

Summary
Hypoxia augments inflammatory responses and osteoclastogenesis by incompletely understood mechanisms. We identified COMMD1 as a cell-intrinsic negative regulator of osteoclastogenesis that is suppressed by hypoxia. In human macrophages, COMMD1 restrained induction of NF-κB signaling and a transcription factor E2F1-dependent metabolic pathway by the cytokine RANKL. Downregulation of COMMD1 protein expression by hypoxia augmented RANKL-induced expression of inflammatory and E2F1 target genes and downstream osteoclastogenesis. E2F1 targets included glycolysis and metabolic genes including CKB that enabled cells to meet metabolic demands in challenging environments, as well as inflammatory cytokine-driven target genes. Expression quantitative trait locus analysis linked increased COMMD1 expression with decreased bone erosion in rheumatoid arthritis. Myeloid deletion of Commd1 resulted in increased osteoclastogenesis in arthritis and inflammatory osteolysis models. These results identify COMMD1 and an E2F-metabolic pathway as key regulators of osteoclastogenic responses under pathological inflammatory conditions, and provide a mechanism by which hypoxia augments inflammation and bone destruction.
eTOC blurb
Pathways that promote osteoclastogenesis are well characterized but less is known about negative regulators that suppress pathological bone loss. Murata et al. identify COMMD1 as an inhibitor of osteoclastogenesis that restrains NF-κB and E2F1-CKB-mediated metabolic pathways in macrophages. COMMD1 is inhibited by hypoxia and suppresses bone loss in RA patients and inflammatory osteolysis models. These results provide insights into negative regulation of metabolic pathways important for inflammatory bone loss.
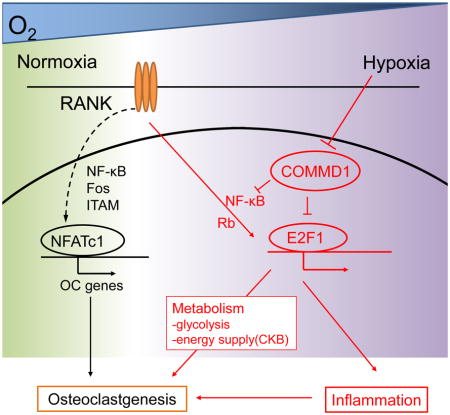
Introduction
Hypoxia occurs in tumors, infections, and inflammatory sites such as the joints of patients with rheumatoid arthritis (RA). Hypoxia induces metabolic stress and increases expression of hypoxia-inducible factors (HIFs) and their target genes important for glycolytic metabolism and angiogenesis (Palazon et al., 2014). Hypoxia also potentiates inflammatory responses, including in arthritis models (Konisti et al., 2012), and contributes to increased bone resorption in inflammatory diseases such as RA by increasing generation of osteoclasts, cells that effectively resorb bone (Schett and Gravallese, 2012). Mechanisms by which hypoxia increases osteoclastogenesis and inflammatory responses are not well understood.
Osteoclasts are large, multinucleated cells that resorb bone. Osteoclast differentiation from myeloid precursors is mediated by the key nonredundant TNF family cytokine RANKL (receptor activator of NF-κB ligand) and its receptor RANK (Nakashima and Takayanagi, 2012). RANK directly activates NF-κB and MAPK-AP-1 signaling pathways and activates calcium signaling in cooperation with ITAM motif-containing immunoreceptors. These canonical osteoclastogenic pathways converge to induce expression and activate the function of transcription factor NFATc1, a master regulator that drives the osteoclast differentiation program. Inflammatory cytokines such as TNF and IL-1 augment these canonical osteoclastogenic pathways (Novack and Teitelbaum, 2008; Schett and Gravallese, 2012), but little is known about how hypoxia promotes bone resorption.
The importance of cellular anabolic metabolism for osteoclastogenesis is emerging. Effective osteoclastogenesis requires both glycolytic and oxidative phosphorylation (OX-PHOS) branches of energy- and ATP-generating metabolism (Chang et al., 2008; Indo et al., 2013; Ishii et al., 2009; Ivashkiv, 2015; Nishikawa et al., 2015; Wei et al., 2010; Zeng et al., 2015). Increased glycolysis appears to be mediated by transcription factor HIF-1α (Indo et al., 2013; Palazon et al., 2014), although the connections between RANKL signaling and HIF-1α have not been clarified. Increased oxidative phosphorylation is mediated at least in part by increased mitochondrial biogenesis, which is induced by RANKL via noncanonical NF-κB signaling and transcriptional coactivator PGC-1β (Ishii et al., 2009; Wei et al., 2010; Zeng et al., 2015). In addition, osteoclastogenesis requires signaling by the mTORC1 pathway, which promotes protein, fatty acid and nucleotide synthesis (Cejka et al., 2010; Indo et al., 2013). Current thinking is that increased RANKL-stimulated flux through these anabolic pathways is required to meet the high energy/ATP demands of osteoclastogenesis, and to generate metabolites important for osteoclast differentiation. The importance of select metabolic pathways in osteoclastogenesis in vivo is supported by genetic and therapeutic studies in which deletion or inhibition of DNMT3A, mTORC1, or creatine kinase B (CKB) alleviates bone loss (Cejka et al., 2010; Chang et al., 2008; Nishikawa et al., 2015). However, in in vitro mechanistic studies it has been difficult to dissect specific effects of metabolism on osteoclast differentiation from nonspecific energy requirements of actively proliferating osteoclast precursors, which may differ from those of nonproliferating osteoclast precursors in vivo (Chiu et al., 2012; Mizoguchi et al., 2009; Muto et al., 2011; Yao et al., 2006). Thus, the role of metabolic pathways in osteoclast differentiation has not been fully clarified.
In addition to upregulating HIF, hypoxia results in strongly diminished mitochondrial respiration and OX-PHOS, and suppresses mTORC1 activity (Wilson and Hay, 2011; Wouters and Koritzinsky, 2008). This raises the important question of how hypoxia can promote osteoclastogenesis despite downregulation of metabolic pathways required for this process. We reasoned that hypoxia induces pathways that promote osteoclastogenesis under conditions of anaerobic metabolism, and investigated mechanisms by which hypoxia synergizes with RANKL. We discovered that RANKL has the potential to induce metabolic pathways mediated by transcription factor E2F, glycolysis genes, and creatine kinase B (CKB), but these pathways are restrained under normoxic conditions by negative regulator COMMD1. Hypoxia suppressed COMMD1 expression at the protein level, thereby releasing metabolic pathways from repression. Genetic and expression quantitative trait locus (eQTL) analysis of allelic variants linked elevated COMMD1 expression with decreased bone erosions in RA patients, and conditional deletion of Commd1 in the myeloid lineage resulted in strikingly increased osteoclastogenesis and bone erosion in mouse arthritis and inflammatory osteolysis models. These results identify a COMMD1-E2F-metabolic pathway important for osteoclastogenesis under inflammatory conditions, and provide a mechanism by which hypoxia augments RANKL-induced osteoclast formation.
Results
COMMD1 pathway is suppressed by hypoxia in RANKL-stimulated human macrophages
We wished to identify mechanisms by which hypoxia enhances osteoclastogenesis. We used nonproliferating human blood-derived macrophages as osteoclast precursors (OCPs), which offers the advantages of studying osteoclastogenesis separately from effects on proliferation, and of using cells directly relevant for human diseases (Sørensen et al., 2007). As expected, hypoxia (2% O2) substantially enhanced RANKL-induced osteoclast differentiation, resorption, and expression of osteoclast marker genes (Figures 1A, 1B and S1A). Hypoxia did not alter expression of NFATc1 mRNA or protein (Figures 1C and 1D), suggesting it regulates alternative osteoclastogenic pathways. RNA sequencing (RNA-seq) (Figures S1B and S1C) and Ingenuity Pathway Analysis (IPA) of RANKL-inducible genes that were superinduced by hypoxia at 48 hr revealed that the 4 most significantly activated upstream regulators (Figure 1E) were in the canonical HIF pathway (VHL-HIF1α-HIF2α-ARNT) (Semenza, 2013). The 3rd most significant upstream regulator (p = 1.2E−12, Figure 1E) was copper metabolism MURR1 domain-containing 1 (COMMD1), which was previously known to mediate copper transport and promote proteasome-mediated degradation of several proteins, most notably NF-κB (Bartuzi et al., 2013) and HIF-1α (Muller et al., 2009b; Sluis et al., 2009; Sluis et al., 2007; van de Sluis et al., 2010). COMMD1 was the only upstream regulator whose downstream pathways were inhibited by hypoxia in RANKL-stimulated macrophages (Figure 1E). These results suggest COMMD1 is negative regulator of osteoclastogenesis that is suppressed by hypoxia to enable enhanced osteoclast differentiation.
Figure 1. COMMD1 pathway is suppressed by hypoxia in RANKL-stimulated human macrophages.

(A) Osteoclastogenesis assays using human macrophages in normoxia or hypoxia (2%). Left, TRAP staining of representative wells. Right, quantitation of pooled data from 9 independent experiments, normalized relative to the number of osteoclasts formed using 10 ng/ml of RANKL under hypoxic conditions (set at 100). Scale bar: 200 μm.
(B) Resorption pit assay. Right panel shows pooled data from 5 experiments with different donors normalized relative to the osteoclasts under hypoxic condition (set as 100%).
(C) RT-qPCR analysis of NFATC1 mRNA in human macrophages cultured with or without RANKL for 48 h and normalized relative to TBP mRNA.
(D) Immunoblot analysis of NFATc1 after stimulation of human macrophages with RANKL for the indicated times. p38 was used as a loading control.
(E) Ingenuity Pathway Analysis (IPA) of RNA-seq data obtained as described in Figure S1 to identify Upstream Regulators of RANKL-inducible genes that were superinduced by hypoxia. HIF-related Upstream Regulators whose pathways were activated by hypoxia are shaded in orange; the only Upstream Regulator whose downstream pathways were inhibited (COMMD1) is shaded in blue.
Bar graphs show means and error bars represent SEM. * p<0.05, **** p<0.0001. The results are representative of 9 (A), 5 (B) or at least 3 (C, D) experiments; (E) represents analysis of two pooled biological replicates. Please see also Figure S1.
COMMD1 is a negative regulator of osteoclastogenesis under normoxic conditions
To test the hypothesis that COMMD1 is a negative regulator of osteoclastogenesis, we siRNA silenced COMMD1 expression. Decreased COMMD1 expression in human macrophages (Figure S2A) resulted in enhanced RANKL-induced osteoclast differentiation, and increased resorption area and actin ring formation in normoxia (Figure 2A–2C), thus phenocopying hypoxia-mediated enhancement of osteoclastogenesis. In addition, silencing of COMMD1 expression resulted in higher expression of osteoclast genes ITGB3 (encoding integrin β3), CTSK (encoding cathepsin K), and CTR (encoding calcitonin receptor) (Figures 2D and S2B–S2E). To obtain additional genetic evidence for a suppressive role of COMMD1 in RANKL-induced osteoclastogenesis, we used a complementary gain-of-function approach and transduced human macrophages with adenoviral particles encoding COMMD1. Overexpression of COMMD1 almost completely suppressed osteoclastogenesis (Figure 2E and 2F) and strongly suppressed osteoclast marker gene expression (Figure 2G). Taken together, these results indicate that COMMD1 is a negative regulator of osteoclastogenesis.
Figure 2. COMMD1 is a negative regulator of osteoclastogenesis.

(A) TRAP staining of human osteoclasts nucleofected with control or COMMD1-specific siRNAs. Right panel shows pooled data from 5 experiments with different donors normalized relative to the number of osteoclasts obtained using COMMD1 siRNA with RANKL (40 ng/ml) (set at 100%).
(B) Resorption pit assay. Right panel shows pooled data from 5 experiments with different donors normalized relative to the number of osteoclasts obtained using COMMD1 siRNA (set at 100%).
(C) Fluorescent images of actin ring staining.
(D) RT-qPCR analysis of ITGB3, CTSK and CTR mRNA normalized relative to TBP mRNA.
(E–G) Human macrophages were transduced with adenoviral particles encoding GFP or COMMD1-HA. Cells then were cultured with M-CSF (20ng/ml) and RANKL (40ng/ml).
(E) Immunoblot of whole cell lysates with HA-antibody. α-tubulin was used as a control.
(F) Osteoclastogenesis assays. Left, TRAP staining of representative wells. Right, quantitation of pooled data from 3 independent experiments, normalized relative to the number of osteoclasts formed using 40 ng/ml of RANKL under adenoviral transduction conditions (set at 100).
(G) Representative RT-PCR results of ITGB3 and CTSK mRNA normalized relative to TBP mRNA.
Bar graphs show means and error bars represent SEM. * p<0.05, ** p<0.01, *** p<0.001. Scale bar, 200 μm. The results are representative of 5 (A, B, C), and at least 3 (D, E, F, G) experiments. Please see also Figure S2.
COMMD1 suppresses pathological bone resorption in RA patients and mouse models
To address the (patho)physiological importance of COMMD1 in regulating bone resorption in vivo, we first tested the relationship between COMMD1 allelic variants that affect gene expression and bone loss in RA patients. After confirming very similar structure of linkage disequilibrium (LD) of the COMMD1 locus in European (Westra et al., 2013) and Japanese populations (Higasa et al., 2016), eQTL analysis revealed that rs10205855 had the strongest cis eQTL association in the Japanese population, and allelic variant rs10205855(C) was significantly associated with increased expression of COMMD1 in a dose-dependent manner (Figures 3A and S3A). To perform functional analysis, we selected rs11125908 in an intron of COMMD1 which was 75bp from and in strong LD with rs10205855 (r2=0.98, Figure S3A). In Japanese RA patients (Terao et al., 2015), the minor allele G at rs11125908, showing significant cis eQTL association with increased COMMD1 expression (Figure 3B), was concurrently associated with lower bone erosions of hand joints (Figure 3C). Furthermore, rs11125908 G alleles demonstrated a strong suppressive association with finger joint destruction, independently of rheumatoid factor (RF) positivity (Figure S3B). This association was still observed in patients positive for RF adjusted for RF amounts (Figure S3C), indicating that rs11125908 G allele is negatively associated with joint destruction in RA, especially finger joint destruction. These results support the model of COMMD1 as a negative regulator of inflammatory bone resorption in human RA patients.
Figure 3. COMMD1 suppresses pathological bone resorption in RA patients and mouse inflammatory arthritis and osteolysis models.

(A) cis-expression quantitative trait loci (eQTL) association of rs10205855 (TT, TC or CC) with COMMD1 expression in Japanese healthy donors.
(B) cis-eQTL association of rs11125908 (AA, AG or GG) with COMMD1 expression in Japanese RA patients.
(C) Total Sharp score (TSS) of hand joints of RA patients in three genotypes for rs1125908. The boxes represent the 25th to 75th percentiles (upper and lower quartiles (UQ and LQ), respectively) and the lines within the boxes represent the median. We computed the inner quartile range (IQR, UQ-LQ). The lines outside the boxes represent the smallest value bigger than 1.5*IQR below LQ and the biggest value smaller than 1.5*IQR above UQ. Symbols indicate outliers.
(D–E) Time course of joint swelling and clinical score of serum-induced arthritis in littermate control and Commd1fl/fl x Lyz2-cre mice (labeled COMMD1ΔM). n = 6 from 2 experiments.
(F–H) TRAP staining of histological sections from calcaneocuboid and tarsometatarsal joints (F), and histomorphometric analysis (G, H) of mice with serum-induced arthritis. n = 6 from 2 experiments.
(I–K) TNF-induced supracalvarial osteolysis model. TRAP staining of histological sections of calvaria (I) and histomorphometric analysis (J, K). n = 7–9 from 3 experiments.
Bar graphs show means and error bars represent SEM. * p<0.05, ** p<0.01, *** p<0.001. Bar, 100 μm. Please see also Figure S3.
To more directly test the causal relationship between COMMD1 expression and bone resorption in vivo, we took a genetic approach in mice and deleted Commd1 specifically in myeloid lineage osteoclast precursors by crossing Commd1 floxed mice with Lysozyme M-Cre mice (termed Commd1fl/fl x Lyz2-cre mice). COMMD1 deficiency did not affect bone mass under homeostatic conditions (Figures S3D and S3E). Therefore, we tested the effects of myeloid COMMD1 deficiency on pathological bone loss in arthritis and inflammatory osteolysis models. Commd1fl/fl x Lyz2-cre mice exhibited minimal differences in joint swelling or inflammation compared to littermate control mice in K/BxN serum-induced arthritis (Figures 3D–3E). In contrast, Commd1fl/fl x Lyz2-cre mice exhibited increased osteoclast number and surface in periarticular bone in K/BxN arthritis (Figures 3F–3H). Substantially increased osteoclast formation and bone erosion were also observed in Commd1fl/fl x Lyz2-cre relative to littermate control mice in the TNF-induced supracalvarial osteolysis model (Figures 3I–3K). Collectively, the results establish a role for COMMD1 in restraining pathological bone loss under inflammatory conditions in vivo.
Hypoxia suppresses COMMD1 expression and nuclear accumulation
We wished to directly test whether hypoxia inactivates COMMD1-dependent pathways and measured the effects of hypoxia on COMMD1 during osteoclastogenesis. Neither RANKL nor hypoxia affected COMMD1 mRNA amounts (Figure 4A), indicating that COMMD1 gene expression was not regulated. COMMD1 has both nuclear and cytosolic functions and shuttles between these compartments (Bartuzi et al., 2013). Immunohistochemistry revealed that RANKL increased nuclear accumulation of COMMD1 in normoxia (Figures 4B and 4C), which is consistent with a negative feedback mechanism. Hypoxia strongly suppressed nuclear accumulation of COMMD1 (Figures 4B and 4C). These results were corroborated using immunoblotting, which showed that hypoxia strongly suppressed both basal and RANKL-induced COMMD1 expression in nuclear and cytoplasmic compartments (Figure 4D and 4E). Suppression of COMMD1 protein expression by hypoxia further supports the model that hypoxia overcomes a COMMD1-mediated inhibitory mechanism to promote osteoclastogenesis.
Figure 4. Hypoxia suppresses COMMD1 expression and RANKL-induced nuclear accumulation.

(A) RT-qPCR of COMMD1 mRNA normalized relative to TBP mRNA.
(B–C) Immunofluorescence microscopy showing the subcellular localization of COMMD1 in macrophages after RANKL stimulation. COMMD1 (green) and nuclear (DAPI, blue) staining is merged in lowest panels. Relative fluorescence intensity of nuclear COMMD1 amounts in 6 experiments with different donors (C).
(D–E) Immunoblot of nuclear and cytoplasmic lysates with COMMD1 antibody. Akt and TBP were used as controls for cytoplasmic and nuclear proteins, respectively. Densitometric quantitation of intensity of bands from 5 experiments with different donors is shown in (E).
Bar graphs show means and error bars represent SEM. * p<0.05, ** p<0.01, *** p<0.001. Bar, 10μm.
(F) Immunoblot of cell lysates from human macrophages treated with DMSO or Torin for 3 hours and cultured with RANKL for two days. p38α was used as a control.
(G) Immunoblot of cell lysates from human macrophages treated with DMSO or MNK inhibitor CGP57380 for 3 hours and cultured with RANKL for two days. α-tubulin was used as a control.
(H) Human macrophages were transduced with adenoviral particles encoding GFP or COMMD1-HA. Cells were stimulated with RANKL (40ng/ml) for additional four days and osteoclastogenesis assays were performed under hypoxic conditions.
The results are representative of at least 3 (A, F–H), 6 (B) and 5 (E) experiments. Please see also Figure S3 and S4.
These results suggested that COMMD1 expression is induced by RANKL at the protein level and that hypoxia suppresses this induction. As hypoxia suppresses mTORC1, a key positive regulator of translation (Wilson and Hay, 2011), we tested whether mTORC1 activity is important for COMMD1 protein expression. Inhibition of mTORC1 abolished COMMD1 protein expression (Figure 4F). mTORC1 regulates translation by increasing the activity of eIF4E, a key translation initiation factor whose activity is positively regulated by the MNK kinases. In accord with a role for eIF4E and translation in mediating COMMD1 expression, inhibition of MNKs suppressed COMMD1 protein expression (Figure 4G). These results support regulation of COMMD1 at the translational level in human macrophages.
siRNA-mediated silencing of residual COMMD1 expressed under hypoxic conditions resulted in increased osteoclastogenesis (Figure S3F). In addition, forced expression of COMMD1 strongly suppressed osteoclastogenesis even under hypoxic conditions (Figure 4H). These results further support the role of COMMD1 as a negative regulator.
COMMD1 and hypoxia reciprocally regulate metabolic and NF-κB target genes in RANKL-stimulated macrophages
To gain insight into which COMMD1 functions and downstream molecules and pathways are important for suppression of osteoclastogenesis, we used RNA-seq to identify genes regulated by COMMD1 in macrophages stimulated with RANKL for 24 or 48 hours (Figure S4A and S4B). IPA analysis of genes whose expression was altered by COMMD1-specific siRNAs versus scrambled siRNA controls showed highly significant activation of cell cycle (which is associated with anabolic metabolism), metabolic, and inflammatory-NF-κB pathways (Figure 5A). Analysis of promoters of genes regulated by COMMD1 showed highly significant enrichment of binding sites for Myc (linked to metabolism), and E2F1 (linked to cell cycle and metabolism with no known function in osteoclastogenesis) (Figure 5B). Gene set enrichment analysis (GSEA) of the hypoxia response showed activation of similar pathways and E2F target genes (Figures S4C–S4F). GSEA identified 24 gene sets significantly enriched (p < 0.05) in COMMD1-regulated genes and 25 gene sets enriched in hypoxia-regulated genes (Figure 5C). 19 gene sets were commonly enriched in both conditions, suggesting that COMMD1 regulates a substantial part of the hypoxic response. Consistent with this view, GSEA showed that previously-identified hypoxia-regulated genes were enriched in our COMMD1-dependent gene set, and COMMD1 silencing resulted in increased expression of hypoxia-related genes (Figures S4G and S4H). Overall, the results show a tight integration, but not complete overlap, of hypoxic and COMMD1 pathways.
Figure 5. COMMD1 and hypoxia reciprocally regulate metabolic, cell cycle, and NF-κB target genes in RANKL-stimulated macrophages.

(A) IPA analysis of RANKL-inducible genes (24 h RANKL treatment) whose expression was superinduced by COMMD1 silencing; this gene set was identified using pooled RNA-seq data from two biological replicates as described in Figure S4A.
(B) Enriched transcription factor binding motifs in region ± 2kb relative to transcription start site in RANKL-inducible COMMD1-dependent genes, by gene set enrichment analysis (GSEA).
(C) GSEA showing Molecular Signatures Database (MsigDB) gene sets significantly enriched in COMMD1-regulated and hypoxia-regulated genes in RANKL-stimulated macrophages. Font colors of labels (right) correspond to metabolic and cell cycle gene sets (green), inflammation-NF-kB signaling gene set (orange) and hypoxia gene set (red).
(D) Immunoblot analysis of IκB after stimulation of human macrophages with RANKL for the indicated times. p38 was used as a loading control.
(E) Immunoblot of nuclear lysate with NF-κB p65 antibody. Human macrophages were stimulated with RANKL for 24 h. TBP was used as a loading control. Lower panel, densitometric quantitation of intensity of nuclear p65 bands from 5 experiments with different donors.
(F–I) Human macrophages were cultured under normoxic or hypoxic conditions for 3 hr prior to RANKL stimulation. RT-qPCR analysis of E2F1 mRNA normalized relative to TBP mRNA (F), immunoblot of E2F1 (G), and immunoblot of pRb and Rb (H). RT-qPCR analysis of E2F1 target gene expression (I). IKK inhibitors BAY11 and IKKi were added 30 min before the addition of RANKL where indicated.
(J–M) Human macrophages were nucleofected with control or COMMD1-specific siRNAs and then cells were stimulated with RANKL. RT-qPCR analysis of E2F1 mRNA normalized relative to TBP mRNA (J), immunoblot of pRB and Rb (K), and RT-qPCR analysis of E2F1 target gene expression (M).
(L) GSEA showing significant enrichment of E2F target genes in the COMMD1-regulated gene set; result from two biological replicates.
Bar graphs (Figures 5E, 5F, 5I, 5J and 5M) show means and error bars represent SEM. The results represent analysis of two pooled biological replicates or are representative of at least 3 (D, G–L) or 5 (E) experiments. Please see also Figure S4 and S5.
The gene sets commonly regulated by hypoxia and COMMD1 (Figure 5C) were notable for anabolic cell metabolism (Myc targets, E2F targets, mTORC1 signaling, glycolysis), which is related to the cell cycle (Myc targets, E2F targets, G2M checkpoint, K-Ras signaling), and inflammatory signaling (TNF signaling via NF-κB). We further tested the regulation of these pathways by COMMD1 and hypoxia. siRNA-mediated silencing of COMMD1 resulted in enhanced RANKL-induced NF-κB activation (Figures 5D and 5E) and increased expression of inflammatory genes (Figure. S5A). In addition, hypoxia and COMMD1 silencing similarly augmented RANKL-induced expression and activity of E2F, a major inducer of anabolic metabolism (Benevolenskaya and Frolov, 2015; Nicolay and Dyson, 2013) (Figures 5F–5M and S5C–S5G). E2F activity was assessed by measuring phosphorylation of its upstream regulator Rb (Figure S5B) and expression of canonical target genes (Figures 5H–5M). These results indicate that COMMD1 silencing and hypoxia similarly superinduce RANKL-mediated activation of the E2F pathway. The NF-κB- and E2F-mediated pathways were functionally inter-related, as IKK inhibitors suppressed the induction of E2F1 and its target genes (Figures 5F and 5I). Neither COMMD1 overexpression nor COMMD1 silencing affected HIF-1α protein expression (Figure S3G and S3H) but did affect expression of a subset of HIF-1α target genes (Figure S3I and S3J). These results reveal that COMMD1 regulates E2F function and support that hypoxia and COMMD1 reciprocally regulate NF-κB, HIF and E2F pathways during osteoclastogenesis.
As expected (Sørensen et al., 2007), terminally differentiated human macrophages did not proliferate substantially in response to RANKL even under hypoxic conditions (Figure S5H). Inhibitors of the cell cycle had no effect on osteoclastogenesis in human OCPs (Figures S5I and S5J), and expression of cell cycle genes subsided after 48 hr of RANKL stimulation, which coincided with induction of the CDK inhibitor p21 (Figure S5K). This suggests that the E2F-mediated transcriptional program functions during the early phase of OCP differentiation to meet metabolic demands rather than inducing cell cycle entry.
E2F1 promotes osteoclastogenesis and glycolytic metabolism
Our results suggested that hypoxia and COMMD1 regulate osteoclastogenesis at least in part by modulating E2F1. We directly tested the role of E2F1 in osteoclastogenesis by silencing E2F1 expression using three different LNAs and siRNAs. E2F1 silencing effectively downregulated expression of canonical E2F target genes (Figure 6A) and suppressed RANKL-induced osteoclastogenesis (Figures 6B, S6A and S6B). Accordingly, expression of osteoclast marker genes ITGB3, CTSK and CTR was suppressed (Figure 6C). Thus, E2F1 is required for effective osteoclastogenesis even under normoxic conditions, and its expression and function are superinduced by hypoxia or COMMD1 silencing. E2F1 silencing broadly suppressed expression of genes encoding proximal components of the glycolytic pathway, which is required for osteoclastogenesis (Indo et al., 2013) (Figures 6D, 6E and S6C). As many of these genes are HIF-1α targets, these results suggest that E2F1 cooperates with HIF-1α to boost their induction. Consistent with our model that COMMD1 negatively regulates E2F1, COMMD1 silencing resulted in the induction of genes encoding early components of the glycolytic pathway (Figure 6F), consistent with the GSEA analysis in Figure 5C. Finally, inhibition of the glycolytic pathway effectively suppressed osteoclastogenesis in nonproliferating human macrophages (Figure 6G). These results suggest a role for E2F1 in meeting the metabolic demands during the early phase of osteoclastogenesis that is dependent on glycolysis.
Figure 6. E2F1 promotes osteoclastogenesis and expression of glycolysis pathway genes.

(A) RT-qPCR analysis of E2F1 and its target genes showing efficacy of E2F1 silencing.
(B) TRAP staining of RANKL-stimulated human macrophages nucleofected with control or E2F1-specific LNA. Right, quantitation of osteoclast numbers from 4 experiments with different donors; the control condition with RANKL (40 ng/ml) for each donor is set as 100%.
(C) RT-qPCR analysis of ITGB3, CTSK and CTR mRNA normalized relative to TBP mRNA from RANKL-stimulated human macrophages nucleofected with control or E2F1-specific LNA.
(D) Schematic of enzymes and metabolites in the glycolysis pathway.
(E–F) Heat maps showing relative expression of glycolysis pathway genes in RANKL-stimulated human macrophages nucleofected with control or E2F1-specific LNAs (E) or control or COMMD1-specific siRNAs (F). Results from pooled data from two biological replicates are shown.
(G) TRAP staining of human osteoclasts treated with 2-deoxy-D-glucose (2DG). Right panel shows pooled data from 3 experiments with different donors, normalized relative to the number of osteoclasts treated with RANKL alone (set at 100%).
Results show means and error bars represent SEM. * p < 0.05, ** p<0.01, *** p<0.001. Scale bar, 200 μm. The results are representative of at least 3 (A, C), 4 (B) and 3 (G) experiments; (E–F) represent analysis of two pooled biological replicates. Please see also Figure S6.
E2F1 induces inflammatory pathways and CKB which augments cellular energy supply during osteoclastogenesis
To gain additional insights into mechanisms by which E2F1 promotes osteoclastogenesis, we performed transcriptomic analysis of macrophages in which E2F1 expression was silenced. GSEA of genes whose induction by RANKL was partially dependent on E2F1 recovered highly significant enrichment of ‘E2F targets’, which validated the approach, and enrichment of metabolic pathways, which further supports a role for E2F1 in metabolic regulation in macrophages (Figure 7A). Genes in TNF and NF-κB signaling, inflammatory response, and cytokine-Jak-STAT signaling pathways were also enriched (Figure 7A), supporting the possibility that E2F1 plays a role in macrophage activation. Expression of TNF-NF-κB target genes that were partially dependent on E2F1 is shown in Figure 7B. Among the genes most strongly downregulated when E2F1 was silenced was CKB (creatine kinase B) (Figure 7C), which had previously been implicated in osteoclastogenesis in vitro and in vivo (Chang et al., 2008). CKB augments the reservoir of high-energy phosphate bonds and is required for proper energy utilization in neurons and osteoclast precursors (Chang et al., 2008; Wyss and Kaddurah-Daouk, 2000). We confirmed that CKB induction by RANKL is partially dependent on E2F1 (Figures 7D, S7A and S7B). As predicted by our model, CKB expression was superinduced under hypoxia or when COMMD1 was silenced (Figures 7D, S7C and S7D). We performed complementary gain- and loss-of-function experiments to obtain functional evidence for the importance of CKB in osteoclast differentiation of human macrophages. Addition of the CKB product phosphocreatine to culture media increased RANKL-induced osteoclastogenesis (Figure 7E), while an inhibitor of creatine kinase, cyclocreatine, inhibited osteoclastogenesis (Figure 7F). These results further support a key role for E2F1 in inducing expression of genes required to meet the metabolic demands of osteoclastogenesis. Integrated regulation of the COMMD1-E2F-metabolic pathway in osteoclastogenesis is depicted in Figure S7E.
Figure 7. E2F1 induces CKB to augment cellular energy supply during osteoclastogenesis.

(A) GSEA of RANKL-inducible E2F1-dependent genes. Pooled data from two biological replicates were analyzed.
(B) Heat map showing relative expression of representative genes from the HALLMARK TNFA_SIGNALING-VIA-NFKB gene set in macrophages treated with E2F1-specific LNAs.
(C) Heat map showing relative expression of RANKL-induced genes that were most highly dependent on E2F1.
(D) RT-qPCR analysis of CKB mRNA normalized relative to TBP mRNA.
(E–F) TRAP staining of RANKL-stimulated human macrophages treated with phosphocreatine (E) or cyclocreatine (F) at the indicated concentrations. Right panels show quantitation of pooled data from 5 (E) or 3 (F) experiments with different donors; the RANKL alone condition for each donor is set as 100%.
Results show means and error bars represent SEM. * p < 0.05. Bar, 200 μm. The results (A, B, C) represent analysis of two pooled biological replicates or are representative of 5 (E), 3 (F) and at least 3 (D) experiments. Please see also Figure S7.
Discussion
The importance of glycolysis in inflammatory macrophage activation, and of several branches of anabolic cellular metabolism, including glycolysis, OX-PHOS and mTORC1-dependent biosynthetic pathways, in osteoclastogenesis is becoming increasing appreciated, but mechanisms that regulate metabolism in macrophages and osteoclast precursors during RANKL responses are not well understood. We investigated mechanisms by which hypoxia, which compromises OX-PHOS and mTORC1 metabolic pathways, is still able to augment RANKL-induced osteoclastogenesis. Our findings uncovered an Rb-E2F1-glycolysis-CKB pathway that promoted inflammatory gene expression and anabolic metabolism but was only weakly activated by RANKL under normoxic conditions because its activation was restrained by COMMD1. This pathway was superinduced under hypoxic conditions to help meet the metabolic demands of osteoclastogenesis under the challenging conditions of an oxygen-depleted environment. Hypoxia induced this E2F1-metabolic pathway by inactivating the upstream negative regulator COMMD1. COMMD1 broadly suppressed the hypoxia-induced transcriptional response in RANKL-stimulated macrophages, including negative regulation of NF-κB signaling, which is important for osteoclastogenesis. Myeloid-specific COMMD1 deletion resulted in increased osteoclastogenesis in arthritis and inflammatory osteolysis models, and RA patients with COMMD1 allelic variants associated with increased COMMD1 expression exhibited decreased bone erosion. These results identify COMMD1 as a negative regulator of inflammatory gene expression, osteoclastogenesis, and pathological bone resorption, and provide new insights into regulation of metabolic pathways during osteoclast differentiation, and into mechanisms by which hypoxia enhances inflammatory responses and osteoclastogenesis.
Negative regulators that restrain osteoclastogenesis help maintain bone mass under physiological conditions, and limit the amount of bone loss in pathological conditions such as inflammatory states. RANKL signaling downregulates the basal expression of important negative regulators such as IRF8, BCL6 and RBP-J to enable effective osteoclastogenesis (Zhao and Ivashkiv, 2011). In contrast, RANKL increased expression and nuclear localization of the negative regulator of COMMD1. Thus, COMMD1 functions not solely as a constitutive inhibitor but is induced as a part of a negative feedback loop that restrains E2F1, NF-κB and likely HIF-1α activity. Our findings introduce the concept that feedback inhibition can also target metabolic pathways, such as glycolysis, in addition to targeting signaling and gene expression mechanisms. Inactivation of COMMD1 by hypoxia enables cells to meet metabolic demands under challenging environmental conditions. Induction of COMMD1 expression coincided with downregulation of the E2F-driven “cell cycle pathway” that occurred at time points later than 48 hours of RANKL stimulation. At these later time points mitochondrial biogenesis and OXPHOS have been induced and can meet metabolic demands, and thus the E2F pathway may no longer be needed.
COMMD1 deficiency did not have an effect on basal bone phenotype under homeostatic conditions, but resulted in dramatic increases in osteoclastogenesis and bone resorption under pathological conditions characterized by hypoxia and inflammatory stress. This supports a model where under non-stress conditions COMMD1 holds in check metabolic pathways that are fully required only under challenging environmental conditions, and thus provide insight into the mechanisms by which hypoxia augments bone resorption in pathologic conditions by downregulating COMMD1. In accord with this model, CKB, which is downstream of COMMD1, did not affect basal bone phenotype, but contributed to pathological bone resorption in disease settings such as arthritis (Chang et al., 2008). This model is consistent with the role of CKB in brain, and also its isoform CKM in muscle, where these enzymes are not required for homeostatic function but are required to meet high energy demands, such as burst activity associated with exercise (van Deursen et al., 1993). Our findings identify multiple components, including COMMD1, E2F and CKB, of a molecular pathway that can be therapeutically targeted to suppress pathological bone loss. The nearly complete suppression of osteoclastogenesis by forced expression of COMMD1 supports therapeutic augmentation of expression or function of this upstream molecule, which would suppress several downstream metabolic and inflammatory pathways. Boosting the function of a negative regulator such as COMMD1 represents a conceptually distinct approach from the current use of inhibitors of positive regulators of inflammation and osteoclastogenesis.
Major known mediators of hypoxic effects on cells are the HIF transcription factors (Palazon et al., 2014). HIF-1α has been implicated in increasing the resorptive functions of osteoclasts in vitro and in ovariectomy-induced bone loss in vivo (Miyauchi et al., 2013). Our results showing overlap between pathways and genes regulated by COMMD1 and hypoxia, in the context of the known role of HIF in mediating hypoxia responses, raises the question of the relationship between COMMD1 and HIF in osetoclastogeensis. In other systems, COMMD1 can physically interact with HIF-1α, destabilize HIF-1α protein in a cell type-specific manner, and inhibit expression of a subset of HIF-1α target genes in a gene-specific manner by incompletely understood mechanisms (Muller et al., 2009a; Sluis et al., 2009; van de Sluis et al., 2010; van de Sluis et al., 2007). Our data suggest that COMMD1 does not regulate HIF-1α protein amounts in macrophages and instead modulates HIF-1α function by an alternative mechanism. One possibility is that COMMD1 regulates distinct transcription factors such as E2F1 that cooperate with HIF to regulate expression of hypoxia-inducible genes in RANKL-stimulated macrophages. Cooperation between E2F1, HIF-1α and other transcription factors regulated by COMMD1 is likely important for determining patterns of gene expression, suggests a plausible explanation for gene-specific effects, and represents an important area for future research.
RANKL-mediated induction of the anabolic metabolic, ‘cell cycle’, and E2F-mediated pathways has not been previously noted. This is likely because previous transcriptomic analyses have been performed under conditions of normoxia, where COMMD1 suppresses these pathways. In addition, most studies have used proliferating mouse OCPs, where RANKL effects may be difficult to discern above background gene expression related to the active cell cycle. Many of the E2F target genes in the ‘cell cycle’ pathways regulated by COMMD1 correspond to anabolic pathway genes that are induced in the early phases of growth factor stimulation as part of a metabolic program that prepares cells to meet the high energy demands of cell division. We propose that in response to RANKL stimulation under hypoxic conditions, macrophages repurpose ‘cell cycle’ anabolic pathways to generate energy and possibly specific metabolites necessary for the energy-intensive and specialized process of osteoclastogenesis. An interesting precedent for this type of metabolic ‘repurposing’ is the switch of innate immune cells to anaerobic glycolysis (Warburg metabolism), even under normoxic conditions, in order to rapidly meet energy demands and generate metabolites important for host defense (Kelly and O’Neill, 2015). In this context, E2F1 functions in human macrophages more as an inducer of anabolic metabolism than as a driver of cell cycle progression. There are 8 E2F family members with context-dependent activating and repressing functions; our findings open an area of research on the function of E2Fs and their target genes in osteoclastogenesis.
In summary, we have identified COMMD1 as a key suppressor of hypoxia-induced pathways under normoxic conditions that functions as a negative regulator of inflammatory gene expression and osteoclastogenesis and pathologic bone resorption, and is itself suppressed by hypoxia. Downregulation of COMMD1 enables RANKL to more effectively induce inflammatory gene expression, and E2F-mediated metabolic pathways including glycolysis and CKB to meet the metabolic demands of osteoclastogenesis under challenging environmental conditions, and promotes bone resorption. These findings identify new regulators and pathways important for hypoxia, osteoclastogenesis and pathologic bone resorption, and provide insights into how hypoxia augments inflammation and osteoclastogenesis.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Lionel B. Ivashkiv (IvashkivL@hss.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
In vivo animal studies
All animal procedures were approved by the Hospital for Special Surgery Institutional Animal Care and Use Committee (IACUC), and Weill Cornell Medical College IACUC. Mice with myeloid-specific deletion of COMMD1 were obtained by crossing COMMD1flox/flox mice (Vonk et al., 2011) with mice with a Lysozyme M promoter-driven Cre transgene on the C57/BL6 background (known as LysMcre or Lyz2-cre; The Jackson Laboratory). COMMD1flox/flox LysMcre(+) mice (referred to as Commd1fl/fl x Lyz2-cre) and littermate control COMMD1+/+LysMcre(+) mice were used for the experiments. Bone phenotypes of 12-week-old male control and Commd1fl/fl x Lyz2-cre mice, including bone volume and architecture was assessed using a Scanco micro-CT-35 instrument (Scanco Medical, Bruttisellen, Switzerland) as previously described (Park-Min et al., 2013). For arthritis experiments, K/BxN serum pools were prepared as described previously (Korganow et al., 1999). Arthritis in 8-week-old female mice was induced by intraperitoneal injection of 100 μl of K/BxN serum on days 0, 2 and 4. The development of arthritis was monitored by measuring the thickness of wrist and ankle joints using dial-type calipers (Bel-Art Products) and scoring the wrist and ankle joints. For each animal, joint thickness was calculated as the sum of the measurements of both wrists and both ankles. Joint thickness was represented as the average for every treatment group. The severity of arthritis was scored in a blinded fashion by three investigators for each paw on a 3-point scale, in which 0 = normal appearance, 1 = localized edema/erythema over one surface of the paw, 2 = edema/erythema involving more than one surface of the paw, 3 = marked edema/erythema involving the whole paw. The scores of all four paws were added for a composite score. For inflammatory osteolysis experiments, we used an established mouse model, the TNF-induced supracalvarial osteolysis model (Kitaura et al., 2005) with minor modifications. In brief, TNF was administrated daily at the dose of 48μg/kg to the calvarial periosteum of mice for five consecutive days. The mice were then sacrificed for the collection of serum and calvarial bones for sectioning. For histopathologic assessment, mice were euthanized, and the hind paws and calvarial bones were harvested and fixed in 4% paraformaldehyde for 2 days. These samples were decalcified with 10% neutral buffered EDTA (Sigma-Aldrich) and embedded in paraffin. To assess osteoclastogenesis and bone resorption, sections were stained with TRAP (tartrate-resistant acid phosphatase) and hematoxylin for osteoclast visualization. All measurements were performed using the Osteometric software (Osteomeasure) using standard procedures (Parfitt et al., 1987) and were confined to the secondary spongiosa and restricted to an area between 400 and 2000 μm distal to the growth plate metaphyseal junction of the distal femur. Osteoclasts were identified as TRAP+ cells that were multinucleated and adjacent to bone.
For human studies
We analyzed 340 patients with RA from the Kyoto University Rheumatoid Arthritis Management Alliance (KURAMA) cohort at Kyoto University, as previously described (Terao et al., 2015). DNA was available from 176 RA patients among the 340 patients in the Kyoto University Rheumatoid Arthritis Management Alliance (KURAMA) cohort. 88.1 % of patients were female, and they had mean age of 62.2 and disease duration of 13.9 years. 84.7% and 81.3% of patients were rheumatoid factor (RF) and anticitrullinated peptide antibody (ACPA) positive, respectively. This study was approved by the local ethics committee at each institution, and written informed consent was obtained from all participants. Details of the cohort were described before (Terao et al., 2015). Genotyping for rs11125908 was performed using Taqman SNP genotyping assay (Thermo Fisher Scientific). Details for scoring of total Sharp score were described previously (Terao et al., 2015). Age, disease duration, positivity of RF and usage of MTX and biological agents were used as covariates for generalized linear regression model to assess the association between rs11125908 and SHS scores. Since previous study showed that finger joint destruction may have unique correlates including RF in comparison with non-finger joint destruction, next we divided SHS scores in the hand into finger joint scores and non-finger joint scores. To analyze the association between rs11125908 and finger joint destruction, non-finger joint destruction in addition to the variables described above were used as covariates. To further assess the association between rs11125908 and finger joint destruction independently of RF levels, next we extracted only RF-positive RA patients. We performed generalized linear regression analysis with maximum levels of RF and the same covariates of SHS.
For healthy donor eQTL, we obtained eQTL associations showing p-values less than 1.0 x10−2 with COMMD1 expression from the Blood eQTL browser based on the data of previously published large eQTL study in the European population (Westra et al., 2013). In Japanese subjects, we analyzed gene expression data previously measured in whole blood from the 298 healthy Japanese individuals (Higasa et al., 2016). For this study, cis eQTL associations with A_19_P00806260 probe were used. Expression data were normalized across the individuals and probes as previously described (Higasa et al., 2016). LocusZoom (Pruim et al., 2011) was used to draw the figure.
Cell Culture
Peripheral blood mononuclear cells were obtained from blood leukocyte preparations purchased from the New York Blood Center by density gradient centrifugation with Ficoll (Thermo Fisher Scientific, Fair Lawn, NJ) using a protocol approved by the Hospital for Special Surgery Institutional Review Board (IRB #93145). Monocytes were obtained from peripheral blood, using anti-CD14 magnetic beads, as recommended by the manufacturer (Miltenyi Biotec, Auburn, CA). Purity of monocytes was >97%, as verified by flow cytometric analysis. Cells were added to 96-well plates in triplicate at a seeding density of 1 × 105 cells per well. Macrophages that were used as osteoclast precursors were obtained by overnight culture of CD14+ monocytes with 20 ng/ml of M-CSF (Peprotech) in alpha modified essential medium (α-MEM) (Thermo Fisher Scientific) supplemented with 10% Hyclone fetal bovine serum (GE Healthcare Life Sciences, Logan, UT) and 1% glutamine (200 mM, Thermo Fisher Scientific). Macrophages were incubated with 20 ng/ml of M-CSF and 40 ng/ml of human soluble RANKL for two additional days. Cytokines were replenished every 2 days. For hypoxia experiments, cells were placed into a hypoxia chamber (2% oxygen, Biospherix, Parish, NY) 3 h before RANKL was added. Cells were fixed and stained for TRAP using the Acid Phosphatase Leukocyte diagnostic kit (Sigma Aldrich, Carlsbad, CA) as recommended by the manufacturer. Multinucleated (>3 nuclei), TRAP-positive osteoclasts were counted in triplicate wells. In vitro resorption activity of osteoclasts was measured using Osteo Assay Surface Plate (Corning, Corning, NY). Cells were seeded in Osteo Assay Surface Plates and cultured in the presence of M-CSF and RANKL. After three days of culture with RANKL, cells were incubated with 10% bleach solution for 5 minutes at room temperature. The wells were washed twice with distilled water and allowed to dry at room temperature for 3 to 5 hours. Individual pits were observed and evaluated using Osteomeasure software (Osteometrics, Atlanta, GA). 2-deoxyglucose (2DG) was added when cells were seeded onto plates with M-CSF. Methotrexate, hydroxyurea, phosphocreatine, and 2-Imino-1-imidazolidineacetic acid (cyclocreatine) were added at the same time as RANKL stimulation.
METHOD DETAILS
Quantitative Real-Time PCR
Total RNA was extracted from cells using RNeasy Mini kit (QIAGEN), and 500 ng of total RNA was reverse transcribed using the RevertAid First Strand cDNA Synthesis kit (Fermentas). Real-time PCR was performed in triplicate with Fast SYBR Green Master Mix and 7500 Fast Real-time PCR system (Applied Biosystems). Primer sequences are provided in the Table S1.
RNA-sequencing
Total RNA was extracted using RNeasy mini kit (Qiagen). True-seq RNA Library preparation kits (Illumina) were used to purify poly-A+ transcripts and generate libraries with multiplexed barcode adaptors following the manufacturer’s instructions. All samples passed quality control analysis on a Bioanalyzer 2100 (Agilent). Paired-end reads were obtained for hypoxia samples (51 base pairs, ~100×106 reads per sample) and single-end reads were obtained for COMMD1 and E2F1 silencing samples (51 base pairs, 20 – 40 ×106 reads per sample) on an Illumina HiSeq 2500 in the Weill Cornell Medical College Genomics Resources Core Facility or the Weill Cornell Epigenomics Core Facility. The TopHat program (v0.9.0) was used to align the reads to the UCSC Hg19 human reference genome, and the Cuffdiff program (v2.2.1) was used for the measurements of transcript abundance (represented by Fragments Per Kilobase of exon model per Million mapped reads (FPKM)) with Galaxy (Goecks et al., 2010). All FPKMs represent combined results, using Cuffdiff, from two biological replicates. Genes and transcripts with FPKM values less than 5 were not included in the analysis. Pearson correlation analysis and heat maps were generated using R (v3.0.2). Two biological replicates were used and the concordance between replicates was very high (R2 range, 0.866–0.9796).
Gene Ontology Analysis and Motif Identification
To identify RANKL-inducible genes that were superinduced by hypoxia for pathway analysis, we selected genes that were induced by RANKL (> 1.5 fold) in pooled data from two donors (except when stated) in both normoxia and hypoxia, and superinduced by hypoxia (> 1.5 fold) at 48 hr after RANKL stimulation (Figure S1C and Figure 1E). When the 24 h time point of RANKL stimulation was analyzed, the same cut offs were used. In the analyses of COMMD1 silencing, the cut offs were 1.2 fold (Figures 5A–5C). E2F1-regulated genes were those induced by RANKL (> 1.2 fold, in control LNA) and suppressed by E2F1 silencing (>1.2 fold, in RANKL stimulation condition) (Figure 7A). These gene sets were analyzed by IPA (Figures 1E, 5A, S4C, S4E) and gene set enrichment analysis (GSEA) (Figures 5B, 5C 7A and Figure S4D) (Subramanian et al., 2005). In Figure S3I, heatmap was created from the expression of HIF1αtarget genes (Benita et al., 2009). In Figure 7C, E2F1-regulated genes (FPKMs > 20) were sorted by the ratio of control relative to E2F1 silencing conditions. For the calculation of gene enrichment score (Mootha et al., 2003; Subramanian et al., 2005), non-pooled FPKMs from individual 2 donors were used (Figure 5F and Figures S4F and S4G). MSigDB pathways by GSEA were ranked based on the p-values.
Flow Cytometry
Proliferation of macrophages was measured using CFSE Cell Division Tracker Kit (BioLegend) according to the manufacturer’s protocol. CD14+ monocytes were suspended at 10 × 106 cells/mL with PBS containing 5 μM CFSE, incubated for 20 min at 37°C and quenched with cell culture medium containing 10% FBS. Macrophages were prepared by culture with M-CSF (20 ng/ml) and stimulated with soluble RANKL for the indicated times in either normoxia or hypoxia. Cells were also stained using human PE-conjugated CD14 antibody (BD Bioscience). A FACSCalibur flow cytometer with CellQuest™ software (BD Bioscience) was used for analysis.
RNA Interference
For RNA interference (RNAi) experiments, primary human macrophages (107 cells) were nucleofected with 0.2 –0.4 nmol of siRNA oligonucleotides using a Nucleofector kit (Lonza) as previously described (Park-Min et al., 2014). Human Monocyte Nucleofector buffer (Lonza Cologne) and the AMAXA Nucleofector System program Y001 for human monocytes were used according to the manufacturer’s instructions. We tested three different sets of siRNAs or antisense LNAs, using at least three donors unless otherwise stated. COMMD1-, and E2F1-specific and control siRNAs were obtained from Thomas Fisher Scientific. COMMD1- and E2F1- specific and control antisense LNAs were purchased from Exiqon. Sequences are shown in Table S1.
Adenoviral Transduction
Recombinant adenoviral particles encoding human COMMD1-HA and control adenoviral particles encoding green fluorescent protein (Ad-CMV-GFP) were from Vector Biolabs (Malvern, PA). For adenoviral transduction, human monocytes were incubated for 6 days with M-CSF (40ng/ml) on 6 well plates in α-MEM medium supplemented with 10 % of fetal bovine serum (GE Healthcare Life Sciences, Logan, UT). Cells were washed and incubated for one hour in low-serum media (0.5% (vol/vol) FBS). Then, cells were cultured with adenoviral particles (50 particles per cell) for 12 hr. Transduction efficiency was monitored by the fluorescence of green fluorescent protein and was typically greater than 85%.
QUANTIFICATION AND STATISTICAL ANALYSIS
In all experiments, data are presented as mean ± SEM. Statistical tests were selected based on appropriate assumptions with respect to data distribution and variance characteristics. All statistical analyses were performed with Graphpad Prism 5.0 software or R using the two-tailed, unpaired t-test (two conditions), one-way or two-way ANOVA for multiple comparisons (more than two conditions) with post hoc Bonferroni’s correction for multiple comparisons. Statistical significance was defined as p < 0.05. For all experiments, *p < 0.05, **p < 0.01, ***p < 0.001. Sample sizes were chosen using the literature or previous experience in the lab to guide power calculations. Number of samples and animals was indicated as “n.” or described in figure legends.
DATA AND SOFTWARE AVAILABILITY
RNA-seq data for this project have been deposited at NCBI’s Gene Expression Omnibus (GEO) with GSE numbers GSE99987.
Supplementary Material
Highlights.
COMMD1 is a negative regulator of osteoclast differentiation
COMMD1 suppresses bone loss in RA and inflammatory arthritis and osteolysis models
COMMD1 negatively regulates E2F1-dependent metabolic pathways in macrophages
Hypoxia suppresses COMMD1 expression to augment osteoclastogenesis
Acknowledgments
We thank Lyudmila Lukashova for micro-CT analysis, Drs. Steven Goldring, Matthew B. Greenblatt, and Jae-Hyuck Shim for helpful discussions, and Baohong Zhao for critical review of the manuscript. This study was supported by the Kyoto University Foundation to K.M., Grant-in-Aid from the Ministry of Education of Japan to F.M., NIAMS grants to K.-H.P.-M., and NIH grants to L.B.I. Department of Advanced Medicine for Rheumatic Diseases is supported by Nagahama City, Shiga, Japan and four pharmaceutical companies (Mitsubishi Tanabe Pharma Co., Chugai Pharmaceutical Co. Ltd, UCB Japan Co. Ltd, and AYUMI Pharmaceutical Co.). The study contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. No authors have any financial interest related to this work.
Footnotes
AUTHOR CONTRIBUTIONS
K.M. conceived, designed and performed most of the experiments, analyzed data, and wrote the manuscript. K.M. and E.G. performed bioinformatic data analysis. C.F., Y.J.L., M.J.L., S-H.M., S.B., R.Y., and Y.Q. contributed to experiments. H.I., K.O., S.M., and T.M. recruited the RA patients. F.M. organized the RA patient data. M.F. recruited the RA patients and performed the TSS scoring. C.T. performed the analysis and interpretation of the RA patient data. K.-H.P.-M. and L.B.I. conceived, designed, and oversaw the project and wrote the manuscript. No authors have any financial interest related to this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bartuzi P, Hofker MH, Van de Sluis B. Tuning NF-κB activity: A touch of COMMD proteins. Biochimica et Biophysica Acta - Molecular Basis of Disease. 2013;1832:2315–2321. doi: 10.1016/j.bbadis.2013.09.014. [DOI] [PubMed] [Google Scholar]
- Benevolenskaya EV, Frolov MV. Emerging Links between E2F Control and Mitochondrial Function. Cancer Research. 2015;75:619–623. doi: 10.1158/0008-5472.CAN-14-2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benita Y, Kikuchi H, Smith AD, Zhang MQ, Chung DC, Xavier RJ. An integrative genomics approach identifies Hypoxia Inducible Factor-1 (HIF-1)-target genes that form the core response to hypoxia. Nucleic Acids Research. 2009;37:4587–4602. doi: 10.1093/nar/gkp425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cejka D, Hayer S, Niederreiter B, Sieghart W, Fuereder T, Zwerina J, Schett G. Mammalian target of rapamycin signaling is crucial for joint destruction in experimental arthritis and is activated in osteoclasts from patients with rheumatoid arthritis. Arthritis and Rheumatism. 2010;62:2294–2302. doi: 10.1002/art.27504. [DOI] [PubMed] [Google Scholar]
- Chang EJ, Ha J, Oerlemans F, Lee YJ, Lee SW, Ryu J, Kim HJ, Lee Y, Kim HM, Choi JY, et al. Brain-type creatine kinase has a crucial role in osteoclast-mediated bone resorption. Nature medicine. 2008;14:966–972. doi: 10.1038/nm.1860. [DOI] [PubMed] [Google Scholar]
- Chiu YH, Mensah KA, Schwarz EM, Ju Y, Takahata M, Feng C, McMahon LA, Hicks DG, Panepento B, Keng PC, Ritchlin CT. Regulation of human osteoclast development by dendritic cell-specific transmembrane protein (DC-STAMP) Journal of Bone and Mineral Research. 2012;27:79–92. doi: 10.1002/jbmr.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goecks J, Nekrutenko A, Taylor J. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome biology. 2010;11:R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higasa K, Miyake N, Yoshimura J, Okamura K, Niihori T, Saitsu H, Doi K, Shimizu M, Nakabayashi K, Aoki Y, et al. Human genetic variation database, a reference database of genetic variations in the Japanese population. J Hum Genet. 2016:1–7. doi: 10.1038/jhg.2016.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indo Y, Takeshita S, Ishii KA, Hoshii T, Aburatani H, Hirao A, Ikeda K. Metabolic regulation of osteoclast differentiation and function. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2013;28:2392–2399. doi: 10.1002/jbmr.1976. [DOI] [PubMed] [Google Scholar]
- Ishii K-a, Fumoto T, Iwai K, Takeshita S, Ito M, Shimohata N, Aburatani H, Taketani S, Lelliott CJ, Vidal-Puig A, Ikeda K. Coordination of PGC-1β and iron uptake in mitochondrial biogenesis and osteoclast activation. Nature Medicine. 2009;15:259–266. doi: 10.1038/nm.1910. [DOI] [PubMed] [Google Scholar]
- Ivashkiv LB. Metabolic-epigenetic coupling in osteoclast differentiation. Nature Medicine. 2015;21:212–213. doi: 10.1038/nm.3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly B, O’Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell research. 2015;25:771–784. doi: 10.1038/cr.2015.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitaura H, Zhou P, Kim HJ, Novack DV, Ross FP, Teitelbaum SL. M-CSF mediates TNF-induced inflammatory osteolysis. Journal of Clinical Investigation. 2005;115:3418–3427. doi: 10.1172/JCI26132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konisti S, Kiriakidis S, Paleolog EM. Hypoxia--a key regulator of angiogenesis and inflammation in rheumatoid arthritis. Nature reviews Rheumatology. 2012;8:153–162. doi: 10.1038/nrrheum.2011.205. [DOI] [PubMed] [Google Scholar]
- Korganow AS, Hong J, Mangialaio S, Duchatelle Vr, Pelanda R, Martin T, Degott C, Kikutani H, Rajewsky K, Pasquali JL, et al. From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity. 1999;10:451–461. doi: 10.1016/s1074-7613(00)80045-x. [DOI] [PubMed] [Google Scholar]
- Miyauchi Y, Sato Y, Kobayashi T, Yoshida S, Mori T, Kanagawa H, Katsuyama E, Fujie A, Hao W, Miyamoto K, et al. HIF1 α is required for osteoclast activation by estrogen deficiency in postmenopausal osteoporosis. Proceedings of the National Academy of Sciences. 2013;110:16568–16573. doi: 10.1073/pnas.1308755110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizoguchi T, Muto A, Udagawa N, Arai A, Yamashita T, Hosoya A, Ninomiya T, Nakamura H, Yamamoto Y, Kinugawa S, et al. Identification of cell cycle-arrested quiescent osteoclast precursors in vivo. The Journal of cell biology. 2009;184:541–554. doi: 10.1083/jcb.200806139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nature genetics. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Muller PA, van de Sluis B, Groot AJ, Verbeek D, Vonk WI, Maine GN, Burstein E, Wijmenga C, Vooijs M, Reits E, Klomp LW. Nuclear-cytosolic transport of COMMD1 regulates NF-kappaB and HIF-1 activity. Traffic. 2009a;10:514–527. doi: 10.1111/j.1600-0854.2009.00892.x. [DOI] [PubMed] [Google Scholar]
- Muller PAJ, de Sluis Bv, Groot AJ, Verbeek D, Vonk WIM, Maine GN, Burstein E, Wijmenga C, Vooijs M, Reits E, Klomp LWJ. Nuclear-Cytosolic Transport of COMMD1 Regulates NF-κB and HIF-1 Activity. Traffic. 2009b;10:514–527. doi: 10.1111/j.1600-0854.2009.00892.x. [DOI] [PubMed] [Google Scholar]
- Muto A, Mizoguchi T, Udagawa N, Ito S, Kawahara I, Abiko Y, Arai A, Harada S, Kobayashi Y, Nakamichi Y, et al. Lineage-committed osteoclast precursors circulate in blood and settle down into bone. Journal of Bone and Mineral Research. 2011;26:2978–2990. doi: 10.1002/jbmr.490. [DOI] [PubMed] [Google Scholar]
- Nakashima T, Takayanagi H. New regulation mechanisms of osteoclast differentiation. 2012;1240:13–18. doi: 10.1111/j.1749-6632.2011.06373.x. [DOI] [PubMed] [Google Scholar]
- Nicolay BN, Dyson NJ. The multiple connections between pRB and cell metabolism. Current Opinion in Cell Biology. 2013;25:735–740. doi: 10.1016/j.ceb.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa K, Iwamoto Y, Kobayashi Y, Katsuoka F, Kawaguchi S-i, Tsujita T, Nakamura T, Kato S, Yamamoto M, Takayanagi H, Ishii M. DNA methyltransferase 3a regulates osteoclast differentiation by coupling to an S-adenosylmethionine–producing metabolic pathway. Nature Medicine. 2015 doi: 10.1038/nm.3774. [DOI] [PubMed] [Google Scholar]
- Novack DV, Teitelbaum SL. The osteoclast: friend or foe? Annual review of pathology. 2008;3:457–484. doi: 10.1146/annurev.pathmechdis.3.121806.151431. [DOI] [PubMed] [Google Scholar]
- Palazon A, Goldrath AW, Nizet V, Johnson RS. Review HIF Transcription Factors, Inflammation, and Immunity. Immunity. 2014;41:518–528. doi: 10.1016/j.immuni.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parfitt AM, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 1987;2:595–610. doi: 10.1002/jbmr.5650020617. [DOI] [PubMed] [Google Scholar]
- Park-Min KH, Lim E, Lee MJ, Park SH, Giannopoulou E, Yarilina A, van der Meulen M, Zhao B, Smithers N, Witherington J, et al. Inhibition of osteoclastogenesis and inflammatory bone resorption by targeting BET proteins and epigenetic regulation. Nature communications. 2014;5:5418. doi: 10.1038/ncomms6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park-Min KH, Lee EY, Moskowitz NK, Lim E, Lee SK, Lorenzo JA, Huang C, Melnick AM, Purdue PE, Goldring SR, Ivashkiv LB. Negative regulation of osteoclast precursor differentiation by CD11b and β2 integrin-B-cell lymphoma 6 signaling. Journal of Bone and Mineral Research. 2013;28:135–149. doi: 10.1002/jbmr.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, Boehnke M, Abecasis GR, Willer CJ, Frishman D. LocusZoom: Regional visualization of genome-wide association scan results. Bioinformatics. 2011;27:2336–2337. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schett G, Gravallese E. Bone erosion in rheumatoid arthritis: mechanisms, diagnosis and treatment. Nature reviews Rheumatology. 2012;8:656–664. doi: 10.1038/nrrheum.2012.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. The Journal of clinical investigation. 2013;123:3664–3671. doi: 10.1172/JCI67230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluis BVD, Groot AJ, Vermeulen J, Wall EVD, Diest PJV, Klomp LW, Vooijs M. COMMD1 Promotes pVHL and O 2 -Independent Proteolysis of HIF-1 a via HSP90/70. 2009;4 doi: 10.1371/journal.pone.0007332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluis BVD, Muller P, Duran K, Chen A, Groot AJ, Klomp LW, Liu PP, Sluis BVD, Muller P, Duran K, et al. Increased Activity of Hypoxia-Inducible Factor 1 Is Associated with Early Embryonic Lethality in Commd1 Null Mice. Increased Activity of Hypoxia-Inducible Factor 1 Is Associated with Early Embryonic Lethality in Commd1 Null Mice 2007 [Google Scholar]
- Sørensen MG, Henriksen K, Schaller S, Henriksen DB, Nielsen FC, Dziegiel MH, Karsdal MA. Characterization of osteoclasts derived from CD14+ monocytes isolated from peripheral blood. Journal of Bone and Mineral Metabolism. 2007;25:36–45. doi: 10.1007/s00774-006-0725-9. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Subramanian A, Tamayo P, Tamayo P, Mootha VK, Mootha VK, Mukherjee S, Mukherjee S, Ebert BL, Ebert BL, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terao C, Yamakawa N, Yano K, Markusse IM, Ikari K, Yoshida S, Furu M, Hashimoto M, Ito H, Fujii T, et al. Rheumatoid factor is associated with the distribution of hand joint destruction in rheumatoid arthritis. Arthritis and Rheumatology. 2015;67:3113–3123. doi: 10.1002/art.39306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Sluis B, Mao X, Zhai Y, Groot AJ, Vermeulen JF, van der Wall E, van Diest PJ, Hofker MH, Wijmenga C, Klomp LW, et al. COMMD1 disrupts HIF-1alpha/beta dimerization and inhibits human tumor cell invasion. J Clin Invest. 2010;120:2119–2130. doi: 10.1172/JCI40583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Sluis B, Muller P, Duran K, Chen A, Groot AJ, Klomp LW, Liu PP, Wijmenga C. Increased activity of hypoxia-inducible factor 1 is associated with early embryonic lethality in Commd1 null mice. Mol Cell Biol. 2007;27:4142–4156. doi: 10.1128/MCB.01932-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Deursen J, Heerschap A, Oerlemans F, Rultenbeek W, Jap P, ter Laak H, Wieringa B. Skeletal muscles of mice deficient in muscle creatine kinase lack burst activity. Cell. 1993;74:621–631. doi: 10.1016/0092-8674(93)90510-w. [DOI] [PubMed] [Google Scholar]
- Vonk WI, Bartuzi P, de Bie P, Kloosterhuis N, Wichers CG, Berger R, Haywood S, Klomp LW, Wijmenga C, van de Sluis B. Liver-specific Commd1 knockout mice are susceptible to hepatic copper accumulation. PLoS One. 2011;6:e29183. doi: 10.1371/journal.pone.0029183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Wang X, Yang M, Smith LC, Dechow PC, Sonoda J, Evans RM, Wan Y. PGC1beta mediates PPARgamma activation of osteoclastogenesis and rosiglitazone-induced bone loss. Cell metabolism. 2010;11:503–516. doi: 10.1016/j.cmet.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westra HJ, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J, Christiansen MW, Fairfax BP, Schramm K, Powell JE, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nature genetics. 2013;45:1238–1243. doi: 10.1038/ng.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer. 2011;11:393–410. doi: 10.1038/nrc3064. [DOI] [PubMed] [Google Scholar]
- Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nature reviews Cancer. 2008;8:851–864. doi: 10.1038/nrc2501. [DOI] [PubMed] [Google Scholar]
- Wyss M, Kaddurah-Daouk R. Creatine and Creatinine Metabolism. Physiological reviews. 2000:1107–1213. doi: 10.1152/physrev.2000.80.3.1107. [DOI] [PubMed] [Google Scholar]
- Yao Z, Li P, Zhang Q, Schwarz EM, Keng P, Arbini A, Boyce BF, Xing L. Tumor necrosis factor-α increases circulating osteoclast precursor numbers by promoting their proliferation and differentiation in the bone marrow through up-regulation of c-Fms expression. Journal of Biological Chemistry. 2006;281:11846–11855. doi: 10.1074/jbc.M512624200. [DOI] [PubMed] [Google Scholar]
- Zeng R, Faccio R, Novack DV. Alternative NF-κB Regulates RANKL-Induced Osteoclast Differentiation and Mitochondrial Biogenesis via Independent Mechanisms. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2015;30:2287–2299. doi: 10.1002/jbmr.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Ivashkiv LB. Negative regulation of osteoclastogenesis and bone resorption by cytokines and transcriptional repressors. Arthritis Research & Therapy. 2011;13:234. doi: 10.1186/ar3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.