

Abstract
Brain biopsy in patients presenting with subacute encephalopathyis never straightforward and only undertaken when a ‘treatable condition’ is a realistic possibility.
This 63 year old right handed, immunocompetent Caucasian woman presented with an 8 month history of rapidly progressive right-sided hearing impairment, a 4 month history of intermittent headaches, tinnitus, ‘dizziness’, dysphagia, nausea and vomiting, with the subsequent evolution of progressive gait ataxia and a subacute global encephalopathy. The possibility of CJD was raised. Brain biopsy was carried out. Western blot for prion protein was negative. She died 9 days later and autopsy brain examination confirmed widespread subacute infarction due to an EBV positive atypical NK/T-cell infiltrate with positivity for CD3, CD56, granzyme B, perforin and EBER with absence of CD4, CD5 and CD8 expression. Molecular studies for T-cell clonality were attempted but failed due to insufficient DNA quality. Serology was consistent with past EBV infection (EBV VCA and EBNA IgG Positive). There was no evidence of disease outside the CNS. Primary central nervous system NK/T-cell lymphoma is extremely rare. The rare reported cases all present with a discrete intracranial mass, unlike the diffuse infiltrative pattern in this case. Whilst the diffuse interstitial pattern is reminiscent of chronic active EBV infection (CAEBV) seen in other organ systems such as the liver and bone marrow, the clinical presentation and epidemiologic profile are not typical for CAEBV.
1. Introduction
Rapidly progressive dementia (RPD) is typically acute to subacute in onset over days to weeks with a rate of progression that is faster than one would expect in the more common neurodegenerative conditions [1]. The differential diagnosis often includes CJD, with varying degrees of clinical likelihood ranging from extremely unlikely to virtually certain [2]. The commonest malignancies which may masquerade as prion disease are CNS lymphoma and intravascular lymphoma, but both are rare and very challenging to diagnose without brain biopsy [2].
Primary CNS lymphomas (PCNSL) are rare, accounting for only 2–6% of all primary brain tumours. They are almost always Non-Hodgkin in type and B-cell (98%) in origin [3]. Primary CNS diffuse large B-cell lymphoma may be EBV-positive, particularly in the setting of HIV infection [4]. EBV infection results in a spectrum of disease with the host’s immune response playing a key role in shaping the clinical manifestations [5]. Infectious mononucleosis, the proteotypical infection is usually acquired orally with the virus infecting epithelial cells and B-cells with a T-cell response. Following primary infection EBV, like all herpes virus, establishes persistent latent infection for the lifetime of the host. Rare individuals infected with EBV may present with chronic active EBV (CAEBV) with persistent or recurring infectious mononucleosis (IM) like symptoms including fever, hepatosplenomegaly, lymphadenopathy and high EBV- DNA load in the peripheral blood [6,7].
EBV may also infect T-cells and NK-cells but in a much less efficient manner than it’s targeting of B-cells. The precise mechanism by which EBV induces T-cell or NK-cell proliferation is unknown [7]. T-cell PCNL are much rarer than B-cell lymphoma and account for < 5% of all PCNSL’s [3]. Most primary CNS T-cell lymphomas are peripheral T cell lymphoma not otherwise specified (PTCL, NOS). NK/T -cell lymphoma is extremely rare in Western countries and usually nasal in location with only six cases previously reported in the CNS [8–13]. The majority of these NK/T-cell lymphomas have occurred in immunocompetent hosts, and by definition are EBV-related. MRI finding are variable, but previously reported cases have presented with a discrete mass unlike the diffuse infiltrative pattern seen in this case.
2. Case history
This 63 year old right handed lady woman had a background history of hyperlipidaemia, recurrent sinusitis, presumed benign paroxysmal positional vertigo, cholecystectomy, and a lumbar micro-discectomy, followed by left upper thoracic shingles treated with oral ‘anti-viral medications’. She subsequently developed rapidly progressive, asymmetrical hearing impairment over one month, approximately 8 months prior to transfer to our hospital, with troublesome post-herpetic neuralgia, but she was able to return to work. Four months prior to transfer, she reported a persistent sensation of ‘dizziness’, intermittent aching headaches, nausea, vomiting, tinnitus, dysphagia for solids, reduced appetite and unquantified weight loss. She deteriorated 3 months prior to transfer, with progressive slowing of speech and ataxia over 2 weeks; she rested in bed over the following 6 weeks and required the assistance of two people to mobilise. One month before transfer, she deteriorated further with progressive dysarthria, she was speaking out of context, with reduced memory for recent events and right facial weakness. Over the following weeks, she became more withdrawn, drowsy, globally encephalopathic, incontinent and she was unable to feed herself. She was sedated to facilitate a brain MRI and CSF examination at the referring hospital, but her GCS dropped to 6/15 necessitating intubation, sedation and ventilation and remained between 3 and 7/15 thereafter. Her family noted 2 very brief myoclonic types leg jerks for a second, with no other myoclonus.
Examination on admission to our university teaching hospital revealed a GCS of 7/15 whilst sedated. Funduscopy did not reveal papilledema. Pupils were asymmetric, sluggishly reactive to light, with disconjugate eye movements at rest and in response to oculocephalic reflex testing. Corneal reflexes were reduced, and she only coughed on deep suctioning. Cranial nerve examination was otherwise impossible. She had asymmetric limb spasticity, localised to painful stimuli, with asymmetric brisk reflexes. Plantar responses were extensor on the right and flexor on the left. Assessment of sensation was not possible.
Laboratory investigations revealed a normocytic anaemia with a haemoglobin of 8.8 g/dl (normal > 11.5), a neutrophilia of 8.1 × 109/L (normal < 7.5), lymphopenia of 0.5 × 109/L (normal > 1.5), with normal haematological investigations otherwise. Sodium was reduced to 128 mmol/L (normal > 135), with albumin 25 g/L (normal > 35), Gamma GT 256 IU/L (normal < 40), alanine aminotransferase 118 IU/L (normal < 35), but normal renal and liver profiles and CRP otherwise. She had biochemical features consistent with a syndrome of inappropriate ADH secretion, LDH was mildly elevated at 243–278 IU/L (normal < 220), with ferritin levels of 1773 ng/ml (normal < 200) attributed an acute phase response. ANA, ANCA, rheumatoid factor, anti-NMDA receptor, anti-voltage gated potassium channel complex (anti-VGKC), anti-voltage gated calcium channel, and anti-Hu, -Yo, Ri, Ma 1, Ma 2, CV2/CRMP5, amphiphysin, Sox-1, Zic-4 and Tr antibodies were negative. Serology was also negative for HIV, hepatitis B, hepatitis C, treponema pallidum, borrelia burgdorferi, cryptococcal antigen, and positive for measles IgG Serology was negative for EBV IgM, but positive for EBV VCA IgG and EBV IgG EBV Cerebrospinal fluid (CSF) examination was repeated after transfer to our hospital after the patient had received a 5 day course of 1 g daily of intravenous methylprednisolone followed by 60 mg of prednisolone daily. This revealed a CSF white cell count of 5, 3, 13 (99% mononuclear cells and b1% polymophonuclear cells), CSF red cell count of 54, 61, 56; CSF protein of 35 mg/dl; CSF glucose of 3.2 mmol/L with simultaneous plasma glucose of 6.1 mmol/L. CSF gram stain was negative, and CSF microscopy and culture were negative for mycobacteria. CSF oligoclonal bands and CSF anti-Hu, Yo, Ri,-VGKC complex, and anti-NMDA receptor antibodies were not detected. CSF cytology showed lymphocytes but no neoplastic cells. CSF PCR for HSV I and II, VZV, JC virus DNA, and enterovirus RNA was negative. CSF PCR for EBV DNA was positive with 93,988 copies/ml.
Axial FLAIR and T2-weighted MRI of brain two months prior to transfer showed bilateral frontal and parietal periventricular, deep white matter, and subcortical hyperintensity, with high signal change in the peri-insular cortices, right temporal cortex, left outer thalamus, left thalamo-capsular junction, and both middle cerebellar peduncles. DWI sequence was normal. Gradient echo sequence did not show any microhaemorrhages. Serial MR imaging of brain showed the multifocal hyperintense changes in the above areas becoming more confluent on FLAIR and T2-weighted sequences, mainly involving the subcortical and deep white matter but also extending towards the cortex, and subsequently involving the mesial temporal lobes, anterior occipital lobes, right caudate nucleus, right thalamus and extending into and swelling the midbrain and the pons bilaterally (Fig. 1). DWI now showed some patchy diffusion restriction. T1-weighted MRI without gadolinium showed isointense to some hypo-intense signal change, without any enhancement following administration of gadolinium.
Fig. 1.

FLAIR axial images (A–D) and T2 axial images (E–H) demonstrate a diffuse infiltrative process involving parasaggital, lateral frontal, insular, and anterior occipital cortices, midbrain, and pons. T1 post gadolinium, diffusion, and MR angiography sequences (not shown) were normal.
CT of thorax, abdomen and pelvis revealed pneumonia, pleural effusions but no mass lesions, pathological lymphadenopathy or hepatosplenomegaly.
She was referred to the National Neurosurgical Department at Beaumont Hospital where she underwent a right frontal brain biopsy. She died 9 days later.
3. Results
Western blot for prion protein was negative on the biopsy sample. The biopsy showed a diffuse parenchymal infiltrate composed of predominantly medium sized lymphoid cells with mild to moderate cytologic atypia.
Examination of the brain at autopsy showed it to be oedematous but not enlarged weighing 1490 g. Flattening of the gyri and narrowing of the sulci were noted but there was no evidence of herniation. The leptomeningeal coverings were clear apart from an area of haemorrhage related to the biopsy (Fig. 2).
Fig. 2.

Macroscopic examination of the brain showing flattening of the sulci and narrowing of the gyri together with a focal area of haemorrhage related to biopsy.
Sectioning the brain showed no discrete lesions (Fig. 3). General autopsy examination showed no evidence of disease in the paranasal sinuses, skin, bone marrow, liver or spleen.
Fig.3.
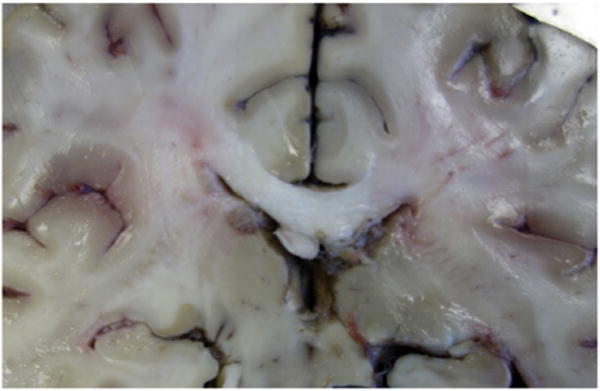
Coronal section of the brain at the level of the hippocampi, with no evidence of mass lesions.
The autopsy brain was sampled extensively and all areas (cortex, subcortical white matter, basal ganglia, thalamus and brainstem) showed a similar appearance with a diffuse interstitial atypical lymphoid infiltrate (Fig. 4). Despite her initial presentation with vertigo, followed by subsequent asymmetrical hearing impairment, dizziness, tinnitus, dysphagia, dysarthria and ataxia, there was no evidence of more extensive tumour burden in the brain stem. There was a slight accentuation of the infiltrate near the perivascular and subpial regions. The infiltrate was composed of mostly medium-sized lymphoid cells with slightly irregular nuclei and clumped chromatin. Fewer admixed small lymphocytes were also present. Widespread small areas of subacute infarction were present. Discrete tumour mass or perivascular infiltration was not identified. Neither was there any evidence of microglial nodules, neuronophagia, or vasculitis.
Fig. 4.
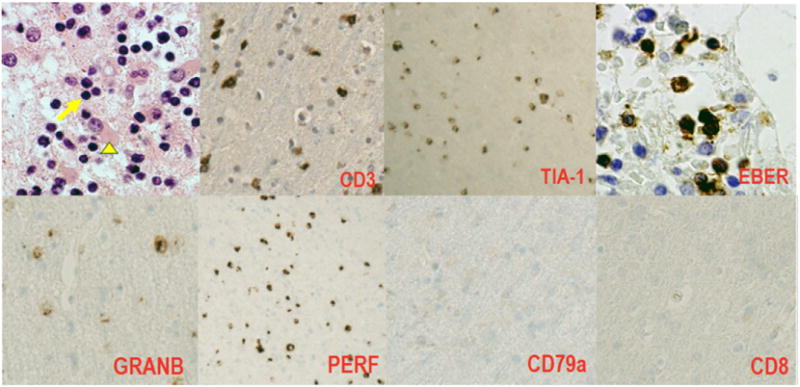
Right frontal brain biopsy: Parenchymal Infiltrate of medium sized lymphoid cells with mild to moderate cytologic atypia (arrow) with reactive astrocytosis (arrowhead). No evidence of microglial nodules, neuronophagia, vasculitis, or necrosis to suggest an encephalitic process, H&E, 40×. Immunohistochemistry demonstrates the atypical lymphoid cells are of NK derivation (CD3+, TIA-1+, EBV+, Granzyme B+ Perforin+, CD79a− CD8−).
The atypical cells showed a phenotype of NK cells with positivity for CD3, CD56, granzyme B, perforin and EBER with absence of CD4, CD5 and CD8 expression (Fig. 2). There were rare scattered B-cells on CD20 and PAX-5 stainingon the autopsy material. There were no B-cells demonstratedon CD20 and CD79a stainingon the ante-mortem biopsy. Molecular studies for T-cell clonality were attempted but failed due to insufficient DNA quality. EBV was positive in nearly all cells within the atypical cellular infiltrate. Serology was consistent with past EBV infection (EBV VCA & EBNA IgG Positive). On the basisof thesefindinga neuropathological diagnosis of an NK/T-cell lymphoproliferative disorder was established.
4. Discussion
This case illustrates a number of very interesting clinical and neuroimaging features, and highlights the challenges of establishing a definite clinical and neuropathological diagnosis of NK/T-cell lymphoproliferative disease of the CNS. The development of fulminant NK/T-cell lymphoproliferative disease of NK cell-derivation in an adult, immunocompetent woman is very rare. Only one previous case has been described in a female [13]; the remainder have been in males [8– 12]. Serological testing was consistent with prior EBV infection, but CSF findings revealed active EBV DNA replication within the CNS, thus highlighting the importance of CSF PCR testing for EBV in patients with suspected primary CNS lymphoma. Unlike prior reported cases in the literature, she did not have a discrete mass lesion on MRI of brain. The diffuse CNS involvement accounted for her global encephalopathy with good clinical-radiological-pathological correlation, with no imaging or pathological evidence of disease outside the CNS.
Her subacute presentation raised the remote possibility of CJD. From a practical viewpoint, when CJD is mentioned in the differential diagnosis of a subacute encephalopathy both infection control and diagnostic issues arise [14,15]. These include taking a biopsy sample for western blot analysis of prion protein, and disposal or quarantining of all instruments until the diagnosis has been positively excluded [16]. Image guidance, often used in brain biopsies to improve diagnostic yield is not possible as none of our currently available image guidance systems are disposable. Therefore the standard approach by the neurosurgeon in these cases, as in this case, is often a large biopsy of non dominant frontal lobe [17]. This can prove a diagnostic challenge for the reporting pathologist, compounded in this case because the patient had been on intravenous and oral steroids prior to biopsy which are known to have a lytic effect on lymphocytes [18]. This made interpretation of the biopsy and autopsy tissue more difficult and may have exacerbated our inability to obtain material for clonality studies.
Primary infection with EBV usually occurs in early childhood and is typically asymptomatic. Adolescents and young adults may develop infectious mononucleosis which is usually benign and self limiting, characterised by a polyclonal B-cell response with control of the infection mediated by cytotoxic T-cells. Sustained T-cell infection by EBV occurs only rarely raising the possibility that T-cell infection and proliferation are due in part to defective immune surveillance [19]. In immunocompetent individuals the development of an acute T-cell LPD suggests a specific vulnerability to EBV, which appears to be more prevalent in individuals of Asian descent. An acute T-cell LPD has been described in the setting of acute EBV infection and less commonly in CAEBV infection and it has been suggested that both the acute and chronic EBV related cases appear to share a defect in immune surveillance [19].
CAEBV infection is characterised by clonal expansion of EBV infected T cells or NK cells in individuals without apparent immunodeficiency [6,7]. The geographic distribution is uneven, with most cases described in Japan and other East Asian countries It occurs when the virus affects T-cells or NK cells, with the T-cell phenotype carrying a less favourable prognosis [7]. It is characterised by persistent or recurrent infectious mononucleosis like symptoms, an unusual pattern of anti EBV antibodies with raised anti- VCA or anti EA and a chronic illness that cannot be explained by other disease processes [6]. This is not the case here. Although CAEBV has been described in the CNS this was part of a widespread disease process. Among the T-cell lymphomas associated with EBV are some that have occurred in the context of CAEBV infection [19,20].
Diagnosis of T-cell/NK-cell lymphoproliferative disorders can be challenging pathologically as the neoplastic cells are small to medium in size. The diagnosis was particularly difficult in this case because apart from subacute infarction and gliosis, the other helpful features such as a mass lesion and perivascular cuffing were absent. Most primary CNS mature T and NK-cell lymphomas are PTCL NOS, with EBV-positive NK/T-cell lymphoma being extremely rare [3]. Of the 6 previously reported cases all have been highly aggressive, presenting with discrete intracranial masses (87% supratentorial). Histologically the most common phenotype has been CD2+, surface CD3 −, cytoplasmic CD3+ and CD56+ and EBV positivity as in this case. Fewer than 3% of cases of primary nasal NKTCL have secondary involvement of the CNS [12,21,22]. In this case there was no evidence of disease outside the CNS on detailed radiological and autopsy examination.
In conclusion, this interesting case adds to the very limited literature on this topic, and illustrates that a fulminant, diffuse EBV- driven NK/T-cell neoplasm may occur in the CNS alone in an immunocompetent, adult Caucasian woman with serological evidence of prior EBV infection, but with active EBV DNA replication within the CNS. Although it is not possible to prove clonality in this NK-cell derived process, the aggressive and ultimately fatal clinical course and diffusely infiltrative nature of the disease support a malignant derivation.
Abbreviations
- EBV VCA
Epstein Barr virus viral capsular antigen
- EBV NA
nuclear antigen
- CAEBV
chronic active EBV infection
- LPD
lymphoproliferative disease
- PCNSL
primary CNS lymphoma
References
- 1.Bucelli RC, Ances BM. Diagnosis and evaluation of a patient with rapidly progressive dementia. Mo Med. 2013;110:422–428. [PMC free article] [PubMed] [Google Scholar]
- 2.Murray K. Creutzfeldt-Jakob disease mimics or how to sort out the subacute encephalopathy patient. Pract Neuriol. 2011;11:19–28. doi: 10.1136/jnnp.2010.235721. [DOI] [PubMed] [Google Scholar]
- 3.Menon MP, Nicolae A, Meeker H, et al. Primary CNS T-cell lymphomas: a clinical, morphologic, immunophenotypic, and molecular analysis. Am J Surg Pathol. 2015;39:1719–1729. doi: 10.1097/PAS.0000000000000503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oyama T, Ichimura K, Suzuki R, et al. Senile EBV+ B-cell lymphoproliferative disorders: a clinicopathologic study of 22 patients. Am J Surg Pathol. 2003 Jan;27:16–26. doi: 10.1097/00000478-200301000-00003. [DOI] [PubMed] [Google Scholar]
- 5.Jenson HB. Epstein-Barr virus. Pediatr Rev. 2011;32:375–383. doi: 10.1542/pir.32-9-375. [DOI] [PubMed] [Google Scholar]
- 6.Okano M, Kawa K, Kimura H, et al. Proposed guidelines for diagnosing chronic active Epstein-Barr virus infection. Am J Hematol. 2005;80:64–69. doi: 10.1002/ajh.20398. [DOI] [PubMed] [Google Scholar]
- 7.Fujiwara S, Kimura H, Imadome K, et al. Current research on chronic active Epstein-Barr virus infection in Japan. Pediatr Int. 2014;56:159–166. doi: 10.1111/ped.12314. [DOI] [PubMed] [Google Scholar]
- 8.Ng SB, Lai KW, Murugaya S, et al. Nasal-type extranodal natural killer/T-cell lymphomas: a clinicopathologic and genotypic study of 42 cases in Singapore. Mod Pathol. 2004;17:1097–1107. doi: 10.1038/modpathol.3800157. [DOI] [PubMed] [Google Scholar]
- 9.Kaluza V, Rao DS, Said JW, et al. Primary extranodal nasal-type natural killer/T-cell lymphoma of the brain: a case report. Hum Pathol. 2006;37:769–772. doi: 10.1016/j.humpath.2006.01.032. [DOI] [PubMed] [Google Scholar]
- 10.Cobo F, Talavera P, Busquier H, et al. NK/T-cell brain lymphoma associated with Epstein-Barr virus in a patient with AIDS. Neuropathology. 2007;27:396–402. doi: 10.1111/j.1440-1789.2007.00784.x. [DOI] [PubMed] [Google Scholar]
- 11.Liu JK, Sayama C, Chin SS, Couldwell WT, et al. Extranodal NK/T-cell lymphoma presenting as a pituitary mass. Case report and review of the literature. J Neurosurg. 2007;107:660–665. doi: 10.3171/JNS-07/09/0660. [DOI] [PubMed] [Google Scholar]
- 12.Guan H, Huang Y, Wen W, et al. Primary central nervous system extranodal NK/T-cell lymphoma, nasal type: case report and review of the literature. J Neuro-Oncol. 2011;103:387–391. doi: 10.1007/s11060-010-0384-5. [DOI] [PubMed] [Google Scholar]
- 13.Prajapati HJ, Vincentelli C, Hwang SN, et al. Primary CNS natural killer/T-cell lymphoma of the nasal type presenting in a woman: case report and review of the literature. Clin Oncol. 2014;32:e26–e29. doi: 10.1200/JCO.2012.47.6796. [DOI] [PubMed] [Google Scholar]
- 14.Brown P, Farrell M. A practical approach to avoiding iatrogenic Creutzfeldt-Jakob disease (CJD) from invasive instruments. Infect Control Hosp Epidemiol. 2015;36:844–848. doi: 10.1017/ice.2015.53. [DOI] [PubMed] [Google Scholar]
- 15.Loftus T, Chen D, Looby S, et al. CJD surveillance in the Republic of Ireland from 2005–2015- a suggested algorithm for referrals. Clin Neuropathol Mar. 23 doi: 10.5414/NP301016. [DOI] [PubMed] [Google Scholar]
- 16.http://www.hse.gov.uk/aboutus/meetings/committees/acdp.
- 17.Schott JM, Reiniger L, Thom M, et al. Brain biopsy in dementia: clinical indications and diagnostic approach. Acta Neuropathol. 2010;120:327–341. doi: 10.1007/s00401-010-0721-y. [DOI] [PubMed] [Google Scholar]
- 18.Weller M. Glucocorticoid treatment of primary CNS lymphoma. J Neuro-Oncol. 1999;43:237–239. doi: 10.1023/a:1006254518848. [DOI] [PubMed] [Google Scholar]
- 19.Quintanilla-Martinez L, Kumar S, Fend F, et al. Fulminant EBV+ T-cell lymphoproliferative disorder following acute/chronic EBV infection: a distinct clinicopathologic syndrome. Blood. 2009;96:443–451. [PubMed] [Google Scholar]
- 20.Ohshima K, Kimura H, Yoshino T, CAEBV Study Group et al. Proposed categorization of pathological states of EBV-associated T/natural killer-cell lymphoproliferative disorder (LPD) in children and young adults: overlap with chronic active EBV infection and infantile fulminant EBV T-LPD. Pathol Int. 2008;8:209–217. doi: 10.1111/j.1440-1827.2008.02213.x. [DOI] [PubMed] [Google Scholar]
- 21.Luther N, Greenfield JP, Chadburn A, et al. Intracranial nasal natural killer/T-cell lymphoma: immunopathologically-confirmed case and review of literature. J Neuro-Oncol. 2005;75:185–188. doi: 10.1007/s11060-005-1862-z. [DOI] [PubMed] [Google Scholar]
- 22.Kim SJ, SY Oh, Hong JY, et al. When do we need central nervous system prophylaxis in patients with extranodal NK/T-cell lymphoma, nasal type? Ann Oncol. 2010;21:1058–1063. doi: 10.1093/annonc/mdp412. [DOI] [PubMed] [Google Scholar]