

Abstract
The impermeability of the plasma membrane towards large, hydrophilic biomolecules is a major obstacle in their use and development against intracellular targets. To overcome such limitations, protein transduction domains (PTDs) have been used as protein carriers, however they often require covalent fusion to the protein for efficient delivery. In an effort to develop more efficient and versatile biological vehicles, a series of PTD-inspired polyoxanorbornene-based synthetic mimics with identical chemical compositions but different hydrophobic/hydrophilic segregation were used to investigate the role of sequence segregation on protein binding and uptake into Jurkat T cells and HEK293Ts. This series was composed of a strongly segregated block copolymer, an intermediately segregated gradient copolymer, and a non-segregated homopolymer. To assess how protein isoelectric point, the study was extended to other proteins (bovine serum albumin, avidin, and streptavidin). Among the series, the block copolymer maximized both protein binding and translocation efficiencies, closely followed by the gradient copolymer, resulting in two protein transporter molecules more efficacious than currently commercially available agents. These two polymers were also used to deliver the biologically active Cre recombinase into a loxP-reporter T cell line. Since exogenous Cre must reach the nucleus and retain its activity to induce gene recombination, this in vitro experiment better exemplifies the broad applicability of this synthetic system. This study shows that increasing segregation between hydrophobic and cationic moieties in these polymeric mimics improves non-covalent protein delivery, providing crucial design parameters for the creation of more potent biological delivery agents for research and biomedical applications.
Keywords: Cell penetrating peptides, protein transduction domain mimics, sequence segregation, non-covalent protein delivery, protein binding, Pep-1
Graphical Abstract
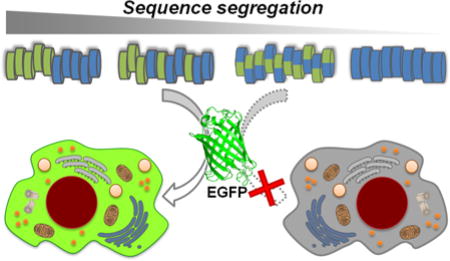
Increased hydrophobic/hydrophilic segregation in a class of polymeric mimics of protein transduction domains generates more efficient vehicles for non-covalent protein delivery.
1. Introduction
Intracellular delivery of bioactive proteins constitutes a convenient and valuable alternative to DNA and siRNA transfections. The large number of recombinant proteins currently available offer many opportunities to study cellular mechanisms and to discover new therapeutic candidates for intracellular targets.1,2 The various intracellular compartments, however, are often inaccessible to many exogenous biomolecules. The low permeability of the cell membrane and endosomal entrapment generally limit intracellular availability, thus posing major challenges to their wide-spread use. As a consequence, there has been rising interest in the development of more efficient delivery systems for mammalian cells capable of overcoming such challenges. Among the different strategies employed in the last few decades, the use of protein transduction domains (PTDs) is one of the most promising solutions.3,4
PTDs are a class of short peptides, mostly cationic, that can traverse the cell membrane. Although their exact mechanism of entry has not been fully elucidated,5 they have been shown to transport a wide range of biomolecules (plasmid DNA, siRNA, proteins, and peptides) into a variety of cell types and in vivo models.6–9 In the majority of cases PTDs, like TAT, require covalent attachment to their cargo for protein delivery.10 Although covalent conjugation offers stability advantages for in vivo applications, it presents several limitations including the risk of altering the biological activity of the cargo.11 Additionally, they can be time consuming and experimentally restrictive12 since every new cargo needs to be developed into a fusion protein to be used. To this end, a non-covalent protein delivery strategy is often preferred, in which the cargo is delivered as part of a supramolecular complex with the PTD.13,14 This non-covalent strategy was first introduced by Heitz, Divita, and coworkers who developed a short primary amphiphilic peptide, Pep-1, able to form stable nanoparticles with proteins.15 Pep-1 is a chimera obtained by the fusion of a hydrophilic translocation moiety, the nuclear localization sequence (NLS) of the SV40 large T-antigen (KKKRKV), to a tryptophan-rich hydrophobic motif through a short linker.16 The NLS is required for solubility and intracellular trafficking, while the tryptophan-rich domain promotes both protein complexation and membrane interactions. Although Pep-1 has been used in a variety of cell types and has become a popular commercially available agent for protein delivery (Chariot™ by Active Motif, Inc.), challenges still remain with generally hard to transfect cell types such as primary and suspension cells. Thus, there is still a real need for more effective transporters that work well against the most challenging cell types. Following the discovery that peptide chirality and H-bonding is not critical for delivery,17,18 several research groups have developed synthetic abiotic oligomers to mimic PTD function by incorporating key functional groups onto alternative backbones.19–22 This provides access to a much larger number of synthetic structures, compared to peptides, and the study of their activity will allow for the creation of new, more effective transporters.
In this context, we previously developed a highly efficient set of synthetic PTD mimics (PTDMs) using the polyoxanorbornene backbone,23 that combined the essential characteristics of Pep-1 and TAT. Similar to the chimeric Pep-1, they contain hydrophobic and cationic domains in a sequential arrangement (block copolymers). The cationic segment is guanidinium-rich, thus mimicking TAT or polyarginine. The hydrophobic region is composed of phenyl-functionalized repeat units, which have proved to impart better cell penetrating properties with respect to other aromatic or aliphatic groups.24,25 We have exploited these PTDMs for the intracellular delivery of siRNA and proteins.25,26 Recently, we investigated the correlation between hydrophobic content and protein delivery efficiency of these PTDMs.25,27 Now, in this paper, we use constitutional macromolecular isomers28 to determine the role of hydrophobic/hydrophilic segregation in non-covalent protein delivery.
We first compared PTDMs of identical chemical compositions but different degrees of hydrophobic/hydrophilic segregation (Figure 1b) based on their ability to interact with and deliver enhanced green fluorescent protein (EGFP) into Jurkat T cells, a non-trivial suspension human leukemic T cell line. PTDM-mediated cellular protein delivery efficiency was also compared with popular commercially available agents. Additionally, HEK293T cells were chosen as a representative adherent cell type, in contrast to the suspension Jurkat T cells. Bovine serum albumin (BSA), avidin, and a recombinant neutral form of streptavidin were also tested to evaluate the influence of protein isoelectric point (pI). Furthermore, the ability of our PTDMs to deliver biologically active proteins was demonstrated with the enzyme Cre recombinase, thus confirming the potential of these novel molecules as protein transporters for biological applications. The diversity of cell lines and cargoes used highlights the versatility of this approach. By exploring how sequence segregation impacts PTDM’s non-covalent protein binding and delivery efficiencies, the importance of this structural design property for the development of more potent delivery solutions was determined.
Figure 1. PTDMs series used in this study.

a) PTDM 1 chemical structure; b) cartoon of PTDM sequence variation analyzed in this study. Hydrophobic (phenyl-containing) units are represented in green and cationic (guanidinium-containing) groups are represented in blue.
2. Materials and methods
2.1. Materials
EGFP was purchased from BioVision. Chariot™, Xfect™, and SAINT-PhD™ were purchased from Active Motif, Clontech, and Synvolux Therapeutics, respectively. Cre recombinase (HNC form) was received from Excellgen. Cre/LoxP reporter human T cell line (ABP-RP-CGFPLoxT) was obtained from Allele Biotech.
2.2. PTDM synthesis
The synthesis of each monomer and corresponding PTDM was reported by us [27]. Briefly, the monomers were obtained by ring-opening a Diels-Alder anhydride adduct with a desired alcohol to obtain a mono-functional intermediate, followed by EDC coupling with another equivalent of alcohol, the same type or different, to introduce the second functionality. The desired polymers were subsequently obtained by ring-opening metathesis polymerization (ROMP) using the Grubbs’ third generation catalyst29 in dichloromethane (CH2Cl2), with polydispersity indices under 1.1 (Ð = Mw/Mn). The final products were purified by dialysis against RO water and recovered by lyophilization. Further characterization of these polymers, including the reaction kinetics elucidating the random character of PTDM 2 is available in our previous report.28
2.3. EGFP fluorescence titration
In a 96-well plate, increasing concentrations of PTDM were added to 200 nM of EGFP in 200 µl PBS at pH 7.2 and left undisturbed for 30 minutes at room temperature in order for the components to bind and reach equilibrium. Samples with buffer only or EGFP only were included for background measurements. Changes in fluorescence were measured at 25 °C using a fluorescence plate reader (Biotek SynergyMx) at 507 nm following excitation at 488 nm. Binding parameter values (Kd and ΔG) were obtained by fitting the titration curves to the following equations:30,31
| (1) |
| (2) |
in which F is the relative fluorescence intensity of the protein/PTDM complex, F0 is the fluorescence intensity of free protein; Fs is the fluorescence intensity at saturation; P is the fixed concentration of protein; c is the concentration of PTDM at each point and Kd is the dissociation constant of protein-ligand complex. R and T are the ideal gas constant and temperature, respectively.
2.4. Non-covalent protein internalization into Jurkat T cells
Varying amounts of protein and PTDMs were mixed in 200 µL PBS and left undisturbed for 30 min at room temperature to allow complex formation. The mixture was then added drop-wise to Jurkat T cells (4 × 105 cells) in either complete (with 10% FBS) or serum free RPMI 1640 in a 12-well plate (1 mL final volume/well). Commercially available reagents were used as suggested by vendors. After four hours of incubation at 37 °C, cells were harvested and washed three times with a solution of heparin in PBS (20 U/mL) then suspended in 300 µL of FACS buffer (0.2% BSA in PBS) to be analyzed by flow cytometry (Becton Dickinson LSRII, BD Biosciences). The fluorescence signal was collected at 530 nm for 10,000 cells after excitation at 488 nm. Fluorescence intensity of the sample treated with protein alone was subtracted from the intensities measured in samples treated with the corresponding protein-carrier complex. Cell viability after treatment was also assessed on these samples using flow cytometry by adding the membrane impermeable, DNA-binding, 7-amino-actinomycin D (7-AAD) viability dye (BD Biosciences). Only viable cells were taken into account.
2.5. Cre delivery into Cre/LoxP reporter human T cells
Increasing concentrations of Cre-recombinase (from 50 to 200 nM) were mixed with PTDM at a molar ratio of 10/1 (PTDM/Cre) in PBS (200 µL total volume) and left undisturbed for 30 min at room temperature to allow complex formation. The complex solution was then added dropwise to 1 × 105 Cre reporter human T cells in serum free media (1 mL final volume/well in 12-well plate). Cells were incubated for two hours at 37 °C before the medium was exchanged with complete growth medium containing 10% serum. Cells were cultured for additionally 72 h with regular medium change, before gene recombination was analyzed using flow cytometry on 10,000 viable cells (λex: 488 nm, λem: 530 nm).
3. Results and discussion
3.1. PTDM Design
For this study, we used a series of synthetic PTDMs specifically designed to be constitutional macromolecular isomers, having identical elemental composition but different hydrophobic/hydrophilic functional group distribution along the backbone (PTDMs 1–3, Figure 1) which have been previously studied for their ability to interact with and cross cell membranes.28 Each polymer was composed of dual-functional repeat units carrying the same or different functional groups (phenyl or guanidinium) on a polyoxanorbornene di-ester scaffold (Figure 1a and Supplemental Figure S1). The choice of functional groups was based on the findings that the substitution of guanidinium and phenyl with amine and indole, respectively, decreased the protein delivery efficiency.25 The phenyl to guanidinium ratio was fixed at 1:1 throughout this series. Additionally, PTDM 4, lacking the phenyl component (see Supplemental Figure S1 for chemical structure), was also included as a synthetic analogue of TAT and a control for the hydrophobic domain.
All polymers were synthesized using ROMP, allowing for controlled, facile synthesis of block copolymer 1, gradient copolymer 2, and homopolymers 3 and 4. All polymers had comparable molecular weights of around 6 kDa, degrees of polymerization with a total of 10 repeat units on average, and polydispersity indices under 1.1 (Figure 1).28 PTDM 1 was composed of dual-functional repeat units containing either two guanidinium or two phenyl groups polymerized in blocks, thus generating the strongest sequence segregation possible. The same repeat units were copolymerized together to give a partially segregated sequence for PTDM 2, with a gradual change in composition along the chain from one moiety to the other.28 PTDM 3 was obtained by the polymerization of one monomer containing both hydrophobic and cationic side chains, therefore minimizing segregation and homogenously distributing hydrophobes and charges along the sequence.
3.2. PTDM/EGFP binding and delivery studies into Jurkat T cells
In the non-covalent delivery strategy, the carrier is required to form a complex with the cargo. The protein binding properties of each PTDM were therefore evaluated using fluorescence spectroscopy titration. We chose EGFP as the model protein for this study because of its inherent autofluorescence, which does not require additional labeling. EGFP and its derivatives have been widely used in biochemistry and cell biology studies as reporters in gene expression, protein localization, and high throughput screening.32,33 The environmental and conformational change that EGFP undergoes upon binding of a ligand often causes a measurable change in its fluorescence emission, making it a good reporter for this binding assay.34,35
Titration of EGFP with each PTDM led to fluorescence quenching, indicative of protein/PTDM binding (Figure 2) [34]. Curve fitting for EGFP quenching by block copolymer 1 yielded an equilibrium dissociation constant value (Kd) of 1.62 ± 0.20 µM. Similarly, PTDM 2, with gradual sequence segregation, had a Kd of 1.59 ± 0.24 µM. In contrast, the dissociation constants for the homopolymer PTDM 3, with homogenous hydrophobic/hydrophilic distribution, and the control PTDM 4, without hydrophobic residues, were about 7 fold higher, suggesting weaker interactions with this protein (the full list of binding parameter values can be found in Supplemental Table S1). Surprisingly, titration of EGFP with Pep-1 did not alter EGFP fluorescence (Supplemental Figure S2), therefore no Kd could be determined. Using the same assay, it has been previously reported that Pep-1 binds the monomeric form of the analogous red fluorescent protein [34].
Figure 2. PTDM interaction with EGFP.

200 nM EGFP was titrated with increasing concentrations of each PTDM. Changes in fluorescence emission were monitored at 507 nm following excitation at 488 nm. Results correspond to the average of three separate experiments.
To test whether these interactions were associated with cell internalization efficiencies, 2 µg (60 nM) of EGFP were complexed with PTDMs 1–4 at 5/1, 10/1, 20/1 and 40/1 molar ratios (PTDM/EGFP) and added to Jurkat T cells in the presence of serum (Supplemental Figure S3). Due to the auto fluorescent nature of EGFP, the amount of protein internalized was quantified by flow cytometry after the cells had been extensively washed with a heparin solution to remove any surface-bound complexes.36,37 Increasing the PTDM/EGFP ratio increased protein internalization, reaching an optimal efficiency at 20/1 (Supplemental Figure S3). A higher ratio (40/1) did not further improve efficiency; therefore the 20/1 ratio was used for all subsequent EGFP delivery experiments in this paper. Figure 3a shows a representative overlay of the cell fluorescence distribution after treatment, which corresponds to EGFP delivery efficiency. EGFP alone was not able to enter the cells (Figure 3a black line) thus confirming the impermeability of the cell membrane. Upon EGFP uptake by the cells, the histogram shifts to the right of the blank population. This shift is categorized in two ways: using a gate to determine the percentage of cells that shift to the right of the blank and the median fluorescence intensity (MFI) of the total population. The most segregated block copolymer 1 (Figure 3a red line) yielded the highest median fluorescence intensity of the cell population (MFI = 2540), indicative of significant protein uptake per cell, closely followed by the gradient copolymer 2 (Figure 3a blue line, MFI = 1644).
Figure 3. EGFP delivery into Jurkat T cells.

a) Representative overlay of histograms obtained from flow cytometric analysis showing different degrees of EGFP internalization into Jurkat T cells mediated by PTDMs 1–4 (60nM EGFP; 20/1 molar ratio PTDM/EGFP). b) Protein uptake per cell expressed as MFI after 4 h treatment of Jurkat T cells with increasing concentration of PTDM/EGFP complex (PTDM/EGFP molar ratio was held constant at 20/1). Data points represent mean ± SEM (standard error of mean) of at least three independent experiments. n.s.(p>0.05), **(p<0.01), ***(p<0.0001) of PTDM 1 vs. PTDM 2, as calculated by two-way ANOVA followed by Bonferroni post-test.
The delivery efficiencies of PTDM 1 and 2 were concentration-dependent, and increasing PTDM/EGFP complex concentration magnified the efficiency disparity between them (Figure 3b, red and blue lines). The efficiencies at higher concentrations directly reflect the ability of these polymers to bind to EGFP, suggesting that better binding facilitates protein delivery. At very high concentrations block copolymer 1 and random copolymer 2 delivered the most protein into the cells, which corresponds with having the lowest Kd. The simpler and versatile synthetic procedure for the PTDM 2, in addition to its satisfactory protein delivery, could make gradient copolymers a preferred choice in certain cases. While delivering EGFP using both PTDM 1 and 2 led to fluorescence increase in the entire population, indicative of homogenous protein internalization, only 30% of the cell population showed an increase in fluorescence when EGFP was delivered by the homopolymer PTDM 3 (Figure 3a green line). Increasing PTDM 3/EGFP complex concentration had no effect. Almost no protein internalization was observed with the control PTDM 4 (Figure 3a purple line), compared to an untreated sample, which was not surprising since TAT and its analogues are only able to deliver covalently-attached cargo. No decrease in cell viability was detected for any of the conditions tested (Supplemental Figure S4). The matching trends observed in both the biophysical and biological assays indicated a correlation between protein binding and delivery efficiencies and highlighted the importance of sequence segregation. In both cases, the strong and moderate sequence segregation characteristic of PTDM 1 and 2, respectively, yielded better performance than the less segregated PTDM 3.
Because of their better performance, delivery efficiencies of PTDMs 1 and 2 were compared to those of other representative and widely-used commercially available delivery agents: the peptide Chariot™, the polymeric Xfect™, and the lipid-based SAINT-PhD™ (Figure 4). PTDM 1 and 2 showed significantly better performance in Jurkats than the other delivery agents in the presence of serum. When using serum-free conditions, only Xfect™ showed moderate EGFP uptake; however, it was still lower than the uptake achieved with our PTDMs under the same conditions (Supplemental Figure S5). This comparison of PTDM delivery efficiency in presence of serum to that in serum-free conditions showed that PTDM activity in Jurkats was serum-insensitive.
Figure 4. Protein delivery comparison between PTDM 1–2 and commercially available reagents in Jurkat T cells in serum-containing media.

Percentage of EGFP positive cells after 4 h of treatment with EGFP complexed with either PTDM 1–2 or one of the commercially available reagents (60 nM EGFP; complete media). Each commercial reagent was used at the company recommended ratio. Values and error bars represent the mean ± SEM of at least three independent experiments.
To examine the possibility of cell-specific behavior, we also tested the protein carrier properties of our PTDM series with adherent HEK293T cells (Figure 5). As observed for Jurkats, PTDM 1 and 2 delivered protein to the whole cell population, while only about 40% of the cell population internalized EGFP when treated with PTDM 3. As expected, PTDM 4 did not deliver any EGFP. These results confirm that some degree of sequence segregation is required for efficient EGFP delivery since the more segregated block copolymer outperformed the less segregated homopolymer in both cell types, while the gradient copolymer showed intermediate performance. This also suggests that sequence segregation is a universal parameter and not cell type-specific.
Figure 5. EGFP uptake into HEK293T cells.

Representative histograms overlay from flow cytometric analysis of sample treated with 4µg EGFP complexed with PTDMs 1–4 (20/1 molar ratio). Complexes were added to subconfluent adherent cells in serum-containing media. After a 4 h incubation, cells were trypsinized and washed three times with a heparin solution (20 U/mL in PBS) to remove membrane-bound protein.
EGFP is a 33 kDa protein with a pI of about 5, therefore negatively charged at physiological pH 7.4. To better understand if the trends observed were independent from the protein’s overall charge and to gain some insight into the interaction between PTDMs and proteins, we studied the uptake of three FITC-labeled proteins with similar molecular weights but different pIs: BSA (pI = 4.7, Life Technologies), avidin (pI = 10.5, Life Technologies) and a recombinant neutral form of streptavidin (pI = 6.8–7.5, Thermo Scientific). Results for all three of these proteins were similar to that of the EGFP study, and demonstrated that sequence segregation is critical for high protein delivery efficiencies regardless of overall protein charge (Figure 6).
Figure 6. Flow cytometric analysis of FITC-labeled proteins uptake.

60 nM of each protein was complexed with each PTDM at 1/20 ratio and added to 4 × 105 Jurkat cells in presence of serum. Uptake was measured after a 4 h incubation. The fluorescence intensity of samples treated with protein alone was subtracted from the one measured in samples treated with the correspondent protein-carrier complex. Data points represent mean ± SEM of three independent experiments.
Taken together, it is clear that functional group segregation is an important and universal parameter for efficient, non-covalent protein delivery vehicles. Moreover, these data confirm that the presence of hydrophobic residues is essential for protein binding, which translates to the polymers ability to deliver protein. Specifically, increasing hydrophobic/hydrophilic segregation improves performance, as demonstrated by the strongly segregated block copolymer PTDM 1, which had the highest overall binding coefficient and delivery efficiencies. In accordance, the gradient copolymer PTDM 2 showed intermediate properties, while the non-segregated homopolymer PTDM 3 was the least effective at protein binding and delivery.
3.3. PTDM-mediated delivery of a biologically active protein: Cre recombinase
To further study the potential of PTDM 1 and 2 as a protein carrier for therapeutic or research purposes, the biological response of a functional protein delivered by either PTDM 1 or 2 was measured and used to determine cellular delivery efficiencies. As the experimental model, we measured loxP-GFP-loxP reporter gene recombination in a transgenic human T cell line subsequent to the PTDM-mediated delivery of the active enzyme Cre recombinase. Cre is largely used for genome alteration both in vitro and in vivo for the generation of conditional transgenic models or gene knock-out studies.38 The Cre-reporter cell type used in this study constitutively expresses GFP and is therefore highly fluorescent. Following gene recombination promoted by Cre at the nuclear level, cells stop producing GFP and gradually lose their fluorescence. Therefore GFP gene knock-out can be used as a measure of efficient PTDM-mediated cell delivery of the functional Cre protein. In the specific recombinant form used here (HNC; His6-NLS-Cre), Cre has an additional N-terminal histidine tag and NLS sequence (PKKKRKV) to better assist intracellular trafficking once in the cytosol.25,39,40
Flow cytometry was used to measure the reduction of cell fluorescence as an indication of gene recombination. As shown in Figure 7a, Cre alone was only able to induce GFP depletion in 5% of the cell population at the concentration used, despite the presence of the NLS. This is consistent with other studies.41 However, when the same amount of Cre was used in combination with PTDM 1, a significant decrease of fluorescence intensity was observed after 72 hours in up to 70% of the cell population, depending on the amount of protein delivered (Figure 7a and b). A similar trend was observed with PTDM 2 (Supplemental Figure S6). These results indicate that proteins delivered via either PTDM 1 or 2 retain their function, are able to reach their intracellular targets, and in the case of Cre penetrate into the nucleus. Biological applications of this technology are currently under evaluation.
Figure 7. PTDM 1-mediated delivery of functional Cre Recombinase into LoxP-GFP reporter human T cell line.

a) Overlay of histograms from flow cytometric analysis showing human reporter T cells treated with 150 nM of Cre Recombinase alone (black line) or in a non-covalent complex with 1 (red line) and 2 (blue line). Quantification of target gene recombination induced by increasing concentration of active Cre delivered into the reporter T cells by b) PTDM 1 and c) PTDM 2. The molar ratio used for complex formation was 10/1 (PTDM/Cre). Delivery was conducted for 2h in serum-free media and cells were analyzed by flow cytometry for gene recombination 72h after treatment and compared to an untreated sample. Values and error bars represent the mean ± SEM of at least three independent experiments.
4. Conclusions
Non-covalent protein delivery into cells has enormous potential for both enhancing basic cell biology research and enabling new therapies. Currently, robust systems remain elusive and there is strong interest in novel, more effective carriers. PTDs have shown considerable promise and the development of synthetic mimics will offer new opportunities to generate more efficient carriers. The superior nature of the PTDMs presented here compared to their peptide analogue Pep-1, confirms the importance of using de novo design in discovering more efficient delivery agents.
In the present study, we provided evidence that: a hydrophobic domain is required for formation and uptake of non-covalent complexes; the distribution of such hydrophobic residues along the PTDM backbone is important; and a strong sequence segregation characteristic of block-type architectures is indeed preferred for the efficient non-covalent protein delivery strategy. We demonstrated superior protein binding and delivery to multiple cell types using block copolymer PTDM 1 when compared to its homopolymer isomer 3. The gradient copolymer PTDM 2 also showed protein binding and delivery efficiencies comparable to that of PTDM 1 and is simpler to synthesize. In conclusion, this work establishes sequence segregation as an important design parameter for PTDM development.
Supplementary Material
Acknowledgments
This work was primarily supported by NSF (CHE-0910963). Joel Sarapas is acknowledged for contributing to the manuscript preparation. Authors would like to give special thanks to Christina Kuksin for copious valuable discussions.
References
- 1.Lien S, Lowman HB. Trends Biotechnol. 2003;21(12):556–562. doi: 10.1016/j.tibtech.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 2.Leader B, Baca QJ, Golan DE. Nat. Rev. Drug Discov. 2008;7(1):21–39. doi: 10.1038/nrd2399. [DOI] [PubMed] [Google Scholar]
- 3.Torchilin V. Drug Discov. Today Technol. 2008;5(2):e95–e103. doi: 10.1016/j.ddtec.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 4.Fonseca SB, Pereira MP, Kelley SO. Adv. Drug Deliv. Rev. 2009;61(11):953–964. doi: 10.1016/j.addr.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 5.Bechara C, Sagan S. FEBS Lett. 2013;587:1693–1702. doi: 10.1016/j.febslet.2013.04.031. [DOI] [PubMed] [Google Scholar]
- 6.Heitz F, Morris MC, Divita G. Br. J. Pharmacol. 2009;157(2):195–206. doi: 10.1111/j.1476-5381.2009.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. Science (80-.) 1999;285(5433) doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- 8.Stewart KM, Horton KL, Kelley SO. Org. Biomol. Chem. 2008;6:2242–2255. doi: 10.1039/b719950c. [DOI] [PubMed] [Google Scholar]
- 9.Snyder EL, Dowdy SF. Expert Opin. Drug Deliv. 2005;2(1):43–51. doi: 10.1517/17425247.2.1.43. [DOI] [PubMed] [Google Scholar]
- 10.Fawell S, Seery J, Daikh Y, Moore C, Chen LL, Pepinsky B, Barsoum J. Proc. Natl. Acad. Sci. U. S. A. 1994;91(2):664–668. doi: 10.1073/pnas.91.2.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Juliano R, Rowshon Alam M, Dixit V, Kang H. Nucleic Acids Res. 2008;36(12):4158–4171. doi: 10.1093/nar/gkn342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Becker-Hapak M, McAllister SS, Dowdy SF. Methods. 2001;24(3):247–256. doi: 10.1006/meth.2001.1186. [DOI] [PubMed] [Google Scholar]
- 13.Lo SO, Wang S. Macromol. Rapid Commun. 2010;31:1134–1141. doi: 10.1002/marc.200900934. [DOI] [PubMed] [Google Scholar]
- 14.Loudet A, Han J, Barhoumi R, Pellois J-P, Burghardt RC, Burgess K. Org. Biomol. Chem. 2008;6(24):4516. doi: 10.1039/b809006h. [DOI] [PubMed] [Google Scholar]
- 15.Morris MC, Depollier J, Mery J, Heitz F, Divita G. Nat. Biotechnol. 2001 Dec;19:1173–1176. doi: 10.1038/nbt1201-1173. [DOI] [PubMed] [Google Scholar]
- 16.Deshayes S, Morris M, Heitz F, Divita G. Adv. Drug Deliv. Rev. 2008;60(4):537–547. doi: 10.1016/j.addr.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 17.Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. Proc. Natl. Acad. Sci. U. S. A. 2000;97(24):13003–13008. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mitchell DJ, Kim DT, Steinman L, Fathman CG, Rothbard JB. J. Pept. Res. 2000;56:318–325. doi: 10.1034/j.1399-3011.2000.00723.x. [DOI] [PubMed] [Google Scholar]
- 19.Bang E-K, Gasparini G, Molinard G, Roux A, Sakai N, Matile S. J. Am. Chem. Soc. 2013;135(6):2088–2091. doi: 10.1021/ja311961k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tezgel aÖ, Telfer JC, Tew GN. Biomacromolecules. 2011;12:3078–3083. doi: 10.1021/bm200694u. [DOI] [PubMed] [Google Scholar]
- 21.Cooley CB, Trantow BM, Nederberg F, Kiesewetter MK, Hedrick JL, Waymouth RM, Wender PA. J. Am. Chem. Soc. 2009;131(45):16401–16403. doi: 10.1021/ja907363k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kolonko EM, Pontrello JK, Mangold SL, Kiessling LL. J. Am. Chem. Soc. 2009;131(21):7327–7333. doi: 10.1021/ja809284s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sgolastra F, Deronde BM, Sarapas JM, Som A, Tew GN. Acc. Chem. Res. 2013;46:2977–2987. doi: 10.1021/ar400066v. (Xx) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Som A, Reuter A, Tew GN. Angew. Chemie - Int. Ed. 2012;51:980–983. doi: 10.1002/anie.201104624. [DOI] [PubMed] [Google Scholar]
- 25.Tezgel AÖ, Jacobs P, Backlund CM, Telfer JC, Tew GN. Biomacromolecules. 2017 doi: 10.1021/acs.biomac.6b01685. acsbiomac.6b01685. [DOI] [PubMed] [Google Scholar]
- 26.Tezgel aÖ, Gonzalez-Perez G, Telfer JC, Osborne Ba, Minter LM, Tew GN. Mol. Ther. 2012;21(1):201–209. doi: 10.1038/mt.2012.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Backlund CM, Sgolastra F, Otter R, Minter LM, Takeuchi T, Futaki S, Tew GN. Polym. Chem. 2016 doi: 10.1039/C6PY01615D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sgolastra F, Minter LM, Osborne Ba, Tew GN. Biomacromolecules. 2014;15:812–820. doi: 10.1021/bm401634r. [DOI] [PubMed] [Google Scholar]
- 29.Love JA, Morgan JP, Trnka TM, Grubbs RH. Angew. Chemie Int. Ed. 2002;41(21):4035–4037. doi: 10.1002/1521-3773(20021104)41:21<4035::AID-ANIE4035>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 30.Copeland, R. A. 0–471.
- 31.Heitz Frederic, Morris May C, Fesquet Didier, Cavadore Jean-Claude, Dorée Marcel, Divita G. 1997 doi: 10.1021/bi962349y. [DOI] [PubMed] [Google Scholar]
- 32.Chalfie M, Tu Y, Euskirchen G, Ward W, Prasher D. Science (80-.) 1994;263(5148):802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 33.Changsen C, Franzblau SG, Palittapongarnpim P. Antimicrob. Agents Chemother. 2003;47(12):3682–3687. doi: 10.1128/AAC.47.12.3682-3687.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deo SK, Daunert S. Anal. Biochem. 2001;289(1):52–59. doi: 10.1006/abio.2000.4909. [DOI] [PubMed] [Google Scholar]
- 35.Kurzawa L, Pellerano M, Morris MC. Biochim. Biophys. Acta - Biomembr. 2010;1798(12):2274–2285. doi: 10.1016/j.bbamem.2010.02.027. [DOI] [PubMed] [Google Scholar]
- 36.McNaughton BR, Cronican JJ, Thompson DB, Liu DR. Proc. Natl. Acad. Sci. U. S. A. 2009;106(15):6111–6116. doi: 10.1073/pnas.0807883106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Veldhoen S, Laufer SD, Trampe A, Restle T. Nucleic Acids Res. 2006;34(22):6561–6573. doi: 10.1093/nar/gkl941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nagy A. Genesis. 2000;26:99–109. [PubMed] [Google Scholar]
- 39.Zou Y-R, Müller W, Gu H, Rajewsky K. Curr. Biol. 1994;4(12):1099–1103. doi: 10.1016/s0960-9822(00)00248-7. [DOI] [PubMed] [Google Scholar]
- 40.Peitz M, Pfannkuche K, Rajewsky K, Edenhofer F. Proc. Natl. Acad. Sci. U. S. A. 2002;99(7):4489–4494. doi: 10.1073/pnas.032068699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin Q, Jo D, Grebre-Amlak KD, Ruley HE. BMC Biotechnol. 2004;4(1):25. doi: 10.1186/1472-6750-4-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.