

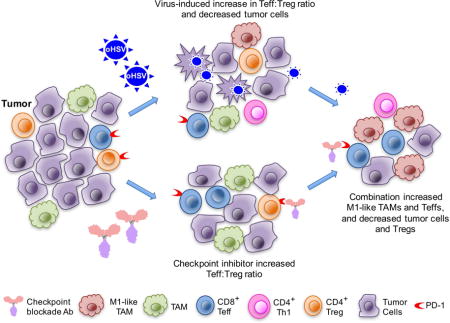
Summary
Glioblastoma is an immunosuppressive, fatal brain cancer that contains glioblastoma stem-like cells (GSCs). Oncolytic herpes simplex virus (oHSV) selectively replicates in cancer cells, while inducing anti-tumor immunity. OHSV G47Δ expressing murine IL-12 (G47Δ-mIL12), antibodies to immune checkpoints (CTLA-4, PD-1, PD-L1), or dual combinations modestly extended survival of a mouse glioma model. However, the triple combination of anti-CTLA-4, anti-PD-1, and G47Δ-mIL12 cured most mice in two glioma models. This treatment was associated with macrophage influx and M1-like polarization, along with increased T effector to T regulatory cell ratios. Immune cell depletion studies demonstrated that CD4+ and CD8+ T cells as well as macrophages are required for synergistic curative activity. This combination should be translatable to the clinic and other immunosuppressive cancers.
Keywords: oncolytic virus, HSV, cancer stem cells, immunotherapy, glioma
eTOC blurb/In brief
Saha et al. show that the combination of an oncolytic virus expressing IL-12 with two immune checkpoint inhibitors, anti-CTLA-4 and anti-PD1 antibodies, can eradicate glioma in two mouse models. The therapeutic efficacy of the combination treatment depends on CD4+ and CD8+ T cells as well as macrophages.
Introduction
Cancer immunotherapy harnesses the patient’s immune system, both innate and adaptive, to attack and destroy tumors. It has elicited impressive therapeutic responses in some patients, but efficacy is significantly obstructed by immune suppression, tolerance, and ineffective activation. Major cellular players in immune suppression are T regulatory cells (Tregs), typically expressing FoxP3; innate tumor associated suppressive myeloid cells, expressing CD11b and Gr1; and M2 polarized tumor-associated macrophages (TAMs) (Ugel et al., 2015). T effector cell (Teff) function is regulated by co-stimulatory and co-inhibitory receptors and ligands. Cytotoxic T lymphocyte-associated protein-4 (CTLA-4), an immune checkpoint co-inhibitory receptor expressed on T cells, competes with the co-stimulatory receptor CD28 for binding of their ligands CD80 and CD86, decreasing activation of T helper cells and Teffs while stimulating Tregs (Sharma and Allison, 2015; Topalian et al., 2015). Programmed death-1 (PD-1), another immune checkpoint receptor, is expressed on activated T cells, B cells, natural killer (NK) cells, and myeloid cells. PD-1 is often upregulated in the tumor microenvironment, while its ligands PD-L1 (CD274, B7-H1), which also binds CD80, and PD-L2 (CD273, B7-DC) are upregulated in activated leukocytes and myeloid cells, as well as many cancer cells (Topalian et al., 2015). Thus, PD-1 and PD-L1 blocking antibodies are thought to act predominantly within the tumor. However, anti-tumor activity is dependent upon tumor antigen presentation, T cell activation, and tumor cell recognition and killing. Anti-CTLA-4 and anti-PD-1 antibodies regulate different inhibitory pathways on immune cells, promoting anti-tumor immunity through distinct and non-redundant immune evasion mechanisms (Curran et al., 2010; Topalian et al., 2015). Antibody blockers of immune checkpoints have demonstrated striking and durable clinical responses in a subset of solid tumors, but only in a fraction of patients (Sharma and Allison, 2015). Thus, it is critical to develop strategies that will improve response rates and expand the range of cancers that can be effectively treated.
Glioblastoma (GBM) is the most common primary malignant brain tumor and is almost always fatal (Carlsson et al., 2014). Despite advances in molecular understanding and therapies, clinical benefits have remained limited and life expectancy of GBM patients has only been extended to approximately 15 months (Carlsson et al., 2014). GBM stem-like cells (GSCs), or tumor initiating cells, are thought to be responsible for tumor maintenance, progression, recurrence, and resistance to therapy, and thus critical targets for therapy (Lathia et al., 2015). A major dilemma for immunotherapy is GBM-induced immunosuppression, including expression of immune checkpoint receptors in T cells, PD-L1 in tumors, and immunosuppressive molecules (ie., TGF-β, IDO, IL-10), and accumulation of Tregs and M2-like TAMs (Perng and Lim, 2015; Preusser et al., 2015b). There are over 10 clinical trials of checkpoint inhibitors for GBM (Preusser et al., 2015b). However, efficacy has been limited, while the combination of nivolumab (anti-PD-1) and ipilimumab (anti-CTLA-4) caused significant toxicity (Curry and Lim, 2015).
There are a relatively limited number of mouse syngeneic glioma models, many of which have immunogenic phenotypes (Maes and Van Gool, 2011; Oh et al., 2014). Studies have demonstrated therapeutic efficacy with immune checkpoint blockade (anti-CTLA-4, -PD-1, and -PD-L1) in orthotopic GBM models, all in immunogenic GL261 (Agarwalla et al., 2012; Reardon et al., 2016; Vom Berg et al., 2013; Wainwright et al., 2014; Zeng et al., 2013), except for one in SMA-560 (Fecci et al., 2007), and many involved combination therapies. We recently described a mouse immunocompetent GSC model (005), which forms intracranial tumors in syngeneic C57Bl/6 mice (Cheema et al., 2013). Mouse 005 GSCs were isolated from gliomas after lentiviral transduction of brains with activated H-Ras and Akt, in Trp53+/− mice, and have stem-like properties including self-renewal and differentiation into more mature phenotypes such as endothelial-like cells (Marumoto et al., 2009; Soda et al., 2011). Mouse 005 GSCs are highly tumorigenic and relatively non-immunogenic, lacking expression of co-stimulatory molecules (CD80 and CD86) and MHC I, which can be induced with IFNγ (Cheema et al., 2013). Brain tumors derived from mouse 005 GSCs are histologically similar to human GBM, with characteristics of tumor heterogeneity, invasiveness, vascularity, and an immunosuppressive microenvironment (Cheema et al., 2013). CT-2A mouse glioma cells isolated from a carcinogen-induced tumor also have a GSC-like phenotype (Binello et al., 2012; Oh et al., 2014; Seyfried et al., 1996).
Oncolytic viruses are a distinct class of anti-cancer agents with unique mechanisms of action: selectively replicating in and killing cancer cells (oncolysis), including GBM, spreading in the tumor while sparing normal tissue, and inducing anti-tumor immune responses (Saha et al., 2015). Replication-competent oncolytic herpes simplex viruses (oHSVs) are engineered for oncolytic activity and safety (Peters and Rabkin, 2015). OHSV talimogene laherparepvec (T-Vec) expressing GM-CSF produced durable responses in patients with advanced melanoma, similar to that seen with individual checkpoint inhibitors (Robert et al., 2015), but with a benign toxicity profile, and was recently approved by the FDA (Ott and Hodi, 2016). G47Δ is similar to T-Vec but without GM-CSF and with an additional mutation that makes it safe in the brain (Todo et al., 2001). G47Δ is efficacious against human GSCs (Wakimoto et al., 2009) and is in a clinical trial for recurrent GBM in Japan. While G47Δ was unable to effectively treat 005 intracerebral tumors, 2 injections of G47Δ expressing murine IL-12 (G47Δ-mIL12) significantly improved anti-tumor efficacy (Cheema et al., 2013). IL-12 is one of the more potent inducers of anti-tumor immunity and IFNγ, yet toxic when systemically administered to patients (Tugues et al., 2015). G47Δ-mIL12 treatment was associated with a reduction of Tregs within the tumor and enhanced T-cell-mediated anti-tumor immune responses; however, only a small proportion of G47Δ-mIL12 treated mice were cured (Cheema et al., 2013). We hypothesized that treatment with G47Δ-mIL12, which induces antitumor immune responses, would synergize with checkpoint blockade. With immunocompetent GBM models in hand, we explored the efficacy and immune changes occurring after treatment with cytokine-expressing oHSV and immune checkpoint inhibitor combinations.
Results
PD-L1 expression in mouse and human GSCs and G47Δ-mlL12 treatment
005 GSCs, cultured as spheres in serum-free media with growth factors, have stem-like properties (Cheema et al., 2013), including expression of CD133 (Prom1), about 30% of single cells proliferating and forming spheres, and transdifferentiation into vascular-like cells (data not shown). To explore GBM immunovirotherapy in 005 GSC-derived tumors, we first characterized the expression of PD-L1, an important immunosuppressive molecule, and the effects of G47Δ-mIL12 on tumor infiltrating immune cells. PD-L1 was only expressed on a low number of cells (18%), but was induced by IFNγ in almost all 005 GSCs in vitro (Figure 1A), reflective of the dynamic expression in patients (Mellman et al., 2016). G47Δ-E and G47Δ-mIL12 did not alter PD-L1 expression in vitro (Figure 1B). Both primary (Figure 1C, left panels) and recurrent (Figure 1C, right panels) human GSCs express PD-L1, with expression varying from 12% (in MGG4 or MGG8) to nearly 100% (in MGG31) of cells. MHC II was not expressed on 005 GSCs or induced by IFNγ (Figure S1A).
Figure 1. Characterization of mouse 005 and human GSCs.
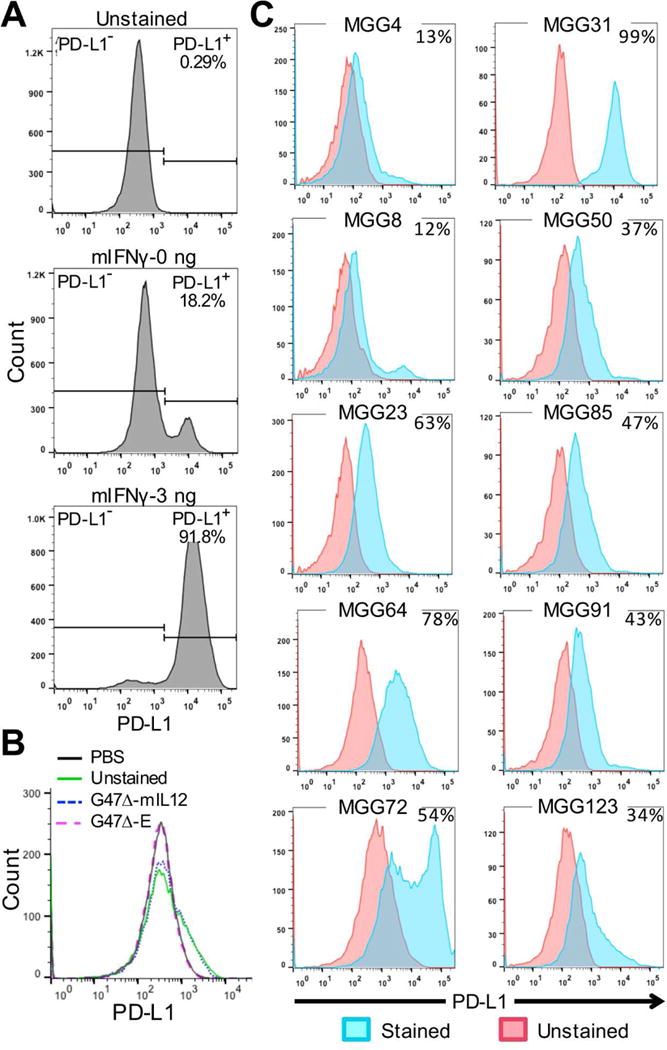
A. 005 GSCs cultured for 24 hr with or without murine IFNγ (mIFNγ: 0 or 3 ng/ml), stained for PD-L1, and analyzed by flow cytometry. B. 005 GSCs infected with G47Δ-E or G47Δ-mIL12 at MOI=1 for 24 hr, stained for PD-L1, and analyzed by flow cytometry. C. Human primary (left) and recurrent (right) GSCs cultured for 24 hr, stained for PD-L1 (cyan), and analyzed by flow cytometry. Percent of PD-L1+ cells indicated in upper right. See also Figure S1.
005 GSCs grow rapidly in vivo; however, in line with their stem-like properties, in vitro differentiation of 005 GSCs with serum prior to injection reduced the ability of these cells to propagate tumors in vivo (Figure S1B). G47Δ-mIL12 infection of GSC-derived brain tumors in vivo led to an almost 2-fold increase in the number of tumor infiltrating CD3+ lymphocytes (Figure 2A, B) and increased M1-like TAMs, as measured by iNOS+ and pSTAT1+ cells (Murray et al., 2014). G47Δ-mIL12 infection did not affect the number of total TAMs (CD68+), CD8+ T cells, granzyme B+ activated T cells, PD-L1+ cells, Ki67+ proliferating cells, or cleaved caspase 3+ apoptotic cells (Figure 2A, B, S1C, D). While IFNγ induced PD-L1 expression in 005 cells in vitro, G47Δ-mIL12 did not increase PD-L1 expression in vivo, and virus mediated changes were limited to increased proinflammatory markers and lymphocytes.
Figure 2. Immunohistochemical staining of immune cell markers after G47.
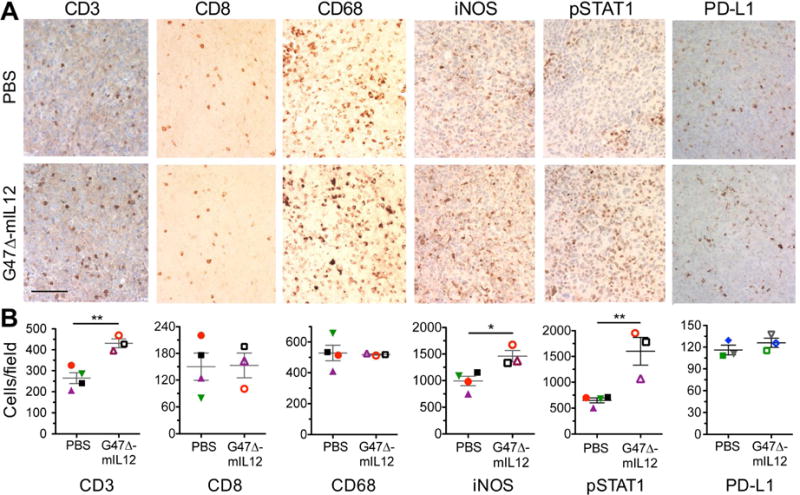
(A–B) Δ-mIL12 treatment. (A–B) 005 GSCs (2 × 104) implanted on day 0, injected intratumorally (IT) with G47Δ-mIL12 (1 × 105 pfu; n=3) or PBS (n=4) on days 18 and 24, animals sacrificed on day 25, and brains collected. Brain tumor sections were stained as indicated. For PD-L1 brain sections, 005 GSCs (2 × 104) implanted on day 0, injected IT with G47Δ-mIL12 (5 × 105 pfu; n=3) or PBS (n=3) on days 17, animals sacrificed on day 24, and brain tumor sections stained with anti-PD-L1 antibody. Representative images are presented; scale bar=100 μm (A). The number of positive cells from 3–9 fields/tumor section (1 section/mouse, except 3 for CD3) were counted (B). Average number of positive cells from each individual mouse is identified by symbol and color. The mean ± SEM of all mice is presented. Data were assessed by Student’s t test; *p<0.05; **p<0.001. See also Figure S1.
Individual immune checkpoint inhibitors only minimally extend survival in 005 GBM
To evaluate the activity of immune checkpoint inhibitors in the 005 GSC-derived brain tumor model, we administered anti-PD-1, anti-PD-L1, or anti-CTLA-4 antibodies intraperitoneally in mice with established orthotopic 005 tumors. Despite limited PD-L1 expression on 005 cells in vitro (Figure 1A), anti-PD-1 treatment, as well as anti-PD-L1 and anti-CTLA-4 treatments, modestly, but significantly, improved median survival compared to mock isotype control antibody treatment by 16, 25, and 20% respectively (Figure 3A–C). One explanation for the limited efficacy of anti-PD-1 or anti-PD-L1 treatment might be the inability of the antibodies to cross the blood-brain/tumor barrier and gain access to the tumor. However, anti-PD-1 and anti-PD-L1 antibodies were detected in 005 tumors 3 hr after antibody administration, but not in the surrounding normal brain (Figure 3D). Similar observations were made for anti-PD-L1 antibody in CT-2A tumors (Figure S2A). Despite curative outcomes from individual checkpoint inhibitors in other glioma models (Reardon et al., 2016), checkpoint inhibition only modestly improved survival in 005 tumors.
Figure 3. Treatment of 005 GSC-derived tumors with systemic immune checkpoint inhibitors, intratumoral G47 Δ-mIL12, or the combination prolongs survival.
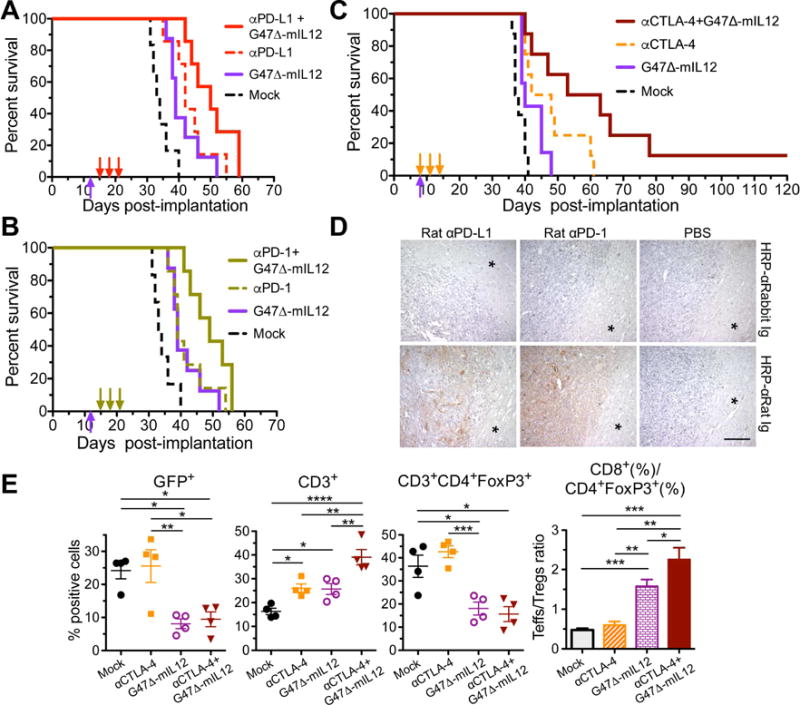
(A–B) Mice implanted with 2 × 104 005 GSCs on day 0, treated with G47Δ-mIL12 (5 × 105 pfu) or PBS injected IT on day 12 (upward arrow) and isotype control IgG (10 mg/kg), anti-(α)PD-L1 antibody (A), or anti-(α)PD-1 antibody (B) injected IP on days 15, 18 and 21 (downward arrows). Values from a single experiment, with Mock (treated with PBS and IgG) and G47Δ-mIL12 groups the same in A and B. Median survival of Mock (33.5 days; n=6) was compared to anti-PD-1 (39 days; n=8, p=0.02), anti-PD-L1 (42 days; n=7, p=0.003), or G47Δ-mIL12 (39 days; n=8, p=0.01) by Log-rank analysis. Similarly, G47Δ-mIL12 was compared to the combination of G47Δ-mIL12 with anti-PD-1 (49 days; n=7, p=0.02) or -PD-L1 (50 days; n=7, p=0.03), and antibodies were compared to the combination of G47Δ-mIL12 with anti-PD-1 (p=0.053) or anti-PD-L1 (p=0.08). Experiment was conducted once (A) or twice (B). C. Mice implanted with 2 × 104 005 GSCs on day 0 and treated with G47Δ-mIL12 or PBS injected IT on day 8 and anti-(α)CTLA-4 antibody or isotype control IgG (5 mg/kg) injected IP on days 8, 11 and 14 (n=8/group, except for G47Δ-mIL12 n=7). Median survival of Mock (37.5 days) was compared to anti-CTLA-4 (45 days; p=0.002) or G47Δ-mIL12 (40 days; p=0.03) alone by Log-rank analysis. Similarly, combination of G47Δ-mIL12 with anti-CTLA-4 (58 days) was compared to anti-CTLA-4 (p=0.05) or G47Δ-mIL12 (p=0.008). Experiment was conducted 2 times. D. Mice implanted with 2 × 104 005 GSCs on day 0, treated with PBS (right; n=2), rat anti-(α)PD-1 antibody (middle; 200 μg/mouse; n=2), or rat anti-(α)PD-L1 antibody (left; 200 μg/mouse; n=2) injected IP on day 25, and sacrificed 3 hr later. Antibodies were detected with HRP-conjugated anti-(α)rat Ig (brown; right) or control HRP-conjugated anti-(α)rabbit Ig (left). * normal brain adjacent to tumor. Scale bar=100 μm. E. 005 GSCs (1.5 × 105) implanted on day 0, treated with PBS or G47Δ-mIL12 (5 × 105 pfu) IT on day 11 and IgG or anti-CTLA-4 antibody (5 mg/kg) injected IP on days 11, 14, and 17 (n=4/group; Mock=PBS/IgG), and mice sacrificed on day 18. Tumors were harvested, cells stained with fluorochrome-conjugated anti-mouse antibodies, and multicolor FACS performed. Scatter plot (each animal 1 point) of the percentages of live sorted positive cells. The ratio of Teff to Treg ratio is presented in a bar graph. Mean ± SEM. Data were assessed by Student’s t test between indicated groups; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. See also Figure S2 and S3.
Combination of G47Δ-mIL12 with individual immune checkpoint inhibitors modestly improves efficacy
Similar to immune checkpoint inhibitors, G47Δ-mIL12, administered as a single intratumoral injection, had only a modest effect on 005 GSC-derived tumors (Figure 3A–3C). OHSV induces anti-tumor immune responses, which should enhance the activity of immune checkpoint inhibitors. Therefore, we examined the combination of G47Δ-mIL12 with individual immune checkpoint inhibitors. Mice bearing 005 GSC-derived tumors were treated with G47Δ-mIL12 on day 12, followed by systemic administration of anti-PD-L1 (Figure 3A), anti-PD-1 (Figure 3B), or isotype control antibodies on days 15, 18 and 21. The combination extended survival, but not significantly versus antibodies alone. With anti-CTLA-4, we initiated treatment earlier: G47Δ-mIL12 on day 8 and anti-CTLA-4 on days 8, 11 and 14 (Figure 3C), to provide more time for an adaptive immune response, in case that was a limitation in the prior experiment. Here, the combination significantly extended survival (p ≤ 0.05) and resulted in 1 long-term survivor, which had no evidence of tumor (Figure S2B). The different checkpoint inhibitors when combined with G47Δ-mIL12 resulted in similar modest extensions in survival.
G47Δ-mIL12 treatment reduces the percentage of 005 cells and Tregs
To assess the immune responses of combination anti-CTLA-4 and G47Δ-mIL12 treatment, the most effective combination, we collected tumor-containing brain quadrants 24 hr after the last antibody injection and performed multicolor flow cytometry of the brain tissue. An example of sequence and gating for flow cytometry of individual cell subtypes is illustrated in Figure S3A. At this time point, anti-CTLA-4 treatment alone did not alter the percentage of 005 GSCs (GFP+), while G47Δ-mIL12 treatment alone significantly reduced them (Figure 3E). This suggests that the effect of anti-CTLA-4 on tumor growth is limited in the first week, in contrast to virus, despite a somewhat longer overall survival. The virus both alone and in combination significantly reduced Tregs (CD4+FoxP3+; Figure 3E), as we reported previously (Cheema et al., 2013), and total FoxP3+ T cells (CD3+; Figure S3B). In contrast to what has been reported in melanoma (Simpson et al., 2013), anti-CTLA-4 alone did not reduce Tregs in the brain tumors (Figure 3E). The percentage of T cells (CD3+) was increased by each treatment alone (Figure 3E), as seen with G47Δ-mIL12 by IHC (Figure 2A, B), and then further with the combination (Figure 3E). An important parameter in immunotherapy success (Quezada et al., 2011) and increased survival in GBM (Sayour et al., 2015) is the ratio of Teffs (CD8+) to Tregs (CD3+/CD4+FoxP3+). This was increased by G47Δ-mIL12 alone and further in combination with anti-CTLA-4 (Figure 3E, S3C). There was no change in leukocytes (CD11b+) or activated T cells (CD3+CD69+) (Figure S3B). Though G47Δ-mIL12 alone and in combination with anti-CTLA-4 increased the Teff/Treg ratio, this was not sufficient to cure most of the mice.
Triple combination of anti-mPD-1 and -mCTLA-4 antibodies with G47Δ-mIL12 cures mouse GBM
Though anti-PD-1 or -CTLA-4 antibodies enhanced the antitumor effects of G47Δ-mIL12 in the 005 GSC-derived GBM model, the effects were limited and not long lasting (Figure 3A–C). Because anti-CTLA-4 and -PD-1 promote anti-tumor immunity through two independent immune pathways (Twyman-Saint Victor et al., 2015), we hypothesized that individual checkpoint inhibitors were insufficient to overcome GBM immunosuppression and the combination was necessary to unleash effective immunity. Using the same conditions as with the anti-CTLA-4 experiment (Figure 3C), we treated established 005 brain tumors with G47Δ-mIL12 or PBS plus anti-PD-1 and anti-CTLA-4, or isotype control antibodies (Figure 4A). Dual checkpoint inhibitors alone were more efficacious than either antibody alone, increasing median survival by 37% compared to 20% for anti-CTLA-4. However, the addition of G47Δ-mIL12 with anti-PD-1 and anti-CTLA-4 antibodies now resulted in 89% long-term survivors (Figure 4A). In 2 additional experiments, the cure rate for triple therapy was 4/6 and 5/7 mice (data not shown). The addition of checkpoint inhibitors did not alter early virus spread (Figure S4A) or increase toxicity (Figure S4B). The cured mice were then re-challenged on day 183 with a 5-fold increased number of 005 GSCs in the contralateral hemisphere and none developed tumors (Figure 4B, S4C), demonstrating immunologic memory for at least 6 months post-treatment, as well as no recurrence of the original tumor. This also supports the safety of the combination, as all mice were healthy at time of sacrifice, about 9 months after treatment. All age-matched naive mice challenged with 005 GSCs succumbed to tumor (Figure 4B, S4C). This demonstrates that curative therapy requires multiple therapeutic interventions (oHSV, IL12, anti-PD-1, and anti-CTLA-4).
Figure 4. Triple combination therapy in 005 and CT-2A glioma models.
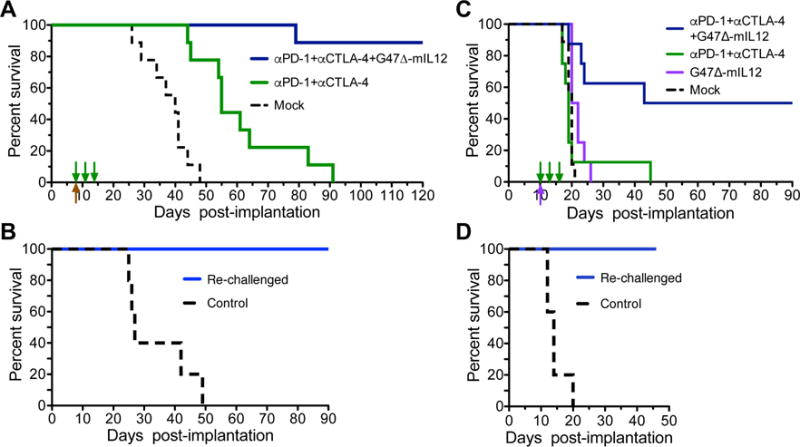
A. Mice implanted with 2 × 104 005 GSCs on day 0 and treated with G47Δ-mIL12 (5 × 105 pfu) or PBS injected IT on day 8 (upward arrow) and anti-CTLA-4 (5 mg/kg) and anti-PD-1 (10 mg/kg) or isotype control IgG (5 mg/kg hamster and 10 mg/kg rat IgG) IP on days 8, 11 and 14 (n=9/group; downward arrows). Median survival of Mock (PBS and IgG; 40 days) was compared to the combination of anti-PD-1 and anti-CTLA-4 (55 days; p=0.0002) by Log-rank analysis. Similarly, triple combination treatment with virus (89% of mice surviving long-term) was compared to the combination of anti-PD-1 and anti-CTLA-4 (p<0.0001) or Mock (p<0.0001). Experiment was conducted 3 times. B. Cured mice (n=8) from the triple combination experiment in (A) were re-challenged on day 183 with 5-fold increased number of 005 GSCs (1 × 105) in the contralateral hemisphere (re-challenged). Age matched (8 months) naive mice were implanted as controls (n=5). All re-challenged mice were alive at day 96 post-challenge without tumor; controls had a median survival of 27 days post-challenge (p=0.0001; Log-rank analysis). C. CT-2A cells (1 × 104) implanted in C57Bl/6 mice on day 0, injected IT with G47Δ-mIL12 (1 × 105 pfu) or PBS on day 10 and anti-CTLA-4 antibody and anti-PD-1 antibody or isotype control IgG (5 mg/kg hamster IgG and 10 mg/kg rat IgG) injected IP on days 10, 13 and 16. Median survival of Mock (PBS/IgG) treated mice (20 days; n=9) was compared to G47Δ-mIL12 (21 days; n=8) by Log-rank analysis (p=0.007). Median survival of mice treated with triple combination (66.5 days; 50% of mice surviving long-term) was compared to anti-PD-1 and -CTLA-4 (19 days; n=8, p=0.005) or G47Δ-mIL12 (p=0.01). D. Cured mice (n=4) from the triple combination experiment in (C) were re-challenged on day 109 with 5-fold increased number of CT-2A cells (5 × 104). Similar age naive mice (~6 months) were implanted as controls (n=5). All re-challenged mice were alive at day 46 post-challenge without tumor, and compared to control by Log-rank analysis (p=0.005). See also Figure S4.
We then examined triple combination therapy in another mouse syngeneic glioma model, CT-2A (Oh et al., 2014). Tumors derived from CT-2A glioma cells are more aggressive than those from 005 GSCs, with median survival of mock-treated mice only 20 days after intracranial implantation of 1 × 104 CT-2A cells (Figure 4C). After CT-2A implantation, mice were treated with G47Δ-mIL12 on day 10 and antibodies on days 10, 13, 16 (Figure 4C). Dual antibody (anti-PD-1 and anti-CTLA-4) treatment had no effect on median survival (day 19) compared to mock (day 20) or G47Δ-mIL12 (day 21), as opposed to what was seen with 005, possibly due to more rapid CT-2A tumor growth. However, triple combination treatment significantly extended survival (p < 0.01), with half the mice surviving long term (Figure 4C). These long-term survivors were then re-challenged on day 109 with a 5-fold increased number of CT-2A glioma cells in the contralateral hemisphere and none succumbed to tumors (Figure 4D, S4D), demonstrating immunologic memory, as well as no recurrence of the original tumor. All age-matched naive mice challenged with CT-2A glioma cells succumbed to tumor by day 20 (Figure 4D, S4D). These synergistic combination results in the CT-2A model, a more aggressive GBM tumor that is non-responsive to individual treatments, validate those in 005.
Effect of triple combination therapy on tumor-associated immune cells
To evaluate the phenotypes of tumor infiltrating cells, we used 2 complementary approaches; multicolor flow cytometry of cells harvested from the tumor-bearing quadrant of the brain and immunohistochemistry of cells present within the tumor. The number of live CD45+ cells in the non-tumor bearing quadrant was <0.3% of those in the tumor bearing quadrant. Dual antibody treatment (anti-PD-1 and anti-CTLA-4) significantly increased CD8+ T cells (Figure 5A) and the Teff to Treg ratio (Figure 5B, S5A), although there was no decrease in the proportion of Tregs (Figure 5B), as also seen with anti-CTLA-4 alone (Figure 3E). Dual antibody therapy also increased infiltration of CD11b+Gr1+ cells (neutrophils, myeloid-derived suppressor cells) (Figure S5A) and CD11b+CD45hi cells (peripheral macrophages), and decreased CD11b+CD45lo cells (microglia) (Figure 5C) (Badie and Schartner, 2000), without significantly changing overall CD11b+ cells (leukocytes/myeloid cells) (Figure 5A).
Figure 5. FACS analysis of treatment effects of triple combination therapy.
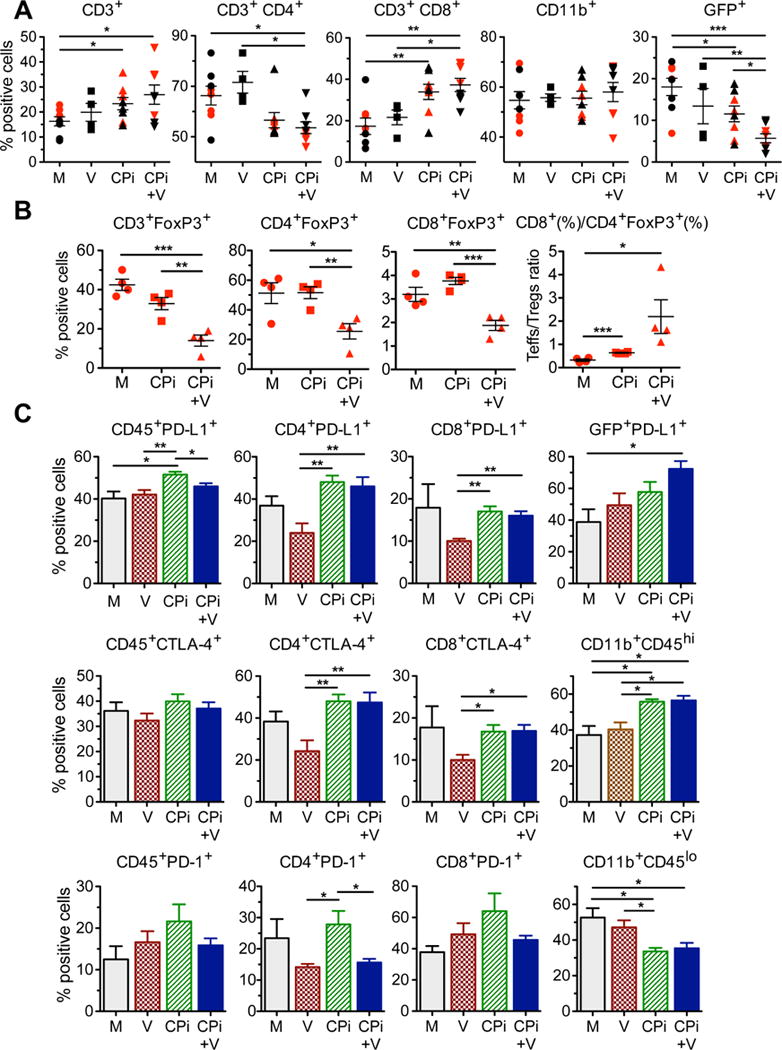
(A–C) Mice implanted with 005 GSCs (1.5 × 105) on day 0, treated with G47Δ-mIL12 (5 × 105 pfu) or PBS injected IT on day 11, and anti-PD-1 and anti-CTLA-4 antibody or rat and hamster IgGs (Mock) (5 mg/kg hamster IgG and 10 mg/kg rat IgG) injected IP on days 11, 14, and 17 (n=4), and sacrificed on day 18. Tumors harvested, dissociated cells stained with fluorochrome-conjugated anti-mouse antibodies, and multicolor FACS was performed. Percentages of live CD3+ cells, CD3+ sorted CD4+ and CD8+ subsets, live CD11b+ (monocytes, macrophages, NK, DC), and live CD45− GFP+ 005 cells from 2 independent experiments (red and black symbols) for M, CPi, V, and CPi+V groups (symbols are individual mice) were analyzed (A). The tumors from the experiment with red symbols in (A) were analyzed for CD3+FoxP3+ subtypes and ratio of CD8+ to CD4+FoxP3+ (B). The tumors from the experiment with black symbols in (A) were analyzed for immune checkpoint expression (PD-L1, CTLA-4, PD-1) on CD45+CD3+CD4+ and CD8+ subsets, CD45−GFP+, and CD11b+CD45hi and lo cells (C). M, Mock (PBS/IgG); V, virus (G47Δ-mIL12); CPi, checkpoint inhibitors (anti-PD-1+anti-CTLA-4); CPi+V, virus + checkpoint inhibitors (G47Δ-mIL12+anti-PD-1+anti-CTLA-4). Data are mean ± SEM and assessed by Student’s t test between indicated groups; *p<0.05, **p<0.01, ***p<0.001. See also Figure S5.
The triple combination (dual antibodies and G47Δ-mIL12) led to a decrease in CD4+ T cells, especially Tregs, and an increase in CD8+ T cells (Figure 5A, B), resulting in an increase in the Teff to Treg ratio of 6.9-fold and 3.4-fold compared to mock (p<0.05) and dual antibody (not significant), respectively (Figure 5B). CD8+FoxP3+ (suppressor or regulatory) T cells, which are not well characterized (Le et al., 2011; Peng et al., 2012), were also decreased, but they represent a much smaller proportion of CD8+ compared to CD4+ T cells (Figure 5B). Virus treatment reduced, but not significantly, PD-L1 and CTLA-4 expression on T cells, which was then significantly increased when combined with checkpoint blockade (Figure 5C). PD-L1+ 005 cells were significantly increased by triple combination treatment. PD-1 expression on immune cells varied compared to mock, but not significantly (Figure 5C), except for CD11b+ cells (Figure S5A). Antibody treatment alone reduced the number of GFP+ 005 cells (1.6-fold vs mock), while the addition of G47Δ-mIL12 further reduced it (3.2-fold vs mock) (Figure 5A). The number of GSC-selective IFNγ+ splenocytes was almost doubled after dual antibody treatment, possibly indicative of systemic blockade effects, which was not further increased after combination treatment with G47Δ-mIL12 (Figure S5B). Interestingly, splenocytes from triple combination treatment were more active against serum-differentiated 005 cells in vitro (Figure S5C). Overall, these results indicate that the major change engendered by triple combination therapy, compared to dual checkpoint inhibitors or G47Δ-mIL12, was an increase in the Teff/Treg ratio and a decrease in tumor cells.
We evaluated a different set of immune cell markers expressed in the tumor immunohistochemically. CD68+, pSTAT1+, iNOS+, and Ki67+ cells were all significantly increased in the triple combination group (Figure 6A, B), while cleaved caspase 3+, granzyme B+, CD3+, and CD8+ cells were increased but non-significantly due to large variability between mice (Figure S6A, B). In contrast, there was a large decrease in PD-L1+ cells in both the dual checkpoint inhibitors and triple combination groups (Figure 6A, B), but not virus alone (Figure 2B). Dual and triple combinations resulted in a pronounced increase in M1-like TAMs at the tumor periphery (Figure S6C, D). CD4+ T cells were broadly distributed in tumors, increasing after triple combination therapy (Figure S6E), as were T-bet+ cells, a marker for Th1 cells (Figure S6F, G). Ki67 and pSTAT1 can be expressed in tumor and immune cells. To distinguish which cell type, we performed double staining for Ki67 and CD3 (for proliferating T cells) and pSTAT1 and CD68 (for M1-like TAMs) (Figure 6C). In the triple combination, 67% of CD68+ cells were also pSTAT1+ (Figure 6D), similar to the percent of pSTAT1+ cells that were CD68+ (74%) (data not shown), indicating that the increase in pSTAT1, and likely iNOS, seen after triple combination therapy was mostly due to M1-type TAMs. 46% of CD3+ cells were Ki67+ (Figure 6D), similar to the percent of Ki67+ cells that were CD3+ (45%) (data not shown), indicating that the increase in Ki67 was largely due to activated proliferating T cells. Overall, these immunohistochemistry results revealed that triple therapy induced T cell activation and an influx of TAMs and M1-like polarization.
Figure 6. Immunohistochemical staining of tumor infiltrating cells.
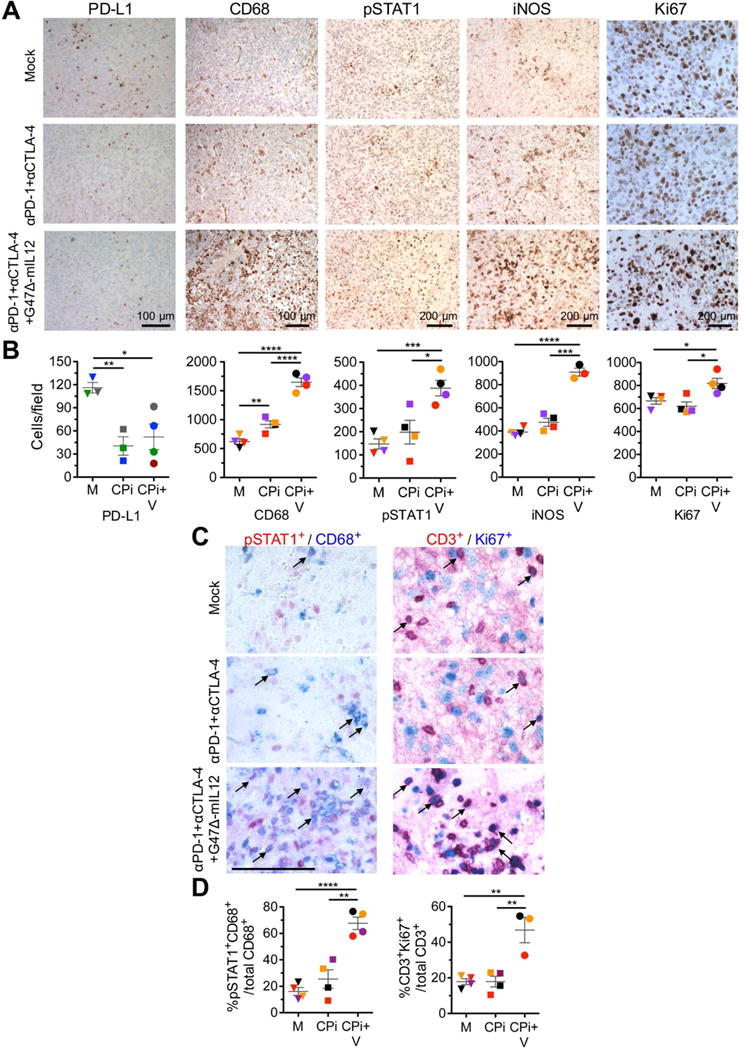
(A–D) Mice implanted with 005 GSCs (2 × 104) on day 0, treated with G47Δ-mIL12 (5 × 105 pfu) or PBS injected IT on day 17, and checkpoint inhibitors anti-PD-1 antibody and anti-CTLA-4 antibody or rat and hamster IgGs (5 mg/kg hamster IgG and 10 mg/kg rat IgG) injected IP on days 17, 20, and 23 (n=4), sacrificed on day 24, and brain tumor sections stained as indicated (Mock; PBS/IgG). Representative images with positive cells stained brown are presented (A). PD-L1 staining was from a separate experiment, but same treatment as above (n=3 or 4). Scale bars=100 or 200 μm as indicated. Number of positive cells per field in (A) were counted (3–5 fields/section/mouse for CD68 and Ki67; 8–10 fields/section/mouse for pStat1 and iNOS; 4 fields/section and 2 sections/mouse for PD-L1), and individual mice in each group are identified by color (B). Representative images with positive cells stained red (pSTAT1 +, CD3+), blue (CD68+, Ki67+), and red/blue colocalized (examples indicated with arrows) are presented (C). Scale bar=100 μm. Brain sections were incubated sequentially with primary (pSTAT1 or CD3 rabbit antibody) and secondary antibodies (AP-conjugated anti-rabbit Ig), followed by red color development. The same sections were then incubated with primary (CD68 or Ki67; rabbit antibody) and secondary antibodies (AP-conjugated anti-rabbit Ig), followed by blue color development. Number of positive cells per field were counted (8–10 fields/section/mouse) and percent of double positive cells plotted (D) with individual mice identified by color. M, Mock (PBS/IgG); CPi, checkpoint inhibitors (anti-PD-1+anti-CTLA-4); CPi+V, checkpoint inhibitors + virus (G47Δ-mIL12+anti-PD-1+anti-CTLA-4). Data are mean ± SEM, assessed by Student’s t test between indicated groups; *p<0.05, ***p<0.001, ****p<0.0001. See also Figure S6.
Depletion/Inhibition of immune cell subtypes
To address whether the changes seen in tumor infiltrating immune cells are necessary to elicit triple therapy efficacy, we performed antibody depletion studies of CD4+ and CD8+ cells (Figure S7), clodronate liposome (clodrosome) depletion of peripheral macrophages (Figure S7), and inhibition of CSF-1R to target TAMs (infiltrating macrophages and resident microglia, and MDSCs) using brain penetrant BLZ945 (Pyonteck et al., 2013). Both clodrosomes and BLZ945 similarly blocked most triple therapy efficacy (Figure 7A), resulting in extended survival akin to single agent treatment groups (Figure 3), indicating that infiltrating macrophages are key contributors to therapy. Both clodrosomes and BLZ945 reduced F4/80+ TAMs to levels of mock treatment, while CD68+ TAMs were not significantly affected compared to triple therapy (Figure 7B). Interestingly, clodrosomes increased CD8+ tumor infiltrating lymphocytes (TILs) well above triple therapy levels, while BLZ945 decreased CD8+ TILs, even below that seen upon mock treatment (Figure 7B). Depletion of CD8+ cells reduced efficacy to that of single agents (Figure 7A). Surprisingly, anti-CD4 antibodies completely abrogated all efficacy (Figure 7A). In addition to target cell depletion, anti-CD8 antibodies also reduced CD4+ TILs and F4/80+ TAMs to the mock level, and increased CD68+ TAMs (Figure 7B). Depletion of CD4 also reduced TAMs, with CD68+ cells down to mock levels and F4/80+ cells significantly lower than mock (Figure 7B). This confirms the complexity of the response necessary to cure GBM and indicates that CD4+ cells play an essential role in the efficacy observed with all agents. The impact of immune cell depletion on other cell types signifies the interconnectedness of the responding cells and confounds interpretation of mechanism.
Figure 7. Depletion/inhibition of immune cell subtypes abrogates triple combination therapy.
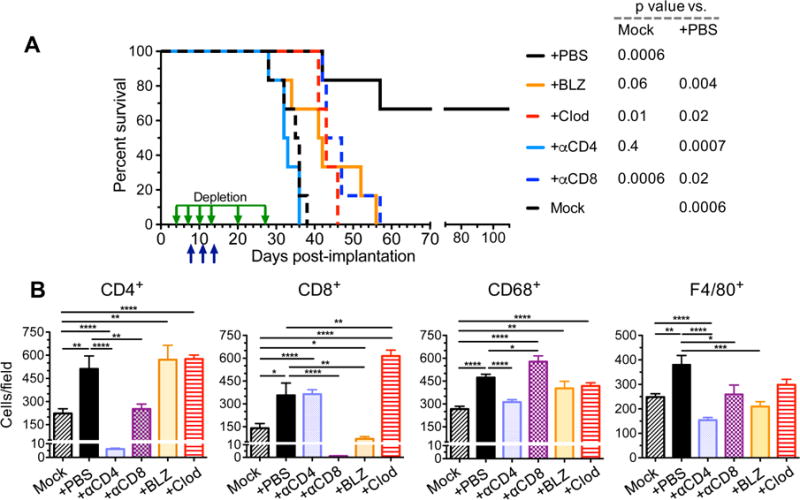
A. C57Bl/6 mice implanted with (2 × 104) 005 GSCs on day 0 and treated with G47Δ-mIL12 or PBS injected IT on day 8 and anti-CTLA-4 and anti-PD-1 antibodies or isotype control IgG (5 mg/kg hamster IgG and 10 mg/kg rat IgG) injected IP on days 8, 11 and 14 (n=6/group; upward arrows). Depletion antibodies against CD4 or CD8 (10 mg/kg) or clodronate liposomes (Clod; first injection 50 mg/kg followed by 25 mg/kg) were injected IP on days 4, 7, 10, 13, 20, and 27 (downward arrows), or BLZ945 (BLZ; 200 mg/kg) was gavaged for two cycles from days 6–10 and days 12–16 in triple therapy mice. Median survival of mice was determined: Mock (PBS/IgG/liposome/20% captisol), 35.5 days or triple therapy +αCD4 antibody, 32.5 days; +BLZ, 41.5 days; +αCD8 antibody, 45 days; +Clod, 43 days; +PBS (IgG/liposome/20% captisol). +PBS was compared to +BLZ (p=0.004), +Clod (p=0.02), +αCD4 (p=0.0007), +αCD8 (p=0.02), or Mock (p=0.0006) by Log-rank analysis. Similarly, Mock was compared to +PBS (p=0.0006), +BLZ (p=0.06), +Clod (p=0.01), +αCD4 (p=0.4), or +αCD8 (p=0.0006). B. C57Bl/6 mice implanted with 005 GSCs (2 × 104) on day 0 and treated with G47Δ-mIL12 (5 × 105 pfu) or PBS injected IT on day 18 and anti-CTLA-4 and anti-PD-1 antibodies or isotype control IgGs (5 mg/kg hamster IgG and 10 mg/kg rat IgG) injected IP on days 18, 21 and 24 (n=2/group). Depletion antibodies (αCD4, αCD8; 10 mg/kg) or Clod (first injection 50 mg/kg followed by 25 mg/kg) were injected IP on days 14, 17, 20, and 23, or BLZ (200 mg/kg) gavaged from days 16–20 and 22–25. Twenty-four hr after the last immune checkpoint injection or 8 hr after the last BLZ gavage, animals were sacrificed on day 25 and brains collected. Brain tumor sections (2 sections/mouse, at least 200 μm apart from each other) were stained for CD4, CD8, CD68, and F4/80. Positive cells were counted (5 fields/section for CD4+ and CD8+, 8 fields/section for CD68+, and 6 fields/section for F4/80+) and presented as mean ± SEM. Data were assessed by Student’s t test between indicated groups *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Only significant differences between Mock or triple therapy and other treatments indicated. See also Figure S7.
Discussion
GSCs contribute to local and systemic immunosuppression of GBM, including downregulation of MHC I, expression of immunosuppressive factors, inhibition of T cell proliferation, and recruitment and M2-like polarization of TAMs (Di Tomaso et al., 2010; Wu et al., 2010; Zhou et al., 2015). This systemic and local immunosuppression has contributed to limited improvements in GBM patient outcomes, especially after vaccine or immunotherapy trials (Curry and Lim, 2015). In order to model GSC-mediated immunosuppression and to evaluate therapeutic countermeasures, we used the immunocompetent 005 GSC-derived GBM model, which recapitulates GBM hallmarks (Cheema et al., 2013). In this model, multiple agents (G47Δ-mIL12, anti-CTLA-4 and anti-PD-1 antibodies) were required to cure a majority of mice of established tumors.
There are currently limited prognostic indicators for checkpoint inhibitor efficacy. High mutational load with increased neoantigens or virus-associated cancers (Nghiem et al., 2016) correlate strongly with checkpoint inhibitor efficacy in patients, while those with low mutational loads, like GBM, exhibit little response (Topalian et al., 2016). The recent failures of nivolumab in the CheckMate-143 trial in recurrent GBM and CheckMate 026 trial as first-line therapy in non-small-cell lung cancer highlights checkpoint limitations, even in potentially responsive tumors. Thus, GBM responsiveness may be facilitated by the expression of virus antigens and release of cellular antigens from oHSV-infected cells. PD-L1+ tumors have also been associated with better responses to anti-PD-1 and -PD-L1 antibodies, although many patients with low PD-L1 expression also respond (Topalian et al., 2016). PD-L1 is expressed in most GBMs; however, there are conflicting results about the proportion of positive tumor cells (~3% vs 40%) (Preusser et al., 2015a). IL-12 is a strong inducer of IFNγ expression and IFNγ is a strong inducer of MHC I and PD-L1 expression in tumor cells (Mandai et al., 2016). However, G47Δ-mIL12 did not induce PD-L1 expression in 005 GSCs, in vitro or in vivo. Decreases in Tregs, with associated increases in the Teff to Treg ratio, have been associated with checkpoint blockade responses (Quezada et al., 2011; Simpson et al., 2013). We previously showed that G47Δ treatment alone of 005 tumors reduced tumoral Tregs, which were further reduced by IL-12 expression (Cheema et al., 2013). Here, a single intratumoral injection of G47Δ-mIL12, but not anti-CTLA-4 treatment, significantly reduced Tregs and increased the Teff to Treg ratio, but efficacy remained limited, suggesting that other immune effects were necessary for curative activity.
Preclinical immunotherapy studies in GBM have been hampered by the lack of representative murine models. Almost all successes reported to date with checkpoint blockade have used the GL261 model (Agarwalla et al., 2012; Reardon et al., 2016; Vom Berg et al., 2013; Wainwright et al., 2014; Zeng et al., 2013). However, the lack of similar robust responses in the clinic suggests that the 005 model, which is more difficult to treat, may be more representative. GL261, which expresses MHC I (Agarwalla et al., 2012), is immunogenic, whereas 005 GSCs are not. GL261 was carcinogen-induced, leading to a high mutation load, while 005 contains only 2 known somatic mutations. Also, the 005 model is enriched in GSCs, which express immunosuppressive factors and generate morphologically heterogeneous tumors. Differences between mouse and human tumors could also underlie some of the disparities in response. Mouse tumors are less necrotic than human tumors, and their small size makes delivery of virus or antibodies easier. The sequencing of virus and antibody administration is also likely to affect therapy, as described for oncolytic vaccinia virus (Liu et al., 2017; Rojas et al., 2015). Here, we mostly administered oHSV at the time of initial antibody treatment. Because anti-CTLA-4 affects immune priming, this may be appropriate. However, anti-PD-1 acts on activated immune cells (Curran et al., 2010; Topalian et al., 2015) and thus might benefit from delayed treatment after virus.
Single checkpoint inhibitors have been tested in combination with oncolytic viruses other than oHSV in mouse tumor models, although not in the brain, and have been shown to improve antitumor activity (Liu et al., 2017; Rajani et al., 2016; Rojas et al., 2015; Shen et al., 2016; Woller et al., 2015; Zamarin et al., 2014). A phase Ib clinical trial was recently completed combining intratumoral FDA approved oHSV T-Vec with ipilimumab (anti-CTLA-4) in unresectable melanoma (Puzanov et al., 2016). Importantly, the incidence of grade 3/4 adverse events was similar to that for ipilumumab alone, while efficacy was greater than for historical controls for either monotherapy (objective and durable response rates) (Puzanov et al., 2016). The unique immune characteristics and additional safety issues associated with therapy in the brain complicate combination approaches with virus. The safety of T-Vec in the brain is unclear, so safer oHSV such as G47Δ will likely be necessary for GBM trials. Dual blockade of two immune checkpoints has been much more efficacious than monotherapy in preclinical tumor models (Curran et al., 2010; Lussier et al., 2015), including in a VSV vaccine study for glioma (Cockle et al., 2016), and in clinical trials, although associated with more frequent and serious adverse events (Antonia et al., 2016; Larkin et al., 2015). In the 005 GSC model, dual checkpoint blockade was more efficacious as well, but the effects were not long lasting, while in CT-2A they did not produce any survival advantage. This was not due to the inability of checkpoint antibodies to cross the blood-brain/tumor-barrier to reach 005 or CT-2A tumors and suggests that checkpoint blockade alone will be insufficient in patients.
The triple combination of G47Δ-mIL12 with anti-CTLA-4 and anti-PD-1 antibodies acted synergistically and cured most of the mice. Importantly, none of the cured re-challenged mice exhibited any pathological symptoms up to almost 9 months after treatment. This synergy may permit decreased dosing of the checkpoint inhibitors to potentially reduce toxicity in the clinic. Depletion of CD8+ T cells or peripheral macrophages abrogated triple combination synergy, while CD4+ depletion blocked all treatment effects. Treatment-mediated decreases in Tregs suggest a key role for CD4+ effector cells, while the lack of MHC II expression on 005 GSCs in vitro suggests indirect effects. A growing number of studies describe CD4+ T cell mediated tumor destruction (Haabeth et al., 2014), including indirect killing of MHCII− tumor cells after antigen presentation by MHCII+ TAMs, CD4+ cytokine activity and TAM induction (Corthay et al., 2005; Perez-Diez et al., 2007), and CD4+ Th17 activation of CD8+ T cells (Martin-Orozco et al., 2009). Interestingly, CD4+ T cell depletion also blocked the therapy-mediated increase in TAMs.
G47Δ-mIL12 induces M1-like polarization (iNOS+ and pSTAT1+) in TAMs, possibly because of IL-12 induced IFNγ expression, which is further increased with triple combination therapy due to an influx of TAMs (CD68+). TAMs are typically activated to undergo polarization into opposite phenotypes, classically activated or M1, associated with inflammation and immunity (anti-tumoral), and alternatively activated or M2, associated with repair and immune-suppression (pro-tumoral) (Mantovani and Allavena, 2015). This nomenclature is an oversimplification because of macrophage plasticity in response to signals in the microenvironment and the diversity of phenotypes falling between the 2 extremes but reflects the counteracting activities (Murray et al., 2014). Typically, TAMs have been considered “bad players”, correlating with poor prognosis in cancer patients (Noy and Pollard, 2014). In GBM, TAMs, including microglia, are the most prevalent immune cells in the tumor and are predominantly M2-like (Kennedy et al., 2013). GSCs induce and recruit M2-like TAMs (Wu et al., 2010; Zhou et al., 2015). Microenvironment signals, such as IFNγ, can skew TAM polarization from M2 to M1-like, with tumor suppressive and anti-angiogenic properties (Rolny et al., 2011; Yuan et al., 2015). Attempts have been made to pharmacologically reduce TAMs in glioma, for example with CSF-1R inhibitors that decreased M2-like TAMs and increased survival in mouse transgenic and human xenograft GBM models (Pyonteck et al., 2013). Unfortunately, CSF-1R inhibitor PLX3397 had no effect in a phase II clinical trial in recurrent GBM (Butowski et al., 2016). We found that BLZ945, another CSF-1R inhibitor, nullified the effect of triple combination therapy. This might be due to effects on TAMs, tumoral macrophage-DCs or MDSCs, or the large reduction in CD8+ TILs. However, it suggests that CSF-1R inhibition combined with immunotherapy might not be beneficial in the clinic.
In conclusion, we demonstrate that a triple combination of oHSV expressing IL-12 and two immune checkpoint inhibitors, anti-PD-1 and anti-CTLA-4 antibodies, was necessary to safely overcome the immune suppressive microenvironment and eliminate tumors using a representative immunocompetent model of GBM, derived from GSCs, that recapitulates human disease. This suggests that multiple distinct immunotherapeutic strategies (i.e., oncolytic viruses, immunomodulatory cytokines, and multiple checkpoint inhibitors) will likely be required to significantly impact GBM in the clinic, and possibly other less immunogenic tumors. The major cellular hallmarks of this combination therapy were an increase in M1-like polarized TAMs and total dependence on CD4+ cells, in addition to an increase in Teffs and decrease in Tregs. This indicates that strategies that increase the number of M1-like TAMs and boost CD4+ T cell activity will be important to effectively treat GBM, and likely other checkpoint inhibitor non-responding tumors outside the brain.
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Samuel D. Rabkin (Rabkin@mgh.harvard.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
Mouse Glioma Cells
Mouse 005 GSCs (no information about sex is available) are GFP+ (Marumoto et al., 2009). They were cultured as spheres in serum-free stem cell medium composed of advanced DMEM/F12 medium (ThermoFisher Scientific), supplemented with 2 mM L-glutamine (Corning), 1% N2 supplement (ThermoFisher Scientific), 2 μg/mL heparin (Sigma-Aldrich), 0.5% penicillin G-streptomycin sulfate-amphotericin B complex (Corning), recombinant human EGF (20 ng/mL; R&D systems), and recombinant human FGF-basic (20 ng/ml; Peprotech) (Cheema et al., 2013). Spheres were dissociated with Accutase (Innovative Cell Technologies) for passaging. Mouse CT-2A glioma cells (no information about sex is available) were cultured on plastic in DMEM-10% fetal calf serum. Cells were dissociated with 0.05% Trypsin/0.53 mM EDTA. Both 005 GSCs and CT-2A were cultured at 37°C in 5% CO2. Only early passages since cells were obtained were used and these were mycoplasma-free (LookOut mycoplasma kit; Sigma).
Human Primary and Recurrent GSCs
Human primary (MGG4, MGG8, MGG23, MGG64, and MGG72) and recurrent (MGG31, MGG50, MGG85, MGG91, and MGG123) GSCs (no information about sex is available) were isolated from discarded GBM specimens according to a protocol approved by the Institutional Review Board. Briefly, mechanically minced tissues were digested with 0.1% Trypsin (Invitrogen) and 10 U/mL of DNaseI (Promega) at 37°C for 45 min. After washing, tissues were triturated by pipetting and passed through a 100 μm cell strainer. Both primary and recurrent GSCs were cultured at 37°C in 5% CO2 as spheres in EF20 stem cell medium composed of neurobasal medium (ThermoFisher Scientific), supplemented with 3 mM L-glutamine, 1% B27 supplement (ThermoFisher Scientific), 0.5% N2 supplement, 2 μg/mL heparin, 0.5% penicillin G-streptomycin sulfate-amphotericin B complex, recombinant human EGF (20 ng/mL), and recombinant human basic-FGF (20 ng/ml), and dissociated with Accutase or NeuroCult™ Chemical Dissociation Kit (Stemcell Technologies). Only early passage cells were used and these were mycoplasma-free (LookOut mycoplasma kit; Sigma). The characteristics of some GSCs were described previously (Nigim et al., 2015; Wakimoto et al., 2009; Wakimoto et al., 2012).
In Vivo Mouse Studies
Female C57Bl/6 mice (aged 8–9 weeks) were obtained from the NCI (Frederick, MD) and housed under BL2 conditions. All mouse procedures were approved by the subcommittee on research animal care at Massachusetts General Hospital.
Intracranial Tumor Models
Dissociated 005 GSCs (2 × 104 cells) or CT-2A cells (1 × 104 cells) in 3 μl PBS were implanted stereotactically into the striatum (2.2 mm lateral from Bregma and 2.5 mm deep) to generate intracranial tumors. On indicated days after tumor implantation (8 or 12 for 005 or 10 for CT-2A), mice were randomly divided into groups and intratumorally (IT) injected (same stereotactic coordinates) with G47Δ-mIL12 or PBS in 2 μl. Immune checkpoint inhibitor antibodies anti-mPD-1 (rat clone RMP1-14; 10 mg/kg), anti-mPD-L1 (rat clone 10F.9G2; 10 mg/kg), or anti-mCTLA-4 (Syrian hamster clone 9H10; 5 mg/kg), and isotype control antibodies rat IgG2a (clone LTF-2) or Syrian hamster IgG, or combinations (same amounts of each antibody) were from BioXcell and administered 3 times intraperitoneally (IP). Mice surviving long-term were re-challenged with an increased dose of cells (1 × 105 for 005 or 5 × 104 for CT-2A) in the contralateral hemisphere. For survival depletion/inhibition studies, C57Bl/6 mice were implanted with (2 × 104) 005 GSCs on day 0 and treated with G47Δ-mIL12 or PBS injected IT on day 8 and anti-CTLA-4 and anti-PD-1 antibodies or isotype control IgG (5 mg/kg hamster IgG and 10 mg/kg rat IgG) injected IP on days 8, 11 and 14. Depletion antibodies (αCD4, αCD8, or rat IgG; 10 mg/kg), clodronate liposomes or control liposomes (first injection 50 mg/kg, followed by 25 mg/kg) were injected IP on days 4, 7, 10, 13, 20, and 27, or CSF-1R inhibitor BLZ945 dissolved in 20% captisol (Captisol®) or only 20% captisol were gavaged orally for two cycles from days 6–10 and 12–16. Mice were followed for neurological symptoms and euthanized before becoming moribund. Animal caretakers were blinded to the treatment. Tumor growth or absence was evaluated in sacrificed mice macroscopically or after histological staining of sections.
METHOD DETAILS
Viruses
G47Δ-mIL12 was constructed from G47Δ (deletions in γ34.5 and α47 genes and inactivating insertion of LacZ into ICP6 (Todo et al., 2001)) by insertion of CMV IE promoter driven mouse IL-12 cDNA (p35 and p40 separated by bovine elastin motifs) into the ICP6 gene (Cheema et al., 2013). G47Δ-E was constructed as G47Δ-mIL12 except it is lacking the IL12 transgene (Cheema et al., 2013). Viruses were grown in Vero cells after low multiplicity infection and purified after freeze-thaw-sonication by benzonase treatment, low-speed centrifugation, filtration, high-speed centrifugation and resuspension in PBS/10% glycerol.
GSC Differentiation
To differentiate GSCs, cells were grown in advanced DMEM/F12 media containing 10% heat-inactivated fetal calf serum (HyClone) on plastic for 3 passages.
Histochemistry
Sections were deparaffinized in xylene twice 10 min each, followed by gradual rehydration using 100%, 90% and 70% ethanol treatment (5 min each). Sections were left in distilled water for 5 min, followed by dipping sections in hematoxylin container for 1 min, washing in tap water for 5 min, dipped into Eosin Y (1% alcoholic) for 30 s, followed by gradual dehydration using 95% ethanol (twice 5 min each) and 100% ethanol (twice 5 min each), treated with xylene twice for 10 min each, and mounted in xylene-based media (Cytoseal XYL (Thermo Scientific)).
To examine virus (LacZ) distribution, mice were implanted with 005 (5 × 105) on day 0. G47Δ-mIL12 (1 × 106 pfu/mouse) or PBS was injected IT and anti-CTLA-4 and anti-PD-1 or isotype control IgGs (5 mg/kg hamster IgG and 10 mg/kg rat IgG) were injected IP on day 19 (n=2/group). 36 hr after virus/antibody injections, mice were perfused with ice-cold PBS, followed by brain fixative (3% paraformaldehyde + 5 mM EGTA + 2 mM MgCl2 in 0.1M Pipes; pH 7.3). Brains were removed and placed in brain fixative for 1 hr, submerged in X-gal substrate solution (X-gal 1 mg/ml + 5 mM potassium ferricyanide + 5 mM potassium ferrocyanide + 2 mM MgCl2 + 0.01% sodium deoxycholate + 0.02% NP-40 in PBS; pH 7.3) for 3 hr at 34°C, washed in PBS, and submerged in 30% sucrose/2 mM MgCl2 in PBS overnight. Frozen brains in OCT were sectioned (20–30 μm) on a cryostat and mounted on slides. Slides were dried at room temperature for 30 min, post-fixed with 0.2% glutaraldehyde / 2% paraformaldehyde in PBS for 10 min, followed by 2 washes in PBS. Sections were incubated with X-gal substrate solution at 37°C for 4 hr in a humidified chamber, washed with tap water, counter-stained with hematoxylin, dehydrated, and mounted in CYTOSEAL™ XYL.
Immunohistochemistry for Antibody Detection
To determine whether antibodies cross the blood brain/tumor barrier and enter the tumor, mice were implanted with 005 GSCs (2 × 104) or CT-2A cells (1 × 104) on day 0, injected IP with PBS, rat anti-mPD-1 (200 μg/mouse), or rat anti-mPD-L1 (200 μg/mouse) on day 25, or rat anti-mPD-L1 (500 μg/mouse) on day 20 for CT-2A, and perfused 3 hr later with ice-cold PBS (to clear blood) followed by 4% paraformaldehyde. Brains were collected, post-fixed in 10% formalin overnight, saturated with 30% sucrose at 4°C for 48 hr, embedded in Tissue-Tek OCT, and frozen at −80°C. Ten μm sections were air-dried, followed by wash and rehydration in PBS, incubation with 3% H2O2, washed in PBS, incubated with 5% bovine serum albumin and 10% goat serum, and then with HRP-conjugated anti-rat Ig, control HRP-conjugated anti-rabbit Ig, or PBS, followed by DAB staining (DAKO). Sections were incubated with a drop of DAB (prepared by adding 1 drop of chromogen in 1 ml of DAB substrate) until developed brown color, rinsed with distilled water, counter-stained with 1:3 diluted hematoxylin for 20 s, washed in running tap water for 5 min, dehydrated with series of graded ethanol and cleaned with xylene, and mounted in CYTOSEAL™ XYL.
Immunohistochemistry for Infiltrating Immune Cells
To analyze tumor infiltrating immune cells, mice were implanted with 005 GSCs (2 × 104) on day 0, injected IT with PBS or G47Δ-mIL12 (1 × 105 pfu) on days 18 and 24, and sacrificed 24 hr later. For combination therapy, tumor-bearing mice were treated with PBS or G47Δ-mIL12 (5 × 105 pfu) injected IT on day 17 and rat and hamster IgGs or anti-mPD-1 (10 mg/kg) and anti-mCTLA-4 (5 mg/kg) injected IP on days 17, 20, and 23, and sacrificed 24 hr later. For depletion/inhibition studies, mice were implanted with 005 GSCs (2 × 104) on day 0 and treated with G47Δ-mIL12 (5 × 105 pfu) or PBS injected IT on day 18 and anti-CTLA-4 and anti-PD-1 antibodies or isotype control IgGs (5 mg/kg hamster IgG and 10 mg/kg rat IgG) injected IP on days 18, 21 and 24. Depletion antibodies (anti-CD4, anti-CD8, or rat IgG; 10 mg/kg), clodronate liposomes or control liposomes (Standard Macrophage Depletion kit) (first injection 50 mg/kg, followed by 25 mg/kg) injected IP on days 14, 17, 20, and 23, or CSF-1R inhibitor BLZ945 (200 mg/kg) dissolved in 20% captisol (Captisol®) or only 20% captisol were gavaged orally for two cycles from days 16–20 and 22–25, and sacrificed 24 hr after the last immune checkpoint injection or 8 hr after the last BLZ945 gavage. Brains were removed and fixed in 10% formalin, embedded in paraffin, and 5 μm sections subjected to immunohistochemistry with antibodies (Key Resources Table, IHC), followed by incubation with appropriate secondary antibodies (Vector Laboratories). Cell counts were from at least three random fields/tumor section. Counter was blinded to the treatment.
For double IHC, the brain sections were incubated sequentially with primary antibodies (pSTAT1 or CD3; rabbit antibody), then secondary antibodies (AP-conjugated anti-rabbit Ig), followed by the development of red color using ImmPACT Vector Red Alkaline Phosphatase Substrate Kit (Vector Laboratories). Next, the same brain sections were incubated with primary antibodies (CD68 or Ki67; rabbit antibody), then secondary antibodies (AP-conjugated anti-rabbit Ig), followed by the development of blue color using Vector Blue Alkaline Phosphatase Substrate Kit (Vector Laboratories). Cell counts were from at least three random fields/tumor section. Counter was blinded to the treatment.
Flow Cytometry
To examine PD-L1 expression in GSCs, 005 GSCs were cultured for 24 hr with or without murine IFNγ (3 ng/ml; PeproTech), or with G47Δ-E (no transgene (Cheema et al., 2013)) or G47Δ-mIL12 at multiplicity of infection (MOI) 1. 005 and human GSCs were spun, counted, re-suspended in FACS buffer (2% inactivated fetal calf serum in PBS), incubated with 7-AAD (BD Biosciences) and APC-conjugated anti-mouse or anti-human PD-L1 antibodies for 30 min, washed, fixed in 4% paraformaldehyde, washed, re-suspended in FACS buffer, and analyzed by LSRII flow cytometer (BD Biosciences).
For 10-color flow cytometric analysis, brain tumor quadrants were harvested, minced, incubated with Accutase and DNase I (10 U/ml; Promega), triturated, passed through a 40 μm screen, resuspended in FACS buffer, and stained with fluorochrome-conjugated anti-mouse antibodies from BioLegend (Key Resources Table), as well as appropriate isotype control antibodies. Zombie UV™ fixable viability Kit (BioLegend) was used to stain dead cells. We followed a ‘no-wash’ sequential staining protocol (BioLegend) to stain dead cells and for surface staining. Intracellular FoxP3 staining was performed following the FoxP3 intracellular staining protocol (BioLegend). Fluorescent minus one (FMO) controls were included for each color, e.g. fluorescent minus CD3ε by staining cells with all colors except APC/Cy7 anti-mouse CD3ε. For single-color compensation controls, UltraComp eBeads (eBioscience) were used and stained with each of eight fluorescently conjugated antibodies according to manufacturer instructions. For Zombie UV and GFP, cells from the non-tumor and tumor quadrant, respectively were used as single color compensation controls. All samples were run in a LSRII flow cytometer. Data was analyzed with FlowJo software (v.10.1; Tree Star). Technician acquiring and gating the data was blinded to the treatments.
Mouse IFNγ ELISPOT assay
Mouse IFNγ ELISPOT assay was performed according to the manufacturer instructions (R&D Systems) with minor modifications. Briefly, 005 GSCs (1 × 105) in 10% RPMI were irradiated (35 Gy) after overnight culture. Splenocytes (1–2 × 106/well) harvested from treated mice (as in (Cheema et al., 2013)) were co-cultured with 1 × 105 irradiated (35Gy) 005 GSCs or FCS-differentiated 005 cells in 12 well plate for 48 hr at 37°C. Stimulated splenocytes (1–2 × 10 5) were plated onto 96-well PVDF-backed microplates coated with monoclonal antibody specific for mouse IFNγ (Mouse IFN-gamma ELISpot kit; R&D Systems). After 24 hr of incubation at 37°C, plates were washed three times, incubated with detection antibody concentrate overnight at 4°C, washed and incubated with streptavidin-AP for 2 hr at room temperature. After washing, the signal was developed with BCIP/NBT Chromogen (R&D Systems) for one hour at room temperature. Spots were identified and counted on an AID Version 3.1.1 ELISPOT reader.
QUANTIFICATION AND STATISTICAL ANALYSIS
Survival data were analyzed by Kaplan Meier survival curves, and comparisons were performed by Log Rank test. Flow cytometric data and immunohistochemistry counts were compared using an unpaired Student’s t test (two-tailed). P values of less than 0.05 were considered significant. All statistical analyses were performed using Prism 6 software version 6.0d.
Supplementary Material
Significance.
Glioblastoma is one of the most immunosuppressive and lethal tumors. Immunotherapy clinical trials, including with immune checkpoint blockade, have not been successful so far. Here, we reveal the synergistic interaction between oncolytic herpes simplex virus expressing IL-12 and two checkpoint inhibitors (anti-CTLA-4 and anti-PD-1) in curing most glioblastoma and inducing immunological memory. Any dual combination only extended survival of mice with GSC-derived tumors, suggesting likely ineffectiveness in the clinic. Therapy curing most mice targeted multiple pathways: glioblastoma stem-like cell oncolysis, inhibition of immune-suppression, induction of anti-tumor immunity requiring CD4+ T cells, and, significantly, macrophage infiltration and M1-like polarization. This highlights a combination strategy to treat tumors that are minimally responsive to immunotherapy.
Highlights.
oHSV expressing IL-12 and 2 checkpoint inhibitors synergistically cure glioblastoma
Immune checkpoint antibodies access GSC-derived brain tumors
Triple combination increases intratumoral M1-like macrophages and Teff:Treg ratio
CD4+ and CD8+ T cells and macrophages are required for triple combination efficacy
Acknowledgments
Funded in part by grants from NIH (R01NS032677 to RLM and R01CA160762 to SDR) and The Thomas A. Pappas Chair in Neurosciences (SDR). We thank Dr. I. Verma (Salk Institute, San Diego CA) for providing the 005 GSCs and Dr. T. Seyfried (Boston College, MA) for the CT-2A cells. We thank Laura Prickett-Rice, Kat Folz-Donahue, Meredith Weglarz, Maris Handley, and Amy Galvin from the HSCI CRM Flow Cytometry Core for their technical assistance, Dr. H. Wakimoto for training and providing GSCs, and M. Humphrey for assistance with surgeries. SDR and RLM are inventors on patents relating to oHSV owned by Georgetown University and Massachusetts General Hospital that have been licensed to Amgen, for which they receive royalties.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
D.S. designed and performed experiments. R.L.M., and S.D.R. designed overall study, supervised the work and obtained funding. D.S., and S.D.R. analyzed data, wrote the manuscript, and made figures. D.S., R.L.M., and S.D.R. reviewed and edited the manuscript.
References
- Agarwalla P, Barnard Z, Fecci P, Dranoff G, Curry WT., Jr Sequential immunotherapy by vaccination with GM-CSF-expressing glioma cells and CTLA-4 blockade effectively treats established murine intracranial tumors. J Immunother. 2012;35:385–389. doi: 10.1097/CJI.0b013e3182562d59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonia SJ, Lopez-Martin JA, Bendell J, Ott PA, Taylor M, Eder JP, Jager D, Pietanza MC, Le DT, de Braud F, et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): a multicentre, open-label, phase 1/2 trial. Lancet Oncol. 2016 doi: 10.1016/S1470-2045(16)30098-5. [DOI] [PubMed] [Google Scholar]
- Badie B, Schartner JM. Flow cytometric characterization of tumor-associated macrophages in experimental gliomas. Neurosurgery. 2000;46:957–961. doi: 10.1097/00006123-200004000-00035. discussion 961–952. [DOI] [PubMed] [Google Scholar]
- Binello E, Qadeer ZA, Kothari HP, Emdad L, Germano IM. Stemness of the CT-2A Immunocompetent Mouse Brain Tumor Model: Characterization In Vitro. J Cancer. 2012;3:166–174. doi: 10.7150/jca.4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butowski N, Colman H, De Groot JF, Omuro AM, Nayak L, Wen PY, Cloughesy TF, Marimuthu A, Haidar S, Perry A, et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro Oncol. 2016;18:557–564. doi: 10.1093/neuonc/nov245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson SK, Brothers SP, Wahlestedt C. Emerging treatment strategies for glioblastoma multiforme. EMBO Mol Med. 2014;6:1359–1370. doi: 10.15252/emmm.201302627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheema TA, Wakimoto H, Fecci PE, Ning J, Kuroda T, Jeyaretna DS, Martuza RL, Rabkin SD. Multifaceted oncolytic virus therapy for glioblastoma in an immunocompetent cancer stem cell model. Proc Natl Acad Sci U S A. 2013;110:12006–12011. doi: 10.1073/pnas.1307935110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockle JV, Rajani K, Zaidi S, Kottke T, Thompson J, Diaz RM, Shim K, Peterson T, Parney IF, Short S, et al. Combination viroimmunotherapy with checkpoint inhibition to treat glioma, based on location-specific tumor profiling. Neuro Oncol. 2016;18:518–527. doi: 10.1093/neuonc/nov173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corthay A, Skovseth DK, Lundin KU, Rosjo E, Omholt H, Hofgaard PO, Haraldsen G, Bogen B. Primary antitumor immune response mediated by CD4+ T cells. Immunity. 2005;22:371–383. doi: 10.1016/j.immuni.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010;107:4275–4280. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curry WT, Lim M. Immunomodulation: checkpoint blockade etc. Neuro Oncol. 2015;17(Suppl 7):vii26–vii31. doi: 10.1093/neuonc/nov174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Tomaso T, Mazzoleni S, Wang E, Sovena G, Clavenna D, Franzin A, Mortini P, Ferrone S, Doglioni C, Marincola FM, et al. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin Cancer Res. 2010;16:800–813. doi: 10.1158/1078-0432.CCR-09-2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fecci PE, Ochiai H, Mitchell DA, Grossi PM, Sweeney AE, Archer GE, Cummings T, Allison JP, Bigner DD, Sampson JH. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clin Cancer Res. 2007;13:2158–2167. doi: 10.1158/1078-0432.CCR-06-2070. [DOI] [PubMed] [Google Scholar]
- Haabeth OA, Tveita AA, Fauskanger M, Schjesvold F, Lorvik KB, Hofgaard PO, Omholt H, Munthe LA, Dembic Z, Corthay A, Bogen B. How Do CD4(+) T Cells Detect and Eliminate Tumor Cells That Either Lack or Express MHC Class II Molecules? Front Immunol. 2014;5:174. doi: 10.3389/fimmu.2014.00174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy BC, Showers CR, Anderson DE, Anderson L, Canoll P, Bruce JN, Anderson RC. Tumor-associated macrophages in glioma: friend or foe? J Oncol. 2013;2013:486912. doi: 10.1155/2013/486912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev. 2015;29:1203–1217. doi: 10.1101/gad.261982.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le DT, Ladle BH, Lee T, Weiss V, Yao X, Leubner A, Armstrong TD, Jaffee EM. CD8(+) Foxp3(+) tumor infiltrating lymphocytes accumulate in the context of an effective anti-tumor response. Int J Cancer. 2011;129:636–647. doi: 10.1002/ijc.25693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Ravindranathan R, Kalinski P, Guo ZS, Bartlett DL. Rational combination of oncolytic vaccinia virus and PD-L1 blockade works synergistically to enhance therapeutic efficacy. Nat Commun. 2017;8:14754. doi: 10.1038/ncomms14754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lussier DM, Johnson JL, Hingorani P, Blattman JN. Combination immunotherapy with alpha-CTLA-4 and alpha-PD-L1 antibody blockade prevents immune escape and leads to complete control of metastatic osteosarcoma. J Immunother Cancer. 2015;3:21. doi: 10.1186/s40425-015-0067-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes W, Van Gool SW. Experimental immunotherapy for malignant glioma: lessons from two decades of research in the GL261 model. Cancer Immunol Immunother. 2011;60:153–160. doi: 10.1007/s00262-010-0946-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandai M, Hamanishi J, Abiko K, Matsumura N, Baba T, Konishi I. Dual Faces of IFNgamma in Cancer Progression: A Role of PD-L1 Induction in the Determination of Pro- and Antitumor Immunity. Clin Cancer Res. 2016;22:2329–2334. doi: 10.1158/1078-0432.CCR-16-0224. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Allavena P. The interaction of anticancer therapies with tumor-associated macrophages. J Exp Med. 2015;212:435–445. doi: 10.1084/jem.20150295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Orozco N, Muranski P, Chung Y, Yang XO, Yamazaki T, Lu S, Hwu P, Restifo NP, Overwijk WW, Dong C. T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity. 2009;31:787–798. doi: 10.1016/j.immuni.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marumoto T, Tashiro A, Friedmann-Morvinski D, Scadeng M, Soda Y, Gage FH, Verma IM. Development of a novel mouse glioma model using lentiviral vectors. Nat Med. 2009;15:110–116. doi: 10.1038/nm.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellman I, Hubbard-Lucey VM, Tontonoz MJ, Kalos MD, Chen DS, Allison JP, Drake CG, Levitsky H, Lonberg N, van der Burg SH, et al. De-Risking Immunotherapy: Report of a Consensus Workshop of the Cancer Immunotherapy Consortium of the Cancer Research Institute. Cancer Immunol Res. 2016;4:279–288. doi: 10.1158/2326-6066.CIR-16-0045. [DOI] [PubMed] [Google Scholar]
- Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nghiem PT, Bhatia S, Lipson EJ, Kudchadkar RR, Miller NJ, Annamalai L, Berry S, Chartash EK, Daud A, Fling SP, et al. PD-1 Blockade with Pembrolizumab in Advanced Merkel-Cell Carcinoma. N Engl J Med. 2016;374:2542–2552. doi: 10.1056/NEJMoa1603702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigim F, Cavanaugh J, Patel AP, Curry WT, Jr, Esaki S, Kasper EM, Chi AS, Louis DN, Martuza RL, Rabkin SD, Wakimoto H. Targeting Hypoxia-Inducible Factor 1alpha in a New Orthotopic Model of Glioblastoma Recapitulating the Hypoxic Tumor Microenvironment. J Neuropathol Exp Neurol. 2015;74:710–722. doi: 10.1097/NEN.0000000000000210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49–61. doi: 10.1016/j.immuni.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh T, Fakurnejad S, Sayegh ET, Clark AJ, Ivan ME, Sun MZ, Safaee M, Bloch O, James CD, Parsa AT. Immunocompetent murine models for the study of glioblastoma immunotherapy. J Transl Med. 2014;12:107. doi: 10.1186/1479-5876-12-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott PA, Hodi FS. Talimogene Laherparepvec for the Treatment of Advanced Melanoma. Clin Cancer Res. 2016;22:3127–3131. doi: 10.1158/1078-0432.CCR-15-2709. [DOI] [PubMed] [Google Scholar]
- Peng LS, Zhuang Y, Shi Y, Zhao YL, Wang TT, Chen N, Cheng P, Liu T, Liu XF, Zhang JY, et al. Increased tumor-infiltrating CD8(+)Foxp3(+) T lymphocytes are associated with tumor progression in human gastric cancer. Cancer Immunol Immunother. 2012;61:2183–2192. doi: 10.1007/s00262-012-1277-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Diez A, Joncker NT, Choi K, Chan WF, Anderson CC, Lantz O, Matzinger P. CD4 cells can be more efficient at tumor rejection than CD8 cells. Blood. 2007;109:5346–5354. doi: 10.1182/blood-2006-10-051318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perng P, Lim M. Immunosuppressive Mechanisms of Malignant Gliomas: Parallels at Non-CNS Sites. Front Oncol. 2015;5:153. doi: 10.3389/fonc.2015.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters C, Rabkin SD. Designing Herpes Viruses as Oncolytics. Mol Ther Oncolytics. 2015;2 doi: 10.1038/mto.2015.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preusser M, Berghoff AS, Wick W, Weller M. Clinical Neuropathology mini-review 6-2015: PD-L1: emerging biomarker in glioblastoma? Clin Neuropathol. 2015a;34:313–321. doi: 10.5414/NP300922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preusser M, Lim M, Hafler DA, Reardon DA, Sampson JH. Prospects of immune checkpoint modulators in the treatment of glioblastoma. Nat Rev Neurol. 2015b;11:504–514. doi: 10.1038/nrneurol.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puzanov I, Milhem MM, Minor D, Hamid O, Li A, Chen L, Chastain M, Gorski KS, Anderson A, Chou J, et al. Talimogene Laherparepvec in Combination With Ipilimumab in Previously Untreated, Unresectable Stage IIIB-IV Melanoma. J Clin Oncol. 2016;34:2619–2626. doi: 10.1200/JCO.2016.67.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19:1264–1272. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quezada SA, Peggs KS, Simpson TR, Allison JP. Shifting the equilibrium in cancer immunoediting: from tumor tolerance to eradication. Immunol Rev. 2011;241:104–118. doi: 10.1111/j.1600-065X.2011.01007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajani K, Parrish C, Kottke T, Thompson J, Zaidi S, Ilett L, Shim KG, Diaz RM, Pandha H, Harrington K, et al. Combination Therapy With Reovirus and Anti-PD-1 Blockade Controls Tumor Growth Through Innate and Adaptive Immune Responses. Mol Ther. 2016;24:166–174. doi: 10.1038/mt.2015.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon DA, Gokhale PC, Klein SR, Ligon KL, Rodig SJ, Ramkissoon SH, Jones KL, Conway AS, Liao X, Zhou J, et al. Glioblastoma Eradication Following Immune Checkpoint Blockade in an Orthotopic, Immunocompetent Model. Cancer Immunol Res. 2016;4:124–135. doi: 10.1158/2326-6066.CIR-15-0151. [DOI] [PubMed] [Google Scholar]
- Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. 2015;372:2521–2532. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- Rojas JJ, Sampath P, Hou W, Thorne SH. Defining Effective Combinations of Immune Checkpoint Blockade and Oncolytic Virotherapy. Clin Cancer Res. 2015;21:5543–5551. doi: 10.1158/1078-0432.CCR-14-2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolny C, Mazzone M, Tugues S, Laoui D, Johansson I, Coulon C, Squadrito ML, Segura I, Li X, Knevels E, et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell. 2011;19:31–44. doi: 10.1016/j.ccr.2010.11.009. [DOI] [PubMed] [Google Scholar]
- Saha D, Ahmed SS, Rabkin SD. Exploring the Antitumor Effect of Virus in Malignant Glioma. Drugs Future. 2015;40:739–749. doi: 10.1358/dof.2015.040.11.2383070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayour EJ, McLendon P, McLendon R, De Leon G, Reynolds R, Kresak J, Sampson JH, Mitchell DA. Increased proportion of FoxP3+ regulatory T cells in tumor infiltrating lymphocytes is associated with tumor recurrence and reduced survival in patients with glioblastoma. Cancer Immunol Immunother. 2015;64:419–427. doi: 10.1007/s00262-014-1651-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyfried TN, el-Abbadi M, Ecsedy JA, Bai HW, Yohe HC. Influence of host cell infiltration on the glycolipid content of mouse brain tumors. J Neurochem. 1996;66:2026–2033. doi: 10.1046/j.1471-4159.1996.66052026.x. [DOI] [PubMed] [Google Scholar]
- Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205–214. doi: 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Patnaik MM, Ruiz A, Russell SJ, Peng KW. Immunovirotherapy with vesicular stomatitis virus and PD-L1 blockade enhances therapeutic outcome in murine acute myeloid leukemia. Blood. 2016;127:1449–1458. doi: 10.1182/blood-2015-06-652503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. 2013;210:1695–1710. doi: 10.1084/jem.20130579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soda Y, Marumoto T, Friedmann-Morvinski D, Soda M, Liu F, Michiue H, Pastorino S, Yang M, Hoffman RM, Kesari S, Verma IM. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc Natl Acad Sci U S A. 2011;108:4274–4280. doi: 10.1073/pnas.1016030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todo T, Martuza RL, Rabkin SD, Johnson PA. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc Natl Acad Sci U S A. 2001;98:6396–6401. doi: 10.1073/pnas.101136398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–461. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16:275–287. doi: 10.1038/nrc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tugues S, Burkhard SH, Ohs I, Vrohlings M, Nussbaum K, Vom Berg J, Kulig P, Becher B. New insights into IL-12-mediated tumor suppression. Cell Death Differ. 2015;22:237–246. doi: 10.1038/cdd.2014.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, Benci JL, Xu B, Dada H, Odorizzi PM, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. 2015;520:373–377. doi: 10.1038/nature14292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugel S, De Sanctis F, Mandruzzato S, Bronte V. Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest. 2015;125:3365–3376. doi: 10.1172/JCI80006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vom Berg J, Vrohlings M, Haller S, Haimovici A, Kulig P, Sledzinska A, Weller M, Becher B. Intratumoral IL-12 combined with CTLA-4 blockade elicits T cell-mediated glioma rejection. J Exp Med. 2013;210:2803–2811. doi: 10.1084/jem.20130678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, Tobias A, Cheng Y, Kim JW, Qiao J, Zhang L, et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res. 2014;20:5290–5301. doi: 10.1158/1078-0432.CCR-14-0514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakimoto H, Kesari S, Farrell CJ, Curry WT, Jr, Zaupa C, Aghi M, Kuroda T, Stemmer-Rachamimov A, Shah K, Liu TC, et al. Human glioblastoma-derived cancer stem cells: establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res. 2009;69:3472–3481. doi: 10.1158/0008-5472.CAN-08-3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakimoto H, Mohapatra G, Kanai R, Curry WT, Jr, Yip S, Nitta M, Patel AP, Barnard ZR, Stemmer-Rachamimov AO, Louis DN, et al. Maintenance of primary tumor phenotype and genotype in glioblastoma stem cells. Neuro Oncol. 2012;14:132–144. doi: 10.1093/neuonc/nor195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woller N, Gurlevik E, Fleischmann-Mundt B, Schumacher A, Knocke S, Kloos AM, Saborowski M, Geffers R, Manns MP, Wirth TC, et al. Viral Infection of Tumors Overcomes Resistance to PD-1-immunotherapy by Broadening Neoantigenome-directed T-cell Responses. Mol Ther. 2015;23:1630–1640. doi: 10.1038/mt.2015.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu A, Wei J, Kong LY, Wang Y, Priebe W, Qiao W, Sawaya R, Heimberger AB. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 2010;12:1113–1125. doi: 10.1093/neuonc/noq082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan A, Hsiao YJ, Chen HY, Chen HW, Ho CC, Chen YY, Liu YC, Hong TH, Yu SL, Chen JJ, Yang PC. Opposite Effects of M1 and M2 Macrophage Subtypes on Lung Cancer Progression. Sci Rep. 2015;5:14273. doi: 10.1038/srep14273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P, Merghoub T, Wolchok JD, Allison JP. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med. 2014;6:226ra232. doi: 10.1126/scitranslmed.3008095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng J, See AP, Phallen J, Jackson CM, Belcaid Z, Ruzevick J, Durham N, Meyer C, Harris TJ, Albesiano E, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys. 2013;86:343–349. doi: 10.1016/j.ijrobp.2012.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, Wu L, Sloan AE, McLendon RE, Li X, et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol. 2015;17:170–182. doi: 10.1038/ncb3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.