

Abstract
Purpose
To prepare mixed polymeric micelles that can carry two different drugs, doxorubicin (DOX) and 17-hydroxyethylamino-17-demethoxygeldanamycin (GDM-OH), for combination cancer chemotherapy.
Methods
The pH-sensitive micelles were prepared from poly (ethylene glycol)-poly(aspartate hydrazide) block copolymers to which either DOX or GDM-OH is conjugated through acid-labile hydrazone bond (individual micelles). Mixed micelles were formed not only by simply mixing two different individual micelles in aqueous solutions (aqueous mixed micelles) but also by evaporating organic solvents from the organic/aqueous mixed solvents in which two block copolymers possessing different drugs were dissolved homogeneously (organic mixed micelles). Particle size measurements, pH-dependent drug release tests, cytotoxicity assays and western blot analysis were subsequently conducted.
Results
Individual and aqueous/organic mixed micelles showed clinically relevant particle size (<100 nm) and pH-dependent drug release patterns. Mixed polymer micelles suppress cancer cell growth effectively in a drug concentration, mixing method and schedule-dependent way.
Conclusion
Combination chemotherapy using polymeric micelles seems to minimize a schedule-dependent change in combination drug efficacy in comparison to drug combination using DMSO formulations.
Keywords: combination chemotherapy, drug delivery, mixed micelles, pH-controlled release, polymeric micelles
INTRODUCTION
Experimental and clinical studies have shown that the disruption of a single molecular target is not sufficient enough to completely cure cancers due to the complex cell survival mechanisms (1). In an effort to sensitize these drug-resistant cancer cells, recent efforts in cancer research have been paid to the development of effective combination chemotherapy that suppresses multiple intra-cellular therapeutic targets (2,3). Among a large number of therapeutic targets identified so far, heat shock protein 90 (HSP90) appears to play an important role in cancer cell survival mechanisms, regulating interactive network of multiple signaling pathways (4–7). Despite encouraging results, the development of dosage forms for HSP90 inhibitors and other poorly water-soluble anticancer drugs is still challenging (8,9).
When it comes to the development of drug dosage forms, systemic toxicity and tumor specificity are considered the major two issues that need to be resolved (10). Toxicity is generally induced by both anticancer drugs and injection vehicles (e.g. DMSO, Cremophor EL, and other surfactants). DMSO is dominantly used as an injection vehicle for conventional drug infusion formulations despite its toxicity. In addition, since it is an organic solvent diffusing easily into tissues, DMSO has inherent limitations in controlling pharmacokinetic (PK) profiles of anticancer drugs in a tumor-specific manner. For these reasons, DMSO is used in animal models and for 17-AAG, a potent HSP90 inhibitor, in clinical trials, but it is not FDA approved yet. Situations with other drug vehicles from low-molecular-weight surfactants are not different in improving PK profiles. As alternatives to these conventional formulations, drug delivery systems (DDS) using polymer-based carriers have recently emerged as promising paradigms to reduce toxicity and improve tumor-targeting efficiency of therapeutic agents (11–13). Particularly, polymeric micelles have shown that various types of toxic therapeutic agents for imaging, diagnosis and treatment can be selectively delivered to tumor tissues with markedly reduced systemic toxicity (14–16). Preferential accumulation of macromolecules in tumor tissues, called the enhanced permeability and retention effect, explains the mechanism behind tumor-specific accumulation of polymeric micelles. From these perspectives, one can readily imagine that polymeric micelles would provide delivery platforms for a combination of drugs to accumulate in tumor tissue at the identical PK patterns in a concurrent or sequential manner.
In this study, a potential application of polymeric micelles as an effective drug delivery formulation for combination chemotherapy is evaluated using mixed pH-sensitive polymeric micelles. We have previously shown that conceptual multi-drug-loaded polymer micelle platforms can be prepared, retaining mono-disperse spherical nanostructures (<100 nm) at fixed drug mixing ratios (17). This approach is also exploited in this study to design mixed pH-sensitive polymeric micelles for HSP90-mediated combination chemotherapy (Fig. 1). Polymer backbones are prepared from poly(ethylene glycol)-poly(aspartate hydrazide) block copolymers. The hydrazide groups on the side chain are used as pH-labile binding linkers for drugs possessing ketone groups. Doxorubicin (DOX) and geldanamycin (GDM) analogues are used as our model anticancer drug and HSP90 inhibitor, respectively. The GDM analogues were 17-hydroxyethylamino-17-demethoxygel-danamycin (GDM-OH), which was previously synthesized to introduce a hydroxyl group for further chemical modification without deteriorating the biological activity of GDM (18,19). The block copolymers are conjugated with DOX possessing a ketone group at its C13 position directly, while a spacer was introduced to GDM-OH.
Fig. 1.

pH-Sensitive polymeric micelles for the delivery of DOX and GDM-OH.
HSP90 is a molecular chaperone that refolds more than 300 proteins, called client proteins, protecting cells from cell death induced by external stress such as heat, light, and chemicals (20–22). Topoisomerase II (TOPOII), involving cell death triggered by DOX-DNA intercalation, is known as one of the HSP90 client proteins. Studies have suggested that combined use of DOX and HSP90 inhibitors is promising at optimized therapeutic schedules (23). Nevertheless, cytotoxic effects of combined drugs are also known to be dependent on drug concentrations and mixing ratios. In consideration of tumor targeting and the follow-up antitumor actions, it is hypothesized that combination drug effects of DOX and HSP90 inhibitors would change when drug formulations are changed. Concentration- and schedule-dependent cytotoxicity of the mixed micelles is extensively evaluated in this study using a human breast cancer cell line MCF-7. Obtained results provide a valuable insight into the optimal combination settings of mixed polymeric micelles for future preclinical and clinical applications.
MATERIALS AND METHODS
Materials and Cell Culture
α-Methoxy-ω-amino poly(ethylene glycol) (PEG, MW= 12,266) was purchased from NOF Corporation (Japan). Acetic anhydride (AA), L-aspartic acid β-benzyl ester, anhydrous hydrazine, benzene, chloroform (CH3Cl), N,N′-diisopropylcarbodiimide (DIC), diethyl ether, Dulbecco’s Modified Eagle’s Medium (DMEM), 4-(dimethylamino) pyridine (DMAP), anhydrous N,N-dimethylformamide (DMF), dimethylsulfoxide (DMSO), dimethylsulfoxide-d6 (DMSO-d6), doxorubicin hydrochloride (DOX), ethanol-amine, anhydrous hexane, levulinic acid, methanol (MeOH), resazurin sodium salt, anhydrous tetrahydrofuran (THF), and triphosgene were purchased from Sigma-Aldrich (USA). Sephadex LH-20 was obtained from Amersham Pharmacia Biotech (Sweden). Regenerated cellulose dialysis tube with molecular weight cut-off (MWCO) ranging from 6–8,000 and Slide-A-Lyzer® dialysis cassettes with 20,000 MWCO were purchased from Fisher Scientific (USA). Amicon-Ultra centrifugal ultrafiltration devices with MWCO 30,000 were purchased from Millipore (USA). A human breast cancer MCF-7 cell line was obtained from American Type Culture Collection. Phosphate-buffered saline (PBS), fetal bovine serum (FBS), and trypsin-EDTA (0.25% trypsin and 2.21 mM EDTA) were purchased from Cellgro (USA). MCF-7 cells were cultured in DMEM containing 10% FBS in a humidified atmosphere with 5% CO2 at 37°C.
Synthesis of Block Copolymers
A poly(ethylene glycol)-poly(aspartate hydrazide) [PEG-p (Asp-Hyd)] block copolymer was synthesized as reported elsewhere (24). Briefly, PEG-p(Asp-Hyd) was obtained by a three-step reaction: 1) Synthesis of β-benzyl L-aspartate N-carboxy-anhydride (BLA-NCA): 5 g of L-aspartic acid β-benzyl ester (22.40 mmol, MW=223.23) and 2.88 g of triphosgene (9.71 mmol, MW=296.75) were reacted in 80 mL of anhydrous THF at 40°C until the reaction solution was completely clear. BLA-NCA was precipitated by adding 200 mL of anhydrous hexane, followed by further purification by recrystallization. 2) Polymerization of poly(ethylene glycol)-poly(β-benzyl L-aspartate) (PEG-PBLA): 1 g of PEG (81.53 μmol, MW=12,266) and 1.02 g of BLA-NCA (4.09 mmol, MW=249.22) were dissolved in 20 mL of anhydrous DMSO at 40°C. Ring-opening polymerization of BLA-NCA was allowed to proceed for 48 h, and PEG-PBLA was precipitated in ice-cold diethyl ether, followed by freeze drying from benzene. 3) Modification of the side-chains of PEG-PBLA: in order to prevent side reactions, the N-terminal amino group of PEG-PBLA was protected by AA prior to the aminolysis reaction. PEG-PBLA was reacted with anhydrous hydrazine in DMF at 40°C for 1 h. The amount of hydrazine varied according to the composition of the polymer, yet the substitution ratio was adjusted to 75–85% with respect to the number of BLA repeating units. Unreacted benzyl ester groups were removed by hydrolysis, using a 0.1 N NaOH solution. The final product was treated with 0.025% NH3 aqueous solution, dialyzed against deionized (DI) water using MWCO 6–8,000 membrane, and collected through freeze drying.
GDM-OH was synthesized by a method reported previously with slight modification (25). GDM was first obtained by fermentation of Streptomyces hygroscopicus subsp. geldanus (26). One-hundred mg of GDM (178.37 μmol, MW=560.64) were then reacted with 110 μL of ethanol-amine in 20 mL of CHCl3 at 25°C until the reaction was completed, judged by TLC (CHCl3:MeOH=95:5). Purple GDM-OH was purified by extraction, followed by freeze drying. In order to introduce a ketone group for pH-sensitive drug binding, GDM-OH was chemically modified further with levulinic acid (GDM-Lev). Fifty mg of GDM-OH (84.79 μmol, MW=589.69) were reacted with 20 mg of levulinic acid (172.24 μmol, MW=116.12) in 10 mL of CHCl3 at 25°C for 3 h in the presence of 45 mg of DIC (356.58 μmol, MW=126.20) and 5 mg of DMAP (40.93 μmol, MW=122.17). GDM-Lev was purified by extraction and silica gel chromatography, followed by freeze drying. 1H-NMR (GDM-OH, DMSO-d6, 25°C) δ 0.72(14-CH3), 0.96(10-CH3), 1.38(13-CH2), 1.59(8-CH3, 14-CH), 1.84(2-CH3, 15-CH2), 2.18–2.41(10-CH, 12-CH, 28-CH3), 3.03–3.05(17-OCH3, 25-CH2), 3.22(6-OCH3, 12-OCH3, 26-CH2), 3.24(22-CH2), 3.99(11-CH), 4.19 (23-CH2), 4.32(6-CH), 4.82(7-CH), 5.43(9-CH), 5.77(5-CH), 6.22–7.14(3-CH, 4-CH, 19-CH).
Preparation of pH-sensitive Polymeric Micelles Loading DOX and GDM-OH
Each polymeric micelle was prepared from drug-conjugated block copolymers as follows: PEG-p(Asp-Hyd) block copolymers were conjugated with either DOX or GDM-OH. Fifty mg of PEG-p(Asp-Hyd) were mixed with either drug in DMSO (100 mg/mL) at a two-fold molar ratio with respect to hydrazide groups. The conjugation reaction was allowed to proceed for three days, and the reaction solution was precipitated in ice-cold ether. Precipitation process was repeated at least three times until supernatent became clear. Sediments were redissolved in MeOH and purified further using Sephadex LH-20 gel chromatography to completely remove unbound drug. The solvent was evaporated, and drug-conjugated block copolymers were collected by freeze drying.
For preparation of the polymeric micelles, 30 mg of drug-conjugated block copolymers were dissolved in 3 mL of DMSO. This solution was titrated to 1 L of Tris-HCl buffer (10 mM, pH 7.4), followed by filtration using 0.45 μm filter. The filtered solution was concentrated by centrifugal ultrafiltration devices with 30,000 MWCO. Organic solvent was completely removed by repeating ultrafiltration to obtain clear polymeric micelle solutions. Drug loading content of the polymeric micelles was determined by UV-Vis colorimetric analysis at 485 nm and 337 nm for DOX and GDM-OH, respectively. Each polymeric micelle solution was diluted into the same drug concentration, and the sample aliquots were stored at 4°C until use.
Preparation of Mixed pH-Sensitive Micelles
Micelles were prepared either as an individual or a mixed micelle formulation. Individual micelles were prepared by dissolving DOX- and GDM-OH-conjugated polymers separately in buffers (pH 5 or 7.4) at 5 mg/mL concentration. Micelle solutions were sonicated for 10 min prior to loading the samples into dialysis cassette. In contrast, two methods were tested to prepare mixed micelles. One method was to prepare individual DOX-loaded micelles (DM) and GDM-OH-loaded micelles (GM) first and simply mix these micelle solutions together into one solution to make the total polymer-drug concentration become 5 mg/mL (2.5 mg/mL for each micelle). This type of micelle is described as ‘aqueous mixed micelles (AMM)’. Another method was to dissolve in acetonitrile the block copolymers to which each drug (either DOX or GDM-OH) was already conjugated, add 3 mL of DI water, and evaporate the organic solvent. To remove acetonitrile completely, rotatory evaporation was repeated at least three times, while the final concentration was adjusted to be 5 mg/mL by adding buffers (pH 5 or 7.4) prior to drug release experiments. These micelles are denoted as ‘organic mixed micelles (OMM).’ In this way, drug mixing ratios of DOX and GDM-OH were precisely controlled in both AMMs and OMMs. Particle size of the micelles was determined by dynamic light scattering measurement (Zetasizer Nano ZS, Malvern, UK).
Drug Release Study
Drug release profiles of DM, GM, AMM and OMM were evaluated by the dialysis method. The release profile was evaluated under sink conditions at pH 5 and 7.4 using acetate buffer and phosphate buffer, respectively. Buffers were prepared according to USP 24 guidelines. 2.5 mL of micelle solutions were loaded into Slide-A-Lyzer® dialysis cassette with 20,000 MWCO. Three cassettes were used in each experiment (n=3). The cassettes were placed in 2.5 L of buffer, which was changed every 3 h, controlling the temperature at 37°C. The sampling time intervals were 0, 0.5, 1, 2, 3, 6, 9, 12, and 24 h, while 150 μL of samples were collected from each cassette and replaced with an equal volume of fresh buffer at each time point. Drugs released were quantified by HPLC first to determine the remaining amount of drug molecules attached to the polymer. This analysis method minimizes errors in quantitative measurements, which are often attributed to hypochromicity/hyperchromicity or fluorescence quenching of drug molecules conjugated to polymers. HPLC analysis conditions were as follows: The system was Shimadzu Prominence HPLC series equipped with SPD-M20A Photodiode Array Detector. Twenty μL samples were injected into ZORBAX SB-C8 (4.6 mm×75 mm, 10 micron, Agilent Technologies) column at 40°C. The mobile phase with a flow rate 1 ml/min consisted of an isocratic mixture of 10 mM ammonium hydroxide (pH 9) and acetonitrile at 55:45 mixing ratio. Detection of GDM-OH and DOX was done at 337 nm and 480 nm, respectively.
Cytotoxicity of DOX and GDM-OH as Free Drugs or as Polymeric Micelles
Samples are distinguished by acronyms such as D, G, DM, GM, AMM, and OMM, denoting DOX, GDM-OH, DOX-loaded micelle, and GDM-OH-loaded micelle, aqueous mixed micelle loading DOX and GDM-OH, and organic mixed micelle loading DOX and GDM-OH, respectively.
MCF-7 cells were seeded in 96-well plates (3,000–5,000 cells/well) in 90 μL of DMEM containing 10% FBS. After 24 h, 10 μL of drug solution with varying concentrations were added to each well. At this stage, D and G or DM and GM were added either simultaneously or with a 24 h interval for schedule-dependent combination treatments. Free drugs or micelles were prepared as stock concentrations. The drug solutions alone were made in 100% DMSO and diluted in 1.5 mL microtubes to give a final DMSO concentration of <0.1% in each well. In the case of micelles, no DMSO was used in preparing samples. Drug-addition schedules for free drugs (or polymeric micelles) are described as follows: D(DM): add D(DM) alone; G(GM): add G(GM) alone; D/G(DM/GM): add D(DM) and G (GM) simultaneously; DG(DMGM): add D(DM) first and G (GM) after 24 h; GD(GMDM): add G(GM) first and D (DM) after 24 h; AMM: add AMM; and OMM: add OMM alone. Cells were cultured at 37°C for 72 h total. Cell viability was determined by a resazurin dye assay, which is based on the ability of living cells to convert an indicator redox dye (resazurin) into a measurable fluorescent end point (resorufin). The assay is characterized by a single addition of the dye directly to the cells in serum-containing medium without either cell washing or removal of medium for measurements reducing technical errors. Most importantly, resazurin dye is non-toxic so that the viability can be measured while cells are alive. For the measurement of cell viability, 10 μL of resazurin solution (60 μM in PBS) were added to each well. After incubation at 37°C for 4 h, the fluorescence at 560ex/590em was measured. The drug dose-response data (mean±SD, n=4) were fitted to a variable of Hill Slope to determine the inhibitory concentrations for 50% cell viability (IC50). Experiments were performed by three researchers in triplicate.
Western Blotting
MCF-7 cells were seeded in 6-well plates at 200,000 cells per well in 2 ml of MEM supplemented with 10% FBS and 1% penicillin/streptomycin. The cells were incubated with drugs at different mixing ratios and concentrations, followed by further 48 h incubation at 37°C. Drug-treated cells were then washed with cold PBS twice, detached from the place and centrifuged. Cell pellets were incubated with cell lysis buffers (25 mM Tris-HCl pH7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, 1% EDTA) containing diluted protease inhibitor cocktail (Pierce, IL, USA). Protein concentration was determined using Bio-Rad protein assay, and equal amounts of protein lysate were used for electrophoresis and SDS-PAGE. The membranes were then incubated overnight at 4°C with the following antibodies (all purchased from Santa Cruz Biotech, CA, USA): rabbit polyclonal anti-TOPOIIα at 1:200 (sc-13058), mouse monoclonal anti-HSP90 at 1:200 (sc-69703), and mouse monoclonal anti-α tubulin at 1:200 (sc-58667). The membranes were then washed with PBST (PBS 0.1% Tween-20) three times, each for 10 min, and further incubated with the corresponding secondary antibodies: goat anti-mouse HRP at 1:10000 (sc-2005), and goat anti-rabbit HRP at 1:10000 (sc-2004) at room temperature for 1 h. The membranes were washed again with PBST three times. ECL Western Blotting Substrate (Pierce, IL, USA) was then added onto the membranes and which were exposed to Kodak x-ray films.
Statistics
Statistical significance for the growth inhibitory effects between regimens was determined by one-way ANOVA using Microsoft Office 2007 Excel Solver. Both α=0.05 and 0.01 were used for statistical analysis. Actual p-values are shown with corresponding results.
RESULTS
Synthesis of Block Copolymers
Detailed methods for characterization of block copolymers with hydrazide drug-binding block copolymers are reported elsewhere (27). Briefly, 1H-NMR measurements (DMSO-d6, 25°C) confirmed that the number of BLA repeating units in the polymer backbone PEG-PBLA (MW=20,064) was 38, which was calculated from the ratio between the integrated peak areas for PEG and benzyl groups at 3.5 ppm and 7.3 ppm, respectively. Eighty-four percent of benzyl esters were replaced by hydrazide groups. Drug loading yields were determined by UV-VIS colorimetric analysis at 485 nm and 337 nm for DOX and GDM-OH, respectively. Each block copolymer chain appeared to contain 13–14 drug molecules. The absence of unbound drugs was confirmed by separation of polymer fractions on Sephadex LH20 column. These results correspond well with our previous reports on DOX-loaded pH-sensitive polymeric micelles (28).
Preparation of pH-Sensitive Polymeric Micelles
Individual polymeric micelles (DM and GM) were readily prepared by dialyzing single drug-conjugated block copolymers against aqueous solutions. Reconstitution of the drug-polymer conjugates in water failed to prepare polymer micelles with homogeneous size distribution. In comparison, polymer micelles that were dialyzed and freeze-dried were successfully reconstituted in water, forming polymer micelles with identical particle size. Mixed micelles were prepared by mixing either different individual micelle solutions (AMM) or dialyzing drug-polymer conjugates dissolved in organic solvents in advance against water (OMM). As shown in Fig. 2, the particle size of DM, GM, AMM and OMM was clinically relevant to preferentially accumulate in tumor tissues (<200 nm). No time-dependent change in particle size was observed in DM and GM, which were incubated in capped sample vials with deionized water at room temperature for 24 h. These observations indicate that neither secondary aggregation nor precipitation of polymer micelles took place for GDM-OH as well as DOX. Mixed polymer micelles (AMM and OMM) also showed <100 nm particle size, yet the particle size slightly increased over time. Rearrangement of polymer chains between polymer micelles is implicated, although the possibility of intermicellar fusion forming onion-like structures cannot be excluded. In every case, results indicate that both individual and mixed polymeric micelles seem to sequester drug molecules in aqueous solutions effectively.
Fig. 2.

Average particle size of micelles after 0.5 h (a) and 24 h (b) incubation.
Drug Release Study
Figure 3 shows drug release patterns of individual micelles (DM and GM) at different pH conditions. DM shows favorable stability at pH 7.4, while it releases drugs preferentially at pH 5 (Fig. 3a). It is noticeable that GM was less stable than DM at pH 7.4, and its drug release was further accelerated at pH 5 (Fig. 3b). In comparison to individual micelles, OMM released more DOX at pH 7.4, while its DOX release at pH 5.0 was similar to that of DM (Fig. 4a). The release profile of GDM-OH from OMM was significantly suppressed at pH 7.4 in contrast to GM (Fig. 4b). Interestingly, GDM-OH release from OMM at pH 5.0 was accelerated. There was no significant difference between release patterns of AMM and individual micelles, suggesting that AMM could be a simple mixture of individual micelles solutions (data not shown).
Fig. 3.

Drug release patterns of DM (a) and GM (b) at pH 5 and 7.4 under sink conditions (37°C, n=3). DM and GM denote DOX-loaded and GDM-OH-loaded micelles, respectively.
Fig. 4.

Release of DOX (a) and GDM-OH (b) from OMM at pH 5 and 7 under sink conditions (37°C, n=3). OMM is an abbreviation of organic mixed micelles.
Cytotoxicity of DOX and GDM-OH as Free Drugs or Various Micelle Formulations
Figure 5 and Table 1 summarize in vitro cytotoxicity of DOX and GDM-OH against human breast cancer cell line MCF-7 in different drug formulations and schedules. The experiments were performed by three different researchers in triplicate (n=4).
Fig. 5.

Inhibitory concentrations for suppressing 50% viability (IC50) of MCF-7 cells in different schedules (mean±SD, n=4, triplicate experiments). Schedules for free drugs (or polymeric micelles) are described as follows: D(DM): D(DM) alone; G(GM): G(GM) alone; D/G(DM/GM): add D(DM) and G(GM) simultaneously; DG(DMGM): add D(DM) first and G(GM) after 24 h; GD(GMDM): add G(GM) first and D(DM) after 24 h; AMM: AMM alone; and OMM: OMM alone.
Table 1.
Median Inhibitory Concentrations (IC50) and P-values (α=0.05* and 0.01**)
| IC50±SD (nM) | Comparison Groups and P-Values | ||||
|---|---|---|---|---|---|
| Group A | GDM | GDM-OH | |||
| GDM | 178.58±45.37 | ||||
| GDM-OH | 721.08±215.85 | 0.0027*,** | |||
| GDM-Lev | 709.8±169.65 | 0.0009*,** | 0.9372 | ||
| Group B | DOX | GDM-OH | D/G | DG | |
| DOX | 117.51±25.04 | ||||
| GDM-OH | 721.08±215.85 | ||||
| D/G | 337.08±48.06 | 0.0002*,** | 0.0133* | ||
| DG | 425.68±33.91 | 0.0236* | |||
| GD | 1432.25±275.94 | 0.005*,** | 0.0063*,** | ||
| Group C | DM | GM | DM/GM | DMGM | |
| DM | 620.3±165.72 | ||||
| GM | 1236.5±191.99 | ||||
| DM/GM | 866.28±135.08 | 0.061 | 0.0197* | ||
| DMGM | 1030.7±244.66 | 0.2839 | |||
| GMDM | 914.55±127.38 | 0.6217 | 0.432 | ||
| Group D | AMM | ||||
| AMM | 866.28±135.08 | ||||
| OMM | 593.25±127.86 | 0.0261* | |||
Since therapeutic effects are dependent on drug activity as well as release patterns, biological activity of GDM-OH was first confirmed (Fig. 5a). GDM-OH appeared to show approximately four-folds lower activity than a parent drug GDM. There is no significant difference in drug activity between GDM-OH and GDM-Lev (Group A in Table 1). These results indicate that the introduction of amino ethanol to GDM reduced drug activity of the parent drug significantly, yet further chemical modification of GDM-OH with levulinic acid induced no notable adverse effects on the drug analogue. Therefore, GM is confirmed to release therapeutically active GDM analogues. No chemical modifications were made to DOX for the conjugation with block copolymers, since the drug possesses a ketone group at C13 position. Thus, combination drug treatment was subsequently made.
As shown in Fig. 5b, when cells were treated with DOX and GDM-OH formulated in DMSO, a significant schedule-dependent change in growth patterns was observed. DOX alone was the most potent among samples tested. Combined use of DOX with GDM-OH showed promising therapeutic effects, which changed schedule dependently. D/G, which indicates that both drugs are given concurrently, showed the lowest IC50 value compared to other combination settings (DG or GD). Interestingly, cells were more sensitive to the sequential drug combination where DOX was added first and GDM-OH was given after 24 h (DG). The changing of drug treatment sequence, adding GDM-OH before DOX, induced antagonistic drug action (GD). Group B in Table 1 shows that the schedule-dependent change in cytotoxicity among drug combination settings is statistically significant (p <0.05). Yet, data analysis at 1% significance level suggests that D/G and DG show no statistical significance, presumably due to relatively low activity of GDM-OH compared to DOX.
Figure 5c shows cytotoxic effects of polymeric micelles alone and in combination. Although IC50 values were variable, statistical analysis indicate that combination drug treatment using polymeric micelle formulations induced no significant difference in cell response (Group C in Table 1). It is also intriguing that cytotoxicity of mixed micelles (DM/GM) was similar to those of individual micelles alone (DM and GM) and in combination (DMGM and GMDM). These results imply that concurrent delivery of multiple drugs using polymeric micelles would be promising to achieve effective combination chemotherapy with the minimal change in cytotoxicity. However, it must be noticed that overall IC50 values of polymeric micelle formulations were five-to-ninefolds higher compared to DMSO formulations. Nevertheless, the prominent difference between formulations was found in GD and GMDM, indicating that adverse effects of GD were markedly reduced in polymeric micelle formulations (Table 1).
Figure 5d compares cytotoxicity between AMM and OMM. Statistical analysis for Group D in Table 1 (p<0.05) confirmed that OMM would be a more potent mixed micelle formulation than AMM that is prepared by simply mixing different polymeric micelle solutions at fixed drug concentration ratios. At the significance level at α=0.01, cytotoxic effects of AMM and OMM are statistically identical, showing that co-entrapment of multiple drugs into one polymeric micelle has no therapeutic advantages over a formulation using two different single drug-loaded polymeric micelles as a mixture. Therefore, more conservative estimation for the optimal mixed polymer micelle formulations would be that AMM and OMM show statistically identical therapeutic effects under our experimental conditions for in vitro cytotoxicity assays.
Expression Levels of TOPOII and HSP90
Mechanisms of cellular response to DOX and GDM co-treatment is subsequently investigated by measuring expression levels of TOPOIIα and HSP90 in MCF-7 cells exposed to drugs at various mixing ratios with concentration ranges where IC50 values were observed (Fig. 6). Expression levels of TOPOIIα were initially low at 10 nM, increased as drug concentrations (DOX and GDM) increased to 100 nM, and were completely suppressed in the presence of DOX alone or in combination with GDM at 1000 nM. GDM induced neither suppression nor complete removal of TOPOIIα alone. However, when cells were exposed to GDM along with DOX, TOPOIIα expression levels were significantly reduced and completely removed at 100 nM and 1000 nM, respectively. These results implicate that HSP90 inhibition seems to crucially influence the expression levels of TOPOIIα in the presence of DOX, lowering thresholds of TOPOIIα-mediated toxicity. To the contrary, expression of HSP90 showed no change under all experimental settings tested in this study. It is suggested that controlled release of multiple drugs from the micelles in the cells would have achieved a long-term effective combination therapy, and thus, drug combination using polymeric micelles appeared to be less schedule-dependent than drug treatment based on the conventional DMSO formulation.
Fig. 6.
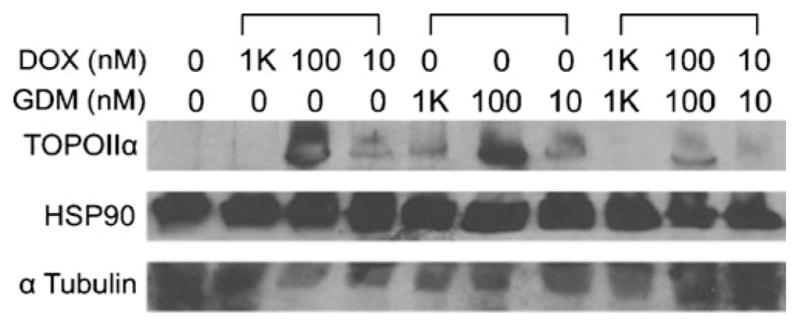
Expression levels of TOPOIIα and HSP90 at various drug combination settings.
DISCUSSION
Combination chemotherapy appears to be inevitable for cancer treatment to avoid undesirable side effects of toxic anticancer drugs and to sensitize drug-resistant cancers (29). From pharmaceutical aspects, co-administration of multiple drugs is challenging due to the toxicity of vehicles and variable pharmacokinetic profiles of each drug molecule in vivo. Therefore, development of drug infusion formulations for combination cancer chemotherapy is highly needed in order to use potent therapeutic agents developed in the laboratory for patients in bed side. Nevertheless, there are no safe, facile and versatile drug delivery systems available so far which can achieve concurrent delivery of multiple drugs to tumor tissues.
Based on these rationales, we prepared and tested pH-sensitive polymeric micelles that can potentially carry multiple drugs selectively to tumor tissues at the identical pharmacokinetic profile (Fig. 1). These micelles are capable of entrapping drug molecules through acid-labile hydrazone linkers that can remain stable at the physiological condition (pH 7.4) yet undergo hydrolysis degradation in the acidic environment such as intratumoral tissues (pH <7) and intracellular endosomal and lysosomal compartments (pH <6). Successful development of such polymeric micelles, therefore, would lead to tumor tissue-specific combination drug delivery for improved drug synergy and reduced systemic side effect. DOX and GDM were used in this research as model drugs, based on their drug action in the cell inducing TOPOII-mediated DNA damage and HSP90 inhibition (30,31).
Prepared polymeric micelles were characterized with unimodal size distributions having clinically relevant particle size (<100 nm) that enables the micelles to accumulate preferentially in tumor tissues (Fig. 2). Drug loading efficiency was fairly high (>31 wt% of polymer) for both DOX and GDM-OH. Drug-loaded micelles (DM and GM) showed pH-controlled drug release patterns (Fig. 3). Based on our previous achievements with DOX-loaded pH-sensitive polymeric micelles, we expect that these micelles would achieve tumor-specific delivery of DOX and GDM-OH in combination. However, unexpectedly rapid drug release at pH 7.4 from GM suggests that further optimization of hydrazone linkage between GDM-Lev and block copolymers might be necessary. Considering the fact that ester bond is generally more stable than hydrazone bond in acidic conditions, rapid drug release of GM at pH 7.4 implies that the hydrazone linkage between polymers and the acetyl group of GDM-Lev is unstable. The hydrazone linkage between GDM-Lev and block copolymers is composed of acetyl and hydrazide, which is different from the hydrazone bond consisting of 2-hydroxyethanone and hydrazide between DOX and block copolymers, respectively. Stability of the hydrazone bond is known to be dependent crucially on electron distributions of chemical structures (32,33). Although there is only a small difference between hydrazone linkages of DOX and GDM analogues, other factors (e.g. unreacted carboxyl acids and spacers for drug conjugation) might have influenced the stability of hydrazone bond in the micelle core. We hypothesize that the numbers of free carboxyl groups of the block copolymer backbone, drug-unbound hydrazide groups, and drug molecules would cooperatively determine drug release patterns from the micelles. This hypothesis is supported by altered drug release patterns of mixed micelles which were prepared from the same drug-polymer conjugates as shown in Fig. 4.
Stability of micelles would be beneficial to in vivo applications of mixed micelles, aiming for identical pharmacokinetics for multiple anticancer agents in the plasma and tumor tissues. In terms of feasibility in preparation, AMM is the simplest approach for the combination delivery of multiple drugs because DM and GM can be prepared separately and simply mixed together right before administration. To the contrary, OMM requires a time- and effort-consuming process. Although AMM and OMM might show different in vivo performance in terms of antitumor activity and biodistribution, it is plausible to conclude that mixed micelles would show no difference in size distribution and stability irrespective of distinct preparation process. These results exhibit that unstable GDM-OH conjugation might have been effectively protected in the core of OMM in the presence of DOX-conjugated block copolymers that prepare more stable micelle core, inducing favorable stability at pH 7.4 and drug release at pH 5. Suppressed GDM-OH release from OMM also evidences that DOX- and GDM-OH-conjugated polymers are mixing together to form a single micelle formation. Therefore, it is concluded that OMM formation is a facile and efficient way to protect unstable drug conjugate with another drug conjugate of higher stability with no fundamental or additional chemical redesigning of the carriers or drug analogues.
Subsequent in vitro cytotoxicity assays demonstrate that polymeric micelle formulations would achieve the least variable effects of combination chemotherapy (Fig. 5). Although the current research is focused on in vitro cytotoxicity evaluations, we expect that combination chemotherapeutic efficacy would be more significant in vivo, taking advantage of characteristic properties of polymer micelles such as pH-controlled drug release, prolonged blood circulation and tumor-specific drug delivery. Nevertheless, Table 1 indicates that combination effects of DOX and GDM-OH seem still controversial in terms of schedule-dependent cytotoxic effects. Thus, further investigation in animal models would provide a better understanding. Combined use of DOX and GDM-OH is based on molecular mechanism of each drug. Studies showed promising combination effects of DOX along with HSP90 inhibitors (34). Considering cell-protecting action of HSP90, we initially anticipated that the drug schedule in which cells were exposed to geldanamycin first and then DOX (GD) would be the most potent compared to other combination settings such as concurrent (D/G) or reversely scheduled (DG) drug action. Our results show that cells treated with DOX before HSP90 inhibitors are more sensitive to the drug combination.
It is of interest that polymeric micelles showed statistically identical cytotoxic effects irrespective of combination settings. These results are clearly different from DMSO formulations which showed a schedule-dependent change in drug efficacy. One possible explanation of this distinct therapeutic outcome of polymeric micelles is that drug release patterns of mixed polymeric micelles may retain drug concentration ratios where the maximal combination effects can be obtained over a prolonged time period. This explanation is supported by our Western blotting results that show expression levels of TOPOII and HSP90 with DOX and GDM alone or in combination. TOPOII has two isoforms, α and β. Anthracyclines including DOX are known to mainly stabilize complexes between DNA and TOPOII isoform α (TOPOIIα), which lead to TOPOII-mediated apoptosis. On the other hand, TOPOIIβ is more ubiquitious, while its intracellular functions are still largely unknown (35). For these reasons, our particular attention has been paid to TOPOIIα expression in this study to elucidate a potential mechanism for DOX/GDM combination effects. Figure 6 confirms that TOPOIIα expression in MCF-7 is fairly low at low drug concentration (10 nM). This is consistent with the fact that MCF-7 cells do not overexpress HER2, which is a surrogate marker of TOP-OII. Previous studies revealed that TOPOII expression is related to breast cancer response to anthracycline-based therapy. Higher TOPOII expression is generally linked to higher sensitivity of cancer to anthracycline drugs (36, 37). This is presumably why MCF-7 was insensitive to DOX at this concentration range. Noticeably, TOPOIIα expression levels increased as DOX dose increased (100 nM). This result is also corresponding well with previous findings that showed TOPOIIα is activated in response to genotoxic stress by drugs binding to DNA (38). At the highest concentration tested in this study (1000 nM), TOPOIIα was completely depleted as DOX activity increased, following the general mechanism of stabilization of TOPOIIα-DNA complexes by anthracycline drugs.
When it comes to cellular response to GDM, the change in TOPOIIα expression at 10 and 100 nM drug concentrations was similar to what was observed in DOX, yet TOPOIIα was still observed at 1000 nM of GDM. These results implicate that a different mechanism would have been involved if TOPOIIα expression increased in the presence of GDM. It is hypothesized that TOPOIIα was released from these TOPOIIα-HSP90 complexes, rather than newly produced in the cell, as GDM inhibits HSP90 at 100 nM (34). It is surprising that TOPOIIα expression levels decreased at high concentrations (1000 nM) where GDM is expected to inhibit HSP90 more effectively. This suggests that GDM is involved in neither degradation nor re-stabilization of TOPOIIα-HSP90 complexes directly but indirectly through other HSP90 client proteins. Although further investigations are necessary, our literature research suggests that the proapoptotic kinase protein kinase Cδ (PKCδ) might have been involved in lowering TOPOIIα expression levels as HSP90 inhibition proceeds. PKCδ is previously identified as one of the intracellular signal pathways that interact with TOPOIIα and increase its expression following DNA damage in an S-phase-specific manner (39). Inhibition of PKCδ appeared to attenuate DNA damage-induced activation of TOPOIIα. Subsequent studies showed that HSP90 forms complexes with not only TOPOIIα but also PKCδ. Therefore, HSP90 inhibition at high GDM concentrations could have induced aberrant PKCδ that eventually suppresses TOPOIIα expression.
Our observations clearly demonstrated that GDM alone fails to induce complete removal of TOPOIIα. To the contrary, TOPOIIα expression was significantly suppressed at 100 nM, and no TOPOIIα bands were observed at 1000 nM only when cells were exposed to DOX and GDM concurrently. These findings demonstrate DOX and GDM function cooperatively when used in combination. It is implicated that HSP90 inhibition may suppress production of new TOPOIIα, while DOX led to depletion of TOPOIIα, which is either already existing or released from TOPOIIα-HSP90 complexes, by stabilizing TOPOIIα-DNA complexes. Taken together, this intriguing link between HSP90 and TOPOIIα indicates that GDM seems to reduce the threshold of TOPOIIα-mediated apoptosis, while DOX stabilizes TOPOIIα-DNA complexes, facilitating consumption of its therapeutic target enzymes in the cell. Therefore, it is concluded that combination use of DOX and GDM would be effective to sensitize cancer cells to anthracycline-based cancer chemotherapy, while polymer micelle formulations can improve such combination efficacy. It must be emphasized again that polymer micelles that can continuously release both drugs in the cell for a prolonged time period were observed to show a less schedule-dependent change in DOX/GDM combination efficacy compared to free drug formulations.
In the meantime, HSP90 expression levels remained the same, irrespective of drug types or doses. These results are obviously different from TOPOIIα expression patterns that changed in a concentration-dependent manner for both DOX and GDM. Chemicals including anticancer drugs are known to induce heat shock response (40,41). Our observations also showed that GDM as well as DOX induced heat shock response, explaining why HSP90 expression levels did not decrease when cells were exposed to both drugs separately or concurrently. Therefore, it is expected that HSP90 inhibitors, other than GDM, that induce no heat shock response could lower the threshold of not only TOPOIIα but also HSP90 more effectively so that they can sensitize cancer cells. Such HSP90 inhibitors would improve combination efficacy with anthracycline drugs in the future. Importantly, the combination approach we proposed, inhibiting multiple therapeutic targets for a prolonged period of time with controlled drug delivery systems, can be exploited for drugs that target any of HSP90 client proteins other than TOPOII. In such a way, conventional therapeutic difficulties involved with cell-cycle schedule-dependent cell response to chemotherapy will be possibly overcome.
CONCLUSIONS
We have prepared mixed pH-sensitive polymeric micelles that would concurrently deliver multiple drugs for the simultaneously targeted delivery of HSP90 and TOPOII inhibitors to tumor tissues. The combination of mixed micelles results in high potency against MCF-7 breast cancer cells in comparison to the use of pH-sensitive polymeric micelle for a single drug (DOX or GDM-OH). The results generated in this study point to the promise of combining a HSP90 inhibitor and conventional anticancer drugs targeting one of HSP90 client proteins. We envision that co-inhibition of HSP90 and its client proteins is a promising strategy for selective suppression of tumor growth with lowered side-effects, reducing amounts of drugs used. In particular the study illustrates the ease with which combining multiple therapeutic agents can be accomplished as mixed polymeric micelles for combination chemotherapy, permitting drug co-solubilization, concurrent drug administration, and perhaps prolonged combination therapeutic efficacy selectively in tumors.
Acknowledgments
This work was partially supported by National Institutes of Health grant R01 AI-43346-08. Y.B. acknowledges research support provided by the Kentucky Lung Cancer Research Program.
Contributor Information
Younsoo Bae, Pharmaceutical Sciences, College of Pharmacy, University of Kentucky, 789 South Limestone, Lexington, Kentucky 40536-0596, USA.
Adam W. G. Alani, Division of Pharmaceutical Sciences, School of Pharmacy, University of Wisconsin-Madison, 777 Highland Avenue, Madison, Wisconsin 53705-2222, USA
Nicole C. Rockich, Division of Pharmaceutical Sciences, School of Pharmacy, University of Wisconsin-Madison, 777 Highland Avenue, Madison, Wisconsin 53705-2222, USA
T. S. Z. Chung Lai, Division of Pharmaceutical Sciences, School of Pharmacy, University of Wisconsin-Madison, 777 Highland Avenue, Madison, Wisconsin 53705-2222, USA
Glen S. Kwon, Division of Pharmaceutical Sciences, School of Pharmacy, University of Wisconsin-Madison, 777 Highland Avenue, Madison, Wisconsin 53705-2222, USA
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Smalley KSM, Haass NK, Brafford PA, Lioni M, Flaherty KT, Herlyn M. Multiple signaling pathways must be targeted to overcome drug resistance in cell lines derived from melanoma metastases. Mol Cancer Ther. 2006;5:1136–44. doi: 10.1158/1535-7163.MCT-06-0084. [DOI] [PubMed] [Google Scholar]
- 3.Scripture CD, Figg WD. Drug interactions in cancer therapy. Nat Rev Cancer. 2006;6:546–58. doi: 10.1038/nrc1887. [DOI] [PubMed] [Google Scholar]
- 4.Xu W, Neckers L. Targeting the molecular chaperone heat shock protein 90 provides a multifaceted effect on diverse cell signaling pathways of cancer cells. Clin Cancer Res. 2007;13:1625–9. doi: 10.1158/1078-0432.CCR-06-2966. [DOI] [PubMed] [Google Scholar]
- 5.Yao Q, Weigel B, Kersey J. Synergism between etoposide and 17-AAG in leukemia cells: critical roles for hsp90, FLT3, topoisomerase II, Chk1, and Rad51. Clin Cancer Res. 2007;13:1591–600. doi: 10.1158/1078-0432.CCR-06-1750. [DOI] [PubMed] [Google Scholar]
- 6.Beliakoff J, Whitesell L. Hsp90: an emerging target for breast cancer therapy. Anticancer drugs. 2004;15:651–62. doi: 10.1097/01.cad.0000136876.11928.be. [DOI] [PubMed] [Google Scholar]
- 7.Goetz MP, Toft DO, Ames MM, Erlichman C. The hsp90 chaperone complex as a novel target for cancer therapy. Ann Oncol. 2003;14:1169–76. doi: 10.1093/annonc/mdg316. [DOI] [PubMed] [Google Scholar]
- 8.Atkins JH, Gershell LJ. Selective anticancer drugs. Nat Rev Cancer. 2002;2:645–6. doi: 10.1038/nrd842. [DOI] [PubMed] [Google Scholar]
- 9.Hande KR. Clinical applications of anticancer drugs targeted to topoisomerase II. Biochim Biophys Acta. 1998;1400:173–84. doi: 10.1016/s0167-4781(98)00134-1. [DOI] [PubMed] [Google Scholar]
- 10.Senter PS, Kopecek J. Drug carriers in medicine and biology. Mol Pharm. 2004;1:395–8. doi: 10.1021/mp040010x. [DOI] [PubMed] [Google Scholar]
- 11.Duncan R. Polymer conjugates as anticancer nanomedicines. Nat Rev Cancer. 2006;6:688–701. doi: 10.1038/nrc1958. [DOI] [PubMed] [Google Scholar]
- 12.Vicent MJ, Greco F, Nicholson RI, Paul A, Griffiths PC, Duncan R. Polymer therapeutics designed for a combination therapy of hormone-dependent cancer. Angew Chem Int Ed. 2005;44:4061–6. doi: 10.1002/anie.200462960. [DOI] [PubMed] [Google Scholar]
- 13.Kopecek J, Kopeckova P, Minko T, Lu ZR. HPMA copolymer–anticancer drug conjugates: design, activity, and mechanism of action. Eur J Pharm Biopharm. 2000;50:61–81. doi: 10.1016/s0939-6411(00)00075-8. [DOI] [PubMed] [Google Scholar]
- 14.Bae Y, Kataoka K. Intelligent polymeric micelles from functional poly(ethylene glycol)-poly(amino acid) block copolymers. Adv Drug Deliver Rev. 2009;61:768–84. doi: 10.1016/j.addr.2009.04.016. [DOI] [PubMed] [Google Scholar]
- 15.Nishiyama N, Bae Y, Miyata K, Fukushima S, Kataoka K. Smart polymeric micelles for gene and drug delivery. Drug Discov Today: Technologies. 2005;2:21–6. doi: 10.1016/j.ddtec.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 16.Kataoka K, Kwon GS, Yokoyama M, Okano T, Sakurai Y. Block copolymer micelles as vehicles for drug delivery. J Control Release. 1993;24:119–32. [Google Scholar]
- 17.Bae Y, Diezi TA, Zhao A, Kwon GS. Mixed polymeric micelles for combination cancer chemotherapy through the concurrent delivery of multiple chemotherapeutic agents. J Control Release. 2007;122:324–30. doi: 10.1016/j.jconrel.2007.05.038. [DOI] [PubMed] [Google Scholar]
- 18.Forrest ML, Zhao A, Won CY, Malick AW, Kwon GS. Lipophilic prodrugs of hsp90 inhibitor geldanamycin for nanoencapsulation in poly(ethylene glycol)-b- poly(caprolactone) micelles. J Control Release. 2006;116:139–49. doi: 10.1016/j.jconrel.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 19.Bae Y, Buresh RA, Williamson TP, Chen THH, Furgeson DY. Intelligent biosynthetic nanobiomaterials for hyperthermic combination chemotherapy and thermal drug targeting of HSP90 inhibitor geldanamycin. J Control Release. 2007;122:16–23. doi: 10.1016/j.jconrel.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 20.Workman P, Powers MV. Chaperoning cell death: a critical dual role for hsp90 in small-cell lung cancer. Nat Chem Biol. 2007;3:455–7. doi: 10.1038/nchembio0807-455. [DOI] [PubMed] [Google Scholar]
- 21.Pick E, Kluger Y, Giltnane JM, Moeder C, Camp RL, Rimm DL, et al. High hsp90 expression is associated with decreased survival in breast cancer. Cancer Res. 2007;67:2932–7. doi: 10.1158/0008-5472.CAN-06-4511. [DOI] [PubMed] [Google Scholar]
- 22.Young JC, Moarefi I, Hartl FU. Hsp90: a specialized but essential protein-folding tool. J Cell Biol. 2001;154:267–73. doi: 10.1083/jcb.200104079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barker CR, McNamara AV, Rackstraw SA, Nelson DE, White MR, Watson AJM, et al. Inhibition of hsp90 acts synergistically with topoisomerase II poisons to increase the apoptotic killing of cells due to an increase in topoisomerase II mediated DNA damage. Nucleic Acids Res. 2006;34:1148–57. doi: 10.1093/nar/gkj516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bae Y, Jang WD, Nishiyama N, Fukushima S, Kataoka K. Multifunctional polymeric micelles with folate-mediated cancer cell targeting and pH-triggered drug releasing properties for active intracellular drug delivery. Mol BioSyst. 2005;1:242–50. doi: 10.1039/b500266d. [DOI] [PubMed] [Google Scholar]
- 25.Kasuya Y, Lu ZR, Kopeckova P, Minko T, Tabibi SE, Kopecek J. Synthesis and characterization of HPMA copolymer-aminopropylgeldanamycin conjugates. J Control Release. 2001;74:203–11. doi: 10.1016/s0168-3659(01)00318-2. [DOI] [PubMed] [Google Scholar]
- 26.DeBoer C, Meulman PA, Wnuk RJ, Peterson DH. Geldanamycin, a new antibiotic. J Antibiot. 1970;23:442–7. doi: 10.7164/antibiotics.23.442. [DOI] [PubMed] [Google Scholar]
- 27.Bae Y, Fukushima S, Harada A, Kataoka K. Design of environment-sensitive supramolecular assemblies for intracellular drug delivery: polymeric micelles that are responsive to intracellular pH change. Angew Chem Int Ed. 2003;42:4640–3. doi: 10.1002/anie.200250653. [DOI] [PubMed] [Google Scholar]
- 28.Bae Y, Nishiyama N, Fukushima S, Koyama H, Matsumura Y, Kataoka K. Preparation and biological characterization of polymeric micelle drug carriers with intracellular pH-triggered drug release property: Tumor permeability, controlled subcellular drug distribution, and enhanced in vivo antitumor efficacy. Bioconjugate Chem. 2005;16:122–30. doi: 10.1021/bc0498166. [DOI] [PubMed] [Google Scholar]
- 29.Xiao L, Rasouli P, Ruden DM. Possible effects of early treatments of hsp90 inhibitors on preventing the evolution of drug resistance to other anti-cancer drugs. Curr Med Chem. 2007;14:223–32. doi: 10.2174/092986707779313372. [DOI] [PubMed] [Google Scholar]
- 30.Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, et al. A high affinity conformation of hsp90 confers tumour selectivity on hsp90 inhibitors. Nature. 2003;425:357–9. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- 31.Fortune JM, Osheroff N. Topoisomerase II as a target for anticancer drugs: when enzymes stop being nice. Prog Nucleic Acid Res Mol Biol. 2000;64:221–53. doi: 10.1016/s0079-6603(00)64006-0. [DOI] [PubMed] [Google Scholar]
- 32.Alani AWG, Bae Y, Rao D, Kwon GS. Polymeric micelles for the pH-dependent controlled, continuous low dose release of paclitaxel. Biomaterials. 2010;31:1765–72. doi: 10.1016/j.biomaterials.2009.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.West KR, Otto S. Reversible covalent chemistry in drug delivery. Curr Drug Discov Technol. 2005;2:123–60. doi: 10.2174/1570163054866882. [DOI] [PubMed] [Google Scholar]
- 34.Barker C, Hamlett J, Pennington SR, Burrows F, Lundgren K, Lough R, et al. The topoisomerase II-hsp90 complex: a new chemotherapeutic target. Int J Cancer. 2006;118:2685–93. doi: 10.1002/ijc.21717. [DOI] [PubMed] [Google Scholar]
- 35.Kersting G, Tzvetkov MV, Huse K, Kulle B, Hafner V, Brockmoller J, et al. Topoisomerase II beta expression level correlates with doxorubicin-induced apoptosis in peripheral blood cells. N-S Arch Pharmacol. 2006;374:21–30. doi: 10.1007/s00210-006-0091-0. [DOI] [PubMed] [Google Scholar]
- 36.Martin-Richard M, Munoz M, Albanell J, Colomo L, Bellet M, Rey MJ, et al. Serial topoisomerase II expression in primary breast cancer and response to neoadjuvant anthracycline-based chemotherapy. Oncology. 2004;66:388–94. doi: 10.1159/000079487. [DOI] [PubMed] [Google Scholar]
- 37.Burgess D, Doles J, Zender L, Xue W, Ma B, McCombie W, et al. Topoisomerase levels determine chemotherapy response in vitro and in vivo. PNAS. 2008;105:9053–8. doi: 10.1073/pnas.0803513105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chang JT, Lu Y-C, Chen Y-J, Tseng C-P, Chen Y-L, Fang C-W, et al. hTERT phosphorylation by PKC is essential for telomerase holoprotein integrity and enzyme activity in head neck cancer cells. Brit J Cancer. 2006;94:870–8. doi: 10.1038/sj.bjc.6603008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoshida K, Yamaguchi T, Shinagawa H, Taira N, Nakayama KI, Miki Y. Protein kinase C δ activates topoisomerase IIδ to induce apoptotic cell death in response to DNA damage. Mol Cell Biol. 2006;26:3414–31. doi: 10.1128/MCB.26.9.3414-3431.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Powers MV, Workman P. Inhibitors of the heat shock response: biology and pharmacology. FEBS Lett. 2007;581:3758–69. doi: 10.1016/j.febslet.2007.05.040. [DOI] [PubMed] [Google Scholar]
- 41.Biship SC, Burlison JA, Blagg BSJ. Hsp90: a novel target for the disruption of multiple signaling cascades. Curr Cancer Drug Tar. 2007;7:369–88. doi: 10.2174/156800907780809778. [DOI] [PubMed] [Google Scholar]