

Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a syndrome of severe immune activation and dysregulation resulting in extreme and often life-threatening inflammation. HLH has been well recognized in pediatric populations and most current diagnostic and therapeutic guidelines are based on pediatric HLH. Recently there has been recognition of HLH in adults, especially secondary to immune deregulation by an underlying rheumatologic, infectious, or malignant condition. In this review we focus on malignancy-associated HLH (M-HLH). In M-HLH possible mechanisms of pathogenesis include severe inflammation, persistent antigen stimulation by the tumor cells, and loss of immune homeostasis due to chemotherapy, hematopoietic stem cell transplant or infection. Previously considered rare, M-HLH may occur in up to 1% of patients with hematologic malignancies. M-HLH is often missed or diagnosed late in most published studies, and associated with a poor median survival of less than 2 months. Identification of the clinical and laboratory features specific to M-HLH in adults may allow early detection, consultation with HLH experts, and intervention. Improved management of adult M-HLH with optimal combinations of T-lympholytic and immunosuppressive agents and incorporation of novel agents based on the pediatric experience will hopefully improve outcomes in M-HLH in adults.
Keywords: hemophagocytosis, lymphohistiocytosis, malignancy, adults
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a syndrome of severe immune activation and deregulation characterized by hyperactive macrophages and lymphocytes, pro-inflammatory cytokine hypersecretion, tissue infiltration, hemophagocytosis, and organ damage1, 2. An excessive deregulated inflammatory response plays a central role in the pathogenesis of HLH including (1) hyperactivation of CD8+ T lymphocytes and macrophages, (2) proliferation and infiltration of these inflammatory cells in organs, and (3) uncontrolled production of Type 1 helper cell (Th1) cytokines. Severe, and often life-threatening inflammation and immune mediated organ damage are the clinical hallmarks of HLH1, 3, 4. HLH occurs either as primary HLH characterized by genetic defects in cytotoxic immune function, or as secondary or acquired HLH characterized by reactive immune overstimulation to an aberrant non-self antigen5-7. “Secondary” HLH may also have “primary” co-factors as demonstrated by so called hypomorphic mutations in HLH-associated gene loci8.
Historically, most diagnostic guidelines, international databases, and treatment trials in HLH have focused on pediatric populations. However, HLH is not a pediatric-specific disease and may occur at any age. A nationwide survey in 300 Japanese hospitals noted that 40% of the HLH cases occurred in adults9. HLH in adults is frequently secondary to an underlying stimulus and has been associated with dismal outcomes. The data regarding HLH in the setting of malignancy in adults is very limited. Recent retrospective studies have shown that this entity may occur in up to 1% of patients with underlying malignancies at diagnosis, during therapy, or even after control of the underlying malignancy, and may be more common than previously estimated10. This review was conceptualized and formulated by a group of experts in adult and pediatric HLH to update the current knowledge of M-HLH with a focus on clinical aspects that would help academic and community hematology-oncology specialists consider, diagnose, and manage this entity.
Primary (Genetic) and Secondary (Reactive) HLH
Primary HLH (also called familial or genetic HLH) is an autosomal recessive disease with an incidence of 1/50,000-1/100,000 live-born children11, 12. Patients often have a clear familial inheritance or an identifiable genetic mutation. These are most frequently bi-allelic mutations in genes encoding for perforin, syntaxin 11, Munc 18-2, Munc 13-4 and other proteins involved in cytotoxic granule activation, polarization, priming, fusion, or function1, 13. In many circumstances no clear immune trigger is identifiable14. Primary HLH carries a high morbidity and mortality, and is associated with a median survival of less than 2 months without treatment15, 16. The development of effective treatment protocols and a concerted international effort have significantly improved the recognition and long-term survival (>50%) in primary HLH17. Primary HLH has traditionally been considered a disease of pediatric populations. Of note, systematic genetic evaluation in adolescent and adult patients with HLH identified hypomorphic mutations (PRF1, MUNC13-4, STXBP2, STX11, SH2D1A, BIRC4) in 7-14% of the patients suggesting that late-onset primary HLH occurs more commonly than previously suspected8, 18.
Secondary HLH includes adults and older children who lack a family history or a known genetic cause for HLH. The list of triggers associated with secondary HLH is extensive19. Secondary HLH is the more frequent presentation of HLH seen in adults20 and comprises two main groups: malignancy-associated HLH (or M-HLH) and non malignancy-associated HLH. Frequently noted malignancy triggers for adult M-HLH include hematological malignancies such as lymphomas, T/NK-cell disorders, acute leukemias, lymphoproliferative diseases, and myelodysplastic syndrome10, 21-33. Non malignancy-associated secondary HLH is further sub-classified as infection-related HLH [especially virally induced by EBV, CMV, or human immunodeficiency virus (HIV), but also bacterial, protozoal or mycotic infection induced]34-38, autoimmune disease-related HLH (usually classified as macrophage activation syndrom (MAS) most comonly triggered by systemic lupus erythematosus, systemic juvenile idiopathic arthritis, polymyosistis, vasculitis)37, 39, 40, spontaneous or iatrogenic immune suppression related HLH, and post hematopoietic or solid organ transplant HLH41, 42. Secondary HLH in adults frequently manifests as an aggressive disease with mortality rates ranging from 8% in MAS complicating systemic juvenile idiopathic arthritis (sJIA)43 to >80% in M-HLH32, 33.
It is also important to note that secondary HLH in adults is often multifactorial with more than one etiology contributing to the immune dysregulation. In either case efforts should be made to promptly identify the underlying trigger and expediently initiate therapy if the diagnosis of HLH is suspected, regardless of the classification.
Physiology of HLH
Patients with HLH have deregulation of the immune system including predisposing immunodeficiency, immune activation and immune activation mediated organ damage1, 44. Jordan et al used perforin-deficient mouse models to gain insight into the pathophysiology of primary HLH44. Perforin-deficient mice did not spontaneously develop HLH. However, upon exposure to lymphocytic choriomeningitis virus (LCMV) they manifested all of the clinical features of HLH including fever, splenomegaly, pancytopenia, hypertriglyceridemia, hypofibrinogenemia, and elevation of multiple serum cytokine levels, and histologic evidence of hemophagocytosis in many tissues (Figure 1). They demonstrated that in murine models CD8+ T-cells, but not NK-cells played a central role in the evolution of HLH. Cytokine neutralization studies revealed that interferon-gamma (IFN-gamma) was uniquely essential and was produced as a result of increased antigen presentation to CD8+ T-cells.
Figure 1.
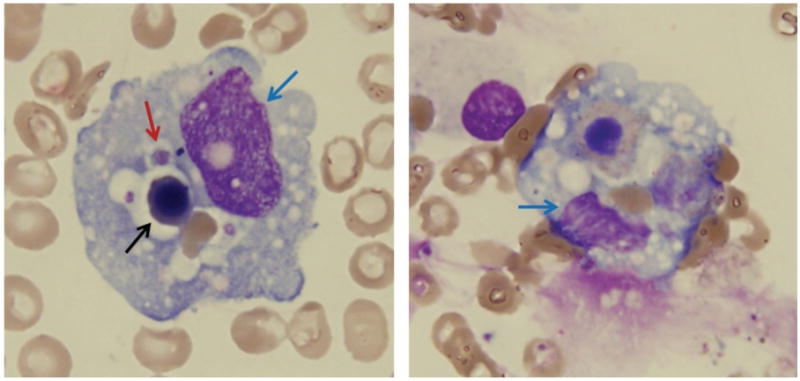
Morphologic features of hemophagocytosis on bone marrow aspirate smear (Wright-Giemsa, original magnification × 500); blue arrow, WBC; black arrow, nucleated RBC; red arrow, platelet.
Another recently published study investigated the regulatory T (Treg) and CD8+ T cells homeostasis in perforin knockout and wild-type mice during LCMV mediated inflammation. The perforin knockout mice had disease that clinically resembled HLH. T-cell interrogation in these mice demonstrated a major reduction of the Treg compartment in spleen and lymph nodes and a 2-fold decreased expression of activation marker CD25 on the Treg cell surface. On the other hand, CD8+ T cells exhibited a greater expression of CD25 (14-fold) and other T-cell activation markers such as CD69. The serum soluble CD25 (soluble interleukin 2 α (s-IL2rα) in the perforin deficient mice was also significantly higher. These data suggest that HLH is a disorder of Treg-CD8+ balance and IL-2 network homeostasis where CD8+ T cells are excessively activated and successfully outcompete Treg cells for IL-2 with resultant T-reg hypofunction45.
Similar to primary HLH, immune-activation and immune mediated-pathology likely play a central role in the evolution of secondary HLH1. Biopsies of lymphoid tissues or histological examination of liver tissue from patients with secondary HLH reveal highly activated macrophages and lymphocytes. Secondary HLH is characterized by acute clinical signs and symptoms of immune activation including hepatomegaly, jaundice, adenopathy, rash, seizures, and focal neurologic deficits, as well as laboratory measures such as very high serum levels of numerous cytokines including IFN-gamma, tumor necrosis factor α (TNF-α), interleukin 6 (IL-6), IL-10, and macrophage colony-stimulating factor (M-CSF)46, 47. Among these, IFN-gamma appears to be one of the most sensitive and early indicators of disease activity. High IFN-gamma levels correlated positively with high peripheral leukocyte count and high serum lactate dehydrogenase and negatively with the CD4/CD8 T-cell ratio47. Furthermore, IFN-gamma, but not TNF, was detected just before a relapse marked by spiking fever. Zhang et al noted clinical features consistent with HLH/ MAS in patients with sJIA who were identified to be heterozygous for rare variants of primary HLH-associated genes13. This suggests that the inflammatory stress of the rheumatologic condition unmasked HLH in these predisposed individuals.
Pathology and Role of Hemophagocytosis in M-HLH
As has been previously well described in primary HLH, a finding of hemophagocytosis or lymphohistiocytosis on tissue evaluation is supportive but on its own lacks sensitivity and specificity to diagnose secondary HLH48, 49, including adult M-HLH. In a large series of 162 HLH patients, hemophagocytosis was seen in around 70% of patients with a confirmed diagnosis of HLH; however, it was also observed in ∼40% patients with a similar clinical presentation who did not meet other criteria for HLH and were not considered to have a clinical diagnosis of HLH50. These data suggest that the presence or absence of phagocytes should be considered as one supportive feature to be evaluated in concordance with other clinical and laboratory manifestations during the work-up of M-HLH.
A bone marrow biopsy is frequently performed as a part of the initial work-up in patients with hematologic malignancies (acute or chronic leukemia, lymphoma, myeloma). Hemophagocytosis is frequently not seen on the initial bone marrow as pathologic alterations due to HLH may take days or week to become manifest49. The absence of hemophagocytosis on the initial bone marrow should not delay therapy in cases with an otherwise high suspicion for hemophagocytosis. Initial bone marrows in more fulminant cases of HLH or follow-up bone marrow evaluations in a patient with HLH may show increased small lymphocytes, predominantly T cells; as well as histiocyte infiltrate. The histiocytes are composed predominantly of mature macrophages, which often show engulfed red blood cells, nucleated erythrocytes or neutrophils. In addition to bone marrow evaluations, changes consistent with HLH may be seen in liver, lymph node, or splenic tissue. In liver biopsy specimens hemophagocytosis, when apparent, typically manifests as perivascular and portal lymphohistiocytic infiltrates. Lymph nodes show a preserved nodal architecture with a sinusoidal infiltration of bland histiocytes containing erythrocytes, admixed with occasionally lymphocytes and neutrophils. Splenectomy is rarely performed but when available in a patient with HLH, splenic tissue often shows increased macrophages including phagocytes in the red pulps.
CD163 functions well in paraffin-embedded tissue specimens (bone marrow, lymph node, liver tissue) to highlight increased macrophages and is frequently used in the work-up of tissue specimens in patients suspected to have HLH51. In addition to macrophages, increased megakaryocytes and erythroid hyperplasia as a result of compensatory hematopoiesis may be noted. Mild reticulin fibrosis and dysplastic changes in erythrocytes and granulocytes as tissue manifestations of HLH have also been described. In patients with a high clinical suspicion or otherwise confirmed HLH available tissue could be used to identify potential triggers for M-HLH such as T/NK cell neoplasm, B cell lymphoproliferative disease, early myelodysplastic syndrome or aplastic anemia by pathologic evaluation by an experienced hemato-pathologist, directed flow-cytometry, TCR clonality and immunohistochemistry stains or for infectious triggers by performing EBER in situ hybridization, CMV and HSV immunohistochemistry, and viral PCRs.
Malignancy Associated Secondary HLH
The vast majority of malignancies triggering secondary HLH in children and adults are hematological malignancies including lymphomas and leukemia. M-HLH is more frequently diagnosed in adults20. Machaczka et al noted that the prevalence of M-HLH was 0.9% in adults with hematological cancers, but could be as high as 20% in patients with specific rare types of B-cell lymphomas (intravascular B-cell lymphoma or B-cell lymphoma without peripheral adenopathies) and T-cell lymphoma (nasal natural-killer-cell or panniculitis-like subtypes)10.
Pathophysiology of M-HLH
M-HLH may manifest during the treatment of a known malignancy or as the presenting feature of a yet undiagnosed malignancy. A number of possible mechanisms of pathogenesis have been identified. First, it is postulated that the hyperinflammation is triggered by the neoplasm due to an excessive secretion of pro-inflammatory cytokines and persistent antigen stimulation by the tumor cells. Second, inherited immune disorders that predispose to both HLH and malignancy (e.g. X-linked lymphoproliferative diseases) may further predispose patients to the development of M-HLH. Third, M-HLH may also occur during chemotherapy. The combined immunodeficiency generated by the underlying malignancy and the loss of immune homeostasis due to chemotherapy (or hematopoietic stem cell transplant or infection) further aggravates T-cell dysfunction that lowers the threshold for triggering HLH in these patients. Fourth, malignancy-induced immunodeficiency combined with tumor directed therapy predisposes to infections, which may act as independent triggers of M-HLH in these patients52. Delavigne et al identified M-HLH in 32 patients with AML receiving induction therapy. A potential infectious trigger for HLH was found in 24 patients (75%) and included a mixture of bacterial, viral, and fungal infections highlighting the importance of proactively evaluating for infectious etiologies in adult patients with suspected M-HLH31.
More recently, symptoms similar to HLH have been described in patients receiving immunotherapies. These are due to proinflammatory cytokine overproduction by T-cell activating immunotherapies used in the treatment of leukemia/lymphoma and solid tumors (e.g. bispecific monoclonal antibody blinatumomab, chimeric antigen receptor (CAR) T-cell therapies, dendritic vaccines, combinations with checkpoint inhibitors, immunomodulatory drugs (IMiDs) such as lenalidomide and thalidomide4, 24, 25, 53. The cytokine release syndrome (CRS) with these agents bears a clinical and immunological signature similar to HLH and often responds to therapies used in HLH54, 55.
Clinical Course of M-HLH
The clinical course of M-HLH is often rapidly progressing and characterized by poor outcomes (Table 1). Lehmberg et al identified M-HLH in 21 pediatric and adolescent patients, most of who had T- (n=12) or B-cell neoplasms (n=7), with EBV as a co-trigger in five patients. An additional 8 patients had chemotherapy-induced HLH. The median survival for M-HLH was 1.2 months25. Machaczka et al noted that 8 out of 887 (0.9%) patients diagnosed with hematological malignancies developed M-HLH10. Six of the eight patients received HLH-directed therapy and 3 achieved remission. The median overall survival was 2.4 months; only one remission was durable. Shabbir et al identified 18 adults with HLH diagnosed at one institution, of whom 6 had M-HLH (4 patients with an underlying hematologic malignancy, 2 patients post-autologous stem cell transplant). Corticosteroids and/or cyclosporine were most frequently used to treat patients24. Other agents used were etoposide, intravenous immunoglobulin, cyclophosphamide and chemotherapy indicating heterogeneous treatment approaches to M-HLH. The 6-month mortality rate was 72% and median survival 1.2 months. Parikh et al reviewed patients treated at the Mayo Clinic over a 15 year period (1996 – 2011) and noted that among 250 adults in whom a diagnosis of HLH was suspected 62 met the HLH 2004 diagnostic criteria23 and were included in the final analysis. Thirty-two of the 62 patients (52%) had M-HLH. The median survival of the entire cohort was 2.1 months; the median survival of patients with M-HLH was 1.4 months compared with 22.8 months for adults with non-malignancy-associated HLH. On a multivariate analysis, the presence of a malignant tumor and hypoalbuminemia were significant predictors of inferior survival. Another study by Otrock et al on the management of adult HLH reported that 29% (21/73) of the adult HLH cases in their institution were malignancy associated. Adult patients with M-HLH had a worse survival compared to patients with non-malignancy-associated HLH (median overall survival 1.13 versus 46.53 months, respectively; P < 0.0001)32.
Table 1. Summary of M-HLH clinical outcomes across different institutions.
| Study | Number of patients | Most frequent associated malignancy | 6-month survival rate | Median survival (months) |
|---|---|---|---|---|
| Tamamyam et al.33 (2016) | 35 | AML/MDS (n=13), T-lymphoma (n=10), DLBCL (n=6), HL (n=6), CLL (n=4), CML (n=2), Follicular lymphoma (n=2) | 30% | 2.0 |
| Parikh et al.23 (2014) | 32 | T-lymphoma (n=19), DLBCL (n=6), EBV-associated PTLD (n=3), HL (n=1), CMML (n=1), hemangioendothelioma (n=1), systemic histiocytosis (n=1) | Not reported | 1.4 |
| Otrock et al.32 (2015) | 21 | B-neoplasms (n=10), T-neoplasms (n=6), HL (n=3), AML (n=1), MDS (n=1) | 20% | 1.1 |
| Lehmberg et al.25 (2015) | 21 | T-neoplasms (n=12), B-neoplasms (n=7) | 67% | 1.2 |
| Machaczka et al.10 (2011) | 8 | HL (n=2), MM (n=2), 1 each of B-CLL, PTCL, AILD, WM, ATL | 38% | 2.4 |
| Shabbir et al.24 (2011) | 6 | AML (n=2), T-cell lymphoma (n=2), post auto-SCT for MM (n=2) | Not reported | 1.2 (entire adult HLH cohort, n=18) |
AML, acute myeloid leukemia, MDS, myelodysplastic syndrome, DLBCL, diffuse large B-cell lymphoma, CLL, chronic lymphocytic leukemia, CML, chronic myeloid leukemia, HL, hodgkins lymphoma, MM, multiple myeloma, PTCL, peripheral T-cell lymphoma, AILD, angioimmunoblastic lymphadenopathy, WM, Waldenstorms macroglobulinemia, ATL, adult T-cell leukemia, SCT, stem cell transplant, PTLD, post-transplant lymphoproliferative disorder, CMML, chronic myelomonocytic leukemia.
Tamamyam et al retrospectively identified pathologic hemophagocytosis and/or lymphohistiocytosis in 61 adults with underling malignancies treated between 1991-2001 at M D Anderson Cancer Center33. Thirteen of 61 (21%) patients met the HLH-2004 diagnostic criteria. To identify potentially missed cases of HLH, they reviewed the published literature and selected additional variables known to be associated with adult HLH, resulting in an extended diagnostic criteria list of 18 variables. Thirty-five patients had additional systemic variables consistent with HLH. The median survival was 2 months among patients treated with HLH-directed therapy and 2 months among those not treated with HLH-directed therapies. Seventeen of 33 (52%) patients died within 8 weeks from diagnosis and 23 (70%) died within 6 months from diagnosis33.
Diagnosis of M-HLH
Traditional Diagnostic Criteria
The initial HLH guidelines in 1991 included 5 features: (1) fever, (2) splenomegaly, (3) cytopenias affecting at least 2-3 lineages in the peripheral blood, (4) hypertriglyceridemia and/or hypofibrinogenemia, and (5) hemophagocytosis in bone marrow, spleen or lymph nodes56. The 2004 guidelines included 3 additional criteria: (6) low or absent natural killer-cell activity, (7) hyperferritinemia and (8) high levels of s-IL2rα. Five of eight criteria must be fulfilled to make a diagnosis of secondary HLH2, 19, 56. However, patients with a molecular diagnosis of HLH do not need to fulfill the diagnostic criteria to diagnose primary HLH in the appropriate clinical scenario. It is important to note that the pathologic finding of hemophagocytosis is not pathognomonic for HLH, a common cause for missed or delayed diagnosis among treating physicians.
Of note, the 1991 and 2004 guidelines were developed using pediatric populations. Information and diagnostic guidelines specific to adult HLH are limited, and current diagnostic and treatment approaches of HLH in adults are extrapolations from retrospective databases and clinical trials in childhood HLH. These knowledge gaps have led to under-diagnosis or delayed diagnosis of M-HLH and non-malignancy-associated HLH in adults, further worsening their outcomes23, 32, 57. Recent efforts focusing on developing adult- and malignancy- specific HLH consensus recommendations, in order to improve our ability to rapidly identify and manage this entity are underway30.
Need for Adult M-HLH Diagnostic Criteria
The first critical step in successful treatment of M-HLH is considering the diagnosis. A greater awareness of M-HLH among oncologists and a growing incidence of the entity, especially in wake of the increased use of immune activating and modulating agents, will lead more hematologists and oncologists to consider and initiate a workup for HLH in adults. Unfortunately at present there remains significant uncertainty regarding the best approach to diagnose M-HLH in adults 4, 23, 58. The HLH-2004 criteria require 5 of 8 criteria to be met including two criteria (sIL2rα and natural killer-cell activity) that typically require send-out specialty labs and are difficult to obtain in a timely manner in smaller institutions or community hospitals. These tests require up to 5-8 days to be finalized, even in larger institutions. Non-availability or a delayed availability of these tests may delay confirmation of the diagnosis or referral of adults with HLH to tertiary care facilities. This may further worsen poor outcomes in adult HLH and M-HLH.
The above constraints serve as the impetus to identify and include additional diagnostic variables easily obtained by physical examination and local routine laboratories to allow for more rapid suspicion and referral of possible adult M-HLH patients to specialized centers until prospectively validated definitive adult M-HLH criteria are developed and validated. We recently proposed one such schema for adult M-HLH that incorporated more rapidly and broadly available physical examination and laboratory variables thereby allowing for possible earlier consideration and referral to tertiary centers for the work-up and treatment of adult M-HLH33, especially from smaller community institutions. We reviewed the published literature and expert opinions and identified 18 variables closely associated with HLH in peer-reviewed manuscripts. Sensitivity analysis suggested that patients meeting any 5 of these 18 criteria could be considered to have a high likelihood of M-HLH (Figure 2). We then used the panel of 18 variables to identify patients who would have been diagnosed to have HLH using these extended criteria from among the 61 patients who had a known pathologic finding of hemophagocytosis or lymphohistiocytosis from a pathology database for years 2001-2014 at our institution. Thirty-five of the 61 (57%) patients who manifested 5 or more additional findings from our proposed extended criteria, beyond pathologic hemophagocytosis or lymphohistiocytosis were more likely based on our criteria to have had a systemic M-HLH rather than just a reactive hemophagocytosis or lymphohistiocytosis in the presence of a malignancy. It is noteworthy that 13 of these 35 patients (38%) also met standard HLH-2004 criteria but the other 22 did not. There was no significant difference in overall survival among the 13 patients who met standard HLH-2004 criteria (median overall survival = 1.43 months) and the 22 patients who did not meet standard HLH-2004 criteria but met our proposed extended 18-point HLH criteria (median overall survival = 1.76) (P=0.34). The 26 remaining patients from the 61 (43%) who showed hemophagocytosis or lymphohistiocytosis on pathology but met NEITHER standard HLH-2004 nor our extended 18-point HLH criteria had a significantly improved overall survival (median overall survival = 17.2 months, P<0.05). The inferior survival in patients who met standard HLH-2004 criteria or our extended 18-point HLH criteria but not among patients who had pathologic evidence of hemophagocytosis/lymphocytosis but met neither criterion suggests that both criteria may reasonably identify patients who likely have a more aggressive systemic process requiring directed therapy. It must be noted that our extended 18-point criteria were developed using retrospective data and it is quite possible that a number of patients who met our criteria would have met the HLH-2004 criteria if HLH specific labs such as triglycerides, sIL2r levels, NK-cell activity were performed but these were not done in a number of patients. The intent of our analysis was not to replace existing HLH standard criteria but rather to highlight that inclusion of additional diagnostic variables, especially variables that are easily and quickly assessed by routine laboratory or physical examination, may promote early suspicion and work-up for HLH in community centers.
Figure 2.

Adult Hemophagocytic Histiocytosis characteristics and Diagnostic Variables. Each closed circle ((x025CB)) represents a negative result. Blanks represent missing information. Each row is one patient and each column is a variable. Numbers show percent positive patients (for each characteristic) or percent positive characteristics (for each patient) excluding missing information. Variables evaluated (listed in order of columns) included BM/lymph node/spleen hemophagocytosis per pathology evaluation, fever, splenomegaly (clinically palpable spleen), hepatomegaly (clinically palpable liver), anemia (hemoglobin < 9.0 g/L), thrombocytopenia (platelets < 100 × 109/L), neutropenia (absolute neutrophil count (ANC) < 1.0 × 109/L), monocytosis (absolute monocyte count (AMC) > 1.0 × 109/L), renal failure (≥ 50% increase in creatinine over baseline), elevation of hepatic enzymes (≥ 2.5× upper limit of normal), hypofibrinogenemia (fibrinogen ≤ 150mg/dL), hyperferritinemia (ferritin ≥ 500micrograms/L), coagulopathy (PT ≥ 1.5× upper limit of normal and/or PTT ≥ 1.5× upper limit of normal and/or D-dimer ≥ 10.0mcg/mL), hypoalbuminemia (< 3.5g/dL), elevated LDH (≥ 2.5× upper limit of normal), hypertriglyceridemia (≥ 265 mg/dL), elevated b2-microglobulin (≥ 2mg/L), and elevated soluble IL-2 receptor (CD25) ≥ 2400U/mL. Abbreviations: Hb, hemoglobin; ULN, upper limit of normal; LDH, lactate dehydrogenase; HLH 2004, Hemophagocytic lymphohistiocytosis 2004 diagnostic criteria; MDS, myelodysplastic syndromes; PTLD, post-transplant lymphoproliferative disorder; CML, chronic myeloid leukemia; DLBCL, diffuse large B-cell lymphoma; CLL, chronic lymphocytic leukemia; XLP, X-linked lymphoproliferative disease; AML, acute myeloid leukemia; HL, Hodgkin's lymphoma.
These criteria are being prospectively evaluated and compared to standard HLH-2004 criteria in our ongoing clinical trial for M-HLH (ClinicalTrials.gov Identifier: NCT02385110). Ideally, these efforts would need to be conducted on a multicenter, international scale involving patients with suspected M-HLH to efficiently generate reproducible clinically applicable data. In the interim, a conservative approach is suggested and a diagnosis and work-up of M-HLH should be considered in adult patients with malignancy who have hemophagocytosis in the bone marrow or tissue with unexplained fever under broad antimicrobial treatment, and/or a sudden increases in serum ferritin and/or unexpected non-tumor progression associated pancytopenia.
Therapy of M-HLH
Traditional HLH Therapy
In addition to the limited awareness and lack of specific diagnostic criteria in adult M-HLH, a third major hurdle to the successful management of M-HLH in adults is the development and validation of an effective multidisciplinary treatment strategy. Furthermore, adult HLH is not a homogeneous disease and therapy must be tailored to the underlying trigger, performance status of the patient, organ functions, and concomitant therapies59. This is even more critical in the management of M-HLH, wherein patients are already functionally and immunologically compromised by the underlying malignancy and chemotherapy.
Traditionally, the initial goal of therapy in HLH has been to suppress the overactive immune system, thus preventing immune-mediated organ damage. Induction therapy is often followed by allogeneic stem cell transplant in most cases of primary HLH if a suitable donor is available1, 2 (level of evidence IIB). The pediatric HLH-94 protocol included an 8-week regimen with etoposide, dexamethasone and intrathecal methotrexate60 (level of evidence IIA). Etoposide is administered as 150 mg/m2 initially twice a week and then weekly, dexamethasone is given orally or intravenously starting at 20 mg/m2 and tapered every subsequent week, and weekly intrathecal methotrexate and hydrocortisone are given to children with evidence of CNS involvement. This systematic therapeutic approach has improved the outcomes and survival of patients with pediatric (predominantly genetic) HLH. The HLH-2004 protocol (modified from HLH-94) starts cyclosporine at the beginning of induction and adds hydrocortisone to the intrathecal therapy (level of evidence IIA). The HLH-2004 protocol has completed accrual with study publication pending.
Another therapeutic approach has been geared to suppress macrophages and CD8 T-lymphocytes with antithymocyte globulins in combination with steroids, cyclosporine A, and intrathecal chemotherapy61 (level of evidence IIA). Under this treatment protocol, newborns or toddlers receives ATG, with a total dosage of 50 mg/kg or 25 mg/kg (varies according to the severity of disease over the course of 5 days). Methylprednisolone 4 mg/kg/day is administered with ATG for 5 days then tapered. Intrathecal methotrexate and corticosteroids are given at various dosages determined by patient age and at various intervals according to the severity of CNS involvement. Cyclosporine is added to reach a plasma concentration of 150 ng/mL prior to hematopoetic stem cell transplant. Mahlaoui et al evaluated this regimen in 38 consecutive children and reported encouraging response rates (complete response 73%, partial response 24%, no response 1 patient)62. Sixteen of 19 responding patients who underwent allogeneic stem cell transplant early after response were cured, and 60% of patients achieved long-term survival. To improve the existing HLH regimens, a phase 2 multicenter trial that combines elements of both standard induction regimens discussed for HLH including ATG, etoposide, and dexamethasone in newly diagnosed patients up to 18 years with HLH was recently completed and the results are awaited (Clinicaltrials.gov Identifier: NCT01104025).
Need for Adult M-HLH Specific Therapeutic Approaches
With greater awareness and targeted laboratory evaluations, many patients who would have been diagnosed with conditions such as hepatic or renal failure of unknown etiology, sudden onset multi-organ failure, culture-negative sepsis, or encephalopathy of unknown etiology may be now identified to have HLH. At our centers, we have identified numerous such patients to have HLH including patients receiving frontline and salvage therapy for underlying hematologic malignancies. The mortality among patients manifesting M-HLH has been high (median survival < 2.0 months). A recent analysis from our center showed that less than 50% of adults with M-HLH received HLH-directed therapy due to lack of awareness and missed diagnosis of this condition in adult patients with malignancies. This might be attributable to a number of factors. First, limited awareness and recognition resulted in consultation or presentation of patients with M-HLH at an advanced stage with irreversible multiorgan failure in intensive care setting when it is no longer feasible to initiate lympholytic or cytotoxic HLH therapy. Second, patients with M-HLH are already myelosuppressed and immunocompromised; the addition of further cytotoxic therapy with etoposide or ATG-based regimens carries additional risks and a high mortality. Third, it is debated whether the focus should be on therapy of the underlying malignancy or the pathologic inflammation. While treatment of the underlying disorder intuitively appears to be the correct approach, we have frequently noted a very aggressive and rapid progression of HLH in our patients with M-HLH, with a majority of patients dying from HLH within 2-4 weeks in spite of continued treatment of the underlying malignancy. We believe that the secondary but uncontrolled inflammation as well as the underlying malignancy must be addressed, often in a sequential manner. In some cases it may be possible to address both entities simultaneously and if possible this would be ideal.
Frequently, HLH induces cytokine-dependent cytopenias, HLH-dependent cholestatic icterus, pulmonary infiltrates, encephalopathy, or renal failure not allowing the initiation of malignancy directed (immuno)-chemotherapy. As soon as the organ damage is recognized as HLH-triggered, one must consider the application of lympholytic agents despite formal contraindications. In those conditions, we suggest a two-step approach (level of evidence III), which
Targets the cytokine storm and T-cell proliferation using etoposide (75-100 mg/m2), corticosteroids, polyvalent immunoglobulins (therapeutic dosing) and, when the first hit is unsuccessful, salvage regimens such as DEP (liposomal doxorubicin, etoposide, methylprednisolone), alemtuzumab-based therapy, or cytokine adsorption using therapeutic cytokine adsorption columns or plasmapheresis63-65,
And
-
2
Targets neoplastic disease by specific treatment as soon as organ function is re-established or at the least improved to an acceptable degree.
It must be noted that this is an expert opinion as no standard of care or prospective validation of therapy for M-HLH in adult patients has been conducted. Treatment suggestions as suggested above are targeted to suppress hyperinflammation that is central to the pathogenesis of this entity. The suggestions are taken from the pediatric protocols HLH-94 and HLH-2004 with adjusted dosing taking into consideration reduced bone marrow reserve and pre-existing comorbidities in adults2, 19 (level of evidence III). Furthermore, since multiple factors may precipitate HLH in adults with malignancies a thorough work-up to identify potential triggers in patients with M-HLH is suggested. An example of such a work-up developed by the authors is provided (Table 2)
Table 2. Suggested adult M-HLH baseline work-up.
|
LDH, lactate dehydrogenase, CRP, C-reactive protein, CBC, complete blood count, CMP, comprehensive metabolic panel, NK, natural killer, DAT, direct antiglobulin testing, ANA, anti-nuclear antibody, HFE, human factors engineering, HLH, hemophagocytic lymphohistiocytosis
No adult-specific frontline M-HLH prospective trials have been conducted in the United States. A recent report from China described the use of combination chemotherapy with DEP (doxorubicin, etoposide, methylprednisolone) as a salvage therapy for adult patients with refractory HLH (level of evidence IIA). The DEP regimen resulted in complete remissions in 27% and partial remissions in 49% of the 63 patients treated for refractory HLH65. These are encouraging results, but the DEP regimen is difficult to use in already myelosuppressed and immunocompromised adults with M-HLH. The incorporation of novel non-cytotoxic agents that are less prone to add cumulative toxicity and myelosuppression to chemotherapy for the underlying malignancy may help alleviate the problem.
Novel Therapies for HLH
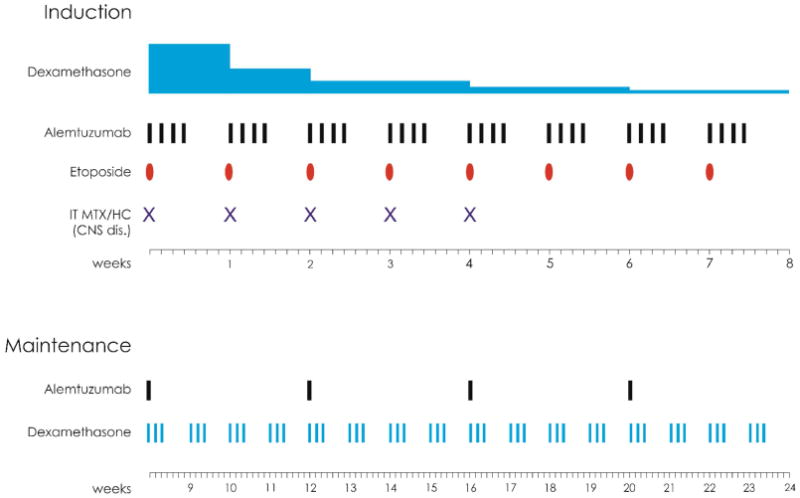
Recently, alemtuzumab has been shown to be an effective salvage therapy for refractory HLH, leading to improved response rates and a higher rate of transition to allogeneic stem cell transplant in pediatric patients66. A clinical trial of alemtuzumab up-front in combination with dexamethasone and etoposide has been developed for the treatment of adults with HLH and is ongoing (ClinicalTrials.gov Identifier: NCT02385110) (Figure 3).
Figure 3. Alemtuzumab in combination with Etoposide and Dexamethasone for the treatment of Adult Patients with Secondary Hemophagocytic Lymphohistiocytosis.
Another novel HLH therapy is IFN-gamma inhibitor NI-0501, which could be a major breakthrough in the therapy of secondary and M-HLH67. NI-0501 is a humanized anti-IFN-gamma monoclonal antibody (mAb) that binds to and neutralizes IFN-gamma. NI-0501 is in clinical trials for primary HLH as the first targeted monoclonal therapy for HLH. Thirteen children with primary HLH have been treated with NI-0501 (12 second-line and 1 first-line) (level of evidence IIA). Significant improvement in HLH parameters was noted in 9 of the 13 patients, and 7 patients proceeded to allogeneic stem cell transplant. Eleven of 13 patients were alive at 8 weeks, and IFN-gamma neutralization was demonstrable. Therapy was well tolerated with none of the typical short- or long-term toxicities reported with etoposide-based regimens. NI-0501 appears to be a non-myelosuppressive drug that may have the potential to improve responses and tolerability in adult M-HLH as frontline or salvage therapy.
Ruxolitinib is a JAK 1/2 inhibitor that has shown activity in inflammatory conditions. It is FDA approved for the treatment of myelofibrosis and polycythemia vera, and has shown activity in corticosteroid-resistant acute graft-versus-host disease68. Ruxolitinib showed efficacy in reducing immunopathology and in prolonging survival in murine models of HLH69, 70. In murine models, treatment with ruxolitinib significantly lessened the clinical and laboratory manifestations of HLH, including weight loss, organomegaly, anemia, thrombocytopenia, hypercytokinemia, and tissue inflammation. The investigators further demonstrated that in vivo exposure to ruxolitinib inhibited STAT activation, suppressed CD8+ T-cell expansion, and reduced proinflammatory cytokines and concluded that JAK-STAT inhibition may be a novel approach to mitigate cytokine mediated hyperinflammation in HLH. It is plausible that ruxolitinib could be used alone in M-HLH without causing the same degree of myelosuppression or immunosuppression as with etoposide or alemtuzumab-based therapies. Alternatively this agent may be effective in combination with standard HLH lympholytic therapies to improve response rates or time to response to induction therapy or as a maintenance therapy to reduce relapse, especially among patients who are not candidates for allogeneic stem cell transplantation for HLH. An ongoing pilot study of ruxolitinib in secondary HLH may help better define its role in secondary HLH, including M-HLH (ClinicalTrials.gov Identifier: NCT02400463) (level of evidence III).
An international collaborative effort to improve the awareness, diagnosis, early referral and therapy of M-HLH is needed. An improved awareness and use of emerging novel therapies such as inhibitors of IFN-gamma and JAK-STAT pathways in rationally developed clinical trials either alone or in combination with T-lympholytic and immunosuppressive agents may allow us to improve outcomes in this difficult condition.
Acknowledgments
Funding: This study was funded in part by the MD Anderson Cancer Centre Support Grant (CCSG) CA016672. No other sources of funding.
Footnotes
COI: The authors have no relevant conflict of interest to disclose.
Author Contributions: ND, KM, CA, SP, ZO, CRH, BB, SW, MM, MJ, PLR and HK collected and reviewed the data and wrote the paper. All authors participated in the discussion, have reviewed the final manuscripts, have added comments or suggestions, and approved the current version of the manuscript.
References
- 1.Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL. How I treat hemophagocytic lymphohistiocytosis. Blood. 2011;118:4041–4052. doi: 10.1182/blood-2011-03-278127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Henter JI, Horne A, Arico M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–131. doi: 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- 3.Rosado FG, Kim AS. Hemophagocytic lymphohistiocytosis: an update on diagnosis and pathogenesis. Am J Clin Pathol. 2013;139:713–727. doi: 10.1309/AJCP4ZDKJ4ICOUAT. [DOI] [PubMed] [Google Scholar]
- 4.Schram AM, Berliner N. How I treat hemophagocytic lymphohistiocytosis in the adult patient. Blood. 2015;125:2908–2914. doi: 10.1182/blood-2015-01-551622. [DOI] [PubMed] [Google Scholar]
- 5.Henter JI, Arico M, Elinder G, Imashuku S, Janka G. Familial hemophagocytic lymphohistiocytosis. Primary hemophagocytic lymphohistiocytosis. Hematol Oncol Clin North Am. 1998;12:417–433. doi: 10.1016/s0889-8588(05)70520-7. [DOI] [PubMed] [Google Scholar]
- 6.Janka G, Imashuku S, Elinder G, Schneider M, Henter JI. Infection- and malignancy-associated hemophagocytic syndromes. Secondary hemophagocytic lymphohistiocytosis. Hematol Oncol Clin North Am. 1998;12:435–444. doi: 10.1016/s0889-8588(05)70521-9. [DOI] [PubMed] [Google Scholar]
- 7.Lehmberg K, Ehl S. Diagnostic evaluation of patients with suspected haemophagocytic lymphohistiocytosis. Br J Haematol. 2013;160:275–287. doi: 10.1111/bjh.12138. [DOI] [PubMed] [Google Scholar]
- 8.Zhang K, Jordan MB, Marsh RA, et al. Hypomorphic mutations in PRF1, MUNC13-4, and STXBP2 are associated with adult-onset familial HLH. Blood. 2011;118:5794–5798. doi: 10.1182/blood-2011-07-370148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ishii E, Ohga S, Imashuku S, et al. Nationwide survey of hemophagocytic lymphohistiocytosis in Japan. Int J Hematol. 2007;86:58–65. doi: 10.1532/IJH97.07012. [DOI] [PubMed] [Google Scholar]
- 10.Machaczka M, Vaktnas J, Klimkowska M, Hagglund H. Malignancy-associated hemophagocytic lymphohistiocytosis in adults: a retrospective population-based analysis from a single center. Leuk Lymphoma. 2011;52:613–619. doi: 10.3109/10428194.2010.551153. [DOI] [PubMed] [Google Scholar]
- 11.Niece JA, Rogers ZR, Ahmad N, Langevin AM, McClain KL. Hemophagocytic lymphohistiocytosis in Texas: observations on ethnicity and race. Pediatr Blood Cancer. 2010;54:424–428. doi: 10.1002/pbc.22359. [DOI] [PubMed] [Google Scholar]
- 12.Henter JI, Elinder G, Soder O, Ost A. Incidence in Sweden and clinical features of familial hemophagocytic lymphohistiocytosis. Acta Paediatr Scand. 1991;80:428–435. doi: 10.1111/j.1651-2227.1991.tb11878.x. [DOI] [PubMed] [Google Scholar]
- 13.Zhang L, Zhou J, Sokol L. Hereditary and acquired hemophagocytic lymphohistiocytosis. Cancer Control. 2014;21:301–312. doi: 10.1177/107327481402100406. [DOI] [PubMed] [Google Scholar]
- 14.Henter JI, Ehrnst A, Andersson J, Elinder G. Familial hemophagocytic lymphohistiocytosis and viral infections. Acta Paediatr. 1993;82:369–372. doi: 10.1111/j.1651-2227.1993.tb12699.x. [DOI] [PubMed] [Google Scholar]
- 15.Janka G. Hemophagocytic Lymphohistiocytosis: When the Immune System Runs Amok. Klinische Padiatrie. 2009;221:278–285. doi: 10.1055/s-0029-1237386. [DOI] [PubMed] [Google Scholar]
- 16.Janka GE. Familial Hemophagocytic Lymphohistiocytosis. Eur J Pediatr. 1983;140:221–230. doi: 10.1007/BF00443367. [DOI] [PubMed] [Google Scholar]
- 17.Trottestam H, Horne A, Arico M, et al. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood. 2011;118:4577–4584. doi: 10.1182/blood-2011-06-356261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang YN, Wang Z, Zhang J, et al. Genetic Features of Late Onset Primary Hemophagocytic Lymphohistiocytosis in Adolescence or Adulthood. PLoS One. 2014;9 doi: 10.1371/journal.pone.0107386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Henter JI, Arico M, Egeler RM, et al. HLH-94: a treatment protocol for hemophagocytic lymphohistiocytosis. HLH study Group of the Histiocyte Society. Med Pediatr Oncol. 1997;28:342–347. doi: 10.1002/(sici)1096-911x(199705)28:5<342::aid-mpo3>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 20.Ramos-Casals M, Brito-Zeron P, Lopez-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. Lancet. 2014;383:1503–1516. doi: 10.1016/S0140-6736(13)61048-X. [DOI] [PubMed] [Google Scholar]
- 21.Tong H, Ren Y, Liu H, et al. Clinical characteristics of T-cell lymphoma associated with hemophagocytic syndrome: comparison of T-cell lymphoma with and without hemophagocytic syndrome. Leuk Lymphoma. 2008;49:81–87. doi: 10.1080/10428190701713630. [DOI] [PubMed] [Google Scholar]
- 22.Majluf-Cruz A, Sosa-Camas R, Perez-Ramirez O, Rosas-Cabral A, Vargas-Vorackova F, Labardini-Mendez J. Hemophagocytic syndrome associated with hematological neoplasias. Leuk Res. 1998;22:893–898. doi: 10.1016/s0145-2126(98)00083-6. [DOI] [PubMed] [Google Scholar]
- 23.Parikh SA, Kapoor P, Letendre L, Kumar S, Wolanskyj AP. Prognostic factors and outcomes of adults with hemophagocytic lymphohistiocytosis. Mayo Clin Proc. 2014;89:484–492. doi: 10.1016/j.mayocp.2013.12.012. [DOI] [PubMed] [Google Scholar]
- 24.Shabbir M, Lucas J, Lazarchick J, Shirai K. Secondary hemophagocytic syndrome in adults: a case series of 18 patients in a single institution and a review of literature. Hematol Oncol. 2011;29:100–106. doi: 10.1002/hon.960. [DOI] [PubMed] [Google Scholar]
- 25.Lehmberg K, Sprekels B, Nichols KE, et al. Malignancy-associated haemophagocytic lymphohistiocytosis in children and adolescents. Br J Haematol. 2015;170:539–549. doi: 10.1111/bjh.13462. [DOI] [PubMed] [Google Scholar]
- 26.Shimazaki C, Inaba T, Okano A, et al. Clinical characteristics of B-cell lymphoma-associated hemophagocytic syndrome (B-LAHS): comparison of CD5+ with CD5- B-LAHS. Intern Med. 2001;40:878–882. doi: 10.2169/internalmedicine.40.878. [DOI] [PubMed] [Google Scholar]
- 27.Dhote R, Simon J, Papo T, et al. Reactive hemophagocytic syndrome in adult systemic disease: report of twenty-six cases and literature review. Arthritis Rheum. 2003;49:633–639. doi: 10.1002/art.11368. [DOI] [PubMed] [Google Scholar]
- 28.Han AR, Lee HR, Park BB, et al. Lymphoma-associated hemophagocytic syndrome: clinical features and treatment outcome. Ann Hematol. 2007;86:493–498. doi: 10.1007/s00277-007-0278-6. [DOI] [PubMed] [Google Scholar]
- 29.Takahashi N, Miura I, Chubachi A, Miura AB, Nakamura S. A clinicopathological study of 20 patients with T/natural killer (NK)-cell lymphoma-associated hemophagocytic syndrome with special reference to nasal and nasal-type NK/T-cell lymphoma. Int J Hematol. 2001;74:303–308. doi: 10.1007/BF02982065. [DOI] [PubMed] [Google Scholar]
- 30.Lehmberg K, Nichols KE, Henter JI, et al. Consensus recommendations for the diagnosis and management of hemophagocytic lymphohistiocytosis associated with malignancies. Haematologica. 2015;100:997–1004. doi: 10.3324/haematol.2015.123562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Delavigne K, Berard E, Bertoli S, et al. Hemophagocytic syndrome in patients with acute myeloid leukemia undergoing intensive chemotherapy. Haematologica. 2014;99:474–480. doi: 10.3324/haematol.2013.097394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Otrock ZK, Eby CS. Clinical characteristics, prognostic factors, and outcomes of adult patients with hemophagocytic lymphohistiocytosis. Am J Hematol. 2015;90:220–224. doi: 10.1002/ajh.23911. [DOI] [PubMed] [Google Scholar]
- 33.Tamamyan GN, Kantarjian HM, Ning J, et al. Malignancy-associated hemophagocytic lymphohistiocytosis in adults: Relation to hemophagocytosis, characteristics, and outcomes. Cancer. 2016;122:2857–2866. doi: 10.1002/cncr.30084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rouphael NG, Talati NJ, Vaughan C, Cunningham K, Moreira R, Gould C. Infections associated with haemophagocytic syndrome. Lancet Infect Dis. 2007;7:814–822. doi: 10.1016/S1473-3099(07)70290-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ohshima K, Shimazaki K, Sugihara M, et al. Clinicopathological findings of virus-associated hemophagocytic syndrome in bone marrow: association with Epstein-Barr virus and apoptosis. Pathol Int. 1999;49:533–540. doi: 10.1046/j.1440-1827.1999.00921.x. [DOI] [PubMed] [Google Scholar]
- 36.Fardet L, Lambotte O, Meynard JL, et al. Reactive haemophagocytic syndrome in 58 HIV-1-infected patients: clinical features, underlying diseases and prognosis. AIDS. 2010;24:1299–1306. doi: 10.1097/QAD.0b013e328339e55b. [DOI] [PubMed] [Google Scholar]
- 37.Tseng YT, Sheng WH, Lin BH, et al. Causes, clinical symptoms, and outcomes of infectious diseases associated with hemophagocytic lymphohistiocytosis in Taiwanese adults. J Microbiol Immunol Infect. 2011;44:191–197. doi: 10.1016/j.jmii.2011.01.027. [DOI] [PubMed] [Google Scholar]
- 38.Ahn JS, Rew SY, Shin MG, et al. Clinical significance of clonality and Epstein-Barr virus infection in adult patients with hemophagocytic lymphohistiocytosis. Am J Hematol. 2010;85:719–722. doi: 10.1002/ajh.21795. [DOI] [PubMed] [Google Scholar]
- 39.Fukaya S, Yasuda S, Hashimoto T, et al. Clinical features of haemophagocytic syndrome in patients with systemic autoimmune diseases: analysis of 30 cases. Rheumatology (Oxford) 2008;47:1686–1691. doi: 10.1093/rheumatology/ken342. [DOI] [PubMed] [Google Scholar]
- 40.Lambotte O, Khellaf M, Harmouche H, et al. Characteristics and long-term outcome of 15 episodes of systemic lupus erythematosus-associated hemophagocytic syndrome. Medicine (Baltimore) 2006;85:169–182. doi: 10.1097/01.md.0000224708.62510.d1. [DOI] [PubMed] [Google Scholar]
- 41.Takagi S, Masuoka K, Uchida N, et al. High incidence of haemophagocytic syndrome following umbilical cord blood transplantation for adults. Br J Haematol. 2009;147:543–553. doi: 10.1111/j.1365-2141.2009.07863.x. [DOI] [PubMed] [Google Scholar]
- 42.Karras A, Thervet E, Legendre C, Groupe Cooperatif de transplantation d'Ile de F Hemophagocytic syndrome in renal transplant recipients: report of 17 cases and review of literature. Transplantation. 2004;77:238–243. doi: 10.1097/01.TP.0000107285.86939.37. [DOI] [PubMed] [Google Scholar]
- 43.Minoia F, Davi S, Horne A, et al. Clinical features, treatment, and outcome of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a multinational, multicenter study of 362 patients. Arthritis Rheumatol. 2014;66:3160–3169. doi: 10.1002/art.38802. [DOI] [PubMed] [Google Scholar]
- 44.Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood. 2004;104:735–743. doi: 10.1182/blood-2003-10-3413. [DOI] [PubMed] [Google Scholar]
- 45.Humblet-Baron S, Franckaert D, Dooley J, et al. IL-2 consumption by highly activated CD8 T cells induces regulatory T-cell dysfunction in patients with hemophagocytic lymphohistiocytosis. J Allergy Clin Immunol. 2016;138:200–209 e208. doi: 10.1016/j.jaci.2015.12.1314. [DOI] [PubMed] [Google Scholar]
- 46.Akashi K, Hayashi S, Gondo H, et al. Involvement of interferon-gamma and macrophage colony-stimulating factor in pathogenesis of haemophagocytic lymphohistiocytosis in adults. Br J Haematol. 1994;87:243–250. doi: 10.1111/j.1365-2141.1994.tb04905.x. [DOI] [PubMed] [Google Scholar]
- 47.Ohga S, Matsuzaki A, Nishizaki M, et al. Inflammatory cytokines in virus-associated hemophagocytic syndrome. Interferon-gamma as a sensitive indicator of disease activity. Am J Pediatr Hematol Oncol. 1993;15:291–298. [PubMed] [Google Scholar]
- 48.Ho C, Yao X, Tian L, Li FY, Podoltsev N, Xu ML. Marrow assessment for hemophagocytic lymphohistiocytosis demonstrates poor correlation with disease probability. Am J Clin Pathol. 2014;141:62–71. doi: 10.1309/AJCPMD5TJEFOOVBW. [DOI] [PubMed] [Google Scholar]
- 49.Gupta A, Tyrrell P, Valani R, Benseler S, Weitzman S, Abdelhaleem M. The role of the initial bone marrow aspirate in the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2008;51:402–404. doi: 10.1002/pbc.21564. [DOI] [PubMed] [Google Scholar]
- 50.Riviere S, Galicier L, Coppo P, et al. Reactive hemophagocytic syndrome in adults: a retrospective analysis of 162 patients. Am J Med. 2014;127:1118–1125. doi: 10.1016/j.amjmed.2014.04.034. [DOI] [PubMed] [Google Scholar]
- 51.Lau SK, Chu PG, Weiss LM. CD163: a specific marker of macrophages in paraffin-embedded tissue samples. Am J Clin Pathol. 2004;122:794–801. doi: 10.1309/QHD6-YFN8-1KQX-UUH6. [DOI] [PubMed] [Google Scholar]
- 52.Celkan T, Berrak S, Kazanci E, et al. Malignancy-associated hemophagocytic lymphohistiocytosis in pediatric cases: a multicenter study from Turkey. Turk J Pediatr. 2009;51:207–213. [PubMed] [Google Scholar]
- 53.Hijiya N, Metzger ML, Pounds S, et al. Severe cardiopulmonary complications consistent with systemic inflammatory response syndrome caused by leukemia cell lysis in childhood acute myelomonocytic or monocytic leukemia. Pediatr Blood Cancer. 2005;44:63–69. doi: 10.1002/pbc.20192. [DOI] [PubMed] [Google Scholar]
- 54.Lee DW, Gardner R, Porter DL, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124:188–195. doi: 10.1182/blood-2014-05-552729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Teachey DT, Rheingold SR, Maude SL, et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood. 2013;121:5154–5157. doi: 10.1182/blood-2013-02-485623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Henter JI, Elinder G, Ost A. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL Study Group of the Histiocyte Society. Semin Oncol. 1991;18:29–33. [PubMed] [Google Scholar]
- 57.Hejblum G, Lambotte O, Galicier L, et al. A web-based delphi study for eliciting helpful criteria in the positive diagnosis of hemophagocytic syndrome in adult patients. PLoS One. 2014;9:e94024. doi: 10.1371/journal.pone.0094024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Padhi S, Varghese RG, Ramdas A, Phansalkar MD, Sarangi R. Hemophagocytic lymphohistiocytosis: critical reappraisal of a potentially under-recognized condition. Front Med. 2013;7:492–498. doi: 10.1007/s11684-013-0292-0. [DOI] [PubMed] [Google Scholar]
- 59.La Rosee P. Treatment of hemophagocytic lymphohistiocytosis in adults. Hematology-American Society of Hematology Education Program. 2015:190–196. doi: 10.1182/asheducation-2015.1.190. [DOI] [PubMed] [Google Scholar]
- 60.Henter JI, Samuelsson-Horne A, Arico M, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100:2367–2373. doi: 10.1182/blood-2002-01-0172. [DOI] [PubMed] [Google Scholar]
- 61.Stephan JL, Donadieu J, Ledeist F, Blanche S, Griscelli C, Fischer A. Treatment of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins, steroids, and cyclosporin A. Blood. 1993;82:2319–2323. [PubMed] [Google Scholar]
- 62.Mahlaoui N, Ouachee-Chardin M, de Saint Basile G, et al. Immunotherapy of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins: a single-center retrospective report of 38 patients. Pediatrics. 2007;120:e622–628. doi: 10.1542/peds.2006-3164. [DOI] [PubMed] [Google Scholar]
- 63.Strout MP, Seropian S, Berliner N. Alemtuzumab as a bridge to allogeneic SCT in atypical hemophagocytic lymphohistiocytosis. Nat Rev Clin Oncol. 2010;7:415–420. doi: 10.1038/nrclinonc.2010.40. [DOI] [PubMed] [Google Scholar]
- 64.Frimmel S, Schipper J, Henschel J, Yu TT, Mitzner SR, Koball S. First description of single-pass albumin dialysis combined with cytokine adsorption in fulminant liver failure and hemophagocytic syndrome resulting from generalized herpes simplex virus 1 infection. Liver Transpl. 2014;20:1523–1524. doi: 10.1002/lt.24005. [DOI] [PubMed] [Google Scholar]
- 65.Wang Y, Huang W, Hu L, et al. Multicenter study of combination DEP regimen as a salvage therapy for adult refractory hemophagocytic lymphohistiocytosis. Blood. 2015;126:2186–2192. doi: 10.1182/blood-2015-05-644914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Marsh RA, Allen CE, McClain KL, et al. Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatr Blood Cancer. 2013;60:101–109. doi: 10.1002/pbc.24188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jordan MB, Locatelli F, Allen C, et al. A Novel Targeted Approach to the Treatment of Hemophagocytic Lymphohistiocytosis (Hlh) with Ni-0501, an Anti-Interferon Gamma Monoclonal Antibody. Journal of Clinical Immunology. 2016;36:282–282. [Google Scholar]
- 68.Spoerl S, Mathew NR, Bscheider M, et al. Activity of therapeutic JAK 1/2 blockade in graft-versus-host disease. Blood. 2014;123:3832–3842. doi: 10.1182/blood-2013-12-543736. [DOI] [PubMed] [Google Scholar]
- 69.Das R, Guan P, Sprague L, et al. Janus kinase inhibition lessens inflammation and ameliorates disease in murine models of hemophagocytic lymphohistiocytosis. Blood. 2016;127:1666–1675. doi: 10.1182/blood-2015-12-684399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Maschalidi S, Sepulveda FE, Garrigue A, Fischer A, de Saint Basile G. Therapeutic effect of JAK1/2 blockade on the manifestations of hemophagocytic lymphohistiocytosis in mice. Blood. 2016;128:60–71. doi: 10.1182/blood-2016-02-700013. [DOI] [PubMed] [Google Scholar]