

Abstract
Functionalized per- and poly-fluoroarenes are important building blocks, with many industrially and medicinally important molecules containing them. Nucleophilic aromatic substitution can be employed as a quick and straightforward way to synthesize these building blocks. While many methods to derivatize fluoroarenes exist which use heteroatom centered nucleophiles, there are fewer methods that use carbon centered nucleophiles, and of those many are poorly defined. This work presents the SNAr reaction of nucleophiles generated from nitroalkanes with a variety of fluorinated arenes. Given that the products are versatile, accessing polyfluorinated arene building blocks in substantial scale is important. This method is highly regioselective, and produces good to moderate yields on a large scale, sans chromatography, and thus fulfills this need. In addition, the regioselectivity of the addition was probed using both DFT calculations and experimentally via halogen exchange.
Graphical abstract
Introduction
The bioefficacy of small molecules developed for use in medicinal and agricultural products has been shown repeatedly to be significantly augmented by the introduction of a fluorine substituent, or in many cases multiple fluorines (Figure 1).1–4 While some elegant solutions5–7 to introduce a single fluorine have recently been published, if a polyfluorinated moiety is desired, these strategies of installing fluorines one-at-a-time can become prohibitively unwieldy because the prerequisite starting materials are a synthetic challenge in their own right. For this reason, the commercial availability of, or synthetic access to polyfluorinated precursors is relatively limited compared to that of monofluorinated analogs. This presents the need for a complementary set of synthetic methodologies that will allow for the simple, high yielding syntheses of diverse, functionalized polyfluorinated molecules that will enable rapid incorporation into larger compounds. Accomplishing this goal will allow for more complete evaluations of the decisive role fluorine plays within functional molecules.
Figure 1.

The importance of (poly)fluorinated arenes in medicinal and agricultural chemicals
While individual replacement of hydrogens on an arene can be complex, complete replacement of the hydrogen content of an arene with fluorine is synthetically straightforward,8–10 through methods such as perchlorination followed by nucleophilic halogen exchange (halex) sequences (Scheme 1). As a result, many important perfluorinated arene motifs are commercially available. Congruent with strategies championed by others,19–21 rather than selectively installing fluorine on each of the desired carbons, our goal is to develop syntheses that begin with inexpensive commercially available perfluorinated starting materials and realize polyfluorination patterns through selective functionalizion13,22–25 or reduction18,26 of the undesired C–F bonds under mild conditions. As evidenced by the sophistication of and number of recent C–F functionalization reactions,11–18 there is an increasing need to functionalize the inchoate highly fluorinated arene cores so that they may be easily incorporated into more elaborate molecules with interesting properties.
Scheme 1.

Strategies to achieve aryl fluorination
Uncatalyzed nucleophilic substitution reactions of fluoroarenes are well established.8,27 Though the literature predominantly features heteroatom nucleophiles, examples are present that utilize carbon nucleophiles such as Grignard reagents,28 and carbanions29,30 directly. The advantage of utilizing a nucleophile generated from deprotonation of a nitroalkane (i.e. a nitronate) in a reaction with polyfluorinated arene motif is that in one step a highly versatile benzylic nitro group is installed, which can allow further elaboration. This versatility is due in part to its acidifying effect as well as the variable oxidation state of the nitro group. As a synthetic handle, a benzylic nitro group can be readily employed in the Michael reaction, the nitro-Henry reaction, the Nef reaction, or reduced to an amine (vide infra), among many other transfromations.31 We present herein conditions for the substitution of a C–F bond of polyfluoroarenes with nitroalkanes. The method efficiently yields the monoarylated nitroalkane, is regioselective, highly scalable, and can be performed largely without the use of column chromatography.
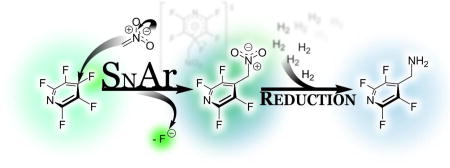
In 2012, Sandford and Cobb32 noted the expected importance of the polyfluorinated heteroaromatic amines and approached this goal through a similar strategy as that presented here, speculating that it could be reached through perfluoroarylation of nitromethanate followed by reduction, but rather produced the bipyridyl species (2f, Figure 2), which likely arises through a subsequent substitution of the initial product. While the desired monoarylated product was not obtained under these conditions, Sandford’s work does provide strong evidence that the nitronate is sufficiently nucleophilic to undergo substitution and highlights the complicating chemistry with which we would need to contend.
Figure 2.

A hypothetical exploitation of the SNAr mechanism for the formation of the monoarylated SNAr product of pentafluoropyridine in contrast to Sandford and Cobb's 2012 work.
In 2012, Vaidyanathaswamy showed that nitromethane could be used as a nucleophile in the addition to both pentafluorobenzonitrile and methyl pentafluorobenzoate, also indicating the feasibility of the reaction.33 In their work, the addition was accomplished using 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as the base, diethyl ether as the solvent, and required a different isolation technique for every product. Given our intention of scaling the reaction to provide substantial quantities, we were concerned about the generality of the reaction, the cost of DBU and its removal from the product, the ability to use diethyl ether on scale (which is banned in all Sanofi34 processes), as well as the unique isolation techniques. We sought to develop a general method with greater scope, higher yield, lower overall cost, and which was more amenable to scaling.
A simple analysis of the generally accepted SNAr mechanism (Figure 2) led us to suspect that it might be possible to favor the formation of the monoaryl product, 2e.
The addition of the nitromethanate to an aryl group gives rise to a new pronucleophile, (2c), which was expected to be deprotonated under any conditions capable of deprotonating nitromethane, given the acidifying effect of the highly fluorinated aryl group. This was expected to give rise to the nitronate 2d, which is also able to participate in a subsequent substitution, leading to 2f. Likely this pathway leads to the diarylated species isolated by Sandford and Cobb. However, the nitronate 2d was expected to be considerably less nucleophilic35 than 2b, due to not only the difference in pKa of the respective conjugate acids, but also due to the increased steric demand of 2d. Therefore, we suspected that we could take advantage of the decreased nucleophilicity to effect the desired selectivity for the monoarylated product, 2e. Nonetheless, in the presence of additional fluoroarene, the subsequent arylation was expected take place (2f, 2g).
We speculated that it might be possible to outcompete the relatively slow second arylation event of 2d by ensuring that the starting fluoroarene 2a was completely consumed by reaction with nitromethanate 2b before a subsequent arylation event could occur. This could be realized by lowering the amount of thermal energy present in the system, and through the addition of another equivalent of base, which would generate a superstoichiometric amount of the intended nucleophile, 2b.
Results and Discussion
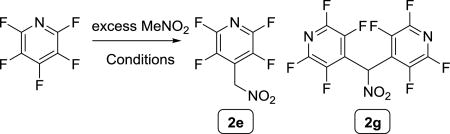
With a hypothesis developed, we began probing the reaction conditions, starting with the reaction of nitromethanate and pentafluoropyridine, (Table 1). Initial screenings of reaction conditions produced only the biaryl product when triethylamine was used as the base (entries 1). Some of the intended product resulted following the use of DBU to generate nitromethanate 2b (entry 2). Next, 1,1,3,3-tetramethylguanidine (TMG) was tried and gave comparable or slightly better results than DBU (entry 3 vs. 2). Given the cost of TMG being ca. 1/4 the cost of DBU36, we opted to utilize TMG for further study (entries 3–13). Increasing the amount of TMG effected complete conversion of the starting arene, and a substantial increase in the amount of the intended product (entry 4). It was found that the use of TMG necessitated a drop in temperature in order avoid the addition of TMG to the perfluoroarene (entries 3, 5, and 6). The first drop in temperature from room temperature to 0 °C increased the selectivity of the reaction toward monoarylation by reducing the available energy of the system to facilitate the apparently more sluggish second arylation event, as evidenced by the decrease in the formation of the biaryl product (2g). Further decreasing the temperature of the reaction to −25 °C led to substantially fewer side products, but also a diminishment of the overall conversion (entry 6). Nonetheless, the majority of the mass balance was now either the desired product (2e) or starting material. The quantity of the nucleophile generated was then investigated, which showed a steady increase in conversion which varied directly with the equivalents of TMG used (entries 6–10). It was not until the equivalents of base, and therefore nitromethanate 2b, exceeded 2.0 equivalents that the amount of other side products declined sharply in entry 11. Further increasing the amount of TMG to 2.5 equivalents (entry 12) had a deleterious effect on the reaction, however. Further decreasing the temperature was found to have completely suppressed the arylation of the TMG, and gave complete conversion to the intended product 2e (entry 13).
Table 1.
Optimization of the SNAr reaction of pentafluoropyridine with nitromethanate
| ||||||
|---|---|---|---|---|---|---|
| Entry | Temp. | Base | Equiv. Base |
Conversion to monoaryl product 2e |
Conversion to biaryl product 2g |
Conversion |
| 1 | rt | TEA | 1.1 | 0% | 36% | 66% |
| 2 | rt | DBU | 1.0 | 3% | 63% | 95% |
| 3 | rt | TMG | 1.0 | 6% | 28% | 82% |
| 4 | rt | TMG | 1.5 | 21% | 53% | 100% |
| 5 | 0°C | TMG | 1.0 | 18% | 1% | 72% |
| 6 | −25 °C | TMG | 1.0 | 57% | <1% | 66% |
| 7 | −25 °C | TMG | 1.25 | 63% | <1% | 71% |
| 8 | −25 °C | TMG | 1.5 | 79% | <1% | 83% |
| 9 | −25 °C | TMG | 1.75 | 79% | <1% | 93% |
| 10 | −25 °C | TMG | 2.0 | 84% | <1% | 96% |
| 11 | −25 °C | TMG | 2.1 | 98% | 0% | 99% |
| 12 | −25 °C | TMG | 2.5 | 92% | 0% | 97% |
| 13 | −35 °C | TMG | 2.1 | >99% | 0% | >99% |
All reactions run in excess nitromethane; TEA = triethylamine; NMR scale; Conversion determined by relative integration of NMR peaks. Products other than 2E and 2G were not characterized fully.
Results of an exploration of the scope of fluoroarenes that were investigated are shown in Figure 3. In reactions in which the perfluoroarene contained a strong electron withdrawing group (2e, 3b–3d), it was found that the reactions were complete in a very short time, i.e. less than 5 minutes after completion of the addition of the fluoroarene. With less activated fluoroarenes (3e–3i), nitromethanate underwent noticeably slower substitution. Consequently, TMG addition was competitive and only became more problematic as the reaction temperature was increased. Thus, a change to the more expensive but less nucleophilic DBU to generate the nucleophile was required.
Figure 3.

Scope of the SNAr reaction of nitromethane with fluoroarenes
Isolated yields, a 20 g scale, b 1 g scale. c DBU instead of TMG, rt.
Initially, workup of the reaction mixtures consisted of quenching the reaction with aqueous acid in order to protonate the anion of the product (2e, Figure 2) and any remaining base. 19F NMR analysis of the aqueous layer revealed that the products were, to varying degrees, soluble in water. It was also found that although it was possible to triturate the materials in order to purify them, substantial loss of material resulted.
In order to mitigate this loss, the crude reaction mixtures were acidified following the reaction with a sodium chloride saturated acidic solution. Immediately, better mass recovery was observed. Presumably, the increase in the ionic strength of the aqueous layer discouraged dissolution of the product therein, resulting in improved partitioning of the product into the ethyl acetate organic layer and increasing the overall recovery. The resultant mixture was then separated and the organic layer washed with a small amount of dilute aqueous acid which was not saturated with sodium chloride. In the case of the less activated arenes 3f–3h, which utilized DBU it was found that the standard workup conditions did not free the product completely from the base, and it was necessary to extract the resultant mixtures with a nonpolar solvent following the above workup conditions in order to obtain pure product. These workup conditions resulted in a simple, chromatography-free, easily scalable workup that afforded analytically pure product from the reaction mixture.
With reaction and workup conditions in hand, we attempted to scale up the reaction. This was accomplished on a 20 g scale of the reaction of pentafluoropyridine, which proceeded cleanly to a single product (2e) in a 91% yield. However, upon scaling we found that a cosolvent which could help dissipate the heat was necessary. Initially, diethyl ether was used which is not an ideal solvent for industrial processes, so it was additionally run on a 1 g scale using heptane as the solvent, but otherwise the same. Though the reaction was biphasic, and the nucleophile salt solidified while cooling, at −35 °C it proceeded to a single product in 96% yield, though the reaction time had to be increased to 4 hours. It is important to note that care was taken to slowly add the fluoroarene to the pregenerated nitromethanate, as the reaction generates significant heat, and increases in temperature were found to lead to the formation of unintended side products, namely that of the substitution of TMG.
The selectivity of the SNAr reaction with 1,2,3-trifluoro-4-nitrobenzene (F3NB) to produce 3b37–39 is interesting when compared to pentafluoronitrobenzene40 (PFNB) because it demonstrates the impact the fluorination pattern has on the regioselectivity of substitution with a highly reactive, yet sterically small nucleophile such as nitromethanate (4b, and 4c, Scheme 2). It has been posited27 that in reference to the site of substitution, ortho- and meta-fluorines facilitate the addition of nucleophiles inductively, whereas, ortho- and para-fluorines hinder the addition by destabilization of the Meisenheimer intermediate via resonance. Thus, the major product of addition to F3NB occurs with excellent selectivity ortho to the nitro group (eqns. 1, 2, Scheme 2). Attack by the nitromethanate at the C2 position could potentially maximize both the inductive and resonance effects because both the electron withdrawing nitro group and the fluorine ortho and meta to the site of substitution promote it. The directing effect of fluorine is dramatically observed when this result is compared with the addition to PFNB, which results in a poorly selective addition (eqns. 3,4). In fact, the major product (albeit only slightly) switches to the para-substituted product. This switch in selectivity can be rationalized by the aforementioned reasons. First, para substitution to the nitro group is favored by two additional ortho and meta fluorines. Secondly, addition ortho to the nitro group is retarded by the presence of a fluorine located in the position para to the site of substitution. The net effect is that both products are produced. It should be noted that that this selectivity is notably lower than other carbonucleophiles, which give the para addition in ca. 86:14 ratio.13,41
Scheme 2.

Effects of aryl hydrogen substiuents on the regioselectivity of the SNAr reaction
a Percent conversion of arene to products. Relative concentration of products 4a : 4b determined by 19F NMR integrations, with complete conversion of starting arene.
Hoping to probe the nuances of this mechanism, the reaction was the repeated with 1-chloro-2,3,5,6-tetrafluoro-4-nitrobenzene (1-Cl-F4NB) which recently became accessible.42 Nucleophilic aromatic substitution reactions are generally thought to have two transition states. The first is the attack of the nucleophile, which leads to formation of the Meisenheimer complex, and the second is the breakdown of that complex. Commonly, formation is the slow step, but the latter has been observed to be the rate-determining.43 Thus, we posited that replacing the 4-fluoro substituent would allow us to probe the reversibility of the first step. If the breakdown of the Meisenheimer complex was the rate-limiting step following a reversible addition of the nucleophile, one could expect that the incorporation of the para chlorine would lead to exclusive substitution at the chlorinated position because the fragmentation of the chloride should be more exothermic. This is not observed, however. With nitromethanate, the substitution of 1-Cl-F4NB occurs exclusively at the ortho position (eqns. 5, 6, 4c, Scheme 2), suggesting that attack of the nucleophile on the arene is not reversible and other forces are responsible for the observed selectivity.
In order to elucidate the factors responsible for the shift in selectivity (4c vs 4b), DFT calculated molecular electrostatic potential energy surfaces44,45 (MESPs) were generated for the three substrates (Figure 4A). While the relative size of the LUMO does not change appreciably across the substrates, the MESP is more supportive of the experimentally observed selectivity. For F3NB, the MESP shows significant partial positive electrostatic anisotropy at the position ortho to the nitro group, suggesting that the initial Coulombic attraction of the nucleophile contributes to the selective formation of the product 3b.
Figure 4.

A: LUMO surfaces (upper), and MESP surfaces (lower).
B: Vertical cross section through electron density surface. Incoming nuceophile added for illustrative purposes.
In the reaction of PFNB with nitromethanate which leads to products 4a (ortho substitution) and 4b (para substitution) in 44% and 56% conversion respectively, this hypothesis of electrostatic attraction of the nucleophile is further supported by the DFT calculations. The MESP surface illustrates that the potential energy is more positive at the para position than that at the ortho position. The observed selectivity can potentially be rationalized, however, by the fact that because there are two ortho positions at which substitutions may occur, there is a greater statistical probability of substitution at these positions compared to the para position, leading to an essentially unselective substitution in which the para position is favored, albeit only slightly.
In the substitution of 1-Cl-F4NB, the reaction is completely selective for the position ortho to the nitro group. The MESP surface for this substrate indicates that the most electropositive carbon has shifted in relation to that of the PFNB, from the position para to the nitro group to the ortho position.
An additional factor that may contribute to the change in the selectivity may be due to the fact that the chlorine is significantly larger than a fluorine substituent. A side-view of a cross section of the electron density surface (through C1 and C4 perpendicular to the molecular plane) indicates that attack of the nucleophile at C1 may be somewhat occluded by the attached chlorine approaching at the Bürgi–Dunitz angle,46 (Figure 4B). Curiously, this is in contrast to our previous observation,41 in which the addition of an oxazolone enolate to 4-chlorotetrafluoropyridine gave substitution at the 4 position as the major product, which is the same as the fluorinated analog. Of all the factors discussed, they all seem to support the observed selectivity of the reaction, however, the relative contribution of each is still unclear.
While we initially focused on the addition of nitromethane, which generates a highly versatile compound that can be easily manipulated, there are numerous ways to make nitroalkanes and they are versatile building blocks in their own right. Therefore, we wanted to try our reaction conditions on more complex nitroalkanes, (Figure 5). Vaidyanathaswamy showed that nitroethane could be used with pentafluorobenzonitrile and methyl pentafluorobenzoate, and similarly, we found it to work under our conditions with pentafluoropyridine (5a). Reactions with other, more complex nitroalkanes were also found to take place (5b and 5c), albeit with modest to poor yields. The majority of the mass balance was presumed to be addition of the base, though the other products were not fully characterized. It should be noted that no further optimization was performed which may have increased yields. Nonetheless, these reactions do indicate that the reaction can work with other enolizable nitroalkanes and can rapidly give rise to highly elaborated polyfluorinated arenes.
Figure 5.

Investigation of the scope of the nucleophile
Isolated yields; a reacted at −25 °C; b reacted at 0 °C.
Polyfluorinated benzylic amines are a useful and biologically important moiety, as can be seen in Figure 1. The structures of cabotegravir and pimavanserin are particularly interesting because, after reduction of the nitro group, the products of Figure 3 could conceivably serve as uninvestigated, fluorinated analogs with improved properties. In order for this to happen, we first had to find conditions that would facilitate the reduction of the aliphatic nitro group to an amine, (Table 2). One complicating factor is that the resultant amine itself could act as a nucleophile and undergo subsequent addition to the substrate, especially at the elevated temperatures frequently used in nitro-reductions.
Table 2.
Reduction of nitromethyl arenes to amines
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Isolated yield, ca. 500 mg scale; isolated as the hydrochloride salt | |||||||||
| Entry | Reductant | Catalyst | Time (hrs) |
Solvent | Acid (equiv.) | Temp. | Pressure | SM remaining |
Conversion to Intended Producta |
| 1 | HC1 | Fe | 144 | EtOH | HC1 | 100 °C | – | 0% | 28% |
| 2 | H2 | Pd/C | 42 | MeOH | AcOH (4.4) | 80 °C | – | 0% | 0% |
| 3 | H2 | Pd/C | 72 | MeOH | CHOOH (2) | 60 °C | 5.5 atm | 0% | 5% |
| 4 | H2 | Pd/C | 72 | MeOH | CHOOH (2) | 100 °C | 6.8 atm | 0% | 5% |
| 5 | H2 | Pd/C | 72 | MeOH | AcOH (10) | 100 °C | 6.8 atm | 0% | trace |
| 6 | H2 | Raney Ni | 72 | MeOH | – | 40 °C | 6.8 atm | 0% | 0% |
| 7 | H2 | Raney Ni (W6) | 1 | EtOH | AcOH (3) | rt | 6.8 atm | 0% | 0% |
| 8 | H2 | Raney Ni (W6) | 1 | EtOH | AcOH (3) | 50 °C | 6.8 atm | 0% | 7% |
| 9 | H2 | Raney Ni (W6) | 1 | EtOH | AcOH (3) | 100 °C | 6.8 atm | 0% | 8% |
| 10 | H2 | Raney Ni (W6) | 1 | EtOH | AcOH (3) | 100 °C | 21 atm | 0% | 13% |
| 11 | H2 | Raney Ni (W6) | 1 | EtOH | AcOH (3) | 150 °C | 21 atm | 0% | 56% |
| 12 | H2 | Raney Ni (W6) | 1 | EtOH | AcOH (6) | 150 °C | 21 atm | 0% | 78% |
| 13 | H2 | Raney Ni (W6) | 1 | EtOH | AcOH (9) | 150 °C | 21 atm | 0% | 72% |
| 14 | H2 | Raney Ni (W6) | 3 | EtOH | AcOH (6) | 150 °C | 21 atm | 0% | 98% |
Conversion to intended product determined by relative integration of 19F peaks
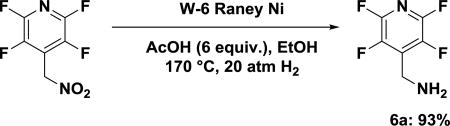
In order to reduce the aliphatic nitro group, a variety of catalytic reductions of 2e was carried out under acidic conditions (Table 2). We hoped the presence of an acid would protonate the product amine immediately upon formation, thus preventing further nucleophilic substitution. Refluxing of 2e with iron dust and concentrated HCl yielded some of the intended product, albeit very slowly (entry 1). We then focused on a catalytic palladium reduction with a hydrogen atmosphere. The reaction did not reduce the nitro group under an atmosphere of hydrogen (balloon pressure, entry 2), but upon increasing the H2 pressure in a pressure reactor in the presence of formic acid (entries 3, 4), slowly gave the desired product. The same reaction conditions but with acetic, instead of formic, acid did not result in a detectable amount of the intended product (entry 5). We next focused on Raney nickel reductions. Reaction with off-the-shelf activated Raney Ni in water resulted in none of the intended amine (entry 6). Questioning the activity of the pre-activated Raney Ni, W-6 Raney Ni was prepared according to literature.47 Screening the activity of the W-6 Raney Ni with 3 equivalents of acetic acid for only 1 hour at room temperature, 50 °C, and 100 °C resulted in 0%, 7%, and 8% of the intended amine, (entries 7, 8, and 9, respectively). Increasing the pressure of hydrogen gas resulted in better yields, with 13% (entry 10). At this point, increasing the temperature to 150 °C led to a substantial increase in the yield to (56%, entry 11). Increasing the acetic acid to 6 equivalents, further improved the yield to 78% (entry 12). However, further increases (9 equiv) proved slightly detrimental to the yield (entry 13). Returning to the conditions in entry 19 but extending the reaction time to 3 hours resulted in 98% conversion (entry 14).
We wanted to avoid the use of a column to isolate the product and to be able to isolate it in such a way that it would be stable. The following workup allowed us to both isolate it and safely store it as the HCl salt. After filtration and evaporation of the solvent, the heterogeneous mixture showed a greenish discoloration and the NMR signals were degraded, potentially due to the presence of nickel salts. Both the discoloration and NMR signal degradation resolved upon extraction with acidic water and washing with ether. Increasing the pH of the aqueous layer and extraction of the aqueous layer into ether led to the isolation of the amine, which was precipitated as the hydrochloride salt from anhydrous ethereal hydrochloric acid.
Conclusion
We have demonstrated a highly scalable method to quickly and easily functionalize and replace a C–F bond in a variety of perfluorinated and polyfluorinated arenes utilizing inexpensive reagents and mild reaction conditions. This will successively help facilitate C–F functionalization as a viable strategy for the synthesis of multifluorinated arenes. Furthermore, we were able to demonstrate for the first time the elusive monoarylation of nitromethane with pentafluoropyridine and provided conditions for its reduction to the benzylic amine. Thus, by using this method, we have shown that more complex molecules can be produced in short order from commercially available poly- and perfluorinated arenes, adding to the collective knowledge for the synthesis of versatile building blocks which will enable the synthesis of many useful highly fluorinated arenes.
Experimental
NMR spectra were obtained on a Bruker A400 instrument. High resolution mass spectra were obtained on a ThermoFisher LTQ-OrbitrapXL using electrospray ionization. Chemicals were obtained from commercial sources and used as received, except as noted. 19F DOSY performed according to Sibi and Jasperse’s technique.48 1 gram scale means that the general procedures were scaled ad valorem 1 gram of the intended product with 100% theoretical yield.
General procedures for the SNAr–F Arylation of nitromethane
General Procedure A
To a clean, dry round bottom flask, 12 equivalents of nitromethane were added. The round bottom flask was fitted with a magnetic stir bar, rubber septum, internal thermometer, and a ventiliation needle. The solution was degassed with with bubbling argon for 10 minutes. While under continuous argon flow, 2.1 equivalents of TMG were added and the solution was stirred for an additional 20 minutes. The solution and the arene to be added were then cooled to −35 °C. Then, the chilled fluoroarene was added dropwise. The addition of the fluoroarene was done such that the reaction temperature did not rise more than 5 °C. The reaction was complete in less than 5 minutes following the completion of the addition, and was then quenched with a solution of 1 M HCl (2.3 equivalents) that had been saturated with NaCl. The resultant crude mixture was extracted with ethyl acetate (3 × 5 reaction volumes), the combined organic layer was washed with successive small amounts (~0.5 reaction volumes) of HCl (0.1 M), separated, and the organic layer was stripped of solvent, in vacuo. Generally purity was sufficient so as to negate the need for further purifiaction. Variations are noted per substrate.
General Procedure B
To a clean, dry round bottom flask, 12 equivalents of nitromethane were added. The round bottom flask was fitted with a magnetic stir bar. Then, 2.1 equivalents of DBU were added and the solution was stirred for an additional 10 minutes. Next, the fluoroarene was added dropwise, and allowed to stir for 1 hour. The reaction was then quenched with a solution of 1 M HCl (2.3 equivalents) that had been saturated with NaCl. The resultant crude mixture was extracted twice with ethyl acetate (2 × 5 reaction volumes), the combined organic layer was washed with a small amount (~0.5 reaction volume) of HCl (0.1 M), separated, and the combined organic layer was stripped of solvent in vacuo. The product was extracted from the resultant oily material (5 × ~20 mL hexanes) and carefully decanted, leaving the denser, colored layer behind each time. The combined hexanes layer was stripped of solvent in vacuo. Generally purity was sufficient so as to negate the need for further purifiaction. Variations are noted per substrate.
2e: 2,3,5,6-tetrafluoro-4-(nitromethyl)pyridine
General procedure A was followed in a 500 mL RB, pentafluoropyridine (95.2 mmol, 10.45 mL), TMG (200.0 mmol, 25.10 mL), MeNO2 (1142.4 mmol, 61.10 mL), NaCl saturated-1M HCl (210 mL), diethyl ether (100 mL). Deviation from standard reaction conditions: After the reaction, the crude material was ground by mortar and pestle and washed with approximately 800 mL 0.1 M HCl over vacuum filtration until no further lightening of coloration was observed. Resulted in a pale yellow-orange crystalline solid, mass 18.197 g, 91% yield. MP 69–71 °C. 19F NMR (376 MHz, Chloroform-d) δ −87.79 (dq, J = 28.2, 13.1 Hz, 2F), −141.45 – −141.72 (m, 2F). 1H NMR (400 MHz, Chloroform-d) δ 5.70 (s, 2H). 13C{1H} NMR (101 MHz, Chloroform-d) δ 143.4 (app. dddd, J=251.3, 15.5, 12.2, 3.3 Hz, 2C), 140.7 (app. dd, J=264.6, 35.5 Hz, 2C), 121.3 (tt, J = 15.6, 3.0 Hz, 1C), 65.4 (s, 1C). 19F DOSY mass: calcd. 210; found 216. HRMS (m/z): [M − H]− calcd for [C6HF4N2O2]− 208.9979; found 208.9966
2e: 2,3,5,6-tetrafluoro-4-(nitromethyl)pyridine: (in heptane)
as in the above procedure, except on a 1 g scale with 10 mL heptane instead of diethyl ether, and reaction time was increased to 4 hours. The initial temperature was −35 °C and was allowed to slowly increase to 0 °C over the course of the reaction. Workup was the same as the standard conditions, but the solidified material was ground in a mortar and pestle and washed with ~ 50 mL aq. HCl (0.1 M) over vacuum filtration. 0.9551 g, 96% yield. 19F & 1H NMR spectra agreed with characterization above.
3b: 1,2-difluoro-4-nitro-3-(nitromethyl)benzene
General Procedure A was followed on a 1 g scale, using 4-Nitro-1,2,3-trifluorobenzene (4.58 mmol, 0.53 mL) with TMG (9.63 mmol, 1.21 mL) and nitromethane (55.02 mmol, 2.98 mL), except the resultant material had to be successively washed with 0.1 M aq. HCl to remove the base. 0.753 g pale yellowish crystalline solid, 75% yield. MP 86–91 °C. 19F NMR (376 MHz, Chloroform-d) δ −123.28 (m, 2F), −133.39 (m, 2F). 1H NMR (400 MHz, Chloroform-d) δ 8.16 (ddd, J = 9.3, 4.4, 2.1 Hz, 1H), 7.52 (dt, J = 9.3, 8.1 Hz, 1H), 5.94 (d, J = 1.8 Hz, 2H). 13C{1H} NMR (101 MHz, Chloroform-d) δ 154.0 (dd, J = 262.4, 13.8 Hz), 149.8 (dd, J = 256.8, 14.3 Hz), 143.9, 122.6 (dd, J = 7.9, 3.9 Hz), 119.2 (dd, J = 18.7, 1.8 Hz), 115.9 (dd, J = 14.0, 1.4 Hz), 67.6 (dd, J = 5.9, 1.7 Hz). HRMS (m/z): [M − H]− calcd for [C7H3F2N2O4]− 217.0066; found 217.0053.
3c: 2,3,5,6-tetrafluoro-4-(nitromethyl)benzonitrile
General Procedure A was followed on a 1 g scale, with pentafluorobenzonitrile (4.27 mmol, 0.54 mL), TMG (8.97 mmol, 1.13 mL), and nitromethane (51.26 mmol, 2.78 mL) in a 100 mL RBF, except with 10 mL diethyl ether. The organic layer was washed with 3 mL 0.1 M HCl total, resulting in an peach colored crystalline solid. 0.852 g, 85% yield. MP 67–71 °C (lit. 65–65.5 °C)33 19F NMR (376 MHz, Chloroform-d) δ −87.80 (dq, J = 27.9, 13.1 Hz, 2F), −141.47 – −141.70 (m, 2F). 1H NMR (400 MHz, Chloroform-d) δ 5.70 (s, 2H). 13C{1H} NMR (101 MHz, Chloroform-d) δ 143.6 (dddd, J = 248.1, 17.0, 12.4, 3.3 Hz, 2C), 140.9 (d, J = 264.7 Hz, 2C), 121.5 (tt, J = 15.3, 2.9 Hz, 1C), 65.6 (s, 1C). HRMS (m/z): [M − H]− calcd for [C8HF4N2O2]− 232.9979; found 232.9965.
3d: methyl 2,3,5,6-tetrafluoro-4-(nitromethyl)benzoate
General Procedure A was followed on a 1 gram scale, with methyl pentafluorobenzoate (3.74 mmol, 0.55 mL), TMG (7.86 mmol, 0.99 mL) and nitromethane (44.92 mmol, 2.43 mL) except the instead of washing the organic layers with 0.1 M HCl, crude material was dissolved in methanol and triturated by dripping the solution into DI water, resulting in an amber colored crystalline solid. 0.746 g, 75% yield. MP 35–36 °C (lit. m.p. 30–31 °C)33 19F NMR (376 MHz, Chloroform-d) δ −137.85 – −138.07 (m, 2F), −139.64 – −139.84 (m, 2F). 1H NMR (400 MHz, Chloroform-d) δ 5.58 (s, 2H), 3.95 (s, 3H). 13C{1H} NMR (101 MHz, Chloroform-d) δ 159.4 (s, 1C), 145.4 (ddt, J = 254.0, 14.8, 4.7 Hz, 2C), 144.4 (ddt, J = 258.3, 14.9, 4.7 Hz, 2C), 115.2 (td, J = 16.3, 4.1 Hz, 1C), 111.2 (t, J = 17.6 Hz, 1C), 65.6 (s, 1C), 53.7 (d, J = 2.3 Hz, 1C). HRMS (m/z): [M − H]− calcd for [C9H4F4NO4]− 266.0082; found 266.0071.
3e: 1,2,3,4,5,6,8-heptafluoro-7-(nitromethyl)naphthalene
General procedure B was followed on a 250 mg scale, with octafluoronaphthalene (0.80 mmol, 217.24 mg), DBU (1.68 mmol, 0.25 mL), and nitromethane (9.58 mmol, 0.52 mL), except the reaction time was increased to 3 hours. The product was separated by normal phase liquid chromatography with the product eluting at 5% EtOAc in hexanes. 0.1688 g off white crystalline solid, 68% yield. MP 96–98 °C. 19F NMR (376 MHz, Chloroform-d) δ −118.32 (ddt, J = 67.7, 19.4, 3.9 Hz, 1F), −138.15 (ddq, J = 16.7, 8.1, 4.1 Hz, 1F), −142.39 (dtt, J = 67.8, 16.8, 4.6 Hz, 1F), −144.72 (dddt, J = 58.1, 19.1, 14.9, 2.4 Hz, 1F), −147.07 (dtt, J = 58.1, 18.1, 4.7 Hz, 1F), −149.98 (dddt, J = 20.2, 17.6, 8.4, 4.2 Hz, 1F), −153.57 (ddtd, J = 19.5, 17.1, 7.4, 4.0 Hz, 1F). 1H NMR (400 MHz, Chloroform-d) δ 5.75 (s, 1H). 13C{1H} NMR (101 MHz, Chloroform-d) δ 151.9 (d, J = 263.7 Hz), 145.6 (dd, J = 254.5, 14.3 Hz), 141.8 (d, J = 261.1 Hz), 141.4 (d, J = 255.3 Hz), 140.8 (dt, J = 258.9, 15.1 Hz), 140.6 (d, J = 259.7 Hz), 139.1 (dt, J = 256.3, 14.9 Hz), 112.8, 107.7 (t, J = 13.5 Hz), 107.2 (t, J = 19.2 Hz), 65.9 (p, J = 2.1 Hz). Structure Assigned by 13C : 1H HMBC. HRMS (m/z): [M − H]− calcd for [C11HF7NO2]− 311.9901; found 311.9893.
3f: 1,2,4,5-tetrafluoro-3-(nitromethyl)-6-(trifluoromethyl)benzene
General procedure B was followed on a 1 gram scale, with octafluorotoluene (3.61 mmol, 0.51 mL), DBU (7.58 mmol, 1.13 mL), and nitromethane (43.31 mmol, 2.35 mL) resulting in a colorless crystalline solid. 0.9362 g, 98% yield. MP 46–51 °C. 19F NMR (376 MHz, Chloroform-d) δ −56.16 – −56.95 (m), −138.25 – −139.03 (m). 1H NMR (400 MHz, Chloroform-d) δ 5.67 (s, 2H). 13C{1H} NMR (101 MHz, Chloroform-d) δ 145.7 (d, J = 257.2 Hz, 2C), 144.1 (d, J = 264.6 Hz, 2C), 120.4 (q, J = 275.3 Hz, 1C), 113.0 – 112.3 (m, 2C), 65.4 (t, J = 2.5 Hz, 1C). HRMS (m/z): [M -H]− calcd for [C8HF7NO2]− 275.9901; found 275.9881.
3g: 1,2,3,4,5-pentafluoro-6-(nitromethyl)benzene
General procedure B was followed on a 1 gram scale, with hexafluorobenzene (4.40 mmol, 0.508 mL), DBU (9.25 mmol, 1.38 mL), and nitromethane (52.84 mmol, 2.86 mL), except with reaction time extended to 4 hours. Resulted in a light tan liquid. 0.833 g, 83% yield. 19F NMR (376 MHz, Chloroform-d) δ −140.82 – −140.95 (m, 2F), −149.28 (tt, J = 20.8, 3.3 Hz, 1F), −160.56 – −160.89 (m, 2F). 1H NMR: (400 MHz, Chloroform-d) δ 5.51 (t, J=1.50, 2H). 13C{1H} NMR (101 MHz, Chloroform-d) δ 146.1 (dddt, J = 252.7, 11.4, 7.6, 4.1 Hz), 143.2 (dtt, J = 258.8, 13.3, 5.3 Hz, 1C), 137.9 (d, J = 253.2, Hz, 2C), 104.1 (td, J = 17.3, 4.2 Hz, 2C), 65.5 (s, 1C). HRMS (m/z): [M − H]− calcd for [C7HF5NO2]− 225.9933; found 225.9916.
3h: 1,2,4,5-tetrafluoro-3-(nitromethyl)benzene
General procedure B was followed on a 2 gram scale, with pentafluorobenzene (9.56 mmol, 1.06 mL), DBU (20.09 mmol, 3.00 mL), and nitromethane (114.78 mmol, 6.22 mL), but the reaction time was lengthened to 18 hours, resulting in a light tan liquid., 1.852 g, 93% yield. 19F NMR: (400 MHz, Chloroform-d) δ −140.87 (m, 2F), −149.28 (tt, J=20.85 Hz, 3.33 Hz 1F), −160.73 (m, 2F). 1H NMR: (400 MHz, Chloroform-d) δ 5.51 (t, J=1.50, 2H). 13C{1H} Proton decoupled NMR (101 MHz, CDCl3) δ 146.1 (dddt, J=252.73 Hz, 11.35 Hz, 7.62 Hz, 4.10 Hz, 2C), δ 143.7 (m, 1C), δ 137.9 (m, 2C), δ 104.1 (td, J=17.26 Hz, 4.18 Hz, 1C), δ 65.5 (s, 1C). HRMS (m/z): [M − H]− calcd for [C7H2F4NO2]− 208.0027; found 208.0022.
3i: 2,2',3,3',5,5',6,6'-octafluoro-4,4'-bis(nitromethyl)-1,1'-biphenyl
General procedure B was followed on a 50 mg scale, with decafluorobiphenyl (0.12 mmol, 40.1 mg), DBU (0.25 mmol, 40 µL), and nitromethane (1.44 mmol, 80 µL), except the reaction was conducted at 60 °C. Following concentration, the material was purified by normal phase flash chromatography, with the product eluting at 25% ethyl acetate in hexanes. 36 mg yield, 72%. MP 171–173 °C. 19F NMR (376 MHz, Chloroform-d) δ −135.81 – −136.38 (m, 4F), −139.33 – −139.58 (m, 4F). 1H NMR (400 MHz, Chloroform-d) δ 5.65 (t, J = 1.4 Hz, 4H). 13C{1H} NMR (101 MHz, Chloroform-d) δ 145.5 (dd, J = 254.8, 11.7 Hz, 4C), 144.0 (dd, J = 254.9, 15.3 Hz, 4C), 110.9 (t, J = 16.7 Hz, 2C), 109.3 (s, 2C), 65.8 (s, 2C). HRMS (m/z): [M − H]− calcd for [C14H3F8N2O4]− 414.9970; found 414.9944
4a: 1,2,3,4-tetrafluoro-5-nitro-6-(nitromethyl)benzene and 3b: 1,2,4,5-tetrafluoro-3-nitro-6-(nitromethyl)benzene
General Procedure A was followed, resulting in both. Relative integrations were 44% : 56% respectively by 19F NMR, with complete consumption of starting material. Separated by normal phase liquid chromatography on silica gel, ethyl acetate/Hexanes with 2e eluting at 5% ethyl acetate, retention time 15 m. The concentration of ethyl acetate was then increased, and 3b eluted at 10% ethyl acetate after 25 minutes. Both 4a and 4b were somewhat viscous liquids.
4a: 1,2,3,4-tetrafluoro-5-nitro-6-(nitromethyl)benzene
19F NMR (376 MHz, Chloroform-d) δ −135.53 (dddt, J = 21.4, 10.0, 6.2, 1.8 Hz, 1F), −141.91 (ddd, J = 21.8, 10.2, 8.3 Hz, 1F), −144.97 (ddd, J = 21.5, 20.1, 8.3 Hz, 1F), −145.77 (ddd, J = 21.6, 20.0, 6.2 Hz, 1F). 1H NMR (400 MHz, Chloroform-d) δ 5.67 (t, J = 2.2 Hz, 2H). 13C{1H} NMR (101 MHz, Chloroform-d) δ 146.6 (dddd, J = 256.4, 11.5, 4.3, 2.5 Hz, 1C), 143.2 (dddd, J = 264.9, 17.1, 11.9, 3.2 Hz, 1C), 142.6 (d, J = 263.5 Hz, 1C), 135.0 (s, 1C), 129.3 – 126.8 (m, 1C), 109.3 (ddd, J = 16.3, 4.6, 2.0 Hz, 1C), 66.8 (s, 1C). HRMS (m/z): [M − H]− calcd for [C7HF4N2O4]− 252.9878; found 252.9869.
4b: 1,2,4,5-tetrafluoro-3-nitro-6-(nitromethyl)benzene
19F NMR (376 MHz, Chloroform-d) δ −136.27 – −136.45 (m, 2F), −144.49 – −144.88 (m, 2F). 1H NMR (400 MHz, Chloroform-d) δ 5.69 (t, J = 1.5 Hz, 2H). 13C{1H} NMR (101 MHz, Chloroform-d) δ 145.7 (dd, J = 257.4, 23.0 Hz), 140.2 (dd, J = 265.0, 22.1 Hz), 132.0, 112.5 (t, J = 16.9 Hz), 65.4 (p, J = 2.1 Hz). HRMS (m/z): [M − H]− calcd for [C7HF4N2O4]− 252.9878; found 252.9869.
4c: 1-chloro-2,3,6-trifluoro-4-nitro-5-(nitromethyl)benzene
Beginning with starting materials prepared as in literature,42 General Procedure A was followed on a 0.218 mmol scale, with 1-chloro-2,3,5,6-tetrafluoro-4-nitrobenzene, (0.218 mmol, 50 mg), TMG (0.46 mmol, 60 µL), and nitromethane (2.62 mmol, 140 µL) with reaction time extended to 3 hours, resulting in a light brown oil, 31 mg, 53% yield. 19F NMR (376 MHz, Chloroform-d) δ −114.00 – −114.19 (m, 1F), −123.57 – −124.11 (m, 1F), −142.87 – −143.54 (m, 1F). 1H NMR (400 MHz, Chloroform-d) δ 5.68 (s, 2H). 13C{1H} NMR (101 MHz, Chloroform-d) δ 152.5 (d, J = 255.5 Hz, 1C), 148.6 (ddd, J = 262.2, 14.5, 4.3 Hz, 1C), 140.9 (ddd, J = 266.4, 15.7, 4.6 Hz, 1C), 136.7 (m, 1C), 116.4 (dd, J = 23.5, 17.8 Hz, 1C), 107.6 (d, J = 19.5 Hz, 1C), 66.1 (s, 1C). HRMS (m/z): [M − H]− calcd for [C7HClF3N2O4]− 268.9582; found 268.9564.
5a: 2,3,5,6-tetrafluoro-4-(1-nitroethyl)pyridine
General Procedure A was followed on a 1 g scale, with pentafluoropyridine (4.76 mmol, 0.52 mL), TMG (10.00 mmol, 1.25 mL), and nitroethane (57.12 mmol, 4.08 mL), with crude reaction mixture separated by normal phase liquid chromatography over silica gel, eluting at 5% DCM in Hexanes, yielding 0.6652 g pale golden liquid, 66% yield. 19F NMR (376 MHz, Chloroform-d) δ −88.23 – −88.66 (m, 2F), −142.21 – −142.44 (m, 2F). 1H NMR (400 MHz, Chloroform-d) δ 5.88 (q, J = 7.2 Hz, 1H), 2.04 (d, J = 7.2 Hz, 3H). 13C{1H} NMR (101 MHz, Chloroform-d) δ 143.7 (dddd, J = 247.5, 16.5, 12.6, 3.1 Hz, 2C), 140.1 (d, J = 263.0 Hz, 2C), 128.1 (tt, J = 13.9, 2.7 Hz, 1C), 74.8, 17.5 (t, J = 2.4 Hz, 1C). HRMS (m/z): [M − H]− calcd for [C7H3F4N2O2]− 223.0136; found 223.0142.
5b: 2,3,5,6-tetrafluoro-4-(1-nitro-2-phenylethyl)pyridine
General Procedure B was followed on a 0.95 g scale, but 2.2 equiv. (2-nitroethyl)benzene (6.93 mmol, 1.047 g), prepared according to literature, was added to DBU (7.62 mmol, 1.14 mL) in 20 mL diethyl ether, vigorously stirring, followed by 250 mg silica gel. The reaction mixture was cooled to 0 °C, and pentafluoropyridine (3.15 mmol, 0.346 mL) in 5 mL diethyl ether (chilled) was added dropwise. Workup was the same as the general procedure. The resultant mixture was separated from the nitroalkene by reverse phase liquid chromatogaphy, C-18 column, eluting at 45–55% acetonitrile in water after 40 minutes. Fractions were stripped of organic solvent, extracted with DCM, dry loaded onto silica gel, and further purified by normal phase liquid chromatography on a silica gel column, with the product eluting at 5% DCM in hexanes. 0.2001 g viscous yellowish liquid, 21% yield. 19F NMR (376 MHz, Methylene Chloride-d2) δ −89.31 – −89.57 (m, 2F), −141.47 – −141.71 (m, 2F). 1H NMR (400 MHz, Methylene Chloride-d2) δ 7.36 – 7.11 (m, 5H), 6.11 (dd, J = 10.0, 6.4 Hz, 1H), 4.11 (dd, J = 13.9, 6.4 Hz, 1H), 3.56 (dd, J = 13.9, 10.0 Hz, 1H). 13C{1H} NMR (101 MHz, Methylene Chloride-d2) δ 144.1 (dddd, J = 246.4, 16.7, 12.8, 3.0 Hz, 2C), 140.9 (d, J = 262.7 Hz, 2C), 133.9 (s, 1C), 129.7 (s, 2C), 129.4 (s, 2C), 128.6 (s, 1C), 127.0 (tt, J = 13.9, 2.8 Hz, 1C), 80.8 (t, J = 2.0 Hz, 1C), 37.7 (t, J = 2.2 Hz, 1C). HRMS (m/z): [M − H]− calcd for [C13H7F4N2O2]− 299.0449; found 299.0429.
5c: 2,3,5,6-tetrafluoro-4-(2-nitropropan-2-yl)pyridine
General Procedure B was followed on a 4.60 mmol scale, but 2.2 equiv. 2-nitropropane (10.12 mmol, 0.92 mL) was used instead of nitromethane, with pentafluoropyridine (4.60 mmol, 0.51 mL), and DBU (9.66 mmol, 1.44 mL). The reaction mixture was cooled to 0 °C. Workup was the same as the general procedure. Crude material purified by normal phase liquid chromatography over silica gel, with the indended product eluting at 10% EtOAc in Hexanes, 17–20 minutes retention. Evaporation yielded 31.4 mg (3% yield) brownish crystalline solid, MP 124–129 °C. 19F NMR (376 MHz, Chloroform-d) δ −90.16 – −90.46 (m,2F), −162.99 – −163.40 (m, 2F). 1H NMR (400 MHz, Chloroform-d) δ 1.80 – 1.69 (m, 6H). 13C{1H} NMR (101 MHz, Chloroform-d) δ 145.4 – 145.1 (m, 1C), 143.7 (d, J = 241.5 Hz, 2C), 132.7 (d, J = 254.1 Hz, 2C), 91.6 (s, 1C), 23.1 (s, 2C). HRMS (m/z): [M − H]− calcd for [C8H5F4N2O2]− 237.02926; found 237.0278.
Procedure for the reduction of 2e to 6a: (perfluoropyridin-4-yl)methanamine
W-6 Raney Ni was prepared according to literature.47 To a 450 mL glass jacketed stirring pressure reactor was added 100 mL anhydrous ethanol, 1 equivalent 1a, 6 equivalents acetic acid, and 40% m/m Raney Ni. The reactor was then sealed, stirring commenced, and the atmosphere filled and vented with 21 ATM hydrogen gas 5X. The reactor was again filled to 21 ATM and heated to 150 °C. The reaction was stirred at 150 °C for 3 hours. The reactor was then cooled in an ice bath to room temperature, vented, and the opened. The resultant solution was vacuum filtered through celite and stripped of solvent in vacuo. The solids that formed were dissolved in a 1 M aqueous HCl and washed with diethyl ether, the organic phase then being discarded. The aqueous layer was made basic with 2 M aqueous NaOH, at which time, visible white precipitate formed. The aqueous layer was then extracted with diethyl ether. The organic layer was dried with MgSO4, and made acidic with anhydrous ethereal HCl. White solids precipitated and were separated by vacuum filtration, washed with additional anhydrous diethyl ether, and dried over vacuum.
6a: (perfluoropyridin-4-yl)methanamine hydrochloride
slightly off-white solid. 2.23 mmol scale, 452 mg, 93% yield. MP 264 °C (vaporized/decomposed). 19F NMR (376 MHz, Deuterium Oxide) δ −90.92 – −91.20 (m, 2F), −142.13 – −143.11 (m, 2F). 1H NMR (400 MHz, Deuterium Oxide) δ 4.49 (s, 2H). 13C{1H} NMR (101 MHz, DMSO-d6) δ 142.3 (dt, J = 242.9, 15.4 Hz, 2C), 140.6 (d, J = 259.5 Hz, 2C), 127.6 (t, J = 16.4 Hz, 1C), 30.5 (s, 1C). HRMS (m/z): [M + H]+ calcd for [C6H5F4N2]+ 181.0383; found 181.0377.
Computational Methods
Structures were built and optimized at MM2 levels using PerkinElmer Chem3D 15.0.0.106. They were then optimized at the B3LYP theory level with basis set 6–311+G(2d,p), with cube files for LUMO, electron density, and electrostatic potential generated in Gaussian 09.49 The electron density surface was rendered at 0.001 au, and mapped with the electrostatic density cube in UCSF Chimera 1.11.2.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Anuradha Singh for an initial experimental result, and Dr. Chris Fennell for his computational assistance.
This work was funded through the generous support of the NIH NIGMS award 5R01GM115697-02. High-resolution mass spectrometry analyses were performed in the Genomics and Proteomics Center at Oklahoma State University, using resources supported by the NSF MRI and EPSCoR programs (award DBI/0722494).
Molecular graphics and analyses were performed with the UCSF Chimera 1.11.2 package, Inkscape 0.92.0 r15299, FreeCAD 0.16, and GIMP 2.8.20. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311). Inkscape and GIMP are available under GNU General Public License. FreeCad is available under the GNU Lesser General Public License.
Footnotes
Associated Content
Supporting Information
Spectral and Computational Data
References
- 1.Penning TD, Talley JJ, Bertenshaw SR, Carter JS, Collins PW, Docter S, Graneto MJ, Lee LF, Malecha JW, Miyashiro JM, Rogers RS, Rogier DJ, Yu SS, Anderson GD, Burton EG, Cogburn JN, Gregory SA, Koboldt CM, Perkins WE, Seibert K, Veenhuizen AW, Zhang YY, Isakson PC. J. Med. Chem. 1997;40:1347. doi: 10.1021/jm960803q. [DOI] [PubMed] [Google Scholar]
- 2.Purser S, Moore PR, Swallow S, Gouverneur V. Chem. Soc. Rev. 2008;37:320. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]
- 3.Gillis EP, Eastman KJ, Hill MD, Donnelly DJ, Meanwell NA. J. Med. Chem. 2015;58:8315. doi: 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]
- 4.Wang J, Sanchez-Rosello M, Acena JL, del Pozo C, Sorochinsky AE, Fustero S, Soloshonok VA, Liu H. Chem. Rev. 2014;114:2432. doi: 10.1021/cr4002879. [DOI] [PubMed] [Google Scholar]
- 5.Schimler SD, Ryan SJ, Bland DC, Anderson JE, Sanford MS. J. Org. Chem. 2015;80:12137. doi: 10.1021/acs.joc.5b02075. [DOI] [PubMed] [Google Scholar]
- 6.Tang P, Furuya T, Ritter T. J. Am. Chem. Soc. 2010;132:12150. doi: 10.1021/ja105834t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Furuya T, Ritter T. Org. Lett. 2009;11:2860. doi: 10.1021/ol901113t. [DOI] [PubMed] [Google Scholar]
- 8.Brooke GM. J. Fluorine Chem. 1997;86:1. [Google Scholar]
- 9.Campbell MG, Ritter T. Chem. Rev. 2015;115:612. doi: 10.1021/cr500366b. [DOI] [PubMed] [Google Scholar]
- 10.Chambers RD, Hutchinson J, Musgrave WKR. J. Chem. Soc. 1964:3573. [Google Scholar]
- 11.Arora A, Weaver JD. Acc. Chem. Res. 2016;49:2273. doi: 10.1021/acs.accounts.6b00259. [DOI] [PubMed] [Google Scholar]
- 12.Senaweera S, Weaver JD. J. Am. Chem. Soc. 2016;138:2520. doi: 10.1021/jacs.5b13450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Senaweera SM, Weaver JD. J. Org. Chem. 2014;79:10466. doi: 10.1021/jo502075p. [DOI] [PubMed] [Google Scholar]
- 14.Singh A, Fennell CJ, Weaver JD. Chem. Sci. 2016;7:6796. doi: 10.1039/c6sc02422j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weaver JD. Synlett. 2014;25:1946. [Google Scholar]
- 16.LaBerge NA, Love JA. In: Organometallic Fluorine Chemistry. Braun T, Hughes RP, editors. Springer International Publishing; Cham: 2015. p. 55. [Google Scholar]
- 17.Hu J-Y, Zhang J-L. In: Organometallic Fluorine Chemistry. Braun T, Hughes RP, editors. Springer International Publishing; Cham: 2015. p. 143. [Google Scholar]
- 18.Senaweera S, Weaver JD. Aldrichimica Acta. 2016;49:45. [PMC free article] [PubMed] [Google Scholar]
- 19.Kiplinger JL, Richmond TG, Osterberg CE. Chem. Rev. 1994;94:373. [Google Scholar]
- 20.Ahrens T, Kohlmann J, Ahrens M, Braun T. Chem. Rev. 2015;115:931. doi: 10.1021/cr500257c. [DOI] [PubMed] [Google Scholar]
- 21.Amii H, Uneyama K. Chem. Rev. 2009;109:2119. doi: 10.1021/cr800388c. [DOI] [PubMed] [Google Scholar]
- 22.Senaweera SM, Weaver JD. J. Am. Chem. Soc. 2016;138:2520. doi: 10.1021/jacs.5b13450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Singh A, Kubik JJ, Weaver JD. Chem. Sci. 2015;6:7206. doi: 10.1039/c5sc03013g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Senaweera SM, Singh A, Weaver JD. J. Am. Chem. Soc. 2014;136:3002. doi: 10.1021/ja500031m. [DOI] [PubMed] [Google Scholar]
- 25.Singh A, Kubik JJ, Weaver JD. Chemical Science. 2015;6:7206. doi: 10.1039/c5sc03013g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Senaweera SM, Singh A, Weaver JD. J. Am. Chem. Soc. 2014;136:3002. doi: 10.1021/ja500031m. [DOI] [PubMed] [Google Scholar]
- 27.Chambers RD, Martin PA, Sandford G, Williams DLH. J. Fluorine Chem. 2008;129:998. [Google Scholar]
- 28.Sun Y, Sun H, Jia J, Du A, Li X. Organometallics. 2014;33:1079. [Google Scholar]
- 29.Chambers RD, Hassan MA, Hoskin PR, Kenwright A, Richmond P, Sandford G. J. Fluorine Chem. 2001;111:135. [Google Scholar]
- 30.Beyki K, Haydari R, Maghsoodlou MT. SpringerPlus. 2015;4:757. doi: 10.1186/s40064-015-1495-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ono N. The Nitro Group in Organic Synthesis. Wiley-VCH; 2001. [Google Scholar]
- 32.Colgin N, Tatum NJ, Pohl E, Cobb SL, Sandford G. J. Fluorine Chem. 2012;133:33. [Google Scholar]
- 33.Vaidyanathaswamy R, Radha K, Dharani M, Raguraman TS, Anand R. J. Fluorine Chem. 2012;144:33. [Google Scholar]
- 34.Prat D, Pardigon O, Flemming H-W, Letestu S, Ducandas V, Isnard P, Guntrum E, Senac T, Ruisseau S, Cruciani P, Hosek P. Organic Process Research & Development. 2013;17:1517. [Google Scholar]
- 35.Bug T, Lemek T, Mayr H. J. Org. Chem. 2004;69:7565. doi: 10.1021/jo048773j. [DOI] [PubMed] [Google Scholar]
- 36.Prices for DBU and TMG retrieved from Sigma-Aldrich website. at http://www.sigmaaldrich.com/catalog/product/aldrich/139009 and http://www.sigmaaldrich.com/catalog/product/aldrich/241768 respectively.
- 37.Lee L, Kreutter KD, Pan W, Crysler C, Spurlino J, Player MR, Tomczuk B, Lu T. Bioorg. Med. Chem. Lett. 2007;17:6266. doi: 10.1016/j.bmcl.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 38.Parikh VD, Fray AH, Kleinman EF. J. Heterocyclic Chem. 1988;25:1567. [Google Scholar]
- 39.Pesti JA, LaPorte T, Thornton JE, Spangler L, Buono F, Crispino G, Gibson F, Lobben P, Papaioannou CG. Org. Process Res. Dev. 2014;18:89. [Google Scholar]
- 40.Brooke GM, Burdon J, Tatlow JC. J. Chem. Soc. 1961:802. [Google Scholar]
- 41.Teegardin KA, Weaver JD. Chemical Communications. 2017 doi: 10.1039/c7cc01606a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Senaweera S, Weaver JD. Currently unpublished work under review. 2017 [Google Scholar]
- 43.Shipilov AI, Zolotkova EE, Igumnov SM. Russian Journal of Organic Chemistry. 2004;40:1117. [Google Scholar]
- 44.Politzer P, Murray JS, Clark T. Halogen Bonding I. Springer International Publishing; Switzerland: 2014. p. 19. 2014; Vol. Volume 358 of the series Topics in Current Chemistry. [Google Scholar]
- 45.Politzer P, Laurence PR, Jayasuriya K. Environmental Health Perspectives. 1985;61:191. doi: 10.1289/ehp.8561191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bürgi HB, Dunitz JD, Lehn JM, Wipff G. Tetrahedron. 1974;30:1563. [Google Scholar]
- 47.Billica HRA, Homer Org. Synth. 1949;29:24. [Google Scholar]
- 48.Subramanian H, Jasperse CP, Sibi MP. Org. Lett. 2015;17:1429. doi: 10.1021/acs.orglett.5b00297. [DOI] [PubMed] [Google Scholar]
- 49.R C, Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery J JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09. Gaussian, Inc.; Wallingford CT: 2010. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.