

Abstract
S K-edge X-ray Absorption Spectroscopy (XAS) was used to study the [Fe4S4] clusters in the DNA repair glycosylases EndoIII and MutY to evaluate the effects of DNA binding and solvation on Fe-S bond covalencies (ie. the amount of S 3p character mixed into the Fe 3d valence orbitals). Increased covalencies in both iron-thiolate and iron-sulfide bonds would stabilize the oxidized state of the [Fe4S4] clusters. The results are compared to those on previously studied [Fe4S4] model complexes, ferredoxin (Fd), and to new data on high potential iron-sulfur protein (HiPIP). A limited decrease in covalency is observed upon removal of solvent water from EndoIII and MutY, opposite to the significant increase observed for Fd where the [Fe4S4] cluster is solvent exposed. Importantly, in EndoIII and MutY, a large increase in covalency is observed upon DNA binding which is due to the effect of its negative charge on the iron-sulfur bonds. In EndoIII, this change in covalency can be quantified and makes a significant contribution to the observed decrease in reduction potential found experimentally in DNA repair proteins, enabling their HiPIP like redox behavior.
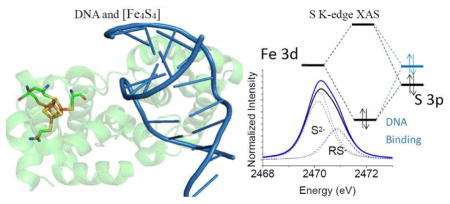
Graphical Abstract
Introduction
Iron-sulfur clusters occur in a wide range of proteins with roles in electron transfer, catalysis, and regulation.1 Those involved in electron transfer are the rubredoxins and ferredoxins. Rubredoxin contains a mononuclear iron center with thiolate ligands, while ferredoxins contain multinuclear iron-sulfide clusters which includes [Fe2S2], [Fe3S4] and [Fe4S4] sites.2 The [Fe4S4] proteins can be further divided into two categories, the low-potential ferredoxins (Fd), which utilize the [Fe4S4]2+/1+ redox couple with a potential as low as −600 mV, and high potential iron proteins (HiPIP), which have an [Fe4S4]3+/2+ redox couple with a reduction potential as high as +350 mV.1 From X-ray crystallography3, Extended X-Ray Absorption Fine Structure (EXAFS)4 and resonance Raman5 spectroscopic results, the [Fe4S4]2+ sites in the Fds and HiPIPs are almost identical. They have similar bond distances and angles, as well as similar vibrational modes. The major difference is in their protein environments. The [Fe4S4] site in Fd is solvent exposed while this cluster in HiPIP is buried.6 Lyophilization of Fd and unfolding of HiPIP lead to significant changes in their S K-edge XAS spectra, showing that the H-bonds from solvent water change the Fe-S bond covalency.7 The higher covalency in HiPIP stabilizes the oxidized state over the reduced state, and this significantly contributes to their [Fe4S4] clusters utilizing different redox couples from Fd.1 The [Fe4S4]2+ in Fd is less covalent and activated toward reduction, while the cluster in HiPIP is more covalent, thus tuned toward oxidation.
Endonuclease III (EndoIII) and MutY are DNA glycosylases present in bacteria to humans that excise oxidized bases or their mispaired base partners as initiating events in the base excision repair (BER) pathway. Specifically, EndoIII removes oxidized pyrimidines while MutY removes adenine from A:oxoG mispairs (where oxoG = 8-oxo-7,8-dihydroguanine).8–9 The importance of these enzymes is highlighted by the correlation of inherited defects in human MutY (MUTYH) and early onset colorectal cancer, referred to as MUTYH-associated polyposis (MAP).10 EndoIII and MutY both contain an [Fe4S4]2+ cluster, which is redox inactive in solution in the absence of DNA.11 The [Fe4S4]2+ cluster in these enzymes has been shown to be essential for substrate binding and catalysis, but it is not required for the global structural integrity of the enzyme.9 However, electrochemical experiments carried out with EndoIII and MutY on DNA-modified gold electrodes showed reversible redox signals at potentials ranging from 60–95 mV versus NHE, suggesting that DNA binding was able to activate these proteins for redox activity. A role for DNA in activating the cluster for redox activity was confirmed by electrochemistry on highly oriented pyrolytic graphite (HOPG) electrodes in the presence and absence of DNA; these experiments revealed that upon binding to DNA, the reduction potential of the [Fe4S4]3+/2+ couple in EndoIII shifted ~ −200 mV into the physiological range corresponding to a significantly increased DNA binding affinity of the oxidized form of the protein.12–13 From electrochemical and in vivo experiments, A model has been proposed in which long-distance DNA-mediated electron transfer between two [Fe4S4] proteins with similar DNA-bound redox potentials facilitates the search for damage across a vast genome.8, 14–15 If there is no DNA damage between the two binding sites, the inter-protein charge transfer (CT) can proceed efficiently, and one of the DNA-bound proteins is reduced, thus its affinity for DNA is decreased. This protein can then dissociate and diffuse to another region of the genome. However, if there is a mismatch or lesion that disrupts the π-stacking of DNA, charge transfer will be attenuated and both proteins would remain bound to the DNA in the vicinity of the lesion. While the CT signaling model has been strongly supported in numerous studies, the origin of the large DNA-induced potential shift that makes such a damage search possible has remained puzzling. Electrostatic effects remain the most likely explanation, given that no significant structural changes are apparent between the free and DNA-bound forms of EndoIII and MutY. Unfortunately, the electrochemical techniques used in earlier work cannot reveal the fundamental molecular-scale changes involved, making an alternative approach necessary to elucidate the origin of this DNA induced potential shift.
S K-edge X-ray Absorption Spectroscopy (XAS) has been developed and applied to experimentally determine the covalency (α2) of sulfur-metal bonds (i.e. the amount of S 3p character mixed into the metal 3d antibonding (Ψ*) valence orbitals: .16 The S 1s orbital is localized on the S atom and the S 1s → S 3p transition is electric dipole allowed, thus the intensity of the 1s →Ψ* pre-edge transition reflects the covalency of this sulfur-metal bond. The energy of the pre-edge transition reflects the energy of the unoccupied or partially occupied Fe d-orbitals, which depends on the effective nuclear charge (Zeff) of the metal and the nature of the ligand field.16 The pre-edge transition energy also depends on the charge of the S; sulfide has a lower Zeff than thiolate, thus the sulfide donor orbitals are at higher energy than those of the thiolate, and their associated S 1s →Ψ* pre-edge transitions are at lower energy.16
The intensity of a pre-edge peak D0 is given by
| (Equation 1) |
where c is a constant, r is the dipole operator, α2 is the bond covalency (i.e. sulfur p character mixed into the metal d orbitals), N is the total number of sulfurs bound to the metal, h is the total number of d electron holes, and Is is the S 1s → 3p electric dipole integral, which is also dependent on the Zeff of the S ligand and has been experimentally determined for thiolate and sulfide ligands in previous work17. Our previous XAS studies on [Fe4S4] sites showed that the contributions of thiolate and sulfide can be distinguished at the pre-edge, with the μ3-sulfide pre-edge ~0.7 eV lower than the thiolate.6, 18 Quantitatively, for the [(RS)4Fe4S4]2+ clusters, there are 9 α and 9 β holes (ie. unoccupied valence orbitals), which have mainly Fe 3d and S 3p character6, thus the maximum covalency value possible is 1800%. In particular, 1 unit of sulfide intensity corresponds to 30.6% S character per Fe-S bond, while 1 unit of thiolate corresponds to 70.8% S character in unoccupied valence orbitals. This difference reflects the different number of bonds to each Fe (3 from sulfides and 1 from thiolate) and the difference in dipole integral due to the Zeff of sulfide relative to thiolate S (6.54 vs 8.47).17 Note that only the sulfur atoms bound to the Fe contribute to the pre-edge feature in the XAS spectra, while all sulfur atoms including the free Cys and Met residues contribute to the edge. Thus the pre-edge intensity directly reflects the covalency of the thiolate-Fe and bridging sulfide-Fe bonds, but the edge normalization must be corrected for the total number of sulfurs in the protein.
The total pre-edge intensity, and therefore S covalency, in HiPIP is significantly higher than in Fd. The pre-edge intensity in a relevant alkyl thiolate [Fe4S4] model complex is even slightly higher than in HiPIP.7 Importantly, there is a direct correlation between the total S covalency and the redox potential of the [Fe4S4] clusters, with an increase of 1% of total S covalency corresponding to ~3.3 mV decrease in redox potential7 which reflects the fact that higher covalency stabilizes the oxidized over the reduced state of the [Fe4S4] cluster. With respect to DNA repair proteins, the power of S K-edge XAS to monitor changes in [Fe4S4] cluster covalency in different environments and to correlate these changes to redox potential results in an ideal method to elucidate the origin of the DNA-induced potential shift at a molecular level.
In the present study, S K-edge XAS is applied to experimentally measure the Fe-S bond covalency of the [Fe4S4] clusters in EndoIII and MutY, both in the absence of and bound to DNA, and with and without solvent water. The effect of DNA binding on the S K-edge intensity and hence covalency is correlated to the reduction potential of the cluster according to a relationship defined in previous XAS studies on [Fe4S4] clusters7, and is in agreement with the electrochemically observed reduction potential decrease upon DNA binding.12 The solvent effect is compared to those previously observed for Fd7 and for the HiPIP proteins in this study. This work provides direct molecular evidence for the proposal that the negative charge of bound DNA tunes the potential of [Fe4S4] clusters and shows that this involves a change in the covalency of the cluster that enables the [Fe4S4]3+/2+ redox couple when EndoIII (and MutY) are bound to DNA.
Experimental
Expression and purification of EndoIII
WT E. coli EndoIII was overexpressed in BL21star-(DE3)pLysS cells containing a pET11-ubiquitin-His6-nth construct and purified as detailed previously19, with the exception that the final buffer contained 10% glycerol, rather than 20% glycerol (20 mM sodium phosphate, pH 7.5, 0.5 mM EDTA, 150 mM NaCl, 10% glycerol, Buffer A). EndoIII is less stable in the absence of glycerol; therefore glycerol was not removed until the day of sample preparation. Glycerol was removed from half the volume of protein solution using HiPrep 26/10 desalting column (GE Healthcare) equilibrated with Buffer A lacking glycerol. Next, the protein solutions either containing 10% glycerol or no glycerol were separately concentrated first with 10,000 MWCO (molecular weight cutoff) Amicon Ultra 15 mL centrifugation filter units (Millipore) and then with 10,000 MWCO Amicon Ultra 0.5 mL centrifugation filter units (Millipore) until the protein solutions were very dark colored, to approximately 300 μL each if using an entire protein preparation from 6 L of bacterial culture.
Expression and purification of MutY
WT MutY was expressed as an N-terminal fusion with MBP (Maltose Binding Protein) to increase the solubility at the concentrations needed for XAS experiments. Of note, the MBP tag has additional Met residues that can complicate XAS data analysis. The MBP-MutY protein was overexpressed in BL21 DE3 competent cells and purified as detailed previously13, with the following modification. Pelleted cells from overexpression were re-suspended in resuspension buffer (20 mM sodium phosphate, pH 7.5, 200 mM NaCl, 1mM EDTA, 10 % glycerol, 1 mM PMSF), sonicated on ice and centrifuged at 12,000 RPM for 15 minutes. The supernatant was saved in a conical tube on ice, and the pellet was re-suspended and underwent a repeat of sonification and centrifugation. The supernatant was batch bound to amylose resin (New England BioLands) for one hour, poured over a PD10 column and washed with amylose wash buffer (20 mM sodium phosphate, pH 7.5, 200 mM NaCl, 1mM EDTA) and eluted in amylose elutant buffer; amylose wash buffer with the addition of 10 mM maltose). The elutant was concentrated using an ultrafiltration cell with a 10,000 MWCO filter with stirring at 4°C. Protein was then diluted 10-fold in heparin buffer A (20 mM sodium phosphate, pH 7.5, 1 mM EDTA, 5% glycerol). The sample was applied to a Pharmacia Hi-trap heparin column on an AKTApurifier FPLC system, and eluted using a 10% linear gradient in heparin buffer A to 100% heparin buffer B and buffer A with the addition of 1 M NaCl). Fractions corresponding to MBP-MutY were combined and concentrated using an ultrafiltration cell with a 10,000 MWCO filter with stirring at 4 °C, to approximately 10mL. Purity of MBP-MutY samples were confirmed via 12 % SDS page stained with SYPRO orange and 7.5 % acetic acid.
DNA preparation
DNA strands for EndoIII studies were purchased from Integrated DNA Technologies (a 20-mer mixed sequence strand: 5′-GTGAGCTAACGTGTCAGTAC-3′ and its complement). DNA strands (5 μmol) were resuspended in MilliQ water (200 μL), and purified by ethanol precipitation. The purified strands were resuspended in EndoIII Buffer A or Buffer A lacking glycerol and quantified based on calculated ε260 values for the strands (Integrated DNA Technologies) of 197,800 M−1cm−1 for the 20-mer strand and 190,200 M−1cm−1 for its complement. Annealing of the strands in either Buffer A or Buffer A without glycerol was accomplished by combining equimolar amounts of the single-stranded DNAs, heating at 90°C for 5 minutes, and slowly cooling to room temperature.
DNA strands for MutY studies containing oxoG or FA (2′-fluoro-adenine) were synthesized at the University of Utah DNA and Peptide Synthesis Core Facility and unmodified strands were purchased from Integrated DNA Technologies. Two lengthed DNA duplexes were used, a 15-mer (5′-GGAGCCAXGAGCTCC-3′ and its compliment 5′-GGAGCTCYTGGCTCC -3′), and a 30-mer (5′-CGATCATGGAGCCACXAGCTCCCGTTACAG-3′ and its compliment 5′-CTGTAACGGGAGCTYGTGGCTCCATGATCG-3′) where X = G or oxoG and Y = C or FA. Oligonucleotides containing the central oxoG or FA were deprotected and cleaved from the solid support by incubation in NH4OH, with the addition of 2-mercaptoethanol to oxoG samples to prevent further oxidation. The cleaved DNA substrates were dissolved in H2O, filtered with a 0.2 μm filter, and HPLC purified using a Beckman Gold Nouveau system with a Waters AP1DEAE 8HR column with a 10–100% gradient of 90:10 H2O/acetonitrile with 2 M NH4Ac. Isolated fractions were lyophilized and de-salted with a SEP-PAK C18 column, and DNA integrity was confirmed using MALDI-MS. The purified strands were resuspended in MBP-MutY concentration buffer with or without glycerol and quantified based on calculated ε260 values for the strands (Integrated DNA Technologies) of 145,800 M−1cm−1 for the 15-mer strand with central G and 131,700 M−1cm−1 for its complement, 136,000 M−1cm−1 for the 15-mer strand with central oxoG and 137,900 M−1cm−1 for its complement, 285,200 M−1cm−1 for the 30-mer strand with central G and 280,800 M−1cm−1 for its complement, and lastly 279,600 M−1cm−1 for the 30-mer strand with central oxoG and 287,000 M−1cm−1 for its complement. Annealing of the strands in either MBP-MutY concentration buffer with or without glycerol was accomplished by combining equimolar amounts of the single-stranded DNAs, heating at 90°C for 5 minutes, and allowed to slowly anneal overnight to 4°C.
XAS sample preparation
To prepare the EndoIII XAS samples, concentrated solutions of EndoIII protein were mixed with the DNA duplex at a ratio of 1 mol EndoIII: 20 mol base-pairs DNA. An equivalent volume of buffer was added to EndoIII alone solutions so that identical EndoIII concentrations were obtained both with and without DNA. Mixtures were allowed to incubate on wet ice for 30 minutes to allow for binding before freezing in liquid nitrogen. The no glycerol samples were lyophilized. Both samples were placed on dry ice and sent to Stanford University for measurement. The EndoIII [4Fe4S] cluster loading was calculated using the protein concentration determined by UV-visible absorbance at 410 nm that is specific to [4Fe4S] clusters (ε410 = 17,000 M−1cm−1) relative to the protein concentration determined by Braford Assay; samples were typically 70–75% loaded with cluster.
To prepare the MutY XAS samples, purified MBP-MutY samples were buffer exchanged in concentration buffer, with the final buffer consisting of 20 mM sodium phosphate, pH 7.5, 150 mM NaCl, 1mM EDTA, 10% glycerol for samples to remain in buffer, or concentration buffer lacking glycerol for samples to be lyophilized for XAS. The protein sample was concentrated to a final concentration of 1.2mM. 125 uL of the purified MBP-MutY was incubated with 25uL of the 6mM annealed DNA duplex at 25 °C for 30 minutes to afford a final concentration of 1mM for both protein and DNA. Following lyophilization or immediately for samples in buffer, samples were snap frozen in liquid nitrogen, stored at −80 °C or on dry ice, and were thawed on wet ice just prior to XAS experiments. The MutY [4Fe4S] cluster loading was determined using the UV-visible absorbance at 410 nm (ε410 = 17,000 M−1cm−1) and at 280 nm (ε280 = 143,240 M−1cm−1); samples were typically 65–75% loaded with cluster.
S K-edge XAS
Sulfur K-edge XAS data were measured at the Stanford Synchrotron Radiation Lightsource on the unfocussed 20-pole, 2.0-Tesla wiggler Beam Line 4–3, under SPEAR3 storage ring parameters of 3 GeV and 500 mA. A Ni-coated, flat, bent pre-monochromator mirror was used for harmonic rejection and vertical collimation. A Si(111) double crystal monochromator was used for energy selection. The energy calibration, data reduction and error analysis follow the methods described in reference.20 Solid samples were ground into a fine powder and dispersed as thinly as possible on Kapton tape to minimize potential self-absorption effects. The solution samples were loaded into 50μL teflon cells, with Kapton tape as the back window, and 6 μm-thick, sulfur-free polypropylene film as front window. A shutter was inserted automatically during each monochromator move to minimize photoreduction. The photon energy was calibrated to the maximum of the first pre-edge feature of Na2S2O3·5H2O at 2472.02 eV. At least three scans were measured for each sample to ensure reproducibility. Raw data were calibrated and averaged using MAVE in the EXAFSPAK package.21 Using the PySpline program22, the background was removed from all spectra by fitting a second-order polynomial to the pre-edge region and subtracting this from the entire spectrum. Normalization of the data was accomplished by fitting a straight line to the post-edge region and normalizing the edge jump to 1.0 at 2490.0 eV. The error from background subtraction and normalization is less than 3%. Intensities of the pre-edge features were quantified by fitting the data with pseudo-Voigt line shapes with a fixed Lorentzian to Gaussian ratio of 1:1, using the EDG_FIT program.21 Pre-edge energies and widths of single peaks were locked based on the previously published results on [Fe4S4] models and proteins (Supporting Info)6. The error from the fitting procedure is less than 3%. The fitted sulfide and thiolate intensities were converted to %S 3p character according to reference17.
Note that all the perturbations (lyophilization, mixing with DNA, and the combination) on the EndoIII and MutY were done on aliquots of the same protein sample. This means that all the data on each protein presented in this study have the same loading ratio, thus the differences observed upon DNA binding and lyophylization are independent of the loading.
DFT calculations
DFT calculations with broken symmetry spin polarization were performed using Gaussian 0923, with the pure functional BP86, and with 6–311G(d) basis sets on Fe and S, and 6–31G(d) basis sets on C and H. This functional and basis set were chosen to be consistent with previous studies.7 The α-carbons of the 4 Cys ligands were fixed to their crystal structure positions24 during geometry optimizations. To qualitatively evaluate the electrostatic effect of DNA binding, a point charge was placed at 5 Å from either a sulfide or a thiolate S atom in the [Fe4S4] cluster, and the electronic structure was reoptimized.
Results and Analysis
1) EndoIII without and with DNA
Sulfur K-edge XAS spectra of EndoIII in the absence and presence of a 20-mer mixed sequence DNA duplex (5′-GTGAGCTAACGTGTCAGTAC-3′ and its complement) at a ratio of 1 mol protein to 20 mol base-pairs were measured in both solution and lyophilized forms. Protein concentrations at 1 mM or greater were used for high quality XAS data. Equal concentrations of EndoIII were compared without and with the DNA 20-mer. The data were first normalized to 1 sulfur, then corrected for non-bonding sulfur atoms (6 in EndoIII and 18 in MutY) and the iron-sulfur cluster loading ratio, calculated as described in the Experimental section. Cluster loading ratios for EndoIIII were 70% or greater. The spectra were then multiplied by 8 to evaluate the total contribution of 4 sulfide and 4 thiolate sulfur atoms.
The normalized S-K edge XAS spectra of the pre-edge region are shown in Figure 1A; background (rising edge) subtracted spectral fits are given in Figure 1B. The energies and intensities obtained from the fits included in Figure 1B are given in Table 1. The peak assignments and the pre-edge shapes used in these fits are based on our previous results on [Fe4S4] clusters.6–7 All pre-edge spectra have two major features. The lower energy feature at ~2470.2 eV is due to the μ-3 sulfide S 1s → Fe 3d transitions, and the higher energy feature at ~2470.9 eV is due to the thiolate S 1s → Fe 3d transitions. Both gain intensity through S 3p mixing into Fe 3d orbitals. For both unbound and DNA bound EndoIII, lyophilization decreases the pre-edge intensity. For both solution and lyophilized data sets, DNA binding increases the pre-edge intensity, indicating that the Fe-S bonds become more covalent. DNA binding in solution increases the total S covalency from 573% to 631% (Table 1). Based on our past studies, each % increase of total S covalency corresponds to ~3.3 mV decrease in redox potential.7 Thus the covalency increase upon DNA binding in Figure 1 and Table 1 corresponds to a decrease of the reduction potential of the [Fe4S4]3+/2+ couple in EndoIII by ~190 mV, which would activate this cluster for oxidation. Critically, the DNA-induced potential shift measured by XAS is very close to that measured electrochemically.9
Figure 1.

S K-edge XAS of EndoIII without and with DNA and upon lyophilization (A) and the fits of the pre-edge region (B) using two peaks, one for the sulfides at ~2470.1 eV and one for the thiolates at ~2470.9 eV.
Table 1.
S K-edge XAS Data for [Fe4S4]2+ Models and Proteins
| Sample name | Energy (eV) | Area | Covalency (% per S) | Total Covalency | |
|---|---|---|---|---|---|
| [Fe4S4(SEt)4]2− | Sulfide | 2470.1 | 1.433 | 43.8 | 690±30 |
| model | Thiolate | 2470.9 | 0.578 | 41.0 | |
|
| |||||
| Fd | Sulfide | 2470.1 | 1.145 | 35.0 | 555±23 |
| Solution | Thiolate | 2470.9 | 0.481 | 34.0 | |
|
| |||||
| Fd | Sulfide | 2470.1 | 1.276 | 39.0 | 620±27a |
| Lyophilized | Thiolate | 2470.9 | 0.523 | 37.0 | |
|
| |||||
| HiPIP | Sulfide | 2470.1 | 1.253 | 38.3 | 618±17 |
| Solution | Thiolate | 2470.9 | 0.558 | 39.5 | |
|
| |||||
| HiPIP | Sulfide | 2470.1 | 1.194 | 36.5 | 588±16a |
| Lyophilized | Thiolate | 2470.9 | 0.503 | 35.6 | |
|
| |||||
| EndoIII | Sulfide | 2470.2 | 1.185 | 36.2 | 573±28 |
| Solution | Thiolate | 2470.9 | 0.488 | 34.5 | |
|
| |||||
| EndoIII | Sulfide | 2470.1 | 1.056 | 32.3 | 523±23a |
| Lyophilized | Thiolate | 2470.9 | 0.477 | 33.8 | |
|
| |||||
| EndoIII+DNA | Sulfide | 2470.2 | 1.293 | 39.5 | 631±31a |
| Solution | Thiolate | 2470.9 | 0.553 | 39.2 | |
|
| |||||
| EndoIII+DNA | Sulfide | 2470.1 | 1.106 | 33.8 | 542±26 a |
| Lyophilized | Thiolate | 2470.9 | 0.482 | 34.1 | |
|
| |||||
| MutY | Sulfide | 2470.1 | |||
| Solutionb | Thiolate | 2470.9 | |||
|
| |||||
| MutY | Sulfide | 2470.1 | |||
| Lyophilizedb | Thiolate | 2470.9 | |||
|
| |||||
| MutY+DNA | Sulfide | 2470.2 | |||
| Solutionb | Thiolate | 2470.9 | |||
|
| |||||
| MutY+DNA | Sulfide | 2470.1 | |||
| Lyophilizedb | Thiolate | 2470.9 | |||
Peak widths, energies and intensity ratios were locked in the fitting, with parameters based on those obtained in the fitting of the data represented in the row above, given that they are similar systems with similar noise level. Errors were estimated based on the relative error of the fit results in the row above.
Only the pre-edge energies are listed as they could be reasonably estimated based on the 2nd derivatives of the pre-edge regions. The error in the intensities are too large to quantitatively analyze these due to the large number of non-bound sulfur atoms.
2) S K-edge XAS of MutY
S K-edge XAS of MutY without and with DNA are shown in Figure 2A. Cluster loading ratios for MutY were also 70%±5%, and all the protein samples used in a series of XAS experiment have the same cluster loading. Similar to EndoIII, the pre-edge intensity increases upon DNA binding, and decreases upon lyophilization. This is consistent with the fact that the N-terminal domain of MutY is similar to EndoIII in structure in the presence of DNA (Figure 3A). Notably, while MutY and EndoIII are structurally homologous, MutY shows specificity for adenine mispaired with 8-oxoguanine, whereas EndoIII targets a range of oxidized substrates. Incorporating 2-fluoroadenine (FA) in place of A across from an oxoG site generates a non-hydrolysable substrate mimic for MutY that exhibits high affinity for MutY.25 Thus, to assess differences due to binding of MutY to its target DNA, the S K-edge XAS were measured both in the presence of specific (oxoG:FA) and non-specific DNA and with different DNA pair lengths under the same conditions as for EndoIII. One difference between MutY and EndoIII is that MutY has an extra domain (Figure 3A) that plays an important role in OG recognition and proper engagement of MutY on the oxoG:A substrate mispair.26 Figure 2B shows that in the case of MutY only the presence of specific DNA leads to the pre-edge intensity increase, and that the length of the DNA strand (15 and 30 base pairs) did not impact these intensity changes. This may be a consequence of differences in affinity and conformation of MutY with specific versus nonspecific DNA. MutY has much higher affinity for substrate-like DNA over nonspecific DNA and MutY also induces dramatic remodeling of its substrate. However, there are more non-bound sulfur atoms in MutY than in EndoIII contributing to the background and precluding quantification of the changes in Figure 2.
Figure 2.

S K-edge XAS of MutY without and with non-specific DNA and upon lyophilization (A), and S-K edge spectra showing impact of different DNA length (15 or 30) and of specific (OGFA) as well as non-specific (GC) binding on MutY (B)
Figure 3.


(A) Overlay of G. stearothermophilus (Gs) EndoIII (magenta, PDB 1ORN) and Gs MutY (cyan, PDB 5DPK), with [Fe4S4] cluster (orange for S and yellow for Fe atoms) and key Arg residues highlighted (red and blue respectively). Bound DNA in both structures are in green. Comparison of Ec EndoIII without DNA (pink, PDB 2ABK) in (B) and Gs EndoIII (magenta, PDB 1ORN) bound to DNA (green) in (C) showing the molecular surroundings of the [Fe4S4] cluster, including critical Arg residues (red). This comparison highlights the overall structure similarity between Gs EndoIII in complex with DNA to that of the Ec homolog without DNA. Small structural differences (RMSD100=1.4 Å) between the two homologs are attributed to their 43% sequence similarity.24
Figure 3A shows that MutY and EndoIII are not only similar in conformation, but have the conserved Arg residuals (Figure 3A, red and blue), that are responsible for DNA binding. Figure 3B and 3C show that these Arg locations are not perturbed by the DNA in EndoIII, although these two structures are for two homologs with 43% sequence similarity.
3) Solvent effect on HiPIP type proteins
From our previous studies, lyophilization perturbs the H-bond environment around the [Fe4S4] cluster sites. For the studies described below, the spectroscopic features and protein activity were measured upon re-dissolving the lyophilized proteins to make sure that the lyophilization process did not lead to irreversible denaturation (Figure S1 and S2). Figure 1 and Table 1 show that upon lyophilization, the sulfur covalency of EndoIII decreases by ~50% total sulfur character out of ~570%, which is opposite to the solvent effect observed for [Fe4S4] Bacillus thermoproteolyticus ferredoxins (Bt Fd).7 The pre-edge intensity of the [Fe4S4] cluster in Bt Fd increases by ~65% total sulfur character out of ~555% upon removal of solvent water. Unlike Bt Fd, where the [Fe4S4] cluster is exposed at the surface, EndoIII has a buried [Fe4S4] cluster, similar to MutY and Chromatium vinosum HiPIP. The XAS spectra of EndoIII (Figure 1), MutY (Figure 2) and C. vinosum HiPIP (Figure S4) all show a decrease in S pre-edge intensity with lyophilization (~30% out of a total of 618% sulfur character for HiPIP).
As shown in Figure 4A, the [Fe4S4] cluster in C. vinosum HiPIP (PDB code 1CKU) has five H-bonds from the protein environment to the thiolate sulfurs within 3.5 Å. All are from the amide backbone. The higher S K-edge intensity in HiPIP relative to Fd and its decrease in S covalency upon lyophilization indicate that relative to [Fe4S4] Fd, the H-bonds to the [Fe4S4] cluster in HiPIP are weak. Removal of the solvent in HiPIP would lead to a more compact site, increasing the backbone H-bonds to the cluster. Also, three of the amides are solvent exposed, which could be directly affected by lyophilization (boxed). Furthermore, there are waters H-bonded to the carbonyls that are conjugated to the amide N atoms, and removal of these water would enhance their N-H…S hydrogen bonds.
Figure 4.



Local H-bond network of the Fe4S4 cluster for (A) HiPIP (PDB code: 1CKU), (B) EndoIII without DNA bound (PDB code: 4UNF), and EndoIII with DNA (PDB code: 1ORN) illustrating some of the H-bonds that could be affected by solvent water. The H-bonds are shown in dashed green, and the Arg and His are labeled. Schemes of the boxed region are shown in (D). In (A), an amide backbone H-bond to the thiolate S bound to Fe and an accessible solvent is shown in the box, and in (B) and (C), an Arg residue H-bond to the thiolate bound to Fe and accessible to solvent is shown in the box.
Figure 4B shows that in the unbound form of EndoIII (PDB code 4UNF), the [Fe4S4] cluster has six H-bonds to thiolate sulfurs within 3.5 Å: one from arginine, one from histidine, and four from the amide backbone. Figure 4C shows a similar H-bond pattern around the cluster for the EndoIII with DNA bound (PDB code: 1ORN). In EndoIII, the increased number of H-bonds and the stronger H-bond from the positively charged Arg are consistent with the somewhat lower S covalency of EndoIII relative to that of HiPIP (Table 1 Solution: 573% vs 618%; lyophilized: 523% vs 588% for EndoIII and HiPIP, respectively). In addition to the more compact site that would result from lyophilization, the Arg, His and amides are all at the surface of the protein and would also be directly impacted by loss of H2O (red dots in Figure 3 and boxed region in Figure 4B and 4C). The larger number of surface exposed H-bonds is consistent with the larger decrease in S edge intensity in EndoIII relative to HiPIP upon lyophilization (50% vs 30% respectively). MutY has similar DNA binding domain structure as EndoIII, and a surface Arg is shown in blue in Figure 3A. Finally, as displayed in Figure 1 and Table 1, lyophilization of DNA bound EndoIII also leads to an intensity decrease (somewhat larger than for the unbound EndoIII). Thus even when bound to DNA, there is solvent access to the [Fe4S4] cluster in EndoIII.
Discussion
Both EndoIII and MutY contain [Fe4S4] clusters which are redox inert in solution, but their reduction potentials decrease by ~200 mV upon DNA binding to turn on their function in DNA mismatch recognition. In nature, a large number of proteins also have [Fe4S4] clusters, with a wide range of reduction potentials (Fd: −700 to −300 mV; HiPIP: 100 to 400 mV) (Scheme 1).1 S K-edge XAS has been found to be a powerful technique to evaluate the contributions of H-bonds and electrostatics to the reduction potentials of these [Fe4S4] clusters.27 From our past studies on Fd and HiPIP, the redox properties of these [Fe4S4] clusters are highly related to their local protein environment.7 In Fd, the [Fe4S4] cluster is at the surface of the protein. There are strong H-bonds to the cluster thiolate sulfurs from solvent water, thus these sulfurs donate less electron density to Fe and the Fe-S bonds are less covalent. This raises the reduction potential of the +2 state and results in the [Fe4S4]2+/1+ redox couple being accessible in the physiological potential range. Upon lyophilization, the S covalency significantly increases due to the loss of H-bonds from solvent to the [Fe4S4] cluster and the covalency becomes similar to that of HiPIP. In contrast, HiPIP, whose cluster is not solvent exposed, has only weak H-bonds from the backbone amides to the iron-sulfur cluster, which results in greater sulfur electon density donation to iron, and thus more covalent Fe-S bonds. In HiPIP, the [Fe4S4]3+/2+ redox couple is activated in the physiological potential range (Scheme 1). In the present study, lyophilization of HiPIP leads to an inverse effect on the Fe-S bond covalency relative to Fd (ie. the covalency decreases). Loss of solvent would lead to a more compact site and increase the H-bonding to the [Fe4S4] cluster from backbone. Loss of water H-bonds to the surface amides would further strengthen these H-bonds to the thiolates. Lyophilization results in a similar effect on EndoIII and MutY.
Scheme 1.

Fds have strong H-bonds to S (5 amide-thiolate H-bonds, 3 amide-sulfide H-bonds, as shown in Figure S6, and more importantly, H-bonds to surface exposed thiolate from solvent water), and thus both type of S donate less electron density to Fe, stabilizing the reduced state. (Redox potential range −700 to −300 mV) HiPIPs have only weak H-bonds to S (5 amide-thiolate H-bonds, as shown in Figure 4A), thus these S donate more electron density to Fe, stabilizing the oxidized state. (Redox potential range 100 to 400 mV) EndoIII/MutY have moderate H-bonding (A few amide-thiolate H-bonds, and importantly, an Arg-thiolate H-bond, as shown in Figure 4 and Figure S6), thus are redox inert. However, binding to DNA introduces negative charge, thus stabilizing the oxidized state.
Both EndoIII and MutY have a strong Arg H-bond to their [Fe4S4]2+ cluster, which is consistent with their lower covalency relative to HiPIP, and making [Fe4S4]2+/3+ couple less accessible. (Table 1). Overall, this behavior correlates with their lack of redox activity in the absence of DNA.28–29 S K-edge XAS experiments show that DNA binding to EndoIII and MutY significantly increases their Fe-S bond covalency, while in EndoIII DNA binding lowers its [Fe4S4]3+/2+ potential by ~200 mV into the physiologically accessible range. From aligning the unbound and DNA bound structures of EndoIII (Figure S5), there is no significant distortion of the protein (RMSD100=1.4 Å; note that these EndoIII proteins are homologs with sequence similarity of 43%), thus DNA binding does not appear to structurally affect its [Fe4S4] site. DNA binding, although at a distance of ~15 Å from the [Fe4S4] cluster, does introduce significant negative charge into the system. For the iron-sulfur cluster, the negative charge would destabilize the occupied S p orbital energies, and result in more S character donated into the unoccupied valence Fe 3d based molecular orbitals (Scheme 2). This increases the Fe-S bond covalency of the cluster, stabilizing the oxidized over the reduced state, and thus decreasing the reduction potential. Simple DFT models using a point negative charge to represent the effect of the DNA qualitatively reproduce this covalency increase and show that it dominantly involves an increase in electron donation from the S oriented toward the negative charge with some limited compensation by the remote sulfurs. (See Supporting Info.)
Scheme 2.

Adding negative charges such as DNA in the proximity of the [Fe4S4] cluster destabilizes the S 3p orbital energy and increases the S 3p character (α2) in Ψ*. This increase in the Fe-S covalency stabilizes the oxidized more than the reduced state of the cluster and decreases the reduction potential.
Conclusion
S K-edge XAS studies on iron sulfur proteins both in the absence of and bound to DNA, and with and without solvent water show that the local environments of the [Fe4S4] clusters affect the covalencies of their Fe-S bonds and thus tune the potentials of the clusters. In Fd, the strong H-bonds from solvent reduce the covalency and stabilize the reduced form of the cluster, while in HiPIP, the cluster is buried and displays only weak amide H-bonds, leading to higher covalency and stabilizing the oxidized form. In EndoIII and MutY the cluster is also buried but with an additional Arg H-bond that reduces the covalency relative to HiPIP and makes the HiPIP couple less accessible. However binding to DNA introduces negative charge that increases the [Fe4S4] covalency for both EndoIII and MutY. Previous correlations between [Fe4S4] covalency and redox potential indicate that this covalency decrease corresponds to an approximately 200 mV negative shift in reduction potential and would enable the [Fe4S4]2+/3+ redox couple that is observed experimentally. Overall, the results obtained by XAS are in agreement with electrochemical studies carried out with EndoIII in the presence and absence of DNA, indicating that the physical mechanism of the potential shift is a charge-induced increase in sulfur-iron bond covalency within the [Fe4S4] cluster. It is these molecular-level changes that are responsible for the ability of otherwise redox-inert repair proteins to communicate with each other over vast distances using reversible DNA-mediated charge transfer, making the daunting task of searching an entire genome for damage possible on a physiologically relevant time scale.
Supplementary Material
Acknowledgments
This work was supported by the NIH grant (GM040392, E.I.S; GM103393, K.O.H; GM120087 J.K.B; CA069875 S.S.D). J.K.B also thanks the Moore foundation. A.R.A. was supported by the National Institute of Aging of the NIH on a predoctoral NRSA (F31AG040954). Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource (SSRL), a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (P41GM103393).
Footnotes
Associated Content
Computational results and analysis and addition figures for XAS spectra and protein structure comparison are available free of charge on the ACS Publications website http://pubs.acs.org
References
- 1.Holm RH, Kennepohl P, Solomon EI. Chem Rev. 1996;96:2239–2314. doi: 10.1021/cr9500390. [DOI] [PubMed] [Google Scholar]
- 2.Stiefel EI, George GN. Ferredoxins, Hydrogenases, and Nitrogenases: Metal-Sulfide Proteins. In: Bertini I, editor. Bioinorganic Chemistry. University Science Books; Mill Valley: 1994. pp. 365–453. [Google Scholar]
- 3.Backes G, Mino Y, Loehr TM, Meyer TE, Cusanovich MA, Sweeney WV, Adman ET, Sanders-Loehr J. J Am Chem Soc. 1991;113:2055–2064. [Google Scholar]
- 4.Teo BK, Shulman RG, Brown GS, Meixner AE. J Am Chem Soc. 1979;101:5624. [Google Scholar]
- 5.Czernuszewicz RS, Macor KA, Johnson MK, Gewirth A, Spiro TG. J Am Chem Soc. 1987;109:7178–7187. [Google Scholar]
- 6.Dey A, Glaser T, Couture MMJ, Eltis LD, Holm RH, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 2004;126:8320–8328. doi: 10.1021/ja0484956. [DOI] [PubMed] [Google Scholar]
- 7.Dey A, Jenney FE, Jr, Adams MWW, Babini E, Takahashi Y, Fukuyama K, Hodgson KO, Hedman B, Solomon EI. Science. 2007;318:1464–1468. doi: 10.1126/science.1147753. [DOI] [PubMed] [Google Scholar]
- 8.Boal AK, Yavin E, Lukianova OA, O’Shea VL, David SS, Barton JK. Biochemistry. 2005;44:8397–8407. doi: 10.1021/bi047494n. [DOI] [PubMed] [Google Scholar]
- 9.Lukianova OA, David SS. Curr Op Chem Biol. 2005;9:145–151. doi: 10.1016/j.cbpa.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 10.Manlove AH, Nuñez NN, David SS. The GO Repair Pathway: OGG1 and MUTYH. In: DMW III, editor. The Base Excision Repair Pathway. World Scientific; New Jersey: 2016. pp. 63–115. [Google Scholar]
- 11.Cunningham RP, Asahara H, Bank JF, Scholes CP, Salerno JC, Surerus K, Munck E, McCracken J, Peisach J, Emptage MH. Biochemistry. 1989;28:4450–4455. doi: 10.1021/bi00436a049. [DOI] [PubMed] [Google Scholar]
- 12.Gorodetsky AA, Boal AK, Barton JK. J Am Chem Soc. 2006;128:12082–12083. doi: 10.1021/ja064784d. [DOI] [PubMed] [Google Scholar]
- 13.Boon EM, Livingston AL, Chmiel NH, David SS, Barton JK. Proc Natl Acad Sci. 2003;100:12543–12547. doi: 10.1073/pnas.2035257100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boal AK, Yavin E, Barton JK. J Inorg Biochem. 2007;101:1913–1921. doi: 10.1016/j.jinorgbio.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boon EM, Pope MA, Williams SD, David SS, Barton JK. Biochemistry. 2002;41:8464–8470. doi: 10.1021/bi012068c. [DOI] [PubMed] [Google Scholar]
- 16.Solomon EI, Hedman B, Hodgson KO, Dey A, Szilagyi RK. Coord Chem Rev. 2005;249:97–129. [Google Scholar]
- 17.Sarangi R, George SD, Rudd DJ, Szilagyi RK, Ribas X, Rovira C, Almeida M, Hodgson KO, Hedman B, Solomon EI. J Am Chem Soc. 2007;129:2316–2326. doi: 10.1021/ja0665949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glaser T, Rose K, Shadle SE, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 2001;123:442–454. doi: 10.1021/ja002183v. [DOI] [PubMed] [Google Scholar]
- 19.Pheeney CG, Arnold AR, Grodick MA, Barton JK. J Am Chem Soc. 2013;135:11869–11879. doi: 10.1021/ja4041779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shadle SE, Hedman B, Hodgson KO, Solomon EI. Inorg Chem. 1994;33:4235–4244. [Google Scholar]
- 21.George GN. EXAFSPAK. Stanford Synchrotron Radiaton Laboratory; Menlo Park, CA: 1990. [Google Scholar]
- 22.Tenderholt AL, Hedman B, Hodgson KO. PySpline: A Modern, Cross-Platform Program for the Processing of Raw Averaged XAS Edge and EXAFS Data. AIP conference Proceedings; 2006. pp. 105–107. [Google Scholar]
- 23.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision B.01. Wallingford CT: 2009. [Google Scholar]
- 24.Fromme JC, Verdine GL. EMBO J. 2003;22:3461–3471. doi: 10.1093/emboj/cdg311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chepanoske CL, Porello SL, Fujiwara T, Sugiyama H, David SS. Nucleic Acids Res. 1999;27:3197–3204. doi: 10.1093/nar/27.15.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chmiel NH, Golinelli MP, Francis AW, David SS. Nucleic Acids Res. 2001;29:553–564. doi: 10.1093/nar/29.2.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hadt RG, Sun N, Marshall NM, Hodgson KO, Hedman B, Lu Y, Solomon EI. J Am Chem Soc. 2012;134:16701–16716. doi: 10.1021/ja306438n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chepanoske CL, Golinell MP, Williams SD, David SS. Arch Biochem Biophys. 2000;380:11–19. doi: 10.1006/abbi.2000.1890. [DOI] [PubMed] [Google Scholar]
- 29.Kuo CF, McRee DE, Fisher CL, O’Handley SF, Cunningham RP, Tainer JA. Science. 1992;258:434–440. doi: 10.1126/science.1411536. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.