

Abstract
Background
GPIHBP1, a glycolipid-anchored protein of capillary endothelial cells, binds lipoprotein lipase (LPL) in the interstitial spaces and transports it to the capillary lumen. GPIHBP1 deficiency prevents LPL from reaching the capillary lumen, resulting in low intravascular LPL levels, impaired intravascular triglyceride processing, and severe hypertriglyceridemia (chylomicronemia). A recent study showed that some cases of hypertriglyceridemia are caused by autoantibodies against GPIHBP1 (“GPIHBP1 autoantibody syndrome”).
Objective
Our objective was to gain additional insights into the frequency of the GPIHBP1 autoantibody syndrome in patients with unexplained chylomicronemia.
Methods
We used ELISAs to screen for GPIHBP1 autoantibodies in 33 patients with unexplained chylomicronemia and then used western blots and immunocytochemistry studies to characterize the GPIHBP1 autoantibodies.
Results
The plasma of one patient, a 36-year-old man with severe hypertriglyceridemia, contained GPIHBP1 autoantibodies. The autoantibodies, which were easily detectable by western blot, blocked the ability of GPIHBP1 to bind LPL. The plasma levels of LPL mass and activity were low. The patient had no history of autoimmune disease but his plasma was positive for antinuclear antibodies.
Conclusions
One of 33 patients with unexplained chylomicronemia had the GPIHBP1 autoantibody syndrome. Additional studies in large lipid clinics will be helpful for better defining the frequency of this syndrome and for exploring the best strategies for treatment.
Keywords: Chylomicrons, endothelial cells, lipids, intravascular lipolysis, triglycerides
Introduction
GPIHBP1 is a glycolipid-anchored protein of capillary endothelial cells that binds lipoprotein lipase (LPL) in the subendothelial spaces and shuttles it across endothelial cells to its site of action in the capillary lumen.1 In the absence of GPIHBP1, LPL remains stranded in the interstitial spaces where it is unable to process triglyceride-rich lipoproteins (TRLs) in the plasma.1-3 Homozygosity for mutations in GPIHBP1, like homozygosity for mutations in LPL, causes a chylomicronemia syndrome4 associated with a substantial risk of acute pancreatitis. A variety of different GPIHBP1 mutations have been described,5-14 and most have been missense mutations that result in a mutant GPIHBP1 protein that lacks the ability to bind LPL. A hallmark of genetic forms of GPIHBP1 deficiency is low levels of LPL in the pre- and post-heparin plasma (consistent with reduced delivery of LPL to the capillary lumen).5-7, 15 Recent studies showed that it is possible to measure GPIHBP1 in human plasma with a monoclonal antibody–based immunoassay.16 In the setting of GPIHBP1 mutations, the plasma levels of GPIHBP1 are very low.16, 17
Beigneux and coworkers recently identified six patients with chylomicronemia caused by autoantibodies against GPIHBP1 (“GPIHBP1 autoantibody syndrome”).17 They demonstrated that GPIHBP1 autoantibodies interfere with the ability of GPIHBP1 to bind LPL. Several patients with the GPIHBP1 autoantibody syndrome had clinical and/or serological evidence of an autoimmune disease (e.g., systemic lupus erythematosus), but others did not. The plasma levels of LPL in patients with the GPIHBP1 autoantibody syndrome were low17 (again reduced delivery of LPL to the capillary lumen). The levels of GPIHBP1 in the plasma were also low, likely because the GPIHBP1 autoantibodies interfere with the detection of GPIHBP1 in an ELISA.17
The frequency of the GPIHBP1 autoantibody syndrome has not been clearly defined. Beigneux and coworkers identified six cases of the GPIHBP1 autoantibody syndrome by screening ∼200 miscellaneous plasma samples, including 130 patients with hypertriglyceridemia.17 In the current studies, we screened for GPIHBP1 autoantibodies in 33 patients with unexplained hypertriglyceridemia from the Academic Medical Center in Amsterdam. Each of these patients had undergone testing for LPL, GPIHBP1, APOC2, LMF1, or APOA5 mutations, and none were identified.
Materials and Methods
Plasma samples
Plasma samples from 33 patients from the Academic Medical Center (Amsterdam) were sent to UCLA for GPIHBP1 autoantibody screening. All patients had been referred because of a suspicion of LPL deficiency; the mean plasma triglyceride level in this group of patients was 1673 ± 2310 mg/dl (range, 171–11,327 mg/dl). All tested negative for mutations in LPL, GPIHBP1, APOC2, LMF1, and APOA5. Samples were taken under a protocol approved by the Academic Medical Center in Amsterdam. Because the plasma samples sent to UCLA were de-identified, the studies were exempt from IRB approval by the UCLA Office of Human Use Protection.
Two GPIHBP1 autoantibody ELISAs
Recombinant human GPIHBP1 (rhGPIHBP1) with an amino-terminal urokinase plasminogen activator receptor (uPAR) tag18 were expressed in Drosophila S2 cells.18, 19 The recombinant GPIHBP1 also contained a carboxyl-terminal tag for the mouse GPIHBP1–specific monoclonal antibody (mAb) 11A12.20 To detect GPIHBP1 autoantibodies in plasma samples,17 96-well plates were coated with uPAR-specific mAb (R24)21 and then incubated with GPIHBP1 for 2 h. After washing the plates, human plasma samples (1:500 dilution) were added to the wells and incubated overnight at 4°C. After washing, binding of autoantibodies to GPIHBP1 was detected with an HRP-labeled goat anti-human Ig(G+M). A second ELISA for GPIHBP1 autoantibodies was identical except that the 96-well plates were coated directly with purified, untagged GPIHBP1. To gauge levels of GPIHBP1 autoantibodies, we also added the HRP-labeled goat anti-human Ig(G+M) to wells that had been coated with dilutions of purified human IgG. We also used an ELISA to screen for autoantibodies against other members in the Ly6 protein family (C4.4A, CD59, CD177).17, 18, 22, 23
Detecting GPIHBP1 autoantibodies with western blots
The medium from Drosophila S2 cells expressing rhGPIHBP1 was size-fractioned on 12% Bis-Tris SDS-PAGE gels, then transferred to a nitrocellulose membrane. The membrane was blocked in Odyssey blocking buffer (Li-Cor). Membranes were then incubated with the plasma from patient A1 (20 AU of GPIHBP1 autoantibody/ml). The binding of autoantibodies to GPIHBP1 was detected with an IRDye680–donkey anti-human IgG (1:200). rhGPIHBP1 was also detected with an IRDye800-conjugated monoclonal antibody against human GPIHBP1 (RF4)17 (1:500).
Western blots to detect LPL autoantibodies
The medium from V5-tagged human LPL–transfected CHO cells was size-fractionated by SDS-PAGE under reducing and nonreducing conditions. Gels were then transferred to a nitrocellulose membrane and blocked in Odyssey blocking buffer (Li-Cor). Western blots were performed with the plasma from patient A1 (20 AU GPIHBP1 autoantibodies/ml) followed by IRDye680-labeled goat anti-human IgG (Li-Cor; 1:200) and an IRDye800-labeled antibody against the V5 tag (1:500).
Measurements of LPL, hepatic lipase (HL), endothelial lipase (EL), and GPIHBP1
Plasma levels of LPL, HL, and EL were measured with monoclonal antibody–based sandwich immunoassays.17, 24-28 An ELISA was also used to measure plasma levels of GPIHBP1.16 In the latter assay, 96-well plates were coated with the GPIHBP1-specific mAb RF4. Dilutions of human plasma were then added and incubated at 4°C. After washing the wells, GPIHBP1 was detected with HRP-labeled mAb RE3. The amount of GPIHBP1 in the plasma was measured against a recombinant GPIHBP1 standard curve. Levels of LPL and HL activity in the plasma at baseline and 2, 3, 6, 9, 12, and 15 min after an intravenous injection of heparin (50 IU/kg) were measured as described.24, 25
ELISA to test the ability of GPIHBP1 autoantibodies to prevent LPL binding to GPIHBP1
To test the ability of GPIHBP1 autoantibodies to block the binding of LPL to GPIHBP1, 96-well plates were coated with mAb R24 and then incubated overnight at 4°C with a uPAR-tagged GPIHBP1, either alone or with various dilutions of human plasma samples. On the next day, the plates were washed, and human LPL (200 ng/well) was added to the wells and incubated for 2 h at 4°C. The plates were washed, and an HRP-labeled LPL-specific monoclonal antibody (5D2) was used to measure LPL binding. In other wells, an HRP-labeled goat anti-human Ig(G+M) was added to detect binding of GPIHBP1 autoantibodies to the immobilized GPIHBP1. HRP-labeled mAb 11A12 was used to confirm the presence of GPIHBP1 on the plates. A GPIHBP1-specific monoclonal antibody (RE3) that blocks LPL binding to GPIHBP1 was used as an experimental control.16
Immunocytochemistry
CHO pgsA-745 (2 × 106 cells) were electroporated with 2 μg of plasmid DNA encoding S-protein–tagged versions of human GPIHBP1, GPIHBP1-W109S, or CD59. After 24 h, cells were washed in PBS and incubated with plasma samples for 1 h at 4°C. Cells were washed and incubated for 1 h at 4°C with V5-tagged human LPL (200 ng/well). After washing, cells were fixed with methanol. Immunocytochemistry studies were performed on nonpermeabilized cells with a rabbit antibody against the S-protein tag (0.2 μg/ml) followed by an Alexa Fluor 647–conjugated donkey anti-rabbit IgG (ThermoFisher, 2.5 μg/ml); an Alexa Fluor 568–conjugated mouse anti-V5 antibody (1:50); and an Alexa Fluor 488–conjugated goat anti-human Ig(G+M) (50 ng/ml). Images were taken with an Axiovert 200M microscope and processed with Zen 2010 software (Zeiss).
Results
We used an ELISA with uPAR-tagged human GPIHBP1 to screen plasma samples from 33 patients with unexplained hypertriglyceridemia for autoantibodies against GPIHBP1 (Figure 1A). The plasma samples from 32 patients were negative for autoantibodies, but one (from patient A1) was positive. The presence of GPIHBP1 autoantibodies in the plasma of patient A1 was confirmed in a separate ELISA with untagged, purified GPIHBP1 (Figure 1B). In the latter assay, the level of GPIHBP1 autoantibodies was measured against a standard curve of human IgG, and the concentration was judged to be ∼1 mg of GPIHBP1 autoantibodies/ml. The immunoglobulins in the plasma from patient A1 did not bind to other proteins of the Ly6 protein family (CD177, C4.4A, CD59).
Figure 1. Detecting GPIHBP1 autoantibodies with a solid-phase ELISA.
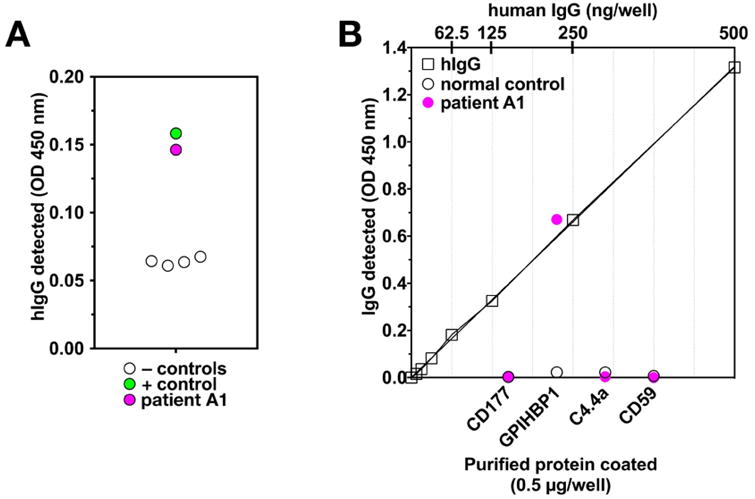
(A) Screening plasma samples for GPIHBP1 autoantibodies. Plasma samples were added to 96-well plates that had been coated with an anti-uPAR mAb and subsequently incubated with conditioned medium from Drosophila S2 cells expressing uPAR-tagged human GPIHBP1. After washing the plates, the binding of GPIHBP1 autoantibodies to immobilized GPIHBP1 was detected with an HRP-labeled anti-human IgG. One patient (patient A1; pink circle) had GPIHBP1 autoantibodies (dilution, 1:500), as did the positive control patient (patient 10217, dilution, 1:12,500) (green circle). Levels of IgG binding were lower in four normal controls (black circles). (B) Testing the binding of plasma samples (1:500) to wells coated with purified, untagged human GPIHBP1 and three other Ly6 proteins (CD177, C4.4A, CD59). The plasma from patient A1 (1:500; pink circle) bound to GPIHBP1 but not to the other Ly6 proteins; the control plasma (1:500; open circles) did not bind to any of the proteins. The level of antibody binding to the GPIHBP1-coated wells was judged according to a human IgG standard curve (top x-axis); wells of a 96-well plate were coated with dilutions of normal human IgG, and the amount of IgG bound to those wells was assessed by quantifying binding of the HRP-labeled anti-human IgG (open squares).
Patient A1 is a 36-year-old male who was born in Eritrea. Screening revealed that he was free of LPL, GPIHBP1, LMF1, APOC2, or APOA5 mutations. He was obese (Body Mass Index, 35.5 kg/m2) and was taking antipsychotic medications that had been prescribed by a psychiatrist. He smoked marijuana daily. In 2011, he had acute pancreatitis; at that time, the plasma triglyceride and cholesterol levels were 2188 mg/dl and 318 mg/dl, respectively. Over the next year, the patient had four additional bouts of pancreatitis. Plasma samples were taken and archived during two hospitalizations for pancreatitis (in 2011 and 2012); the plasma triglyceride levels in those samples were 580 and 1929 mg/dl, respectively. Those samples, both positive for GPIHBP1 autoantibodies, were analyzed at UCLA.
Western blots confirmed the presence of GPIHBP1 autoantibodies in the plasma samples from patient A1. The autoantibodies in the plasma sample detected GPIHBP1 produced in Drosophila S2 cells (Figure 2).
Figure 2.
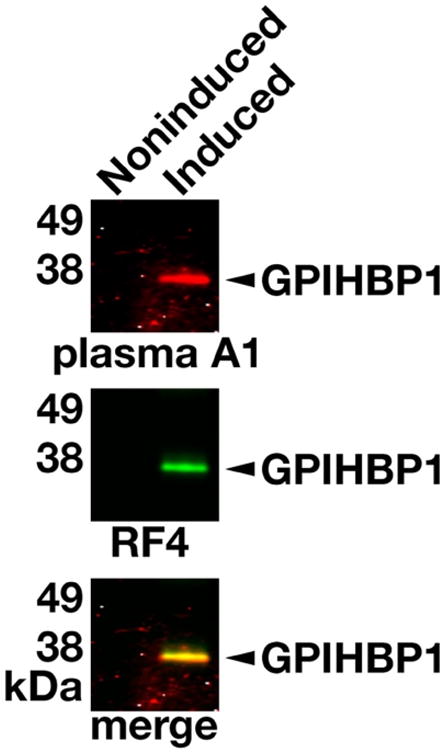
Western blot demonstrating binding of GPIHBP1 autoantibodies in the plasma from patient A1 to human GPIHBP1 in the medium of transfected Drosophila S2 cells. The expression human GPIHBP119 in Drosophila S2 cells was induced with 0.5 mM CuSO4. Proteins in the medium from copper-induced and noninduced cells were size-fractionated by SDS-PAGE and then transferred to a sheet of nitrocellulose for western blots. The membrane was incubated with the plasma from patient A1, followed by a IRDye680-labeled donkey anti-human IgG, and then with IRDye800-labeled human GPIHBP1-specific monoclonal antibody RF4.16 Both RF4 and the IgGs in the plasma of patient A1 bound avidly to GPIHBP1 in the medium of cells that had been induced with CuSO4 (Induced); GPIHBP1 was absent from the medium of noninduced cells (Noninduced).
We suspected that the GPIHBP1 autoantibodies would block the ability of GPIHBP1 to bind LPL. Indeed, the plasma from patient A1 blocked the binding of human LPL to GPIHBP1 in an ELISA whereas control plasma samples lacking autoantibodies did not (Figure 3). Plasma A1 blocked LPL binding to GPIHBP1 on the surface of GPIHBP1-transfected cells by immunocytochemistry, whereas control plasma samples lacking GPIHBP1 autoantibodies did not (Figure 4).
Figure 3. GPIHBP1 autoantibodies in the plasma of patient A1 abolish the ability of GPIHBP1 to bind LPL.
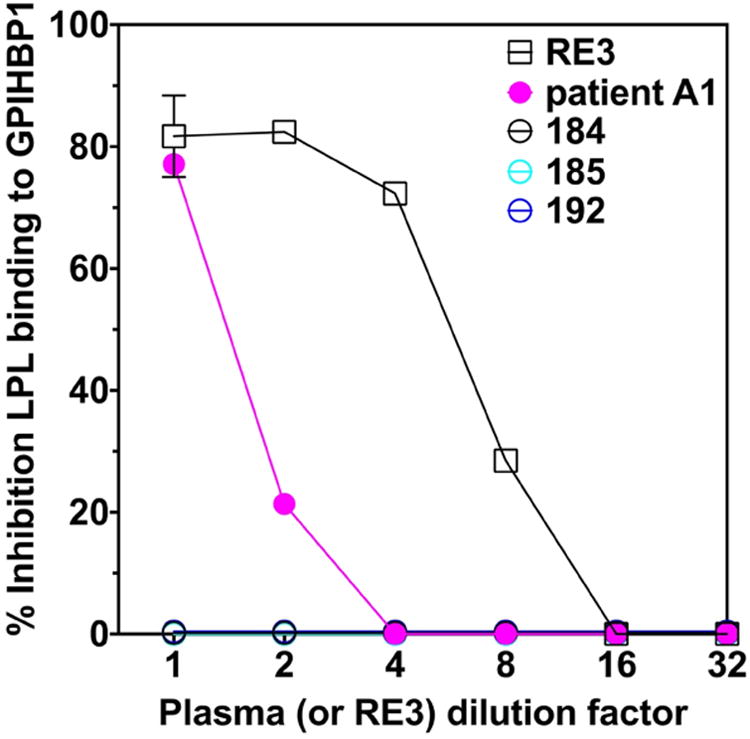
96-well plates were coated with an anti-uPAR mAb and then incubated overnight at 4°C with uPAR-tagged GPIHBP1, either alone or in the presence of dilutions of human plasma. After washing the plates, human LPL (200 ng) was added to each well and incubated for 2 h at 4°C. The plates were then washed, and the binding of LPL was detected with an HRP-labeled LPL-specific monoclonal antibody (5D2). The plasma of patient A1 blocked the binding of LPL to GPIHBP1 (pink solid circles), whereas plasma samples from three controls (184, 185, 192) did not. The GPIHBP1-specific mAb RE3 (black open squares) also blocked binding of LPL to GPIHBP1 (the “1:1 dilution” of mAb RE3 represents 20 μg/ml of purified IgG).
Figure 4.
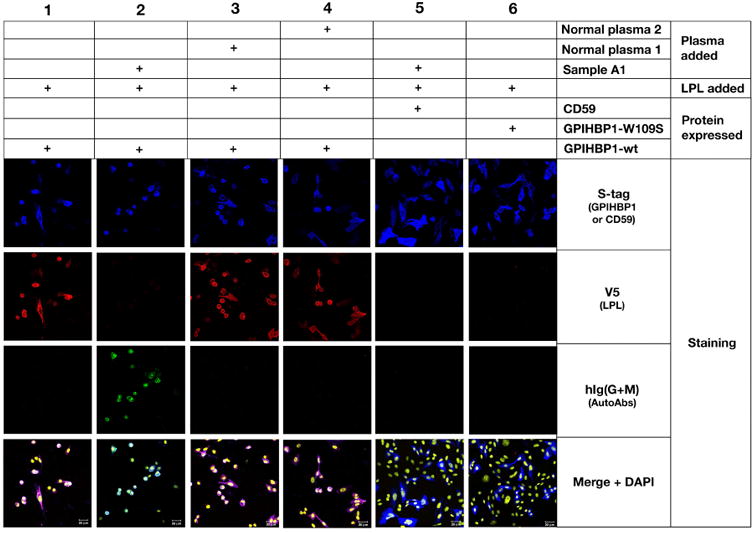
Immunocytochemistry studies showing that the GPIHBP1 autoantibodies in the plasma of patient A1 bind to GPIHBP1 on the surface of GPIHBP1-transfected CHO pgsA-745 cells and abolish binding of LPL. GPIHBP1 autoantibodies (green) in the plasma from patient A1 (1:20 dilution) bound to cells expressing S-protein–tagged human GPIHBP1 (Column 2), but not to cells expressing S-protein–tagged CD59 (Column 5). CD59 and GPIHBP1 expression on cells was detected with an antibody against the S-protein tag (blue). V5-tagged wild-type human LPL (detected with a V5 tag–specific antibody; red) binds avidly and specifically to GPIHBP1-transfected cells (Column 1). The binding of GPIHBP1 autoantibodies (in the plasma of patient A1) to GPIHBP1 abolished LPL binding (Column 2). In contrast, control plasma samples 1 and 2 (which did not contain GPIHBP1 autoantibodies) did not block the binding of LPL to GPIHBP1 (Columns 3 and 4, respectively). Cells expressing a mutant GPIHBP1 (GPIHBP1-W109S)12, 25 did not bind LPL (Column 6). DNA was stained with DAPI (yellow).
We predicted that the GPIHBP1 autoantibodies in patient A1 would be accompanied by low plasma levels of LPL. Indeed, the LPL mass level in the plasma of A1 was very low, 23.1 ng/ml (normal range, 40–156 ng/ml; mean, 84 ng/ml), consistent with reduced LPL binding to GPIHBP1 and reduced delivery of LPL to the intravascular compartment. We suspected that the levels of hepatic triglyceride lipase (HL) and endothelial lipase (EL), which do not bind GPIHBP1,29 would not be low. Indeed, the HL level in the plasma of A1, 59.8 ng/ml, was not low (normal range, 18–136 ng/ml; mean, 60 ng/ml). The EL level, 133.8 ng/ml, was also not low (normal range, 41–141 ng/ml; mean, 77 ng/ml). After an intravenous injection of 5000 IU of heparin, the LPL activity level in patient A1 was less than one-half of those in a pool of six normal control subjects, whereas the HL activity was significantly higher than the activity in the pooled plasma of normal subjects (Figure 5).
Figure 5. Levels of LPL catalytic activity in the post-heparin plasma of patient A1 were low, whereas levels of hepatic lipase activity were high.
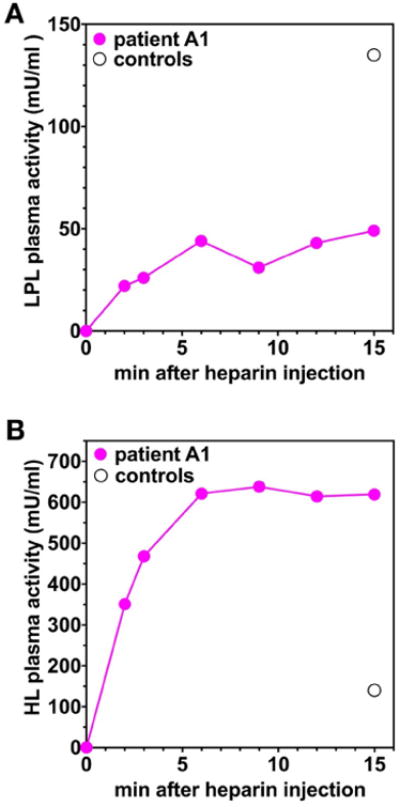
Plasma samples were obtained from patient A1 at baseline and at six time points after an intravenous injection of heparin, and LPL (A) and HL (B) activity levels were measured (pink solid circles). As a control, LPL and HL activity levels were also measured in a pool of plasma samples from six normal control subjects at the 15-min time point (black open circle).
In the recent study by Beigneux et al.,17 several patients with the GPIHBP1 autoantibody syndrome had systemic lupus erythematosus, and their plasma tested positive for antinuclear antibodies. Patient A1 had no clinical evidence of autoimmune disease, but testing revealed that his plasma was positive for antinuclear antibodies. The plasma of patient A1 did not have autoantibodies against LPL, as judged by an ELISA17 or a western blot assay (Figure 6).
Figure 6. The plasma of patient A1 does not contain autoantibodies against LPL.
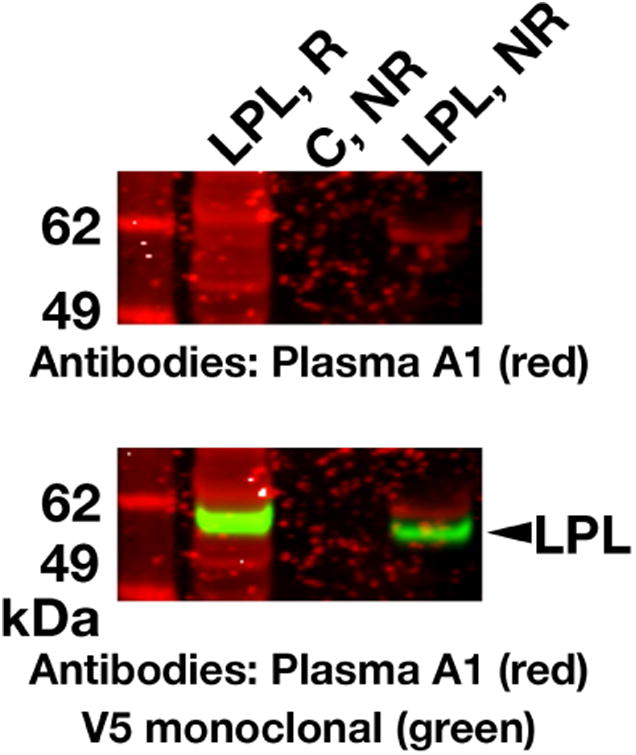
Medium from V5-tagged human LPL– transfected CHO cells was size-fractionated by SDS-PAGE under reducing (R) and nonreducing (NR) conditions. Western blots were performed with plasma from patient A1 (20 AU GPIHBP1 autoantibodies/ml) followed by IRDye680-labeled goat anti-human IgG (red). V5-tagged LPL on the same membrane was detected with an IRDye800-labeled V5 antibody (green). In the western blot with the plasma from patient A1, several bands with a size roughly similar to that of LPL were detected. However, the V5 antibody western blot revealed that the bands in the “plasma A1 western blot” were distinct from authentic V5-tagged LPL. As a negative control (C), we tested medium from nontransfected CHO cells.
The plasma triglycerides in patient A1 were 130 mg/dl in 2014, but no plasma samples were archived and available for analysis. The patient was subsequently lost to follow-up.
Discussion
We recently described autoantibodies against GPIHBP1, the endothelial cell LPL transporter, as a cause of acquired forms of chylomicronemia (“GPIHBP1 autoantibody syndrome”).17 In the current study, in order to increase our understanding of the frequency of this syndrome, we screened 33 patients with unexplained chylomicronemia from the Amsterdam Medical Center. Using a pair of ELISAs, we identified a single patient (patient A1) with the GPIHBP1 autoantibody syndrome. The GPIHBP1 autoantibodies were identified in two plasma samples obtained during 2011 and 2012 hospitalizations for chylomicronemia and acute pancreatitis. The GPIHBP1 autoantibodies in patient A1 interfered with the main function of GPIHBP1, which is to bind LPL and transport it to the capillary lumen. When GPIHBP1 is absent or functionally defective, LPL remains stranded within the interstitial spaces and never reaches the capillary lumen.1
In the initial description of the GPIHBP1 autoantibody syndrome, Beigneux et al.17 identified six patients with GPIHBP1 autoantibodies by screening ∼200 miscellaneous plasma samples, including many with hypertriglyceridemia and a handful from patients with both hypertriglyceridemia and autoimmune diseases. In that study, the information about the patient population, including genetic testing, was minimal. In the current study, we screened 33 hypertriglyceridemic patients in whom extensive genetic screening had failed to identify LPL, GPIHBP1, APOC2, LMF1, or APOA5 mutations. The sole patient identified as having GPIHBP1 autoantibodies (patient A1) did not have any clinical signs of autoimmune disease, but his plasma tested positive for antinuclear antibodies. In the earlier study, four of the six patients with the GPIHBP1 autoantibody syndrome had autoimmune diseases. At this point, the frequency of the GPIHBP1 autoantibody syndrome in patients with acquired forms of chylomicronemia remains incompletely defined, but it would appear that it is not rare—particularly in patients with clinical or serological evidence of autoimmune diseases. Additional studies are needed to gauge the overall frequency of GPIHBP1 autoantibodies in patients with acquired forms of hypertriglyceridemia.
In the study by Beigneux et al.,17 the hypertriglyceridemia in two GPIHBP1 autoantibody syndrome patients (patients 157 and 164) responded to treatment with immunosuppressive therapy (rituximab, mycophenolate mofetil).17, 30, 31 In one of the patients, normalization of plasma triglyceride levels during therapy was accompanied by a complete disappearance of GPIHBP1 autoantibodies.17 The identification of additional patients with the GPIHBP1 autoantibody syndrome will help to define optimal strategies for treatment. More patients would also help to define the relationship, if any, between GPIHBP1 autoantibody titers and plasma triglyceride levels.
The plasma triglyceride levels in patient A1 normalized several years after the hospitalizations for pancreatitis, raising the possibility that the GPIHBP1 autoantibody syndrome can, at least in some cases, resolve spontaneously. The plasma triglyceride levels in another patient with the GPIHBP1 autoantibody syndrome (patient 38 in the study by Beigneux and coworkers17) also normalized in the absence of immunosuppressive drugs. In that case, however, the normalization of plasma triglyceride levels occurred in the setting of suboptimal nutritional status accompanying exocrine pancreatic insufficiency (from recurrent bouts of acute pancreatitis).
A hallmark of both genetic GPIHBP1 deficiency and the GPIHBP1 autoantibody syndrome is low plasma levels of LPL.5-7, 15 In patient A1, both the pre-heparin LPL mass level and the post-heparin LPL catalytic activity levels were low. These findings make sense, given that GPIHBP1 is solely responsible for shuttling LPL to the capillary lumen.1 In the current study, we also examined plasma levels of two related lipases in the same protein family—HL and EL. We suspected that the levels of HL and EL would not be perturbed by GPIHBP1 autoantibodies, given that neither lipase binds to GPIHBP1.29 Indeed, HL and EL mass levels were not reduced in the plasma of patient A1. Also, after an injection of heparin, HL activity levels in plasma A1 were higher than in the normal controls. High levels of HL activity in post-heparin plasma were noted previously in Gpihbp1 knockout mice2 and in a chylomicronemia patient who was homozygous for a p.Q115P missense mutation in GPIHBP1.5 These observations suggest that defective GPIHBP1 function, whether genetic or acquired, does not reduce post-heparin HL activity levels and in some cases may lead to higher-than-normal plasma levels of HL.
Highlights.
One of 33 patients with hypertriglyceridemia had GPIHBP1 autoantibodies.
The patient's plasma was also positive for antinuclear antibodies.
The patient had low LPL mass levels and low post-heparin LPL activity levels.
Levels of hepatic triglyceride lipase (HL) and endothelial lipase (EL) are not reduced.
Acknowledgments
This work was supported by grants from the NHLBI (HL090553, HL087228, and HL125335) and a Transatlantic Network Grant from the Leducq Foundation (12CVD04). Xuchen Hu is supported by a Ruth L. Kirschstein National Research Service Award (T32HL69766), the NIH NIGMS Training Grant (T32GM08042), and the UCLA Medical Scientist Training Program. GKH is supported by a VIDI grant (016.156.445).
Abbreviations
- GPIHBP1
glycosylphosphatidylinositol-anchored high density lipoprotein binding protein-1
- TRLs
triglyceride-rich lipoproteins
- LPL
lipoprotein lipase
- HL
hepatic triglyceride lipase
- EL
endothelial lipase
- mAb
monoclonal antibody, uPAR, urokinase-type plasminogen activator receptor
Footnotes
Author Contributions: Xuchen Hu performed experiments, analyzed the data, wrote the first draft of the manuscript, and edited the manuscript. Geesje M. Dallinga-Thie generated the patient cohort and contributed to scientific discussions. G. Kees Hovingh provided background and insights into the patient with GPIHBP1 autoantibodies. Sandy Y. Chang performed ELISAs. Norma P. Sandoval performed ELISAs. Tiffany Ly P. Dang generated monoclonal antibodies for ELISAs, western blots, and immunocytochemistry studies. Isamu Fukamachi performed ELISAs. Kazuya Miyashita performed ELISAs. Katsuyuki Nakajima performed ELISA analyses and contributed to scientific discussions. Masami Murakami performed ELISA analyses and contributed to scientific discussions. Loren G. Fong provided important scientific insights and edited the paper. Michael Ploug provided important scientific insights, edited the manuscript, and generated purified proteins (including GPIHBP1) for ELISAs. Stephen G. Young analyzed the data and wrote the manuscript. Anne P. Beigneux performed experiments, analyzed the data, and wrote the manuscript. All authors approved the final version of the manuscript.
Disclosures: The authors have no financial interests to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Davies BS, Beigneux AP, Barnes RH, 2nd, et al. GPIHBP1 is responsible for the entry of lipoprotein lipase into capillaries. Cell Metab. 2010;12:42–52. doi: 10.1016/j.cmet.2010.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beigneux AP, Davies B, Gin P, et al. Glycosylphosphatidylinositol-anchored high density lipoprotein–binding protein 1 plays a critical role in the lipolytic processing of chylomicrons. Cell Metab. 2007;5:279–291. doi: 10.1016/j.cmet.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goulbourne C, Gin P, Tatar A, et al. The GPIHBP1–LPL complex is responsible for the margination of triglyceride-rich lipoproteins in capillaries. Cell Metab. 2014;19:849–860. doi: 10.1016/j.cmet.2014.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chait A, Brunzell JD. Chylomicronemia syndrome. Adv Intern Med. 1992;37:249–273. [PubMed] [Google Scholar]
- 5.Franssen R, Young SG, Peelman F, et al. Chylomicronemia with low postheparin lipoprotein lipase levels in the setting of GPIHBP1 defects. Circ Cardiovasc Genet. 2010;3:169–178. doi: 10.1161/CIRCGENETICS.109.908905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olivecrona G, Ehrenborg E, Semb H, et al. Mutation of conserved cysteines in the Ly6 domain of GPIHBP1 in familial chylomicronemia. J Lipid Res. 2010;51:1535–1545. doi: 10.1194/jlr.M002717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Plengpanich W, Young SG, Khovidhunkit W, et al. Multimerization of GPIHBP1 and familial chylomicronemia from a serine-to-cysteine substitution in GPIHBP1's Ly6 domain. J Biol Chem. 2014;289:19491–19499. doi: 10.1074/jbc.M114.558528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ariza MJ, Martinez-Hernandez PL, Ibarretxe D, et al. Novel mutations in the GPIHBP1 gene identified in 2 patients with recurrent acute pancreatitis. J Clin Lipidol. 2016;10:92–100 e101. doi: 10.1016/j.jacl.2015.09.007. [DOI] [PubMed] [Google Scholar]
- 9.Rabacchi C, D'Addato S, Palmisano S, et al. Clinical and genetic features of 3 patients with familial chylomicronemia due to mutations in GPIHBP1 gene. J Clin Lipidol. 2016;10:915–921 e914. doi: 10.1016/j.jacl.2016.03.009. [DOI] [PubMed] [Google Scholar]
- 10.Ahmad Z, Wilson DP. Familial chylomicronemia syndrome and response to medium-chain triglyceride therapy in an infant with novel mutations in GPIHBP1. J Clin Lipidol. 2014;8:635–639. doi: 10.1016/j.jacl.2014.08.010. [DOI] [PubMed] [Google Scholar]
- 11.Buonuomo PS, Bartuli A, Rabacchi C, Bertolini S, Calandra S. A 3-day-old neonate with severe hypertriglyceridemia from novel mutations of the GPIHBP1 gene. J Clin Lipidol. 2015;9:265–270. doi: 10.1016/j.jacl.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 12.De Castro-Oros I, Civeira F, Pueyo MJ, et al. Rare genetic variants with large effect on triglycerides in subjects with a clinical diagnosis of familial vs nonfamilial hypertriglyceridemia. J Clin Lipidol. 2016;10:790–797. doi: 10.1016/j.jacl.2016.02.010. [DOI] [PubMed] [Google Scholar]
- 13.Chokshi N, Blumenschein SD, Ahmad Z, Garg A. Genotype-phenotype relationships in patients with type I hyperlipoproteinemia. J Clin Lipidol. 2014;8:287–295. doi: 10.1016/j.jacl.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 14.Patni N, Brothers J, Xing C, Garg A. Type 1 hyperlipoproteinemia in a child with large homozygous deletion encompassing GPIHBP1. J Clin Lipidol. 2016;10:1035–1039 e1032. doi: 10.1016/j.jacl.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 15.Rios JJ, Shastry S, Jasso J, et al. Deletion of GPIHBP1 causing severe chylomicronemia. J Inherit Metab Dis. 2012;35:531–540. doi: 10.1007/s10545-011-9406-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu X, Sleeman MW, Miyashita K, et al. Monoclonal antibodies that bind to the Ly6 domain of GPIHBP1 abolish the binding of LPL. J Lipid Res. 2017;58:208–215. doi: 10.1194/jlr.M072462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beigneux AP, Miyashita K, Ploug M, et al. Autoantibodies Against GPIHBP1 as a Cause of Hypertriglyceridemia. N Engl J Med. 2017;376:1647–1658. doi: 10.1056/NEJMoa1611930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gårdsvoll H, Hansen LV, Jorgensen TJ, Ploug M. A new tagging system for production of recombinant proteins in Drosophila S2 cells using the third domain of the urokinase receptor. Protein Expr Purif. 2007;52:384–394. doi: 10.1016/j.pep.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 19.Beigneux AP, Fong LG, Bensadoun A, et al. GPIHBP1 Missense Mutations Often Cause Multimerization of GPIHBP1 and Thereby Prevent Lipoprotein Lipase Binding. Circ Res. 2014;116:624–632. doi: 10.1161/CIRCRESAHA.116.305085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beigneux AP, Gin P, Davies BSJ, et al. Highly conserved cysteines within the Ly6 domain of GPIHBP1 are crucial for the binding of lipoprotein lipase. J Biol Chem. 2009;284:30240–30247. doi: 10.1074/jbc.M109.046391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gardsvoll H, Jacobsen B, Kriegbaum MC, et al. Conformational regulation of urokinase receptor function: impact of receptor occupancy and epitope-mapped monoclonal antibodies on lamellipodia induction. J Biol Chem. 2011;286:33544–33556. doi: 10.1074/jbc.M111.220087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hansen LV, Gardsvoll H, Nielsen BS, et al. Structural analysis and tissue localization of human C4.4A: a protein homologue of the urokinase receptor. Biochem J. 2004;380:845–857. doi: 10.1042/BJ20031478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mysling S, Kristensen KK, Larsson M, et al. The acidic domain of the endothelial membrane protein GPIHBP1 stabilizes lipoprotein lipase activity by preventing unfolding of its catalytic domain. Elife. 2016;5:e12095. doi: 10.7554/eLife.12095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Surendran RP, Visser ME, Heemelaar S, et al. Mutations in LPL, APOC2, APOA5, GPIHBP1 and LMF1 in patients with severe hypertriglyceridaemia. J Intern Med. 2012;272:185–196. doi: 10.1111/j.1365-2796.2012.02516.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kastelein JJ, Jukema JW, Zwinderman AH, et al. Lipoprotein lipase activity is associated with severity of angina pectoris. REGRESS Study Group. Circulation. 2000;102:1629–1633. doi: 10.1161/01.cir.102.14.1629. [DOI] [PubMed] [Google Scholar]
- 26.Ishida T, Miyashita K, Shimizu M, et al. ELISA system for human endothelial lipase. Clin Chem. 2012;58:1656–1664. doi: 10.1373/clinchem.2012.187914. [DOI] [PubMed] [Google Scholar]
- 27.Machida T, Miyashita K, Sone T, et al. Determination of serum lipoprotein lipase using a latex particle-enhanced turbidimetric immunoassay with an automated analyzer. Clin Chim Acta. 2015;442:130–135. doi: 10.1016/j.cca.2015.01.016. [DOI] [PubMed] [Google Scholar]
- 28.Miyashita K, Kobayashi J, Imamura S, et al. A new enzyme-linked immunosorbent assay system for human hepatic triglyceride lipase. Clin Chim Acta. 2013;424:201–206. doi: 10.1016/j.cca.2013.06.016. [DOI] [PubMed] [Google Scholar]
- 29.Gin P, Beigneux AP, Voss C, et al. Binding preferences for GPIHBP1, a glycosylphosphatidylinositol-anchored protein of capillary endothelial cells. Arterioscler Thromb Vasc Biol. 2011;31:176–182. doi: 10.1161/ATVBAHA.110.214718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blom DJ, Marais AD. Severe hypertriglyceridemia in a patient with lupus. Am J Med. 2005;118:443–444. doi: 10.1016/j.amjmed.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 31.Ashraf AP, Beukelman T, Pruneta-Deloche V, Kelly DR, Garg A. Type 1 hyperlipoproteinemia and recurrent acute pancreatitis due to lipoprotein lipase antibody in a young girl with Sjogren's syndrome. J Clin Endocrinol Metab. 2011;96:3302–3307. doi: 10.1210/jc.2011-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beigneux AP, Davies BSJ, Tat S, et al. Assessing the role of the glycosylphosphatidylinositol-anchored high density lipoprotein-binding protein 1 (GPIHBP1) three-finger domain in binding lipoprotein lipase. J Biol Chem. 2011;286:19735–19743. doi: 10.1074/jbc.M111.242024. [DOI] [PMC free article] [PubMed] [Google Scholar]