

SUMMARY
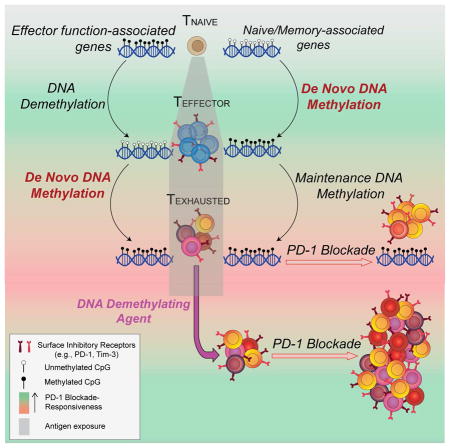
Immune-checkpoint blockade (ICB)-mediated rejuvenation of exhausted T cells has emerged as a promising approach for treating various cancers and chronic infections. However, T cells that become fully exhausted during prolonged antigen exposure remain refractory to ICB-mediated rejuvenation. We report that blocking de novo DNA methylation in activated CD8 T cells allows them to retain their effector functions despite chronic stimulation during a persistent viral infection. Whole-genome bisulfite sequencing of antigen-specific murine CD8 T cells at the effector and exhaustion stages of an immune response identified progressively acquired heritable de novo methylation programs that restrict T-cell expansion and clonal diversity during PD-1 blockade treatment. Moreover, these exhaustion-associated DNA-methylation programs were acquired in tumor-infiltrating PD-1hi CD8 T cells, and approaches to reverse these programs improved T cell responses and tumor control during ICB. These data establish de novo DNA-methylation programming as a regulator of T-cell exhaustion and barrier of ICB-mediated T-cell rejuvenation.
eTOC
de novo DNA methylation programs promote T-cell exhaustion, and blocking these programs can enhance T cell rejuvenation, aiding tumor control by immune checkpoint blockade.
INTRODUCTION
Antigen-driven clonal expansion and differentiation of naive CD8 T cells initially instills the cells with an effector program that facilitates their ability to directly and indirectly kill antigen-presenting cells (Kaech and Cui, 2012). However, prolonged stimulation of the cells, as occurs during chronic infection and cancer, results in a progressive suppression of the cell’s effector function, commonly referred to as “exhaustion” (Wherry, 2011). During acquisition of the exhausted state, T-cell functional impairment follows a hierarchical loss in expression of the effector cytokines interleukin-2 (IL-2), tumor necrosis factor alpha (TNFα), and interferon γ (IFNγ), respectively. Accompanying the impairment in cytokine expression is a decline in the cells’ cytolytic ability, proliferative capacity, and sustained upregulated expression of surface inhibitory receptors (IRs), including programmed cell death 1 (PD-1) (Wherry, 2011) The discovery that IRs serve as an immune checkpoint regulating T cell effector function was rapidly translated into therapeutic approaches that block these checkpoints for the treatment of various cancers (Sharma and Allison, 2015a).
While immune checkpoint blockade (ICB) therapy has yielded striking clinical responses, its success is unfortunately limited to a minority of patients with cancer (Sharma and Allison, 2015a). The mechanism(s) underlying ICB therapy non-responsiveness remains a major challenge for a broader success of this therapy. Recent efforts to identify antigen-specific CD8 T cells that retain the potential to respond to ICB therapy have established that the cumulative expression of multiple inhibitory receptors progressively restricts the cells’ ability to be rejuvenated (Jin et al., 2010). Commensurate with the progressive upregulation of IRs and reduced responsiveness to ICB therapy, it has become evident that aspects of the T-cell exhaustion gene-expression program are reinforced, resulting in stable maintenance of exhaustion-associated features even if the antigen levels are reduced or cleared (Angelosanto et al., 2012; Utzschneider et al., 2013; Youngblood et al., 2011).
Given that exhaustion-associated gene expression programs can be maintained in the absence of antigen, we sought to better understand the heritable nature of T-cell-intrinsic transcriptional programming and how it impacts ICB therapy. During cellular differentiation, recruitment and/or eviction of specific transcription factors to regions of accessible chromatin facilitate cell-type-specific gene expression programming (Vaquerizas et al., 2009). Long-term maintenance of transcription factor accessibility to gene regulatory elements is controlled in part by covalent modifications to histones and DNA that affect chromatin structure, resulting in an “epigenetic memory” of gene expression programs in a dividing population of cells (Bintu et al., 2016; Bird, 2002). Consistent with prior reports showing that exhaustion-associated gene expression programs become reinforced, a recent study reported that T cell exhaustion is associated with a general increase in chromatin accessibility, and that many of these accessible regions are retained after PD-1 blockade therapy (Pauken et al., 2016). While a variety of transcription factors and epigenetic modifications are associated with changes in chromatin accessibility, recent evidence supports the notion that DNA methylation is a critical epigenetic mechanism for establishing stable gene-silencing programs (Bintu et al., 2016; Cedar and Bergman, 2009). We report here that de novo DNA methylation acquired during and after the peak of an effector CD8 T cell response to a chronic viral infection or tumor challenge is critical for establishing T-cell exhaustion. Our study identifies genome-wide de novo DNA-methylation programs that promote terminal differentiation of exhausted T cells and reveals that these programs persist after PD-1 blockade therapy. In contrast, CD8 T cells lacking the acquisition of such methylation programs resist functional exhaustion and retain a greater potential for expansion after ICB. These data establish Dnmt3a-mediated de novo DNA methylation as a mechanism restricting the efficacy of ICB therapy and have broad implications for novel approaches to enhance T cell-based immunotherapies.
RESULTS
Post-effector De Novo DNA-Methylation Programming Accompanies T-cell Exhaustion
Recent studies demonstrated that the phenotypic and functional changes that occur during the naïve-to-effector stage of CD8 T cell differentiation are accompanied by genome-wide changes in DNA-methylation programming (Scharer et al., 2013); however, the role of these changes in regulating the functional state of the cell are largely unknown. To determine if T-cell post-effector adaptation is also accompanied by additional epigenetic modifications, and to assess the consequence of these epigenetic programs on the development of T-cell exhaustion, we measured the quantity and function of antigen-specific wild-type (WT) or mutant CD8 T cells that lack the ability to acquire de novo DNA-methylation programs during persistent LCMV infection in mice. Inhibition of de novo methylation programming was achieved by conditionally deleting the de novo DNA methyltransferase Dnmt3a (Dnmt3a-cKO; hereafter referred to as cKO mice) using a Cre recombinase under the control of the granzyme b promoter (Figures S1A and S1B). WT and cKO mice were initially depleted of CD4 T cells and then infected with the chronic strain of LCMV (Figures 1A and 1B). Longitudinal tracking of virus-specific CD8 T cells in the blood of chronically infected WT mice revealed a progressive decline in gp33-specific (a dominant LCMV epitope) CD8 T cells (Figure 1C) as well as a reduction in the total quantity of polyclonal LCMV-specific CD8 T cells (Figure S1C). In contrast, the contraction of virus-specific cKO CD8 T cells after the peak of the effector response was modest, and the quantity of cKO virus-specific CD8 T cells that survived the contraction stage of the immune response was maintained at a much greater level relative to the WT CD8 T cells (Figures 1C and S1C). We next assessed whether the retained cKO CD8 T cells also maintained their effector function despite persistent antigen exposure and maintenance of elevated expression of PD-1 (Figures S1E and S1G). As expected, WT antigen-specific CD8 T cells isolated from mice after two months of chronic infection were severely impaired in their ability to produce IFNγ and IL-2 in response to peptide stimulation (Figure 1D). In contrast, cKO CD8 T cells retained a substantial capacity to co-produce both cytokines (Figures 1D and S1F). Thus, CD8 T cells lacking the ability to acquire new DNA methylation programs retain a heightened capacity to recall effector cytokines despite prolonged TCR stimulation and sustained PD-1 expression.
Figure 1. Whole-Genome methylation profiling of LCMV-specific WT and Dnmt3a-Deficient CD8 T Cells.

(A) Experimental setup; CD4 T cells were depleted in WT and Dnmt3a-cKO mice prior to infection with LCMV clone 13.
(B) Longitudinal summary graph of WT and Dnmt3a cKO mice serum viral titers during chronic LCMV infection.
(C) Summary graph of gp33-specific CD8 T cell quantity in the peripheral blood of WT and cKO mice during chronic LCMV infection.
(D) Representative FACS analysis of PD-1 expression on gp33-specific CD8 T cells from the spleens of WT and cKO mice after 2 months of chronic infection, gated on CD8+ T cells. Representative intracellular FACS analysis of IFNγ and IL-2 expression from gp33 peptide-stimulated CD44hi CD8 T cell splenocytes of chronically infected WT and cKO mice.
(E) Experimental setup for performing whole-genome bisulfite sequencing (WGBS) methylation analysis of virus-specific CD8 T cells.
(F) The number of newly methylated regions in gp33-specific effector or exhausted WT CD8 T cells.
(G) Heat map showing cluster analysis of the top 3000 differentially methylated regions (DMRs) among naïve and LCMV-specific WT and cKO CD8 T cells at the effector and chronic stages of the immune response. Color intensity scales from red (methylated region) to blue (non-methylated region).
(H) Summary table of ingenuity pathway analysis showing putative upstream regulators for Dnmt3a-targeted genes.
(I) Nucleotide-resolution methylation profiling of the IFNγ and Myc loci in naïve and LCMV-specific CD8 T cells. Vertical blue and red lines indicate CpG positions in the loci. The ratio of blue to red indicates the percentage of unmethylated versus methylated reads, respectively, in the WGBS.
N= 3–5 mice per group of two or more independent experiments. Error bars indicate SEM.
To identify de novo DNA-methylation programs associated with progressive commitment to T-cell exhaustion, we next measured genome-wide DNA-methylation changes in WT and cKO virus-specific CD8 T cells at the effector (8 days post-infection—8 dpi, Figure S1H) and exhaustion (35 dpi) stages of the immune response (Figure 1E). Initial assessment of CpG methylation levels across the entire genome of all samples demonstrated that differentiation of both WT and cKO day 8 antigen-specific effector CD8 T cells was coupled to ~ 10,000 demethylated regions relative to naïve CD8 T cells (Figures S1I and S1J). In addition to DNA demethylation, WT CD8 T cells acquired ~ 980 de novo programs, the majority of which were not acquired in the cKO effector CD8 T cells (Figure 1F and S1K). We next examined both DNA demethylation and de novo methylation events in WT CD8 T cells during the effector-to-exhaustion stage of T cell development. In contrast to the striking level of DNA demethylation that occurred during the effector stage of the immune response, only 280 additionally demethylated regions were detected (Figures S1J). Moreover, we found that WT CD8 T cells acquired an additional 1200 Dnmt3a-dependent DNA methylation events during the effector-to-exhaustion transition (Figure 1F). These data indicate that, during the effector-to-exhaustion transition, WT CD8 T cells continue to undergo epigenetic changes that are predominantly de novo events. To further characterize when WT and Dnmt3a cKO CD8 T cells acquire divergent DNA methylation programs, we performed an unsupervised principle-component analysis (PCA) and generated a dendrogram of the WGBS datasets based on of the methylation status of the top 3000 most variable CpG sites in all WT and cKO antigen-specific CD8 T cells. Our analysis revealed that the DNA methylation status of WT CD8 T cells segregated from the methylation status of cKO CD8 T cells during the effector-to-exhaustion transition (Figure S1L). Importantly, we found that several of the chronic-associated de novo epigenetic events were acquired at sites that were initially methylated in naïve CD8 T cells (Figure 1G).
We next performed a gene ontology (GO) biological process analysis of the exhaustion-associated Dnmt3a-targeted loci to determine if Dnmt3a-targeted genes were involved in biological processes known to be impacted during T-cell exhaustion. Importantly, the analysis of Dnmt3a-targets in exhausted WT CD8 T cells identified several immune-related functions among the top ranked GO biological processes (Figure S1M). In addition, Biocarta pathway enrichment analysis of all Dnmt3a-targets showed statistical enrichment of immune-related signaling pathways, such as TCR-signaling and IL-7/IL-2-signaling pathways (Figure S1N). We further characterized these de novo programs using ingenuity-pathway analysis and observed that several transcription factors and signaling molecules, known to regulate immune-related pathways, were identified as putative regulators of the Dnmt3a-targeted loci (Figures 1H). Specifically among the list of Dnmt3a-targeted loci were IFNγ, Myc (Figure 1I), Tcf7, Ccr7, T-bet, and Eomesodermin (Eomes) (Figure S2A). These data document the DNA-methylation programs acquired during and after the peak of the effector response that are coupled to the progressive decline in CD8 T cell effector functions.
Chronic T cell Stimulation Induces Post-effector De Novo DNA-Methylation Programming
Our data demonstrate that de novo DNA-methylation programs are progressively acquired during the development of T-cell exhaustion and that specific DMRs serve as an epigenetic signature for exhausted T cells. However, it is unclear whether these post-effector DNA-methylation programs are acquired due to persistent stimulation or if they are acquired due to a hard-wired differentiation program that continues regardless of additional TCR stimulation. Therefore, we next sought to examine whether exhaustion-associated de novo DNA-methylation programs were also acquired in highly functional WT memory CD8 T cells generated during an acute viral infection. We designed loci-specific bisulfite sequencing primers to assess the methylation status of an exhaustion-associated DMR in the IFNγ locus in naïve and virus-specific CD8 T cells isolated from chronically infected and infection matched immune WT and cKO mice (Figure 2A). Both memory T cells from immune mice and cKO CD8 T cells from chronically infected mice retain high expression of IFNγ (Figure 2B), and accordingly remained demethylated at the IFNγ DMR, while only WT exhausted T cells from chronically infected animals remethylated this region (Figures 2C and S2B). Thus, acquisition of the post-effector de novo DNA-methylation program at the IFNγ locus is not simply due to slow accumulation of DNA-methylation marks over time in the aged cells, rather results from chronic stimulation. We next asked if naïve and memory-associated genes that are downregulated during the effector stage of the immune response acquire and maintain de novo DNA methylation under conditions of persistent antigen stimulation. To do this we measured the methylation level of the DMR in the Ccr7 locus in naïve and virus-specific WT and cKO CD8 T cells isolated 8 dpi (effector cells) and 60 dpi from chronic LCMV-infected mice and compared it to that of functional memory WT CD8 T cells isolated 60 dpi from acute infected mice. Indeed, downregulation of CCR7 in effector (at 8 dpi) CD8 T cells accompanied de novo methylation of the locus (Figure S2B). Functional memory cells had a reduction in this de novo program consistent with a subset of functional memory CD8 T cells expressing CCR7. However, the Ccr7 effector-associated DMR underwent further methylation in the exhausted WT cells (Figures 2D and S2B). These data suggest that the transcriptional repression of genes during the effector stage of the immune response may become epigenetically reinforced during or shortly after the effector stage.
Figure 2. Exhaustion-Associated De Novo DNA Methylation Programs Are not Acquired during a Primary Acute Viral Infection.

(A) Experimental setup for loci-specific methylation analysis of exhaustion-associated DMRs in functional versus exhausted virus-specific CD8 T cells.
(B) Representative intracellular FACS analysis and summary graph of mean fluorescence intensity (MFI) for IFNγ and IL-2 expression among ex vivo stimulated (gp33-peptide) WT and cKO CD44hi CD8 T cells isolated 2 months post-acute (Armstrong strain) or chronic (Clone 13 strain) LCMV infection.
(C) Schematic of CpG sites in the IFNγ and (D) Ccr7 loci. Loci-specific bisulfite sequencing analysis of the IFNγ and Ccr7 DMRs in naïve and tetramer+ CD8 T cells isolated from acutely and chronically infected WT and cKO mice. Horizontal lines represent individual sequenced clones from the pool of FACS-purified tetramer+ CD8 T cells. Filled circles, methylated cytosine; open circles, nonmethylated cytosine.
(E) Nucleotide-resolution view of DNA methylation programs at IFNγ and Ccr7 loci from WGBS analyses of exhausted and functional memory WT CD8 T cells.
(F) Venn diagram of WGBS data showing overlap between exhaustion-specific Dnmt3a- targeted loci and functional memory-specific demethylated loci among WT CD8 T cells.
Error bars indicate SEM.
To more broadly define the de novo DNA methylation programs that are unique to T cell exhaustion, we performed WGBS methylation profiling of WT memory CD8 T cells isolated 35 days after acute LCMV infection and compared the methylation profile with the profile of exhausted WT CD8 T cells to identify DMRs. Importantly, more than 71% of the unmethylated DMRs in functional memory WT CD8 T cells relative to exhausted WT CD8 T cells were also identified as Dnmt3a-targets in exhausted cells (e.g., IFNγ and Ccr7 loci) (Figures 2E, 2F, S3A, and S3B). We next asked if these programs are only acquired in the absence of CD4 T cell help. Interestingly, we found that even in the presence of CD4 T cell help, WT exhausted CD8 T cells still acquire similarly high levels of DNA methylation at several of these regions (Figure S3C). Taken together, the lack of exhaustion-associated de novo methylation programs at a genome-wide level in functional memory CD8 T cells further suggests that these programs result from the chronic stimulation of exhausted T cells.
De Novo DNA-Methylation Programming Regulates Development of Fully Exhausted T Cells
Progressive adaptation of antigen-specific CD8 T cells to chronic stimulation occurs asynchronously among the total pool of antigen-experienced cells and results in heterogeneous populations of CD8 T cells with varying degrees of T-cell exhaustion. Coupled to this adaptation is the differential expression of the T-box transcription factors T-bet and Eomes (Speiser et al., 2014; Wherry, 2011). In line with these prior findings, we identified exhaustion-associated de novo programs in the T-bet and Eomes loci among our WGBS data (Figures S4A and S2A). Correspondingly, Dnmt3a-deficient CD8 T cells had significantly higher T-bet and lower Eomes expression compared to the levels observed in exhausted WT cells (Figures S4C and S4D). Furthermore, the Eomeslo cells were predominantly Tim-3− and T-bet+ (Figures S4C, S4E, and S4F). Recent studies have also linked Tcf7 expression to retention of T-cell function during chronic infection (Im et al., 2016; Utzschneider et al., 2016). Consistent with these reports, we identified an effector–associated de novo DNA-methylation program in the Tcf7 locus (Figure S2A). Loci-specific methylation analysis of the Tcf7 DMR in virus-specific CD8 T cells isolated from chronically infected WT and cKO mice demonstrated that exhausted WT cells retained the repressive program acquired during the effector stage of the immune response (Figure S4B) whereas cKO cells retained an unmethylated Tcf7 DMR and expressed significantly higher levels of Tcf1 (Figures S4G and S4H). Together, these data further support a model whereby de novo DNA methylation serves as a mechanism for stable epigenetic gene silencing and reinforces commitment to terminal differentiation of exhausted T cells.
Exhaustion-Associated De Novo Methylation Programming Is Coupled to the Limited Proliferative Capacity of Exhausted CD8 T Cells
Preservation of cKO CD8 T-cell quantity during prolonged exposure to high levels of antigen prompted us to assess the epigenetic status of genes associated with the cell’s proliferative potential. c-Myc has been shown to be essential for activation-induced metabolic reprogramming and antigen-driven proliferation of naive CD8 T cells (Wang et al., 2011) and our data revealed that the Myc locus acquires an exhaustion-associated de novo DMR (Figure 1I). Loci-specific methylation analysis of the Myc DMR among naïve, functional memory and exhausted WT virus-specific CD8 T cells confirmed that the Myc locus undergoes striking demethylation during the effector stage of the immune response, followed by remethylation during chronic antigen exposure (Figure S5A). In contrast, memory CD8 T cells retained their demethylated state (Figure S5A). To determine if this epigenetic program was coupled to changes in Myc-regulated expression programs, we measured the surface expression of CD98, a downstream metabolic target of c-Myc, in antigen-specific WT and cKO CD8 T cells during chronic or acute LCMV infection. Demethylation of the Myc DMR in WT effector antigen-specific CD8 T cells was coupled to upregulated CD98 expression (Figure S5B). Additionally, both WT and cKO memory CD8 T cells maintained elevated levels of CD98 expression (Figure S5B). In contrast, CD98 expression was downregulated on the exhausted WT cells but was retained at higher levels on cKO CD8 T cells during chronic infection (Figure S5B), indicating that the de novo methylation acquired during the development of T-cell exhaustion is coupled to Myc-regulated protein expression.
To examine whether these programs were coupled to T-cell proliferation, we stained virus-specific CD8 T cells for Ki67 at the effector and chronic stages of the immune response. At 8 dpi, the majority of both WT and cKO effector CD8 T cells expressed high levels of Ki67 (Figure S5C) but by 60 dpi the proliferation of virus-specific WT CD8 T cells was substantially reduced. In contrast, antigen-specific and polyclonal virus-specific cKO CD8 T cells have significantly higher levels of Ki67 in both Tim-3+ and Tim-3− subsets of PD-1+ cells (Figures S5D–F). These data suggest that the elevated quantity of cKO cells during persistent infection is in part coupled to their retained proliferative capacity.
CD8 T Cell-Intrinsic De Novo Programming Regulates Exhaustion
To determine if the retained effector function and proliferative capacity of Dnmt3a-cKO CD8 T cells exposed to chronic stimulation are due to cell-intrinsic changes in DNA methylation programming rather than extrinsic factors, we generated cKO transgenic P14 cells (transgenic CD8 T cells with a TCR specific to the gp33 epitope) and co-adoptively transferred congenically distinct WT and cKO P14 CD8 T cells into Rag1 KO mice. Use of the chimeric Rag1 KO mouse model enabled us to control for the influence of extrinsic factors such as other activated Gzmb-expressing immune cells—NK and CD4 T cells, undetected differences in viral titers, inflammatory microenvironment, and differences in the depletion of peripheral CD4 T cells among WT versus cKO mice. Chimeric Rag1 KO mice were infected with the chronic strain of LCMV and the P14 cells were tracked longitudinally (Figure 3A). Both WT and cKO P14 cells underwent a similarly robust expansion during the effector stage of the immune response, but following this proliferative burst, WT P14 cells experienced a much greater degree of contraction relative to host-matched cKO P14 cells, resulting in an elevated frequency of cKO P14 cells that was maintained into the chronic stage of the immune response (Figure 3B). The selective preservation of cKO P14 cells during chronic infection was not only observed in the blood and spleen, but also in peripheral non-lymphoid tissues, including the liver and lungs (Figure 3C). We found that the cKO P14 cells retained an enhanced capacity to proliferate, higher Tcf1 expression, and heightened effector cytokine production (Figures 3D and 3E). Locus-specific DNA methylation analysis of a post-effector-associated DMR (e.g., in the IFNγ locus) and an effector-associated DMR (e.g., in the Tcf7 locus) further confirmed selective inhibition of de novo DNA-methylation programming in cKO P14 cells (Figures 3F and 3G). These data confirm that the retained effector properties of cKO CD8 T cells are cell-intrinsic, and collectively suggest that Dnmt3a-mediated DNA-methylation programming is critical for establishing the major hallmarks of T-cell exhaustion.
Figure 3. CD8 T cell-Intrinsic De Novo Programming Regulates Exhaustion.

(A) Experimental setup for tracking congenically distinct WT and cKO P14 CD8 T cells in LCMV infected Rag1 KO mice.
(B) Longitudinal measurement of WT and cKO P14 cell frequencies in the peripheral blood of chimeric Rag1 KO mice during chronic LCMV infection.
(C) Representative FACS analysis and summary graphs of cKO (red) and WT (blue) P14 cell frequency in tissues of chronically infected chimeric Rag1 KO mice at 35 dpi.
(D) Representative FACS analysis and summary graph of IFNγ expression frequency among cKO and WT P14 cells from spleens of chronically infected chimeric Rag1 KO mice.
(E) Representative FACS analysis and summary graphs of intracellular TCF1 and Ki67 levels among cKO and WT P14 cells in spleens from chronically infected chimeric Rag1 KO mice.
(F) Representative post-sort purity FACS analysis of cKO and WT P14 cells from spleens of chronically infected chimeric Rag1 KO mice.
(G) Locus-specific DNA methylation analysis of the IFNγ and Tcf7 DMRs in naïve P14 cells from uninfected mice, and WT and cKO P14 splenocytes from chronically infected chimeric Rag1 KO mice.
N = 3 mice per group of two independent experiments. Error bars indicate SEM.
PD-1 Blockade Therapy Does not Erase Exhaustion-Associated DNA-Methylation Programs in Rejuvenated T cells
Our finding that exhaustion-associated DNA-methylation programs play a critical role in establishing a terminal-exhaustion fate prompted us to next ask whether PD-1 blockade-mediated rejuvenation of T cells results in erasure of these programs (Figure 4A). To address this question we treated chronically infected WT mice with anti-PD-L1, isolated the rejuvenated antigen-specific CD8 T cells, and assessed the methylation status of the exhaustion associated DMRs (Figure 4A). As expected, PD-1 blockade treatment increased the quantity of gp33-specific and total polyclonal CD44hi PD-1+ CD8 T cells in WT mice (Figure 4B). Quite strikingly, we observed no change in the effector and post-effector de novo DNA-methylation programs despite T-cell expansion (Figures 4C and S2C). Preservation of the de novo DNA-methylation programs at the IFNγ, Myc, Tcf7, Ccr7 and Tbx21 loci in the rejuvenated WT CD8 T cells prompted us to more broadly assess the stability of DNA-methylation programming in all Dnmt3a-targeted genes. WGBS was performed on FACS-purified antigen-specific CD8 T cells from the spleens of the PD-1 blockade–treated and untreated WT mice (Figure 4A). Only 5964 DMRs between the treated versus untreated WT CD8 T cells were detected among the WT exhausted and WT rejuvenated WGBS data sets. Among the 5964 DMRs only 84 were Dnmt3a-mediated programs, which accounts for less than 2% reprogramming of the total Dnmt3a-mediated exhaustion programs (Figure 4D). Notably, when we compared PD-1 blockade-associated DNA methylation DMRs with the methylation profiles of functional memory WT CD8 T cells we found that < 1% of these DMRs were present in the functional T cells (Figure 4D). Importantly, exhaustion-associated DNA-methylation programming across the IFNγ, Myc, Tcf7, and Tbx21 loci were unchanged in rejuvenated WT CD8 T cells (Figures 4E and S2C). These data illustrate how remarkably stable the exhaustion-associated DNA-methylation programs are. Given that PD-1 blockade does not erase exhaustion-associated methylation programs, and that they play a causal role in restricting effector function, it raises the question of whether these specific epigenetic programs restrict the therapeutic response of PD-1 blockade treatment.
Figure 4. Exhaustion-Associated De Novo DNA-Methylation Programs are Preserved during PD-L1 Blockade Therapy.

(A) Experimental setup for loci-specific bisulfite sequencing analysis of exhaustion-associated DMRs in virus-specific effector (isolated at 8 dpi with the chronic strain of LCMV), exhausted, and rejuvenated CD8 T cells.
(B) Representative FACS analysis of CD44 expression and gp33 tetramer staining in CD8 T cells from the spleens of chronically infected WT after mock treatment or PD-1 blockade treatment. Summary graph of polyclonal (CD44hi PD-1+) virus-specific CD8 T cell fold expansion in the spleens of chronically infected WT mice after mock treatment (gray bar) or PD-1 blockade (red bar) treatment. Fold expansion was calculated relative to change in mock-treated WT mice. Error bars indicate SD.
(C) Longitudinal loci-specific bisulfite sequencing analysis of de novo DNA methylation programs in antigen-specific WT CD8 T cells. Red bars represent the average percentage of DNA methylation of all CpG sites in the specified DMR at different time points during chronic virus infection. Representative methylation profiles are adjacent to the corresponding bar graphs. Error bars indicate SEM.
(D) Venn diagrams of WGBS data showing overlap between Dnmt3a-mediated exhaustion- specific DMRs and PD-1 blockade–generated DMRs among WT CD8 T cells (top panel), and overlap between functional memory-related DMRs and PD-1 blockade–generated DMRs among WT CD8 T cells (bottom panel).
(E) Nucleotide-resolution view of DNA methylation programs at IFNγ, Myc, Tcf7 and Tbx21 loci from WGBS analyses of exhausted WT CD8 T cells before and after PD-1 blockade.
N= 3–5 mice per group of two independent experiments.
Inhibition of De Novo DNA-Methylation Programming Synergizes with PD-1 Blockade to Enhance Rejuvenation of CD8 T cells
Maintenance of PD-1hi Dnmt3a cKO virus-specific CD8 T cells in an environment with viral loads comparable to chronically infected WT mice provided a unique opportunity to test if Dnmt3a-mediated de novo DNA methylation restricts the ability of cells to respond to PD-1 blockade therapy. Chronically infected WT and Dnmt3a cKO mice were treated with anti-PD-L1 for two weeks and the antigen-specific T cell response was measured (Figure 5A). Quite strikingly, the cKO virus-specific CD8 T cells increased in frequency and quantity during PD-1 blockade to a significantly greater degree compared to WT virus-specific CD8 T cells. The expanded population of cKO T cells was observed not only in the spleen (Figures 5B, S6A, and S6B) but also in non-lymphoid tissues (Figures 5C, 5D, S6C, and S6D). Notably, gp33-specific and np396-specific CD8 T cells, which are known to be less responsive to PD-1 blockade relative to gp276-specific CD8 T cells, underwent a synergistic expansion after PD-1 blockade treatment in cKO mice (Figures 5B, S6C, and S6E). Consistent with our observation that Dnmt3a-deficient CD8 T cells resist terminal differentiation, the expanded virus-specific cKO CD8 T cells retained lower levels of Tim-3 and Eomes and a higher level of T-bet after PD-1 blockade treatment (Figures S6A and S6B). These data demonstrate that de novo DNA methylation programs restrict the ability of exhausted T cells to respond to PD-1 blockade therapy.
Figure 5. Inhibiting De Novo DNA Methylation Synergizes with PD-L1 Blockade.

(A) Experimental setup of anti-PD-L1 treatment in chronically infected WT and Dnmt3a cKO mice.
(B) Representative FACS analysis and summary graphs of gp33-, gp276-, and np396-specific CD8 T cell frequencies in the spleens of treated and untreated chronically infected WT and cKO mice; WT mock-treated = gray bars, WT anti–PD-L1–treated = red bars, cKO mock-treated = black bars, and cKO PD-1 blockade–treated = green bars.
(C) Representative FACS analysis showing the frequencies of gp33-specific CD8 T cells in the lungs and (D) liver of chronically infected WT and cKO mice after mock or PD-1 blockade treatment.
(E) Longitudinal analysis of the gp33-specific TCR repertoire Simpson’s diversity index among WT and cKO CD8 T cells isolated from the peripheral blood before and after PD-1 blockade treatment.
(F) Representative intracellular FACS analysis of IFNγ and IL-2 expression from gp33-stimulated CD44hi CD8 T cell splenocytes of mock or PD-1 blockade-treated chronically infected WT and cKO mice. Summary graphs of fold change in quantity of total IFNγ-expressing or IFNγ and IL-2 co-expressing CD8 T cells from spleens of mock or PD-1 blockade–treated chronically infected WT and cKO mice.
(G) Longitudinal measurement of viral titers in the serum and summary graphs of the viral titers in the spleen or liver of chronically infected WT and cKO mice after mock or PD-1 blockade treatment.
Fold change was calculated relative to mock-treated WT mice. N= 3–5 mice per group of two independent experiments. Error bars indicate SEM.
Because we observed a striking response across multiple LCMV epitope-specific CD8 T-cell populations in cKO mice, we next proceeded to determine if the expanded pool of cKO cells maintained their T cell clonal diversity. GP33-specific CD8 T cells were single-cell sorted from the blood of chronically infected WT and cKO mice before and after PD-L1 blockade, and the TCR β-chain of the individual cells was sequenced. Interestingly, rejuvenated WT CD8 T cells displayed a reduced TCR repertoire diversity among the expanded gp33-specific T cells, whereas the TCR repertoire diversity of gp33-specific cKO CD8 T cells was maintained after PD-1 blockade treatment (Figures 5E and S6G). These data suggest that exhaustion-associated DNA methylation programming restricts the clonal breadth of cells that can proliferate in response to PD-1 blockade therapy.
We next assessed the function of the PD-L1 blockade-expanded population of antigen-specific cKO CD8 T cells by measuring their ability to recall effector cytokines after peptide stimulation. Indeed the expanded population of LCMV-specific cKO CD8 T cells retained a heightened capacity to produce IFNγ and IL-2 (Figures 5F, S6H, and S6I). Given that PD-1 blockade in the cKO mice resulted in expansion of an extensive repertoire of LCMV-specific CD8 T cells that retained effector properties, we next proceeded to determine whether the enhanced therapeutic response impacted the viral load. Strikingly, anti-PD-L1 treatment resulted in a significant reduction in the viral burden in multiple tissues in cKO mice compared to that in anti-PD-L1–treated WT mice (Figure 5G). These data collectively serve as a proof-of-principle that therapeutic approaches designed to erase or block exhaustion-associated DNA-methylation programs may synergize with ICB therapy and enhance the therapeutic response.
DNA Demethylating Agent Treatment Prior to PD-1 Blockade Enhances the Reinvigoration Potential of Exhausted CD8 T Cells
Our finding that de novo DNA methylation programming is causal in reinforcing the development of T-cell exhaustion and establishes a stable cell-intrinsic barrier to PD-1 blockade-mediated rejuvenation of T cells prompted us to ask whether therapeutic use of DNA demethylating agents prior to PD-1 blockade treatment could enhance rejuvenation of WT exhausted CD8 T cells. WT mice chronically infected with LCMV were treated with a low dose of the standard DNA demethylating agent 5-aza-2′-deoxycytidine (decitabine “DAC”) for two weeks followed by PD-1 blockade (Figure 6A). Longitudinal analysis of virus-specific CD8 T cell quantity in the peripheral blood of chronically infected mice revealed a synergistic expansion of gp33-specific T cells in treated mice that received DAC prior to PD-1 blockade therapy compared to those receiving monotherapy (Figures 6B and 6C). Importantly, this increase in antigen-specific and the total polyclonal virus-specific CD8 T cell frequencies was also observed in the spleen, lungs, and liver of chronically infected WT mice receiving sequential combination therapy (Figures 6D, 6F, 6G, S7A, and S7B). Notably, concurrent treatment of DAC with PD-1 blockade did not enhance rejuvenation of WT exhausted T cells (data not shown). The striking expansion of virus-specific T cells observed in both lymphoid and non-lymphoid tissues of sequentially treated mice suggests that DNA demethylating agent treatment may synergize with PD-1 blockade to enhance the rejuvenation.
Figure 6. DNA Demethylating Agent Treatment Prior to PD-L1 Blockade Enhances WT CD8 T-Cell Rejuvenation.

(A) Experimental setup for sequential decitabine and anti-PD-L1 treatment during chronic LCMV infection.
(B) Summary graph showing longitudinal tracking of gp33-specific CD8 T cell quantity in the peripheral blood of chronically LCMV-infected WT mice during mono- or sequentially combined decitabine and anti-PD-L1 treatments.
(C) Summary graph of gp33-specific CD8 T cell fold expansion in the PBMC after mono PD-1 blockade therapy (red bar) or sequential decitabine and PD-1 blockade (blue bar) therapy. Fold change was calculated relative to corresponding cell numbers before anti-PD-L1 treatment.
(D) Representative FACS analysis showing the frequencies of gp33-specific CD8 T cells in the spleen, lung, and liver tissues of chronically infected WT mice after indicated treatments.
(E) Representative FACS analysis showing Ki67 levels in gp33-specific CD8 T cells in the spleens of chronically infected WT mice after indicated treatments.
(F) Summary graph of the quantity of gp33-specific CD8 T cells in spleens of chronically LCMV-infected WT mice after indicated treatments.
(G) Summary graphs of the frequencies of gp33-specific CD8 T cells in the lung and liver tissues of chronically infected WT mice after indicated treatments.
(H) Summary graph of the frequencies of Ki67-expressing gp33-specific CD8 T cells in the spleens of chronically infected WT mice after indicated treatments.
N= 3–5 mice per group of two independent experiments. Error bars indicate SEM.
To determine whether the elevated frequencies of rejuvenated WT CD8 T cells that result from sequential DAC and PD-1 blockade treatment are coupled to enhanced proliferation, we measured Ki67 expression in antigen-specific and polyclonal virus-specific CD8 T cells in chronically infected mice. Interestingly, DAC treatment significantly increased the proliferative capacity of virus-specific CD8 T cells (Figures 6E, 6H, S7C and S7D). Furthermore, combination treatment results in a greater number of cells that could recall the expression of the effector cytokine (Figures S7E, S7F, and S7G), albeit not quite as effective as genetic deletion of the DNA methyltransferase. While the antitumor effect of DNA demethylating agents is commonly attributed to its direct cytotoxicity or immunoregulating influence on tumor cells or microenvironment (Chiappinelli et al., 2016; Li et al., 2014), our data suggest that therapeutic DNA demethylation may also function by reversing exhaustion-associated de novo methylation programs in CD8 T cells, consequently enhancing the T cell’s ability to expand during ICB therapy.
Tumor-Infiltrating CD8 T Cells Acquire Exhaustion-Associated de novo DNA Methylation Programs
Recent advances in ICB therapies have provided a major breakthrough in the treatment of various cancers. Therefore, we sought to determine if our findings, made using a model of chronic viral infection, extended to a tumor setting. To assess the DNA methylation status of tumor-associated CD8 T cells we generated tumor-bearing mice utilizing the well-established murine Tramp-C2 tumor model. This model is characterized by a relatively slow tumor growth that results in prolonged stimulation of T cells (Curran and Allison, 2009). After > 45 days post Tramp-C2 cell inoculation tumor-infiltrating CD8 T cells (TILs) were characterized (Figure 7A). Phenotypic analysis of tumor-specific (Spas1 tetramer+) CD8 T cells demonstrated that they exhibited characteristics of persistent antigen exposure, including co-expression of PD-1 and Tim-3 (Figure 7B). To determine if exhaustion-associated DNA-methylation programs are acquired in phenotypically activated CD8+ TILs, we FACS purified the PD-1hi Tim3+ CD8+ TILs and measured the DNA methylation status of Tcf7, Ccr7, Myc, and IFNγ DMRs (Figure 7C). Additionally, we FACS purified tumor-matched PD-1lo CD8+ TILs to determine if the exhaustion-associated DMRs were specifically acquired in activated TILs. Similar to exhausted LCMV-specific CD8 T cells, the de novo DMRs at the Tcf7, Ccr7, Myc, and IFNγ loci in PD-1hi TILs were heavily methylated, whereas PD-1lo TILs retained demethylated Ccr7 and Tcf7 loci (Figures 7D, 7E, and S7H). Select methylation of the Tcf7 and Ccr7 loci among the PD-1hi TILs demonstrates that the cells underwent de novo programming at these loci, and further indicates that the extensive methylation of the Myc and IFNγ DMRs in the PD-1hi TILs are also de novo programs. These results demonstrate that tumor-infiltrating CD8 T cells acquire de novo DNA-methylation programming associated with reinforcement of T-cell exhaustion.
Figure 7. Tumor-Infiltrating CD8 T Cells Acquire Exhaustion-Associated DNA Methylation Programs.

(A) Experimental setup for generating TRAMP-C2 tumor-bearing mice and loci-specific DNA methylation analysis of tumor-infiltrating CD8 T cells (TILs).
(B) Representative FACS analysis of CD44, PD-1, CD62L, and Tim-3 expression on Tumor-specific Spas1+ TILs harvested after ≥ 45 days post-tumor inoculation.
(C) Representative FACS analysis of PD-1 and Tim-3 expression on total TILs and post-sort purity check of FACS-purified PD-1hi and PD-1low TILs populations.
(D) Loci-specific bisulfite sequencing methylation analysis and summary graphs of individual CpG sites in the Tcf7, Ccr7, (E) Myc and IFNγ loci among PD-1low and PD-1hi populations of TILs. Horizontal lines represent individual sequenced clones from the pool of FACS-purified TILs. Filled circles = methylated cytosine; open circles = nonmethylated cytosine.
(F) Experimental setup of sequential decitabine and anti-PD-L1 treatment of TRAMP-C2 tumor-bearing mice starting at ≥ 1 month post-inoculation.
(G) Representative FACS analysis and summary graphs of Ki67 levels among PD-1hi CD8+ TILs and tumor antigen-specific (Spas1+) TILs after mono PD-1 blockade or sequentially combined decitabine and anti-PD-L1 treatments.
(H) Longitudinal measurement of tumor growth in WT mice receiving mono anti-PD-L1 (red), or sequentially combined decitabine and anti-PD-L1 (blue) treatments. Summary graph of normalized tumor volume after mono anti-PD-L1 (red) or combined decitabine and anti-PD-L1 (blue) treatments.
N= 3–5 mice per group of two independent experiments. Error bars indicate SEM.
Given the critical role exhaustion-associated DNA methylation plays in regulating ICB-responsiveness during chronic virus infection, we sought to further assess the impact these programs have on TIL-rejuvenation during PD-1 blockade. The Tramp-C2 tumor model has been previously shown to be refractory to single PD-1/PD-L1 blockade therapy (Yu et al., 2012), therefore we tested whether primary DAC treatment could enhance rejuvenation of CD8 TILs after PD-L1 blockade. Sequential DAC and PD-L1 blockade treatments were initiated more than 4 weeks and 6 weeks, respectively, after tumor inoculation (Figure 7F). Interestingly, we found that DAC treatment was able to induce proliferation of both polyclonal (PD-1hi) and antigen-specific (Spas-1 tetramer+) tumor-infiltrating CD8 T cells after PD-L1 blockade treatment (Figure 7G). Importantly, the enhanced proliferation of tumor-specific CD8 T cells was coupled to significant control of tumor growth (Figures 7H and S7I). These data suggest that administration of DNA demethylating agents prior to ICB therapy may enhance reinvigoration of antitumor CD8 T cells; providing a rationale for therapeutic strategies that combine epigenetic reprogramming and ICB.
DISCUSSION
Reinvigoration of exhausted CD8 T cells by PD-1 blockade therapy has emerged as one of the most promising frontiers to treat individuals with chronic infections or cancer. However, the stability of exhaustion-specific gene-expression programs suggests that a cell-intrinsic mechanism(s) may restrict the capacity and durability of PD-1 blockade-mediated T-cell rejuvenation. We and others have demonstrated that changes in epigenetic programming are coupled to transcriptional reprogramming during CD8 T cell effector and memory differentiation (Crompton et al., 2015; Russ et al., 2014; Scharer et al., 2013; Youngblood et al., 2011). Yet, whether these reprogramming events play a direct role in regulating the effector properties in functional and exhausted CD8 T cells has remained unclear. Here we show that progressive de novo DNA-methylation programming is critical for establishing T-cell exhaustion. Such DNA-methylation programming reinforces the repression of key genes involved in effector function, proliferation, metabolic activity, and tissue homing in exhausted T cells. We also demonstrate that these long-lived, exhaustion-associated epigenetic programs serve as a major cell-intrinsic barrier limiting the rejuvenation of antigen-specific CD8 T cells during PD-1 blockade therapy. Our findings highlight epigenetic programs among exhausted T-cells as a potential mechanism to explain PD-1 blockade therapeutic failures.
Recent efforts to better understand the mechanism of action for ICB therapy found that the Cxcr5+ subset of PD-1+ T cells remain responsive to PD-1 blockade therapy, while T cells that express both PD-1 and Tim-3, and lack Cxcr5 expression are less responsive to PD-1 blockade therapy (Im et al., 2016; Wherry, 2011). Importantly, when we compared our cKO and WT exhausted CD8 T cell gene expression datasets (Figures S3D–4F) with the recently published gene expression data of Cxcr5+ Tim-3− (PD-1 blockade-responsive) versus Cxcr5− Tim-3+ (PD-1 blockade-non-responsive) subsets of exhausted WT CD8 T cells (Im et al., 2016), we found a significant correlation among gene expression patterns of cKO CD8 T cells and the PD-1 blockade-responsive subset of WT cells (Figure S3G) further highlighting the role de novo DNA methylation plays in regulating PD-1 blockade-responsiveness. These data indicate that progressive development of less differentiated CD8 T cells into terminally differentiated exhausted CD8 T cells is coupled to the acquisition of transcriptionally repressive DNA-methylation programs. Moreover, once established, such epigenetic programs restrict the cell’s reinvigoration potential during PD-1 blockade therapy.
While a durable clinical response has been generated in some patients that experience ICB-mediated reinvigoration of T cells, new evidence suggests that T cell rejuvenation may be transient. In a recent clinical trial utilizing ICB for melanoma treatment, about one-third of patients who initially responded to PD-1 blockade experienced tumor relapse (Ribas et al., 2016). Through efforts to better understand the stable nature of exhaustion-associated transcriptional states it was recently discovered that the short-lived reinvigoration of T cells during PD-1 blockade therapy was associated with preservation of the chromatin landscape (Pauken et al., 2016). The results presented in our current study provide mechanistic insight into the heritable maintenance of exhaustion gene expression program during ICB, and how this may leave patients vulnerable to tumor relapse (Ghoneim et al., 2016; Sharma and Allison, 2015b).
DNA-methylation programming in fully differentiated cells is often thought to be irreversible owing to the fact that DNA methylation can be propagated during DNA replication through a mechanism of maintenance methylation. Yet the recent discovery that cytosine methylation can be erased through a mechanism of TET-mediated hydroxylation has generated great interest in determining if this mechanism can be exploited to de-differentiate fate-committed cells (Pastor et al., 2013); specifically exhausted T cells during ICB therapy. In addition to exploiting natural mechanisms of epigenetic reversal, it may be possible to directly reverse epigenetic modifications with existing chemotherapeutics (Chiappinelli et al., 2016; Roulois et al., 2015; Wang et al., 2015). FDA-approved DNA demethylating agents, such as decitabine, may provide a means to modify exhaustion-associated DNA methylation programs that restrict ICB-responsiveness. Consistent with our results, two recent reports have highlighted the ability of DNA demethylating agents to enhance CTLA-4 blockade-mediated T cell responses (Chiappinelli et al., 2016; Wang et al., 2015). In these studies the effect of DNA demethylation was largely assigned to changes in expression of endogenous retrovirus dsRNA in the tumor, which in turn triggered a type I IFN immune response (Chiappinelli et al., 2016). Our data suggest that decitabine not only modifies the tumor cells, but it may also have a direct effect on antigen-specific CD8 T cells. In conclusion, our study highlights the critical role de novo DNA methylation plays in establishing the ICB-nonresponsive state of T-cell exhaustion and reveals the potential benefit of therapeutic strategies that combine ICB with approaches that reverse or inhibit T cell-intrinsic exhaustion-associated epigenetic programs.
STAR method
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CD8a (clone 53-6.7) | BioLegend | Cat#100732 |
| Anti-mouse/human CD44 (clone IM7) | BioLegend | Cat#103039 |
| Anti-mouse CD62L (clone MEL-14) | BioLegend | Cat#104405 |
| Anti-mouse PD-1 (clone 29F.1A12) | BioLegend | Cat#135216 |
| Anti-mouse Tim-3 (clone RMT3-23) | BioLegend | Cat#119704 |
| Anti-mouse CD98 (clone RL388) | BioLegend | Cat#128207 |
| Anti-mouse T-bet (clone 4B10) | BioLegend | Cat#644806 |
| Anti-mouse Eomes (clone Dan11mag) | eBioscience | Cat#25-4875-82 |
| Anti-mouse/human Tcf1 (clone C63D9) | Cell Signaling | Cat#14456S |
| Anti-mouse/human Ki67 (clone SolA15) | eBioscience | Cat#11-5698-82 |
| Anti-mouse IFNγ (clone XMG1.2) | BioLegend | Cat#505810 |
| Anti-mouse IL-2 (clone JES6-5h4) | BioLegend | Cat#503808 |
| Anti-mouse/human Granzyme b (clone GB11) | BioLegend | Cat#515408 |
| InVivoMAb anti-mouse PD-L1 (B7-H1) | BioXcell | Cat# BE0101 |
| GK1.5 antibody | Harlan Bioproducts | N/A |
| Bacterial and Virus Strains | ||
| Lymphocytic choriomeningitis virus, | Matloubian et al., 1990 | NA |
| Armstrong strain | ||
| Lymphocytic choriomeningitis virus, Clone 13 strain | Matloubian et al., 1990 | N/A |
| XL10-Gold ultracompetent E. coli | Stratagene | Cat#200314 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ghost Dye Violet 510 viability dye | Tonbo Biosciences | Cat#13-0870 |
| Foxp3/Transcription Factor Buffer Set | eBioscience | Cat#00-5523 |
| BD Cytofix/Cytoperm | BD Biosciences | Cat#554714 |
| 5-Aza-2′-deoxycytidine (decitabine) | Sigma-Aldrich | Cat#A3656 |
| LCMV gp33, gp276, and np396 monomers | Yerkes NIH tetramer core facility | NA |
| Spas-1 monomer | Peptide synthesis facility at SJCRH | NA |
| LCMV gp33, gp276, and np396 peptides | Peptide synthesis facility at SJCRH | NA |
| RNAsin | Promega | Cat#N2111 |
| Critical Commercial Assays | ||
| QIAGEN DNeasy kit | QIAGEN | Cat#69506 |
| EZ DNA methylation kit | Zymo Research | Cat#D5002 |
| TruSeq DNA Methylation Kit | Illumina | Cat# EGMK81312 |
| pGEM-T Vector cloning kit | Promega | Cat#A3600 |
| JumpStart Taq ReadyMix | Sigma-Aldrich | Cat#P2893 |
| Directprep 96 Miniprep kit | QIAGEN | Cat#27361 |
| iScript cDNA Synthesis Kit | Biorad | Cat#1708891 |
| Taq polymerase–based PCR kit | QIAGEN | Cat#201225 |
| Deposited Data | ||
| WGBS data | This paper | GEO: GSE99450 |
| Microarray data | This paper | GEO: GSE99449 |
| Experimental Models: Cell Lines | ||
| TRAMP-C2 prostate adenocarcinoma cells | ATCC | CRL-2731 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 | The Jackson Laboratory | JAX: 000664 |
| Mouse: B6.Cg-Tcratm1Mom Tg(TcrLCMV)327Sdz | Taconic | Model#:4138 |
| Mouse: B6;129S4-Dnmt3a < tm3.1Enl > | MTA from Riken Institute | RBRC037313 |
| Mouse: B6, Granzyme b-Cre | SJCRH animal house | NA |
| Oligonucleotides | ||
| Primers for IFNg, Myc, Tbx21, Ccr7, and Tcf7: See Table S1 | This paper | N/A |
| pUC/M13 reverse primer | Promega | Cat#Q5421 |
| Software and Algorithms | ||
| Prism 6 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| FlowJo 9.9.5 | FlowJo | NA |
| Model-based analysis of bisulfite sequencing | Wu et al., 2015 | https://www.ncbi.nlm.nih.gov/pubmed/26184873 |
| LIMMA | Bioconductor | https://bioconductor.org/packages/release/bioc/html/limma.html |
| DAVID 6.8 | David Pathway enrichment analysis tool | https://david.ncifcrf.gov/ |
| Ingenuity network analysis | QIAGEN | https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/ |
| Gene Ontology (GO) enrichment analysis | Enrichr | http://amp.pharm.mssm.edu/Enrichr/enrich |
| Non-redundant GO biological processes analysis | Revigo | http://revigo.irb.hr/ |
| Other | ||
| Fortessa flow cytometer | Flow Cytometry core facility at SJCRH | NA |
| ABI Big Dye sequencer (Applied Biosystem) | Hartwell Center at SJCRH | NA |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ben Youngblood (benjamin.youngblood@stjude.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice and Viral Infections
WT C57BL/6 mice were purchased from Jackson Laboratory. Dnmt3a cKO mice were generated by crossing floxed Dnmt3a mice with mice expressing a Granzyme b–driven recombinase transgene. Equal numbers of male and female 6–8 week old WT and cKO mice were used and littermates of the same sex were randomly assigned to experimental groups. For chronic infection, WT and cKO mice were treated with 0.75 mg GK1.5 antibody (Harlan Bioproducts) 2 days prior to and on the day of infection to deplete CD4 T cells (Ha et al., 2008). CD4-deficient mice were then infected with the LCMV clone 13 (2×106 pfu, i.v.). Serum and tissue virus titers were determined by plaque assay as previously described (Matloubian et al., 1990). For acute infection, equal numbers of male and female 6–8 week old WT and cKO mice were infected with the Armstrong strain of LCMV (2×105 pfu, i.p.) without CD4 T cell depletion. All mice were housed in specific pathogen free conditions prior to use. Prior to the described studies, mice were drug naïve and determined to be healthy by the veterinary staff. All protocols were approved by the St Jude Children’s Research Hospital Institutional Animal Care and use Committee, and all mice were handled per their guidelines.
TRAMP-C2 Tumor Mouse Model
TRAMP-C2 prostate adenocarcinoma cell line was purchased as ATCC® CRL-2731™ and grown as previously described (Foster et al., 1997). 1×106 tumor cells were injected s.c in the right flank of syngeneic 6–8 week old male WT C57BL/6 mice and tumor growth was monitored. Male recipients for tumor growth experiments were used due to the origin of the TRAMP C2 tumor line. All mice were housed in specific pathogen free conditions prior to use. All protocols were approved by the St Jude Children’s Research Hospital Institutional Animal Care and use Committee, and all mice were handled per their guidelines.
METHOD DETAILS
In Vivo Decitabine and PD-1 Blockade Treatment
Under chronic LCMV infection setting
Equal numbers of male and female 6–8 week old WT C57BL/6 mice were chronically infected with LCMV clone 13 after CD4 T cell depletion as described above. Mice were randomly assigned to experimental groups. At ≥ 33 dpi mice were i.p. injected with vehicle or decitabine (Sigma-Aldrich; 1.2 mg/kg) dissolved in sterile PBS every 3 days for 2 weeks, followed by PBS or anti-PD-L1 treatment (BioXcell; 200 μg) every 3 days for 2 weeks.
Under tumor setting
TRAMP-C2 tumor-bearing male C57BL/6 mice (as described above) were randomly assigned to experimental groups. After ≥ 30 post-inoculation, tumor-bearing mice were i.p injected with vehicle or decitabine treatment for 2 weeks as described in the chronic LCMV infection model, followed by anti-PD-L1 treatment (BioXcell; 200 μg) every 3 days for 2 weeks. At indicated time points, tumors were harvested and tumor-infiltrating CD8 T cells were characterized.
Analysis of Antigen-Specific T-Cell Function and Phenotype
Antigen-specific CD8 T cells were identified or purified by FACS using fluorescently labeled H-2Db tetramers bound to LCMV peptides. LCMV and tumor-specific Spas-1 monomers were obtained from the Yerkes NIH tetramer core facility. Phenotypic analysis of the cells was performed using the following fluorescently labeled antibodies: CD8, CD44, CD62L, PD-1, Tim-3, and CD98 (BioLegend), as previously described (Wang et al., 2011; Wherry et al., 2003). The total pool of virus-specific CD8 T cells was defined as CD44hi PD-1+ CD8 T cells. Ki67, Tcf1, T-bet, and Eomes intracellular staining was performed using the eBioscience ICS-staining protocol and fluorescently labeled antibodies against T-bet (clone 4B10; BioLegend), Eomes (clone Dan11mag; eBioscience), Tcf1 (clone C63D9; Cell Signaling), and Ki67 (clone SolA15; eBioscience). Ex vivo stimulation of antigen-specific splenocytes was performed using gp33 peptide, as previously described (Youngblood et al., 2011). Intracellular staining for IFNγ, IL-2, and Granzyme b was performed with fluorescently labeled antibody clones XMG1.2, JES6-5h4, and GB11, respectively (all from BioLegend) and by using BD Cytofix/Cytoperm™ (BD Biosciences) per the manufacturer’s instructions. Cell death frequency was calculated using Ghost Dye Violet 510 viability dye (Tonbo Biosciences) to determine the frequency of live cells in the total lymphocyte-singlet gate.
Genome-Wide and Loci-Specific Methylation Analysis
We sorted viable naïve (CD44low CD62L+) and tetramer+ (gp33+ CD44hi) CD8 T cells from the splenocytes of acutely or chronically infected WT and cKO mice. For loci-specific methylation analysis of TILs, viable PD-1hi and PD-1low subsets of TILs were sorted from TRAMP-C2 tumors at > 45 days post-inoculation. DNA was isolated from the FACS-purified naïve, antigen-specific CD8 T cells, and TILs by using the Qiagen DNeasy kit. Genomic DNA was bisulfite treated using the EZ DNA methylation kit (Zymo Research). Bisulfite-induced deamination of cytosine allows for sequencing-based discrimination of methylated versus non-methylated cytosine (Trinh et al., 2001).
To perform genome-wide methylation analysis, a bisulfite-modified DNA-sequencing library was generated according to Illumina’s instructions accompanying the TruSeq DNA Methylation Kit (Part# EGMK81312). Briefly, Bisulfite converted DNA was annealed with random hexamer with tagging sequence. DNA copy was synthesized using TruSeq DNA Methyl Polymerase. Then Exonuclease I was used to digest excess random primer. The 3′ ends of the strands are then selectively tagged with a second specific sequence tag to generate di-tagged DNA using DNA polymerase. The di-tagged DNA with known sequence tags at both ends was cleaned up by the AMPureXP Beads. The dsDNA library was generated by PCR amplification with Illumina primers for 10 cycles and library fragments of ~350 bp (insert plus adaptor and PCR primer sequences). The library was purified by the AMPureXP Beads. The purified DNA was captured on an Illumina flow cell for cluster generation. Libraries were sequenced on the HiSeq 2500 following the manufacturer’s protocols.
To perform loci-specific methylation analysis, the bisulfite-modified DNA was PCR amplified with locus-specific primers (Supplemental Table 1). The PCR amplicon was cloned into the pGEM-T TA cloning vector (Promega) and then transformed into XL10-Gold ultracompetent E. coli bacteria (Stratagene). Individual bacterial colonies were grown overnight over Luria-Bertani (LB) agar containing ampicillin (100 mg/L), X-gal (80 mg/L), and IPTG (20 mM). White colonies were selected and subcultured into LB broth with ampicillin (100 mg/L) overnight; the cloning vector was purified; and the genomic insert was sequenced.
Microarray gene expression analysis
Microarray gene expression analysis was performed in FACS-purified naïve and LCMV-specific CD8 T cells from chronically infected WT and cKO mice at 35 dpi. The microarray data were measured by Affymetrix Mouse Gene 2.0 ST arrays and were processed with Partek Genomics Suite 6.6 by the RMA method. To compare gene expression patterns of WT and cKO CD8 T cells to those of CXCR5+ Tim-3− and CXCR5− Tim-3+ subsets of exhausted WT CD8 T cells published in (Im et al., 2016), we downloaded their normalized microarray data from GSE84105 and compared Tim-3+ with CXCR5+. Genes that were in GSE84105 but were not measured in our array were removed. We identified 524 upregulated genes in Tim-3+ and 667 downregulated genes in Tim-3+ using fdr<=0.05 and log2 fold at least 1 as the cutoff. The enrichment of the 2 gene sets in our KO vs. WT comparison was done using CAMERA. The differential gene expression analysis and CAMERA analysis were done using LIMMA package 3.24.5 in Bioconductor.
TCR Repertoire Analysis
LCMV gp33-specific T cells were stained with specific tetramer, resuspended in freshly made sort buffer (PBS containing 0.1% BSA (Gibco) and 200 U RNAsin/ml (Promega)) and filtered prior to sorting. Several hundreds of tetramer-positive T cells were single cell sorted into the wells of a 96-well PCR plates (Eppendorf) that had been preloaded with 2.5 ul of reverse transcription mixture (0.5 μl 5X iScript reaction mix, 0.5 μl iScript reverse transcriptase (Biorad), and 0.1% Triton X-100 (Sigma-Aldrich)) using a iCyt Synergy cell sorter (Sony). The parameters used for sorting are: Multi-drop sort OFF, Multi-drop exclude OFF, Division 10, Center sort%: 90. The last two columns of the plate were left unsorted to serve as negative controls for the PCR. Following sorting, the plates were sealed immediately using plate sealer film (MicroAmp, Applied Biosystems) and centrifuged at 500g for 3 minutes prior to storing at −80°C until reverse transcription and PCR.
The CDR3β region of individual cells were amplified and sequenced using a nested, single-cell, multiplex PCR approach (Dash et al., 2011). Briefly, cDNA was synthesized directly from single cells as per the manufacturer’s instructions with minor modifications. The cDNA synthesis was followed by two rounds of PCR with a Taq polymerase–based PCR kit (Qiagen) and a cocktail of TCR β specific primers to amplify the CDR3β transcripts from each cell in a 25-μl reaction volume. The PCR products were visualized on a 2% agarose gel, then purified using exonuclease/Shrimp alkaline phosphatase enzymes (Dash et al., 2015) and sequenced using TRBC reverse primer, using an ABI Big Dye sequencer (Applied Biosystem) at the Hartwell Center of St. Jude Children’s Research Hospital.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
N = 3–5 per group of at least two independent experiments. Statistical analysis was performed using Prism 6 (GraphPad) software and Statistical significance was determined if P-value is less than 0.05. Comparisons were made using Mann-Whitney U test (for pairwise comparisons in Figures 1–4, 5B, 5F, 6, and 7); ANOVA test (for multiple comparisons in Figure 5G); unpaired t test with Welch’s correction (for CpG methylation comparisons); or parametric two-tailed paired t test for paired analysis of Simpson’s diversity index of TCR repertoire data.
Whole-Genome Bisulfite DNA Sequencing Analysis
WGBS data were aligned to the mm10 mouse genome using BSMAP2.74. CpG methylation was performed using model-based analysis of bisulfite sequencing (Wu et al., 2015). Differentially methylated regions (DMRs) were identified using Bioconductor package DSS and custom R scripts with a threshold of 20% change in methylation ratio and p-value ≤ 0.01 as a cutoff. The minimum length for DMR is 50 bps and the minimum number of CpG sites for DMR is 3. DMRs were annotated using mm10 RefSeq in the following order: promoter from −1kb to 1kb of TSS (transcription start site), 5′ distal from −50kb to −1 kb of TSS, downstream from −1 kb to 1kb of TTS (transcription termination site), 3′ distal from 1kb to 50 kb of TTS, exon, intron, and any other genomic regions were defined as intergenic. The M-value, the measurement of CpG methylation status, was used for principle component analysis (PCA) and dendrogram. For the dendrogram, complete linkage method with Euclidean distance measurement based on M-value was used. The M-value is calculated as the log2 ratio of the number of methylated reads versus unmethylated reads. To avoid zero read count, 0.5 is added to the read count. The top 3000 most variable CpG sites with >5X coverage were selected to do the PCA and clustering analysis.
Simpson’s Diversity Index Analysis
TCR repertoire sequencing data were analyzed using a custom-built macro-enabled excel sheet in conjunction with an IMGT web interface to derive CDR3β nucleotide and amino acid sequences with corresponding TRBV-TRBJ family usage (Dash et al., 2015; Lefranc et al., 2009). Simpson’s Diversity Index (D) was calculated on the population from each mouse as previously described (Thomas et al., 2013), with D = Σi[(ni(ni − 1))/(N(N − 1))], where ni is the number of sequences in the ith clonotype and N is the total number of sequences in the whole population.
DATA AND SOFTWARE AVAILABILITY
The accession number for the WGBS datasets and microarray data reported in this paper are GSE99450 and GSE99449 respectively.
Supplementary Material
(A) Representative histogram analysis of granzyme B expression in naïve and gp33-specific CD8 T cells from chronically infected WT (top panel) and cKO (bottom panel) mice at 8 dpi.
(B) Representative PCR gel showing intact and recombined Dnmt3a floxed regions in genomic DNA from naïve (CD44low CD62L+) and tetramer+ cKO CD8 T cells respectively. Both cell types were sorted from the same infected mice 8 days after chronic LCMV infection. PCR amplification using the same primers was performed in sorted naïve (CD44low CD62L+) and tetramer+ WT CD8 T cells as a control at 8 dpi showing the wild type allele with no recombination.
(C) Longitudinal tracking of polyclonal CD44hi PD-1+ LCMV-specific CD8 T cells in the peripheral blood of WT and Dnmt3a cKO mice during chronic LCMV infection.
(D) Summary graph of CD44 MFI on gp33-specific WT and cKO CD8 T cells at 2 months post LCMV infection.
(E) Summary graph of the frequency of PD-1+ gp33-specific WT and cKO CD8 T cell splenocytes after 2 months of chronic LCMV infection.
(F) Summary graph of the frequency of IL-2-producing cells among total IFNγ+ WT and cKO CD8 T cell splenocytes after 5 hours of ex vivo stimulation with the gp33 peptide after 2 months of chronic LCMV infection.
(G) Summary graph of TCR MFI on gp33-specific WT and cKO CD8 T cells after 2 months of chronic LCMV infection.
(H) Representative intracellular FACS analysis of IFNγ and IL-2 expression in CD44hi CD8 T cells from the spleens of WT and cKO CD8 T cells after 5 hours of ex vivo stimulation with the gp33 peptide at 8 dpi.
(I) Violin plot showing the methylation distribution (number of methylated CpGs per total number of CpGs) across the genomes of naïve and LCMV-specific WT and cKO CD8 T cells at the effector and chronic stages of chronic LCMV infection.
(J) Bar graph shows the number of demethylated regions in WT and cKO antigen-specific CD8 T cells from chronically infected mice during naïve-to-effector (naïve-to-day 8 p.i; WT: black bar, cKO: black dashed bar) and effector-to-exhausted (day 8-to-day 35 p.i; WT: gray bar, cKO: gray dashed bar) stages of the immune response.
(K) Bar graph shows the number of newly methylated regions in antigen-specific cKO CD8 T cells from chronically infected mice at the effector (black dashed bar) and chronic (gray dashed bar) stages of the immune response.
(L) Principle component analysis (PCA) of DNA methylation profiles in naïve and LCMV-specific WT and cKO CD8 T cells at the effector and chronic stages of chronic LCMV infection.
(M) Graph-based visualization of statistically enriched (P < 0.001) non-redundant gene ontology (GO) biological processes regulated by Dnmt3a-targeted genes in exhausted WT CD8 T cells. Bubble size indicates the frequency of the GO term in the GO annotation database, and color intensity indicates −log P-value. Highly similar GO terms are linked by edges in the graph, where the line width indicates the degree of similarity. The network was generated using Cytoscape.
(N) Bar graph of statistically enriched (P < 0.05) Biocarta pathways regulated by Dnmt3a-targeted genes in exhausted WT CD8 T cells. Statistical enrichment was calculated using the David Pathway enrichment analysis tool.
N= 3–5 mice per group of two independent experiments. Comparisons were made using the Mann-Whitney U test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. Error bars are the mean ± SEM.
(A) WGBS nucleotide-resolution view of Dnmt3a-mediated de novo DNA-methylation programs at the Tcf7, Tbx21, Ccr7, and Eomes loci in WT and cKO tetramer+ CD8 T cells from chronically infected mice at 8 dpi or 35 dpi. Vertical blue and red lines indicate CpG positions in the loci. The ratio of blue to red indicates the percentage of unmethylated versus methylated reads, respectively, in the WGBS.
(B) Loci-specific bisulfite sequencing analysis summary graphs of methylation status at individual CpG sites in the DMRs of the Ccr7, Tcf7, IFNγ, and Tbx21 loci in naïve, and tetramer+ CD8 T cells isolated during the effector (at 8 dpi of mice infected with the chronic strain of LCMV) or post-effector stages from chronically infected WT and cKO mice and the memory stage of acutely infected WT mice.
(C) Loci-specific bisulfite sequencing summary graphs of methylation status at individual CpG sites in the DMRs of the Ccr7, Tcf7, IFNγ, and Tbx21 loci in naïve, and tetramer+ CD8 T cells isolated after PD-1 blockade treatment of chronically infected WT mice.
N= 3–5 pooled samples from independent experiments. Error bars are the mean ± SEM.
(A) Scatter plot demonstrates correlation between Dnmt3a-mediated DMRs in exhausted WT cells versus functional memory-related DMRs.
(B) Cluster dendrogram analysis of naïve and LCMV-specific CD8 T cells from acutely infected WT mice at 35 dpi and chronically infected WT and cKO mice at 8 and 35 dpi.
(C) Loci-specific bisulfite sequencing analysis of exhaustion-associated DMRs in the Myc, IFNγ, Tbx21, Tcf7 and Ccr7 loci among LCMV-specific WT CD8 T cells isolated from chronically LCMV-infected WT mice at ≥ 35 dpi, with or without CD4 T cell help. Horizontal lines represent individual sequenced clones from the pool of FACS-purified TILs. Filled circles, methylated cytosine; open circles, nonmethylated cytosine.
(D & E) Heat maps show representative differences in gene expression between WT and cKO CD8 T cells.
(F) Scatter plot showing statistically significant inverse relationship between gene expression changes and DNA methylation changes in WT exhausted cells. Y-axis shows log2 fold change between WT vs cKO antigen-specific CD8 T cells from chronically infected mice at ≥ 35 dpi, and x-axis shows differential DNA methylation changes in WT vs cKO antigen-specific CD8 T cells from chronically infected mice at 35 dpi with 20% cutoff (≥ 0.2).
(G) Graph-based visualization of statistical enrichment between differentially expressed genes in CXCR5+ subset versus Tim-3+ subset of exhausted LCMV-specific WT CD8 T cells (using microarray data deposited in GSE84105, with FDR ≤ 0.05 and log2 fold change ≥ 1 as the cutoff) and gene expression patterns in our microarray data of cKO versus WT LCMV-specific CD8 T cells in chronically infected mice. The enrichment of the 2 gene sets in our KO versus WT comparison was done using CAMERA.
(A) Schematic of CpG sites at exhaustion-associated DMRs in the Tbx21 and (B) Tcf7 loci. Loci-specific bisulfite sequencing analysis of the DMRs in the Tbx21 and Tcf7 loci in naïve and tetramer+ CD8 T cells isolated from chronically infected WT and Dnmt3a cKO mice. Horizontal lines represent individual sequenced clones from the pool of FACS-purified tetramer+ CD8 T cells. Filled circles, methylated cytosine; open circles, nonmethylated cytosine.
(C) Representative FACS analysis of T-bet and Eomes co-expression among gp33-specific CD8 T cells from the spleens of chronically WT and cKO mice 2 months post-infection.
(D) Summary graph of T-bet and Eomes co-expression among gp33-specific CD8 T cells from the spleens of chronically WT and cKO mice 2 months post-infection.
(E) Representative FACS analysis of Tim-3 and Eomes co-expression among gp33-specific CD8 T cells from the spleens of chronically WT and cKO mice 2 months post-infection.
(F) Summary graphs of Tim-3 and Eomes co-expression among gp33-specific CD8 T cells from the spleens of chronically WT and cKO mice 2 months post-infection.
(G) Representative FACS analysis of intracellular levels of TCF1 and Ki67 in gp33-specific CD8 T cells from spleens of chronically infected WT and cKO mice 2 months p.i.
(H) Summary graph of the frequency of TCF1 expression among gp33-specific CD8 T cells from spleens of chronically infected WT and cKO mice 2 months p.i.
N= 3–5 mice per group of two independent experiments. Comparisons were made using the Mann-Whitney U test. *P ≤ 0.05, ***P ≤ 0.001. Error bars are the mean ± SD.
(A) Schematic of CpG sites in the Myc locus exhaustion-associated DMR. Loci-specific bisulfite sequencing analysis and summary graphs of the methylation percentage of individual CpG sites in the DMR of the Myc locus in naïve, effector, and virus-specific CD8 T cells isolated from acutely or chronically infected WT and cKO mice.
(B) Representative histogram analysis and summary graphs of CD98 expression on naïve and tetramer+ CD8 T cells in spleens from WT and cKO mice during acute (2 months post-infection) or chronic (8 days or 2 months post-infection) LCMV infection.
(C) Representative FACS analysis and summary graph of the number of Ki67+ cells among Tim-3 +/− PD-1+ gp33-specific CD8 T cell subsets from WT and cKO mice after 8 days of chronic infection.
(D) Representative FACS analysis and summary graph of the number of Ki67+ cells among Tim-3 +/− PD-1+ gp33-specific CD8 T cell subsets from WT and cKO mice after 2 months of chronic infection.
(E) Representative FAC analysis of Ki67 levels among the total PD-1+ Tim-3+ and PD-1+ Tim-3− polyclonal CD8 T cells after 2 months of chronic infection.
(F) Summary graphs of the frequency and number of Ki67+ cells among the total PD-1+ Tim-3+ and PD-1+ Tim-3− polyclonal CD8 T cells after 2 months of chronic infection.
N= 3–5 mice per group of two independent experiments. Comparisons were made using the Mann-Whitney U test. *P ≤ 0.05, **P ≤ 0.01. Error bars are the mean ± SEM (panel B) or the mean ± SD (panels C, D, and F).
(A) Representative FACS analysis of PD-1 and Tim-3 expression on gp33-specific CD8 T cells and summary graphs of the fold change in the quantities and the frequencies of Tim-3 −/+ PD-1+ gp33-specific CD8 T cell subsets in the spleens of mock-treated (gray bars) or anti–PD-L1–treated (red bars) chronically infected WT and cKO mock-treated (black bars) or PD-1 blockade–treated (green bars) mice. Fold change was calculated relative to change in mock-treated WT mice.
(B) Representative intracellular FACS analysis of T-bet and Eomes expression in gp33-specific CD8 T cells and summary graphs of the frequencies of T-bethi Eomeslow and T-betint Eomeshi T-cell subsets in the spleens of chronically infected WT and cKO mice after mock or PD-1 blockade treatment.
(C) Summary graphs of gp33-, gp276-, and np396-specific CD8 T cell frequencies in the spleens of chronically infected WT and cKO mice after mock or PD-1 blockade treatment.
(D) Summary graphs of the frequencies of gp33-specific CD8 T cells in the lung or liver of chronically infected WT and cKO mice after mock or PD-1 blockade treatment.
(E) Summary graphs of the fold change in gp33-, gp276-, np396-specific, and total IFNγ+ (after ex vivo stimulation with the gp33 peptide) CD8 T cell quantities in the spleens of chronically infected WT (red) and cKO (green) mice after PD-1 blockade treatment compared to corresponding mock-treated mice.
(F) Summary graph of the frequency of dead cells among total np396-specific CD8 T cells and representative histogram analysis of dead np396-specific CD8 T cells in the spleens of chronically infected WT and cKO mice after mock or PD-1 blockade treatment.
(G) Summary graph of longitudinal Simpson’s diversity index analysis of gp33-specific TCR repertoire among WT and cKO CD8 T cells isolated from peripheral blood by single-cell sorting before and after mock treatment.
(H) Representative intracellular FACS analysis of IFNγ expression in CD44hi CD8 T cells from the spleens of mock or PD-1 blockade–treated chronically infected WT and cKO mice after 5 hours of ex vivo stimulation with the gp33 peptide.
(I) Summary graphs of the frequency of total IFNγ-expressing CD44hi CD8 T cells and the ratio of IFNγ+, IL-2+ co-producing to total IFNγ-producing CD8 T cells after 5 hours of ex vivo stimulation with the gp33 peptide in splenocytes of mock or PD-1 blockade–treated chronically infected WT and cKO mice. Summary graphs of the MFI of IL-2 or IFNγ in the IFNγ+, IL-2+ coexpressing or the total IFNγ-producing CD8 T cells respectively.
N= 3–5 mice per group of two independent experiments. Comparisons were made using the Mann-Whitney U test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. Error bars are the mean ± SEM.
(A) Representative FACS analysis showing the frequencies of polyclonal CD44hi PD-1+ LCMV-specific CD8 T cells in the spleen, lung, and liver tissues of mock plus mock-treated, mock plus anti–PD-L1–treated, decitabine plus mock-treated, or decitabine plus PD-1 blockade–treated chronically infected WT mice.
(B) Summary graphs show the frequencies of polyclonal CD44hi PD-1+ LCMV-specific CD8 T cells in the spleen, lung, and liver tissues of mock plus mock-treated (gray bars), mock plus anti–PD-L1–treated (red bars), decitabine plus mock-treated (black bars), or decitabine plus PD-1 blockade–treated (blue bars) chronically infected WT mice.
(C) Representative FACS analysis showing Ki67 levels in polyclonal CD44hi PD-1+ LCMV-specific CD8 T cells in the spleens of mock plus mock-treated, mock plus anti–PD-L1–treated, decitabine plus mock-treated, or decitabine plus PD-1 blockade–treated chronically infected WT mice.
(D) Summary graphs show the frequencies of Ki67 levels in polyclonal CD44hi PD-1+ LCMV-specific CD8 T cells in the spleens of mock plus mock-treated (gray bars), mock plus anti–PD-L1–treated (red bars), decitabine plus mock-treated (black bars), or decitabine plus PD-1 blockade–treated (blue bars) chronically infected WT mice.
(E) Representative intracellular FACS analysis of IFNγ expression among CD44hi CD8 T cells in the spleens of mock plus mock-treated, mock plus anti–PD-L1–treated, decitabine plus mock-treated, or decitabine plus PD-1 blockade–treated chronically infected WT mice.
(F) Summary graphs of the frequency and number of IFNγ-producing cells among CD44hi CD8 T cells in the spleens of mock plus mock-treated, mock plus anti–PD-L1–treated, decitabine plus mock-treated, or decitabine plus PD-1 blockade–treated chronically infected WT mice.
(G) Graph shows longitudinal measurement of serum LCMV titers in chronically infected WT mice during mono- or sequentially combined decitabine and anti-PD-L1 treatments.
(H) Representative FACS analysis of PD-1 and Tim-3 expression among TILs, and representative histogram analysis and summary bar graphs of intracellular TCF1 levels among PD-1hi (red) or PD-1low (black) tumor-infiltrating CD8 T cells after mono PD-1 blockade (top panel) or sequentially combined decitabine and anti-PD-L1 (bottom panel) treatments.
(I) Longitudinal measurement of Tramp C2 tumor growth in WT mice receiving mock (gray) or single decitabine (black) treatment.
N= 3–5 mice per group of two independent experiments. Comparisons were made using the Mann-Whitney U test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001. Error bars are the mean ± SEM (panels B–F, H–I) or the mean ± SD (panel G).
Supplemental Table 1. Primer Sequences for Loci-Specific DNA Methylation Analysis.
HIGHLIGHTS.
Post-effector de novo DNA methylation programs promote terminal T-cell exhaustion
Exhaustion-associated DNA methylation programs are preserved during PD-1 blockade
Blocking de novo methylation enhances PD-1 blockade-mediated T cell-rejuvenation
PD-1hi TILs acquire exhaustion-associated DNA-methylation programs
Acknowledgments
We thank Drs. Richard Cross and Greig Lennon in the St. Jude Flow Cytometry & Cell Sorting Shared Resource for cell sorting and the staff in the St. Jude Hartwell Center for all sequencing services. We also thank Dr. Angela McArthur at St. Jude for scientific editing and Dr. Rafick Sekaly for his critical comments. This work was supported by the National Institutes of Health grant 1R01AI114442 and ALSAC (to BAY) and the SJCRH Garwood Postdoctoral Fellowship (to AM).
Footnotes
AUTHOR CONTRIBUTIONS
HEG and BAY designed the experiments and supervised the study. HEG, YF, RC, GN, and BAY collected WGBS and microarray data, analyzed and interpreted results. HAA, PD, and AM collected data. HEG, PD, WA, and PGT contributed to the design of TCR repertoire experiments, collected and analyzed data, and interpreted results. All authors contributed to the preparation of the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Angelosanto JM, Blackburn SD, Crawford A, Wherry EJ. Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection. Journal of virology. 2012;86:8161–8170. doi: 10.1128/JVI.00889-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bintu L, Yong J, Antebi YE, McCue K, Kazuki Y, Uno N, Oshimura M, Elowitz MB. Dynamics of epigenetic regulation at the single-cell level. Science. 2016;351:720–724. doi: 10.1126/science.aab2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A. DNA methylation patterns and epigenetic memory. Genes & development. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nature reviews Genetics. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A, et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell. 2016;164:1073. doi: 10.1016/j.cell.2015.10.020. [DOI] [PubMed] [Google Scholar]
- Crompton JG, Narayanan M, Cuddapah S, Roychoudhuri R, Ji Y, Yang W, Patel SJ, Sukumar M, Palmer DC, Peng W, et al. Lineage relationship of CD8 T cell subsets is revealed by progressive changes in the epigenetic landscape. Cellular & molecular immunology. 2015 doi: 10.1038/cmi.2015.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran MA, Allison JP. Tumor vaccines expressing flt3 ligand synergize with ctla-4 blockade to reject preimplanted tumors. Cancer research. 2009;69:7747–7755. doi: 10.1158/0008-5472.CAN-08-3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash P, McClaren JL, Oguin TH, 3rd, Rothwell W, Todd B, Morris MY, Becksfort J, Reynolds C, Brown SA, Doherty PC, et al. Paired analysis of TCRalpha and TCRbeta chains at the single-cell level in mice. The Journal of clinical investigation. 2011;121:288–295. doi: 10.1172/JCI44752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash P, Wang GC, Thomas PG. Single-Cell Analysis of T-Cell Receptor alphabeta Repertoire. Methods Mol Biol. 2015;1343:181–197. doi: 10.1007/978-1-4939-2963-4_15. [DOI] [PubMed] [Google Scholar]
- Foster BA, Gingrich JR, Kwon ED, Madias C, Greenberg NM. Characterization of prostatic epithelial cell lines derived from transgenic adenocarcinoma of the mouse prostate (TRAMP) model. Cancer research. 1997;57:3325–3330. [PubMed] [Google Scholar]
- Ghoneim HE, Zamora AE, Thomas PG, Youngblood BA. Cell-Intrinsic Barriers of T Cell-Based Immunotherapy. Trends in molecular medicine. 2016;22:1000–1011. doi: 10.1016/j.molmed.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha SJ, Mueller SN, Wherry EJ, Barber DL, Aubert RD, Sharpe AH, Freeman GJ, Ahmed R. Enhancing therapeutic vaccination by blocking PD-1-mediated inhibitory signals during chronic infection. The Journal of experimental medicine. 2008;205:543–555. doi: 10.1084/jem.20071949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Nasti TH, Sharpe AH, et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature. 2016 doi: 10.1038/nature19330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin HT, Anderson AC, Tan WG, West EE, Ha SJ, Araki K, Freeman GJ, Kuchroo VK, Ahmed R. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:14733–14738. doi: 10.1073/pnas.1009731107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nature reviews Immunology. 2012;12:749–761. doi: 10.1038/nri3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefranc MP, Giudicelli V, Ginestoux C, Jabado-Michaloud J, Folch G, Bellahcene F, Wu Y, Gemrot E, Brochet X, Lane J, et al. IMGT, the international ImMunoGeneTics information system. Nucleic acids research. 2009;37:D1006–1012. doi: 10.1093/nar/gkn838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Chiappinelli KB, Guzzetta AA, Easwaran H, Yen RW, Vatapalli R, Topper MJ, Luo J, Connolly RM, Azad NS, et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget. 2014;5:587–598. doi: 10.18632/oncotarget.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matloubian M, Somasundaram T, Kolhekar SR, Selvakumar R, Ahmed R. Genetic basis of viral persistence: single amino acid change in the viral glycoprotein affects ability of lymphocytic choriomeningitis virus to persist in adult mice. The Journal of experimental medicine. 1990;172:1043–1048. doi: 10.1084/jem.172.4.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor WA, Aravind L, Rao A. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nature reviews Molecular cell biology. 2013;14:341–356. doi: 10.1038/nrm3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, Drake AM, Chen Z, Sen DR, Kurachi M, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science. 2016;354:1160–1165. doi: 10.1126/science.aaf2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A, Hamid O, Daud A, Hodi FS, Wolchok JD, Kefford R, Joshua AM, Patnaik A, Hwu WJ, Weber JS, et al. Association of Pembrolizumab With Tumor Response and Survival Among Patients With Advanced Melanoma. Jama. 2016;315:1600–1609. doi: 10.1001/jama.2016.4059. [DOI] [PubMed] [Google Scholar]
- Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, Han H, Liang G, Jones PA, Pugh TJ, et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell. 2015;162:961–973. doi: 10.1016/j.cell.2015.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russ BE, Olshanksy M, Smallwood HS, Li J, Denton AE, Prier JE, Stock AT, Croom HA, Cullen JG, Nguyen ML, et al. Distinct Epigenetic Signatures Delineate Transcriptional Programs during Virus-Specific CD8 T Cell Differentiation. Immunity. 2014;41:853–865. doi: 10.1016/j.immuni.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharer CD, Barwick BG, Youngblood BA, Ahmed R, Boss JM. Global DNA Methylation Remodeling Accompanies CD8 T Cell Effector Function. J Immunol. 2013 doi: 10.4049/jimmunol.1301395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015a;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015b;161:205–214. doi: 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speiser DE, Utzschneider DT, Oberle SG, Munz C, Romero P, Zehn D. T cell differentiation in chronic infection and cancer: functional adaptation or exhaustion? Nature reviews Immunology. 2014;14:768–774. doi: 10.1038/nri3740. [DOI] [PubMed] [Google Scholar]
- Thomas PG, Handel A, Doherty PC, La Gruta NL. Ecological analysis of antigen-specific CTL repertoires defines the relationship between naive and immune T-cell populations. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:1839–1844. doi: 10.1073/pnas.1222149110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh BN, Long TI, Laird PW. DNA methylation analysis by MethyLight technology. Methods (San Diego, Calif. 2001;25:456–462. doi: 10.1006/meth.2001.1268. [DOI] [PubMed] [Google Scholar]
- Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, Danilo M, Alfei F, Hofmann M, Wieland D, et al. T Cell Factor 1-Expressing Memory-like CD8(+) T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity. 2016;45:415–427. doi: 10.1016/j.immuni.2016.07.021. [DOI] [PubMed] [Google Scholar]
- Utzschneider DT, Legat A, Fuertes Marraco SA, Carrie L, Luescher I, Speiser DE, Zehn D. T cells maintain an exhausted phenotype after antigen withdrawal and population reexpansion. Nature immunology. 2013;14:603–610. doi: 10.1038/ni.2606. [DOI] [PubMed] [Google Scholar]
- Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM. A census of human transcription factors: function, expression and evolution. Nature reviews Genetics. 2009;10:252–263. doi: 10.1038/nrg2538. [DOI] [PubMed] [Google Scholar]
- Wang L, Amoozgar Z, Huang J, Saleh MH, Xing D, Orsulic S, Goldberg MS. Decitabine Enhances Lymphocyte Migration and Function and Synergizes with CTLA-4 Blockade in a Murine Ovarian Cancer Model. Cancer immunology research. 2015;3:1030–1041. doi: 10.1158/2326-6066.CIR-15-0073. [DOI] [PubMed] [Google Scholar]
- Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35:871–882. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ. T cell exhaustion. Nature immunology. 2011;12:492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. Journal of virology. 2003;77:4911–4927. doi: 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Xu T, Feng H, Chen L, Li B, Yao B, Qin Z, Jin P, Conneely KN. Detection of differentially methylated regions from whole-genome bisulfite sequencing data without replicates. Nucleic acids research. 2015;43:e141. doi: 10.1093/nar/gkv715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngblood B, Oestreich KJ, Ha SJ, Duraiswamy J, Akondy RS, West EE, Wei Z, Lu P, Austin JW, Riley JL, et al. Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8+ T cells. Immunity. 2011;35:13. doi: 10.1016/j.immuni.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu P, Steel JC, Zhang M, Morris JC, Waitz R, Fasso M, Allison JP, Waldmann TA. Simultaneous inhibition of two regulatory T-cell subsets enhanced Interleukin-15 efficacy in a prostate tumor model. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:6187–6192. doi: 10.1073/pnas.1203479109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Representative histogram analysis of granzyme B expression in naïve and gp33-specific CD8 T cells from chronically infected WT (top panel) and cKO (bottom panel) mice at 8 dpi.
(B) Representative PCR gel showing intact and recombined Dnmt3a floxed regions in genomic DNA from naïve (CD44low CD62L+) and tetramer+ cKO CD8 T cells respectively. Both cell types were sorted from the same infected mice 8 days after chronic LCMV infection. PCR amplification using the same primers was performed in sorted naïve (CD44low CD62L+) and tetramer+ WT CD8 T cells as a control at 8 dpi showing the wild type allele with no recombination.
(C) Longitudinal tracking of polyclonal CD44hi PD-1+ LCMV-specific CD8 T cells in the peripheral blood of WT and Dnmt3a cKO mice during chronic LCMV infection.
(D) Summary graph of CD44 MFI on gp33-specific WT and cKO CD8 T cells at 2 months post LCMV infection.
(E) Summary graph of the frequency of PD-1+ gp33-specific WT and cKO CD8 T cell splenocytes after 2 months of chronic LCMV infection.
(F) Summary graph of the frequency of IL-2-producing cells among total IFNγ+ WT and cKO CD8 T cell splenocytes after 5 hours of ex vivo stimulation with the gp33 peptide after 2 months of chronic LCMV infection.
(G) Summary graph of TCR MFI on gp33-specific WT and cKO CD8 T cells after 2 months of chronic LCMV infection.
(H) Representative intracellular FACS analysis of IFNγ and IL-2 expression in CD44hi CD8 T cells from the spleens of WT and cKO CD8 T cells after 5 hours of ex vivo stimulation with the gp33 peptide at 8 dpi.
(I) Violin plot showing the methylation distribution (number of methylated CpGs per total number of CpGs) across the genomes of naïve and LCMV-specific WT and cKO CD8 T cells at the effector and chronic stages of chronic LCMV infection.
(J) Bar graph shows the number of demethylated regions in WT and cKO antigen-specific CD8 T cells from chronically infected mice during naïve-to-effector (naïve-to-day 8 p.i; WT: black bar, cKO: black dashed bar) and effector-to-exhausted (day 8-to-day 35 p.i; WT: gray bar, cKO: gray dashed bar) stages of the immune response.
(K) Bar graph shows the number of newly methylated regions in antigen-specific cKO CD8 T cells from chronically infected mice at the effector (black dashed bar) and chronic (gray dashed bar) stages of the immune response.
(L) Principle component analysis (PCA) of DNA methylation profiles in naïve and LCMV-specific WT and cKO CD8 T cells at the effector and chronic stages of chronic LCMV infection.
(M) Graph-based visualization of statistically enriched (P < 0.001) non-redundant gene ontology (GO) biological processes regulated by Dnmt3a-targeted genes in exhausted WT CD8 T cells. Bubble size indicates the frequency of the GO term in the GO annotation database, and color intensity indicates −log P-value. Highly similar GO terms are linked by edges in the graph, where the line width indicates the degree of similarity. The network was generated using Cytoscape.
(N) Bar graph of statistically enriched (P < 0.05) Biocarta pathways regulated by Dnmt3a-targeted genes in exhausted WT CD8 T cells. Statistical enrichment was calculated using the David Pathway enrichment analysis tool.
N= 3–5 mice per group of two independent experiments. Comparisons were made using the Mann-Whitney U test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. Error bars are the mean ± SEM.
(A) WGBS nucleotide-resolution view of Dnmt3a-mediated de novo DNA-methylation programs at the Tcf7, Tbx21, Ccr7, and Eomes loci in WT and cKO tetramer+ CD8 T cells from chronically infected mice at 8 dpi or 35 dpi. Vertical blue and red lines indicate CpG positions in the loci. The ratio of blue to red indicates the percentage of unmethylated versus methylated reads, respectively, in the WGBS.
(B) Loci-specific bisulfite sequencing analysis summary graphs of methylation status at individual CpG sites in the DMRs of the Ccr7, Tcf7, IFNγ, and Tbx21 loci in naïve, and tetramer+ CD8 T cells isolated during the effector (at 8 dpi of mice infected with the chronic strain of LCMV) or post-effector stages from chronically infected WT and cKO mice and the memory stage of acutely infected WT mice.
(C) Loci-specific bisulfite sequencing summary graphs of methylation status at individual CpG sites in the DMRs of the Ccr7, Tcf7, IFNγ, and Tbx21 loci in naïve, and tetramer+ CD8 T cells isolated after PD-1 blockade treatment of chronically infected WT mice.
N= 3–5 pooled samples from independent experiments. Error bars are the mean ± SEM.
(A) Scatter plot demonstrates correlation between Dnmt3a-mediated DMRs in exhausted WT cells versus functional memory-related DMRs.
(B) Cluster dendrogram analysis of naïve and LCMV-specific CD8 T cells from acutely infected WT mice at 35 dpi and chronically infected WT and cKO mice at 8 and 35 dpi.
(C) Loci-specific bisulfite sequencing analysis of exhaustion-associated DMRs in the Myc, IFNγ, Tbx21, Tcf7 and Ccr7 loci among LCMV-specific WT CD8 T cells isolated from chronically LCMV-infected WT mice at ≥ 35 dpi, with or without CD4 T cell help. Horizontal lines represent individual sequenced clones from the pool of FACS-purified TILs. Filled circles, methylated cytosine; open circles, nonmethylated cytosine.
(D & E) Heat maps show representative differences in gene expression between WT and cKO CD8 T cells.
(F) Scatter plot showing statistically significant inverse relationship between gene expression changes and DNA methylation changes in WT exhausted cells. Y-axis shows log2 fold change between WT vs cKO antigen-specific CD8 T cells from chronically infected mice at ≥ 35 dpi, and x-axis shows differential DNA methylation changes in WT vs cKO antigen-specific CD8 T cells from chronically infected mice at 35 dpi with 20% cutoff (≥ 0.2).
(G) Graph-based visualization of statistical enrichment between differentially expressed genes in CXCR5+ subset versus Tim-3+ subset of exhausted LCMV-specific WT CD8 T cells (using microarray data deposited in GSE84105, with FDR ≤ 0.05 and log2 fold change ≥ 1 as the cutoff) and gene expression patterns in our microarray data of cKO versus WT LCMV-specific CD8 T cells in chronically infected mice. The enrichment of the 2 gene sets in our KO versus WT comparison was done using CAMERA.
(A) Schematic of CpG sites at exhaustion-associated DMRs in the Tbx21 and (B) Tcf7 loci. Loci-specific bisulfite sequencing analysis of the DMRs in the Tbx21 and Tcf7 loci in naïve and tetramer+ CD8 T cells isolated from chronically infected WT and Dnmt3a cKO mice. Horizontal lines represent individual sequenced clones from the pool of FACS-purified tetramer+ CD8 T cells. Filled circles, methylated cytosine; open circles, nonmethylated cytosine.
(C) Representative FACS analysis of T-bet and Eomes co-expression among gp33-specific CD8 T cells from the spleens of chronically WT and cKO mice 2 months post-infection.
(D) Summary graph of T-bet and Eomes co-expression among gp33-specific CD8 T cells from the spleens of chronically WT and cKO mice 2 months post-infection.
(E) Representative FACS analysis of Tim-3 and Eomes co-expression among gp33-specific CD8 T cells from the spleens of chronically WT and cKO mice 2 months post-infection.
(F) Summary graphs of Tim-3 and Eomes co-expression among gp33-specific CD8 T cells from the spleens of chronically WT and cKO mice 2 months post-infection.
(G) Representative FACS analysis of intracellular levels of TCF1 and Ki67 in gp33-specific CD8 T cells from spleens of chronically infected WT and cKO mice 2 months p.i.
(H) Summary graph of the frequency of TCF1 expression among gp33-specific CD8 T cells from spleens of chronically infected WT and cKO mice 2 months p.i.
N= 3–5 mice per group of two independent experiments. Comparisons were made using the Mann-Whitney U test. *P ≤ 0.05, ***P ≤ 0.001. Error bars are the mean ± SD.
(A) Schematic of CpG sites in the Myc locus exhaustion-associated DMR. Loci-specific bisulfite sequencing analysis and summary graphs of the methylation percentage of individual CpG sites in the DMR of the Myc locus in naïve, effector, and virus-specific CD8 T cells isolated from acutely or chronically infected WT and cKO mice.
(B) Representative histogram analysis and summary graphs of CD98 expression on naïve and tetramer+ CD8 T cells in spleens from WT and cKO mice during acute (2 months post-infection) or chronic (8 days or 2 months post-infection) LCMV infection.
(C) Representative FACS analysis and summary graph of the number of Ki67+ cells among Tim-3 +/− PD-1+ gp33-specific CD8 T cell subsets from WT and cKO mice after 8 days of chronic infection.
(D) Representative FACS analysis and summary graph of the number of Ki67+ cells among Tim-3 +/− PD-1+ gp33-specific CD8 T cell subsets from WT and cKO mice after 2 months of chronic infection.
(E) Representative FAC analysis of Ki67 levels among the total PD-1+ Tim-3+ and PD-1+ Tim-3− polyclonal CD8 T cells after 2 months of chronic infection.
(F) Summary graphs of the frequency and number of Ki67+ cells among the total PD-1+ Tim-3+ and PD-1+ Tim-3− polyclonal CD8 T cells after 2 months of chronic infection.
N= 3–5 mice per group of two independent experiments. Comparisons were made using the Mann-Whitney U test. *P ≤ 0.05, **P ≤ 0.01. Error bars are the mean ± SEM (panel B) or the mean ± SD (panels C, D, and F).
(A) Representative FACS analysis of PD-1 and Tim-3 expression on gp33-specific CD8 T cells and summary graphs of the fold change in the quantities and the frequencies of Tim-3 −/+ PD-1+ gp33-specific CD8 T cell subsets in the spleens of mock-treated (gray bars) or anti–PD-L1–treated (red bars) chronically infected WT and cKO mock-treated (black bars) or PD-1 blockade–treated (green bars) mice. Fold change was calculated relative to change in mock-treated WT mice.
(B) Representative intracellular FACS analysis of T-bet and Eomes expression in gp33-specific CD8 T cells and summary graphs of the frequencies of T-bethi Eomeslow and T-betint Eomeshi T-cell subsets in the spleens of chronically infected WT and cKO mice after mock or PD-1 blockade treatment.
(C) Summary graphs of gp33-, gp276-, and np396-specific CD8 T cell frequencies in the spleens of chronically infected WT and cKO mice after mock or PD-1 blockade treatment.
(D) Summary graphs of the frequencies of gp33-specific CD8 T cells in the lung or liver of chronically infected WT and cKO mice after mock or PD-1 blockade treatment.
(E) Summary graphs of the fold change in gp33-, gp276-, np396-specific, and total IFNγ+ (after ex vivo stimulation with the gp33 peptide) CD8 T cell quantities in the spleens of chronically infected WT (red) and cKO (green) mice after PD-1 blockade treatment compared to corresponding mock-treated mice.
(F) Summary graph of the frequency of dead cells among total np396-specific CD8 T cells and representative histogram analysis of dead np396-specific CD8 T cells in the spleens of chronically infected WT and cKO mice after mock or PD-1 blockade treatment.
(G) Summary graph of longitudinal Simpson’s diversity index analysis of gp33-specific TCR repertoire among WT and cKO CD8 T cells isolated from peripheral blood by single-cell sorting before and after mock treatment.
(H) Representative intracellular FACS analysis of IFNγ expression in CD44hi CD8 T cells from the spleens of mock or PD-1 blockade–treated chronically infected WT and cKO mice after 5 hours of ex vivo stimulation with the gp33 peptide.
(I) Summary graphs of the frequency of total IFNγ-expressing CD44hi CD8 T cells and the ratio of IFNγ+, IL-2+ co-producing to total IFNγ-producing CD8 T cells after 5 hours of ex vivo stimulation with the gp33 peptide in splenocytes of mock or PD-1 blockade–treated chronically infected WT and cKO mice. Summary graphs of the MFI of IL-2 or IFNγ in the IFNγ+, IL-2+ coexpressing or the total IFNγ-producing CD8 T cells respectively.
N= 3–5 mice per group of two independent experiments. Comparisons were made using the Mann-Whitney U test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. Error bars are the mean ± SEM.
(A) Representative FACS analysis showing the frequencies of polyclonal CD44hi PD-1+ LCMV-specific CD8 T cells in the spleen, lung, and liver tissues of mock plus mock-treated, mock plus anti–PD-L1–treated, decitabine plus mock-treated, or decitabine plus PD-1 blockade–treated chronically infected WT mice.
(B) Summary graphs show the frequencies of polyclonal CD44hi PD-1+ LCMV-specific CD8 T cells in the spleen, lung, and liver tissues of mock plus mock-treated (gray bars), mock plus anti–PD-L1–treated (red bars), decitabine plus mock-treated (black bars), or decitabine plus PD-1 blockade–treated (blue bars) chronically infected WT mice.
(C) Representative FACS analysis showing Ki67 levels in polyclonal CD44hi PD-1+ LCMV-specific CD8 T cells in the spleens of mock plus mock-treated, mock plus anti–PD-L1–treated, decitabine plus mock-treated, or decitabine plus PD-1 blockade–treated chronically infected WT mice.
(D) Summary graphs show the frequencies of Ki67 levels in polyclonal CD44hi PD-1+ LCMV-specific CD8 T cells in the spleens of mock plus mock-treated (gray bars), mock plus anti–PD-L1–treated (red bars), decitabine plus mock-treated (black bars), or decitabine plus PD-1 blockade–treated (blue bars) chronically infected WT mice.
(E) Representative intracellular FACS analysis of IFNγ expression among CD44hi CD8 T cells in the spleens of mock plus mock-treated, mock plus anti–PD-L1–treated, decitabine plus mock-treated, or decitabine plus PD-1 blockade–treated chronically infected WT mice.
(F) Summary graphs of the frequency and number of IFNγ-producing cells among CD44hi CD8 T cells in the spleens of mock plus mock-treated, mock plus anti–PD-L1–treated, decitabine plus mock-treated, or decitabine plus PD-1 blockade–treated chronically infected WT mice.
(G) Graph shows longitudinal measurement of serum LCMV titers in chronically infected WT mice during mono- or sequentially combined decitabine and anti-PD-L1 treatments.
(H) Representative FACS analysis of PD-1 and Tim-3 expression among TILs, and representative histogram analysis and summary bar graphs of intracellular TCF1 levels among PD-1hi (red) or PD-1low (black) tumor-infiltrating CD8 T cells after mono PD-1 blockade (top panel) or sequentially combined decitabine and anti-PD-L1 (bottom panel) treatments.
(I) Longitudinal measurement of Tramp C2 tumor growth in WT mice receiving mock (gray) or single decitabine (black) treatment.
N= 3–5 mice per group of two independent experiments. Comparisons were made using the Mann-Whitney U test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001. Error bars are the mean ± SEM (panels B–F, H–I) or the mean ± SD (panel G).
Supplemental Table 1. Primer Sequences for Loci-Specific DNA Methylation Analysis.