

Abstract
The affinity of cytokine-receptor complexes on the cell surface is often poorly predictive of functional potency. To address this conundrum, we explored the inter-relationships of receptor binding to a wide range of downstream functional metrics for a prototypical cytokine, Interleukin-13 (IL-13), through structure-based engineering of agonists covering a spectrum of binding strengths for IL-13Rα1. Surprisingly, engineered IL-13 agonists spanning a broad affinity range exhibited similar potencies of STAT6 phosphorylation, while delays in STAT6 activation and nuclear translocation were only apparent for ligands with dramatically lower affinities. From this data, we developed a mechanistic model that quantitatively reproduced the kinetics of STAT6 phosphorylation for the entire spectrum of binding affinities. Receptor endocytosis plays a key role in buffering STAT6 phosphorylation potencies, while the lifetime of signaling complexes at the plasma membrane determines the long-term functional potency. The surprisingly complex inter-relationships between extracellular ligand binding and function highlight the importance of feedback mechanisms in modulating receptor responsiveness, and suggest new mechanism-based strategies to enhance the therapeutic efficacy of cytokine therapy.
INTRODUCTION
Cytokines exert their effects through receptor dimers on the cell surface that activate the JAK/STAT pathway (1–5). Although there are many variations on this theme, cytokine engagement generally occurs through a two site binding paradigm that orients the receptors into homo- or heterodimeric signaling complexes (6). Signaling does not appear to be an “all or none” phenomenon, and is influenced by parameters including the structure of the dimeric complex (7) as well as the interaction parameters between the cytokine and the receptors (5). For example, cytokine-receptor systems that utilize shared receptors to engage multiple cytokines can elicit differential cellular responses to different ligands. Examples of such functional receptor plasticity include the type I IFN family (8–10), the IL-2/IL-15 system (11–13), and the IL-4/IL-13 system (14–16). Moreover, viral cytokines that differentially activate the IL-10 receptor have been reported (17). Differential cellular responses by these cytokines have been linked to variations in the affinity and stability of ligand interaction with the receptor subunits (15, 18–22) and to cell surface receptor densities (23, 24), suggesting that the spatiotemporal dynamics of signaling complex formation plays a central role in regulating signaling specificity.
A notable feature of these systems is the poor correlation between receptor binding affinity and signal activation potencies (5, 9, 25). This phenomenon has recently been highlighted by engineering cytokines with increased receptor binding affinities and/or specificities (6). For example, an interferon-α2 mutant (IFNα2-YNS) binds the Interferon Receptor 1 (IFNAR1) more than 60-fold better than IFNα2 and exhibits a much stronger anti-tumor response (26). Yet, it activates the JAK/STAT pathway to the same extent as wild-type IFNα2 (26). More recently, interleukin-4 (IL-4) variants were engineered to bind either the type I (IL-4Rα/γc subunits) or the type II (IL-4Rα/IL-13Rα1 subunits) IL-4 receptors with high affinity and specificity (27). Although these mutants bound with very different affinities to their specific receptors, they exhibited similar signal activation potencies (27). These observations suggest that cells can mount a robust signaling response elicited by low affinity ligands, yet the mechanism remains unresolved. This issue has important practical implications for immune-cytokine therapy, which historically has been limited by dose-dependent off-target toxicity. A more granular understanding of the mechanisms governing cytokine potency at lower, less toxic doses, could potentially rescue such approaches. For example, recently low dose IL-2 has shown great promise as a therapeutic for cancer, diabetes and other autoimmune diseases (28–30). It is important to determine the parameters relating cytokine affinity to function in order to understand if low dose approaches that preserve efficacy but limit toxicity could be generalized to other cytokine systems.
Here we systematically explored the role of ligand-receptor complex affinity and stability for cytokine receptor signaling using IL-13 as a model system. IL-13 forms a Type II receptor complex with IL-13Rα1 and IL-4Rα receptor subunits, activates STAT6 and contributes to the regulation of the innate immune response by promoting dendritic cells and macrophages differentiation (3, 31–34). Deregulation of IL-13 leads to the development of asthma and fibrosis, making this cytokine a very attractive pharmacological target (32, 35, 36). Using yeast surface display, we isolated a series of IL-13 agonists with different IL-13Rα1 binding affinities, spanning a range of nearly 6 orders of magnitude. This broad bandwidth of binding affinities allowed us to study signal activation including receptor dimerization and endocytosis, STAT6 phosphorylation and nuclear translocation as well as long-term cellular responses. Based on correlations between binding constants and potencies of IL-13 agonists at different levels of signal transduction we developed a quantitative model matching the experimental data. Our results highlight a complex interplay between ligand binding kinetics and feedback by receptor endocytosis and begin to explain the apparent non-linear correlations between the extracellular cytokine-receptor binding parameters and functional output. The practical implications of these results is that the toxicity that is commonly seen in cytokine therapies could be a byproduct of dosing strategies that are much higher than necessary, given that substantially lower affinity variants retain full biological activity. With this understanding, it also supports concepts about engineering cytokines for cell-type selectivity through fine-tuning their affinity to differentially target cell populations based on expression levels of shared receptors.
RESULTS
Engineering IL-13 agonists with a range of binding affinities
IL-13 engages the Type II receptor first through binding to IL-13Rα1, followed by binding of the IL-13/IL-13Rα1 complex to IL-4Rα. To study the role that cytokine-receptor complex stability plays in signaling and function, we used yeast surface display to engineer a series of IL-13 agonists with a wide range of binding affinities to IL-13Rα1. IL-13 was displayed on the yeast surface and biotinylated ectodomains of IL-13Rα1 and IL-4Rα coupled to streptavidin-AlexaFluor 647 were used as selection reagents. We first confirmed that IL-13 displayed on the yeast surface was functional. Yeast-displayed IL-13 could bind IL-13Rα1 (Fig. 1A, second panel) and bound IL-4Rα only in the presence of IL-13Rα1 (Fig. 1A, third and fourth panels), in accordance with the previously described cooperativity exhibited by these two chains (15).
Figure 1. Structure-based engineering of IL-13 agonists.
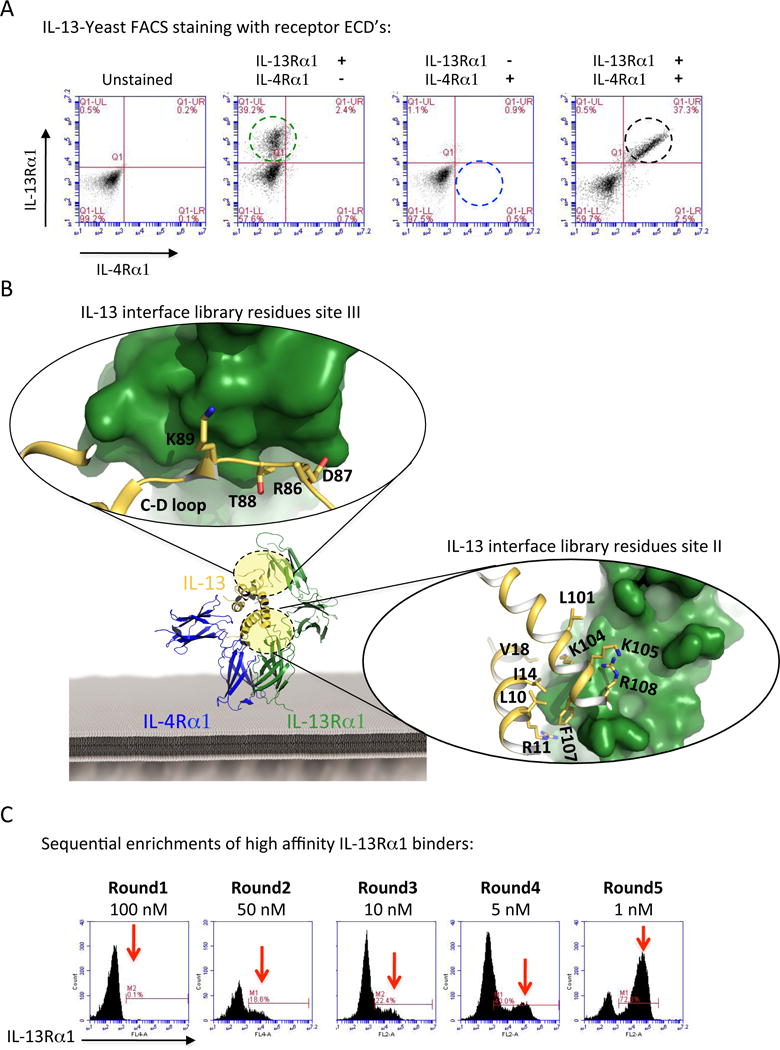
(A) Surface staining of IL-13-displaying yeast with biotinylated IL-13Rα1 and IL-4Rα ECDs. IL-13Rα1 binds IL-13 displayed on yeast independent of IL-4Rα (second panel from the left), whereas IL-4Rα on the other hand requires the presence of IL-13Rα1 to bind IL-13 recapitulating the cooperativity that has been observed previously 15 (right two panels). (B) Crystal structure of the IL-13 ternary complex (IL-13Rα1 in green, IL-4Rα in blue, and IL-13 in gold). Magnifications of the site II (A–D helixes) and site III (C–D loop) binding interfaces highlighting the amino acids on IL-13 involved in IL-13Rα1 binding are shown. (C) Sequential enrichment of IL-13 agonists. The site-specifically mutagenized library was selected against decreasing concentrations of IL-13Rα1 for five rounds. Clones were selected from each round to isolate variants with a range of IL-13Rα1 binding affinities.
The IL-13/IL-13Rα1 binding interface is composed of two binding sites, one to the receptor D2D3 domains called Site-2 and the other between the top of IL-13 and the receptor D1 domain called Site-3 (Fig. 1b). Helices A and D (site II) and the helix C and D connecting loop (loop C–D) (site III) on IL-13 contribute to the formation of these two binding interfaces. We created a Site-2/Site-3 focused library and introduced a ‘soft randomization’ on 13 amino acids in helices A and D and loop C–D (Fig. 1B). The theoretical diversity of this library was 108 variants. We carried out selections by enriching the library for IL-13Rα1 binders through five rounds of selection in which the IL-13Rα1 concentration was gradually decreased from 100 nM to 1 nM (Fig. 1C). Eighteen clones were selected based on their on-yeast binding titration curves for IL-13Rα1 (Fig. 2A), which were subsequently confirmed by surface plasmon resonance (SPR) experiments (Fig. 2B). A11, B2, A7, A6, A8, A5, IL-13. B4 and B6 mutants binding KD were calculated using the kinetic model; C10, C11, C12, C2, C3, C4, C7, C9 and D7 mutants binding KD were calculated using the steady-state equilibrium model due to their extremely fast on- and off- binding rates (fig. S1). These engineered agonists exhibited binding affinities ranging from 80-fold higher to 10000-fold lower binding to IL-13Rα1 compared to wild-type IL-13 (Fig. 2, B and C). The increase in binding affinity was mainly achieved by a decrease in the dissociation rate constant koff, i.e. by increasing the half-life of the complex, while the association rate constants kon were similar between the different IL-13 agonists. For IL-13 mutants with binding affinities in the higher nanomolar to micromolar regime, koff could not be quantified due to the limited time resolution of SPR. However, further increased koff roughly proportional to the change in KD can be assumed for these mutants.
Figure 2. Biophysical characterization of selected IL-13 agonists.
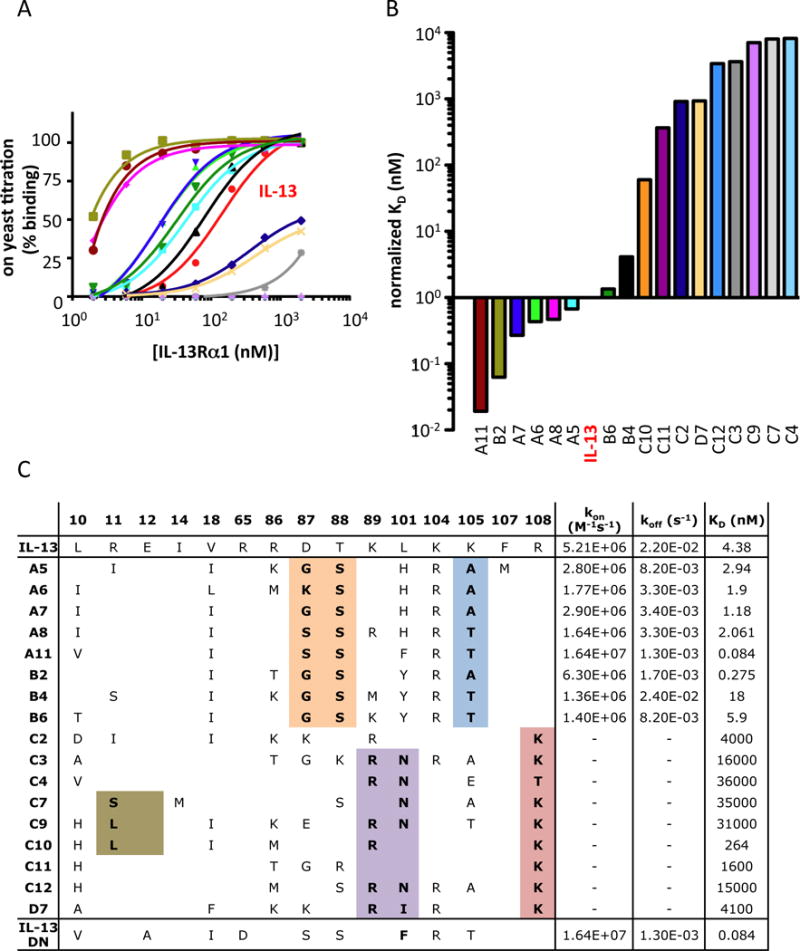
(A) Binding isotherms of the IL-13 agonists displayed on the surface of yeast. Yeast-displayed IL-13 agonists were incubated with the indicated concentrations of biotinylated IL-13Rα1 ECD, stained with Alexa-647-coupled Streptavidin, and assessed via flow cytometry. Sigmoidal curves were fitted using Prism software (GraphPad). (B) Normalized KD binding affinities of the recombinant IL-13 agonists determined via surface plasmon resonance (SPR). The IL-13 KD was fixed to one and other agonists were normalized accordingly. (C) Summary of the amino acids mutated in the IL-13 agonists as well as the kon, koff and KD binding parameters values obtained from SPR experiments.
Sequencing of these IL-13 agonists showed that while the loss of IL-13Rα1 binding affinity occurred by mutations in either the Site-2 or Site-3 binding interface, gain of affinity mainly occurred through mutations in Site-3 (Fig. 2C). IL-13 agonists with increased binding affinity for IL-13Rα1 share common mutations in Site-3, i.e. D87/G,S; T88/S, suggesting a common mechanism for affinity maturation.
Structure of IL-13 high affinity agonist complexed with IL-13Rα 1 and IL-4Rα
The overall receptor-ligand binding geometry of the IL-13 agonists is an important factor to consider when interpreting differences in signal activation (7, 37). To address this, we solved the structure of the ternary complex formed by the high affinity IL-13 agonist A11 and the IL-13Rα1 and IL-4Rα receptors chains to 3.0 Å resolution (Fig. 3A). Superposition of the ternary complexes formed by A11 and IL-13 showed no major perturbations in cytokine-receptor architecture (RMSD: 0.738) (Fig. 3B). In the IL-13 A11-IL-13Rα1 binding interface, side chain densities were clear for both helix D and the C–D loop of A11 (fig. S2). The Site-2 binding interface on A11 closely resembles that of IL-13 wt, with amino acids Phe107 and Arg108 predominantly mediating binding (Fig. 3, C and D). The Site-3 binding interfaces, however, differ significantly between the IL-13 and A11 ternary complexes. The gain in binding affinity achieved by the A11 agonist seems to occur through a new network of interactions created in the Site-3 binding interface (Fig. 3, C and D). Mutation of the amino acids Asp87 and Thr88 to the smaller amino acids Gly or Ser allows Trp65 to assume a different rotamer on IL-13Rα1, enabling this residue to form extensive contacts with Trp35 and Arg86 on IL-13 (Fig. 3D). Overall these structural data suggest that the A11 agonist has increased its binding affinity by optimizing the Site-3 rather than the Site-2 binding interface network. Since the IL-13 agonists are highly related sequences, it is reasonable to assume that they recruit IL-13Rα1 and IL-4Rα with similar geometries and therefore any signaling changes observed during our study can most likely be attributed to changes in binding affinities and complex stabilities.
Figure 3. Structural characterization of the high affinity IL-13 A11 agonist.
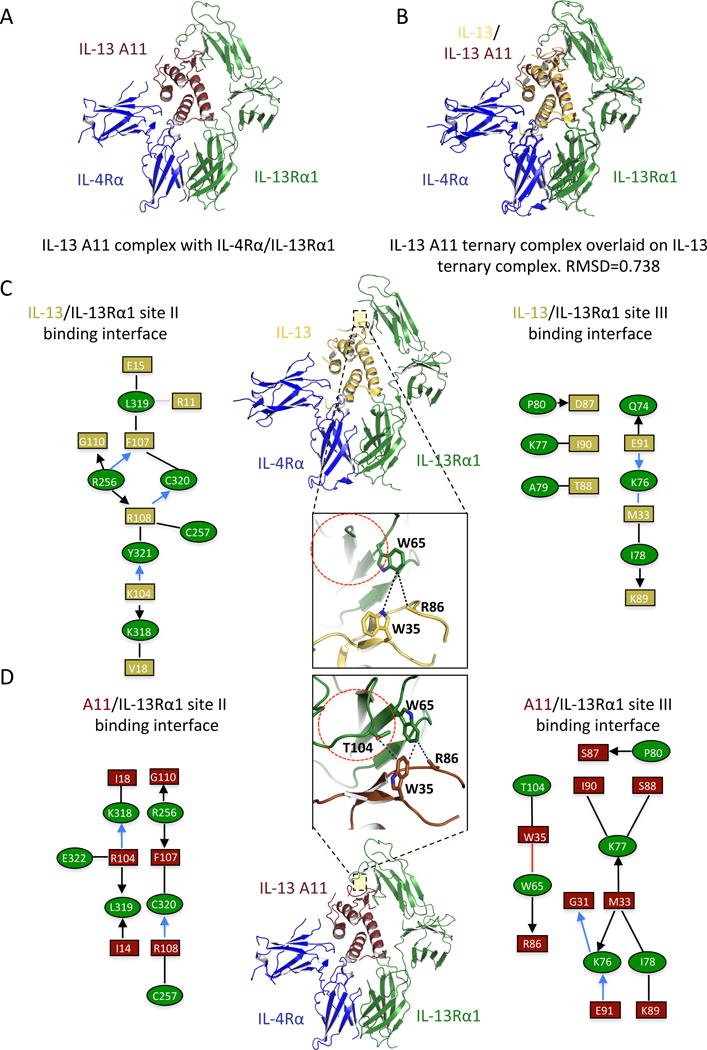
(A) Crystal structure of the IL-13 agonist A11 (brown) in complex with IL-13Rα1 (green) and IL-4Rα (blue). (B) Overlay of the IL-13 wt and IL-13 A11 ternary complex crystal structures. Note that IL-13 A11 engages the two receptor chains with identical geometry to that of IL-13 wt (RMSD=0.738). (C–D) Schematic representation of ligand-receptor contact interaction of IL-13Rα1 with IL-13 (top half) versus IL-13 A11 (bottom half). Black lines and arrows represent van der Waals interactions, blue lines represent hydrogen bonds and red lines designate aromatic amino acid interactions.
Receptor dimerization and spatiotemporal dynamics
Initiation of signal activation by IL-13 is known to require simultaneous interaction of the ligand with IL-13Rα1 and IL-4Rα, but the spatial and temporal organization of these receptor subunits in the plasma membrane before and after ligand binding still remains ill-defined. In order to interrogate the effects of ligand binding affinity on the spatiotemporal dynamics of the IL-13 signaling complex at physiological receptor concentrations on the cell surface, we devised quantitative ligand binding and receptor dimerization assays based on single-molecule fluorescence imaging (fig. S3). Using TIRF microscopy, we probed binding of IL-13 A11, which was site-specifically labeled with the photostable fluorophore DY647 (DY647IL-13 A11) with a degree of labeling >90%. Specific binding of DY647IL-13 A11 to endogenous receptor at the surface of HeLa cells could be detected at a concentration of 2 nM, which is sufficient to saturate all IL-13Rα1 (Video S1 and fig. S3A). At moderate excitation power, individual diffraction-limited signals were observed, which were randomly distributed and continuously diffusing in the membrane (Video S1). Single-step photobleaching at elevated laser power confirmed that each signal corresponded to individual IL-13 A11 molecules rather than clusters (Video S1, Video S2 and fig. S3B). An average density of 0.14 IL-13Rα1 molecules/μm2 was determined from binding experiments with DY647IL-13 A11, while a significant lower level of 0.07 IL-4Rα molecules/μm2 was obtained for binding experiments with DY647IL-4 (fig. S3C).
For quantifying receptor dimerization at these relatively low cell surface expression levels we employed dual color single molecule imaging. HeLa cells were transfected with IL-13Rα1 and IL-4Rα extracellular domains fused to the HaloTag and the SNAPf-tag, respectively (Fig. 4A). These tags were employed for selective post-translational labeling with tetramethylrhodamine (TMR) and DY647, respectively, which ensure high brightness and photostability. Integrity of ligand recognition by HaloTag-IL-13Rα1 and SNAPf-IL-4Rα with respect to complex stability was confirmed by binding experiments with DY647IL-13 wt and DY647IL-4, respectively, and chasing with unlabeled ligand (fig. S3, D and E). Using dual-color TIRF microscopy, individual IL-13Rα1 and IL-4Rα receptor subunits at the plasma membrane could be detected and followed in real-time for extended periods (Video S3). Single step photobleaching at elevated laser power confirmed random distribution of IL-13Rα1 and IL-4Rα in the plasma membrane rather than co-organization into clusters (Video S4). Individual receptor subunits were localized beyond the diffraction limit with an average precision of 20 nm, allowing for tracking and co-localization with high spatial resolution. Single molecule co-locomotion analysis revealed that significant receptor dimerization could only be discerned following stimulation with the ligand (fig. S3G), in accord with a two-step ternary complex assembly mechanism. A representative co-trajectory corresponding to an individual ternary complex is shown in Fig. 4, A to C and Video S5, with IL-13Rα1 and IL-4Rα exhibiting co-locomotion for more than 500 frames (≈16 s).
Figure 4. Assembly and dynamics of the ternary complexes formed by IL-13 agonists.
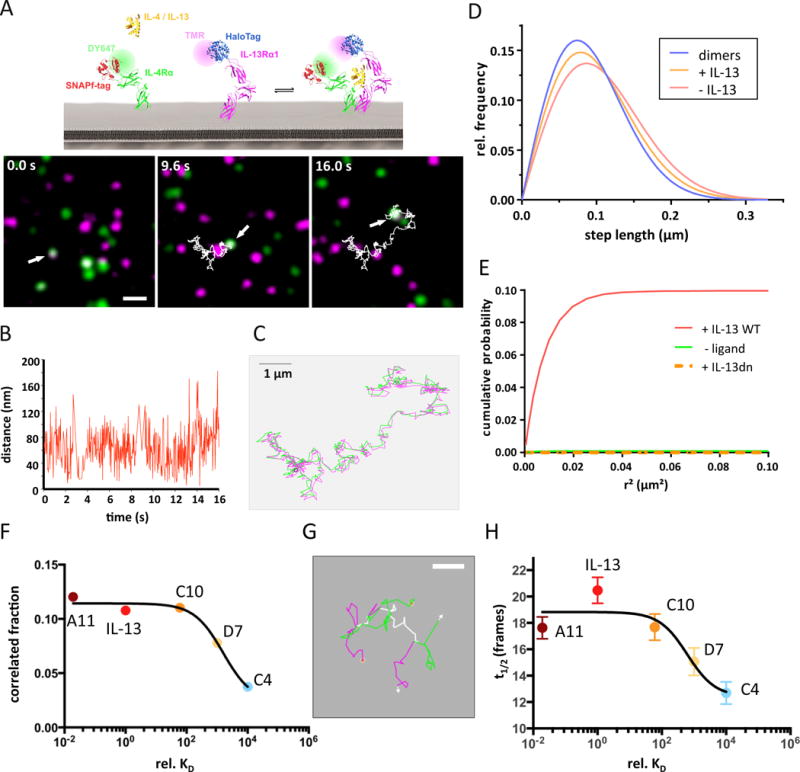
(A–C) IL-13 receptor dimerization detected by dual color single molecule imaging and co-locomotion-analysis. Images identifying an individual IL-13Rα1/IL-4Rα dimer in the presence of 200 nM IL-13 at different time points of a 500 frame (16 s) movie (A), the distance between the two molecules in each frame (B) and an overlay of the individual trajectories of IL-13Rα1 (magenta) and IL-4Rα (green) (C) are presented. Scale bars: 1μm in panels A and C. (D) Diffusion properties represented as step length distribution obtained for IL-13Rα1 in the absence (red) and presence (orange) of IL-13. For comparison, the step length distribution obtained for receptor dimers identified by co-locomotion analysis is shown (blue). (E) Cumulative probability of detecting an IL-13Rα1 molecule in the vicinity of an IL-4Rα molecule as a function of the squared distance, corrected for the statistical probability, in the presence and absence of IL-13 wt. For comparison, the cumulative correlation probability obtained in the presence of IL-13 DN is shown. (F) Correlation fraction for the IL-13Rα1 and IL-4Rα receptor subunit as a function of IL-13 agonist affinity. (G) Assembly and dissociation of an individual IL-13Rα1/IL-4Rα dimer in presence of agonist D7. Overlaying trajectories are shown in white. Scale bar 0.5 μm. (H) Average lifetime of the IL-13Rα1/IL-4Rα dimer as a function of agonist affinity.
For all IL-13 agonists that were subjected to these studies (wt, A11, C10, D7 and C4), substantial ligand-induced receptor co-locomotion was confirmed (fig. S4). In contrast, no co-locomotion of IL-13Rα1 and IL-4Rα was detectable for a dominant negative mutant of IL-13 (IL-13DN), which binds to IL-13Rα1 with high affinity, but not to IL-4Rα (Fig. 2C and fig. S3G). Accordingly, stimulation with IL-13DN did not yield STAT6 phosphorylation (fig. S6B). The local diffusion properties were quantified by a step-length distribution analysis (fig. S5), revealing a small, yet significant decrease in receptor mobility upon ternary complex formation (Fig. 4D), which could be caused by increased friction due to the increased diameter of the dimerized receptor or by further interactions of the receptor due to downstream signaling. A deconvolution of the step-length distribution observed for the receptor subunits after binding of IL-13 suggested that ≈50% of the available IL-13Rα1 was recruited into ternary complexes at saturating IL-13 concentrations.
Similar dimerization efficiencies over a broad spectrum of receptor affinities
To quantify the spatial co-organization of IL-13Rα1 and IL-4Rα in the absence and presence of ligand we employed single molecule image analysis by particle image cross-correlation spectroscopy (PICCS) (35) as schematically illustrated in fig. S3F. For this purpose, cells expressing a similar density of IL-13Rα1 and IL-4Rα at a level of ~1 molecule/μm2 were chosen. No significant cross-correlation was observed in the absence of agonist or in the presence of IL-13DN (correlated fraction α ≈ 0), excluding transient receptor pre-organization prior to ligand binding (Fig. 4E). Upon stimulation with IL-13 wt at saturating concentration, cross-correlation was detectable with a correlation fraction α ≈ 0.1 (Fig. 4E), confirming ligand-induced dimerization of IL-13Rα1 and IL-4Rα. Based on control experiments with a transmembrane helix fused to both the HaloTag and the SNAPf-tag, the correlation fraction α observed after stimulation is consistent with ~50% of IL-13Rα1 being in complex with IL-4Rα, as estimated from the diffusion analysis. This highly efficient dimerization by IL-13 is in stark contrast to a recent study proposing very inefficient dimerization of IL-13Rα1 and IL-4Rα at the plasma membrane (39).
Taking the correlation fraction α as a measure for receptor dimerization, we studied the effects of stabilizing and destabilizing the interaction of IL-13 with IL-13Rα1 (Fig. 4F). Intriguingly, the receptor dimerization efficiency did not change for either the complex-stabilizing A11 or moderately destabilizing C10 agonists, yet dropped significantly for the low affinity binders D7 and C4 agonists, probably because saturated receptor occupancy was not achieved. These results suggested that recruitment of IL-13Rα1 and IL-4Rα by IL-13 on the plasma membrane is very robust across a wide affinity range, tolerating a decrease in affinity by almost two orders of magnitude. Since the engineered IL-13 agonists bind IL-13Rα1 with dramatically altered stabilities, we probed the lifetime of individual complexes, which has been conjectured to play a critical role for functional plasticity of other cytokine receptors. Indeed, formation and dissociation of individual complexes could be observed (Video S6 and Fig. 4G), which is in line with reversible ligand binding to the cell surface receptor. While the length of co-trajectories is limited by tracking fidelity and photobleaching, significant differences in complex lifetime could be observed for the low-affinity IL-13 agonists D7 and C4 (Fig. 4H). The mean lifetime of complexes paralleled the increased dissociation kinetics of these IL-13 agonists as measured by surface plasmon resonance, corroborating the relevance of reversible ligand binding for the spatiotemporal dynamics of ternary complexes. These data highlight the potential to control the signal delivered to a responsive cell by two distinct mechanisms: (1) by regulating the maximum number of complexes formed on the cell surface by a given cytokine, which is determined by the receptor density but independent of the ligand binding affinity; and (2) by regulating the lifetime of individual complexes, which heavily relies on the koff of the ligand that is inducing signal activation.
Kinetics of STAT activation correlates with cytokine-receptor binding stability
We next studied the signaling signatures exhibited by the different IL-13 agonists that we characterized by affinity measurements, structure and single molecule microscopy. We measured pSTAT6 dose/response as well as kinetics of signal activation in the IL-13-responsive cell line A549. A549 cells were stimulated for 15 min with the indicated doses of IL-13 or IL-13 agonists and the levels of pSTAT6 were analyzed by flow cytometry. We observed two groups of agonists based on their abilities to activate STAT6. One group (agonists C2, C3, C4, C7, C9, C11, C12 and D7) activated STAT6 to a significantly lower level than IL-13 (Fig. 5A). The other group (agonists A11, B2, A5, A6, A7, A8, B4, B6 and C10) activated STAT6 with very similar potencies and very similar to IL-13 (Fig. 5A). These observations were confirmed by western blot studies (fig. S6A). Interestingly, the range of IL-13Rα1 binding affinities among the second group varied from 80 pM (A11) to 100 nM (C10), suggesting the existence of a large ‘buffering region’ that allows A549 cells to respond very similarly to agonists exhibiting a wide range of binding affinities. Indeed, about a 1000-fold decrease in binding affinity compared to IL-13 was required to significantly decrease the levels of agonist-induced pSTAT6. When the IL-13Rα1 binding affinities of the IL-13 agonists were plotted against their STAT6 activation potencies, we observe a region comprising agonists from A11 to C10 in which the activation of STAT6 did not differ drastically (Fig. 5B).
Figure 5. STAT6 activation profile in response to IL-13 agonist.
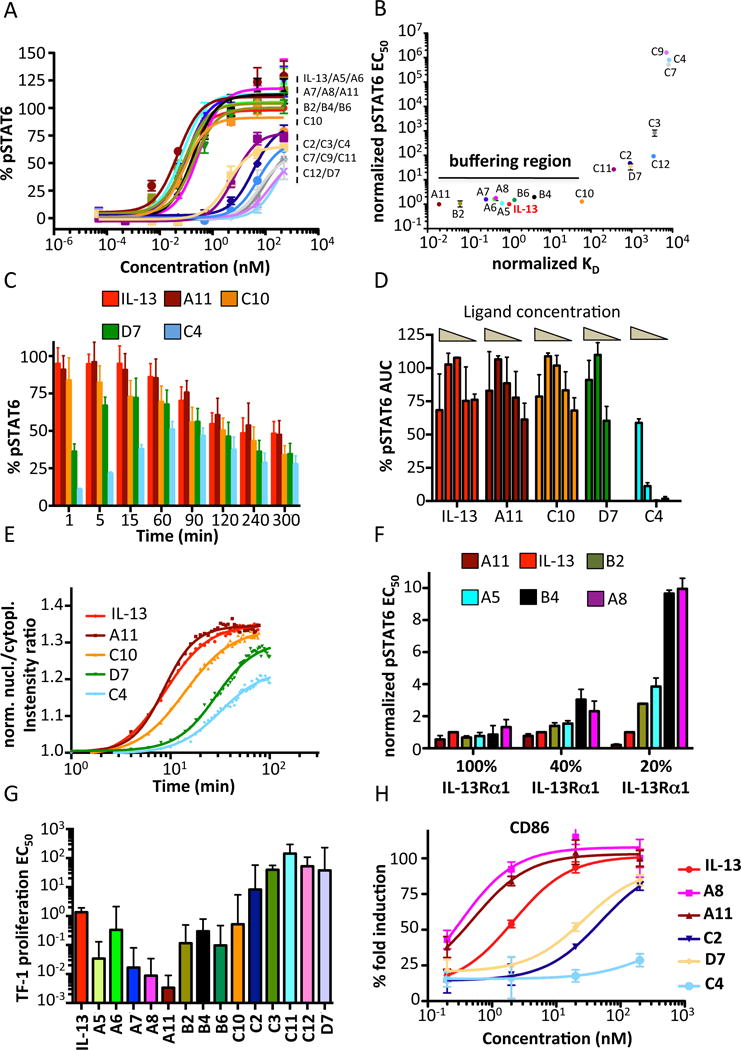
(A) A549 cells were stimulated with the indicated doses of IL-13 wt or IL-13 agonists and the levels of pSTAT6 were analyzed by flow cytometry, using anti-pSTAT6-specific antibodies coupled to fluorescent dyes. Sigmoidal curves were fitted with Prism software (GraphPad) (B) Dot plot graph in which the normalized pSTAT6 EC50 values are plotted against the normalized KD binding values for each IL-13 agonist. IL-13 pSTAT6 EC50 and KD values were set to one and agonist parameter values were normalized accordingly. Agonists binding within 100-fold of IL-13 wt in either direction lead to similar profiles of STAT6 activation, generating the denoted buffering region. (C) A549 cells were stimulated with 200 nM of IL-13 wt or the indicated IL-13 agonists and the kinetics of STAT6 phosphorylation were analyzed by flow cytometry. (D) Total levels of pSTAT6, calculated by measuring Area Under the Curve (AUC) from the kinetics studies shown in Figure S6. Decrease in ligand concentration only marginal affect total amount of pSTAT6 activated. (E) HeLa cells were stimulated for the indicated time-lengths with 200 nM of IL-13 or the indicated agonists and the translocation of mEGFP-STAT6 to the nucleus was measured via fluorescence microscopy. (F) A549 cells were transfected with different concentrations of IL-13Rα1 siRNA to obtain a range of IL-13Rα1 silencing. Cells were then stimulated with IL-13 wt or engineered agonists and pSTAT6 levels were quantified by flow cytometry. (G) TF-1 EC50 proliferation values obtained from fitting sigmoidal profiles to the dose-response curves shown in Fig. S10b. (H) Upregulation of CD86 in monocytes stimulated with the indicated doses of IL-13 wt and IL-13 mutants.
Since changes in binding affinity affect the lifetime of cytokine-receptor complexes formed by different IL-13 agonists, we next asked whether the kinetics of STAT6 activation would be modulated by these changes in half-life. We performed a series of STAT6 activation kinetics studies in A549 cells where the time of onset and decay of phosphorylated STAT6 is measured. As shown in Fig. 5C, agonists that bind very weakly to IL-13Rα1 (D7 and C4) exhibited a delay in STAT6 activation. C4 agonist, which binds IL-13Rα1 8000 times weaker than IL-13, promotes 50% of STAT6 activation when compared to IL-13 after one hour of stimulation. This delayed kinetic of STAT6 activation exhibited by IL-13 D7 and C4 agonists, was also observed for IL-13 wt, A11 and C10 agonists when sub-saturating doses of ligand were used (fig. S6, B to G). Interestingly, the number of STAT6 molecules activated per unit of time, measured as area under the curve (AUC), by all the IL-13 agonist tested was minimally altered by changes in STAT6 activation kinetics and ligand concentrations (Fig. 5D), suggesting that cells have evolved to activate a finite number of STAT molecules.
A functional consequence of STAT6 activation is its translocation to the nucleus where it carries out its transcriptional regulation functions. We next studied how complex stability impacts the kinetics of STAT6 nuclear translocation in HeLa cells stably transfected with mEGFP-STAT6 (fig. S7A). Similarly to the STAT6 activation kinetics, the STAT6 nuclear translocation was further delayed for agonists that weakly bound IL-13Rα1 (Fig. 5E), and a proportionally decreased amplitude corresponding to the maximum pSTAT6 signal was observed. Notably, the STAT6 nuclear translocation induced by the C10 agonist is slower than that induced by IL-13, yet the two ligands exhibited very similar pSTAT6 intensities. Overall these data indicate that while the amount of STAT6 activated by IL-13 is very resistant to changes in binding affinity, the duration of STAT6 activation is more sensitive to changes in affinity and closely correlates with the stability of the ternary complex.
It was previously shown in the Interferon cytokine system that surface receptor density contributes to signal activation potency (23). Thus, we next studied whether changes in the IL-13Rα1 surface density would modulate pSTAT6 activation by the different IL-13 agonists. To this end, we compared the STAT6 activation profile induced by IL-13 agonists in cells in which IL-13Rα1 levels were decreased using siRNA. As shown in Fig. 5F, while minimal differences in the STAT6 activation profile between agonists were observed in cells expressing endogenous levels of IL-13Rα1, the same was not true when IL-13Rα1 levels dropped below 50%. When IL-13Rα1 levels were reduced to 40% of the endogenous level, IL-13 agonists B4 and A8, which bound more weakly to IL-13Rα1, activated STAT6 less potently than the other IL-13 agonists. These differences were further increased when the levels of receptor were reduced by 80%. These data clearly show a close correlation between binding affinities, surface receptor levels, and signal activation.
On- and off- ligand binding rates differentially modulate STAT6 activation and biological potencies
We next studied the impact of complex stability on biological activities induced by IL-13. The TF-1 cell system is commonly used to assess the potency of a large number of cytokines including IL-13. In response to IL-13, these cells proliferate in a dose-dependent manner, readily allowing for the evaluation of IL-13 agonist potency (40). TF-1 cells were stimulated with the indicated doses of various IL-13 agonists for 96 h and cell abundance was estimated by flow cytometry. Surprisingly, we observed a broad range of proliferation potencies, in contrast to the highly similar STAT6 activation profiles induced by different IL-13 agonists (Fig. 5, A and G). While A11 was virtually identical to IL-13 wt in activating STAT6 (Fig. 5A), it promoted a more potent proliferation of TF-1 cells (Fig. 5G and fig. S7B). The levels of cell proliferation induced by the different IL-13 agonists closely matched their binding affinities rather than their respective abilities to activate STAT6.
To further expand on the biological activities of IL-13 agonists, we analyzed the differentiation of monocytes into dendritic cells in vitro. We stimulated monocytes with IL-13 or our engineered agonists for seven days and analyzed the levels of CD86 and CD209, classical dendritic cells markers, by flow cytometry. As shown in (Fig. 5H and fig. S7, C to E), the potency of the different IL-13 agonists correlates with their binding affinities (CD86 R2=0.91; CD209 R2=0.92), consistent with our findings for the TF-1 proliferation response. Here again we observe that A11 is more potent than IL-13 in inducing a late response, i.e. DC differentiation, despite eliciting very similar profiles of STAT6 activation (fig. S7F). We could not correlate DC differentiation EC50 to the kon and koff constants because we could not accurately estimate these kinetic parameters for several of the weakly binding agonists tested in this experiment (C4, C2 and D7). The lack of correlation between complex stability and signaling potencies exhibited by the IL-13 agonists could not be ascribed to receptor levels in the cells used for the experiments or differential receptor downregulation induced by the different IL-13 mutants (fig. S8). Next we asked whether other parameters involved in the IL-13/IL-13Rα1 binding affinity were contributing to the observed bioactivities. The KD binding parameter of a given ligand derives from two kinetic parameters: kon, which reflects the rate at which the ligand binds to its cognate receptor, and koff, which reflects the length of time the ligand remains bound to the receptor. Therefore we studied how these two components correlated with the activation of membrane proximal signaling events, i.e. STAT6 activation, and with the potency of bioactivities induced by IL-13, i.e. TF-1 proliferation. Figure 6 presents a series of linear correlation plots in which the EC50 values of pSTAT6 and of TF-1 proliferation induced by the different IL-13 agonists are plotted against the kon, koff or KD binding values of these mutants. Interestingly, the levels of STAT6 activation induced by the IL-13 agonists correlated reasonably well with their kon binding values (R2=0.52) but poorly with their koff binding values (R2=0.021) (Fig. 6, B and C). For example, while the KD binding constant of IL-13 wt is approximately 80-fold lower than that of the A11 agonist, their kon binding constants are very similar (Fig. 2C). In agreement with their similar kon binding values, these two cytokines elicit very similar levels of STAT6 activation. In contrast, the potency of TF-1 proliferation induced by the different IL-13 agonists correlated more closely with their koff binding values (R2=0.54) and very poorly with their kon binding values (R2=2.8×10−5) (Fig. 6, E and F).
Figure 6. Correlation of receptor binding kinetics with signaling and bioactivities induced by IL-13 agonists.
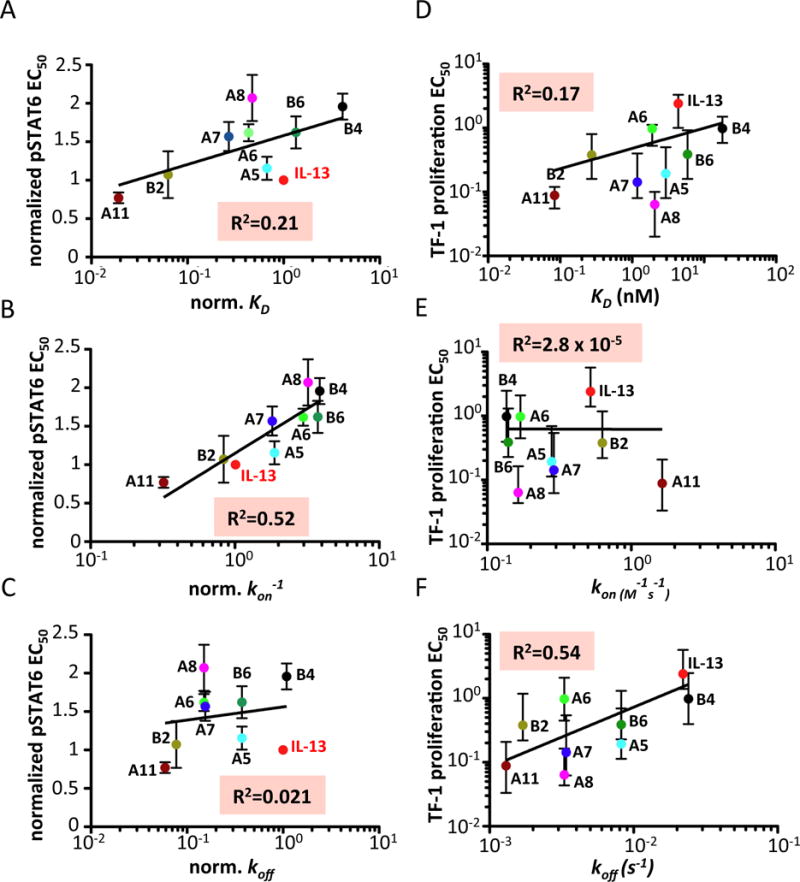
(A–C) Correlation of the pSTAT EC50 induced by IL-13 wt or the indicated IL-13 agonists with their binding parameters KD (top panel), kon (middle panel) and koff (bottom panel). (D–F) Correlation of the TF-1 proliferation EC50 induced by IL-13 wt or the indicated IL-13 agonists with their binding parameters KD (top panel), kon (middle panel) and koff (bottom panel).
Endocytosis specifically enhances activity of low-affinity agonists
The correlation of kinetic binding parameters suggested that downstream cellular processes are involved in regulating STAT6 phosphorylation activity. Since endocytosis was recently proposed to play an important role for IL-4 signaling (39), we hypothesized that the surprisingly high activity of low-affinity agonists may be caused by endocytosis of signaling complexes. We confirmed efficient endosomal uptake of both IL-13 wt and D7 bound to endogenous receptors in HeLa cells (Fig. 7A). We also observed co-localization of HaloTag-IL-13Rα1 and SNAPf-IL-4Rα in intracellular vesicular structures, corroborating that entire signaling complexes are endocytosed (fig. S9A). Endocytosis of IL-13 was considerably reduced upon addition of EHT1864 (Fig. 7B), an inhibitor of Rac family GTPases, which have previously been implicated in IL-2 receptor endocytosis. At the highest concentration of EHT1864, which could be applied without loss of receptor cell surface expression (100 μM), 2-fold reduced endocytosis was observed as quantified from the intensity of intracellular endosomes and the number of ligands bound to the cell surface (Fig. 7C). The functional role of endocytosis was investigated by time-lapse STAT6 phosphorylation assays in the absence and in presence of the endocytosis inhibitor (Fig. 7D). A clear correlation between agonist binding affinity and the role of endocytosis was observed: while no significant reduction in STAT6 phosphorylation kinetics was observed for A11 in the presence of EHT1864, a retardation of the STAT6 phosphorylation was observed for D7 which is a low affinity binder. Importantly, EHT1864 did not affect the pSTAT6 activation EC50 induced by any of the IL-13 mutants, suggesting a specific effect in the kinetics of STAT6 activation (fig. S9B).
Figure 7. Role of receptor endocytosis.
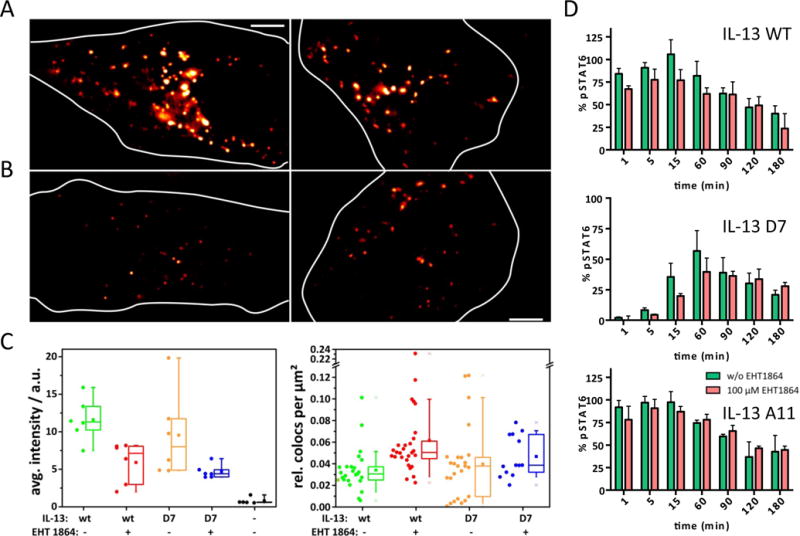
(A) Epifluorescence images showing endocytosis of fluorescent IL-13 wt and D7 bound to the endogenous IL-13 receptor in HeLa cells. (B) Endocytosis of IL-13 wt and D7 bound to the endogenous IL-13 receptor after treatment with 100 μM EHT 1864. (C) Left: quantification of IL-13 wt and D7 in endosomal compartments (left) and at the plasma membrane (right) in absence and in presence of EHT 1864. (d) Kinetics of STAT6 phosphorylation in absence and in presence of EHT1864.
A mechanistic model correlating cytokine binding to functional output
These results identify endocytosis as a key determinant of STAT6 phosphorylation. Based on these results we conceived a simple steady-state model for receptor activation comprising reversible cell surface binding followed by irreversible uptake of functional signaling complexes into endosomes followed by sorting into degradation and recycling pathways (Fig. 8A). Importantly, this model assumes that signaling complexes remain active with respect to STAT6 phosphorylation within endosomes for some time. As a consequence of endosomal uptake, the ligand is considered to remain bound to the receptor because of the high local concentration of individual ligand molecules in the very small volume (0.1–1 mM). Based on this model, we simulated the kinetics of pSTAT6 formation for different IL-13 agonists (cf. Fig. 5C) assuming that the measured concentration of pSTAT6 is proportional to the total number of active signaling complexes in the plasma membrane and in endosomes. Using the experimental binding constants of the different IL-13 agonists, global, realistic rate constants of endocytosis, degradation and recycling were obtained by fitting the experimental data. This simple model could reproduce the observed pattern of STAT6 phosphorylation kinetics as well as the selective effect of inhibited endocytosis on the activity of low-affinity agonists remarkably well (Fig. 8, B and C, and fig. S10A). Strikingly, the correlation of STAT6 phosphorylation intensity with kon (Fig. 6b) was confirmed by the model (Fig. 8D), as well as the characteristic STAT6 phosphorylation kinetics at decreased concentrations of IL-13 agonists (Fig. 8, E and F and fig. S10B). These results highlight that the early signaling events are strongly regulated by an interplay of ligand binding and receptor endocytosis kinetics.
Figure 8. Quantitative model of IL-13 signaling.
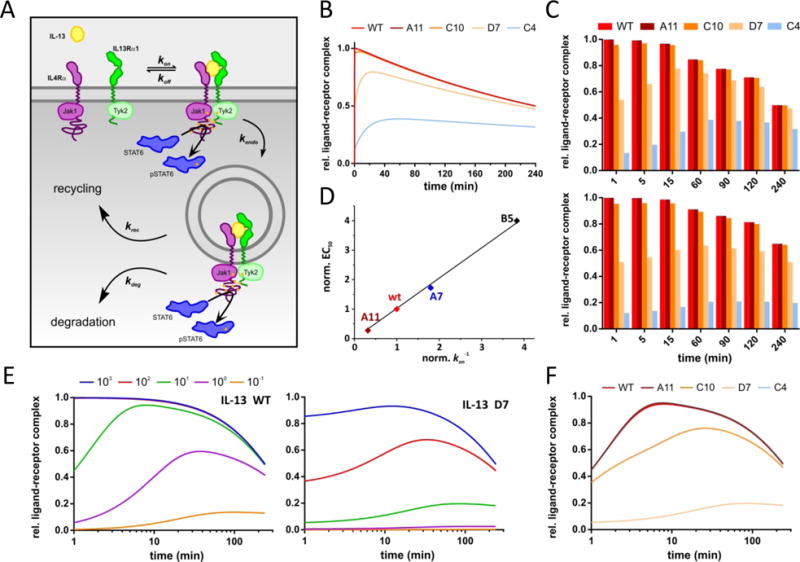
(A) Steady-state model of ligand binding, receptor endocytosis and further endocytic trafficking used for simulations of STAT6 phosphorylation kinetics and dose-response curves. (B) Kinetics of STAT6 phosphorylation obtained for different IL-13 agonists. (C) Comparison of simulated STAT6 phosphorylation in absence (top) and in presence (bottom) of an endocytosis inhibitor. (D) EC50 values from dose-response curves simulated for different IL-13 agonists with different kon. (E) Kinetics of STAT6 phosphorylation obtained for different IL-13 wt concentrations (given in nanomolar). (F) Comparison of simulated STAT6 phosphorylation kinetics for different IL-13 agonists at a concentration of 10 nM.
DISCUSSION
There are several instances of cytokines (e.g. type I IFN, IL-4/IL-13, IL-10/vIL-10, among others) sharing common cell surface receptors and yet eliciting differential activities (5, 9, 25, 32). During the past decade, evidence is accumulating that the stability of the cytokine-receptor complexes formed by the different members of these families correlates with their potency in activating long term cellular responses (e.g. proliferation, differentiation, apoptosis, etc.) (19, 22, 27, 41). Long-lived complexes can promote more potent long-term responses than short-lived complexes (19, 27, 41). However, the same correlation does not hold true for early, membrane-proximal signaling events, where low and high binding affinity ligands activate downstream signaling to the same extent (19, 27). This apparent disconnect between complex stability and signal activation has raised the question: how do cells integrate different receptor binding affinities into similar STAT activation while preserving functional diversity? Our data support a model where fine-tuning of cytokine-cytokine receptor binding kinetics allows cytokines to exhibit robust signal activation at a wide range of binding affinities and yet preserve differential responses. We found that kon and koff binding constants correlate with distinct functions in determining the signaling output of a given cytokine: kon correlates with the amount of STAT that is activated by controlling the number of ligand-receptor complexes formed in the plasma membrane, whereas koff correlates with the kinetics of STAT activation, through the modulation of ligand-receptor complex half-life as a function of endocytosis. A striking observation emerging from our data is the presence of a region in the receptor binding affinity space, herein referred as the “signal buffering region”, where levels of STAT6 activation appear unaffected by large changes in binding affinity: ligands with 100-fold increased (A11) and 100-fold decreased (C10) binding affinities for IL-13Rα1 subunit compared to IL-13 elicited comparable STAT6 activation levels than the wild type cytokine. Similar observations have been made in other systems where affinity maturation of the cognate ligands did not yield stronger signal activation (19, 26, 27, 42). Prominently, the observed 10.000-fold difference in binding affinity between the IL-13 variants resulted from changes in their koff. All IL-13 variants exhibit comparable kon, indicating that STAT activation remains largely unaffected by changes in koff above a certain threshold. Moreover, all IL-13 variants yielded similar levels of surface complex formation, arguing in favor of kon, and not koff, as the main factor determining the number of complexes formed by a given cytokine and generating the signaling buffering region that we observe in our experimental data. A caveat to this interpretation is that since our IL-13 agonists have rather similar kon values, we could not test the impact that large changes in kon could have in complex formation and signal activation. More systematic engineering of kon by manipulating the electrostatic potential (43) will be required to explore whether a further increase in STAT activation potency could be achieved by increasing complex association. Collectively, our data suggest that cytokines can effectively signal given a sufficient kon value, largely independent of affinity. In nature, cytokine signaling would benefit from loose affinity constraints by allowing it to sense and respond to a wide range of binding affinities and complex stabilities.
How cells translate virtually identical STAT activation profiles into different levels of cell proliferation, differentiation or cell fate still remains unclear. Our results show that IL-13 agonists with slower koff persist longer as receptor complexes, promote faster kinetics of signal activation, and induce more potent effector functions (i.e., higher levels of proliferation and DC differentiation) than low affinity ligands, suggesting that cytokines fine-tune their late responses by controlling the half-life of their complexes, and downstream signal activation kinetics. In agreement with this we show that total levels of STAT6 molecules activated by a cytokine-receptor complex remained unaltered through a wide range of complex stabilities and ligand concentrations. Although short-lived complexes and low ligand doses induce delayed STAT6 activation kinetics, the absolute number of molecules activated per unit of time did not change significantly. These observations agree with previous studies by our laboratory in the IL-4 system. We recently described a series of IL-4 mutants with enhanced receptor binding affinity and specificity when compared to IL-4 wt, i.e. super-4, a type I receptor specific mutant; and KFR, a type II receptor specific mutant (27). Despite more than 10.000 fold differences in receptor binding affinity between these two mutants, they activated STAT6 to roughly similar extent. However, here again, their late responses potencies correlated with their complex stability. In view of our new data, we propose that the different responses elicited by super-4 and KFR mutants most likely result from different kinetics of signal activation induced by these mutants.
Our data also suggests that the level of expression of cytokine receptors on the cell surface plays a major role, through mass action, in ‘titrating’ high and low potency ligands with respect to signaling and function. Cells that express high levels of cytokine receptors are relatively insensitive to differences in ligand affinity with respect to membrane proximal STAT activation. In contrast, when cells express lower levels of cytokine receptors, ligands with slower koff gain a functional advantage. Indeed, we observed that a reduction of the IL-13Rα1 levels by siRNA resulted in low affinity IL-13 variants signaling more weakly than high affinity IL-13 variants. Similarly, surface receptor density was shown to determine the differential antiproliferative responses exhibited by low- and high- affinity IFN subtypes (23, 24). Although we observe a clear correlation between koff, receptor complex half-life, signaling kinetics and late responses potency, we still do not fully understand how all these parameters interconnect together and translate into more potent and diverse responses. The impact of negative-feedback mechanisms in signal activation remains largely unexplored. Interestingly, a new negative feedback mechanism acting at the level of ligand-receptor complex formation was recently described for IFN (44). Low affinity IFN subtypes were more sensitive than high affinity IFN subtypes to this regulation. It is thus tempting to speculate that high affinity ligands could survive the negative regulation of these feedback mechanisms by forming more stable complexes, thus eliciting more potent or more diverse late responses.
Our results challenge the equilibrium model for cytokine signal activation. In this model, cytokines recruit their receptor subunits following a two-step mechanism, reaching a dynamic equilibrium between binary and ternary complexes on the plasma membrane (9, 45, 46). Modulation of the on- and off- binding rates for any of the two receptors subunits is predicted to produce a parallel alteration on the number of complexes formed by a given cytokine and on its signal activation potency. However, our data show that 10,000-fold differences in binding affinity resulting from changes in koff only marginally alter STAT activation. This is not a unique feature of IL-13 and it is also found in other cytokines such as the type I IFN family, where more than fifteen IFN subtypes, all sharing the same surface receptor but binding with very different affinities, still activate STAT with very similar potencies (47). By extending the equilibrium ligand binding model into a steady-state model including endocytic trafficking, we could reproduce with high fidelity the kinetics and the potencies of STAT phosphorylation by IL-13 agonists covering a large range of kon and koff. The intricate interplay of complex stability and receptor trafficking has been previously appreciated for explaining the potencies of cytokines and other hormones (48, 49). Here, we applied a minimal model comprising reversible ligand binding to the cell surface receptor followed by irreversible uptake of intact signaling complexes into endosomes and further sorting into degradation and recycling pathways. In this model, increasing ligand binding affinity by increasing the stability of the complex (i.e. decreasing ligand dissociation from the cell surface) enhances signaling only until the koff is exceeded by the rate of endocytosis (depicted as ke in Fig. 8A). Once ke dominates over koff, the final number of signaling complexes is determined by the kon binding value of the ligand and further increases in affinity by changes in koff will not have an effect on signal activation. This model thus explains the existence of a signal buffering region where mutants with similar kon and koff < ke will promote comparable levels of signal activation. Moreover, this model also explains the delay kinetics of signal activation that we observe for low affinity IL-13 variants. Assuming that downstream signaling is maintained by complexes in early endosomes; as irreversible endocytosis constantly removes active signaling complexes from the equilibrium, the number of active signaling complexes increases with ongoing endocytosis. Recently, a critical role of receptor endocytosis for efficient receptor dimerization and signaling exclusively from endosomes was proposed (39), which is not supported by our dimerization assays performed with full-length receptor at physiological expression levels. In contrast, our studies reveal a critical role of endocytosis to control ligand binding to the receptor, thus buffering signaling activity over a large range of ligand binding affinities.
Collectively our results provide additional insight into how cytokines exhibit such functional plasticity, despite activating a limited set of signaling molecules (4 JAKs and 7 STATs). From our analysis, there appears to be a mechanistic rationale for purposeful modulation of on- and off- binding rates by protein engineering to design engineered cytokine variants that preserve efficacy yet are less toxic therapeutics. Low dose IL-2 is showing great promise as a therapeutic for cancer, diabetes and other autoimmune diseases (28–30) and it appears likely that similar low dose approaches with other cytokines, or higher dose treatments with affinity impaired cytokines, rather than affinity enhanced, could rescue cytokine therapies previously limited by dose dependent toxicity.
MATERIAL AND METHODS
Protein expression and purification
Human IL-13, the IL-13Rα1 ectodomain (amino acids 1-310) and the IL-4Rα ectodomain (amino acids 1-202) were cloned into the pAcGP67-A vector (BD Biosciences) in frame with an N-terminal gp67 signal sequence and a C-terminal hexahistidine tag and produced using the baculovirus expression system, as described in (15). Baculovirus stocks were prepared by transfection and amplification in Spodoptera frugiperda (Sf9) cells grown in SF900II media (Invitrogen) and protein expression was carried out in suspension Trichoplusiani (High Five) cells grown in InsectXpress media (Lonza). Following expression, proteins were captured from High Five supernatants after 60 h by nickel-NTA agarose (Qiagen) affinity chromatography, concentrated, and purified by size exclusion chromatography on a Superdex 200 column (GE Healthcare), equilibrated in 10 mM HEPES (pH 7.2) containing 150 mM NaCl. Recombinant cytokines were purified to greater than 98% homogeneity. IL-13 agonists and the IL-13Rα1 used in surface plasmon resonance measurements and cell-based assays were expressed fully glycosylated, as estimated from Coomassie-stained SDS-PAGE. For site-specific fluorescence labeling by phosphopantetheinyl transfer, a ybbR-tag (DSLEFIASKLA) (50) was fused to the N-terminus of IL-13 wt and mutants. Enzymatic protein labeling via a DY647-coenzyme A conjugate was performed as described recently (51). For biotinylated receptor expression, IL-4Rα and IL-13Rα1 ectodomains were cloned into the pAcGP67-A vector with a C-terminal biotin acceptor peptide (BAP)-LNDIFEAQKIEWHW followed by a hexahistidine tag. Receptors were coexpressed with BirA ligase in the presence of excess biotin (10 μM). For crystallization studies, the mutant IL-13 A11 was coexpressed with IL-13Rα1 in the presence of 10 μM Tunicamycin to inhibit glycosylation and IL-4Rα was coexpressed with 1/1000 Endo H virus in the presence of 5 μM Kifunensin to remove its glycosylation. After nickel-NTA purification, the IL-13Rα1-IL-13 A11 binary complex was incubated together with IL-4Rα to form a ternary complex. The proteins were then treated overnight with carboxypeptidase-A (Sigma) and B (Calbiochem) at a w/w ratio of 1:100 and subsequently purified by size exclusion chromatography. Protein was concentrated to 8–20 mg/ml for crystallization. Protein concentrations were quantified by UV spectroscopy at 280 nm using a Nanodrop2000 spectrometer (Thermo Scientific).
Cell lines and media
The IL-13 responsive cell lines A549 and TF-1 were grown in RPMI containing 10% v/v FBS, penicillin-streptomycin, and L-glutamine (2 mM). TF-1 cells were grown in the presence of GM-CSF to promote proliferation and survival. HeLa cells were grown in Eagle’s MEM containing 10% v/v FBS, NaHPO3 (2 g/L), L-glutamine (2 mM), HEPES (10 mM), NEAA and penicillin-streptomycin. All cell lines were maintained at 37°C with 5% CO2. HeLa cells stably transfected with mEGFP-STAT6 were grown in the presence of 0.8 mg/ml G-418 (Invitrogen) to select for and maintain stable transfectants.
Flow cytometry staining and antibodies
For cell surface staining of IL-13 receptors, A549 cells were stained with antibodies against IL-13Rα1 (1:300 (BD Biosciences)) coupled to Fluorescein-5-isothiocyanate (FITC) for 1 hour at 4°C. Cells were then washed and the levels of surface IL-13Rα1 were measured using an Accuri C6 flow cytometer. Intracellular phospho-STAT6 staining was performed after ice-cold methanol (100% v/v) permeabilization. Antibodies to pSTAT6 Alexa488 were purchased from BD Biosciences and used at a 1:50 dilution. The induction of STAT6 phosphorylation was calculated by subtracting the Mean Fluorescence Intensity (MFI) of the stimulated samples from that of the unstimulated sample. The normalized values were plotted against cytokine concentration to yield dose-response curves from which the EC50 values were calculated based on nonlinear least squares regression fit to a sigmoidal curve.
Yeast display of IL-13
General yeast display methodologies are modified from previously described protocols (52). Human IL-13 cDNA was cloned into the yeast display vector pCT302. S. cerevisiae strain EBY100 was transformed with the pCT302_IL-4 vector and grown for two days at 30°C on SDCAA plates. Individual colonies of IL-13-displaying yeast were grown overnight at 30°C in SDCAA liquid media (pH 4.5), followed by induction in SGCAA media (pH 4.5) for 2 days at 20°C. Yeast were stained with biotinylated IL-13Rα1, tetramerized biotinylated IL-4Rα, or biotinylated IL-4Rα in the presence of biotinylated IL-13Rα1. IL-4Rα tetramers were formed by incubating 500 nM of biotinylated IL-4Rα with 125 nM of Streptavidin (SAB)-Alexa647 conjugate for 15 min on ice. Fluorescence was analyzed on an Accuri C6 flow cytometer.
Assembly, transformation, and selection of an IL-13 site-directed library
Assembly PCR was carried out using 6 overlapping primers, three of which contained the randomized codon sequences used for mutation (L10 (LFIV), R11 (RSNHLI), I14 (LFIV), V18 (LFIV), R86 (RKTM), D87 (EDKR), T88 (ITKR), K89 (RKTM), L101 (LFIYHN), K104 (RKTM), K105 (KTAE), F107 (LFIV), R108 (RKTM)). The PCR product was further amplified using the primers:
5′-GTAGCGGTGGGGGCGGTTCTCTGGAAGTTCTGTTCCAGGGTCCGAGCGGCGG ATCCCCAGGCCCTGTGCCTC-3′
5′-AGATCTCGAGCAAGTCTTCTTCGGAGATAAGCTTTTGTTCGCCACCAGAAGCG GCCGCGTTGAACTGTCCCTC-3′
These primers also contained the necessary homology to the pCT302 vector sequence requisite for homologous recombination. Insert DNA was combined with linearized vector backbone pCT302 and electrocompetent S. cerevisiae EBY100 were electroporated and rescued, as previously described, forming a library of 3×108 transformants. Selections were performed on this library using magnetic activated cell sorting (MACS, Miltenyi). The first round of selection was performed with 2×109 cells from the yeast library, approximately 10-fold coverage relative to the number of transformants. Subsequent rounds of selection used 1×107 yeast cells (greater than 10-fold coverage in each round). Naïve and sorted IL-13 libraries were grown fresh overnight at 30°C in SDCAA liquid media (pH 4.5), followed by induction in SGCAA liquid media (pH 4.5) for 2 days at 20°C. Monomeric selection with IL-13Rα1 was performed through sequential binding of IL-13Rα1, 2 μg/ml SAV-Alexa647, and 50 μl Miltenyil anti-Alexa647 microbeads. Fluorescence analysis was performed on an Acurri C6 flow cytometer.
Surface plasmon resonance
SPR experiments were conducted on a Biacore T100 instrument using a Biacore SA sensor chip (GE Healthcare). Biotinylated IL-13Rα1 was captured at a low density (50–100 response units (RU)) and kinetics measurements were conducted at 30 μL/min. An unrelated biotinylated protein was immobilized as a reference surface for the SA sensor chip with matching RU to the experimental surface. All measurements were made using 3-fold serial dilutions of IL-13 agonists in the running buffer (1xHBS-P (GE Healthcare), 0.1 % BSA). The IL-13Rα1 bound to the chip surface was regenerated with 7 mM glycine (pH 3.0) and 250 mM NaCl. Kinetic parameters were determined using 120 s to 190 s of IL-13 agonist association time and 20 s to 1200 s dissociation time. All data fitting was performed using the Biacore T100 evaluation software version 2.0 with a 1:1 Langmuir binding model.
Crystallization and data collection
IL-13 A11/IL-13Rα1/IL-4Rα ternary complex crystals were grown in sitting drops at 25°C by mixing 0.1 μL protein solution (12 mg/ml in 10 mM HEPES (pH 7.2) and 150 mM NaCl) with an equal volume of 0.2 M lithium sulfate, 0.1 M phosphate/citrate (pH 4.2) and 20% PEG 1000. Crystals grew in 2–5 days. They were subsequently flash frozen in liquid nitrogen using mother liquor containing 27% glycerol as a cryoprotectant. A 3.0 Å data set was collected at beamline 8.2.2, Advance Light Source (ALS), University of California, Berkeley. The data set was indexed, integrated and scaled using the HKL2000 software package (Otwisnowski and Minor, 1997). Data processing statistics are presented in Supplementary Table 1.
Structure Determination and Refinement
The IL-13 A11 ternary crystal structure was solved by molecular replacement with the program PHASER using the coordinates of IL-13, IL-13Rα1, and IL-4Rα separately (pdb code 3BPN). After all the domains were placed, the IL-13 wt sequence was converted to IL-13 A11 and iterative rounds of refinement with PHENIX and model adjustment with COOT were used to refine the structures. Ramachandran analysis was conducted using MolProbity (http://molprobity.biochem.duke.edu). All structural figures and overlays were prepared using PyMOL.
Live-cell dual-color single-molecule imaging studies
Single molecule imaging of cytokine-receptor complexes living cells was performed as recently described in detail (56). HeLa cells were grown to 50% confluence on glass cover slides coated with RGD (PLL-PEG-RGD) functionalized poly-L-lysine-graft-poly(ethylene glycol) prepared according to (53). For ligand binding studies, DY647IL-13 agonists or DY647IL-4 were added to a final concentration of 2 nM and incubated for at least 5 min at room temperature before imaging.
For probing receptor dimerization IL-13Rα1 and IL-4Rα were fused to N-terminal HaloTag and SNAPf-tag, respectively. To this end, genes of full length IL-13Rα1 and IL-4Rα, respectively, without the N-terminal signal sequences were inserted into the pDisplay vector (Invitrogen) via BglII and PstI (IL-13Rα1)/XhoI (IL-4Rα) restriction sites. Subsequently, genes coding for the HaloTag and SNAPf-tag, respectively, were inserted via the BglII site. The constructs including the signal sequence of the pDisplay vector (Igκ) were transferred by restriction with EcoRI and XhoI into the pSems-26m vector (Covalys Biosciences) to yield pSems-HaloTag-IL-13Rα1 and pSems-SNAPf-IL-4Rα. HeLa cells seeded on PLL-PEG-RGD coated glass cover slides were transfected with pSems-HaloTag-IL-13Rα1 and pSems-SNAPf-IL-4Rα using calcium phosphate precipitation. 4–5 days after transfection, the cover slides were mounted into home-built microscopy chambers and the receptor molecules were labeled via their respective tags with fluorescent dyes through incubation of 20 nM HTL-TMR (Promega Corp.) and 40 nM SNAP-Surface® 647 (NEB) for 15 min following 5 washing steps with PBS. Labeled receptors are denoted as TMRIL-13Rα1 and DY647IL-4Rα henceforth. Imaging experiments were started immediately after labeling in cell medium supplemented with 1 mM ascorbic acid, 40 μg/ml catalase, 1 mM methylviologen, 4.5 mg/ml glucose (all Sigma-Aldrich) and 0.5 mg/ml glucose-oxidase (Roche) to optimize photostability of DY647 (54). Ternary complex formation was induced by adding 200 nM of the designated IL-13 agonist and images of the same cover slide were acquired for a maximum of 30 min post-stimulation to reduce artifacts that may arise from cellular feedback mechanisms such as receptor endocytosis or stress responses. Individual receptor molecules were simultaneously imaged for 50–300 frames at a frame rate of 31 Hz with an inverse IX71 microscope that was equipped with a triple-line total internal reflection illumination condenser (Olympus), a 150× TIRF objective (UAPO 150×/1.45 TIRFM Olympus), a UNIBLITZ VMM-D4 Shutter (Vincent Associates), an Ixon DU-897 EMCCD camera (512×512 pixel, Andor Technology), and a spectral image splitter (DV2, Optical Insights). The fluorescent dyes were simultaneously excited with a 200 mW 561 nm solid state laser (CL561-200, CrystaLaser) and a 140 mW 642 nm diode laser (LuxX®, Omicron-Laserage). Appropriate dichroic and emission filters were purchased from Semrock and Chroma Technology. The image splitter was calibrated with fluorescent beads that have pixel accuracy and therefore generated images that were further used to calculate a correction-matrix that later enabled the evaluation algorithms to precisely co-localize the particles in both channels with sub-pixel resolution.
Single particle tracking of receptors, ternary complex quantification, and diffusion analysis
Dual-color images were separated into their two respective channels and localization of single molecules with sub-micrometer precision (10–40 nm) as well as tracking of their movement and filtering of immobile particles (non-specifically bound fluorophores) was carried out by self-written software on the basis of algorithms described in (55). Individual TMRIL-13Rα1/DY647IL-4Rα dimers were identified by co-locomotion analysis (Fig. S3f): after frame-by-frame co-localization of molecules both channels with a distance threshold of 2 pixels (214 nm), co-localized molecules were subjected to tracking with a minimum trajectory length of 10 consecutive steps (56). The fraction of receptors in a ternary complex was determined by particle image cross-correlation spectroscopy (PICCS), as described previously (38). To this end, a self-written MATLAB script was used to calculate the correlated fraction of the coordinates of localized particles in both channels in a 25 μm2 region of interest (ROI) in the first 20 frames (to minimize reduction of cross-correlation by photobleaching) of each image stack. Since only those cells that showed similar levels of both receptor chains were analyzed, a single correlated fraction always refers to the channel with the limiting number of molecules. For each IL-13 agonist, all single correlated fraction values from at least 10 cells were histogramized and fitted by a Gaussian distribution to obtain a mean value for the correlation fraction.
The diffusion constants of TMRIL-13Rα1 in absence of ligand (i.e. the binary complex) and of TMRIL-13Rα 1/IL-13 wt/DY647IL-4Rα ternary complexes were determined from a collection of trajectories of the same cell by fitting a step-length histogram (t = 1, frame = 0.032 s) to a two-dimensional Gaussian probability distribution
| Eqn. 1 |
, where P is the probability of finding a molecule with diffusion constant D (in μm2/s) in a distance r (in μm) from its origin at time point t0 = 0 after time t (in s). A similar histogram of TMRIL-13Rα1 trajectories of the same cell after addition of IL-13 wt was fitted with a linear combination of the previously obtained distributions with their parameters as fixed constants and an additional parameter f (Eqn. 2) to get an estimate of the fraction of IL-13Rα1 molecules moving in ternary complexes,
| Eqn. 2 |
siRNA silencing of IL-13Rα1
A549 cells were plated at 3×105 cells per well in 60 mm plates and incubated for 16 h prior to transfection. Cells were transfected with 25 nM, 75 nM or 125 nM of siRNA targeting IL-13Rα1 (Sigma-Proligo) or control siRNA (Sigma-Proligo) per plate using Lipofectamine RNAi MAX (Invitrogen). Forty-eight hours after transfection, cells were plated in a 96-well plate and used for IL-13 stimulation and surface IL-13Rα1 determination.
TF-1 cell proliferation assays
Two thousand TF-1 cells/well were seeded in a 96 well plate and stimulated with the indicated doses of IL-13 wt and the selected IL-13 agonists. After 96 h of stimulation, cells were harvested and cell number was determined using flow cytometry-based counting on an Accuri C6 flow cytometer. The number of cells obtained for each agonist was plotted against the cytokine concentration in order to obtain sigmoidal dose/response curves, from which the TF-1 proliferation EC50 values were calculated.
Dendritic cell differentiation studies
CD14+ monocytes were isolated (>97% purity) from peripheral blood mononuclear cells (PMBCs) obtained from healthy blood donors (Stanford Blood Center) by density centrifugation using a RosetteSep Human Monocyte Enrichment Cocktail (Stem Cell Technologies) followed by magnetic separation with microbeads conjugated to antibodies against CD14 (Miltenyi Biotec). We subsequently cultured 0.5−1 × 106 CD14+ monocytes with 50 ng/ml GM-CSF alone or with the indicated concentrations of the IL-13 variants in 12-well plates (Corning) containing IMDM medium (Gibco) supplemented with 10% v/v human AB serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine, sodium pyruvate, nonessential amino acids and 50 μM 2-mercaptoethanol. Fresh cytokines were added on days 2 and 4. Cells were processed between days 6 and 7 with 5 mM EDTA and subsequently stained with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen), fluorescently labeled antibodies against CD86 (no. 555660) and CD209 (no. 551265), or appropriate fluorescently labeled isotype control antibodies. All antibodies were used at 1:50 dilution. Dendritic cell differentiation was assessed by flow cytometry with a BD LSRII flow cytometer, and the MFI was determined using FlowJo analysis software (Treestar).
Quantification of STAT6 nuclear translocation
HeLa cells stably expressing STAT6 N-terminally fused to mEGFP were imaged at 37°C using an inverted confocal laser scanning microscope FV1000 (Olympus) that was equipped with a 60× oil-immersion objective (NA: 1.35, Olympus) and a temperature-controlled microscope stage system BC-110 (20/20 Technology). An Argon multiline laser GLG3135 (Showa Optronics) was used for excitation of mEGFP at 488 nm. Appropriate dichroic and emission filters were obtained from Olympus. The diameter of the pinhole was set to 80–100 μm and images were captured with FV1000 application software (FV10-ASW, Olympus).
The nuclear translocation of mEGFP-STAT6 was initiated by addition of 200 nM of each IL-13 agonist and single section images were captured every 2 min for a total duration of 80–100 min at 37°C. Mean fluorescence intensities were measured and background was subtracted using ImageJ (NIH) for each time point for both the nucleus and cytoplasm. The nuclear:cytoplasmic fluorescence intensity ratios with respect to time t (in min) were calculated relative to the mean initial ratio at t = 0 and t = 2 min and averaged over >10 imaged cells. The resulting sigmoidal-curves were fitted to
| Eqn. 3 |
to obtain the amplitude n/cend and half-time t½ (in min) of STAT6 translocation for each IL-13 agonist. The Hill coefficient n was fixed at 2 due to the bimolecular homodimerization of STAT6 after activation.
Imaging of receptors in endosomes
HeLa cells were grown on PLL-PEG-RGDcoated glass cover slides and incubated with 2 nM DY647IL-13 wt or DY647IL-13 D7, respectively. After an incubation time of 15 min at 37°C followed by 5 washing steps, endosomes containing large amounts of labeled ligand were imaged by in highly inclined and laminated optical sheet (HILO) mode (57) using the TIRF microscopy setup described above. Endocytosis was blocked/decreased in some experiments by incubation of the cells in presence of 100 μM Rac1 inhibitor EHT1864 (Santa Cruz Biotechnology) for 1h. For quantification of endocytosed ligand, the integral intensity was quantified after background subtraction using a rolling cylinder algorithm (58). For direct co-visualization of receptors in endosomes, HeLa-TMRIL-13Rα1-DY647IL-4Rα cells that were used for dual color single molecule imaging were imaged by laser scanning microscopy 15 min after receptor labeling and incubation at 37°C.
Model simulations
Two MATLAB scripts simulating STAT6 phosphorylation kinetics and pSTAT6 dose response experiments were developed to verify the proposed steady-state model in silico. The simulations are based on the following assumptions:
Ligand binding to the cell surface receptor is reversible and determined by ligand dependent kon (association of ligand and receptor) and koff (dissociation of ligand and receptor) rate constants.
The dissociation rate constant koff determined for the binary IL-13/IL-13Rα1 is decreased by a factor of ≈100 due to simultaneous interaction with IL-4Rα.
Ligand-receptor complexes that are capable of forming intact ternary signaling complexes are taken up irreversibly into endosomes with a rate constant kendo.
Complexes in endosomes are further sorted into degradation and recycling pathways with rate constants kdeg and krec, respectively.
The amount of pSTAT6 is proportional to the number of signaling complexes (i. e. ligand-receptor complexes) at the plasma membrane and in endosomes.
The receptor comprising the IL-13Rα1 and the IL-4Rα subunits was treated as a single entity at a volume concentration of 1 pM. Thus, realistic excess of ligand over cell surface receptor was ensured.
Common parameters for both simulations:
| Name | Value | Description |
|---|---|---|
| dt | 1 ms | Simulation time resolution |
| k_endo | 3 · 10−3 s−1 | Estimated endocytosis rate |
| k_deg | 6 · 10−5 s−1 | Estimated degradation rate |
| k_rec | 6.5 · 10−4 s−1 | Estimated recycling rate |
| cR13_start | 1 pM | IL-13Ra1 concentration at t=0 |
| cBin | 0 at t=0 | Surface binary complex at t |
| cBinEndo | 0 at t=0 | Endosomal binary complex at t |
Common differential equations for both simulations:
| Eqn. 4 |
| Eqn. 5 |
| Eqn. 6 |
a) STAT6 phosphorylation kinetics simulation
Simulations were performed for IL-13 wt and C4, D7, C10, A11 agonists. Since no rate constants for C4, D7 and C10 were available, dissociation rates were calculated based on IL-13 wt koff and measured KD values, kon was set to a common reasonable value. The ligand concentration is kept constant due to pseudo first order reaction conditions. For modeling EHT1864 treatment the endocytosis rate was decreased by 75%.
Parameters:
| Name | Value | Description |
|---|---|---|
| k_off_WT | 2 · 10−2 s−1 | IL-13 wt dissociation rate |
| k_off_C4 | 150 s−1 | IL-13 C4 dissociation rate |
| k_off_D7 | 20 s−1 | IL-13 D7 dissociation rate |
| k_off_C10 | 1 s−1 | IL-13 C10 dissociation rate |
| k_off_A11 | 4 · 10−4 s−1 | IL-13 A11 dissociation rate |
| k_on | 1 · 106 M−1s−1 | Common association rate |
| t_end | 14400 s | Simulation end time, 3h |
| cIL13 | 200 nM | IL-13 concentration |
The simulation main result (black line) is plotted as relative concentration of receptors in signaling competent complexes (sum of binary complexes on surface and in endosomes divided by the initial IL-13Rα1 concentration) over time, which is supposed to correlate with the level of pSTAT6.
b) pSTAT6 dose response simulation
Simulations of STAT6 dose response curves are based on the phosphorylation kinetics simulation described above with the following changes: Curves were modeled for IL-13 wt and A7, B4 A11, A6 agonists. In each run the relative concentration of receptors in signaling competent complexes (surface and endosomes) at t = 15 min is calculated with an increasing initial ligand concentration in the range between 10−13 and 10−7 mol/l and plotted against the applied ligand concentration. Since no pseudo first order reaction conditions are present for low concentrations the changes in ligand concentration during each run is calculated according to an additional differential equation:
| Eqn. 7 |
Parameters:
| Name | Value | Description |
|---|---|---|
| k_off_WT | 2.2 · 10−2 s−1 | IL-13 wt dissociation rate constant |
| k_off_A7 | 3.4 · 10−3 s−1 | IL-13 A7 dissociation rate constant |
| k_off_B4 | 2.4 · 10−2 s−1 | IL-13 B4 dissociation rate constant |
| k_off_A11 | 1.3 · 10−3 s−1 | IL-13 A11 dissociation rate constant |
| k_off_A6 | 3.3 · 10−3 s−1 | IL-13 A6 dissociation rate constant |
| k_on_WT | 5.21 · 106 M−1s−1 | IL-13 wt association rate constant |
| k_on_A7 | 2.90 · 106 M−1s−1 | IL-13 A7 association rate constant |
| k_on_B4 | 1.36 · 106 M−1s−1 | IL-13 B4 association rate constant |
| k_on_A11 | 1.64 · 107 M−1s−1 | IL-13 A11 association rate constant |
| k_on_A6 | 1.77 · 106 M−1s−1 | IL-13 A6 association rate constant |
| t_end | 900 s | Simulation end time, 15 min |
| cIL13_range | 10−13 to 10−7 M | IL-13 concentration range |
Supplementary Material
Supplementary Table 1.
Supplementary Figure 1 to 10.
Supplementary Videos 1 to 6.
Acknowledgments
We thank members of the Garcia and Piehler laboratories for helpful advice and discussion and Kevin Jude, Gabriele Hikade and Hella Kenneweg for technical assistance. This work was supported by the NIH-RO1-AI51321 (K.C.G.), by the Howard Hughes Medical Institute (K.C.G.) and by the SFB 944 (J.P.)
REFERENCES AND NOTES
- 1.Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell. 1990;61:203–212. doi: 10.1016/0092-8674(90)90801-k. [DOI] [PubMed] [Google Scholar]
- 2.Stroud RM, Wells JA. Mechanistic diversity of cytokine receptor signaling across cell membranes. Sci STKE. 2004;2004:re7. doi: 10.1126/stke.2312004re7. [DOI] [PubMed] [Google Scholar]
- 3.Wang X, Lupardus P, Laporte SL, Garcia KC. Structural biology of shared cytokine receptors. Annu Rev Immunol. 2009;27:29–60. doi: 10.1146/annurev.immunol.24.021605.090616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wells JA, de Vos AM. Structure and function of human growth hormone: implications for the hematopoietins. Annual review of biophysics and biomolecular structure. 1993;22:329–351. doi: 10.1146/annurev.bb.22.060193.001553. [DOI] [PubMed] [Google Scholar]
- 5.Moraga I, Spangler J, Mendoza JL, Garcia KC. Multifarious determinants of cytokine receptor signaling specificity. Adv Immunol. 2014;121:1–39. doi: 10.1016/B978-0-12-800100-4.00001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spangler JB, Moraga I, Mendoza JL, Garcia KC. Insights Into Cytokine-Receptor Interactions from Cytokine Engineering. Annu Rev Immunol. 2014 doi: 10.1146/annurev-immunol-032713-120211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moraga I, Wernig G, Wilmes S, Gryshkova V, Richter CP, Hong WJ, Sinha R, Guo F, Fabionar H, Wehrman TS, Krutzik P, Demharter S, Plo I, Weissman IL, Minary P, Majeti R, Constantinescu SN, Piehler J, Garcia KC. Tuning Cytokine Receptor Signaling by Re-orienting Dimer Geometry with Surrogate Ligands. Cell. 2015;160:1196–1208. doi: 10.1016/j.cell.2015.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, Stark GR. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov. 2007;6:975–990. doi: 10.1038/nrd2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Piehler J, Thomas C, Garcia K Christopher, Schreiber G. Structural and dynamic determinants of type I interferon receptor assembly and their functional interpretation. Immunol Rev. 250:317–334. doi: 10.1111/imr.12001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Boxel-Dezaire AH, Rani MR, Stark GR. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity. 2006;25:361–372. doi: 10.1016/j.immuni.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 11.Ring AM, Lin JX, Feng D, Mitra S, Rickert M, Bowman GR, Pande VS, Li P, Moraga I, Spolski R, Ozkan E, Leonard WJ, Garcia KC. Mechanistic and structural insight into the functional dichotomy between IL-2 and IL-15. Nature immunology. 2012;13:1187–1195. doi: 10.1038/ni.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Waldmann TA. The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nat Rev Immunol. 2006;6:595–601. doi: 10.1038/nri1901. [DOI] [PubMed] [Google Scholar]
- 13.Rochman Y, Spolski R, Leonard WJ. New insights into the regulation of T cells by gamma(c) family cytokines. Nat Rev Immunol. 2009;9:480–490. doi: 10.1038/nri2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heller NM, Qi X, Junttila IS, Shirey KA, Vogel SN, Paul WE, Keegan AD. Type I IL-4Rs selectively activate IRS-2 to induce target gene expression in macrophages. Sci Signal. 2008;1:ra17. doi: 10.1126/scisignal.1164795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.LaPorte SL, Juo ZS, Vaclavikova J, Colf LA, Qi X, Heller NM, Keegan AD, Garcia KC. Molecular and structural basis of cytokine receptor pleiotropy in the interleukin-4/13 system. Cell. 2008;132:259–272. doi: 10.1016/j.cell.2007.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Junttila IS, Mizukami K, Dickensheets H, Meier-Schellersheim M, Yamane H, Donnelly RP, Paul WE. Tuning sensitivity to IL-4 and IL-13: differential expression of IL-4Ralpha, IL-13Ralpha1, and gammac regulates relative cytokine sensitivity. J Exp Med. 2008;205:2595–2608. doi: 10.1084/jem.20080452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoon SI, Jones BC, Logsdon NJ, Walter MR. Same structure, different function crystal structure of the Epstein-Barr virus IL-10 bound to the soluble IL-10R1 chain. Structure. 2005;13:551–564. doi: 10.1016/j.str.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 18.Subramaniam PS, Khan SA, Pontzer CH, Johnson HM. Differential recognition of the type I interferon receptor by interferons tau and alpha is responsible for their disparate cytotoxicities. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:12270–12274. doi: 10.1073/pnas.92.26.12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jaitin DA, Roisman LC, Jaks E, Gavutis M, Piehler J, Van der Heyden J, Uze G, Schreiber G. Inquiring into the differential action of interferons (IFNs): an IFN-alpha2 mutant with enhanced affinity to IFNAR1 is functionally similar to IFN-beta. Mol Cell Biol. 2006;26:1888–1897. doi: 10.1128/MCB.26.5.1888-1897.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rao BM, Driver I, Lauffenburger DA, Wittrup KD. Interleukin 2 (IL-2) variants engineered for increased IL-2 receptor alpha-subunit affinity exhibit increased potency arising from a cell surface ligand reservoir effect. Molecular pharmacology. 2004;66:864–869. doi: 10.1124/mol.66.4.. [DOI] [PubMed] [Google Scholar]
- 21.Rao BM, Driver I, Lauffenburger DA, Wittrup KD. High-affinity CD25-binding IL-2 mutants potently stimulate persistent T cell growth. Biochemistry. 2005;44:10696–10701. doi: 10.1021/bi050436x. [DOI] [PubMed] [Google Scholar]
- 22.Yoon SI, Jones BC, Logsdon NJ, Harris BD, Kuruganti S, Walter MR. Epstein-Barr virus IL-10 engages IL-10R1 by a two-step mechanism leading to altered signaling properties. J Biol Chem. 2012;287:26586–26595. doi: 10.1074/jbc.M112.376707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moraga I, Harari D, Schreiber G, Uze G, Pellegrini S. Receptor density is key to the alpha2/beta interferon differential activities. Mol Cell Biol. 2009;29:4778–4787. doi: 10.1128/MCB.01808-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levin D, Harari D, Schreiber G. Stochastic receptor expression determines cell fate upon interferon treatment. Mol Cell Biol. 2011;31:3252–3266. doi: 10.1128/MCB.05251-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walter MR. The molecular basis of IL-10 function: from receptor structure to the onset of signaling. Current topics in microbiology and immunology. 2014;380:191–212. doi: 10.1007/978-3-662-43492-5_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kalie E, Jaitin DA, Abramovich R, Schreiber G. An interferon alpha2 mutant optimized by phage display for IFNAR1 binding confers specifically enhanced antitumor activities. J Biol Chem. 2007;282:11602–11611. doi: 10.1074/jbc.M610115200. [DOI] [PubMed] [Google Scholar]
- 27.Junttila IS, Creusot RJ, Moraga I, Bates DL, Wong MT, Alonso MN, Suhoski MM, Lupardus P, Meier-Schellersheim M, Engleman EG, Utz PJ, Fathman CG, Paul WE, Garcia KC. Redirecting cell-type specific cytokine responses with engineered interleukin-4 superkines. Nat Chem Biol. 2012;8:990–998. doi: 10.1038/nchembio.1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kennedy-Nasser AA, Ku S, Castillo-Caro P, Hazrat Y, Wu MF, Liu H, Melenhorst J, Barrett AJ, Ito S, Foster A, Savoldo B, Yvon E, Carrum G, Ramos CA, Krance RA, Leung K, Heslop HE, Brenner MK, Bollard CM. Ultra low-dose IL-2 for GVHD prophylaxis after allogeneic hematopoietic stem cell transplantation mediates expansion of regulatory T cells without diminishing antiviral and antileukemic activity. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20:2215–2225. doi: 10.1158/1078-0432.CCR-13-3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Posch C, Weihsengruber F, Bartsch K, Feichtenschlager V, Sanlorenzo M, Vujic I, Monshi B, Ortiz-Urda S, Rappersberger K. Low-dose inhalation of interleukin-2 bio-chemotherapy for the treatment of pulmonary metastases in melanoma patients. British journal of cancer. 2014;110:1427–1432. doi: 10.1038/bjc.2014.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu A, Snowhite I, Vendrame F, Rosenzwajg M, Klatzmann D, Pugliese A, Malek TR. Selective IL-2 responsiveness of regulatory T cells through multiple intrinsic mechanisms support the use of low-dose IL-2 therapy in Type-1 diabetes. Diabetes. 2015 doi: 10.2337/db14-1322. [DOI] [PubMed] [Google Scholar]
- 31.Dasgupta P, Keegan AD. Contribution of alternatively activated macrophages to allergic lung inflammation: a tale of mice and men. Journal of innate immunity. 2012;4:478–488. doi: 10.1159/000336025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wills-Karp M. Interleukin-13 in asthma pathogenesis. Immunol Rev. 2004;202:175–190. doi: 10.1111/j.0105-2896.2004.00215.x. [DOI] [PubMed] [Google Scholar]
- 33.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*) Annu Rev Immunol. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, Locati M, Mantovani A, Martinez FO, Mege JL, Mosser DM, Natoli G, Saeij JP, Schultze JL, Shirey KA, Sica A, Suttles J, Udalova I, van Ginderachter JA, Vogel SN, Wynn TA. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang HE, Reinhardt RL, Bando JK, Sullivan BM, Ho IC, Locksley RM. Divergent expression patterns of IL-4 and IL-13 define unique functions in allergic immunity. Nature immunology. 2011;13:58–66. doi: 10.1038/ni.2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, Zhang Y, Elias JA. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 1999;103:779–788. doi: 10.1172/JCI5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Syed RS, Reid SW, Li C, Cheetham JC, Aoki KH, Liu B, Zhan H, Osslund TD, Chirino AJ, Zhang J, Finer-Moore J, Elliott S, Sitney K, Katz BA, Matthews DJ, Wendoloski JJ, Egrie J, Stroud RM. Efficiency of signalling through cytokine receptors depends critically on receptor orientation. Nature. 1998;395:511–516. doi: 10.1038/26773. [DOI] [PubMed] [Google Scholar]
- 38.Semrau S, Holtzer L, Gonzalez-Gaitan M, Schmidt T. Quantification of biological interactions with particle image cross-correlation spectroscopy (PICCS) Biophys J. 2011;100:1810–1818. doi: 10.1016/j.bpj.2010.12.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gandhi H, Worch R, Kurgonaite K, Hintersteiner M, Schwille P, Bokel C, Weidemann T. Dynamics and interaction of interleukin-4 receptor subunits in living cells. Biophys J. 2014;107:2515–2527. doi: 10.1016/j.bpj.2014.07.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McKenzie AN, Culpepper JA, de Waal Malefyt R, Briere F, Punnonen J, Aversa G, Sato A, Dang W, Cocks BG, Menon S, et al. Interleukin 13, a T-cell-derived cytokine that regulates human monocyte and B-cell function. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:3735–3739. doi: 10.1073/pnas.90.8.3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kalie E, Jaitin DA, Podoplelova Y, Piehler J, Schreiber G. The stability of the ternary interferon-receptor complex rather than the affinity to the individual subunits dictates differential biological activities. J Biol Chem. 2008;283:32925–32936. doi: 10.1074/jbc.M806019200. [DOI] [PubMed] [Google Scholar]
- 42.Pearce KH, Jr, Cunningham BC, Fuh G, Teeri T, Wells JA. Growth hormone binding affinity for its receptor surpasses the requirements for cellular activity. Biochemistry. 1999;38:81–89. doi: 10.1021/bi9817008. [DOI] [PubMed] [Google Scholar]
- 43.Selzer T, Albeck S, Schreiber G. Rational design of faster associating and tighter binding protein complexes. Nature structural biology. 2000;7:537–541. doi: 10.1038/76744. [DOI] [PubMed] [Google Scholar]
- 44.Francois-Newton V, de Freitas Almeida G Magno, Payelle-Brogard B, Monneron D, Pichard-Garcia L, Piehler J, Pellegrini S, Uze G. USP18-based negative feedback control is induced by type I and type III interferons and specifically inactivates interferon alpha response. PLoS One. 2011;6:e22200. doi: 10.1371/journal.pone.0022200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kossiakoff AA. The structural basis for biological signaling, regulation, and specificity in the growth hormone-prolactin system of hormones and receptors. Advances in protein chemistry. 2004;68:147–169. doi: 10.1016/S0065-3233(04)68005-3. [DOI] [PubMed] [Google Scholar]
- 46.Whitty A, Borysenko CW. Small molecule cytokine mimetics. Chemistry & biology. 1999;6:R107–118. doi: 10.1016/S1074-5521(99)80034-9. [DOI] [PubMed] [Google Scholar]
- 47.Thomas C, Moraga I, Levin D, Krutzik PO, Podoplelova Y, Trejo A, Lee C, Yarden G, Vleck SE, Glenn JS, Nolan GP, Piehler J, Schreiber G, Garcia KC. Structural linkage between ligand discrimination and receptor activation by type I interferons. Cell. 2011;146:621–632. doi: 10.1016/j.cell.2011.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sarkar CA, Lowenhaupt K, Wang PJ, Horan T, Lauffenburger DA. Parsing the effects of binding, signaling, and trafficking on the mitogenic potencies of granulocyte colony-stimulating factor analogues. Biotechnology progress. 2003;19:955–964. doi: 10.1021/bp020017g. [DOI] [PubMed] [Google Scholar]
- 49.Lauffenburger DA, Fallon EM, Haugh JM. Scratching the (cell) surface: cytokine engineering for improved ligand/receptor trafficking dynamics. Chemistry & biology. 1998;5:R257–263. doi: 10.1016/s1074-5521(98)90110-7. [DOI] [PubMed] [Google Scholar]
- 50.Yin J, Straight PD, McLoughlin SM, Zhou Z, Lin AJ, Golan DE, Kelleher NL, Kolter R, Walsh CT. Genetically encoded short peptide tag for versatile protein labeling by Sfp phosphopantetheinyl transferase. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15815–15820. doi: 10.1073/pnas.0507705102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Waichman S, Bhagawati M, Podoplelova Y, Reichel A, Brunk A, Paterok D, Piehler J. Functional immobilization and patterning of proteins by an enzymatic transfer reaction. Analytical chemistry. 2010;82:1478–1485. doi: 10.1021/ac902608a. [DOI] [PubMed] [Google Scholar]
- 52.Boder ET, Wittrup KD. Yeast surface display for screening combinatorial polypeptide libraries. Nat Biotechnol. 1997;15:553–557. doi: 10.1038/nbt0697-553. [DOI] [PubMed] [Google Scholar]
- 53.VandeVondele S, Voros J, Hubbell JA. RGD-grafted poly-L-lysine-graft-(polyethylene glycol) copolymers block non-specific protein adsorption while promoting cell adhesion. Biotechnol Bioeng. 2003;82:784–790. doi: 10.1002/bit.10625. [DOI] [PubMed] [Google Scholar]
- 54.Vogelsang J, Kasper R, Steinhauer C, Person B, Heilemann M, Sauer M, Tinnefeld P. A reducing and oxidizing system minimizes photobleaching and blinking of fluorescent dyes. Angew Chem Int Ed Engl. 2008;47:5465–5469. doi: 10.1002/anie.200801518. [DOI] [PubMed] [Google Scholar]
- 55.Serge A, Bertaux N, Rigneault H, Marguet D. Dynamic multiple-target tracing to probe spatiotemporal cartography of cell membranes. Nat Methods. 2008;5:687–694. doi: 10.1038/nmeth.1233. [DOI] [PubMed] [Google Scholar]
- 56.Wilmes S, Beutel O, Li Z, Francois-Newton V, Richter CP, Janning D, Kroll C, Hanhart P, Hotte K, You C, Uze G, Pellegrini S, Piehler J. Receptor dimerization dynamics as a regulatory valve for plasticity of type I interferon signaling. The Journal of cell biology. 2015;209:579–593. doi: 10.1083/jcb.201412049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tokunaga M, Imamoto N, Sakata-Sogawa K. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat Methods. 2008;5:159–161. doi: 10.1038/nmeth1171. [DOI] [PubMed] [Google Scholar]
- 58.Lisse D, Richter CP, Drees C, Birkholz O, You C, Rampazzo E, Piehler J. Monofunctional stealth nanoparticle for unbiased single molecule tracking inside living cells. Nano letters. 2014;14:2189–2195. doi: 10.1021/nl500637a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1.
Supplementary Figure 1 to 10.
Supplementary Videos 1 to 6.