

Abstract
The basic-helix-loop-helix Per-Arnt-Sim (bHLH/PAS) family comprises many transcription factors, found throughout all three kingdoms of life; bHLH/PAS members “sense” innumerable intracellular and extracellular “signals” — including endogenous compounds, foreign chemicals, gas molecules, redox potential, photons (light), gravity, heat, and osmotic pressure. These signals then initiate downstream signaling pathways involved in responding to that signal. The term “PAS”, abbreviation for “Per-Arnt-Sim” was first coined in 1991. Although the mouse Arnt gene was not identified until 1991, evidence of its co-transcriptional binding partner, aryl hydrocarbon receptor (AHR), was first reported in 1974 as a “sensor” of foreign chemicals, up-regulating cytochrome P450 family 1 (CYP1) and other enzyme activities that usually metabolize the signaling chemical. Within a few years, AHR was proposed also to participate in inflammation. The mouse [Ah] locus was shown (1973–1989) to be relevant to chemical carcinogenesis, mutagenesis, toxicity and teratogenesis, the mouse Ahr gene was cloned in 1992, and the first Ahr(−/−) knockout mouse line was reported in 1995. After thousands of studies from the early 1970s to present day, we now realize that AHR participates in dozens of signaling pathways involved in critical-life processes, affecting virtually every organ and cell-type in the animal, including many invertebrates.
Keywords: AHR, bHLH/PAS family of transcription factors, cancer, dose-response curve, embryonic stem cells, prostaglandins, eicosanoids, lipid mediators, mouse genetics, pro-inflammatory and post-inflammatory response, “brain-gut-microbiome”
“During the oral defense of my thesis (spring, 1964), a professor commented, ‘Everyone knows that genes in the DNA are transcribed into RNA which is translated into protein. You’re proposing that protein might control DNA? Why, that’s heresy!’ After a very awkward silence — my mentor Professor Howard S. Mason spoke up, ‘And what’s wrong with a little heresy?’ ” -----Daniel W. Nebert
1. Introduction
The first evidence for existence of aryl hydrocarbon receptor (AHR) occurred more than four decades ago. What do we know today about the AHR transcription factor, and in what critical-life processes does AHR participate?
To address these questions, we begin by describing the history of enzyme induction by foreign chemicals and inducible cytochrome P450 (CYP) monooxygenases; earliest studies were carried out in rat liver. Inbred mouse strains, unlike rats, were found to differ quite dramatically in degree of inducibility of certain P450 enzyme activities; this led to comparison of “potency” of inducers such as polycyclic aromatic hydrocarbons (PAHs) vs 2,3,7,8,-tetrachlorodibenzo-p-dioxin (TCDD; called “dioxin” in lay terms). Astonishingly, TCDD was found to be ~36,000 times more potent than PAHs.
A landmark study followed, comparing dose-response curves between TCDD-treated C57BL/6 (B6) and DBA/2 (D2) mice, showing that “inducible-resistant” D2 mice could be “forced” by TCDD to “turn on” their enzyme activity; due to the shape of the dose-response curve, it was concluded that a “receptor must exist that recognizes TCDD and regulates AHH acitivty.” The manuscript was first rejected in 1973 by reviewers, with comments such as “heresy” and “implausible to think that a foreign chemical would bind to an intracellular receptor.” Eventually, after rebuttal letters, the manuscript was accepted for publication; it appeared in 1974.
Next, AHR not only recognized foreign chemicals but was also found to be associated with inflammation — again, a hypothesis rejected by many colleagues. After the mouse Ahr and human AHR genes had been cloned and sequenced, AHR was finally identified as “a member of the PAS domain family of signal sensors.” Shortly thereafter, Ahr(−/−) knockout mouse lines provided strong evidence of the vast importance of AHR in numerous critical-life processes independent of foreign chemical treatment. AHR is now appreciated to function during the cell cycle, cell migration, cell adhesion, and other embryonic stem (ES) cell functions; these findings are consistent with early studies that had shown AHR-dependent birth defects in PAH- and especially TCDD-treated laboratory animals. Finally, it became appreciated that AHR is involved in many signaling pathways that affect various critical-life functions in most organs, tissues and/or cell types — in both vertebrates and invertebrates.
2. History and background
2.1. Inbred mouse strain differences in enzyme induction
The earliest studies of enzyme induction by foreign chemicals — in liver of PAH-treated rats — were conducted by Allan Conney, a graduate student in the Millers’ laboratory (Conney et al., 1956; Conney et al., 1957); as a postdoctoral fellow in the laboratory of Jim Gillette, Conney continued those studies (Conney et al., 1959). Subsequently, induced “benzpyrene hydroxylase” throughout the rat gastrointestinal (GI) tract was described, following oral benzpyrene treatment; highest induced enzyme levels were found in duodenum (Wattenberg et al., 1962). Thus, here was an exciting concept: a novel “signal” is introduced to the animal, or cell; the “response” is to increase enzyme(s) to metabolize that signal. This model was reminiscent of earlier studies in E. coli: the “signal” (addition of tryptophan to tryptophan-deficient culture medium) led to a bacterial “response” of dramatic increases in enzymes in the tryptophan-metabolizing pathway (Newton and Snell, 1965).
Following these studies by Conney and Wattenberg, the original “benzpyrene hydroxylase” name was changed to the broader term “aryl hydrocarbon hydroxylase” (AHH), because the substrate was shown to include any of several PAHs, and several PAHs were shown to be inducers having varying potency; formation of hydroxylated benzo[a]pyrene (nmol/min/mg protein) became the standard AHH assay, which was developed and applied to PAH-treated cultures of fetal hamster cells (Nebert and Gelboin, 1968a; Nebert and Gelboin, 1968b). Induced AHH activity in cell culture was shown to involve both transcription of DNA into mRNA and translation of mRNA into protein (Nebert and Gelboin, 1970). Subsequently, substantial differences in AHH inducibility between PAH-treated B6 and D2 mice were reported (Nebert and Gelboin, 1969); lack of AHH inducibility was then shown to behave usually as an autosomal recessive trait (Gielen et al., 1972; Robinson et al., 1974).
These genetic differences led to a model system far superior to that of PAH-treated vs untreated rats, i.e. an identical dose of the same chemical in genetically different mice results in striking differences, apparently based predominantly on a single gene. This single gene was subsequently found to be largely responsible for PAH-induced cancer of multiple types, mutagenesis, toxicity and birth defects [reviewed in (Nebert, 1989)]. In fact, PAH treatment of a pregnant mouse with a particular genotype, and then observing differences in toxicity and/or teratogenesis in utero among her offspring having different genotypes — became an especially powerful tool for studies in developmental embryology [(Nebert et al., 1972; Shum et al., 1979) & reviewed in (Nebert, 1989)].
With regard to clinical relevance, human AHH activity in placenta — comparing cigarette smokers with nonsmokers during pregnancy — revealed that cigarette smoke induces AHH activity (Welch et al., 1968; Nebert et al., 1969). This finding has important implications for the health of newborns from cigarette-smoking mothers.
2.2. Proof that AHH activity is a P450 monooxygenase
“Cytochrome P-450” was first detected as a “colored pigment in the cell (Strittmatter and Velick, 1957) which — when reduced with NADPH and bound to CO — shows a spectrophotometric Soret peak wavelength at 450 nm” (Omura and Sato, 1962; Omura and Sato, 1964). Soon thereafter at the same symposium, three independent laboratories reported that “microsomal mixed-function oxidase” named for electron spin resonance properties of “microsomal Fex” (Mason et al., 1965), enzymatic functions of microsomal cytochrome P-450 (Omura et al., 1965), and particular steroid hydroxylases (Ernster and Orrenius, 1965) all appeared to be one and the same enzyme or enzyme family.
The enzyme active-site comprises a heme-iron center — with tetrahedral iron tethered to the four nitrogen atoms of the porphyrin ring, cysteine-thiolate in fifth position, and binding of H2O or substrate (hydroxyl group, nitrogen atom, or molecular O2) in the sixth position. The O atom transferred to the substrate is derived from atmospheric diatomic O2 rather than H2O (Hayaishi et al., 1955; Mason et al., 1955; Mason, 1957); hence, the name “monooxygenase” is more suitable for these enzymes.
Because of spectral properties similar to those of mitochondrial cytochromes, P-450 was misnamed a “cytochrome” (Omura and Sato, 1962), an inaccurate label that unfortunately has persisted to this day. A more appropriate term would have been “heme-thiolate monoogenase” (Daiber and Ullrich, 2002); however, the name “cytochrome P450” had become thoroughly entrenched — long before details of the enzyme proteins and functions had been recognized.
After PAH treatment of rats, a second form of liver microsomal cytochrome P-450, called “P-448” could be detected spectrophotometrically (Glaumann et al., 1969; Lu et al., 1971); another lab termed the PAH-inducible enzyme “P1-450” (Parli and Mannering, 1970). It was thus postulated that “AHH activity” was “P-448” or “P1-450.” Therefore, a spectrophotometric assay — to study the height and location of the Soret peak — was carried out in PAH-treated fetal hamster cell cultures; indeed, upon treatment of the cell homogenate with NADPH and CO, a peak developed and was associated with increasing AHH activity as a function of time, during which the Soret peak shifted from 450 to 446 nm (Nebert, 1970). In later studies, it became clear that PAH-inducible AHH activity is associated with both two distinct enzymes, “P1-450” and “P-448” (Atlas et al., 1975; Atlas et al., 1977); ultimately, these were named “CYP1A1” and “CYP1A2,” respectively. The latter represents high-spin iron Fe3+ that causes a hypsochromic shift in the Soret peak of reduced CO-bound heme.
2.3. Genetic differences in mouse AHH induction by TCDD
In clinical studies spearheaded by Ray Suskind before 1970, workers exposed to TCDD in trichlorophenol-processing factories were shown to be at extremely high risk for chloracne and porphyria cutea tarda [reviewed in (Zack and Suskind, 1980)]. This led Alan Poland, using chick egg liver (Poland and Glover, 1973), to show that TCDD was ~36,000 times more potent than any PAH in the induction of δ-aminolevulinic acid synthetase — a key enzyme in porphyrin synthesis.
Then came the first “Aha!” moment: If δ-aminolevulinic acid synthetase activity is strikingly induced by the highly potent TCDD, and heme is a product of porphyrin synthesis, and AHH represents a P450 hemoprotein, would TCDD be superior to PAHs in causing AHH induction? In particular — would the “lack of AHH induction,” seen in PAH-treated D2 mice (Nebert and Gelboin, 1969; Gielen et al., 1972), be overcome by TCDD treatment? Further, would TCDD be able to increase inducible AHH activity in B6 mice to even higher levels? Following a telephone call, these questions were answered by Alan Poland visiting the Nebert laboratory, where the conclusive experiments were performed together (Poland et al., 1974).
Fig. 1 illustrates dose-response curves of B6 vs D2 hepatic AHH activity as a function of TCDD dosage. Earlier studies, with PAH inducer 3-methylcholanthrene (80 mg/kg; 24 h), had shown 5- to 10-fold induction of hepatic AHH activity in B6, but no detectable increases in D2 liver AHH (Nebert and Gelboin, 1969; Gielen et al., 1972); in fact, no further induction of AHH activity is found — even at 800 mg/kg, about the highest dose of 3-methylcholanthrene physically and chemically possible (unpublished). In contrast (Fig. 1), whereas TCDD (1 μg/kg; 24 h) elicits ~6-fold increases in hepatic AHH activity in B6 but no detectable increase in D2 mice, higher TCDD doses (10–100 μg/kg; 24 h) raise D2 hepatic AHH activity to levels similar to those of B6 mice.
Fig. 1.
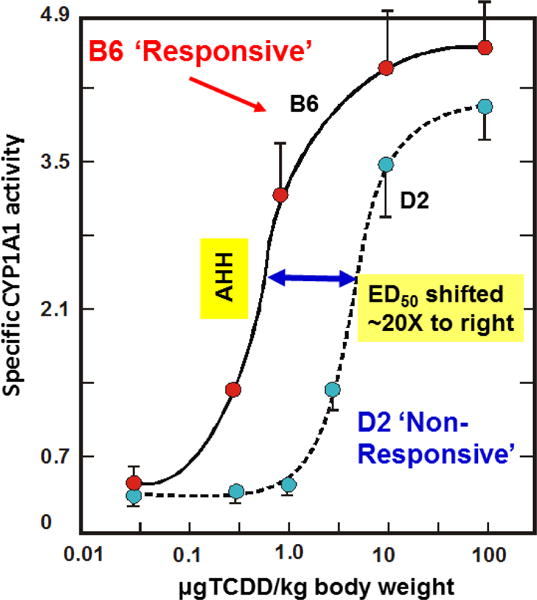
Dose-response curve of induced AHH activity (the CYP1A1 monooxygenase), as a function of intraperitoneal TCDD administered 48 h earlier — comparing B6 (C57BL/6 mouse with high-affinity AHR) with D2 (DBA/2 mouse with poor-affinity AHR). Units on Y-axis denote “nmol of 3-hydroxybenzo[a]pyrene formed per min per mg liver microsomal protein” [redrawn and modified from data in (Poland et al., 1974)].
The ED50 value denotes the “effective dose to reach 50% of maximal response.” When comparing curves in Fig. 1, the ED50 value for D2 is shifted ~20-fold to the right, relative to that of B6 mice. The shape of such a dose-response curve is consistent with a receptor — rather than changes in enzyme itself (Poland et al., 1974). Indeed, subsequent cloning and DNA sequencing studies showed that the amino-acid sequences of B6 “P1-450” (Kimura et al., 1984) and D2 “P2-450” (Kimura and Nebert, 1986) are identical, whereas “P3-450” (P-448) is a product of a different gene (Kimura et al., 1984); the former two are now officially named “CYP1A1,” whereas the latter is “CYP1A2.”
Following treatment with radiolabeled TCDD, hepatic cytosol accumulation of radiolabel was found to be greatest in B6 mice, intermediate in B6D2F1 mice, and least in D2 mice (Poland et al., 1976) — a pattern mirroring strain sensitivity to AHH induction by TCDD, demonstrated two years earlier (Poland et al., 1974). These data are consistent with a small pool of high-affinity binding-sites (estimated Kd equlibrium constant ~0.27 nM) that stereospecifically and reversibly binds TCDD. Thus, these findings strengthened the earlier hypothesis: the hepatic cytosolic TCDD-binding species is a receptor that up-regulates AHH activity. Furthermore, a proposed receptor gene mutation in D2 mice most likely causes the diminished-affinity phenotype for TCDD (Poland et al., 1976). These 1974–76 studies “set the stage” for “the AHR story,” paving the way for future discoveries (Okey, 2007).
2.4 Creation of mutant benzo[a]pyrene-resistant hepatoma cell lines
We would be remiss not to mention the landmark study (Hankinson, 1979) in which the mouse hepatoma line Hepa-1c1c7 was shown to exhibit inducible AHH activity — and from which benzo[a]pyrene-resistant clones were isolated; ultimately, the most important clones included AHR-deficient (c2), ARNT-deficient (c4) and CYP1A1-deficient (c1 & c37). Mutations in the Ahr, Arnt, and Cyp1a1 genes, respectively [vide infra], were shown in each case to be responsible for the phenotype (Legraverend et al., 1982; Hankinson et al., 1985; Kimura et al., 1987b). These mutant clones, compared with the wild-type parent Hepa-1 line, became an invaluable model system for unraveling many of the mysteries of the AHR-CYP1 axis [e.g. (Robertson et al., 1987), among dozens of other publications].
3.0 Early evidence for AHR involvement in inflammation
3.1 Ethanol-induced peritonitis: Ahr allelic differences
As mentioned [vide supra], the high- vs poor-affinity AHR, encoded by B6 Ahrb1 vs D2 Ahrd alleles, respectively, were shown to be pivotal for demonstrating differences in PAH-caused cancer of several types, mutagenesis, birth defects — as well as toxicity of ovary, eye, bone marrow, atherosclerosis and oxidative stress [reviewed in (Nebert, 1989)]. Curiously, in 1979 differences in “ethanol sleep time” were observed in AHR “high- vs poor-affinity” mice, even among pups from the same litter.
The “ethanol sleep time” is actually a misnomer, because the mouse remains alert with eyes open; however, the anterior abdomen is warm to the touch and, upon opening the abdominal cavity, one sees an acute inflammatory response to intraperitoneal ethanol — more severe in “long-sleep” Ahrd than “short-sleep” Ahrb1 mice. This intriguing finding in 1979, manuscript again rejected (as “implausible” and “heresy”), was published a decade later (Bigelow et al., 1989) and ultimately presented at a Nobel Symposium (Nebert, 1994).
Planar polychlorinated biphenyls were also known to induce AHH activity (Poland and Glover, 1977). A study in chick egg liver (Rifkind and Muschick, 1983) reported that the nonsteroidal anti-inflammatory agent benoxaprofen decreases toxicity of planar 3,4,3′,4′-tetrachlorobiphenyl — suggesting involvement of “the arachidonic acid cascade.” Then came another “Aha!” moment. AHR is associated with both planar PCB-caused toxicity in chick egg liver (Rifkind and Muschick, 1983) and ethanol-induced peritonitis in AHR poor-affinity mice (Bigelow et al., 1989); thus, everything was consistent with the proposal (Nebert et al., 1981) that one or more endogenous ligands of AHR might represent AHR-mediated CYP1-generated metabolites present in the arachidonic acid/eicosanoid/lipid-mediator (LM) second-messenger pathway response to inflammation. This hypothesis also makes sense, in terms of the CYP1 enzyme active-site, believed to accommodate chemicals having structural similarities in size and planar shape for both PAHs and many LMs.
3.2 Fertility, fitness and longevity: Ahr allelic differences
Besides the inflammatory response, allelic differences in Ahr as manifested by high- vs poor-affinity AHR — were curiously discovered to be associated with fertility, general health and longevity. Recombinant inbred (RI) lines were developed in the 1970s from progenitor C57BL/6 and C3H/He mice having Ahrb1 (high-affinity) and Ahrb2 (intermediate-affinity) alleles, respectively; between generations 7 and 13, individual female and male RI mice were then crossed with D2 (Ahrd allele; poor-affinity AHR) and followed for several generations. Highest levels of high-affinity AHR in both female and male offspring were found intriguingly to be associated with greater fertility, fitness, and longer life span (Nebert et al., 1984). This improbable finding, difficult to explain at the time, suggested that AHR might be pivotal in one or more critical life functions.
Three decades later, these observations are now much clearer. “Disease tolerance” reflects the host’s ability to lower effects of infection on host fitness; for example, an initial exposure to bacterial lipopolysaccharide (LPS) usually induces a state of refractoriness to further LPS challenge (“endotoxin tolerance”). It was shown that a first LPS exposure of mice activates AHR and the hepatic enzyme TDO (tryptophan 2,3-dioxygenase) — which provides an active ligand for AHR, which in turn down-regulates early inflammatory gene expression. Upon LPS rechallenge, AHR participates in long-term regulation of systemic inflammation only in the presence of IDO1 (indoleamine 2,3-dioxygenase-1); moreover, AHR-complex-associated SRC (SRC proto-oncogene, non-receptor tyrosine kinase) activity enhances IDO1 phosphorylation and signaling. This resulting endotoxin-tolerant state protects mice against immunopathology of both gram-positive and -negative infections, consistent with a role for AHR in contributing to host fitness (Bessede et al., 2014). Also, Ahr(−/−) knockout mice have been shown to be hypersensitive to LPS-induced septic shock (Ichihara et al., 2007).
Bacterial pigmented virulence factors (e.g. naphthoquinone phthiocol of Mycobacterium tuberculosis and phenazines of Pseudomonas aeruginosa) are also known AHR ligands; upon ligand binding, AHR activation leads to virulence-factor degradation and regulated cytokine and chemokine production. The relevance of AHR to host defense was underscored by AHR-deficient mice showing heightened susceptibility to both P. aeruginosa and M. tuberculosis infections (Moura-Alves et al., 2014).
Both of these recent studies (Bessede et al., 2014; Moura-Alves et al., 2014) are consistent with the earlier breeding studies that had been carried out in a nonsterile animal room (Nebert et al., 1984). The data suggest that “resistance to chronic subclinical, low-grade infections” in a “dirty” mouse colony might have been the beneficial reason for high-affinity-AHR mice to have evolved the phenotypes of greater fertility, fitness, and longer life span.
4. AHR gene identified and characterized
4.1. Molecular cloning of the AHR gene
When an AHR ligand enters the cell as a “signal,” it binds to cytoplasmic AHR — which is already bound to 90-kDa heat shock protein (HSP90), a 38-kDa AHR-interacting protein (AIP), and a less well-characterized “p23” protein. Because of such low intracellular concentrations of AHR, cloning of the gene had remained elusive; in fact, attempting to clone Ahr led first to cloning the Arnt gene coding for aryl hydrocarbon receptor translocator (Hoffman et al., 1991). When the NH2-terminal sequence of the mouse AHR protein was finally reported (Bradfield et al., 1991), this led quickly to “fishing out” the B6 high-responsive Ahrb1 cDNA (Burbach et al., 1992; Ema et al., 1992), and, subsequently the mouse Ahr gene (Schmidt et al., 1993) and human AHR cDNA (Dolwick et al., 1993) and gene (Le Beau et al., 1994).
It could then be concluded (Burbach et al., 1992) that AHR is a ligand-activated transcription factor; AHR encodes a basic region/helix-loop-helix (bHLH) protein, similar to many transcription factors that undergo dimerization for function, as well as having extensive sequence similarity to ARNT (Hoffman et al., 1991) and two homologous Drosophila regulatory proteins “single-minded” (SIM) and “periodic” [PER; in vertebrates called “period circadian clock-1” (PER1) (Konopka and Benzer, 1971; Crews et al., 1988). These data were consistent with an earlier study in mouse liver (Tukey et al., 1982), demonstrating that — as TCDD-bound AHR became diminished in cytosol — it appeared in the nucleus and was associated with increases in P1-450 mRNA induction, i.e. intranuclear increases in AHR are correlated with CYP1A1 increases, reflecting induction of AHH activity.
4.2. The bHLH/PAS (per-Arnt-sim) family of signal sensors
Chemoreceptors exist in all phyla of life, comprising two types: transmembrane and cytoplasmic. Cytoplasmic chemoreceptors appear to “sense” the “signal” within the cytoplasm, including signals that enter the cell. These sensors contain diverse signal-input domains — usually located NH2-terminally to the protein segment that defines a chemoreceptor [i.e. so-called “methyl-accepting” (MA) domain]; however, various other NH2-terminal domains also exist. The most common signal-input domain is the PAS domain. It is not rare, however, for some cytoplasmic chemoreceptors to have instead, COOH-terminal domains that function in signal-sensing (Collins et al., 2014).
The term “PAS” was first coined in late 1991 (Nambu et al., 1991). In eubacteria, PAS domains are usually positioned near the NH2-terminus of signaling proteins — such as sensor histidine kinases, cyclic-diGMP synthases/hydrolases, and MA chemotaxis proteins. The PAS domain family in all living organisms forms “structural clades” on the basis of two principal variables: [a] topological location (inside or outside the plasma membrane); and [b] class of small molecules to which they bind. Binding of a chemically diverse range of small-molecule metabolites is a hallmark of the PAS domain family (Fig. 2). PAS ligand-binding proteins function either as a primary cue to initiate an intracellular-signaling response, or they provide a domain having the capacity to respond to secondary physical or chemical signals — such as gas molecules, redox potential, or photons (Henry and Crosson, 2011).
Fig. 2.
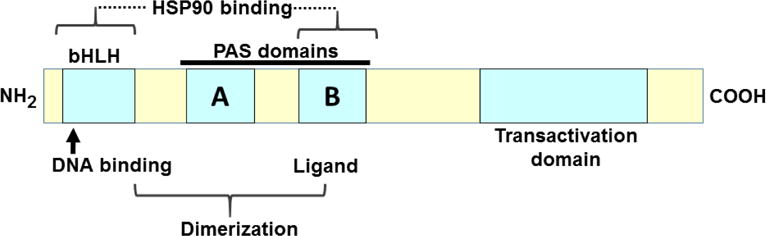
Schematic representation of bHLH/PAS sensor proteins, in which specific functions are localized to particular regions, or modules. Ligands bind to the PAS-B domain; binding to HSP90 includes both the bHLH and PAS-B domains; dimerization with ARNT involves both the bHLH domain and PAS domains; DNA-binding and the nuclear localization signal reside in the basic region of the bHLH domain; the trans-activation function comprises a large segment toward the carboxy (COOH)-terminus. The carboxyl half of AHR protein displays the greatest amino-acid variation across the animal kingdom, whereas bHLH/PAS domains are highly conserved [redrawn and modified from (Okey, 2007) and references therein].
Genes coding for various bHLH-PAS proteins found in eukaryotic metazoans include: vertebrate AHR; ARNT; AHR repressor (AHRR); ARNTL (ARNT-like; also called BMAL); CLOCK (clock circadian regulator); NPAS2 (neuronal PAS protein-2); MIA3 (melanoma inhibitory activity family member-3—first named in Drosohila as Tgo (tango); EGLN1 (egl-9 family hypoxia-inducible factor; previously HIF); SIM1 and SIM2 (single-minded; Sim is a single gene in Drosophila); and several steroid receptor coactivators (SRCs); genes encoding bHLH-PAS proteins in Drosophila, but not vertebrates, include trh (trachealess) (Pecenova and Farkas, 2016) and ss (spineless) (Céspedes et al., 2010), also called aristopaedia (Emmons et al., 1999). Estimated numbers of bHLH/PAS sensor genes are ~30 in the human genome, twelve in Ciona intestinalis (sea squirt), 15 in Drosophila, and nine in the nematode Caenorhabditis elegans (Hahn et al., 2006).
Cryptochromes are not to be confused with bHLH/PAS proteins or cytochromes. Cryptochromes, receptors for blue and ultraviolet UV-A light, participate in circadian rhythmicity of plants and animals; they share sequence similarity to DNA photolyases but have no photolyase activity (Lin and Todo, 2005). However, a number of plant and bacterial photosensor genes (e.g. PLPB, BPLPA) also encode PAS domain-containing receptors that exhibit ligand-binding, dimerization, and signal-transduction qualities (Pellequer et al., 1998).
Similar to AHR, Drosophila spineless functions as a heterodimer with tango, the Drosophila ortholog of ARNT; moreover, spineless participates in genesis of specialized fly neurons involved in color vision. Interestingly, although spineless-tango heterodimers exhibit very similar binding specificities to AHR-ARNT heterodimers found in mammals — interaction of spineless with tango apparently does not require presence of exogenous or endogenous ligands and, as such, represents an instance in which AHR physiological function appears to be independent of ligand-binding-mediated signal cascades [reviewed in (Lindsey and Papoutsakis, 2012)]. AHR also does not appear to require ligand-binding prior to evoking signal-mediated pathways — in innumerable other invertebrates: the nematode Caenorhabditis elegans (Powell-Coffman et al., 1998); the cnidarian, a sea anemone Nematostella vectensis (Reitzel et al., 2014); the inter-tidal copepod Tigriopus japonicus (Kim et al., 2015); the zebra mussel Dreissena polymorphia (Chatel et al., 2015); the pearl oyster Pinctada martesii (Du et al., 2015); and the red flour beetle Tribolium castaneum (Grunwald et al., 2015) — just to name a few recent examples.
Although originally named “HIF1α” and “HIF1β”, the latter was realized to be ARNT; thus, the former became simply “HIF” (official gene name is now EGLN1). Today it is known that two evolutionarily highly conserved HIF genes exist: HIF1A and HIF3A (Hif1a & Hif3a in mouse).
Although there are substantial size-differences between the bHLH-PAS family members, they all share NH2-terminally-located bHLH and PAS domains (Fig. 2). The majority (including AHR) contain “twin-PAS segments,” consisting of PAS-A and PAS-B domains. Several bHLH-PAS members also carry “Gln- and Ser-rich” motifs in COOH-terminal regions (Hahn et al., 2006; Okey, 2007).
As mentioned above, bHLH proteins, like many transcription factors, undergo dimerization before becoming functionable. Thus, vertebrate AHR dimerizes with ARNT before becoming functionally active; neither AHR/AHR nor ARNT/ARNT dimers are ever functional (Wu et al., 2013). Both SIM1 and SIM2 are also nuclear proteins that constitutively complex with the general bHLH/PAS partner, ARNT (Woods and Whitelaw, 2002). SIM proteins, in combination with ARNT, attenuate transcription under hypoxic conditions, by way of hypoxia-inducible response elements (HREs), which are present in promoter/enhancer regions of numerous genes.
Such cross-talk between co-expressed bHLH/PAS factors can occur through competition for ARNT — seen, for example, during SIM-mediated repression of AHR-induced transcription from “xenobiotic-response elements” (XREs; also called “AHREs”). Interestingly, SIM1/ARNT dimers, but not SIM2/ARNT, are able to activate transcription via the mouse Epo enhancer (erythropoietin gene) during normoxia. This finding implies there exists a hypoxic-switch mechanism in cells co-expressing SIM1 vs SIM2 and EGLN1 proteins during normoxia vs hypoxia; thus, up- vs down-regulation causing activation of the EGLN1 gene occurs concomitantly with attenuation of SIM activities (Woods and Whitelaw, 2002). On the other hand, SIM1 and SIM2 do not form homodimers, and individually, neither one of them interacts with AHR (Probst et al., 1997).
In animals, these bHLH transcription factors participate in numerous developmental programs — such as cell migration, cell adhesion, DNA damage control and protection from oxidative stress, environmental adaptation, organogenesis, hematopoiesis, myogenesis, neurogenesis, and sex determination — which exist in all kingdoms of life (Pecenova and Farkas, 2016). In plants, bHLH transcription factors participate in a vast array of signal-sensing; the “signals” detected include diatomic O2, small metabolites (e.g. phytoalexins), and light, which control plant growth and development and plant-stress adaptation responses. For example, the signaling cascade that triggers the phytohormone jasmonate (224.3 Da) — modulates a diverse, but specific, range of aspects of plant growth, development and defense (Goossens et al., 2017).
PAS domain-containing proteins take part in circadian rhythmicity and regulate responses to environmental change (Vogt and Schippers, 2015). Hence, it should come as no surprise that AHR — originally discovered as a “sensor” of 3-methylcholanthrene (268.4 Da), benzo[a]pyrene (252.3 Da) and TCDD (322.0-Da) molecules — should be a member of the bHLH PAS family of transcription factors.
4.3. Generation of knockout and other transgenic mouse lines
4.3.1. Generation of the Ahr knockout mouse
Following publication of the mouse AHR NH2-terminal amino-acid sequence (Bradfield et al., 1991), Ahr(−/−) knockout mouse lines were independently created in several laboratories (Fernandez-Salguero et al., 1995; Lahvis and Bradfield, 1998; Shimizu et al., 2000). Perhaps not too suprising, Ahr(−/−) mice display strong resistance to the exceptionally toxic effects of TCDD (Fernandez-Salguero et al., 1996), protection against PAHs such as benzo[a]pyrene (Shimizu et al., 2000), and impaired DNA-PAH adduct formation caused by CYP1 enzymes (Kondraganti et al., 2003).
Far more interesting to some of us, however, was that Ahr(−/−) mice also exhibited: immune system impairment (Fernandez-Salguero et al., 1995; Lahvis and Bradfield, 1998); lack of closure of ductus venosus in the neonate (Lahvis et al., 2005); hepatic fibrosis (Fernandez-Salguero et al., 1995) and liver necrosis due to formation of numerous arteriovenous (A–V) shunts (Harstad et al., 2006); and impaired fertility, increased risk of embryonic death, multiple-organ dysregulation of organogenesis during in utero development, and stunted postnatal growth [reviewed in (Tohyama, 2014)].
More comprehensive examination has shown that AHR participates in numerous developmental signaling pathways, in many cases consistent with earlier findings in Ahr(−/−) mice, tissue culture, and biochemical experiments. These pathways include: cell-cycle regulation, immediate-early gene induction, cross-talk with the RB1/E2F axis, apoptosis, mitogen-activated protein-kinase cascades, and mobilization of pivotal calcium stores [reviewed in (Puga et al., 2009)]. This information from a decade ago has now been extended to the latest discoveries in Ahr(−/−) mice — including AHR-mediated dysfunctions in: ectoderm transition to epithelium; development of the heart, central nervous system (CNS), inner ear, liver, pancreas, kidney, bone formation, hematopoiesis, immune cell functions, and even glucose homeostasis. These phenotypes will covered in greater detail later in this review.
4.3.2. Generation of Cyp1 knockout mice
Because CYP1 gene induction appears to be the most sensitive to — and heavily reliant on — AHR regulation, details of these genes (CYP1A1, CYP1A2, and CYP1B1) are important to introduce. Whereas no human “knockout” equivalent has been found for AHR, CYP1A1 or CYP1A2, almost 150 CYP1B1 mutations are associated with primary congenital glaucoma [reviewed in (Li et al., 2011)]. Metabolites in the arachidonic acid pathway, cyclooxygenases, and prostaglandins are known to be altered in glaucomatous human ciliary muscle cells, compared with normal HCM cells; these data are consistent with lipid mediator (LM)-controlled regulation of uveoscleral outflow and, hence, glaucoma formation (Husain et al., 2002). Discovery of human CYP1B1 “knockouts” resulted in confirmation in the mouse, in which a defect in the ciliary body (which functions to control intraocular pressure) in Cyp1b1(−/−) mice is enhanced by absence of the tyrosinase gene (Tyr) — indicating pigmentation is important in contributing to the Cyp1b1-null phenotype (Libby et al., 2003). These data indicate that, during embryogenesis, development of the eye’s anterior chamber involves CYP1B1-mediated metabolism of a critical endogenous substrate in the LM second-messenger cascade.
CYP1 enzyme functions deserve to be mentioned. The vertebrate CYP1A1 monoxygenase metabolizes planar substrates — many of which include PAHs, certain halogenated biphenyls and dibenzofurans, β-naphthoflavone, and indole/tryptophan metabolites; CYP1A1 metabolizes only a few drugs. Constitutive CYP1A1 (mRNA or protein) expression in vertebrates is almost always nil (i.e. “promoter activity” only), whereas inducible CYP1A1 activity is ubiquitous, located in virtually every tissue and cell-type of the body. Inducible CYP1A1 is also seen in very early embryogenesis, even in liver of partial-hepatectomized mice (Kimura et al., 1987a), and also without exposure to any foreign chemical [reviewed in (Nebert et al., 2000; Nebert et al., 2004)].
CYP1A2 metabolizes at least two dozen drugs, including caffeine and theophylline; CYP1A2 substrates include many environmental aromatic amines and PAHs. Human CYP1A2 activity is not found in embryo or kidney; AHR-dependent CYP1A2 induction occurs mostly in liver, GI tract, pancreas, nasal epithelium, brain and lung [reviewed in (Nebert et al., 2004)]. CYP1A2 is very likely the result of a gene-duplication event with the older gene CYP1A1 [(Ahokas et al., 1979); reviewed in (Nelson et al., 1996)].
CYP1B1 (like CYP1A1) metabolizes many environmental chemicals — including certain PAHs and biphenyls, but in additiion metabolizes N-heterocyclic amines, arylamines and amino-azo dyes; metabolism can render the products carcinogenic and/or toxic, as well as detoxified, byproducts. Also similar to CYP1A1, CYP1B1 metabolizes only a few drugs. Unlike CYP1A1, however, CYP1B1 often displays substantial basal levels in endocrine tissues and certain cancers. Moreover, inducible CYP1B1 expression is about as ubiquitous as CYP1A1, with regard to expression in almost all tissues and cell-types, but especially in endocrine tissues and immune cells [reviewed in (Nebert et al., 2004)].
Laboratory animal studies show strong correlations between induced CYP1 activities and various types of cancer [reviewed in (Nebert, 1989)]. However, conclusions from innumerable clinical epidemiological studies on “CYP1 polymorphisms and cancers” are all questionable [reviewed in (Nebert and Dalton, 2006)], because it is now well established that CYP1 enzymes participate not only in metabolic activation but also detoxication; “activation” vs “detoxication” varies as a function of dose of carcinogen, route-of-administration, pharmacokinetics of the chemical, target-organ specificity, time, and genotype [reviewed in (Nebert et al., 2013a)].
Most importantly, substrates of all three CYP1 monooxygenases include an unknown number of polyunsaturated fatty acid (PUFA)-derived ω–6 and ω–3 lipid mediators (LMs). The relevance of this topic is discussed later.
4.3.3. Cyp1 gene structures and consequences of being ablated
The CYP1A1_CYP1A2 locus, located on human Chr 15q24.1 and mouse Chr 9 spanning nucleotides 57676937–57703822, contains the two genes oriented head-to-head, sharing a bidirectional promoter. CYP1A1 — on the reverse-strand — is 5′-ward of CYP1A2, with the two genes sharing a 23.3-kb (human) and 14.0-kb (mouse) bidirectional promoter between them. This “intergene-spacer region” contains multiple highly-conserved (AHREs) AHR-binding sites (Corchero et al., 2001), most or all of which presumably function in regulation of one or both CYP1A genes. CYP1B1, located on the reverse-strand of human Chr 2p22.2 and mouse Chr 17 spanning nucleotides 79706953–79715041, is in a subfamily distinct from that of CYP1A genes, because of much less homology and therefore having evolved from the CYP1A locus more than 420 million years ago (Poland et al., 1974; Probst et al., 1997). We can estimate this evolutionary date because fish genomes carry only the CYP1A1 and CYP1B1 genes, but not a CYP1A2 gene (Ahokas et al., 1979); sea vertebrates are known to have diverged evolutionarily from land vertebrates ~420 million years ago (Ahokas et al., 1979; Nelson et al., 1996).
Studies of mouse Ahr gene knockouts and all three Cyp1 single-, plus all three possible double-, and the triple-knockout (TKO) lines have been described. The single- and double-knockouts and cell-type conditional knockouts as experimental models have helped immensely in understanding the role of intestinal CYP1A1 in protection against oral benzo[a]pyrene [reviewed in (Nebert et al., 2013a)].
The eight gene knockout lines (described above) are all viable and able to reproduce — although serious viability problems are seen in the Ahr(−/−) knockout, as mentioned earlier (Fernandez-Salguero et al., 1995; Lahvis and Bradfield, 1998), and in Cyp1a1/1a2/1b1(−/−) TKO mice (Dragin et al., 2008). Interestingly, only about one in 40 TKO F1 newborns survived to adulthood (Dragin et al., 2008); yet, astonishly, although dwarfed in size, they were able to breed and quickly became more normal-in-size and healthier in subsequent generations. Most likely, unknown epigenetic effects are at play (Nebert et al., 2010). These fertile, healthy TKO mice are being shared worldwide with other laboratories.
TKO F1 pups exhibit phenotypes that include incomplete penetrance for: slower weight gain and greater risk of embryolethality before gestational day-11 (GD11); increased risk of hydrocephalus, hermaphroditism and cystic ovaries; and prominent suppression of the immune response to zymosan-induced peritonitis (Dragin et al., 2008). These traits were proposed (Dragin et al., 2008; Nebert and Karp, 2008) to be consistent with absence of (one or more of the three) CYP1 enzymes — leading to perturbations in critical concentrations of one or several of the >150 molecules identified in the LM second-messenger pathway [reviewed in (Serhan et al., 2007; Nebert and Karp, 2008; Nebert et al., 2013b)].
4.3.4. Generation of transgenic “knock-in” mice
Many experiments using “humanized” CYP1 knock-in transgenic mice have been described [(Derkenne et al., 2005; Dragin et al., 2006; Dragin et al., 2007; Jiang et al., 2005; Shi et al., 2008), reviewed in (Cheung and Gonzalez, 2008)] in which widely diverse questions were addressed. Further details are beyond the scope of this review.
4.4. Endogenous ligands of AHR
Throughout the 1970s, several colleagues argued that, if AHR recognizes only foreign chemical ligands, how could AHR also bind endogenous ligands? On the contrary, we and others argued that every endogenous receptor must have arisen during evolution primarily in order to carry out a critical life function, whereas the “capacity also to recognize one or another class of foreign chemicals” must have occurred as an “add-on” function. Indeed — in the entire field of toxicology today — the general realization and accepted rule is that uptake of the vast majority of foreign chemicals and ions into the cell occurs by way of one or another receptor, transporter, membrane moiety, or other subcellular mechanism.
From the mid-1970s to the present, therefore, many laboratories have enthusiastically searched for “the” endogenous ligand of AHR. It had first been proposed long ago (Nebert et al., 1981) that one or more endogenous ligands of AHR might represent CYP1-generated metabolites of the arachidonic-acid cascade, participating in inflammation.
Probably the first intriguing report of AHH activity being induced by something other than PAHs and TCDD was hepatic cell cultures being stimulated by light (Paine, 1977). The “search for an endogenous ligand of AHR” was extended to show AHR-mediated CYP1 induction by ultraviolet photoproducts of tryptophan, tryptophan metabolites, indole-containing compounds, indioids, equilenin, heme metabolites, arachidonic acid metabolites, dietary indoles, and foodstuff containing flavonoids. Other “unusual” stimulants included inflammation (Bigelow et al., 1989; Nebert, 1994), various alterations in cell culture conditions — e.g. changing plated cells to suspended cells, disruption of cell-cell contact, changes in cell shape, modulation by cyclic-AMP, exposure of serum to hydrodynamic shear stress, and modification of LDL levels [elegantly reviewed in (Nguyen and Bradfield, 2008) & later in (Soshilov and Denison, 2014; Hubbard et al., 2015)]. Hence, because of the proposed involvement of AHR and CYP1 in inflammation, and other advances described in the preceding sections, importance of the “AHR-CYP1 axis” was realized [reviewed in (Robertson et al., 1987; Nebert et al., 2004; Nebert and Karp, 2008)].
5. AHR and cell-signaling pathways
5.1. Lipid mediator (LM) second-messenger cascade
At least since the early 1980s, it has been accepted that LM second-messengers participate in the inflammatory process; also, that certain cytochromes P450 participate in the arachidonic acid pathway which represents a substantial portion of the LM-mediated pathway including the inflammatory response. The complexity of the LM cascade has been partially unraveled in recent years, due to advancements in high-pressure liquid chromatography, gas chromatography, tandem-mass spectrometry, and other sophisticated metabololipidomics methodologies.
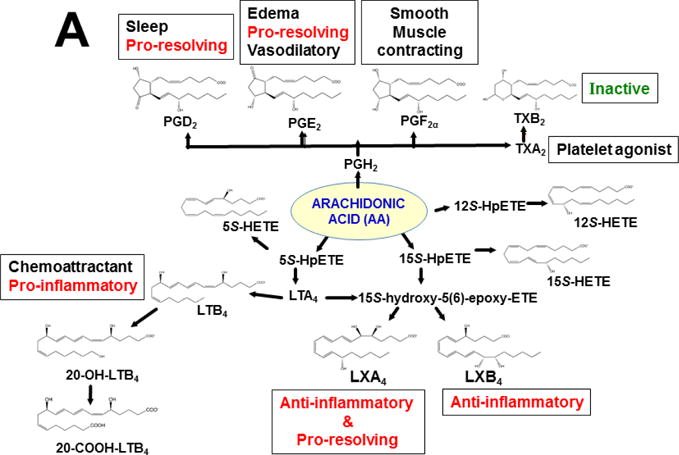
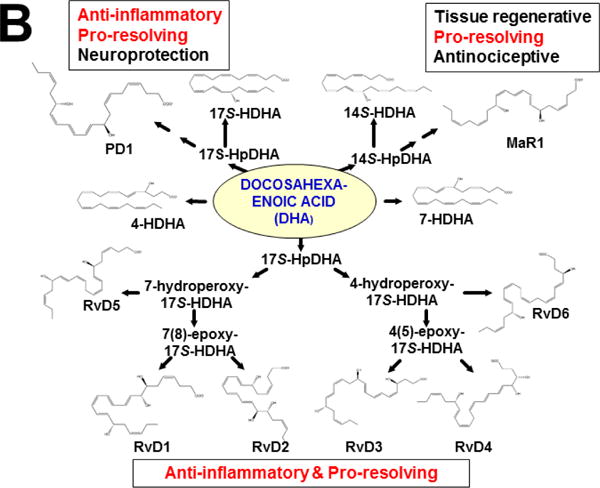
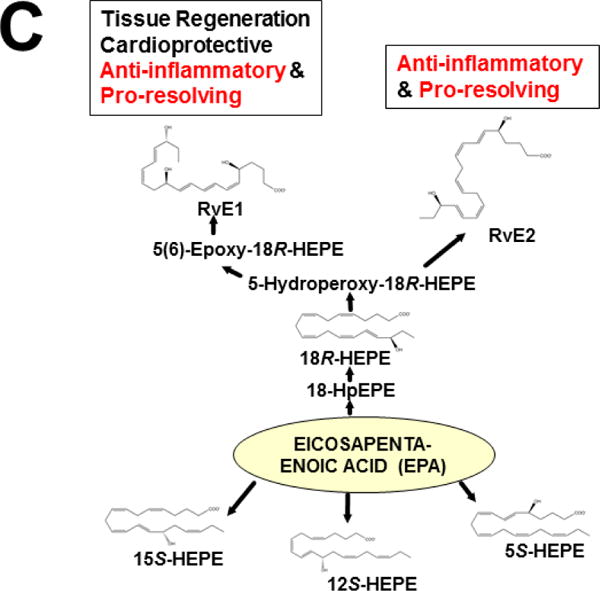
Originally derived from PUFAs — the (ω–6)- and (ω–3)-fatty acid intermediates (Figs. 3 & 4A) — are now known to be further metabolized in an extensive LM second-messenger cascade having potent bioactivities and comprising >150 identified metabolites (Samuelsson, 2012). The ω–6 fatty acid-derived arachidonic acid (AA) is converted [reviewed in (Nebert and Karp, 2008; Campbell et al., 2011; Greene et al., 2011; Nebert et al., 2013b; Hankinson, 2016)] to eleven known classes of LMs (Fig. 3). AA itself was shown long ago to be converted by cytochrome P450 to the EETs (Proctor et al., 1987), some of which are active in inflammation (Panigrahy et al., 2012). On the other hand, the two ω–3 fatty acid-derived (Fig. 3) docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA) are converted to four known classes of LMs (Campbell et al., 2011; Greene et al., 2011; Ji et al., 2011; Serhan et al., 2011; Mas et al., 2012).
Fig. 3.
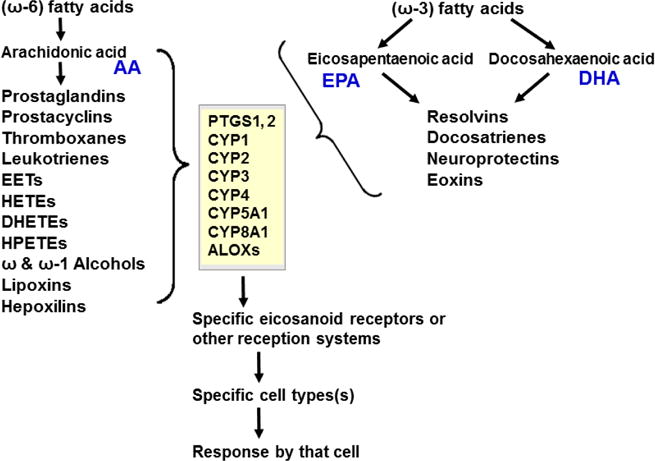
LM second-messenger pathways, derived from (ω–6)- and (ω–3)-fatty acids. The former gives rise to eleven classes of LMs derived from arachidonic acid (AA). The latter gives rise to four classes of LMs derived from eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA). Enzymes involved in the synthesis and degradation of the more than 150 total LMs in the 15 classes include two cyclooxygenases, members from six families of CYP monooxygenases, plus arachidonate lipooxygenases (ALOXs; six in human; seven in mouse). The “active” LMs are then “sensed by” (bind to) appropriate receptors in various organs and cell types, leading downstream to virtually every critical-life response in the organism [modified from (Nebert et al., 2013b)].
Fig. 4.
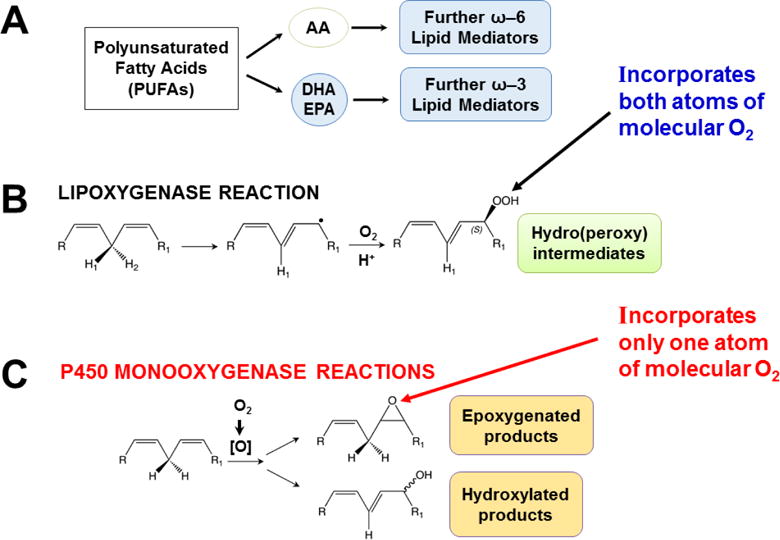
Polyunsaturated fatty acids (PUFAs), lipoxygenase vs monooxygenase mechanisms of reactions, and LM biosynthesis pathways. A, scheme depicting the origin of ω–6 and ω–3 LMs. B, diagram showing mechanism for the lipoxygenase reaction. C, diagram showing mechanisms for the P450-monooxygenase reactions.
5.1.1. LM metabolism by lipoxygenases vs P450 monooxygenases
Oxidative metabolism of LMs is carried out in multiple steps — not only by specific arachidonate lipoxygenases but also by specific P450 monooxygenases in the CYP1, CYP2, CYP3, CYP4, CYP5 and CYP8 families (Fig. 3) [reviewed in (Nelson et al., 2004; Nebert and Karp, 2008; Nebert et al., 2013b)]. Arachidonate lipoxygenases are encoded by six human genes: ALOX5, ALOX12, ALOX12B, ALOX15, ALOX15B, and ALOXE3, and seven mouse genes: Alox5, Alox12, Alox12b, Alox12e, Alox15, Alox8 and Aloxe3. Also, cyclooxygenases PTGS1 and PTGS2 (official names “prostaglandin-endoperoxide synthetases 1 and 2” encoded by the PTGS1 and PTGS2 genes, respectively; commonly misnamed “COX1 and COX2”) participate in the oxidative activation and inactivation of LMs. Although some P450-mediated specific reactions of bioactive LMs have been identified [(Divanovic et al., 2013) & reviewed in (Samuelsson, 2012; Nebert et al., 2013b)], the vast majority remains to be determined; this has been largely due to major technical challenges involved in identifying specific LM chemical structures, particularly with regard to their intricate stereochemistry.
Lipoxygenases (Fig. 4B) insert both atoms (Kühn et al., 1987), whereas P450 monooxygenases (Fig. 4C) insert one atom (Hayaishi et al., 1955; Mason et al., 1955; Mason, 1957), of diatomic oxygen — into substrates to form products. Another important distinction between the two types of reactions is that, although occasionally lipoxygenases can produce epoxides (e.g. leukotriene A4 formation by ALOX5), the major product is a fixed-chirality hydroperoxide; on the other hand, P450 monooxygenases can generate racemic mixtures of internal- and terminal-monohydroxy products, as well as epoxides which, following hydrolysis, often proceed to form racemic mixtures of dihydroxy products (Fig. 4C).
Ultimately, among many other functions [reviewed in (Nebert and Karp, 2008; Nebert et al., 2013b)], AA-derived LMs more likely participate in the pro-inflammatory phase, whereas the DHA- and EPA-derived metabolomes orchestrate the resolution phase of self-limited inflammatory responses. In addition, the location of many of these metabolic reactions is usually highly tissue- and/or cell type-specific (Nebert and Karp, 2008; Campbell et al., 2011; Greene et al., 2011; Nebert et al., 2013b).
5.1.2. LMs and the AHR-CYP1 axis
The first convincing LM identified to act apparently as an AHR ligand was lipoxin A4 (Schaldach et al., 1999), followed later by 12(R)-hydroxy-5,8,10,14-eicosatetraenoic acid [12(R)-HETE] (Chiaro et al., 2008). As a means of dissecting the challenging problems of identifying which CYP enzyme might participate in what step(s) of the LM cascade involved in acute inflammation, one possibility would be to use Cyp1 knockout mouse lines (Dragin et al., 2008) — in combination with the latest advances in metabololipidomics (Dalli and Serhan, 2012) — for separating and identifying as many unique LM metabolites as technically possible.
This approach was recently carried out, using as an endpoint, zymosan-induced peritonitis in the Cyp1a1/1a2/1b1(−/−) TKO mouse model, to see if an inflammatory response would alter the LM metabolite profile (Divanovic et al., 2013). The names and chemical structures of the AA-, DHA- and EPA-derived LMs — which were able to be identified by the metabololipidomics instrumentation methodology used — are illustrated in Figs. 5A, 5B & 5C, respectively; also listed (enclosed in rectangles) are some of the critical-life responses elicited by that particular LM active metabolite (Divanovic et al., 2013).
Fig. 5.
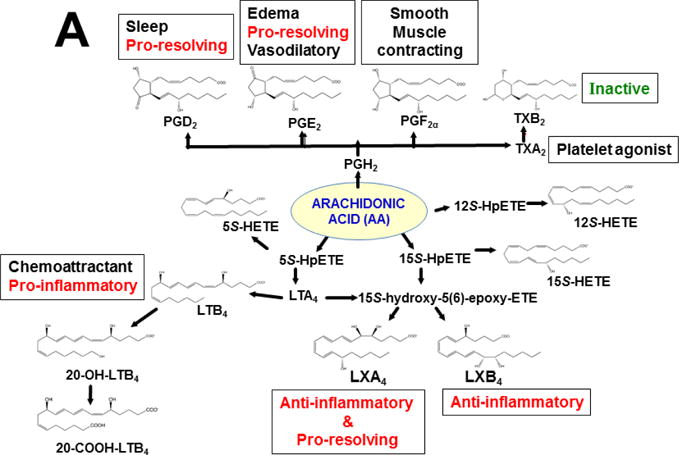
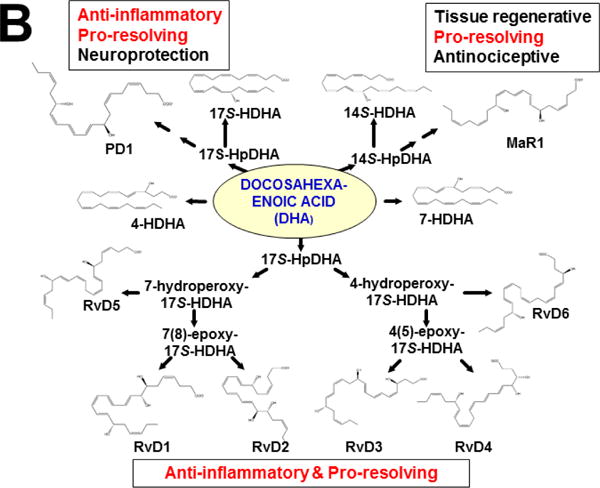
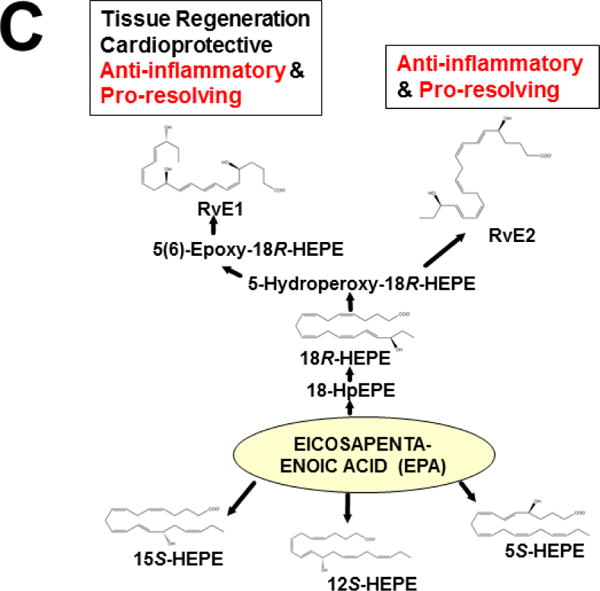
LM metabolites that were able to be identified and quantified by the multiple-reaction monitoring and liquid chromatography-UV coupled with tandem mass spectrometry-based LM metabololipidomics system used (Divanovic et al., 2013). The three LM metabolomes include: A, arachidonic acid (AA)-derived, B, docosahexaenoic acid (DHA)-derived, and C, eicosapentaenoic acid (EPA)-derived LMs. For various metabolites in the pathways, some of the established critical-life processes elicited by specific LMs are depicted in the rectangular boxes. Processes described in red font denote pro-inflammatory and pro-resolving inflammatory functions, i.e. initiation and resolution of inflammation, respectively.
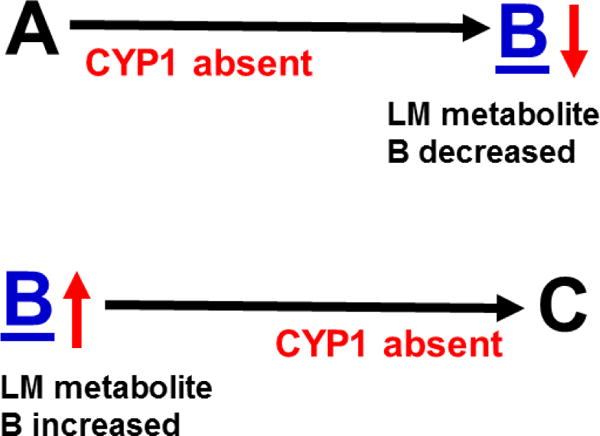
The rationale is explained in Fig. 6. When comparing TKO with wild-type mice, if one (or more) of the three CYP1 enzyme(s) participates in any metabolic step before LM B, then the concentation of B would be decreased (due to ablation of CYP1). In contrast, if one (or more) CYP1 enzyme(s) is involved in any metabolic step after B, then the level of B would be increased (due to ablation of CYP1), when comparing TKO with wild-type mice.
Fig. 6.
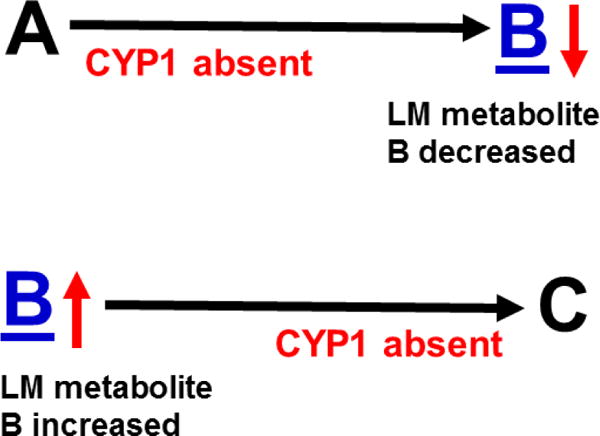
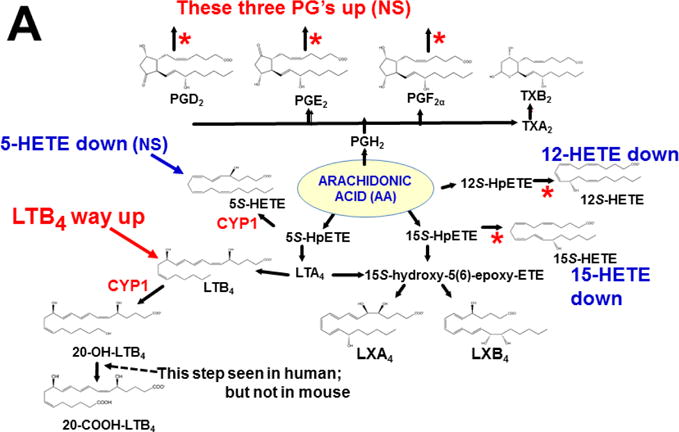
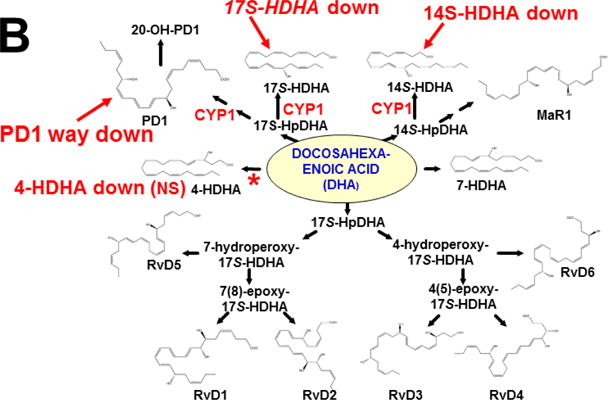
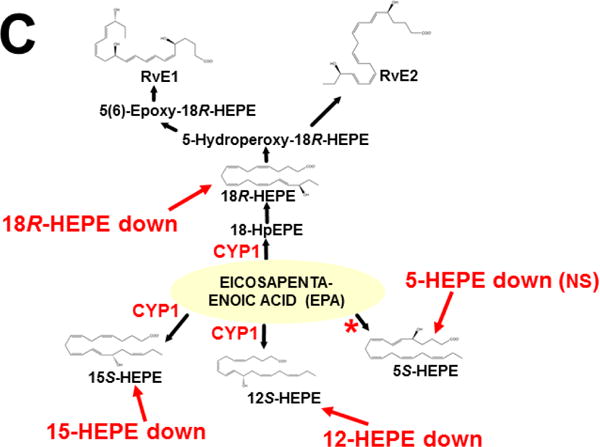
Hypothesis designed for the study of peritoneal exudate of zymosan-treated mice (Divanovic et al., 2013). A, B and C denote metabolites in the LM second-messenger cascade that can be identified and quantified by the multiple-reaction monitoring and liquid chromatography-UV coupled with tandem mass spectrometry-based LM metabololipidomics system (Divanovic et al., 2013). If CYP1A1, CYP1A2 and CYP1B1 monooxygenases are all involved, just two involved, or just one of the three enzymes involved — in a metabolic step between A and B, then metabolite B levels will be decreased in TKO mice, compared with wild-type (WT) mice. If three, two or one of the CYP1 enzymes are(is) involved in a metabolic step between B and C, then metabolite B levels will be increased in TKO mice, compared with WT mice.
The results (Divanovic et al., 2013) are summarized in Figs. 7A, 7B & 7C. When comparing TKO with wild-type mice, those steps altered — are denoted in red as “way up” (P <0.05), simply “up” or “down” (P <0.08 >0.05), and “up (NS)” or “down (NS)” (trend seen; P <0.12 > 0.08). Therefore, zymosan-induced peritonitis appears to be associated with eight substantially altered metabolic steps in TKO, compared with wild-type mice: increases in neutrophil LTB4 levels (P <0.05) and decreases in PD1 were the most striking differences; six other pathways slightly affected (P < 0.08 >0.05) included decreased 5S-HETE, neuroprotectin D1/protectin D1, 17S-HDHA, 14S-HDHA, 18R-HEPE, 15S-HEPE, and 12S-HEPE (Divanovic et al., 2013).
Fig 7.
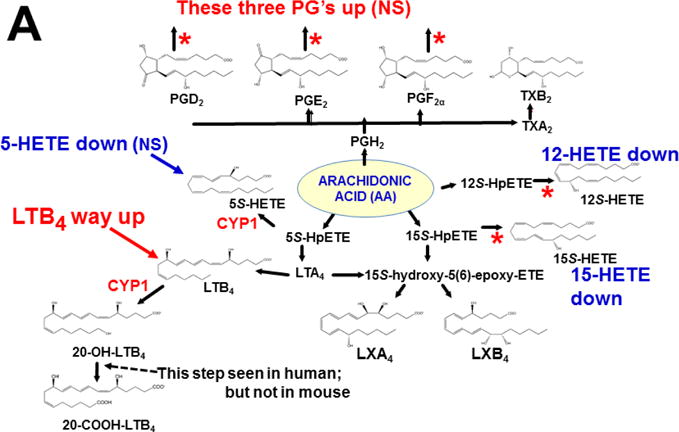
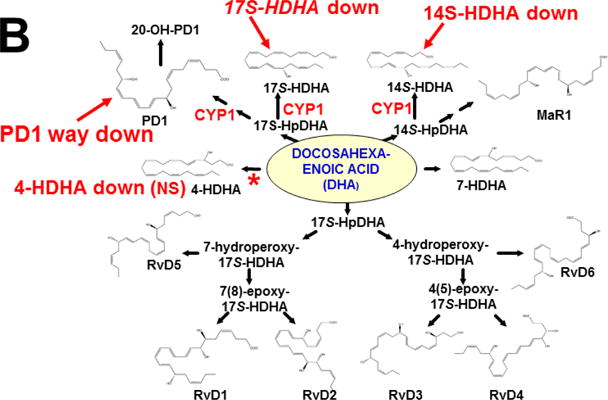
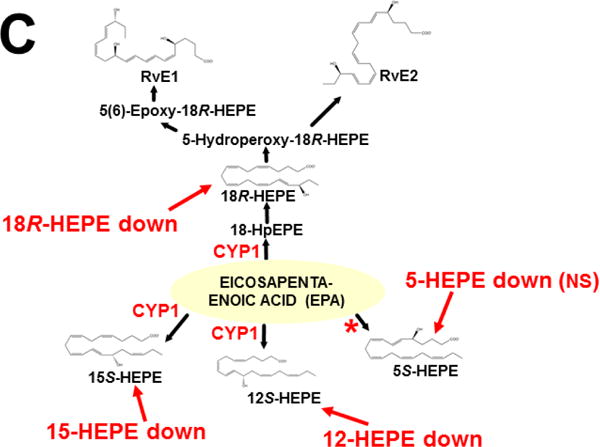
Identification of metabolic steps at which CYP1 enzymes are proposed to participate (Divanovic et al., 2013). A, AA-derived lipoxins, prostaglandins, thromboxanes, and leukotrienes. B, DHA-derived resolvins, protectins and maresins. C, EPA-derived resolvins. CYP1 labels are placed in accordance with the findings in the study, combined with what is known about CYP-mediated LM metabolism: “way up” or “way down” = P <0.05; “up” or “down” = P <0.08 > 0.05; and * denotes “Trend,” = P <0.12 > 0.08. Positions of the (bolded red) CYP1 labels and the bolded red asterisks depict the proposed steps, as determined in the study (Divanovic et al., 2013). In cases where there are two or more steps between the identifiable LM metabolite, the precise step at which CYP1 acts, and which one (or two or three) of the three CYP1 enzymes participates — will require further experiments.
This initial study only screened the possibility that any (or all) of the three CYP1 monooxygenases might affect the LM metabolite profile; these data should be corroborated using larger numbers of animals. Further, the study has the limitation of failing to distinguish whether one (or more) of the three CYP1 enzymes participates in a specific step in the LM cascade; to expand on the initial study, therefore, future experiments should be carried out, using Cyp1 single-knockout mice, compared with Cyp1 double- and triple-knockout mice. Mouse and human enzymes in the LM metabolite pathway are not always absolutely conserved — e.g. the 20-hydroxy-LTB4 conversion to 20-COOH-LTB4 step (Fig. 7A) was found to occur in human, but not mouse, neutrophils (Divanovic et al., 2013). Hence, humanized CYP1 knock-in mice [(Derkenne et al., 2005; Dragin et al., 2006; Dragin et al., 2007; Jiang et al., 2005; Shi et al., 2008), reviewed in (Cheung and Gonzalez, 2008)] should also be studied. Subsequently, various other studies using Cyp2, Cyp3 and Cyp4 single-knockout mice, compared with the (replaced) human orthologous CYP, CYP3, CYP4 gene knocked-in (Scheer and Wolf, 2014; Scheer and Wilson, 2016), should also be carried out.
Results of such studies, including examination of specific cell types — other than neutrophils and peritoneal exudate — would define contributions of specific P450 monooxygenases to the LM second-messenger response that mediates various LM-mediated critical-life processes. Possibly, these findings would ultimately provide potentially useful new therapeutic targets for treating either the pro-inflammatory or the post-inflammatory response.
5.2. AHR-signaling pathways involved in critical-life processes
An appreciation of the vast depth of AHR involvement in many critical life processes has exploded in recent years. A summary of AHR-mediated pathways is listed in Table 1. Further details comprise the remainder of this review.
Table 1.
Summary of organs, systems, cell functions, and developmental biology in which AHR-signaling is involved.
| Location | AHR-signaling pathway involvement |
|---|---|
| Central nervous system | Development of brain and nervous system; Neurogenesis; Neuronal cell development; Cardiorespiratory brainstem development in ventrolateral medulla; “Brain-gut-microbiome” |
| Eye | Ciliary body formation and function; Thyroid-associated eye disease |
| Gastrointestinal tract | Development of GI tract; Rectal prolapse during aging; “Brain-gut-microbiome” |
| Heart | Development of heart organ; Cardiovascular physiology; Atherogenesis; Cardiomyogenesis; Cardiorespiratory function |
| Hematological system | Development of blood cell-forming system; Hematopoiesis; Activation or suppression of erythroid development |
| Immune system | Immune system development; The immune response; Innate immunity; Pro-inflammatory response; Anti-inflammatory response; Immunomodulatory effects |
| Inner ear | Development of the cochlea |
| Kidney | Development of the kidney; Hypertension |
| Liver | Development of liver organ; Hyperlipidemia; Glucose and lipid metabolism; Hepatic steatosis |
| Musculoskeletal system | Transmesoderm–>osteoblast transition; Bone formation; Osteoclastogenesis |
| Pancreas | Development of pancreas; Beta-cell regulation; Pancreatic fibrosis |
| Endocrine system | Serum lowered testosterone levels; Infertility; Mammary gland duct cell epithelial hyperplasia; Degenerative changes in testis; Gerrm-cell apoptosis; Endometriosis |
| Reproductive system | Development of male and female sex organs; Spermatogenesis; Fertility |
| Respiratory tract | Development of respiratory tract; Disruption of GABA-ergic transmission defects; Cardiorespiratory function |
| Vascular system | Angiogenesis; Atherosclerotic plaque formation |
| Skin | Barrier physiology; Atopic dermatitis |
| Cellular functions | Cell migration; Cell adhesion; Circadian rhythmicity |
| DNA changes | DNA synthesis; DNA repair; DNA-adduct formation; Mutagenesis |
| Oxidative stress | Mitochondrial ROS formation; Anti-oxidant protection against ROS formation; Mitochondrial H2O2 production; Crosstalk with hypoxia and HIF-signaling pathways; Transforming growth factor-signaling pathways; MID1-PP2A-CDC25B-CDK1 signalig pathway regulating mitosis |
| Tumor cells | Growth suppression; Tumor initiation; Tumor promotion |
| ES cell basic functions | Ectoderm–>epithelium transition; Cell adhesion; Cell-cycle regulation; Apoptosis; Cavitation during morula–>blastula formation; Activator of Rho/Rac GTPases; WNT-signaling pathways; Homeobox-signaling pathways |
| Other basic functions | Transgenerational inheritance; Epigenetic effects; Chromatin remodeling; Histone modification; Aging-related and degenerative diseases |
AHR-signaling is now known to participate in: cell migration (Mulero-Navarro and Fernandez-Salguero, 2016), epithelial cell development (Ikuta et al., 2009), cytoskeletal/adhesion regulation (Zhang et al., 2016), circadian rhythmicity (Anderson et al., 2013), barrier organ physiology (Esser and Rannug, 2015), cardiovascular and respiratory physiology (Sauzeau et al., 2011b), kidney development (Nanez et al., 2011), inner ear cochlear development in the neonate (Safe and Luebke, 2016), bone formation (Herlin et al., 2013) and osteoclastogenesis (Iqbal et al., 2013; Izawa et al., 2016), GI tract (Hubbard et al., 2015; Schiering et al., 2016), intestinal immunity (Qiu and Zhou, 2013), innate immunity (Cella and Colonna, 2015), hematopoiesis (Lindsey and Papoutsakis, 2012; Fracchiolla et al., 2016), transgenerational inheritance (Baker et al., 2014), reproductive organ development (Mulero-Navarro and Fernandez-Salguero, 2016), regulation of female reproduction (Hernandez-Ochoa et al., 2009), prostate gland development (Schneider et al., 2014), hyperlipidemia, atherogenesis, and hypertension (Xiao et al., 2014), thyroid-associated eye disease (Woeller et al., 2016), eye and ciliary body function (Shichi and Nebert, 1982; Volotinen et al., 2009), hepatic steatosis (Mellor et al., 2016), pancreatic beta-cell regulation (Sabatini and Lynn, 2015), glucose and lipid metabolism (Gooley, 2016), circadian clock and metabolic syndrome disruption (Jaeger and Tischkau, 2016), DNA damage control (Wells et al., 2010), tumor prevention by regulating gut immunity and growth suppression in tumor cells (Ikuta et al., 2016), and protection against oxidative stress (Wölfle et al., 2014).
Is there a “common thread” connecting most — if not all — of these critical life processes? The answer is yes: LM-mediated second-messenger pathways are involved in the regulation of every one of these critical life processes.
5.2.1. AHR and the “brain-gut-microbiome”
Realizing the importance of the “brain-gut-microbione” has greatly expanded during the past decade [reviewed in (Luna et al., 2016; Mazzoli and Pessione, 2016)]. Microbes in the GI tract are now appreciated to provide each host with innumerable beneficial functions (e.g. contributions to vitamin supplementation, food digestion, and defense against pathogens of all kinds). The “microbiome” interacts with the host organism’s genome and physiology — by way of direct contact through surface antigens, as well as soluble molecules produced by bacterial metabolism. There is clearly bi-directional communication between the GI tract and the CNS, which also provides communication pathways between intestinal microbiota and the host’s neural circuitry including the CNS. Modulation of GI tract and CNS function via the microbiome is now recognized to include such ill-defined traits as “behavior,” cognitive functions, “mood,” “suicidal tendencies,” “obsessive-compulsive disorder,” appetite, and autism spectrum disorder. Production of bioactive compounds by microbiota and their potential probiotic activities consists of neuroactive molecules — e.g. histamine, serotonin, catecholamines, and trace amines (Luna et al., 2016; Mazzoli and Pessione, 2016).
Termed “the holobiont,” the microbiome of each host and its organs forms a close functional entity having evolutionarily designed interactions that support nutritional intake, defense mechanisms, and reproduction (Lee et al., 2016). AHR is considered as an evolutionarily highly-conserved “sensor” capable of recognizing endogenous as well as foreign chemicals/signals — including many ligands that are metabolites produced directly, or indirectly, by the microbiome. Thus, AHR-signaling pathways most likely elicit substantial impact on underlying mechanisms in neurodevelopment and neurodegeneration.
The balance — between pro- and anti-inflammatory signaling — is critical for maintaining immune homeostasis under physiological conditions, as well as in responding to inflammation (activation vs suppression) in different pathological settings. Recently identified gene products found to participate in immune system regulation include: PD1 (programmed cell death-1 receptor), CTLA4 (cytotoxic T-lymphocyte antigen-4), galectins, the intracellular enzyme indoleamine 2,3-dioxygenase (IDO), CD69, AHR, and members of the growth-arrest and DNA-damage-inducible-45 (GADD45) family (Fornace, Jr. et al., 1989). These models have emerged as potential targets for regulation of the activation-suppression balance in immune cell function (de la Fuente et al., 2012).
In the GI tract, CYP1A1 expression depends on TOLL-like receptor-2 (TLR2) (Do et al., 2012) — which recognizes bacterial surface antigens, e.g. lipoteichoic acid. Presence of ligands of bacterial origin for TLR2 appears to be pivotal for detoxication of oral PAHs by intestinal CYP1A1. For example, gut epithelial CYP1A1 is silenced in Tlr2(−/−) knockout mice, even when exposed to substantial amounts of benzo[a]pyrene; after dietary daily benzo[a]pyrene for three weeks, Tlr2(−/−) but not their wild-type littermates developed colonic polyps (Do et al., 2012).
These data bring to mind an earlier study using an Arnt(−/−) conditional knockout specific for GI tract epithelium (Ito et al., 2007): CYP1A1 expression (without foreign chemical treatment) was found to be strikingly elevated in virtually all non-GI-tract tissues; high CYP1A1 levels were seen — even in early-stage fetuses in pregnant Arnt(−/−) mice. Curiously, a dramatic induction of the metallothionein-1 gene (Mt1) was seen throughout the GI tract. Intriguingly, CYP1A1 over-expression was lost upon administering a synthetic purified diet. Further, high-CYP1A1 expression was “recovered” by adding the phytochemical indole-3-carbinol to the purified diet. It is possible that these observations (Ito et al., 2007) might involve TLR2 activation in GI tract, as described above (Do et al., 2012).
Finally, dysregulated expression of intestinal CYP1A1 was recently found to deplete the reservoir of naturally-occurring AHR ligands, thereby generating a quasi-AHR-deficient mouse; using a Cyp1a1 reporter model system, Schiering and coworkers identified intestinal epithelial cells as “first-line responders” to dietary AHR ligands (Schiering et al., 2016). Whether CYP1A1 is constitutively expressed throughout the body, or restricted specifically to intestinal epithelial cells — the result was loss of AHR-dependent type-3 innate lymphoid cells and T-helper-17 (Th17) cells, as well as enhanced susceptibility to enteric infection. These striking effects on intestinal immune system function of excessive AHR ligand degradation, could be counter-balanced by increasing dietary AHR ligand levels. These findings indicate that intestinal epithelial cells serve as “gatekeeper” for supply of AHR ligands to the host (Schiering et al., 2016), thereby emphasizing the importance of feedback-control in modulating AHR-signaling pathway activation through the brain-gut-microbiome.
There is a relevant connection between gut microbiota and caspase recruitment domain family member-9 (CARD9). Card9 is a susceptibility gene for inflammatory bowel disease (IBD) and helps recovery from colitis by promoting interleukin-22 production; Card9(−/−) knockout mice are more susceptible to colitis. Microbiota from Card9(−/−) mice, as well as IBD patients cannot metabolize tryptophan efficiently into metabolites that act as AHR ligands. Colitis can be attentuated after inoculation of mice with bacteria capatable of metabolizing tryptophan or by AHR agonist treatment (Lamas et al., 2016).
5.2.2. AHR-signaling in endocrine dysregulation
For at least four decades, accumulating evidence has suggested the AHR-CYP1 axis participates in one or more pathways that elicit anti-estrogenic and anti-androgenic effects, both by TCDD or PAH treatment, as well as by interference or competition with sex hormone and angiogenesis action; female and male reproductive organs — including oocytes, spermatocytes, and fertility — can be affected.
AHR ligands are suggested to be involved in endocrine disruption. A number of studies have implied AHR interactions with estrogen receptor-α and -β (ESR1, 2); however, many of the purported ligands typically induce trans-activation of estrogen-responsive genes and reporter constructs, but their potencies are usually at least 1,000-fold lower than that observed for 17β-estradiol. Moreover, many cell assays are carried out in transformed or malignant cells in culture — which are notoriously polyploid and, thus, gene-expression pathways are far from “normal”. Therefore, risk assessment of estrogenic compounds on the basis of their potencies in simple reporter-gene or binding assays is likely to be inappropriate [reviewed in (Safe et al., 2002)].
Today it is clear, for example, that AHR activation and natural cytotoxicity-triggering receptors NCR1/2/3 activation by endogenous, as yet undefined, ligands contributes to uterine non-killer (NK) cell activation/maturation and angiogenic functions during early- to mid-gestation pregnancy. Major histocompatibility complex (MHC)-independent activation of uterine NK cells also likely provides critical contributions to human pregnancy success [reviewed in (Felker et al., 2013)].
The doses of environmental toxicants employed in laboratory-animal experiments commonly exceed any comprehensible level found in the environment. Hence, the dose of exposure, timing, route-of-administration, and target cell-type must all be scrutinized very carefully [reviewed in (Safe et al., 2002; Kreitinger et al., 2016)].
On the other hand, a recent investigation of Ahr(−/−) mice without foreign chemical treatment (Huang et al., 2016) revealed lowered serum testosterone levels in males and significant growth of mammary ducts with elevated ESR1 expression in the ductal epithelium. Further abnormalities included: decreased expression of Notch1 and Notch3 receptors and their downstream target HES1 (hairy-and-enhancer of split-1); diminished fertility with degenerative changes in the testis; germ-cell apoptosis; and decreased number of early spermatids (Huang et al., 2016).
Comparing women with vs without endometriosis, intriguingly, there is an association between this uterine disorder and up-regulation of AHR and ARNT expression and all three CYP1 target genes in endometriotic tissues and stromal cells (Bulun et al., 2000). This clinical study prompted an investigation in Rhesus monkeys chronically exposed to TCDD and other halogenated PAHs (Rier et al., 2001). TCDD exposure and elevated serum TCDD concentrations were associated with increased serum levels of triglycerides, 1,2,3,6,7,8-hexachlorodibenzofuran, 3,3′,4,4′-tetrachlorobiphenyl (TCB), and 3,3′,4,4′,5-pentachlorobiphenyl (PnCB). Those having elevated serum levels of 3,3′,4,4′-TCB, 3,3′,4,4′,5-PnCB, and increased total serum toxic equivalents — had a high prevalence of endometriosis, and disease severity was correlated with serum concentrations of 3,3′,4,4′-TCB.
In humans, a meta-analysis of four genome-wide association studies, plus four replication studies (which included altogether totals of 25,871 endometriosis cases and 65,356 controls, and combining several ethnic groups), uncovered six statistically significant (P ≤ 5 × 10−8) genome-wide significant loci — which included rs7521902 near WNT4 (P = 1.8 × 10–15) on human Chr 1p36.12 (Rahmioglu et al., 2014). This association is particularly intriguing in this review because of AHR’s involvement in WNT4-signaling pathways [vide infra].
In summary, AHR-signaling involvement in reproductive organs is associated with such diverse clinical disorders as infertility, mammary ductal epithelial cell hyperplasia, and endometriosis. The underlying cause of AHR-associated endometriosis is fascinating and will require further study; etiology remains controversial and could involve: [a] CYP1-mediated metabolic activation of unknown chemicals to toxic intermediates, or ROS formation; [b] endocrine-specific growth-factor or tumor promoter-like activity; [c] dysregulation of the immune response caused by inflammatory signals via certain environmental toxicants; or [d] epigenetic effects comprising histone modification and chromatin remodeling (Sofo et al., 2015).
5.2.3. AHR-CYP1 axis and reactive oxygen species (ROS) formation
Interestingly, administering intraperitoneal TCDD (high dose of 5 μg/kg; three consecutive days) was found to cause a dramatic, protracted oxidative stress response. Hepatic oxidized glutathione levels are sustained for at least 8 weeks; urinary levels of 8-hydroxydeoxyguanosine (a product of DNA-base oxidation and subsequent excision repair) remain ~20-fold higher than controls after 8 weeks — consistent with chronic, potentially promutagenic DNA-base damage (Shertzer et al., 1998). Mitochondrial H2O2 production rises at 1 and 4 weeks, remaining significantly elevated at 8 weeks; enhanced H2O2 production is not due to either increased Mn-superoxide dismutase or decreased mitochondrial glutathione peroxidase activity (Senft et al., 2002a). This was the earliest report suggesting that AHR-signaling pathways are involved in mitochondrial respiration-dependent ROS formation.
The initial hypothesis was that TCDD-induced mitochondrial ROS production most likely would be CYP1-mediated. Using Cyp1a1(−/−) and Cyp1a2(−/−) mice, alone or in combination with Ahr(−/−) knockout mice, however, it was concluded that both constitutive and TCDD-induced mitochondrial ROS formation is AHR-mediated — but independent of CYP1A1/2 metabolism (Senft et al., 2002b).
Also noteworthy is the fact that AHR-signaling includes up-regulation of electrophile-responsive antioxidant enzymes [reviewed in (Nebert et al., 2000)]; for example, Nqo1 (NAD(P)H:quinone oxidoreductase-1) and Aldh3a1 (aldehyde dehydrogenase-3A1) genes carry promoter binding-sites for both AHR and NFE2L1 [nuclear factor (erythroid-derived 2)-like 1; previous name NRF1].
Hence, there appears to be a “yin-yang equilibrium,” controlled by AHR, i.e. both up- and down-regulation of ROS formation. This delicate balance occurs in such seemingly unrelated signaling pathways as: circadian rhythmicity (Patel et al., 2014); enhanced immune response exhibiting increased levels of inflammatory cytokines and decreased Socs2 (suppression of cytokine signaling-2) expression — that had been observed in zymosan-caused peritonitis in liver of Cyp1a1/1a2/1b1(−/−) triple-knockout mice (Nebert and Karp, 2008) — and infectious myocarditis (Barroso et al., 2016); inflammatory plaque formation during atherosclerosis (Marinkovic et al., 2013; Uno et al., 2014; Pernomian and da Silva, 2015); hepatic steatosis (Gooley, 2016; Mellor et al., 2016; Nault et al., 2017); atopic dermatitis associated with a dysfunctional skin barrier (Nomura and Kabashima, 2016); and aging and age-associated diseases (Gasiewicz et al., 2014; Patel et al., 2014).
5.3. AHR-signaling pathways in embryonic stem (ES) cells
5.3.1. Early evidence of AHR-CYP1 signaling in the one-cell zygote
Why does AHR participate in such an abundance of critical-life processes in virtually every organ and cell type of the body? The first hint of “early AHR-CYP1 signaling” during embryogenesis came with sister-chromatid-exchange (SCE) studies; mouse embryo GD7.5 explant cultures in medium containing benzo[a]pyrene and 5-bromodeoxyuridine results in SCEs are associated with AHR-responsive, but not AHR-nonresponsive, mouse lines (Galloway et al., 1980). These data provided direct evidence in 1980 that the AHR-CYP1 axis was functional at an early embryonic stage.
It was then found that AHR-CYP1 signaling is detectable in the mouse oocyte; curiously, within 12 h after fertilization — without any “foreign ligand signal” — a >100-fold increase in Cyp1a1 mRNA levels was found (Dey and Nebert, 1998), following which this disappears by the 2-cell stage (GD1.5), as well as in the blastocyst (GD3.5).
This finding, first submitted as a manuscript in 1988, was another example of being rejected from publication (as “implausible” and “heresy”). Ten years later, however, supported with additional experimental data, a proposed hypothesis became feasible for activation of AHR-CYP1 signaling in the newly formed zygote following fertilization [reviewed in (Nebert et al., 2000)].
AHR was discovered to play pivotal roles in cell-cycle regulation and apoptosis (Fig. 8). AHR interacts directly with the RB1 protein (Ge and Elferink, 1998), and formation of the AHR-RB1 complex prevents normal progression of G1 to S phase during the cell cycle, by blocking E2F-mediated transcription of S-phase genes (Puga et al., 2000). Hence, in the presence of sufficient amounts of any unknown endogenous ligand (or undesirable foreign chemical able to bind to AHR), activated AHR would cause arrest at the G1/S boundary (Fig. 8B). Availability of CYP1A1 monooxygenase therefore appears to be critical in the fertilized egg, because — if AHR-mediated CYP1A1 activity is able to metabolize/detoxify any harmful AHR ligand — substantial amounts of CYP1A1 activity would prevent AHR from blocking progression of the 1-cell embryo from G1 into S phase — without which the result, of course, would be death! Given the details of AHR-CYP1 signaling in the second-messenger LM cascade (vide supra), the unknown endogenous ligand in the zygote is very likely to be one (or more) of the LM active metabolites.
Fig. 8.
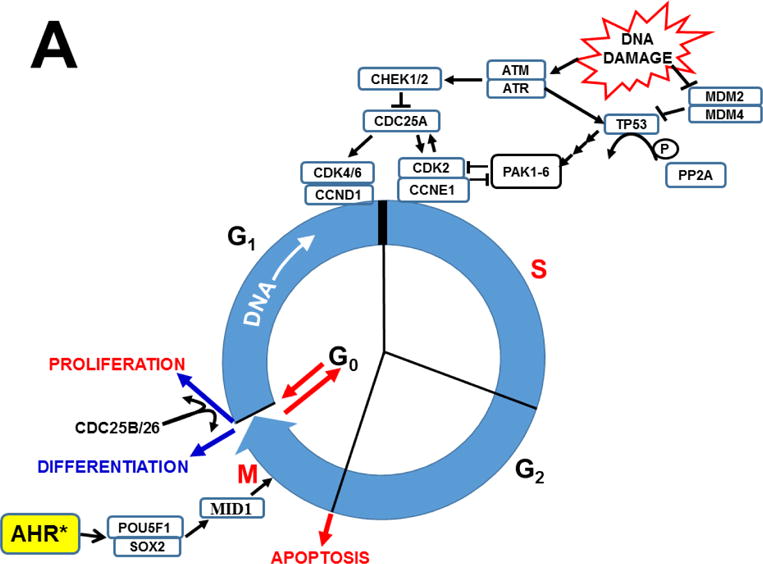
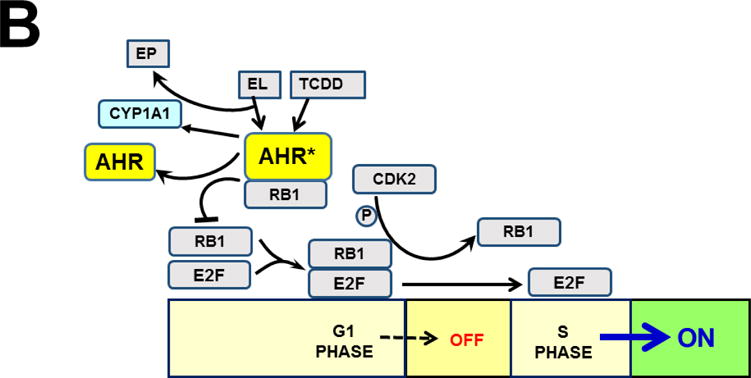
Established effects of AHR and the AHR-CYP1 axis on cell cycle functions. Arrows denote “activation or stimulation” whereas lines with a foot indicate “repression or inhibition.” A, blue circle denotes the cell cycle (G1 –> S –> G2 –> M –> return to the G1 phase). The G1/S cell cycle checkpoint controls commitment of eukaryotic cells to transition through the G1 phase to enter into the DNA synthesis S phase. The G2/M checkpoint precedes the cell’s entrance into mitosis, with locations indicated when cells have specific options (e.g. G0 for the inactive, or resting, phase; versus processes such as apoptosis, differentiation, and proliferation). Two cell cycle kinase complexes — CDK4/6 (cyclin-dependent kinase 4 & 6)-CCND1 (cyclin D1), and CDK2-CCNE1 (cyclin E1) — participate as part of a dynamic transcription complex, to move the cell from G1 to S phase (described further in panel 8B). DNA damage (upper right) activates ATM and ATR (serine-threonine kinases), and also represses MDM2 and MDM4 (MDM proto-oncogenes 2 & 4), thereby releasing their inhibition of TP53 in concert with activation of TP53 by ATM/ATR. Phosphorylation (P denotes inorganic PO4) of TP53 by PP2A (protein phosphatase-2A), via several steps, activates PAK1–6 (p21 (RAC1; CDKN1A)-activated kinases-1 through -6), which results in repression of CDK2. Whereas CDC25A (cell division cycle-25A) activates both CDK4/6 and CDK2, CDC25A is suppressed by CHEK1/2. At the M/G1 boundary (lower left), the CDC25B/26-CDK1 complex induces either differentiation or proliferation. Activated AHR* stimulates the complex of POU5F1 (POU class-5 homeobox-1; formerly OCT4) and SOX2 (SRY-box-2), which in turn up-regulates MID1 (midline-1), thus allowing MID1 to activate mitosis. For the sake of avoiding clutter, many additional factors participating in the cell cycle are not shown. The two cell cycle kinase complexes — CDK4/6-CCND1 (cyclin D1) and CDK2-CCNE1 (cyclin E1)-CDK2 — work in concert to relieve inhibition of the dynamic transcription complex (shown in panel B) that includes RB1 and E2F. B, in G1-phase uncommitted cells, hypo-phosphorylated RB1 binds to the E2F-transcription complex, whereas phosphorylation of RB1 by CDK2 releases RB1, thus allowing E2F-mediated S-phase genes, a requirement for DNA synthesis, to be turned on. At top, TCDD (or other planar foreign chemical) or endogenous ligand (EL) binds to AHR, which activates it to AHR*; this allows it to complex with RB1 which in turn prevents RB1 binding to E2F. AHR*-mediated up-regulation of CYP1 results in metabolic degradation of EL to the endogenous product EP. This removes EL from AHR*, inactivates AHR which releases RB1, making it then able to bind to E2F and promote G1–>S progression. [Portions of panel A were helped by https://www.cellsignal.com/common/content/content.jsp?id=pathways-cc-g1s.]
5.3.2. Early studies of AHR-CYP1 axis signaling in ES cells
It therefore came as no surprise, when the AHR-CYP1 axis was reported to exist in cultured embryonic stem (ES) cells (Kuroda et al., 2005). Three years later, it was shown that mouse ES cells express TCDD-inducible CYP1A1 and CYP1B1 — but not CYP1A2, nor any of three “Phase II” drug-metabolizing enzymes (NQO1, glutathione S-transferase-A1, or UDP glucuronosyltransferase 1A6); Cyp1a1 gene induction was by far the most sensitive, first detected after only 3 h of culture and at “minimal effective” TCDD doses of 1 nM (Neri et al., 2008). [Recall (vide supra) the CYP1A1 and CYP1B1 genes are evolutionarily much earlier than the CYP1A2 gene.]
In an independent study, Cyp1a1 was shown to appear earliest — following which AHR, ARNT, and glucocorticoid receptor mRNAs as well — were detected in differentiating mouse ES cells throughout the culture period (Maezawa et al., 2008). The AHR-signaling pathway and sensitivity to PAH-caused toxicity were also described in human ES cells (Kee et al., 2010). Moreover, inappropriate or sustained activation of AHR by TCDD during neurogenesis was found to interfere with signaling pathways that regulate neuroepithelial stem cell/neural precursor cell proliferation (Latchney et al., 2011).
It then became appreciated that pluripotent stem cells can be generated from mouse somatic cells, using a combination of seven small-molecule compounds. These pluripotent stem cells were found to resemble ES cells — in terms of their gene expression profiles, epigenetic status, and potential for differentiation and germline transmission; this model system opened the way to study “master genes” that are dispensable for cell-fate reprogramming (Hou et al., 2013).
Various tryptophan derivatives — including endogenous 2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl-ester (ITE) — were shown to regulate transcription of the master pluripotency gene POU5F1 (trivial name Oct4). ITE was revealed to enhance AHR binding to the POU5F1 promoter and suppress its transcription. Lowering endogenous ITE levels in cancer cells by tryptophan deprivation, or hypoxia, leads to POU5F1 induction, which can be blocked by synthetic ITE treatment (Cheng et al., 2015). Very recently, ITE-treated hepatoma-derived Huh7 cells/C3A human cells were discovered to induce CYP1A1, CYP1A2, CYP3A4 and CYP1B1 (Zhang et al., 2017); these represent P450 monooxygenases in two of the six P450 families shown to participate in LM-mediated second-messenger signaling pathways (Fig. 3).
5.3.3. Crosstalk between AHR- and hypoxia-inducible factor (HIF)-signaling pathways
Before going further, it must be emphasized that during regulatory expression of numerous genes before becoming functional, AHR and HIF1A/3A both require their binding partner ARNT/ARNT2, the latter being critical for responses to intracellular hypoxia (low pO2). Such competition/crosstalk implicates differences in AHR-signaling capacity during conditions of diminished pO2 [reviewed in (Vorrink and Domann, 2014)].
The story is even more complicated than the transcriptional factors AHR and HIF competing with ARNT at AHRE-binding sites. For example, mouse Cyp1a1 up-regulation is inhibited by hypoxia, whereas AHR activation actually enhances erythropoietin gene (Epo) up-regulation during hypoxic conditions; in this particular instance, the Epo regulatory region was found to contain AHREs upstream of its transcriptional start-site. Thus, Epo can be categorized as “an AHR-target gene” (Chan et al., 1999). Because of these studies, alterations in AHR-signaling pathways must be kept in mind during any study involving hypoxia.
Using ES cell-differentiated embryoid bodies, it was found that BNIP3 (BCL2/adenovirus E1B 19-kD-interacting protein), which is a BH3-only pro-apoptotic protein, is up-regulated during cavitation in a hypoxia-dependent manner (Qi et al., 2012). Silencing of the Bnip3 gene blocks apoptosis of the core cells and delays cavitation. In contrast, Bnip3 up-regulation is regulated by HIF1A. Ablation of either HIF1A or ARNT — partners of AHR in the complex that binds to AHRE regulatory sequences of most, if not all, AHR-target genes — inhibits Bnip3 up-regulation, thereby blocking apoptosis and cavitation (Qi et al., 2012).
Apoptosis-inducing factor (AIF) cooperates with BNIP3 to promote lumen clearance; Bnip3 gene silencing in Aif(−/−) knockout embryoid bodies blocks apoptosis and cavitation. In addition, AIF regulates Bnip3 expression by way of mitochondrial ROS production and consequent EPAS1 (previous name, HIF2A) stabilization. These exciting data demonstrate a mechanism of cavitation during morula–>blastula formation that is very much interconnected with AHR-signaling pathways, via hypoxia-induced apoptosis of the core cells mediated by HIFs, BNIP3, and AIF (Qi et al., 2012).
5.3.4. AHR-signaling in cardiorespiratory functions and cardiomyogenesis
Studies of AHR-signaling pathways and the CYP1 axis in cultured ES cells have exploded today, largely because they offer an excellent model system to study fundamental signaling pathways in undifferentiated, pluripotent cells in culture. For example, comparing Ahr(−/−) and Vav3(−/−) mouse embryonic fibroblasts, it was found that VAV3, activator of Rho/Rac GTPases, is an AHR target and, without AHR present, γ-aminobutyric acid (GABA)-ergic transmission defects occur in the ventrolateral medulla — an important cardiorespiratory brainstem region (Sauzeau et al., 2011a).
Silencing Ahr [by short hairpin (sh)RNA interference] in mouse teratocarcinoma P19 cells allows cells to differentiate into cardiomyocytes when treated with dimethylsulfoxide. When AHR is silenced, cardiomyocyte-specific Gata4 (*encoding GATA4-binding protein) and Nkx2-5 (NK2 homeobox-5) gene expression become elevated, along with increases in Gsk3b (GSK3; glycogen synthase kinase-3β); in contrast, Arnt, Cyp1a1 and Jup (encoding junction plakoglobin; also called β-catenin) genes are concomitantly suppressed (Zhu et al., 2012). These findings suggest AHR silencing enhances P19-cell differentiation by way of AHR- and WNT-signaling pathways (†the WNT family comprises 18 genes in the human and mouse genomes). Jup-encoded plakoglobin plays a pivotal role in intercellular junctions; when Jup is suppressed by AHR — cell adherence and proliferation become dysregulated (Prochazkova et al., 2013). Disruption of AHR-signaling activity thus perturbs cardiomyocyte differentiation; reasons for this perturbation include alterations in expression of homeobox transcription factors (Wang et al., 2013).
If ES cells do not differentiate into cardiomyocytes, AHR-expression prevents cardiogenesis — thereby resulting in commitment to a neuronal cell fate. Any undesirable early expression of AHR in specific cells of the developing early embryo must therefore be repressed, in order to maintain ES cell mitotic progression and to prevent premature loss of pluripotency (Ko et al., 2016).
5.3.5. AHR-signaling in neurogenesis and neuronal cell development
As mentioned earlier, active bHLH/PAS heterodimers comprise a ubiquitous signal-regulated subunit (e.g. HIF1A/3A, AHR) or a tissue-specific subunit (e.g. neuronal PAS-domain proteins NPAS1/3/4, SIM1/2), paired with a general partner protein, ARNT or ARNT2. High ARNT/ARNT2 ratios in P19 embryonic carcinoma cells and ES cells — upon neuronal differentiation — become dramatically reverted to high ARNT2/ARNT ratios, whereas Arnt and Arnt2 mRNA half-lives do not change during neuronal differentiation; the alteration to predominant ARNT2 expression in neurons is proposed to allow specific functions of neuronal bHLH/PAS factors to occur, and/or to evade neuronal bHLH/PAS factors from interfering with AHR/ARNT-signaling functions (Hao et al., 2013).
5.3.6. AHR-signaling in early development of hematopoiesis
HIF1A is known to help hematopoietic stem cells adapt to stress. In Arnt(−/−) conditional knockout mice, ARNT was discovered to be essential for adult and fetal hematopoietic stem-cell viability and homeostasis, but ARNT deficiency is directly HIF-dependent (Krock et al., 2015). Hence, HIF activity likely regulates hematopoietic stem cell homeostasis by way of these pro-survival factors.
Overexpression of RNA-binding protein MSI2 (musashi-2) is known to induce multiple pro-self-renewal phenotypes — including 17-fold increases in short-term blood-progenitor repopulating cells and a net 23-fold ex vivo expansion of long-term repopulating hematopoietic stem cells. Recently it was shown that MSI2 directly attenuates AHR-signaling, via posttranscriptional down-regulation of a canonical AHR-pathway in cord-blood hematopoietic stem cells (Rentas et al., 2016).
5.3.7. AHR-signaling in osteogenesis and bone formation
As mentioned earlier, AHR-mediated pathways are involved during bone formation (Herlin et al., 2013) and osteoclastogenesis (Iqbal et al., 2013). During studies of mouse ES cell culture, between GD0.5 and 3.5 during the period of panmesoderm development, TCDD treatment disrupts expression of genes in the transforming growth factors-β (TGFB1/2/3), bone morphogenetic proteins-2, -4 (BMP2/4), and WNT3A/5A (wingless-type MMTV integration site members-3a and -5a)-signaling pathways; AHR-signaling specifically suppresses secretion of BMP4, WNT3A, and WNT5A. Comparing Ahr(+/+) wild-type and Ahr(−/−) knockout ES cells treated with TCDD, the mitochondrial copy number was found to be enhanced — suggesting mitochondrial stress (ROS formation) and remodeling might occur during this period of panmesoderm development (Wang et al., 2016).
5.3.8. Early embryonic AHR gene-signaling pathways fundamental to development
As the zygote develops, cells that first are totipotent become pluripotent stem cells, differentiating into endoderm, ectoderm and mesoderm — which then proceed to form specific cell-types that include e.g. intestinal stem cells, epithelial stem cells, and mesenchymal stem cells, respectively. As mentioned earlier, AHR-CYP1 axis signaling during the cell cycle will clearly elicit effects at any and all of these steps (Fig. 8). For example, inappropriate or sustained activation of AHR was found to interfere with signaling pathways that regulate neuroepithelial stem cell/neural precursor cell proliferation (Latchney et al., 2011).
In mouse ES cells, interactions between*POU3/4 (OCT3/4), NANOG, SOX2 and polycomb group proteins (PcG) binding to Ahr gene promoter region were discovered to repress Ahr expression. Removal of repressive signals in Ahr promoter-region chromatin leads to release of pluripotency factors and PcG proteins, and binding of†SP factors — which result in establishing open chromatin; this, in turn, engages RNA polymerase-2 to drive full-length RNA transcript elongation. These data indicate that AHR-signaling is required for ES cells to become activated quickly, as needed, during embryonic development (Ko et al., 2014).
5.3.9. AHR-signaling pathways during DNA synthesis and repair
As emphasized earlier, to maintain mitotic progression and pluripotency, ES cells must maintain low or negligible Ahr expression. Repression of AHR-signaling pathways cannot, however, be a completely one-way street. AHR-signaling pathways are often vital and therefore must fluctuate reversibly between expression and repression — with some fraction of cells escaping repression, as needed, to become activated at any one time. Cells that escape Ahr repression (i.e. activated AHR) lead to higher levels of POU5F1 and SOX2 (Fig. 8A, lower left), which then result in activation) of Mid1 (midline-1). Subsequently cells show extended mitotic traverse times, i.e. disruption of the MID1-PP2A-CDC25B/26-CDK1 signaling pathway (Fig. 8A) that regulates mitosis (Ko et al., 2016).
Control of the eukaryotic cell cycle includes passage through “checkpoints” from one phase to the next — requiring a coordinated set of proteins that monitor cell growth and DNA integrity. Obviously, uncontrolled cell division, or propagation of damaged DNA, can lead to genomic instability and disease states — including cancer.
The G1/S checkpoint (Fig. 8) controls progression of cells into S phase (DNA synthesis). As mentioned earlier, during G1 (Fig. 8B), hypo-phosphorylated RB1 blocks E2F action by binding to E2F (E2F includes a family of eight proteins). RB1 phosphorylation, by*cyclin-dependent kinase-2 (CDK2) in late G1, causes dissociation of RB1, allowing E2F-mediated transcription of S-phase-promoting genes to proceed. Also described earlier, formation of an AHR-RB1 binding complex prevents normal progression of G1 to S phase by stalling E2F-mediated transcription of S-phase genes [reviewed in (Nebert et al., 2000)]. Hence, in the presence of sufficient amounts of unknown endogenous ligand — or planar foreign chemical such as TCDD, PAHs or halogenated biphenyls — that bind to AHR, activated AHR would cause arrest at the G1/S boundary; this would be lethal to the 1-cell zygote or early embryo.
DNA damage (Fig. 8A, upper right) activates response pathways through ATM/ATR (gene products of “ataxia telangiectasia-mutated”/“ataxia telangiectasia and RAD3-related”) and checkpoint-1/2 (CHEK1/2) kinases to suppress CDC25A, thereby preventing CDK4/6 and CDK2 activity; this results in cell-cycle arrest at the G1/S boundary, followed by either DNA repair or, at end of the G2 phase, cell death (apoptosis). DNA damage also activates multiple kinases that phosphorylate tumor protein-53 (TP53). MDM2 and MDM4 — acting apparently as non-redundant components of a complex (Shadfan et al., 2012) — are known to be major essential negative regulators of*TP53 in normal cells that suppress TP53 activity. Under conditions of cellular stress, the†MDM2-MDM4 complex must be suppressed so that TP53 is able to respond to that stress. Phosphorylation of TP53 thus stimulates PAK1-6 (p21) to prevent CDK2 action, which arrests the cell at the G1/S boundary, thereby allowing binding of TP53 to DNA.
Protein phosphatase 2A (PP2A) is known to phosphorylate more than 300 substrates involved in the cell cycle (Fig. 8) — controlling virtually all major pathways and cell-cycle checkpoints (Wlodarchak and Xing, 2016). CDC25B/26 (cell division cycle-25B, -26; there are 23 highly conserved CDC genes in vertebrates) is a complex that acts, along with many other regulators of the core cell cycle machinery, in signaling pathways and transcriptional programs controlling cell fate choices — i.e. when to proceed into apoptosis vs when to switch between differentiation and proliferation (Agius et al., 2015).
7. Concluding remarks
“When reaching the great mountaintop, we can behold all mountains in a single glance.” -----Du Fu, prominent Chinese poet (712–770 AD) of the Tang dynasty.
From the mountain top as described in this review, looking back over six decades of scientific history and first evidence of AHR in the early 1970s, what have we learned? AHR is now realized to be among the earliest-discovered “pioneer” members of the bHLH/PAS family, now numbering about 30 in the human and mouse genomes; the only earlier one was the Drosophila “periodic” (per) locus (Konopka and Benzer, 1971). We now know that bHLH/PAS proteins are sensors of both foreign and endogenous “signals” to which the organism, or individual cell, responds. Although the name “PAS” (abbreviation for per-Arnt-sim) for this family was not coined until the 1990s — evidence for AHR, the binding partner of ARNT, was reported almost two decades earlier than ARNT. In that landmark publication (Poland et al., 1974), AHR was proposed to be a sensor of TCDD and PAHs, and an up-regulator of CYP1A1-mediated metabolism of PAHs.
How we propose bHLH/PAS members to operate is shown in Fig. 9A. A particular (foreign or endogenous) signal is “sensed” by the inactive sensor bHLH/PAS protein, causing activation of the sensor, which then stimulates/activates its primary target; this results in regulatory expression of downstream targets in the pathway — resulting in the “response” or “responses.” At any of the downstream steps, it is likely one or more moieties will inactivate the sensor which, in turn, represses the no-longer activated primary target. The inactive sensor then awaits the next signal — before becoming activated again.
Fig. 9.
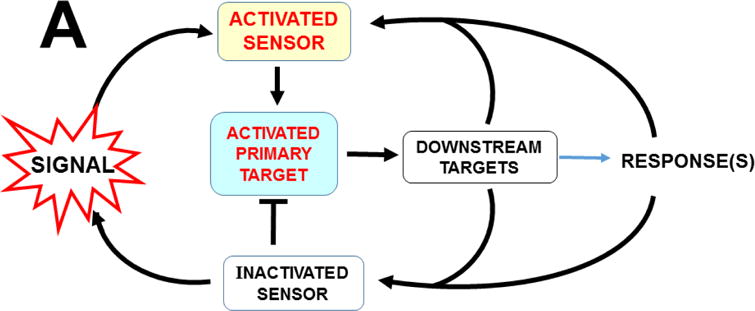
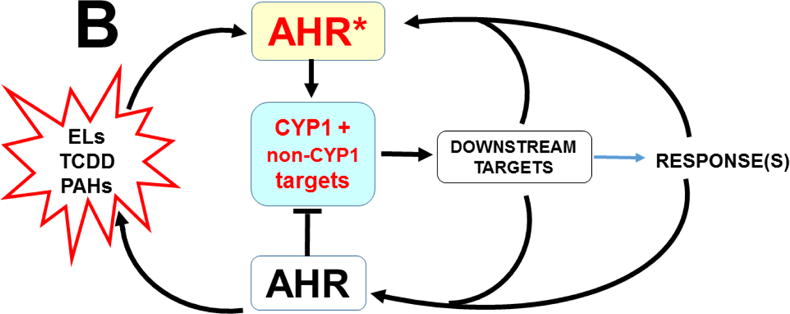
Overall scheme postulated to operate for some, perhaps all, of the bHLH/PAS sensors. A, generalized diagram. B, specific scheme for the AHR-CYP1 axis. ELs, endogenous ligands. See text for details.
Fig. 9B illustrates specifically how the AHR-CYP1 axis operates in signal-sensor functions. In this instance, an endogenous ligand (EL) such as particular tryptophan metabolites, TCDD, or planar foreign chemical (e.g. PAHs or halogenated biphenyls) binds to AHR, causing it to become activated (AHR*); this in turn up-regulates CYP1 enzymes and other non-CYP1 targets, resulting in downstream events leading to the response(s) — which, in this case, includes metabolism (degradation) of the EL (or ELs) as well as many foreign chemicals (metabolic activation or detoxication). Eventually, a downstream moiety, or signal, will inactivate AHR* to AHR, resulting in down-regulation of CYP1 and non-CYP1 targets. Then, inactivated AHR awaits the next signal before becoming activated.
First, foreign chemicals were postulated to be “recognized by” (bound to) AHR (Poland et al., 1974), thereby up-regulating CYP1 in various organs, cell types, and as a function of age (of the embryo or postpartum animal). Within the next six years, it was proposed (Nebert et al., 1981), and then demonstrated (Rifkind and Muschick, 1983; Bigelow et al., 1989), that AHR is likely to be involved also in “recognition” (binding) of endogenous compounds involved in the inflammatory process. In this case, up-regulated CYP1-mediated metabolism (most likely, of LMs) are converted either from active to inactive products, or vice versa. It is also clear that activated AHR must bind to ARNT before becoming the transcriptionally-active heterodimer (Burbach et al., 1992).
These two details (AHR binding of foreign chemicals, as well as endogenous compounds) appear to be common to all vertebrates. Intriguingly, AHR-signaling has also been found in the fly, worm, sea anemone, crustaceans, and even the beetle [vide supra]. With regard to spineless, the Drosophila homolog of AHR — although spineless-tango heterodimers exhibit similar transcriptional properties to those of mammalian AHR-ARNT heterodimers, there appears to be no ligand-binding by spineless before spineless-tango heterodimer interaction [reviewed in (Lindsey and Papoutsakis, 2012)]. Hence, the implication is that perhaps all invertebrate AHR homologs have no ligand-binding properties.
To the knowledge of this author, however, binding to an endogenous ligand has not been ruled out in any invertebrate study of AHR-signaling pathways. Although CYP1 genes evolutionarily are not present in invertebrates, we believe that up-regulation mediated by the AHR homolog of other enzymes remains a possibility among invertebrates.
Furthermore, little is currently known, evolutionarily, as to how extensive LM second-messenger signaling cascades might be in invertebrates. In crustaceans, PUFAs and prostaglandins have been detected. In one intriguing relevant study of the black tiger shrimp (Penaeus monodon) genome (Wimuttisuk et al., 2013), TBLASTX analysis found homologues of ten mammalian prostanoid biosynthesis genes: PLA2 (cytosolic phospholipase A2), HPGDS (hematopoietic prostaglandin D synthase), PGDS (glutathione-dependent prostaglandin D synthase), PGTES2 and PTGES3 (prostaglandin E synthases, 2 and 3), PGE2 (prostaglandin E2), AKR1C3 (prostaglandin F synthase), PGF2A (prostaglandin F2α), TBXAS1 (thromboxane A synthase, also known as CYP5A1), and PTGS1 (cyclooxygenase-1). Thus, for invertebrates, it must be emphasized that additional research will be required — before any further conclusions can be reached about the possibility of any AHR-CYP or AHR-enzyme-homolog signaling pathways (illustrated in general for bHLH/PAS sensors in Fig. 9A).
In summary, the exciting unraveling of all the secrets of AHR, the AHR-CYP1 axis, and AHR-signaling pathways (Table 2) — over more than the past four decades — has been nothing short of incredible. Perhaps this is especially so, because of several of these quantum leaps, along the way, in which the data were considered “heresy” at the time, when first presented to the scientific community.
Table 2.
Timeline of proposed key discoveries, as the exciting “AHR-signaling story” developed.
| Year | Discovery |
|---|---|
| 1956 | “Benzpyrene hydroxylase” activity induced by PAHs in rat liver |
| 1962 | Benzpyrene hydroxylase activity induced throughout rat GI tract by oral benzpyrene |
| 1968 | “AHH activity” assay designed; AHH induction by PAHs in fetal hamster cell cultures |
| 1969 | Differences in AHH inducibility by PAHs between inbred strains of mice |
| 1970 | PAH-induced AHH activity represents a cytochrome P450 (P1-450) |
| 1972 | PAH-induced AHH activity in B6 × D2 crosses exhibits predominantly Mendelian inheritance |
| 1972 | In utero AHH induction in fetuses by treatment of mother with PAHs |
| 1973–1989 | Importance of AHR-mediated CYP1 shown for cancer, mutagenesis, toxicity, and teratogenesis |
| 1974 | Evidence of AHR, based on dose-response curve: AHH induction as a function of TCDD concentration in B6 vs D2 inbred mouse strains |
| 1976 | Radiolabeled TCDD binding assay detects cytosolic AHR |
| 1979 | Development of benzo[a]pyrene-resistant clones in Hepa-1 cells: c1 & c37, c2, and c4 |
| 1980 | AHR-mediated CYP1A1-induced sister-chromatid-exchange detected in GD7.5 embryo |
| 1981, 1989 | AHR-mediated P1-450 induction proposed to be involved in inflammatory process |
| 1982 | Intranuclear appearance of radiolabeled TCDD (AHR) asspcoated with P1-450 induction |
| 1988, 1998 | AHR-mediated CYP1A1 induction demonstrated in GD0.5 one-cell zygote |
| 1991 | Mouse Arnt gene cloned (human ARNT gene shortly thereafter) |
| 1991 | The term “bHLH/PAS” was first coined |
| 1992 | Mouse Ahr gene cloned (human AHR gene shortly thereafter) |
| 1995 | Creation of first of several Ahr(−/−) knockout mouse lines |
| 1996 | Standardized gene nomenclature for CYP genes in all species becomes well established |
| 2005 | AHR-CYP1 axis demonstrated in cultured ES cells |
| 2008, 2013 | Underscoring the importance of AHR-CYP1 axis in LM second-messenger pathways involving innumerable critical life events — in virtually every organ, tissue and cell-type |
Acknowledgments
Throughout this review, we have consistently used standardized gene and gene product nomenclature from the HUGO Gene Nomenclature Committee [http://www.genenames.org/]. We thank our colleagues — especially Drs. Elizabeth Kopras, Zijuan Liu, Ge Zhang, and anonymous reviewers — for careful readings of this review. With regard to graphics, we thank Drs. Jesmond Dalli for his help, early on, with the six panels of Figs. 5 & 7, and Zijuan Liu for her help with other figures. This project was funded, in part, by Grants R01 ES008147, R01 ES014403, and P30 ES06096.
Abbreviations
- AA
arachidonic acid
- AHH
aryl hydrocarbon hydoxylase
- AHR
aryl hydrocarbon receptor
- ARNT
aryl hydrocarbon receptor translocator
- B6
C57BL/6J mice
- bHLH
basic-helix-loop-helix domain in proteins
- Chr
chromosome
- CNS
central nervous system
- CYP
cytochrome P450
- CYP1 family of enzymes
CYP1A1, CYP1A2, and CYP1B1
- D2
DBA/2J mice
- DHA
docosahexaenoic acid
- DHETEs
dihydroxyeicosatrienoic acids
- EETs
epoxyeicosatrienoic acids
- EPA
eicosapentaenoic acid
- GI
gastrointestinal
- HETEs
hydroxyeicosatetraenoic acids
- HIF
hypoxia-inducible factor
- HpETEs
hydroperoxyeicosatetraenoic acids
- LM
lipid mediator
- LPS
lipopolysaccharide
- LTB4
leukotriene B4
- NQO1, NAD(P)H
quinone oxidoreductase-1
- PAH
polycyclic aromatic hydrocarbon
- PAS
per-Arnt-sim
- Per
periodic locus (Drosophila)
- PUFA
polyunsaturated fatty acid
- Sim
single-minded locus (Drosophila)
- TCDD
2,3,7,8,-tetrachlorodibenzo-p-dioxin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
The author declares he has no actual, or potential, conflicts of interest.
GATA is a family of transcription factors that activate or repress erythroid development; they bind to DNA sites having the consensus sequence 5′-[AT]GATA[AG]-3′ within regulatory regions of GATA-responsive genes.
The acronym POU is derived from names of three transcription factors: pituitary-specific PIT1; octamer proteins OCT1/2, and from Caenorhabditis elegans the neural UNC-86. The octamer sequence is ATGCAAAT. There are six POU classes of genes in the mammalian genome. POU3 and POU4 domains are present evolutionarily “as far back” as the common ancestor of sponges and eumetazoans. NANOG protein is a protein evolutionarily having a highly conserved homeodomain motif localized to the nucleus and facilitates DNA-binding. SOX2 is one of 19 members of the SOX family [SRY (sex-determining region Y)-box-1 family of transcription factors known to play pivotal roles in many stages of mammalian development]. Polycomb group (PcG) proteins are epigenetic regulators of transcription essential for stem-cell identity, differentiation, and disease states — functioning within multiprotein complexes, called polycomb repressive complexes (PRCs); PRCs modify histones (and other proteins) and silence target genes.
SP factors represent a family of nine specificity-protein/krüppel-like factor (SP/KLF) transcriptional factors that bind to GC and GT boxes; these are common recurring motifs in promoters, as well as distal regulatory elements of mammalian genes.
CDKs comprise a family of five mammalian protein kinases, known for participation in regulating the cell cycle; they also participate in transcription regulation, mRNA-processing, and differentiation during neurogenesis. CDKs are so highly conserved that yeast cells can divide normally when the human homolog is replaced with the yeast Cdk gene product.
The original term “p53” is encoded by the human TP53 gene, called “transformation protein-53”, and by the mouse Trp53 gene, called “transformation-related protein-53”.
The official term “MDM2” has been variously named MDM2 proto-oncogene, E3 ubiquitin protein ligase, and transformed mouse 3T3 cell double-minute-2. “MDMX” is the previous name for MDM4; the latter is now the official nomenclature.
References
- Agius E, Bel-Vialar S, Bonnet F, Pituello F. Cell cycle and cell fate in the developing nervous system: role of CDC25B phosphatase. Cell Tissue Res. 2015;359:201–213. doi: 10.1007/s00441-014-1998-2. [DOI] [PubMed] [Google Scholar]
- Ahokas JT, Saarni H, Nebert DW, Pelkonen O. The in vitro metabolism and convalent binding of benzo[a]pyrene to DNA catalysed by trout liver microsomes. Chem Biol Interact. 1979;25:103–111. doi: 10.1016/0009-2797(79)90072-3. [DOI] [PubMed] [Google Scholar]
- Anderson G, Beischlag TV, Vinciguerra M, Mazzoccoli G. The circadian clock circuitry and the AHR signaling pathway in physiology and pathology. Biochem Pharmacol. 2013;85:1405–1416. doi: 10.1016/j.bcp.2013.02.022. [DOI] [PubMed] [Google Scholar]
- Atlas AS, Thorgeirsson SS, Boobis AR, Kumaki K, Nebert DW. Differential induction of murine Ah locus-associated monooxygenase activities in rabbit liver and kidney. Biochem Pharmacol. 1975;24:2111–2116. doi: 10.1016/0006-2952(75)90114-8. [DOI] [PubMed] [Google Scholar]
- Atlas SA, Boobis AR, Felton JS, Thorgeirsson SS, Nebert DW. Ontogenetic expression of polycyclic aromatic compound-inducible monooxygenase activities and forms of cytochrome P-450 in rabbit. Evidence for temporal control and organ specificity of two genetic regulatory systems. J Biol Chem. 1977;252:4712–4721. [PubMed] [Google Scholar]
- Baker TR, King-Heiden TC, Peterson RE, Heideman W. Dioxin induction of transgenerational inheritance of disease in zebrafish. Mol Cell Endocrinol. 2014;398:36–41. doi: 10.1016/j.mce.2014.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barroso A, Gualdron-Lopez M, Esper L, Brant F, Araujo RR, Carneiro MB, Avila TV, Souza DG, Vieira LQ, Rachid MA, Tanowitz HB, Teixeira MM, Machado FS. Aryl hydrocarbon receptor modulates production of cytokines and reactive oxygen species and development of myocarditis during Trypanosoma cruzi infection. Infect Immun. 2016;84:3071–3082. doi: 10.1128/IAI.00575-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessede A, Gargaro M, Pallotta MT, Matino D, Servillo G, Brunacci C, Bicciato S, Mazza EM, Macchiarulo A, Vacca C, Iannitti R, Tissi L, Volpi C, Belladonna ML, Orabona C, Bianchi R, Lanz TV, Platten M, Della Fazia MA, Piobbico D, Zelante T, Funakoshi H, Nakamura T, Gilot D, Denison MS, Guillemin GJ, DuHadaway JB, Prendergast GC, Metz R, Geffard M, Boon L, Pirro M, Iorio A, Veyret B, Romani L, Grohmann U, Fallarino F, Puccetti P. Aryl hydrocarbon receptor control of a disease-tolerance-defence pathway. Nature. 2014;511:184–190. doi: 10.1038/nature13323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigelow SW, Collins AC, Nebert DW. Selective mouse breeding for short ethanol sleep time has led to high levels of hepatic aromatic hydrocarbon (Ah) receptor. Biochem Pharmacol. 1989;38:3565–3572. doi: 10.1016/0006-2952(89)90129-9. [DOI] [PubMed] [Google Scholar]
- Bradfield CA, Glover E, Poland A. Purification and NH2-terminal amino acid sequence of the Ah receptor from the C57BL/6J mouse. Mol Pharmacol. 1991;39:13–19. [PubMed] [Google Scholar]
- Bulun SE, Zeitoun KM, Kilic G. Expression of dioxin-related transactivating factors and target genes in human eutopic endometrial and endometriotic tissues. Am J Obstet Gynecol. 2000;182:767–775. doi: 10.1016/s0002-9378(00)70325-5. [DOI] [PubMed] [Google Scholar]
- Burbach KM, Poland A, Bradfield CA. Cloning of the Ah-receptor cDNA reveals a distinctive ligand-activated transcription factor. Proc Natl Acad Sci U S A. 1992;89:8185–8189. doi: 10.1073/pnas.89.17.8185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell EL, Serhan CN, Colgan SP. Antimicrobial aspects of inflammatory resolution in the mucosa: a role for proresolving mediators. J Immunol. 2011;187:3475–3481. doi: 10.4049/jimmunol.1100150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cella M, Colonna M. Aryl hydrocarbon receptor: linking environment to immunity. Semin Immunol. 2015;27:310–314. doi: 10.1016/j.smim.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Céspedes MA, Galindo MI, Couso JP. Dioxin toxicity in vivo results from an increase in the dioxin-independent transcriptional activity of aryl hydrocarbon receptor. PLoS ONE. 2010;5:e15382. doi: 10.1371/journal.pone.0015382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan WK, Yao G, Gu YZ, Bradfield CA. Cross-talk between aryl hydrocarbon receptor and hypoxia inducible factor signaling pathways. Demonstrated competition and compensation. J Biol Chem. 1999;274:12115–12123. doi: 10.1074/jbc.274.17.12115. [DOI] [PubMed] [Google Scholar]
- Chatel A, Faucet-Marquis V, Gourlay-France C, Pfohl-Leszkowicz A, Vincent-Hubert F. Genotoxicity and activation of cellular defenses in transplanted zebra mussels Dreissena polymorpha along the Seine river. Ecotoxicol Environ Saf. 2015;114:241–249. doi: 10.1016/j.ecoenv.2014.03.023. [DOI] [PubMed] [Google Scholar]
- Cheng J, Li W, Kang B, Zhou Y, Song J, Dan S, Yang Y, Zhang X, Li J, Yin S, Cao H, Yao H, Zhu C, Yi W, Zhao Q, Xu X, Zheng M, Zheng S, Li L, Shen B, Wang YJ. Tryptophan derivatives regulate Oct4 transcription in stem-like cancer cells. Nat Commun. 2015;6:7209. doi: 10.1038/ncomms8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung C, Gonzalez FJ. Humanized mouse lines and their application for prediction of human drug metabolism and toxicological risk assessment. J Pharmacol Exp Ther. 2008;327:288–299. doi: 10.1124/jpet.108.141242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiaro CR, Patel RD, Perdew GH. 12(R)-Hydroxy-5(Z),8(Z),10(E),14(Z)-eicosatetraenoic acid [12(R)-HETE], an arachidonic acid derivative, is an activator of aryl hydrocarbon receptor. Mol Pharmacol. 2008;74:1649–1656. doi: 10.1124/mol.108.049379. [DOI] [PubMed] [Google Scholar]
- Collins KD, Lacal J, Ottemann KM. Internal sense of direction: sensing and signaling from cytoplasmic chemoreceptors. Microbiol Mol Biol Rev. 2014;78:672–684. doi: 10.1128/MMBR.00033-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conney AH, Gillette JR, Inscoe JK, Trams ER, Posner HS. Induced synthesis of liver microsomal enzymes which metabolize foreign compounds. Science. 1959;130:1478–1479. doi: 10.1126/science.130.3387.1478. [DOI] [PubMed] [Google Scholar]
- Conney AH, Miller EC, Miller JA. The metabolism of methylated aminoazo dyes. Evidence for induction of enzyme synthesis in the rat by 3-methylcholanthrene. Cancer Res. 1956;16:450–459. [PubMed] [Google Scholar]
- Conney AH, Miller EC, Miller JA. Substrate-induced synthesis and other properties of benzpyrene hydroxylase in rat liver. J Biol Chem. 1957;228:753–766. [PubMed] [Google Scholar]
- Corchero J, Pimprale S, Kimura S, Gonzalez FJ. Organization of the CYP1A cluster on human chromosome 15: implications for gene regulation. Pharmacogenetics. 2001;11:1–6. doi: 10.1097/00008571-200102000-00001. [DOI] [PubMed] [Google Scholar]
- Crews ST, Thomas JB, Goodman CS. The Drosophila single-minded gene encodes a nuclear protein with sequence similarity to the per gene product. Cell. 1988;52:143–151. doi: 10.1016/0092-8674(88)90538-7. [DOI] [PubMed] [Google Scholar]
- Daiber A, Ullrich V. Peroxynitrite reactions with heme and heme-thiolate (P450) proteins. Methods Enzymol. 2002;359:379–389. doi: 10.1016/s0076-6879(02)59200-4. [DOI] [PubMed] [Google Scholar]
- Dalli J, Serhan CN. Specific lipid mediator signatures of human phagocytes: microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood. 2012;120:e60–e72. doi: 10.1182/blood-2012-04-423525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Fuente H, Cibrian D, Sanchez-Madrid F. Immunoregulatory molecules are master regulators of inflammation during the immune response. FEBS Lett. 2012;586:2897–2905. doi: 10.1016/j.febslet.2012.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkenne S, Curran CP, Shertzer HG, Dalton TP, Dragin N, Nebert DW. Theophylline pharmacokinetics: comparison of Cyp1a1(−/−) and Cyp1a2(−/−) knockout mice, humanized hCYP1A1_1A2 knock-in mice lacking either the mouse Cyp1a1 or Cyp1a2 gene, and Cyp1(+/+) wild-type mice. Pharmacogenet Genomics. 2005;15:503–511. doi: 10.1097/01.fpc.0000167326.00411.50. [DOI] [PubMed] [Google Scholar]
- Dey A, Nebert DW. Markedly increased constitutive Cyp1a1 mRNA levels in the fertilized ovum of the mouse. Biochem Biophys Res Commun. 1998;251:657–661. doi: 10.1006/bbrc.1998.9519. [DOI] [PubMed] [Google Scholar]
- Divanovic S, Dalli J, Jorge-Nebert LF, Flick LM, Galvez-Peralta M, Boespflug ND, Stankiewicz TE, Fitzgerald JM, Somarathna M, Karp CL, Serhan CN, Nebert DW. Contributions of the three CYP1 monooxygenases to pro-inflammatory and inflammation-resolution lipid mediator pathways. J Immunol. 2013;191:3347–3357. doi: 10.4049/jimmunol.1300699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do KN, Fink LN, Jensen TE, Gautier L, Parlesak A. TLR2 controls intestinal carcinogen detoxication by CYP1A1. PLoS One. 2012;7:e32309. doi: 10.1371/journal.pone.0032309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolwick KM, Schmidt JV, Carver LA, Swanson HI, Bradfield CA. Cloning and expression of a human Ah receptor cDNA. Mol Pharmacol. 1993;44:911–917. [PubMed] [Google Scholar]
- Dragin N, Dalton TP, Miller ML, Shertzer HG, Nebert DW. For dioxin-induced birth defects, mouse or human CYP1A2 in maternal liver protects, whereas mouse CYP1A1 and CYP1B1 are inconsequential. J Biol Chem. 2006;281:18591–18600. doi: 10.1074/jbc.M601159200. [DOI] [PubMed] [Google Scholar]
- Dragin N, Shi Z, Madan R, Karp CL, Sartor MA, Chen C, Gonzalez FJ, Nebert DW. Phenotype of the Cyp1a1/1a2/1b1(−/−) triple-knockout mouse. Mol Pharmacol. 2008;73:1844–1856. doi: 10.1124/mol.108.045658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragin N, Uno S, Wang B, Dalton TP, Nebert DW. Generation of ‘humanized’ hCYP1A1_1A2_Cyp1a1/1a2(−/−) mouse line. Biochem Biophys Res Commun. 2007;359:635–642. doi: 10.1016/j.bbrc.2007.05.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Liao C, Zhou H, Diao X, Li Y, Zheng P, Wang F. Gene cloning and expression analysis of AHR and CYP4 from Pinctada martensii after exposure to pyrene. Ecotoxicology. 2015;24:1574–1582. doi: 10.1007/s10646-015-1424-x. [DOI] [PubMed] [Google Scholar]
- Ema M, Sogawa K, Watanabe N, Chujoh Y, Matsushita N, Gotoh O, Funae Y, Fujii-Kuriyama Y. cDNA cloning and structure of mouse putative Ah receptor. Biochem Biophys Res Commun. 1992;184:246–253. doi: 10.1016/0006-291x(92)91185-s. [DOI] [PubMed] [Google Scholar]
- Emmons RB, Duncan D, Estes PA, Kiefel P, Mosher JT, Sonnenfeld M, Ward MP, Duncan I, Crews ST. The spineless-aristapedia and tango bHLH-PAS proteins interact to control antennal and tarsal development in Drosophila. Development. 1999;126:3937–3945. doi: 10.1242/dev.126.17.3937. [DOI] [PubMed] [Google Scholar]
- Ernster L, Orrenius S. Substrate-induced synthesis of the hydroxylating enzyme system of liver microsomes. Fed Proc. 1965;24:1190–1199. [PubMed] [Google Scholar]
- Esser C, Rannug A. The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharmacol Rev. 2015;67:259–279. doi: 10.1124/pr.114.009001. [DOI] [PubMed] [Google Scholar]
- Felker AM, Chen Z, Foster WG, Croy BA. Receptors for non-MHC ligands contribute to uterine natural killer cell activation during pregnancy in mice. Placenta. 2013;34:757–764. doi: 10.1016/j.placenta.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Salguero P, Pineau T, Hilbert DM, McPhail T, Lee SS, Kimura S, Nebert DW, Rudikoff S, Ward JM, Gonzalez FJ. Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding AH receptor. Science. 1995;268:722–726. doi: 10.1126/science.7732381. [DOI] [PubMed] [Google Scholar]
- Fernandez-Salguero PM, Hilbert DM, Rudikoff S, Ward JM, Gonzalez FJ. Aryl-hydrocarbon receptor-deficient mice are resistant to 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced toxicity. Toxicol Appl Pharmacol. 1996;140:173–179. doi: 10.1006/taap.1996.0210. [DOI] [PubMed] [Google Scholar]
- Fornace AJ, Jr, Nebert DW, Hollander MC, Luethy JD, Papathanasiou M, Fargnoli J, Holbrook NJ. Mammalian genes coordinately regulated by growth-arrest signals and DNA-damaging agents. Mol Cell Biol. 1989;9:4196–4203. doi: 10.1128/mcb.9.10.4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fracchiolla NS, Annaloro C, Guidotti F, Fattizzo B, Cortelezzi A. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) role in hematopoiesis and in hematologic diseases: A critical review. Toxicology. 2016;374:60–68. doi: 10.1016/j.tox.2016.10.007. [DOI] [PubMed] [Google Scholar]
- Galloway SM, Perry PE, Meneses J, Nebert DW, Pedersen RA. Cultured mouse embryos metabolize benzo[a]pyrene during early gestation: genetic differences detectable by sister chromatid exchange. Proc Natl Acad Sci U S A. 1980;77:3524–3528. doi: 10.1073/pnas.77.6.3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasiewicz TA, Singh KP, Bennett JA. The Ah receptor in stem cell cycling, regulation, and quiescence. Ann N Y Acad Sci. 2014;1310:44–50. doi: 10.1111/nyas.12361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge NL, Elferink CJ. A direct interaction between aryl hydrocarbon receptor and retinoblastoma protein. Linking dioxin signaling to the cell cycle. J Biol Chem. 1998;273:22708–22713. doi: 10.1074/jbc.273.35.22708. [DOI] [PubMed] [Google Scholar]
- Gielen JE, Goujon FM, Nebert DW. Genetic regulation of aryl hydrocarbon hydroxylase induction. Simple Mendelian expression in mouse tissues in vivo. J Biol Chem. 1972;247:1125–1137. [PubMed] [Google Scholar]
- Glaumann H, Kuylenstierna B, Dallner G. Differential distribution of cytochrome P-450 and cytochrome P-448 in liver microsomal membranes. Life Sci. 1969;8:1309–1315. doi: 10.1016/0024-3205(69)90187-8. [DOI] [PubMed] [Google Scholar]
- Gooley JJ. Circadian regulation of lipid metabolism. Proc Nutr Soc. 2016;75:440–450. doi: 10.1017/S0029665116000288. [DOI] [PubMed] [Google Scholar]
- Goossens J, Mertens J, Goossens A. Role and functioning of bHLH transcription factors in jasmonate signaling. J Exp Bot. 2017;68:erw440. doi: 10.1093/jxb/erw440. [DOI] [PubMed] [Google Scholar]
- Greene ER, Huang S, Serhan CN, Panigrahy D. Regulation of inflammation in cancer by eicosanoids. Prostaglandins Other Lipid Mediat. 2011;96:27–36. doi: 10.1016/j.prostaglandins.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunwald S, Niedermeier J, Wenzel U. Hormesis is induced in the red flour beetle Tribolium castaneum through ingestion of charred toast. Eur J Nutr. 2015;54:535–541. doi: 10.1007/s00394-014-0734-8. [DOI] [PubMed] [Google Scholar]
- Hahn ME, Karchner SI, Evans BR, Franks DG, Merson RR, Lapseritis JM. Unexpected diversity of aryl hydrocarbon receptors in non-mammalian vertebrates: insights from comparative genomics. J Exp Zool A Comp Exp Biol. 2006;305:693–706. doi: 10.1002/jez.a.323. [DOI] [PubMed] [Google Scholar]
- Hankinson O. Single-step selection of clones of a mouse hepatoma line deficient in aryl hydrocarbon hydroxylase. Proc Natl Acad Sci USA. 1979;76:373–376. doi: 10.1073/pnas.76.1.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hankinson O. The role of AHR-inducible cytochrome P450s in metabolism of polyunsaturated fatty acids. Drug Metab Rev. 2016;48:342–350. doi: 10.1080/03602532.2016.1197240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hankinson O, Andersen RD, Birren BW, Sander F, Negishi M, Nebert DW. Mutations affecting regulation of transcription of the cytochrome P1-450 gene in the mouse Hepa-1 cell line. J Biol Chem. 1985;260:1790–1795. [PubMed] [Google Scholar]
- Hao N, Bhakti VL, Peet DJ, Whitelaw ML. Reciprocal regulation of the basic helix-loop-helix/PER-ARNT-SIM partner proteins, ARNT and ARNT2, during neuronal differentiation. Nucleic Acids Res. 2013;41:5626–5638. doi: 10.1093/nar/gkt206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harstad EB, Guite CA, Thomae TL, Bradfield CA. Liver deformation in Ahr-null mice: evidence for aberrant hepatic perfusion in early development. Mol Pharmacol. 2006;69:1534–1541. doi: 10.1124/mol.105.020107. [DOI] [PubMed] [Google Scholar]
- Hayaishi O, Katagiri M, Rothberg S. Mechanism of the pyrocatechase reaction. J Am Chem Soc. 1955;77:5450–5451. [Google Scholar]
- Henry JT, Crosson S. Ligand-binding PAS domains in a genomic, cellular, and structural context. Annu Rev Microbiol. 2011;65:261–286. doi: 10.1146/annurev-micro-121809-151631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlin M, Finnila MA, Zioupos P, Aula A, Risteli J, Miettinen HM, Jamsa T, Tuukkanen J, Korkalainen M, Hakansson H, Viluksela M. New insights to the role of aryl hydrocarbon receptor in bone phenotype and in dioxin-induced modulation of bone microarchitecture and material properties. Toxicol Appl Pharmacol. 2013;273:219–226. doi: 10.1016/j.taap.2013.09.002. [DOI] [PubMed] [Google Scholar]
- Hernandez-Ochoa I, Karman BN, Flaws JA. The role of aryl hydrocarbon receptor in the female reproductive system. Biochem Pharmacol. 2009;77:547–559. doi: 10.1016/j.bcp.2008.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman EC, Reyes H, Chu FF, Sander F, Conley LH, Brooks BA, Hankinson O. Cloning of a factor required for activity of the Ah (dioxin) receptor. Science. 1991;252:954–958. doi: 10.1126/science.1852076. [DOI] [PubMed] [Google Scholar]
- Hou P, Li Y, Zhang X, Liu C, Guan J, Li H, Zhao T, Ye J, Yang W, Liu K, Ge J, Xu J, Zhang Q, Zhao Y, Deng H. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science. 2013;341:651–654. doi: 10.1126/science.1239278. [DOI] [PubMed] [Google Scholar]
- Huang B, Butler R, Miao Y, Dai Y, Wu W, Su W, Fujii-Kuriyama Y, Warner M, Gustafsson JA. Dysregulation of Notch- and ESR1-signaling in Ahr(−/−) male mice. Proc Natl Acad Sci U S A. 2016;113:11883–11888. doi: 10.1073/pnas.1613269113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard TD, Murray IA, Perdew GH. Indole and tryptophan metabolism: endogenous and dietary routes to AH receptor activation. Drug Metab Dispos. 2015a;43:1522–1535. doi: 10.1124/dmd.115.064246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husain S, Kaddour-Djebbar I, Abdel-Latif AA. Alterations in arachidonic acid release and phospholipase Cβ1 expression in glaucomatous human ciliary muscle cells. Invest Ophthalmol Vis Sci. 2002;43:1127–1134. [PubMed] [Google Scholar]
- Ichihara S, Yamada Y, Ichihara G, Nakajima T, Li P, Kondo T, Gonzalez FJ, Murohara T. A role for aryl hydrocarbon receptor in regulation of ischemia-induced angiogenesis. Arterioscler Thromb Vasc Biol. 2007;27:1297–1304. doi: 10.1161/ATVBAHA.106.138701. [DOI] [PubMed] [Google Scholar]
- Ikuta T, Kurosumi M, Yatsuoka T, Nishimura Y. Tissue distribution of aryl hydrocarbon receptor in the intestine: implication of putative roles in tumor suppression. Exp Cell Res. 2016;343:126–134. doi: 10.1016/j.yexcr.2016.03.012. [DOI] [PubMed] [Google Scholar]
- Ikuta T, Namiki T, Fujii-Kuriyama Y, Kawajiri K. AHR protein trafficking and function in the skin. Biochem Pharmacol. 2009;77:588–596. doi: 10.1016/j.bcp.2008.10.003. [DOI] [PubMed] [Google Scholar]
- Iqbal J, Sun L, Cao J, Yuen T, Lu P, Bab I, Leu NA, Srinivasan S, Wagage S, Hunter CA, Nebert DW, Zaidi M, Avadhani NG. Smoke carcinogens cause bone loss through aryl hydrocarbon receptor and induction of CYP1 enzymes. Proc Natl Acad Sci USA. 2013;110:11115–11120. doi: 10.1073/pnas.1220919110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Chen C, Satoh J, Yim S, Gonzalez FJ. Dietary phytochemicals regulate whole-body CYP1A1 expression through an aryl hydrocarbon receptor nuclear translocator-dependent system in gut. J Clin Invest. 2007;117:1940–1950. doi: 10.1172/JCI31647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izawa T, Arakaki R, Mori H, Tsunematsu T, Kudo Y, Tanaka E, Ishimaru N. Nuclear receptor AHR controls bone homeostasis by regulating osteoclast differentiation via the RANK/c-FOS Signaling Axis. J Immunol. 2016;197:4639–4650. doi: 10.4049/jimmunol.1600822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger C, Tischkau SA. Role of aryl hydrocarbon receptor in circadian clock disruption and metabolic dysfunction. Environ Health Insights. 2016;10:133–141. doi: 10.4137/EHI.S38343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Xu ZZ, Strichartz G, Serhan CN. Emerging roles of resolvins in the resolution of inflammation and pain. Trends Neurosci. 2011;34:599–609. doi: 10.1016/j.tins.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Dalton TP, Jin L, Wang B, Tsuneoka Y, Shertzer HG, Deka R, Nebert DW. Toward the evaluation of function in genetic variability: characterizing human SNP frequencies and establishing BAC-transgenic mice carrying the human CYP1A1_CYP1A2 locus. Hum Mutat. 2005;25:196–206. doi: 10.1002/humu.20134. [DOI] [PubMed] [Google Scholar]
- Kee K, Flores M, Cedars MI, Reijo Pera RA. Human primordial germ cell formation is diminished by exposure to environmental toxicants acting through the AHR signaling pathway. Toxicol Sci. 2010;117:218–224. doi: 10.1093/toxsci/kfq179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BM, Rhee JS, Hwang UK, Seo JS, Shin KH, Lee JS. Dose- and time-dependent expression of aryl hydrocarbon receptor (AHR) and aryl hydrocarbon receptor nuclear translocator (ARNT) in PCB-, B[a]P-, and TBT-exposed intertidal copepod Tigriopus japonicus. Chemosphere. 2015;120:398–406. doi: 10.1016/j.chemosphere.2014.07.099. [DOI] [PubMed] [Google Scholar]
- Kimura S, Donovan JC, Nebert DW. Expression of the mouse P1450 gene during differentiation without foreign chemical stimulation. J Exp Pathol. 1987a;3:61–74. [PubMed] [Google Scholar]
- Kimura S, Gonzalez FJ, Nebert DW. The murine Ah locus. Comparison of the complete cytochrome P1-450 and P3-450 cDNA nucleotide and amino acid sequences. J Biol Chem. 1984;259:10705–10713. [PubMed] [Google Scholar]
- Kimura S, Nebert DW. cDNA and complete amino acid sequence of mouse P2-450. Nucleic Acids Res. 1986;14:6765–6766. doi: 10.1093/nar/14.16.6765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S, Smith HH, Hankinson O, Nebert DW. Analysis of two benzo[a]pyrene-resistant mutants of the mouse hepatoma Hepa-1 P1450 gene via cDNA expression in yeast. EMBO J. 1987b;6:1929–1933. doi: 10.1002/j.1460-2075.1987.tb02453.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko CI, Fan Y, de GM, Wang Q, Xia Y, Puga A. Repression of aryl hydrocarbon receptor Is required to maintain mitotic progression and prevent loss of pluripotency of embryonic stem cells. Stem Cells. 2016;34:2825–2839. doi: 10.1002/stem.2456. [DOI] [PubMed] [Google Scholar]
- Ko CI, Wang Q, Fan Y, Xia Y, Puga A. Pluripotency factors and polycomb group proteins repress aryl hydrocarbon receptor expression in mouse embryonic stem cells. Stem Cell Res. 2014;12:296–308. doi: 10.1016/j.scr.2013.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondraganti SR, Fernandez-Salguero P, Gonzalez FJ, Ramos KS, Jiang W, Moorthy B. Polycyclic aromatic hydrocarbon-inducible DNA adducts: evidence by 32P-postlabeling and use of Ahr-null knockout mice for mechanisms of metabolic activation in liver. Int J Cancer. 2003;103:5–11. doi: 10.1002/ijc.10784. [DOI] [PubMed] [Google Scholar]
- Konopka RJ, Benzer S. Clock mutants of Drosophila melanogaster. Proc Natl Acad Sci U S A. 1971;68:2112–2116. doi: 10.1073/pnas.68.9.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitinger JM, Beamer CA, Shepherd DM. Environmental immunology: lessons learned from exposure to a select panel of immunotoxicants. J Immunol. 2016;196:3217–3225. doi: 10.4049/jimmunol.1502149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krock BL, Eisinger-Mathason TS, Giannoukos DN, Shay JE, Gohil M, Lee DS, Nakazawa MS, Sesen J, Skuli N, Simon MC. Aryl hydrocarbon receptor nuclear translocator (ARNT) is an essential regulator of mouse hematopoietic stem cell viability. Blood. 2015;125:3263–3272. doi: 10.1182/blood-2014-10-607267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühn H, Wiesner R, Alder L, Fitzsimmons BJ, Rokach J, Brash AR. A Formation of lipoxin B by the pure reticulocyte lipoxygenase via sequential oxygenation of the substrate. Eur J Biochem. 1987;169:593–601. doi: 10.1111/j.1432-1033.1987.tb13650.x. [DOI] [PubMed] [Google Scholar]
- Kuroda M, Oikawa K, Ohbayashi T, Yoshida K, Yamada K, Mimura J, Matsuda Y, Fujii-Kuriyama Y, Mukai K. A dioxin-sensitive gene, mammalian WAPL, is implicated in spermatogenesis. FEBS Lett. 2005;579:167–172. doi: 10.1016/j.febslet.2004.11.070. [DOI] [PubMed] [Google Scholar]
- Lahvis GP, Bradfield CA. Ahr null alleles: distinctive or different? Biochem Pharmacol. 1998;56:781–787. doi: 10.1016/s0006-2952(98)00134-8. [DOI] [PubMed] [Google Scholar]
- Lahvis GP, Pyzalski RW, Glover E, Pitot HC, McElwee MK, Bradfield CA. Aryl hydrocarbon receptor is required for developmental closure of the ductus venosus in the neonatal mouse. Mol Pharmacol. 2005;67:714–720. doi: 10.1124/mol.104.008888. [DOI] [PubMed] [Google Scholar]
- Lamas B, Richard ML, Leducq V, Pham HP, Michel ML, Da CG, Bridonneau C, Jegou S, Hoffmann TW, Natividad JM, Brot L, Taleb S, Couturier-Maillard A, Nion-Larmurier I, Merabtene F, Seksik P, Bourrier A, Cosnes J, Ryffel B, Beaugerie L, Launay JM, Langella P, Xavier RJ, Sokol H. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat Med. 2016;22:598–605. doi: 10.1038/nm.4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latchney SE, Lioy DT, Henry EC, Gasiewicz TA, Strathmann FG, Mayer-Proschel M, Opanashuk LA. Neural precursor cell proliferation is disrupted through activation of aryl hydrocarbon receptor by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Stem Cells Dev. 2011;20:313–326. doi: 10.1089/scd.2009.0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Beau MM, Carver LA, Espinosa R, III, Schmidt JV, Bradfield CA. Chromosomal localization of the human AHR locus encoding the structural gene for the Ah receptor to 7p21–>p15. CytogenetCell Genet. 1994;66:172–176. doi: 10.1159/000133694. [DOI] [PubMed] [Google Scholar]
- Lee HU, McPherson ZE, Tan B, Korecka A, Pettersson S. Host-microbiome interactions: aryl hydrocarbon receptor and the central nervous system. J Mol Med (Berl) 2016;xx doi: 10.1007/s00109-016-1486-0. 2016 Nov 17 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legraverend C, Hannah RR, Eisen HJ, Owens IS, Nebert DW, Hankinson O. Regulatory gene product of the Ah locus. Characterization of receptor mutants among mouse hepatoma clones. J Biol Chem. 1982;257:6402–6407. [PubMed] [Google Scholar]
- Li N, Zhou Y, Du L, Wei M, Chen X. Overview of CYP1B1 gene mutations in patients with primary congenital glaucoma. Exp Eye Res. 2011;93:572–579. doi: 10.1016/j.exer.2011.07.009. [DOI] [PubMed] [Google Scholar]
- Libby RT, Smith RS, Savinova OV, Zabaleta A, Martin JE, Gonzalez FJ, John SW. Modification of ocular defects in mouse developmental glaucoma models by tyrosinase. Science. 2003;299:1578–1581. doi: 10.1126/science.1080095. [DOI] [PubMed] [Google Scholar]
- Lin C, Todo T. The cryptochromes. Genome Biol. 2005;6:220. doi: 10.1186/gb-2005-6-5-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey S, Papoutsakis ET. The evolving role of aryl hydrocarbon receptor (AHR) in normophysiology of hematopoiesis. Stem Cell Rev. 2012;8:1223–1235. doi: 10.1007/s12015-012-9384-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu AY, Kuntzman R, West S, Conney AH. Reconstituted liver microsomal enzyme system that hydroxylates drugs, other foreign compounds and endogenous substrates. Determination of substrate specificity by the cytochrome P-450 and P-448 fractions. Biochem Biophys Res Commun. 1971;42:1200–1206. doi: 10.1016/0006-291x(71)90033-7. [DOI] [PubMed] [Google Scholar]
- Luna RA, Savidge TC, Williams KC. The brain-gut-microbiome axis: what role does it play in autism spectrum disorder? Curr Dev Disord Rep. 2016;3:75–81. doi: 10.1007/s40474-016-0077-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maezawa K, Miyazato K, Matsunaga T, Momose Y, Imamura T, Johkura K, Sasaki K, Ohmori S. Expression of cytochrome P450 and transcription factors during in vitro differentiation of mouse embryonic stem cells into hepatocytes. Drug Metab Pharmacokinet. 2008;23:188–195. doi: 10.2133/dmpk.23.188. [DOI] [PubMed] [Google Scholar]
- Marinkovic N, Pasalic D, Potocki S. Polymorphisms of genes involved in polycyclic aromatic hydrocarbon biotransformation and atherosclerosis. Biochem Med (Zagreb) 2013;23:255–265. doi: 10.11613/BM.2013.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mas E, Croft KD, Zahra P, Barden A, Mori TA. Resolvins D1, D2, and other mediators of self-limited resolution of inflammation in human blood following ω-3 fatty acid supplementation. Clin Chem. 2012;58:1476–1484. doi: 10.1373/clinchem.2012.190199. [DOI] [PubMed] [Google Scholar]
- Mason HS. Mechanisms of oxygen metabolism. Science. 1957;125:1185–1188. doi: 10.1126/science.125.3259.1185. [DOI] [PubMed] [Google Scholar]
- Mason HS, Fowlkes WL, Peterson E. Oxygen transfer and electron transport by the phenolase complex. J Am Chem Soc. 1955;77:2914–2915. [Google Scholar]
- Mason HS, North JC, Vanneste M. Microsomal mixed-function oxidations: the metabolism of xenobiotics. Fed Proc. 1965;24:1172–1180. [PubMed] [Google Scholar]
- Mazzoli R, Pessione E. The neuro-endocrinological role of microbial glutamate and GABA signaling. Front Microbiol. 2016;7:1934. doi: 10.3389/fmicb.2016.01934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellor CL, Steinmetz FP, Cronin MT. Identification of nuclear receptors associated with hepatic steatosis to develop and extend adverse outcome pathways. Crit Rev Toxicol. 2016;46:138–152. doi: 10.3109/10408444.2015.1089471. [DOI] [PubMed] [Google Scholar]
- Moura-Alves P, Fae K, Houthuys E, Dorhoi A, Kreuchwig A, Furkert J, Barison N, Diehl A, Munder A, Constant P, Skrahina T, Guhlich-Bornhof U, Klemm M, Koehler AB, Bandermann S, Goosmann C, Mollenkopf HJ, Hurwitz R, Brinkmann V, Fillatreau S, Daffe M, Tummler B, Kolbe M, Oschkinat H, Krause G, Kaufmann SH. AHR sensing of bacterial pigments regulates antibacterial defence. Nature. 2014;512:387–392. doi: 10.1038/nature13684. [DOI] [PubMed] [Google Scholar]
- Mulero-Navarro S, Fernandez-Salguero PM. New trends in aryl hydrocarbon receptor biology. Front Cell Dev Biol. 2016;4:45. doi: 10.3389/fcell.2016.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nambu JR, Lewis JO, Wharton KA, Jr, Crews ST. Drosophila single-minded gene encodes a helix-loop-helix protein that acts as a master regulator of CNS midline development. Cell. 1991;67:1157–1167. doi: 10.1016/0092-8674(91)90292-7. [DOI] [PubMed] [Google Scholar]
- Nanez A, Ramos IN, Ramos KS. A mutant Ahr allele protects the embryonic kidney from hydrocarbon-induced deficits in fetal programming. Environ Health Perspect. 2011;119:1745–1753. doi: 10.1289/ehp.1103692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nault R, Fader KA, Lydic TA, Zacharewski TR. Lipidomic evaluation of aryl hydrocarbon receptor-mediated hepatic steatosis in male and female mice elicited by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Chem Res Toxicol. 2017;30:1060–1075. doi: 10.1021/acs.chemrestox.6b00430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebert DW. Microsomal cytochromes b5 and P450 during induction of aryl hydrocarbon hydroxylase activity in mammalian cell culture. J Biol Chem. 1970;245:519–527. [PubMed] [Google Scholar]
- Nebert DW. The Ah locus: genetic differences in toxicity, cancer, mutation, and birth defects. Crit Rev Toxicol. 1989;20:153–174. doi: 10.3109/10408448909017908. [DOI] [PubMed] [Google Scholar]
- Nebert DW. Drug metabolism and signal transduction: possible role of Ah receptor and arachidonic acid cascade in protection from ethanol toxicity. EXS. 1994;71:231–240. doi: 10.1007/978-3-0348-7330-7_23. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Brown DD, Towne DW, Eisen HJ. Association of fertility, fitness and longevity with the mouse Ah locus among C57BL/6N × C3H/HeN recombinant inbred lines. Biol Reprod. 1984;30:363–373. doi: 10.1095/biolreprod30.2.363. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Dalton TP. The role of cytochrome P450 enzymes in endogenous signalling pathways and environmental carcinogenesis. Nat Rev Cancer. 2006;6:947–960. doi: 10.1038/nrc2015. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Dalton TP, Okey AB, Gonzalez FJ. Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J Biol Chem. 2004;279:23847–23850. doi: 10.1074/jbc.R400004200. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Eisen HJ, Negishi M, Lang MA, Hjelmeland LM, Okey AB. Genetic mechanisms controlling the induction of polysubstrate monooxygenase (P-450) activities. Annu Rev Pharmacol Toxicol. 1981;21:431–462. doi: 10.1146/annurev.pa.21.040181.002243. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Gálvez-Peralta M, Shi Z, Dragin N. Inbreeding and epigenetics: beneficial as well as deleterious effects. Nat Rev Genet. 2010;11:662. doi: 10.1038/nrg2664-c2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebert DW, Gelboin HV. Substrate-inducible microsomal aryl hydroxylase in mammalian cell culture. I. Assay and properties of induced enzyme. J Biol Chem. 1968a;243:6242–6249. [PubMed] [Google Scholar]
- Nebert DW, Gelboin HV. Substrate-inducible microsomal aryl hydroxylase in mammalian cell culture. II. Cellular responses during enzyme induction. J Biol Chem. 1968b;243:6250–6261. [PubMed] [Google Scholar]
- Nebert DW, Gelboin HV. In vivo and in vitro induction of aryl hydrocarbon hydroxylase in mammalian cells of different species, tissues, strains, and developmental and hormonal states. Arch Biochem Biophys. 1969;134:76–89. doi: 10.1016/0003-9861(69)90253-7. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Gelboin HV. Role of ribonucleic acid and protein synthesis in microsomal aryl hydrocarbon hydroxylase induction in cell culture. The independence of transcription and translation. J Biol Chem. 1970;245:160–168. [PubMed] [Google Scholar]
- Nebert DW, Goujon FM, Gielen JE. Aryl hydrocarbon hydroxylase induction by polycyclic hydrocarbons: simple autosomal dominant trait in the mouse. Nat New Biol. 1972;236:107–110. doi: 10.1038/newbio236107a0. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Karp CL. Endogenous functions of aryl hydrocarbon receptor (AHR): intersection of cytochrome P4501 (CYP1)-metabolized eicosanoids and AHR biology. J Biol Chem. 2008;283:36061–36065. doi: 10.1074/jbc.R800053200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebert DW, Roe AL, Dieter MZ, Solis WA, Yang Y, Dalton TP. Role of aromatic hydrocarbon receptor and [Ahr] gene battery in the oxidative stress response, cell cycle control, and apoptosis. Biochem Pharmacol. 2000;59:65–85. doi: 10.1016/s0006-2952(99)00310-x. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Shi Z, Gálvez-Peralta M, Uno S, Dragin N. Oral benzo[a]pyrene: understanding pharmacokinetics, detoxication, and consequences—Cyp1 knockout mouse lines as a paradigm. Mol Pharmacol. 2013a;84:304–313. doi: 10.1124/mol.113.086637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebert DW, Wikvall K, Miller WL. Human cytochromes P450 in health and disease. Philos Trans R Soc Lond B Biol Sci. 2013b;368:20120431. doi: 10.1098/rstb.2012.0431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebert DW, Winker J, Gelboin HV. Aryl hydrocarbon hydroxylase activity in human placenta from cigarette smoking and nonsmoking women. Cancer Res. 1969;29:1763–1769. [PubMed] [Google Scholar]
- Nelson DR, Koymans L, Kamataki T, Stegeman JJ, Feyereisen R, Waxman DJ, Waterman MR, Gotoh O, Gunsalus IC, Nebert DW. P450 superfamily: update on new sequences, gene mapping, accession numbers, and nomenclature. Pharmacogenetics. 1996;6:1–42. doi: 10.1097/00008571-199602000-00002. [DOI] [PubMed] [Google Scholar]
- Nelson DR, Zeldin DC, Hoffman SM, Maltais LJ, Wain HM, Nebert DW. Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenetics. 2004;14:1–18. doi: 10.1097/00008571-200401000-00001. [DOI] [PubMed] [Google Scholar]
- Neri T, Merico V, Garagna S, Redi CA, Zuccotti M. Expression of phase I and phase II genes in mouse embryonic stem cells cultured in the presence of 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochim Biophys Acta. 2008;1780:826–836. doi: 10.1016/j.bbagen.2008.02.002. [DOI] [PubMed] [Google Scholar]
- Newton WA, Snell EE. Formation and interrelationships of tryptophanase and tryptophan synthetases in Escherichia coli. J Bacteriol. 1965;89:355–364. doi: 10.1128/jb.89.2.355-364.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LP, Bradfield CA. The search for endogenous activators of the aryl hydrocarbon receptor. Chem Res Toxicol. 2008;21:102–116. doi: 10.1021/tx7001965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura T, Kabashima K. Advances in atopic dermatitis in 2015. J Allergy Clin Immunol. 2016;138:1548–1555. doi: 10.1016/j.jaci.2016.10.004. [DOI] [PubMed] [Google Scholar]
- Okey AB. An aryl hydrocarbon receptor odyssey to the shores of toxicology: the Deichmann Lecture, International Congress of Toxicology-XI. Toxicol Sci. 2007;98:5–38. doi: 10.1093/toxsci/kfm096. [DOI] [PubMed] [Google Scholar]
- Omura T, Sato R. A new cytochrome in liver microsomes. J Biol Chem. 1962;237:1375–1376. [PubMed] [Google Scholar]
- Omura T, Sato R. The carbon monoxide-binding pigment of liver microsomes. Evidence for its hemoprotein nature. J Biol Chem. 1964;239:2370–2378. [PubMed] [Google Scholar]
- Omura T, Sato R, Cooper DY, Rosenthal O, Estabrook RW. Function of cytochrome P-450 of microsomes. Fed Proc. 1965;24:1181–1189. [PubMed] [Google Scholar]
- Paine AJ. Comparison of the photochemical and enzymic generation of excited states of oxygen on the induction of benzo[a]pyrene mon-oxygenase activity in liver cell cultures. Chem Biol Interact. 1977;16:309–314. doi: 10.1016/0009-2797(77)90110-7. [DOI] [PubMed] [Google Scholar]
- Panigrahy D, Edin ML, Lee CR, Huang S, Bielenberg DR, Butterfield CE, Barnes CM, Mammoto A, Mammoto T, Luria A, Benny O, Chaponis DM, Dudley AC, Greene ER, Vergilio JA, Pietramaggiori G, Scherer-Pietramaggiori SS, Short SM, Seth M, Lih FB, Tomer KB, Yang J, Schwendener RA, Hammock BD, Falck JR, Manthati VL, Ingber DE, Kaipainen A, D’Amore PA, Kieran MW, Zeldin DC. Epoxyeicosanoids stimulate multiorgan metastasis, and tumor dormancy escape, in mice. J Clin Invest. 2012;122:178–191. doi: 10.1172/JCI58128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parli CJ, Mannering GJ. Induction of drug metabolism. IV. Relative abilities of polycyclic hydrocarbons to increase levels of microsomal 3-methyl-4-methylaminoazobenzene N-demethylase activity and cytochrome P1-450. Mol Pharmacol. 1970;6:178–183. [PubMed] [Google Scholar]
- Patel SA, Velingkaar NS, Kondratov RV. Transcriptional control of antioxidant defense by the circadian clock. Antioxid Redox Signal. 2014;20:2997–3006. doi: 10.1089/ars.2013.5671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecenova L, Farkas R. Multiple functions and essential roles of nuclear receptor coactivators of bHLH-PAS family. Endocr Regul. 2016;50:165–181. doi: 10.1515/enr-2016-0019. [DOI] [PubMed] [Google Scholar]
- Pellequer JL, Wager-Smith KA, Kay SA, Getzoff ED. Photoactive yellow protein: a structural prototype for the three-dimensional fold of the PAS domain superfamily. Proc Natl Acad Sci U S A. 1998;95:5884–5890. doi: 10.1073/pnas.95.11.5884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernomian L, da Silva CH. Current basis for discovery and development of aryl hydrocarbon receptor antagonists for experimental and therapeutic use in atherosclerosis. Eur J Pharmacol. 2015;764:118–123. doi: 10.1016/j.ejphar.2015.06.058. [DOI] [PubMed] [Google Scholar]
- Poland A, Glover E. 2,3,7,8-Tetrachlorodibenzo-p-dioxin: a potent inducer of δ -aminolevulinic acid synthetase. Science. 1973;179:476–477. doi: 10.1126/science.179.4072.476. [DOI] [PubMed] [Google Scholar]
- Poland A, Glover E. Chlorinated biphenyl induction of aryl hydrocarbon hydroxylase activity: study of the structure-activity relationship. Mol Pharmacol. 1977;13:924–938. [PubMed] [Google Scholar]
- Poland A, Glover E, Kende AS. Stereospecific, high affinity binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin by hepatic cytosol. Evidence that the binding species is receptor for induction of aryl hydrocarbon hydroxylase. J Biol Chem. 1976;251:4936–4946. [PubMed] [Google Scholar]
- Poland AP, Glover E, Robinson JR, Nebert DW. Genetic expression of aryl hydrocarbon hydroxylase activity. Induction of monooxygenase activities and cytochrome P1-450 formation by 2,3,7,8-tetrachlorodibenzo-p-dioxin in mice genetically “nonresponsive” to other aromatic hydrocarbons. J Biol Chem. 1974;249:5599–5606. [PubMed] [Google Scholar]
- Powell-Coffman JA, Bradfield CA, Wood WB. Caenorhabditis elegans orthologs of aryl hydrocarbon receptor and its heterodimerization partner, aryl hydrocarbon receptor nuclear translocator. Proc Natl Acad Sci U S A. 1998;95:2844–2849. doi: 10.1073/pnas.95.6.2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probst MR, Fan CM, Tessier-Lavigne M, Hankinson O. Two mouse homologs of the Drosophila single-minded protein that interact with mouse aryl hydrocarbon receptor nuclear translocator protein. J Biol Chem. 1997;272:4451–4457. doi: 10.1074/jbc.272.7.4451. [DOI] [PubMed] [Google Scholar]
- Prochazkova J, Kabatkova M, Smerdova L, Pachernik J, Sykorova D, Kohoutek J, Simeckova P, Hruba E, Kozubik A, Machala M, Vondracek J. Aryl hydrocarbon receptor negatively regulates expression of the plakoglobin gene (jup) Toxicol Sci. 2013;134:258–270. doi: 10.1093/toxsci/kft110. [DOI] [PubMed] [Google Scholar]
- Proctor KG, Falck JR, Capdevila J. Intestinal vasodilation by epoxyeicosatrienoic acids: arachidonic acid metabolites produced by a cytochrome P450 monooxygenase. Circ Res. 1987;60:50–59. doi: 10.1161/01.res.60.1.50. [DOI] [PubMed] [Google Scholar]
- Puga A, Barnes SJ, Dalton TP, Chang C, Knudsen ES, Maier MA. Aromatic hydrocarbon receptor interaction with retinoblastoma protein potentiates repression of E2F-dependent transcription and cell cycle arrest. J Biol Chem. 2000;275:2943–2950. doi: 10.1074/jbc.275.4.2943. [DOI] [PubMed] [Google Scholar]
- Puga A, Ma C, Marlowe JL. Aryl hydrocarbon receptor cross-talks with multiple signal-transduction pathways. Biochem Pharmacol. 2009;77:713–722. doi: 10.1016/j.bcp.2008.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y, Tian X, Liu J, Han Y, Graham AM, Simon MC, Penninger JM, Carmeliet P, Li S. BNIP3 and AIF cooperate to induce apoptosis and cavitation during epithelial morphogenesis. J Cell Biol. 2012;198:103–114. doi: 10.1083/jcb.201111063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Zhou L. Aryl hydrocarbon receptor promotes RORγ+ group 3 ILCs and controls intestinal immunity and inflammation. Semin Immunopathol. 2013;35:657–670. doi: 10.1007/s00281-013-0393-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmioglu N, Nyholt DR, Morris AP, Missmer SA, Montgomery GW, Zondervan KT. Genetic variants underlying risk of endometriosis: insights from meta-analysis of eight genome-wide association and replication datasets. Hum Reprod Update. 2014;20:702–716. doi: 10.1093/humupd/dmu015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitzel AM, Passamaneck YJ, Karchner SI, Franks DG, Martindale MQ, Tarrant AM, Hahn ME. Aryl hydrocarbon receptor (AHR) in the cnidarian Nematostella vectensis: comparative expression, protein interactions, and ligand-binding. Dev Genes Evol. 2014;224:13–24. doi: 10.1007/s00427-013-0458-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentas S, Holzapfel NT, Belew MS, Pratt GA, Voisin V, Wilhelm BT, Bader GD, Yeo GW, Hope KJ. Musashi-2 attenuates AHR signalling to expand human haematopoietic stem cells. Nature. 2016;532:508–511. doi: 10.1038/nature17665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rier SE, Turner WE, Martin DC, Morris R, Lucier GW, Clark GC. Serum levels of TCDD and dioxin-like chemicals in Rhesus monkeys chronically exposed to dioxin: correlation of increased serum PCB levels with endometriosis. Toxicol Sci. 2001;59:147–159. doi: 10.1093/toxsci/59.1.147. [DOI] [PubMed] [Google Scholar]
- Rifkind AB, Muschick H. Benoxaprofen suppression of polychlorinated biphenyl toxicity without alteration of mixed-function oxidase function. Nature. 1983;303:524–526. doi: 10.1038/303524a0. [DOI] [PubMed] [Google Scholar]
- Robertson JA, Hankinson O, Nebert DW. Autoregulation plus positive and negative elements controlling transcription of genes in the [Ah] battery. Chem Scripta. 1987;27A:83–87. [Google Scholar]
- Robinson JR, Considine N, Nebert DW. Genetic expression of aryl hydrocarbon hydroxylase induction. Evidence for involvement of other genetic loci. J Biol Chem. 1974;249:5851–5859. [PubMed] [Google Scholar]
- Sabatini PV, Lynn FC. All-encomPASsing regulation of beta-cells: PAS domain proteins in beta-cell dysfunction and diabetes. Trends Endocrinol Metab. 2015;26:49–57. doi: 10.1016/j.tem.2014.11.002. [DOI] [PubMed] [Google Scholar]
- Safe SH, Pallaroni L, Yoon K, Gaido K, Ross S, McDonnell D. Problems for risk assessment of endocrine-active estrogenic compounds. Environ Health Perspect. 2002;110(Suppl 6):925–929. doi: 10.1289/ehp.02110s6925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safe TM, Luebke AE. Prenatal low-dosage dioxin (TCDD) exposure impairs cochlear function resulting in auditory neuropathy. Hear Res. 2016;331:7–12. doi: 10.1016/j.heares.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuelsson B. Role of basic science in the development of new medicines: examples from the eicosanoid field. J Biol Chem. 2012;287:10070–10080. doi: 10.1074/jbc.X112.351437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauzeau V, Carvajal-Gonzalez JM, Riolobos AS, Sevilla MA, Menacho-Marquez M, Roman AC, Abad A, Montero MJ, Fernandez-Salguero P, Bustelo XR. AHR transcriptional factor controls cardiovascular and respiratory functions by regulating expression of the Vav3 proto-oncogene. J Biol Chem. 2011a;286:2896–2909. doi: 10.1074/jbc.M110.187534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauzeau V, Carvajal-Gonzalez JM, Riolobos AS, Sevilla MA, Menacho-Marquez M, Roman AC, Abad A, Montero MJ, Fernandez-Salguero P, Bustelo XR. Transcriptional factor aryl hydrocarbon receptor (AHR) controls cardiovascular and respiratory functions by regulating expression of the Vav3 proto-oncogene. J Biol Chem. 2011b;286:2896–2909. doi: 10.1074/jbc.M110.187534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaldach CM, Riby J, Bjeldanes LF. Lipoxin A4: a new class of ligand for the Ah receptor. Biochemistry. 1999;38:7594–7600. doi: 10.1021/bi982861e. [DOI] [PubMed] [Google Scholar]
- Scheer N, Wilson ID. A comparison between genetically humanized and chimeric liver humanized mouse models for studies in drug metabolism and toxicity. Drug Discov Today. 2016;21:250–263. doi: 10.1016/j.drudis.2015.09.002. [DOI] [PubMed] [Google Scholar]
- Scheer N, Wolf CR. Genetically humanized mouse models of drug-metabolizing enzymes and transporters and their applications. Xenobiotica. 2014;44:96–108. doi: 10.3109/00498254.2013.815831. [DOI] [PubMed] [Google Scholar]
- Schiering C, Wincent E, Metidji A, Iseppon A, Li Y, Potocnik AJ, Omenetti S, Henderson CJWCR, Nebert DW, Stockinger B. Feedback control of AHR signalling regulates intestinal immunity. Nature. 2016;542:242–245. doi: 10.1038/nature21080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt JV, Carver LA, Bradfield CA. Molecular characterization of the mouse Ahr gene. Organization, promoter analysis, and chromosomal assignment. J Biol Chem. 1993;268:22203–22209. [PubMed] [Google Scholar]
- Schneider AJ, Branam AM, Peterson RE. Intersection of AHR and WNT-signaling in development, health, and disease. Int J Mol Sci. 2014;15:17852–17885. doi: 10.3390/ijms151017852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senft AP, Dalton TP, Nebert DW, Genter MB, Hutchinson RJ, Shertzer HG. Dioxin increases reactive oxygen production in mouse liver mitochondria. Toxicol Appl Pharmacol. 2002a;178:15–21. doi: 10.1006/taap.2001.9314. [DOI] [PubMed] [Google Scholar]
- Senft AP, Dalton TP, Nebert DW, Genter MB, Puga A, Hutchinson RJ, Kerzee JK, Uno S, Shertzer HG. Mitochondrial reactive oxygen production is dependent on aryl hydrocarbon receptor. Free Radic Biol Med. 2002b;33:1268–1278. doi: 10.1016/s0891-5849(02)01014-6. [DOI] [PubMed] [Google Scholar]
- Serhan CN, Fredman G, Yang R, Karamnov S, Belayev LS, Bazan NG, Zhu M, Winkler JW, Petasis NA. Novel proresolving aspirin-triggered DHA pathway. Chem Biol. 2011;18:976–987. doi: 10.1016/j.chembiol.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Lu Y, Hong S, Yang R. Mediator lipidomics: search algorithms for eicosanoids, resolvins, and protectins. Methods Enzymol. 2007;432:275–317. doi: 10.1016/S0076-6879(07)32012-0. [DOI] [PubMed] [Google Scholar]
- Shadfan M, Lopez-Pajares V, Yuan ZM. MDM2 and MDM4: alone and together in regulation of TP53. Transl Cancer Res. 2012;1:88–89. [PMC free article] [PubMed] [Google Scholar]
- Shertzer HG, Nebert DW, Puga A, Ary M, Sonntag D, Dixon K, Robinson LJ, Cianciolo E, Dalton TP. Dioxin causes a sustained oxidative stress response in the mouse. Biochem Biophys Res Commun. 1998;253:44–48. doi: 10.1006/bbrc.1998.9753. [DOI] [PubMed] [Google Scholar]
- Shi Z, Chen Y, Dong H, Amos-Kroohs RM, Nebert DW. Generation of a ‘humanized’ hCYP1A1_1A2_Cyp1a1/1a2(−/−)_Ahrd mouse line harboring the poor-affinity aryl hydrocarbon receptor. Biochem Biophys Res Commun. 2008;376:775–780. doi: 10.1016/j.bbrc.2008.09.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shichi H, Nebert DW. Genetic differences in drug metabolism associated with ocular toxicity. Environ Health Perspect. 1982;44:107–117. doi: 10.1289/ehp.8244107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu Y, Nakatsuru Y, Ichinose M, Takahashi Y, Kume H, Mimura J, Fujii-Kuriyama Y, Ishikawa T. Benzo[a]pyrene carcinogenicity is lost in mice lacking the aryl hydrocarbon receptor. Proc Natl Acad Sci U S A. 2000;97:779–782. doi: 10.1073/pnas.97.2.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shum S, Jensen NM, Nebert DW. The mouse Ah locus: in utero toxicity and teratogenesis associated with genetic differences in benzo[a]pyrene metabolism. Teratology. 1979;20:365–376. doi: 10.1002/tera.1420200307. [DOI] [PubMed] [Google Scholar]
- Sofo V, Gotte M, Lagana AS, Salmeri FM, Triolo O, Sturlese E, Retto G, Alfa M, Granese R, Abrao MS. Correlation between dioxin and endometriosis: an epigenetic route to unravel the pathogenesis of the disease. Arch Gynecol Obstet. 2015;292:973–986. doi: 10.1007/s00404-015-3739-5. [DOI] [PubMed] [Google Scholar]
- Soshilov AA, Denison MS. Ligand promiscuity of aryl hydrocarbon receptor agonists and antagonists revealed by site-directed mutagenesis. Mol Cell Biol. 2014;34:1707–1719. doi: 10.1128/MCB.01183-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter P, Velick SF. The purification and properties of microsomal cytochrome reductase. J Biol Chem. 1957;228:785–799. [PubMed] [Google Scholar]
- Tohyama C. Clarification of molecular targets of dioxin toxicity. Nihon Eiseigaku Zasshi. 2014;69:1–7. doi: 10.1265/jjh.69.1. [DOI] [PubMed] [Google Scholar]
- Tukey RH, Hannah RR, Negishi M, Eisen HJ, Nebert DW. The Ah locus: correlation of intranuclear appearance of inducer-receptor complex with induction of cytochrome P1-450 mRNA. Cell. 1982;31:275–284. doi: 10.1016/0092-8674(82)90427-5. [DOI] [PubMed] [Google Scholar]
- Uno S, Sakurai K, Nebert DW, Makishima M. Protective role of cytochrome P450 1A1 (CYP1A1) against benzo[a]pyrene-induced toxicity in mouse aorta. Toxicology. 2014;316:34–42. doi: 10.1016/j.tox.2013.12.005. [DOI] [PubMed] [Google Scholar]
- Vogt JH, Schippers JH. Setting the PAS, the role of circadian PAS domain proteins during environmental adaptation in plants. Front Plant Sci. 2015;6:513. doi: 10.3389/fpls.2015.00513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volotinen M, Maenpaa J, Kankuri E, Oksala O, Pelkonen O, Nakajima M, Yokoi T, Hakkola J. Expression of cytochrome P450 (CYP) enzymes in human nonpigmented ciliary epithelial cells: induction of CYP1B1 expression by TCDD. Invest Ophthalmol Vis Sci. 2009;50:3099–3105. doi: 10.1167/iovs.08-2790. [DOI] [PubMed] [Google Scholar]
- Vorrink SU, Domann FE. Regulatory crosstalk and interference between xenobiotic- and hypoxia-sensing pathways at the AHR-ARNT-HIF1 α signaling node. Chem Biol Interact. 2014;218:82–88. doi: 10.1016/j.cbi.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Chen J, Ko CI, Fan Y, Carreira V, Chen Y, Xia Y, Medvedovic M, Puga A. Disruption of aryl hydrocarbon receptor homeostatic levels during ES stem cell differentiation alters expression of homeobox transcription factors that control cardiomyogenesis. Environ Health Perspect. 2013;121:1334–1343. doi: 10.1289/ehp.1307297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Kurita H, Carreira V, Ko CI, Fan Y, Zhang X, Biesiada J, Medvedovic M, Puga A. AHR activation by dioxin disrupts activin, BMP, and WNT signals during early differentiation of mouse embryonic stem cells and Inhibits cardiomyocyte functions. Toxicol Sci. 2016;149:346–357. doi: 10.1093/toxsci/kfv246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wattenberg LW, Leong JL, Strand PJ. Benzpyrene hydroxylase activity in the gastrointestinal tract. Cancer Res. 1962;22:1120–1125. [PubMed] [Google Scholar]
- Welch RM, Harrison YE, CONNEY AH, Poppers PJ, Finster M. Cigarette smoking: stimulatory effect on metabolism of 3,4-benzpyrene by enzymes in human placenta. Science. 1968;160:541–542. doi: 10.1126/science.160.3827.541. [DOI] [PubMed] [Google Scholar]
- Wells PG, Lee CJ, McCallum GP, Perstin J, Harper PA. Receptor- and reactive intermediate-mediated mechanisms of teratogenesis. Handb Exp Pharmacol. 2010;196:131–162. doi: 10.1007/978-3-642-00663-0_6. [DOI] [PubMed] [Google Scholar]
- Wimuttisuk W, Tobwor P, Deenarn P, Danwisetkanjana K, Pinkaew D, Kirtikara K, Vichai V. Insights into the prostanoid pathway in ovary development of the penaeid shrimp Penaeus monodon. PLoS One. 2013;8:e76934. doi: 10.1371/journal.pone.0076934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodarchak N, Xing Y. PP2A as a master regulator of the cell cycle. Crit Rev Biochem Mol Biol. 2016;51:162–184. doi: 10.3109/10409238.2016.1143913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woeller CF, Roztocil E, Hammond CL, Feldon SE, Phipps RP. Aryl hydrocarbon receptor and its ligands inhibit myofibroblast formation and activation: implications for thyroid eye disease. Am J Pathol. 2016;186:3189–3202. doi: 10.1016/j.ajpath.2016.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wölfle U, Seelinger G, Bauer G, Meinke MC, Lademann J, Schempp CM. Reactive molecule species and antioxidative mechanisms in normal skin and skin aging. Skin Pharmacol Physiol. 2014;27:316–332. doi: 10.1159/000360092. [DOI] [PubMed] [Google Scholar]
- Woods SL, Whitelaw ML. Differential activities of mouse single-minded 1 (SIM1) and SIM2 on a hypoxic response element. Cross-talk between basic helix-loop-helix/per-Arnt-sim homology transcription factors. J Biol Chem. 2002;277:10236–10243. doi: 10.1074/jbc.M110752200. [DOI] [PubMed] [Google Scholar]
- Wu D, Potluri N, Kim Y, Rastinejad F. Structure and dimerization properties of the aryl hydrocarbon receptor PAS-A domain. Mol Cell Biol. 2013;33:4346–4356. doi: 10.1128/MCB.00698-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L, Zhang Z, Luo X. Roles of xenobiotic receptors in vascular pathophysiology. Circ J. 2014;78:1520–1530. doi: 10.1253/circj.cj-14-0343. [DOI] [PubMed] [Google Scholar]
- Zack JA, Suskind RR. The mortality experience of workers exposed to tetrachlorodibenzodioxin in a trichlorophenol process accident. J Occup Med. 1980;22:11–14. [PubMed] [Google Scholar]
- Zhang X, Lu J, He B, Tang L, Liu X, Zhu D, Cao H, Wang Y, Li L. A tryptophan derivative, ITE, enhances liver cell metabolic functions in vitro. Int J Mol Med. 2017;39:101–112. doi: 10.3892/ijmm.2016.2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Dong S, Wang H, Tao S, Kiyama R. Biological impact of environmental polycyclic aromatic hydrocarbons (ePAHs) as endocrine disruptors. Environ Pollut. 2016;213:809–824. doi: 10.1016/j.envpol.2016.03.050. [DOI] [PubMed] [Google Scholar]
- Zhu C, Chen YL, Wang XJ, Hu XS, Yu ZB, Han SP. shRNA-mediated gene silencing of AHR promotes the differentiation of P19 mouse embryonic carcinoma cells into cardiomyocytes. Mol Med Rep. 2012;6:513–518. doi: 10.3892/mmr.2012.941. [DOI] [PubMed] [Google Scholar]