis that persistent Torso signaling in the absence of GCL is detrimental to PGC formation, we investigated whether mutations in the torso pathway could suppress the gcl mutant phenotype. We reduced torso mRNA expression through in vivo RNAi in gclΔ/Δ embryos and found that this manipulation abrogated the characteristic defect in PGC formation (Fig. 4B). Similarly, introducing tsl mutations completely restored PGC formation and division in gclΔ/Δ embryos (Fig. 4C, D, S4A). These results indicate that it is Torso activity, and not presence of the Torso protein per se, that inhibits PGC formation. The rescued PGCs in gclΔ/Δ, tsl−/− embryos showed the hallmarks of bona fide PGCs: Vasa expression and global transcriptional repression as indicated by the absence of RNAPII pSer2 (Fig. 4E). In gclΔ/Δ, tsl−/− embryos, we observed accumulation of Torso protein at the plasma membrane of Vasa expressing PGCs (Fig. 4F), as well as at the interface between PGCs and the adjacent somatic tissue (Fig. S4B, C). This pattern of expression was not observed in gcl heterozygous control embryos (Fig. 4F, S4B, C), indicating that in the wild-type setting, CRL3GCL actively promotes degradation of Torso at the posterior pole to prevent Torso expression and subsequent activation of the somatic program within PGCs. Consistent with GCL acting upstream of Torso, the developmental defects typically associated with mutations in the Torso pathway were not rescued in gclΔ/Δ, tsl−/− embryos (Fig. S4D, E). Therefore, our results demonstrate that CRL3GCL specifically restricts Torso expression and activity at the posterior pole in order to allow PGCs to form properly.
Figure 4. GCL controls PGC formation by inhibiting Torso activity at the posterior pole.
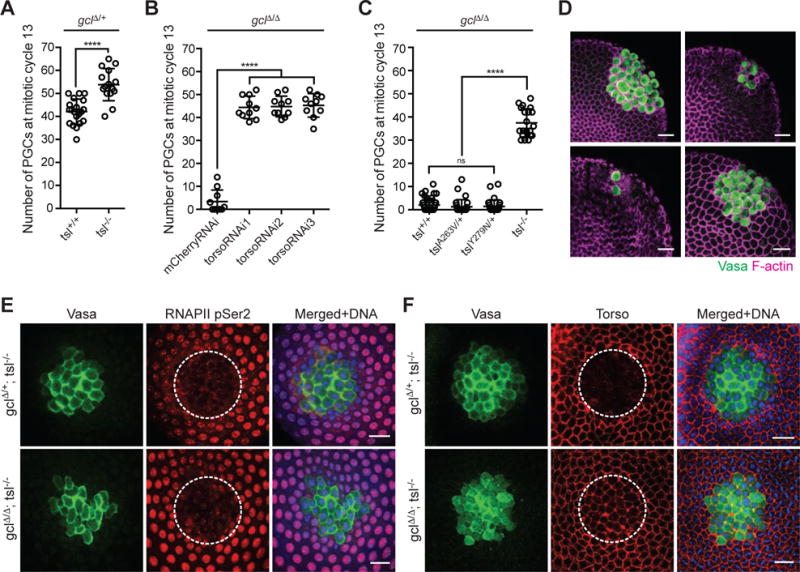
(A) Torso signaling was manipulated in gclΔ/+ control embryos by introducing tsl loss-of-function mutations. Number of PGCs in embryos from mothers of indicated genotype was counted and plotted. Bars represent the mean ± standard deviation. tsl−/− corresponds to tslA263V/Y279N. (n > 15, ****P < 0.0001, Mann-Whitney test)
(B) Torso expression was manipulated by combining gclΔ/Δ mutation with transgenic-based torso knockdown (RNAi) with three individual shRNA lines. The germline-specific driver maternal tubulinGAL4∷VP16 was used to express shRNA. Non-targeting RNAi against mCherry in gclΔ/Δ embryos was used as a control. Number of PGCs in embryos from mothers of indicated genotype was counted and plotted. Bars represent the mean ± standard deviation. (n = 10, ****P < 0.0001, Mann-Whitney test)
(C) Torso signaling was manipulated in gclΔ/Δ embryos by introducing tsl loss-of-function mutations. Number of PGCs in embryos from mothers of indicated genotype was counted and plotted. Bars represent the mean ± standard deviation. Tsl−/− corresponds to tslA263V/Y279N. (n > 20, ****P < 0.0001, Mann-Whitney test)
(D) Representative embryos of indicated maternal genotype. Anti-Vasa (green) immunostaining marks PGCs in the embryos, and F-actin labeled by phalloidin (magenta) outlines both germ and somatic cells. Scale bar = 20μm.
(E) Fixed embryos from mothers of indicated genotype were immunostained with anti-Vasa (green) and anti-RNA PolymeraseII phosphoSer2 (RNAPII pSer2) (red). DNA (blue). To image a uniform layer of PGCs, posterior portion of the embryos was cut, turned, and mounted. Dotted line outlines Vasa-expressing PGCs. Scale bar = 20μm.
(F) Fixed embryos from mothers of indicated genotype were immunostained with anti-Vasa (green) and anti-Torso (red). DNA (blue). Dotted line outlines Vasa-expressing PGCs. In gclΔ/Δ, tsl−/− embryo, but not gclΔ/+, tsl−/−, these cells express Torso at the cell membrane. Scale bar = 20μm.
See also Figures S4 and S5.
Torso activation leads to transcriptional upregulations of tll and hkb through transduction of the Ras/Raf/MEK/MAPK signaling pathway (Duffy and Perrimon, 1994) (Fig. S5A). To determine the role of this canonical pathway in preventing PGC formation, we employed in vivo RNAi against dsor1 (MEK) and rolled (MAPK) mRNAs. We found that reducing dsor1 or rolled mRNAs could not rescue the PGC formation defect in gclΔ/Δ embryos (Fig. S5B), despite the efficient knockdowns as shown by reduction in the dpERK signal (Fig. S5C). These findings suggest the existence of an alternative pathway downstream of the Torso RTK with non-transcriptional outputs that is responsible for counteracting PGC formation. This is consistent with previous findings that the PGC formation defect seen in gclΔ/Δ embryos cannot be rescued by global transcriptional inhibition (Cinalli and Lehmann, 2013) and that PGCs form in the absence of transcription (Lehner, 1992).
Inhibition of the Torso signaling pathway is critical for germ cell lineage development
To identify the sequences in Torso required for CRL3GCL-dependent degradation, we examined the cytosolic portion of Torso by generating a series of truncation and deletion mutants and performing co-immunoprecipitation and immunoblotting analysis (Fig. S6). This analysis revealed a degron motif located between the two split kinase domains that is essential for the interaction between Torso and GCL (Fig. S6). By substituting the critical amino acids within this motif with alanines, we generated a Torso variant that cannot interact with GCL (TorsoDeg) (Fig. S6F). Unlike TorsoWT, TorsoDeg did not coimmunoprecipitate with GCL even with the overexpression of DN CUL3 that would stabilize the substrate-adaptor interaction by promoting the accumulation of the substrates (Fig. 5A).
Figure 5. Deregulated Torso activity at the posterior pole inhibits PGC formation.
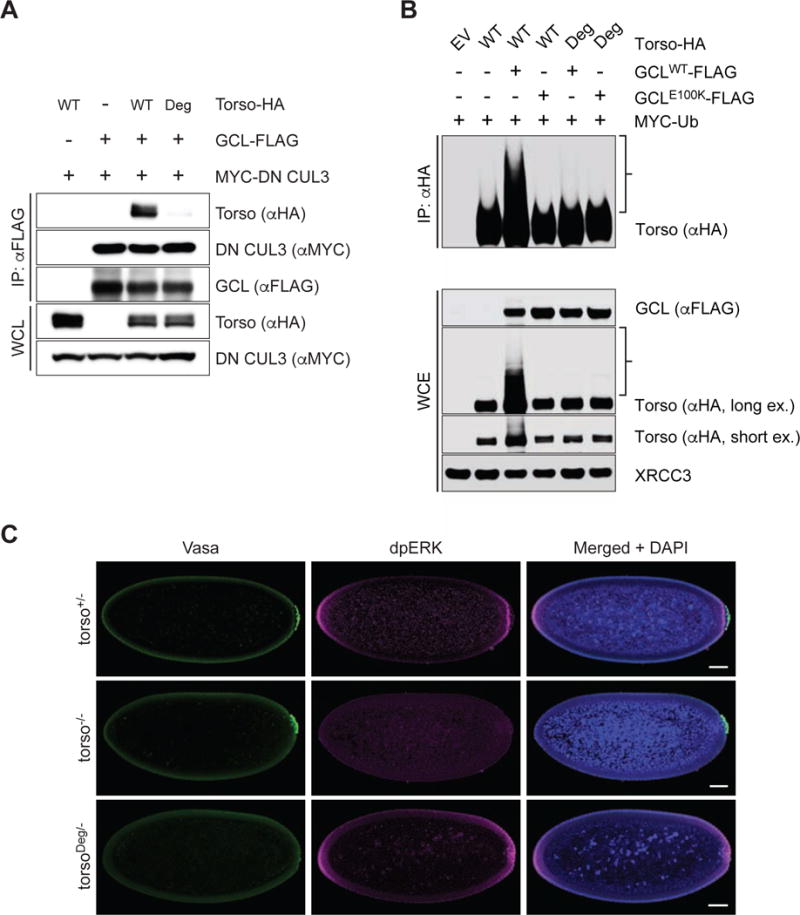
(A) HEK293T cells were transfected with FLAG-tagged GCLWT, HA-tagged Torso (either WT or Deg mutant), and MYC-tagged dominant negative CUL3 (DN CUL3), as indicated. Cell lysates were immunoprecipitated (IP) with anti-FLAG resin, and immunocomplexes were probed with the indicated antibodies. For TorsoDeg amino acids IENIG were mutated to alanines.
(B) HEK293T cells were transfected with MYC-tagged wild-type Ubiquitin (Ub), either HA-tagged Torso or HA-tagged TorsoDeg, and either FLAG-tagged GCLWT or FLAG-tagged GCLE100K. Twenty-four hours post-transfection, cells were treated with MG132 for 3 hours before collection for immunoprecipitation (IP) under denaturing condition and immunoblotted as indicated. The bracket on the right indicates a ladder of bands with a relative molecular mass of >150 kDa which, presumably, are poly-ubiquitylated species of Torso. WCE, whole-cell extract.
(C) Embryos from torso+/− (torso+/HH), torso−/− (torsoHH/WK), or torsoDeg/− (torsoDeg/HH) females were immunostained with antibodies recognizing Vasa (green) to assess PGC formation and dpERK (magenta) to assess Torso signaling activity. Presence of the rounded nuclei (Blue) around the periphery of the embryos was used to select and assess embryos of nuclear cycle 12–13. Arrowhead points at the absence of PGC at posterior pole. Scale bar = 50 μm.
See also Figure S6.
Consistent with the loss of interaction with GCL, this TorsoDeg mutant is insensitive to CRL3GCL-mediated ubiquitylation (Fig. 5B). To address the biological implication of the degron motif, we generated an endogenous torso mutant that carries alanines in lieu of the degron motif using CRISPR/Cas9 (torsoDeg). Embryos carrying one copy of maternally inherited torsoDeg exhibited a dominant PGC formation defect (Fig. 5C). Importantly, this degron mutation did not interfere with Torso RTK function. Unlike a missense mutation within the kinase domain found in previously reported loss-of-function alleles of torso (torsoM522K and torsoG556R) (Schupbach and Wieschaus, 1986), torsoDeg did not affect the catalytic activity as indicated by the appropriate dpERK expression and normal embryonic patterning (Fig. 5C and data not shown). Together, these results demonstrate that CRL3GCL functions as a dedicated E3 ubiquitin ligase for Torso for PGC formation and that inhibition of Torso signaling exclusively at the posterior pole is required for the proper germline development.
GCL regulates Torso during mitosis at the plasma membrane
Physical and genetic interactions suggest that the CRL3GCL and its substrate, the Torso RTK, would co-localize in the same subcellular compartment. However, the prominent locations of Torso and GCL are the plasma membrane and the nucleoplasmic side of the nuclear envelope (nuclear envelope, hereafter), respectively (Casanova and Struhl, 1989, Jongens et al.,, 1994, Robertson et al., 1999). Because PGC formation is coupled to mitotic divisions (Cinalli and Lehmann, 2013), we reasoned that CRL3GCL could reach the plasma membrane transiently and target Torso at the posterior pole during mitosis after nuclear envelope breakdown. Indeed, an early study noted GCL protein at the plasma membrane during the initial stage of “pole bud formation” (Jongens et al.,, 1992). To determine the localization of CRL3GCL during the cell cycle, we analyzed the subcellular distribution of HA-tagged GCL transgenes in relation to Torso protein. We observed the GCLWT transgene at the nuclear membrane during interphase in blastoderm embryos (Fig. 6A). However, following nuclear envelope breakdown during mitosis, we found that GCL localized closer to submembranous F-actin (Fig. 6A). To simultaneously observe GCL and Torso, we examined expression of the transgene, which is unable to target Torso for degradation (see above, Fig. 3), and found that this mutated form of GCL co-localized with Torso at the plasma membrane during mitosis. In interphase, however, showed distinct nuclear localization while Torso was continuously expressed at the plasma membrane throughout all stages of the cell cycle (Fig. 6B). Therefore, the GCL-Torso interaction takes place at the plasma membrane and is regulated by the cell cycle-dependent subcellular changes in GCL localization.
Figure 6. GCL localizes at the cortical membrane after nuclear envelope breakdown and co-localizes with Torso during mitosis.
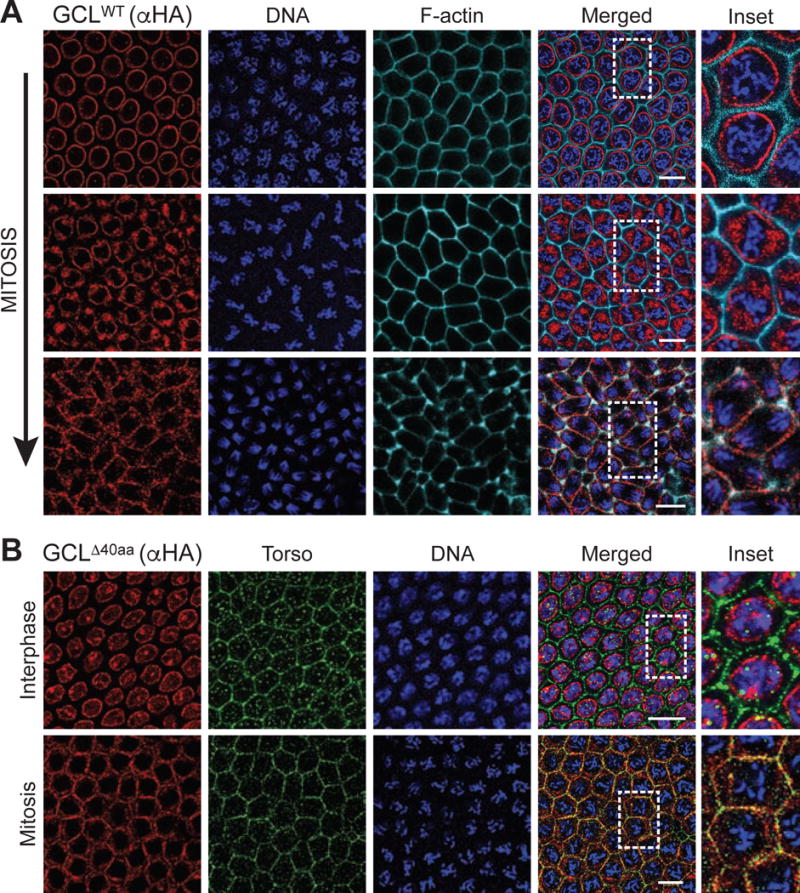
(A) Central region of embryos of nuclear cycle 12–13 expressing FLAG-HA-tagged GCLWT. Fixed embryos were immunostained with anti-HA (red). DNA (blue) was used to determine the cell cycle stage of each embryo, and phalloidin (cyan) labels F-actin and outlines cell membrane. Dashed rectangle highlights the area shown in inset. Scale bar = 10μm.
(B) Central region of embryos of nuclear cycle 12–13 expressing FLAG-HA-tagged during interphase or mitosis. DNA (blue) was used to determine the cell cycle stage of each embryo. Fixed embryos were immunostained with anti-HA (red) and Torso (green) to assess co-localization. Dashed rectangle highlights the area shown in inset. Scale bar = 10μm.
Nuclear membrane localization sequesters GCL
To examine whether membrane localization of GCL is required for Torso-degradation, we generated a GCL transgene with a mutation in the N-terminal glycine required for myristoylation-mediated membrane targeting (GCLG2A). As expected from the role of myristoylation, the HA-tagged GCLG2A variant was nucleoplasmic during interphase and cytoplasmic during mitosis (Fig. 7A). This mislocalization of GCL led to a dramatically reduced interaction with Torso in comparison to the wild type despite similar levels of the bait. As a result, Torso expression remained unaffected in vivo (Fig. 7A, B). Furthermore, GCLG2A did not support PGC formation in a gcl mutant background (Fig. 7F). Together, these results directly support our hypothesis that membrane enrichment of GCL enables the efficient control of Torso protein, an essential step in PGC formation.
Figure 7. Disruption of the nuclear localization signal in GCL results in gain-of-function defects in oogenesis.
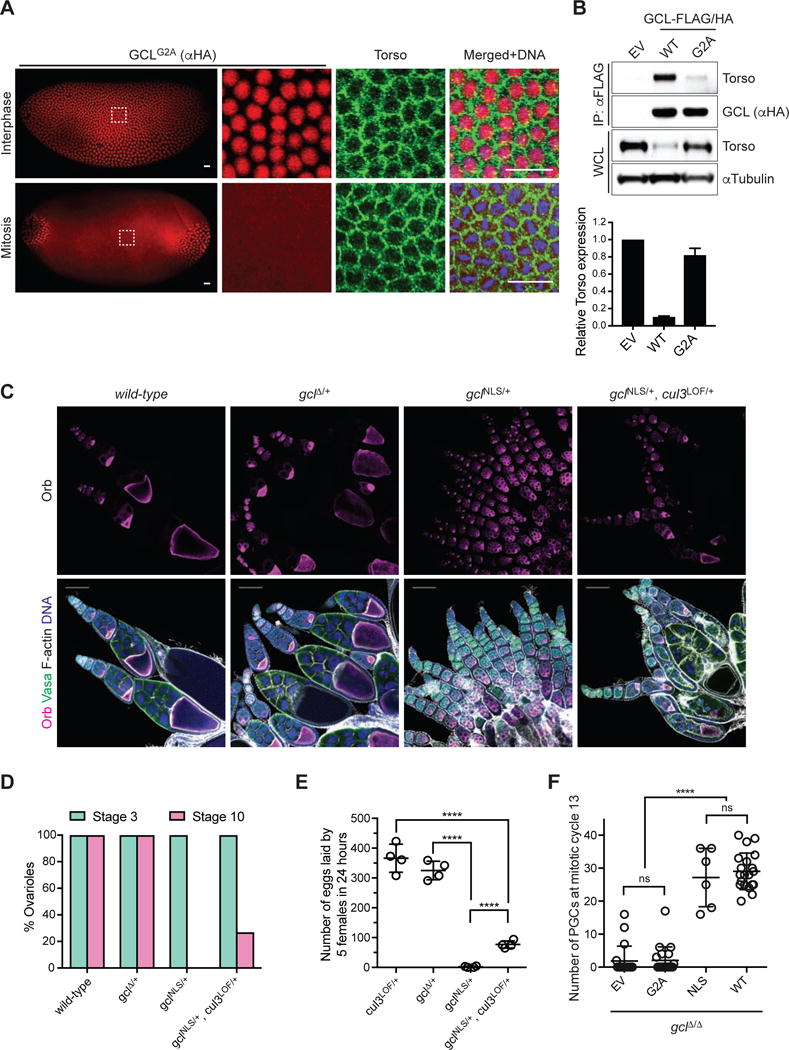
(A) Immunofluorescence analysis in embryos expressing FLAG-HA-tagged GCLG2A with antibodies recognizing HA (red) and Torso (green). DNA (blue) was used to determine the cell cycle stage of each embryo. Scale bar = 20μm. GCL transgenes, generated with UAS promoter and the k10 3′UTR regulatory sequence, were expressed using the germline-specific driver maternal tubulinGAL4∷VP16. Embryos during nuclear cycle 12–13.
(B) (Top) Lysates prepared from 0–2 hours AEL embryos expressing either empty vector (EV) or a FLAG-HA-tagged GCL variant (WT or G2A) using the UAS-GAL4 system were immunoprecipitated (IP) with anti-FLAG resin. Immunocomplexes were probed with antibodies specific to the indicated endogenous proteins. (Bottom) Densitometric scanning quantification of the Torso expression levels in the embryo lysates relative to the corresponding αTubulin (loading control) levels. The averaged relative Torso expression levels from triplicate experiments are plotted with respect to the EV control. Error bars represent standard deviation.
(C) Fixed ovarioles of indicated genotype were immunostained with anti-Orb (magenta), which marks an oocyte in each egg chamber, and Vasa (green). DNA (blue), F-actin (grey). Wild-type (w−1118) ovarioles are used as controls. Images are representative of at least 100 ovarioles analyzed per genotype. Scale bar = 100μm.
(D) Percentage of ovarioles with early stage (Stage 3, green) or late stage (Stage 10, magenta) egg chambers was scored for the ovarioles of indicated genotype. At least 100 ovarioles analyzed per genotype.
(E) Number of eggs laid by 5 females of indicated genotype was counted over the course of 24 hours to assess their fertility. Each circle represents a biological replicate. (****P < 0.0001, Mann-Whitney test)
(F) Number of PGCs in embryos of indicated maternal genotype. Bars represent the mean ± standard deviation. (n=20 for each genotype, except for ΔNLS, ****P < 0.0001, ns=not significant, Mann-Whitney test)
To address the biological significance of GCL’s localization at the nuclear envelope, as oppose to the plasma membrane, we generated a CRISPR/Cas9-induced mutant gcl allele that lacks the nuclear localization signal (NLS, gclNLS). One copy of gclNLS in otherwise wild-type females resulted in unexpected defects during the early stages of oogenesis (Fig. 7C–D) and complete female sterility (Fig. 7E). To determine if these defects were CUL3-dependent, we introduced a single copy of cul3 LOF allele and found that the defects were alleviated. In gclNLS/+,cul3LOF/+ females, a significant number of egg chambers developed to later stages of oogenesis (Fig. 7C–D), also reflected in an increased number of eggs laid (Fig. 7E). The dominant gain-of-function phenotypes suggest that the NLS normally sequesters GCL to the nucleus and prevents GCL from targeting aberrant substrates together with CUL3. Indeed, during oogenesis, wild-type GCL protein is expressed in postmitotic germ cells (nurse cells and oocyte), where it would remain at the interior of the nuclear envelope (Moore et al., 2009). To determine the function of nuclear localization during embryogenesis and PGC formation, we generated a conditional GCL transgene that lacks the nuclear localization signal (GCLNLS) and is expressed under GAL4/UAS control. This allowed us to control the expression level and timing of the dominant active GCLNLS protein during oogenesis using a late oogenesis driver, maternal tubulin-gal4, and to test the role of the NLS in gclΔ/Δ embryos. Like the endogenous mutant allele, expression of this construct also led to sterility in most female flies; however, in rare developing embryos PGC formation was rescued (Fig. 7F). This finding supports the hypothesis that CRL3GCL functions directly with Torso at the plasma membrane and is sequestered at the nuclear membrane for temporal regulation. Altogether, our results demonstrate that cell cycle-dependent localization changes in GCL provide exquisite regulation of its function and that the continuous cytoplasmic activity of GCL throughout the cell cycle is detrimental for normal development.
Discussion
The exact molecular events leading to PGC formation in early Drosophila development and the mechanism of GCL function in this process were largely unknown. Here, we show that GCL is a substrate-specific adaptor for the E3 ubiquitin ligase complex, CRL3GCL. We find that CRL3GCL targets the RTK Torso for degradation in order to inhibit somatic signaling and promote PGC formation. Disruption of the CRL3GCL complex results in Torso protein accumulation at the posterior pole where PGCs would form, thereby activating the downstream somatic program at the expense of the germline program. The interaction between CRL3GCL and Torso is intricately regulated in space and time by the cell cycle-dependent control of GCL’s subcellular localization.
GCL requires a previously uncharacterized but conserved domain, which we have named the “GCL domain,” for its substrate recognition function. Deletion and missense mutations within the GCL domain inhibit GCL from interacting with the RTK substrate and interfere with PGC formation in vivo. Expressing the mammalian homolog of GCL (Gmcl1) can partially rescue the PGC formation defect in Drosophila gcl mutants (Leatherman et al., 2000), suggesting that the substrate-specific adaptor functions of GCL has been retained across great evolutionary distance. The GCL domain is located C-terminal to the BTB domain and is predicted to form β-sheet structures akin to those found in Kelch and MATH domains used as substrate binding motifs in other BTB adaptors (Canning et al.,, 2013). Future experiments including structural analysis will be needed to identify the specific architectural features in association with its substrate in Drosophila and other species.
CRL3GCL is the dedicated E3 ubiquitin ligase complex that targets the Torso RTK for degradation specifically at the posterior pole to promote PGC formation, controlling the downstream somatic signaling pathway. gcl null mutant animals are viable and fertile, but produce healthy embryos that are defective in PGCs formation, suggesting the critical GCL function is restricted to controlling the separation of germ and somatic cell lineages. The PGC formation defect in gclΔ/Δembryos was rescued by genetically inhibiting activation of the Torso signaling pathway. Consistently, a mutant Torso RTK that is resistant to GCL-mediated ubiquitylation and degradation produces a phenotype similar to that of gclΔ/Δembryos. This suggests that Torso is the only biologically relevant target for GCL in PGC formation. In the absence of Torso, GCL appears to be dispensable for PGC formation, indicating that yet unknown factors bring about the morphological changes required to separate the future germ cells from the soma (Cinalli and Lehmann, 2013, Lerit et al., 2017). These factors are germ plasm-enriched, but sensitive to Torso signaling.
Our study illuminates critical aspects of RTK regulation. We find that ubiquitous overexpression of GCL depletes Torso even in regions where Torso is normally inactive suggesting that CRL3GCL promotes Torso degradation independently of receptor activation. This differs from the classic mechanisms for modulating RTK via endocytosis- and lysosome-dependent degradation stimulated upon ligand binding and receptor activation (Haglund and Dikic, 2012, Lloyd et al., 2002, Marmor and Yarden, 2004). The degron motif found in Torso is conserved at the protein domain level in other RTKs with similar split kinase domains, such as Torso’s closest homolog the mammalian PDGF receptor (Li, 2005), suggesting that other RTKs may share Torso’s alternate modes of degradation. Further studies are needed to explore the molecular mechanisms by which a CRL3 complex leads to degradation of a membrane bound protein such as the Torso RTK. For instance, segregase activity of p97 has been shown to extract ubiquitylated substrates from immobile membranes into the cytosol (Kuchay et al., 2017, Rape et al., 2001). In Drosophila embryos, p97, also known as TER94, was found to biochemically and genetically interact with other germ plasm components, such as Tudor and Vasa to promote PGC formation (Thomson et al., 2008).
The inhibition of PGC formation by Torso is likely independent of the canonical signaling cascade that leads to transcription of posterior somatic genes, since the lack of PGC formation in gclΔ/Δembryos is not rescued by loss of MAPK/ERK. Instead, additional downstream signaling pathways may require ligand-induced Torso receptor activation. Candidates include the JAK/STAT pathway, which is activated in dominant gain-of-function alleles of Torso (Li et al., 2002), and the ARF-GEF Steppke, which has been recently identified as a negative regulator of PGC formation (Lee et al., 2015). It will be of interest to explore further whether and how these alternative pathways oppose PGC formation.
PGC formation is achieved by remarkably precise spatiotemporal control of CRL3GCL. Previous studies have shown that GCL protein expression is developmentally coordinated by restricting translation to the germ plasm, the site of PGC formation, in early embryogenesis (Moore et al.,, 2009, Rangan et al., 2009). We now find that cell cycle-dependent subcellular localization changes of GCL further confine the activity of CRL3GCL. GCL co-localizes with its phenotypically critical substrate only during mitosis upon nuclear envelope breakdown. Indeed, the bud furrow constriction that leads to the completion of PGC formation is coupled to a mitotic division that divides a single bud into two daughter cells (Cinalli and Lehmann, 2013). These findings suggest that during bud furrow constriction and subsequent mitotic divisions, Torso is precisely and efficiently degraded only within PGCs, while it continues to be expressed and active in the neighboring cells fated to become somatic cells. These findings are consistent with previous reports suggesting that subcellular localization changes provide an efficient way of controlling accessibility of E3 ubiquitin ligases to their substrates (Skaar et al., 2013). Since protein translation is significantly lower during mitosis than in interphase (Fan and Penman, 1970, Stumpf et al., 2013), it seems likely that GCL would be translated during interphase, but would stay inactive at the nuclear membrane until nuclear envelope breakdown. Indeed, it is not unprecedented that a nuclear lamina protein is found at the plasma membrane (Cartegni et al., 1997). How the change in GCL localization is regulated during mitosis remains unknown. It is possible that cell cycle-specific cues, such as phosphorylation by a cyclin-dependent kinase, instruct GCL to relocate and shuttle between the two positions. Notably, other CRL3 adaptors are dynamically expressed and are essential throughout development in mammals (Jin et al., 2012, Werner et al., 2015), suggesting other CRL3s might use molecular and cellular mechanisms similar to CRL3GCL.
GCL localization is restricted during interphase to the nuclear envelope. However, our observation that GCL co-localizes with its substrate Torso at the plasma membrane suggests that the nuclear envelope is not the subcellular location where GCL promotes PGC formation. In support of this notion, it was previously shown that PGCs could form in the absence of nuclei (Raff and Glover, 1989). Instead, the nuclear envelope may serve to sequester GCL during interphase to prevent CRL3GCL activity from inappropriately targeting cytoplasmic substrates. Consistent with this sequestration model, we found that specifically mutating the nuclear localization signal of GCL caused oogenesis defects unrelated to the normal function of GCL. Nuclear sequestration may allow GCL to be expressed and poised in non-dividing cells. This type of regulation could be especially advantageous in quiescent cells, such as stem cells, in which specific pathways need to be switched on or off promptly upon expansion and differentiation in response to signaling and/or developmental cues. Because the regulatory sequences of GCL characterized in this study are evolutionarily conserved, our study can now bring new insight to the molecular function of GCL from worms to humans.
Germline fate specification in the early embryo has been universally linked to broad transcriptional repression of somatic signatures (Seydoux and Braun, 2006). Examples from many different organisms show that this repression is critical for germ cells to follow their unique potential as carriers of the next generation. In Drosophila and C. elegans, Polar granule component (Pgc) and PIE-1, respectively, globally repress PolII-dependent gene transcription in future germ cells, while a more selective transcriptional silencing of somatic gene expression occurs in mammals (Nakamura and Seydoux, 2008). Here we reveal an alternative, protein degradation-dependent mechanism uniquely suited for the swift and precise control of germ cell lineage by switching off somatic signaling pathways. Indeed, ubiquitin-mediated destruction of a protein substrate is a highly efficient and irreversible way of controlling protein function (Skaar et al.,, 2013). It will be interesting to see if degradation-based mechanisms also play a role in germ cell specification in mammals, which rely on inductive signals from extra-embryonic tissues to initially separate the germline potential from the soma (Lawson et al., 1999, Ohinata et al., 2005, Tam and Zhou, 1996). More broadly, the exquisitely controlled spatiotemporal switch described here could be used to define cell fate at the single cell level and coordinate the development of different cell lineages.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ruth Lehmann (Ruth.Lehmann@med.nyu.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
D. melanogaster
Drosophila were maintained on cornmeal molasses yeast medium at 25°C using standard procedures. A list of all strains used is provided in Key Resources Table.
To generate transgenic strains, expression plasmids were injected into embryos of a strain carrying the attP2 landing site and integrated into the 3L chromosome using phiC31 integrase. Injection was performed by BestGene, Inc.
To generate CRISPR/Cas9-induced mutants, an optimal gRNA and ssODN (indicated in Key Resources Table) were injected into a vasa-cas9 (X) strain (Bloomington #51323) by BestGene Inc. Correctly targeted mutants were identified by PCR screening and sequencing.
Tissue culture Cells
S2 cells were obtained from Drosophila Genomics Resource Center (DGRC) and cultured in Schneider media supplemented with 10% fetal bovine serum (FBS) under standard growth conditions. HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium containing 10% FBS. Transfection of the expression vectors was done with Effectene (Qiagen) according to the manufacturer’s protocol. Where indicated, MG132 (Peptides International, 10μM) and MLN4924 (Active Biochem, 0.5μM) were used.
METHOD DETAILS
Histology
Embryos were collected and fixed according to the standard protocol for hand devitellinization. Briefly, embryos were dechorionated in 50% (v/v) Clorox® for 2 minutes, washed thoroughly with H2O, and fixed in a solution containing 5% formaldehyde/50% heptane for an hour with shaking. Fixed embryos were then manually devitellinized, permeabilized in PBSTB (0.1% w/v bovine serum albumin (BSA), 0.1% Triton X-100 in PBS), and stained with indicated primary antibodies diluted in PBSTB (4°C overnight) and appropriate secondary antibodies (1:500, Room temperature for 2 hours). Embryos were mounted in Vectashield mounting medium (VECTOR Laboratories).
Ovaries were dissected in cold PBS and fixed in 5% formaldehyde in PBS for 20 minutes and permeabilized in PBST (1% Triton X-100 in PBS) for an hour before staining with indicated antibodies. Primary and secondary antibodies were diluted in PBSTB (1% w/v BSA, 0.2% Triton X-100 in PBS). Ovaries were mounted in Vectashield mounting medium containing DAPI (VECTOR Laboratories) prior to imaging.
The following primary antibodies were used at the indicated dilution: rabbit anti-Vasa (Lehmann laboratory, 1:5,000), chicken anti-Vasa (Lehmann laboratory, 1:500), rabbit anti-Torso (a gift from V. Cleghon, 1:1,000), rabbit anti-RNA Polymerase II CTD repeat (Phospho S2) (Abcam ab5095, 1:200), mouse anti-HA.11 (Covance 16B12, 1:1,000), rabbit anti-dpERK (Cell Signaling 4370, 1:250), mouse anti-Orb 4HB (Developmental Studies Hybridoma Bank, AB_528418), mouse anti-γTubulin (Sigma-Aldrich GTU-88, 1:500), rabbit anti-phospho-Histone H3 (Ser 10) (Cell Signaling 9701S, 1:1,000), and Alexa Fluor 633-conjugated Phalloidin (Life Technologies A12379, 1:500).
Biochemical methods
For co-immunoprecipitation assays, Drosophila embryos or cells were collected in ice cold PBS and lysed in a buffer containing 0.2% NP40, 250mM NaCl, 50mM Tris-HCl with cOmplete™ Protease Inhibitor Cocktail (Roche) and Phosphatase Inhibitor Cocktail 2 (Sigma), followed by a brief sonication. In vitro translated proteins were prepared using TnT T7 Quick Coupled Transcription/Translation System (Promega) according to the manufacturer’s protocol. Immunoprecipitation was performed as previously described (Pagan et al., 2015). Briefly, lysates or in vitro translated protein mixes were immunoprecipitated for two hours at 4°C with anti-FLAG M2 affinity gel (Sigma-Aldrich) or anti-HA agarose gel (Sigma-Aldrich), as indicated. Immunoprecipitates were eluted with either LDS sample buffer or FLAG peptide (Sigma-Aldrich).
For total cell lysis, Drosophila embryos or cells were lysed in a denaturing buffer containing 1% SDS and boiled for 10 minutes.
Ubiquitylation assay was previously described (Bloom and Pagano, 2005). Briefly, FLAG-tagged GCL, HA-tagged Torso, and MYC-tagged ubiquitin were overexpressed in HEK293T cells for 24 hours. Three hours prior to collection, MG132 or MLN4924 was added. Cells were harvested and denatured by adding 1% SDS and boiling for 10 minutes. The SDS lysates were diluted 1:10 with lysis buffer, incubated with anti-HA agarose gel (Sigma-Aldrich) for 1 hour at 4°C with rotation. The agarose gel was washed four times with lysis buffer, and the immunoprecipitated proteins were eluted in LDS sample buffer.
For immunoblotting, lysates and/or eluents were separated by SDS-PAGE, transferred onto PVDF membranes, and were probed with the following antibodies: rabbit anti-GCL (a gift from T. Jongens, University of Pennsylvania, Philadelphia, USA; 1:1,000), rabbit anti-Torso (a gift from V. Cleghon, 1:1,000), rabbit anti-MYC (Santa Cruz Biotechnology sc-789-G, 1:10,000), rabbit anti-HA (Bethyl A190-108A, 1:10,000), rabbit anti-FLAG (Sigma-Aldrich F7425, 1:10,000), rabbit anti-CUL3 (Bethyl A301-109A, 1:5,000), rabbit anti-XRCC3 (Novus NB100-165, 1:1,000), and mouse anti-αTubulin (Sigma-Aldrich T6199, 1:10,000). Proteins of interest were detected with Horseradish Peroxidase linked ECL anti-Rabbit IgG from donkey (GE Healthcare Life Sciences, NA934V) or anti-mouse IgG from sheep (GE Healthcare Life Sciences, NA931V), then were visualized with Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific 32106) according to the manufacturer’s protocol.
Mass spectrometry
Lysates from 0–2 hour old embryos expressing FLAG-tagged GCL transgenes were prepared as indicated above in Biochemical methods. GCL-containing complexes were immunoprecipitated using anti-FLAG M2 affinity gel (Sigma Aldrich) and eluted with either 3× FLAG peptide (Sigma Aldrich) or LDS sample buffer. A small fraction of eluents was separated by SDS-PAGE and visualized using SilverQuest Silver Staining Kit (Thermo Fisher) according to the manufacturer’s protocol. Rest of the eluents were reduced with DTT at 57°C, alkylated with iodoacetamide at room temperature in the dark for 45 minutes, and loaded on a NuPAGE® 4–12% Bis-Tris Gel 1.0 mm (Life Technologies). The gel was stained using GelCode Blue Stain Reagent (Thermo Scientific), and the stained bands were excised and destained in 1:1 v/v solution of methanol and 100mM Ammonium Bicarbonate solution. Excised gel pieces were dehydrated with an acetonitrile rinse and in a SpeedVac concentrater. To digest the gel, 300ng of sequencing grade modified trypsin (Promega) was added to each sample, which was then covered with 100mM ammonium biarbonate. Digestion proceeded overnight on a shaker at room temperature. Proteins were extracted by incubating each sample with slurry of R2 20 μm Poros beads (Life Technologies) in 5% formic acid and 0.2% trifluoroacetic acid (TFA) for 3 hours at 4°C with shaking. The beads were loaded onto equilibrated C18 ziptips (Millipore). Peptides were eluted by the addition of 40% acetonitrile in 0.5% acetic acid followed by the addition of 80% acetonitrile in 0.5% acetic acid. Peptides were then concentrated in a SpeedVac concentrator and reconstituted in 0.5% acetic acid. Each sample was analyzed individually by liquid chromatography (LC) separation online with mass spectrometry (MS) using the autosampler of EASY-nLC 1000 (Thermo Scientific). Peptides were gradient eluted from the column directly to Q Exactive mass spectrometer (Thermo Scientific) using a 1-hour gradient. Following each full MS, twenty data-dependent high resolution HCD MS/MS spectra were acquired. MS/MS spectra were searched using a Uniprot Drosophila database using Sequest within Proteome Discoverer (Thermo Scientific). MS was performed and analyzed at NYUMC Proteomics and Genomics Core.
Plasmids
gcl, cul3, or torso cDNA (obtained from Drosophila Genomics Resource Center) was amplified by PCR and subcloned into a variety of pActin5C vectors containing 3xFLAG or 3xHA tags. The transgenic GCL expression constructs were generated by cloning cDNA and a tag containing 3× FLAG and 3× HA into the pWALIUM22 vector (Ni et al., 2011) using Gibson Assembly® master mix (New England Biology). For mammalian expression constructs, gcl or torso cDNA was subcloned into pcDNA3.1 vector (Thermo Fisher) containing either 3xFLAG or 3xHA tag, respectively. All deletion and missense mutants were generated by Q5® site-directed mutagenesis kit (New England Biology) according to the manufacturer’s protocol using primers designed through NEBaseChanger®.
Fluorescent in situ hybridization (FISH)
Embryo samples were prepared and processed according to the previously described protocol for single molecule FISH (smFISH) (Trcek et al., 2015). Briefly, 0 to 1 hour old embryos were dechorionated and fixed in a scintillation vial for 20 minutes at room temperature in a solution containing 5ml of 4% paraformaldehyde diluted in PBS and 5ml heptane. Paraformaldehyde-containing layer was removed and 5ml of 100% methanol was added with vigorous shaking. The devitellinized embryos that fell to the bottom of the vial were collected and washed with 100% methanol three times. Embryos were rehydrated in PBST (PBS with 0.1% Tween 20), post-fixed for 20 minutes in 4% paraformaldehyde in PBST, and washed with PBST three times. Embryos were treated Proteinase K (3μg/ml in PBST) at room temperature for 13 minutes then on ice for an hour and then washed with glycine (2mg/ml) twice. Embryos were post-fixed again in 4% paraformaldehyde in PBST for 20 minutes and washed with PBST thoroughly. Embryos were prehybridized in a solution containing 10% deionized formamide (Thermo Fisher) and 2× SSC for 10 minutes, and then hybridized at 37°C over night. Hybridization solution was prepared as the following:
| Final Concentration | Hybridization mix (μl) | |
|---|---|---|
| Deionized formamide | 10% | 6 |
| Competitior (4mg/ml E.coli tRNA + 5 mg/ml salmon sperm ssDNA) | 1 | |
| Probe 1 | 10ng | 2 |
| Probe 2 | 10ng | 2 |
| 20% Dextran Sulfate | 10% | 30 |
| BSA (20mg/ml) | 2mg/ml | 5 |
| 20× SSC | 2× | 5 |
| Vanadyl Ribonucleoside Complex (200mM) | 10mM | 2.5 |
| ddH2O | 6.5 | |
| Total volume | 60 |
The next day, hybridization solution was removed, and the embryos were washed twice with 10% deionized formamide in 2× SSC for 15 minutes each, followed by two washes in PBS for an hour each. Embryos were stained with DAPI and mounted in ProLong Gold Antifade Reagent (Thermo Fisher) and cured for at least 24 hours before imaging.
Stellaris RNA FISH probes for nanos (Quasar670-conjugated) and gcl (CAL Fluor 590-conjugated) were purchased from Biosearch Technologies, Inc.
RNA expression analysis
Total RNA was isolated from dissected ovaries using TRIzol Reagent (Thermo Fisher) and subsequently treated with a TURBO DNA-free™ Kit (Thermo Fisher) to remove any residual genomic DNA contamination. RNA to cDNA EcoDry™ Premix (Oligo dT) (Clontech) was used to reverse transcribe 1μg of total RNA. Quantitative PCR was carried out using LightCycler® 480 SYBR Green I Master (Roche) with the following primers:
| Forward Primer (5′ to 3′) | Reverse Primer (5′ to 3′) | |
|---|---|---|
| gcl | TGGGCAGGAGGATTGTAGTC | TTTCGTCTGCAGGCTAAGCA |
| dmn (CG8269) | AGACGCCTGGAAGTAAGCAG | GTAAGGCGGCTCAACTTGTC |
Technical triplicates were used for each genotype. Results for gcl were normalized against that for dmn.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics and reproducibility
Statistical calculations were done using GraphPad. Mann-Whitney U test was used to compare data sets. All experiments were performed in three or more biological replicates. Panel images of embryos and ovarioles are representative of at least a hundred visualized and imaged. Data are represented as either absolute numbers or percentages ± standard deviation, as indicated in Figure Legends.
Microscope and analysis
All fluorescent images were acquired with ×20 or ×40 oil objectives on a Zeiss LSM 780 confocal microscope. All post-acquisition analysis was performed using ImageJ (NIH; http://rsb.info.nih.gov/ij/). To quantify dpERK fluorescent intensity in Figure 2G, a rectagular region of interest (ROI) was set across an embryo, and intensity values of dpERK for the ROI were plotted using ImageJ as a function of the distance from left to right. Relative fluorescent intensity was calculated by normalizing against the center of the embryo (50% distance from left to right). Data are represented as an averaged intensity of three biological replicates with standard deviation.
PGC Count
Fixed embryos were stained with an antibody against Vasa (germ cell marker), Phalloidin (F-actin) and DAPI (DNA). Embryos undergoing mitotic cycle 12–13 were used to count the number of PGCs. Posterior pole of the embryos was imaged using Zeiss LSM 780 confocal microscope across multiple z-planes covering the span of the embryo. Images were then processed in ImageJ, and the total number of Vasa-positive PGCs in each embryo was manually counted using the Cell Counter plugin in ImageJ. For each genotype, 10 or more embryos were assessed to be statistically significant, as indicated in Figure Legends. When appropriate, control siblings were studied to ensure consistency of genetic background.
Densitometric quantification of western blots
ImageJ software was used to measure the signal intensity of bands on immunoblots. To calculate the relative protein expression level, band intensity of the protein of interest was normalized against that of α-Tubulin (internal loading control). Data are represented as an averaged intensity of three biological replicates with standard deviation.
Supplementary Material
Table S1. Complete list of mass spectrometry results of GCL immunocomplexes, Related to Figure 2 and S2.
Highlights.
CRL3GCL targets the Torso RTK for ubiquitylation and degradation
Cell cycle-dependent localization change of GCL limits Torso degradation to mitosis
CRL3GCL-mediated degradation of Torso occurs independent of receptor activation
Localized degradation of Torso is critical for proper Primordial Germ Cell formation
Acknowledgments
We thank all members of the Lehmann and the Pagano labs for advice and support. We thank A. Arkov for identification of gcl alleles, B. Ueberheide at NYUMC Proteomics and Genomics Core for mass spectrometry analysis, and A. Soshnev for the illustrated figures. We thank R. Schekman, D. Dominguez-Sola, T. Hurd, F. Teixeira, and J. Pagan for helpful discussions, and L. Barton, T. Trcek Pullsic, J. Abrams, and B. Lin for critical reading of the manuscript. Stocks obtained from the Bloomington Drosophila Stock Center (NIH P40OD018537) were used in this study. This work was supported by NCI Stem Cell Biology Program Training Grant (1T32CA16002) to J.P. Fellowships from the American Italian Cancer Foundation and Associazione Italiana per la Ricerca sul Cancro (AIRC) co-funded by the European Union to A.M. R.L. and M.P. are Howard Hughes Medical Institute investigators.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Conceptualization, J.P., M.P., and R.L.; Investigation and Validation, J.P., R.M.C., A.M., and R.L. Writing, J.P., M.P., and R.L.; Visualization, JP; Supervision and Funding Acquisition, M.P. and R.L.
References
- Bloom J, Pagano M. Experimental tests to definitively determine ubiquitylation of a substrate. Methods Enzymol. 2005;399:249–66. doi: 10.1016/S0076-6879(05)99017-4. [DOI] [PubMed] [Google Scholar]
- Canning P, Cooper CD, Krojer T, Murray JW, Pike AC, Chaikuad A, Keates T, Thangaratnarajah C, Hojzan V, Ayinampudi V, Marsden BD, Gileadi O, Knapp S, von Delft F, Bullock AN. Structural basis for Cul3 protein assembly with the BTB-Kelch family of E3 ubiquitin ligases. J Biol Chem. 2013;288(11):7803–14. doi: 10.1074/jbc.M112.437996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartegni L, di Barletta MR, Barresi R, Squarzoni S, Sabatelli P, Maraldi N, Mora M, Di Blasi C, Cornelio F, Merlini L, Villa A, Cobianchi F, Toniolo D. Heart-specific localization of emerin: new insights into Emery-Dreifuss muscular dystrophy. Hum Mol Genet. 1997;6(13):2257–64. doi: 10.1093/hmg/6.13.2257. [DOI] [PubMed] [Google Scholar]
- Casanova J, Struhl G. Localized surface activity of torso, a receptor tyrosine kinase, specifies terminal body pattern in Drosophila. Genes Dev. 1989;3(12B):2025–38. doi: 10.1101/gad.3.12b.2025. [DOI] [PubMed] [Google Scholar]
- Cinalli RM. Unpublished Doctor of Philosophy. New York University, ProQuest Dissertations Publishing; 2012. Analysis of Drosophila melanogaster pole cell formation. [Google Scholar]
- Cinalli RM, Lehmann R. A spindle-independent cleavage pathway controls germ cell formation in Drosophila. Nat Cell Biol. 2013;15(7):839–45. doi: 10.1038/ncb2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinalli RM, Rangan P, Lehmann R. Germ cells are forever. Cell. 2008;132(4):559–62. doi: 10.1016/j.cell.2008.02.003. [DOI] [PubMed] [Google Scholar]
- Duffy JB, Perrimon N. The torso pathway in Drosophila: lessons on receptor tyrosine kinase signaling and pattern formation. Dev Biol. 1994;166(2):380–95. doi: 10.1006/dbio.1994.1324. [DOI] [PubMed] [Google Scholar]
- Fan H, Penman S. Regulation of protein synthesis in mammalian cells. II. Inhibition of protein synthesis at the level of initiation during mitosis. J Mol Biol. 1970;50(3):655–70. doi: 10.1016/0022-2836(70)90091-4. [DOI] [PubMed] [Google Scholar]
- Gabay L, Seger R, Shilo BZ. MAP kinase in situ activation atlas during Drosophila embryogenesis. Development. 1997;124(18):3535–41. doi: 10.1242/dev.124.18.3535. [DOI] [PubMed] [Google Scholar]
- Gao M, Arkov AL. Next generation organelles: structure and role of germ granules in the germline. Mol Reprod Dev. 2013;80(8):610–23. doi: 10.1002/mrd.22115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genschik P, Sumara I, Lechner E. The emerging family of CULLIN3-RING ubiquitin ligases (CRL3s): cellular functions and disease implications. EMBO J. 2013;32(17):2307–20. doi: 10.1038/emboj.2013.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haglund K, Dikic I. The role of ubiquitylation in receptor endocytosis and endosomal sorting. J Cell Sci. 2012;125(Pt 2):265–75. doi: 10.1242/jcs.091280. [DOI] [PubMed] [Google Scholar]
- Hudson AM, Cooley L. Drosophila Kelch functions with Cullin-3 to organize the ring canal actin cytoskeleton. J Cell Biol. 2010;188(1):29–37. doi: 10.1083/jcb.200909017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Pahuja KB, Wickliffe KE, Gorur A, Baumgartel C, Schekman R, Rape M. Ubiquitin-dependent regulation of COPII coat size and function. Nature. 2012;482(7386):495–500. doi: 10.1038/nature10822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson TK, Henstridge MA, Herr A, Moore KA, Whisstock JC, Warr CG. Torso-like mediates extracellular accumulation of Furin-cleaved Trunk to pattern the Drosophila embryo termini. Nat Commun. 2015;6:8759. doi: 10.1038/ncomms9759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jongens TA, Ackerman LD, Swedlow JR, Jan LY, Jan YN. Germ cell-less encodes a cell type-specific nuclear pore-associated protein and functions early in the germ-cell specification pathway of Drosophila. Genes Dev. 1994;8(18):2123–36. doi: 10.1101/gad.8.18.2123. [DOI] [PubMed] [Google Scholar]
- Jongens TA, Hay B, Jan LY, Jan YN. The germ cell-less gene product: a posteriorly localized component necessary for germ cell development in Drosophila. Cell. 1992;70(4):569–84. doi: 10.1016/0092-8674(92)90427-e. [DOI] [PubMed] [Google Scholar]
- Kimura T, Ito C, Watanabe S, Takahashi T, Ikawa M, Yomogida K, Fujita Y, Ikeuchi M, Asada N, Matsumiya K, Okuyama A, Okabe M, Toshimori K, Nakano T. Mouse germ cell-less as an essential component for nuclear integrity. Mol Cell Biol. 2003;23(4):1304–15. doi: 10.1128/MCB.23.4.1304-1315.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiman SE, Yogev L, Gal-Yam EN, Hauser R, Gamzu R, Botchan A, Paz G, Yavetz H, Maymon BB, Schreiber L, Barzilai S, Amariglio N, Rechavi G, Simon AJ. Reduced human germ cell-less (HGCL) expression in azoospermic men with severe germinal cell impairment. J Androl. 2003;24(5):670–5. doi: 10.1002/j.1939-4640.2003.tb02725.x. [DOI] [PubMed] [Google Scholar]
- Klingler M, Erdelyi M, Szabad J, Nusslein-Volhard C. Function of torso in determining the terminal anlagen of the Drosophila embryo. Nature. 1988;335(6187):275–7. doi: 10.1038/335275a0. [DOI] [PubMed] [Google Scholar]
- Kuchay S, Giorgi C, Simoneschi D, Pagan J, Missiroli S, Saraf A, Florens L, Washburn MP, Collazo-Lorduy A, Castillo-Martin M, Cordon-Cardo C, Sebti SM, Pinton P, Pagano M. PTEN counteracts FBXL2 to promote IP3R3- and Ca2+-mediated apoptosis limiting tumour growth. Nature. 2017 doi: 10.1038/nature22965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson KA, Dunn NR, Roelen BA, Zeinstra LM, Davis AM, Wright CV, Korving JP, Hogan BL. Bmp4 is required for the generation of primordial germ cells in the mouse embryo. Genes Dev. 1999;13(4):424–36. doi: 10.1101/gad.13.4.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leatherman JL, Kaestner KH, Jongens TA. Identification of a mouse germ cell-less homologue with conserved activity in Drosophila. Mech Dev. 2000;92(2):145–53. doi: 10.1016/s0925-4773(99)00335-4. [DOI] [PubMed] [Google Scholar]
- Leatherman JL, Levin L, Boero J, Jongens TA. germ cell-less acts to repress transcription during the establishment of the Drosophila germ cell lineage. Curr Biol. 2002;12(19):1681–5. doi: 10.1016/s0960-9822(02)01182-x. [DOI] [PubMed] [Google Scholar]
- Lee DM, Wilk R, Hu J, Krause HM, Harris TJ. Germ Cell Segregation from the Drosophila Soma Is Controlled by an Inhibitory Threshold Set by the Arf-GEF Steppke. Genetics. 2015;200(3):863–72. doi: 10.1534/genetics.115.176867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehner CF. The pebble gene is required for cytokinesis in Drosophila. J Cell Sci. 1992;103(Pt 4):1021–30. doi: 10.1242/jcs.103.4.1021. [DOI] [PubMed] [Google Scholar]
- Lerit DA, Shebelut CW, Lawlor KJ, Rusan NM, Gavis ER, Schedl P, Deshpande G. Germ Cell-less Promotes Centrosome Segregation to Induce Germ Cell Formation. Cell Rep. 2017;18(4):831–839. doi: 10.1016/j.celrep.2016.12.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WX. Functions and mechanisms of receptor tyrosine kinase Torso signaling: lessons from Drosophila embryonic terminal development. Dev Dyn. 2005;232(3):656–72. doi: 10.1002/dvdy.20295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WX, Agaisse H, Mathey-Prevot B, Perrimon N. Differential requirement for STAT by gain-of-function and wild-type receptor tyrosine kinase Torso in Drosophila. Development. 2002;129(18):4241–8. doi: 10.1242/dev.129.18.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd TE, Atkinson R, Wu MN, Zhou Y, Pennetta G, Bellen HJ. Hrs regulates endosome membrane invagination and tyrosine kinase receptor signaling in Drosophila. Cell. 2002;108(2):261–9. doi: 10.1016/s0092-8674(02)00611-6. [DOI] [PubMed] [Google Scholar]
- Marmor MD, Yarden Y. Role of protein ubiquitylation in regulating endocytosis of receptor tyrosine kinases. Oncogene. 2004;23(11):2057–70. doi: 10.1038/sj.onc.1207390. [DOI] [PubMed] [Google Scholar]
- Martinho RG, Kunwar PS, Casanova J, Lehmann R. A noncoding RNA is required for the repression of RNApolII-dependent transcription in primordial germ cells. Curr Biol. 2004;14(2):159–65. doi: 10.1016/j.cub.2003.12.036. [DOI] [PubMed] [Google Scholar]
- Moore J, Han H, Lasko P. Bruno negatively regulates germ cell-less expression in a BRE-independent manner. Mech Dev. 2009;126(7):503–16. doi: 10.1016/j.mod.2009.04.002. [DOI] [PubMed] [Google Scholar]
- Nakamura A, Seydoux G. Less is more: specification of the germline by transcriptional repression. Development. 2008;135(23):3817–27. doi: 10.1242/dev.022434. [DOI] [PubMed] [Google Scholar]
- Ni JQ, Zhou R, Czech B, Liu LP, Holderbaum L, Yang-Zhou D, Shim HS, Tao R, Handler D, Karpowicz P, Binari R, Booker M, Brennecke J, Perkins LA, Hannon GJ, Perrimon N. A genome-scale shRNA resource for transgenic RNAi in Drosophila. Nat Methods. 2011;8(5):405–7. doi: 10.1038/nmeth.1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohinata Y, Payer B, O’Carroll D, Ancelin K, Ono Y, Sano M, Barton SC, Obukhanych T, Nussenzweig M, Tarakhovsky A, Saitou M, Surani MA. Blimp1 is a critical determinant of the germ cell lineage in mice. Nature. 2005;436(7048):207–13. doi: 10.1038/nature03813. [DOI] [PubMed] [Google Scholar]
- Pagan JK, Marzio A, Jones MJ, Saraf A, Jallepalli PV, Florens L, Washburn MP, Pagano M. Degradation of Cep68 and PCNT cleavage mediate Cep215 removal from the PCM to allow centriole separation, disengagement and licensing. Nat Cell Biol. 2015;17(1):31–43. doi: 10.1038/ncb3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pintard L, Willems A, Peter M. Cullin-based ubiquitin ligases: Cul3-BTB complexes join the family. EMBO J. 2004;23(8):1681–7. doi: 10.1038/sj.emboj.7600186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff JW, Glover DM. Centrosomes, and not nuclei, initiate pole cell formation in Drosophila embryos. Cell. 1989;57(4):611–9. doi: 10.1016/0092-8674(89)90130-x. [DOI] [PubMed] [Google Scholar]
- Rangan P, DeGennaro M, Jaime-Bustamante K, Coux RX, Martinho RG, Lehmann R. Temporal and spatial control of germ-plasm RNAs. Curr Biol. 2009;19(1):72–7. doi: 10.1016/j.cub.2008.11.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rape M, Hoppe T, Gorr I, Kalocay M, Richly H, Jentsch S. Mobilization of processed, membrane-tethered SPT23 transcription factor by CDC48(UFD1/NPL4), a ubiquitin-selective chaperone. Cell. 2001;107(5):667–77. doi: 10.1016/s0092-8674(01)00595-5. [DOI] [PubMed] [Google Scholar]
- Robertson SE, Dockendorff TC, Leatherman JL, Faulkner DL, Jongens TA. germ cell-less is required only during the establishment of the germ cell lineage of Drosophila and has activities which are dependent and independent of its localization to the nuclear envelope. Dev Biol. 1999;215(2):288–97. doi: 10.1006/dbio.1999.9453. [DOI] [PubMed] [Google Scholar]
- Savant-Bhonsale S, Montell DJ. torso-like encodes the localized determinant of Drosophila terminal pattern formation. Genes Dev. 1993;7(12B):2548–55. doi: 10.1101/gad.7.12b.2548. [DOI] [PubMed] [Google Scholar]
- Schupbach T, Wieschaus E. Germline autonomy of maternal-effect mutations altering the embryonic body pattern of Drosophila. Dev Biol. 1986;113(2):443–8. doi: 10.1016/0012-1606(86)90179-x. [DOI] [PubMed] [Google Scholar]
- Seydoux G, Braun RE. Pathway to totipotency: lessons from germ cells. Cell. 2006;127(5):891–904. doi: 10.1016/j.cell.2006.11.016. [DOI] [PubMed] [Google Scholar]
- Skaar JR, Pagan JK, Pagano M. Mechanisms and function of substrate recruitment by F-box proteins. Nat Rev Mol Cell Biol. 2013;14(6):369–81. doi: 10.1038/nrm3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens LM, Frohnhofer HG, Klingler M, Nusslein-Volhard C. Localized requirement for torso-like expression in follicle cells for development of terminal anlagen of the Drosophila embryo. Nature. 1990;346(6285):660–3. doi: 10.1038/346660a0. [DOI] [PubMed] [Google Scholar]
- Stumpf CR, Moreno MV, Olshen AB, Taylor BS, Ruggero D. The translational landscape of the mammalian cell cycle. Mol Cell. 2013;52(4):574–82. doi: 10.1016/j.molcel.2013.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam PP, Zhou SX. The allocation of epiblast cells to ectodermal and germ-line lineages is influenced by the position of the cells in the gastrulating mouse embryo. Dev Biol. 1996;178(1):124–32. doi: 10.1006/dbio.1996.0203. [DOI] [PubMed] [Google Scholar]
- Thomson T, Liu N, Arkov A, Lehmann R, Lasko P. Isolation of new polar granule components in Drosophila reveals P body and ER associated proteins. Mech Dev. 2008;125(9–10):865–73. doi: 10.1016/j.mod.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trcek T, Grosch M, York A, Shroff H, Lionnet T, Lehmann R. Drosophila germ granules are structured and contain homotypic mRNA clusters. Nat Commun. 2015;6:7962. doi: 10.1038/ncomms8962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner A, Iwasaki S, McGourty CA, Medina-Ruiz S, Teerikorpi N, Fedrigo I, Ingolia NT, Rape M. Cell-fate determination by ubiquitin-dependent regulation of translation. Nature. 2015;525(7570):523–7. doi: 10.1038/nature14978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang M, Calabrese MF, Liu J, Waddell MB, Nourse A, Hammel M, Miller DJ, Walden H, Duda DM, Seyedin SN, Hoggard T, Harper JW, White KP, Schulman BA. Structures of SPOP-substrate complexes: insights into molecular architectures of BTB-Cul3 ubiquitin ligases. Mol Cell. 2009;36(1):39–50. doi: 10.1016/j.molcel.2009.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Complete list of mass spectrometry results of GCL immunocomplexes, Related to Figure 2 and S2.