

Abstract
Natural killer (NK) cell responsiveness in the mouse is determined in an education process guided by inhibitory Ly49 and NKG2A receptors binding to MHC class I molecules. It has been proposed that inhibitory signaling in human NK cells involves Abl-1 (c-Abl)-mediated phosphorylation of Crk, lowering NK cell function via disruption of a signaling complex including C3G and c-Cbl, suggesting that NK cell education might involve c-Abl. Mice deficient in c-Abl expression specifically in murine NK cells displayed normal inhibitory and activating receptor repertoires. Furthermore, c-Abl-deficient NK cells fluxed Ca2+ normally after triggering of ITAM receptors, killed YAC-1 tumour cells efficiently and showed normal, or even slightly elevated, capacity to produce IFN-γ after activating receptor stimulation. Consistent with these results, c-Abl deficiency in NK cells did not affect NK cell inhibition via the receptors Ly49G2, Ly49A and NKG2A. We conclude that signaling downstream of murine inhibitory receptors does not involve c-Abl and that c-Abl plays no major role in NK cell education in the mouse, which contrasts with data in humans.
Introduction
Natural Killer (NK) cells are large granular lymphocytes belonging to the group 1 of innate lymphoid cells [1]. NK cells secrete cytokines, such as IFN-γ, and perform cytotoxicity to control viral infections and malignant transformation. From having been considered innate, NK cells were recently demonstrated to exhibit an adaptive, or memory-like, phenotype [2], suggesting that NK cells are regulated in a more complex way than previously thought.
NK cells express a plethora of activating and inhibitory receptors at the cell surface, whose intracellular signals integrate to generate a balanced functional output. Activating receptors recognise ligands induced by stress, malignant transformation and pathogen invasion [3]. Inhibitory receptors, such as Ly49 receptors in mice and KIR (Killer-Immunoglobulin Receptors) in humans, recognise Major Histocompatibility Complex (MHC) class I on surrounding cells [4, 5]. When target cells lack MHC class I molecules, or expresses MHC class I molecules of the wrong type, NK cell may become activated and mediate “missing self” killing [6]. Importantly, MHC class I molecules not only determine target cell killing but also guides a process called ‘NK cell education’, which empowers NK cells with functional capacity [7]. During NK cell education, inhibitory and activating signals are balanced in the NK cells, which sets a triggering threshold in individual NK cells that is balanced against the inhibitory input [7-11]. The molecular signals that control NK cell education are poorly characterised and identifying them may facilitate the development of strategies to modulate NK cell activity, either enhancing or decreasing their activity.
Signaling downstream of the NK cell receptors proceeds through different pathways depending on which receptors that are activated. Most activating receptors use classical ITAM-dependent signaling pathways, but some receptors, such as NKG2D, mediate NK cell activation via direct coupling to the PI3K pathway with the adaptor molecule DAP10 [12]. Irrespective of which pathways are engaged, inhibitory NK cell receptors exert dampening influences by targeting key players in the signaling pathways. The most studied mechanism of NK cell inhibition via Ly49 and KIR receptors is an active dephosphorylation of the guanine nucleotide exchange factor Vav-1, a process catalysed by the inhibitory receptor-associated phosphatase SHP-1 [13]. Other phosphatases, such as SHIP, have been implied in inhibitory NK cell signaling [14] and it is likely that the combined action of SHP-1 and SHIP also targets other substrates than Vav-1 that might contribute to NK cell inhibition. Mice lacking SHP-1 or SHIP specifically in NK cells were recently created. NK cell function was deficient in both these mice, implying non-redundant roles for SHP-1 and SHIP-dependent dephosphorylation events in in NK ell education [10, 11].
A role for the non-receptor tyrosine kinase c-Abl in lymphocyte function was suggested from early work with c-Abl-deficient mice, in which c-Abl was shown to control B and T cell development [15]. Conditional depletion of c-Abl in T cells revealed an involvement of c-Abl in a variety of T cell properties such as actin remodelling and immune synapse formation [16-18]. A role for c-Abl in NK cell function is poorly studied. Fetal liver chimeric mice containing a c-Abl-deficient hematopoietic compartment showed normal development of NK cells and an unaffected capacity of NK cells to kill YAC-1 lymphoma cells and MHC class I-deficient Con A blasts [19]. In contrast, Peterson and Long showed that in human NK cells, c-Abl phosphorylated the adapter molecule Crk, leading to its dissociation from c-Cbl and C3G [20], dampening NK cell activation [21]. Thus, c-Abl was proposed to be actively involved in inhibitory signaling downstream of KIR receptors. We generated mice with a disrupted c-Abl gene in NKp46-positive cells to test if c-Abl was involved in inhibitory signaling, and consequently in NK cell education, also in the mouse. We could not confirm a role for c-Abl in inhibitory signaling downstream of three inhibitory NK cell receptors. Consequently, NK cell education appeared unaltered in mice lacking c-Abl in NK cells. In contrast, c-Abl-deficient NK cells actually showed a small increase in IFN-γ secretion and degranulation after triggering of ITAM-dependent activating receptors, suggesting that c-Abl may impose weak dampening effects on NK cell activation independently from inhibitory receptor signaling.
Material and methods
Mice
All mice were bred and housed at the animal facility at Karolinska Institutet, Huddinge, Sweden. Mice used for experiments were of 6-10 weeks of age. C57Bl/6 (B6) mice were purchased from Jackson laboratory. iNcreCre +/- (Cre +/-) mice were described previously [22], as were c-Abl fl/fl mice [23, 24]. Breedings, experimental procedures, and mouse handling were done according to the animal ethics guidelines prescribed by Karolinska Institutet and the Karolinska University Hospital. The procedures were approved by the Stockholm branch of the national animal ethics committee in Sweden (Stockholm Södra Djurförsöksetiska Nämnd).
Antibodies and flow cytometry analysis
Single cell splenocyte suspensions were incubated with anti-FcyRIII (2.4G2) before staining with surface markers. Phenotypic surface staining was performed at 4°C in PBS supplemented with 1% FCS for 20 minutes. Antibodies used in this study were CD3 (145-2c11), KLRG1 (2F1) from eBiosciences, NK1.1 (PK136), DX5 (DX5), CD27 (LG.3A10), Ly49A (YE1/48.10.6), NKG2A (16I11), DNAM-1 (480.1) from BioLegend, NKp46 (29A1.4), CD11b (M1/70), Ly49I (YLI-90), Ly49G2 (4D11), IFN-γ (XMG 1.2) from BD Pharmingen (Stockholm, Sweden). Anti-Ly49C (4LO3311) hybridoma was a gift from Suzanne Lemieux (INRS–Institut Armand-Frappier, Laval, Quebec). Anti-Ly49C antibodies were biotinylated following DSB-X™ Biotin Protein Labeling Kit (Life Technologies). Dead cells were excluded using LIVE/DEAD® Fixable Aqua Dead Cell Stain Kit (Invitrogen). Flow cytometry analysis was done in LSR Fortessa (BD Biosciences) and data analysis was done by FlowJo (Tree Star).
In vitro antibody/cytokine stimulation assay
24-well plates were coated overnight at 4°C in PBS with 20 μg/ml purified anti-NK1.1 (PK136, eBiosciences) and anti-NKp46 (polyclonal, R&D systems). A single cell splenocyte suspension was subsequently prepared and NK cells were enriched using a negative selection kit (MACS Miltenyi Biotec). NK cells (4×10 to 5×10 cells) were stimulated for 6hours at 37 C in RPMI+10% FCS. CD107a (BD Pharmingen) was added (1:300 dilution) to the culture during stimulation. After one hour of stimulation, GolgiPlug™ Protein Transport Inhibitor diluted 1:1000 (BD Pharmingen) was dispensed into the culture. 1 μg/ml of phorbol-12-myristate-13-acetate (PMA; SIGMA) plus 0.25 μg/ml of ionomycin (SIGMA) was used for positive control and PBS alone for negative control. For cytokine stimulation, IL-18 (1 ng/ml) plus IL-12 (1 ng/ml) was used. After stimulation, staining was performed as described above. For intracellular staining, cells were fixed and then permeabilized using Cytofix/Cytoperm kit (BD Pharmingen), followed by staining with anti-IFN-γ (XMG1.2).
Calcium fluorometry
Freshly enriched splenic NK cells were incubated in vitro with 20 μg/ml purified anti-NK1.1 (PK136). Antibody-coated NK cells were stained at 4°C in HBSS+1%FCS and probenecid with the calcium indicator dye Fluo-4 (Life Technologies). Events were recorded for 30 secs in the flow cytometer, after which purified Goat-Anti-Mouse IgG (H + L) Polyclonal Antibody (Jackson Immunoresearch) (20ug/ml) was added. Measurement of real-time iCa mobilization was subsequently performed for up to 4 mins. Receptor-induced iCa mobilization was quantified by determining the Area Under the Curve (AUC) over time using the FlowJo software.
Chromium release assay
YAC-1 cells were incubated for 1 hr in the presence of Na2O4Cr51 (PerkinElmer Life and Analytical Sciences, Waltham, MA, USA) and then washed in PBS. Isolated Cre+/- c-Ablfl/fl and Cre+/- c-Abl+/fl mice NK cells were then mixed with YAC-1 cells at different E:T ratios. After 4 hrs of E:T cell coincubation at 37°C, cell culture supernatants were analysed on a γ radiation counter (Wallac Oy, Turku, Finland). Specific lysis was calculated according to formula: % specific lysis = (experimental release – spontaneous release)/(maximum release – spontaneous release) × 100. If the difference between spontaneous and maximum release was <2-fold, then experiments were discarded.
Statistics
Statistical significance was done for Mann-Whitney tests and T-test analysis of variance with Bonferroni correction (Prism 5, GraphPad software). The degree of significance is indicated as follows: **P<0.01.
Results
Mice with NK cell-specific deletion of the c-Abl kinase
We crossed iNCRCre+ (Cre) mice, in which the expression of the Cre recombinase is driven by the NK cell-specific NKp46 promoter [22] to mice with a floxed c-Abl gene [16]. We confirmed the specificity of the c-Abl deletion by sorting NK cells and T cells from wild-type B6 mice, Cre+/- c-Abl +/fl and Cre+/- c-Abl fl/fl mice and exposing DNA from these cells to two PCR reactions, one that amplifies only the recombined c-Abl gene (PCR 1) and one that separates the wild-type and non-recombined floxed c-Abl alleles (PCR 2). In c-Abl +/fl mice, T cells showed no band using PCR 1 and two bands using PCR 2, confirming the lack of Cre-mediated deletion (Fig. 1A). In DNA from sorted NK cells, PCR 1 amplified a strong recombined band and PCR 2 amplified a wild-type band only, suggesting a complete recombination of the floxed c-Abl allele (Fig. 1A). A similarly clear pattern was seen in c-Abl fl/fl NK cells (Fig 1A).
Fig. 1.
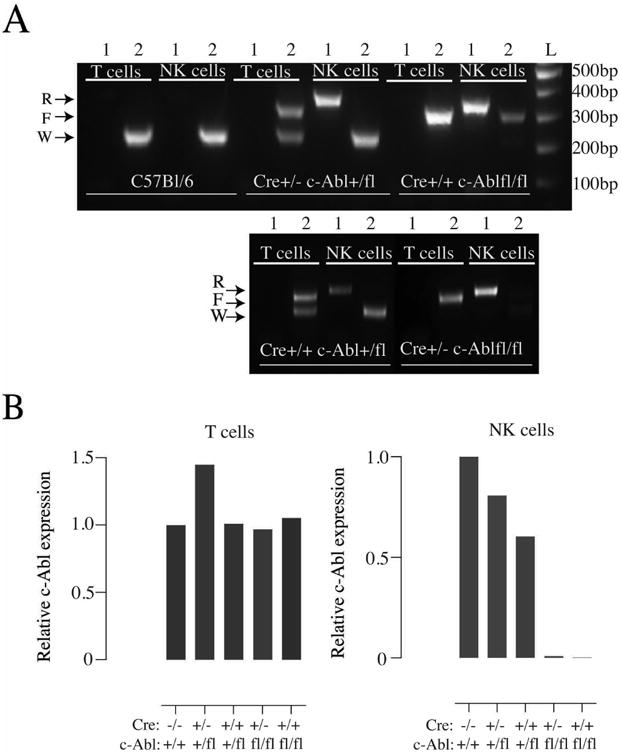
(A) Agarose gel electrophoresis of PCR-amplified DNA extracted from sorted T and NK cells from the spleens of C57Bl/6, Cre+/- c-Abl +/fl, Cre+/+ c-Abl fl/fl, Cre+/+ c-Abl +/fl and Cre+/- c-Abl fl/fl mice. ‘R’ represents the recombined (deleted) c-Abl allele, ‘F’ the floxed but non-recombined c-Abl allele and ‘W’ represents the wild-type c-Abl allele. Lane 1 represent a PCR reaction for the recombined c-Abl allele and lane 2 a PCR reaction that distinguishes wild type and floxed c-Abl alleles. (B) Real-Time Q-PCR analysis of RNA from sorted T and NK cells from the spleens of the same mouse lines. C57Bl/6 is denoted as Cre -/-c-Abl +/+. The values were normalised to the expression level of control C57Bl/6 mice, which was set to 1. Data are representative of five mice generated from one experiment.
To confirm that deletion of the c-Abl gene was followed by lack of c-Abl gene expression, we exposed sorted NK cells and T cells from our mice to a quantitative real-time PCR experiment and looked for c-Abl mRNA expression. Data from T and NK cells from B6 mice were used as controls and was normalised to a value of one. As expected, c-Abl expression was detected in all samples except in NK cells from Cre+/- c-Abl fl/fl mice (Fig. 1B). Notably, c-Abl expression in NK cells from c-Abl +/fl mice was somewhat reduced compared to B6 mice, suggesting a gene-dose effect (Fig. 1B). We did not investigate additional tissues and cell types, but based on our specificity analysis using T cells and on published data using the same Cre-expressing mouse lines conditional deletion of other genes [10, 11, 22], we conclude that NK cell-specific deletion of c-Abl was achieved.
c-Abl deficiency does not affect the number of NK cells or NK cell maturation
To enumerate the percentages of NK cells and other lymphocyte cell populations, we performed a flow cytometric analysis of Cre+/- c-Abl +/fl and Cre+/- c-Abl fl/fl mice. The results revealed similar frequencies of the four major lymphocyte subsets, including NK1.1+CD3+ (NKT) cells, NK1.1-CD3- cells and NK1.1-CD3+ (T) cells (Fig. 2A). There was a trend towards slightly higher numbers of NK1.1+CD3- cells in Cre+/- c-Abl fl/fl mice in comparison to Cre+/- c-Abl +/fl mice, but the difference was not statistically significant (Fig. 2).
Fig. 2.
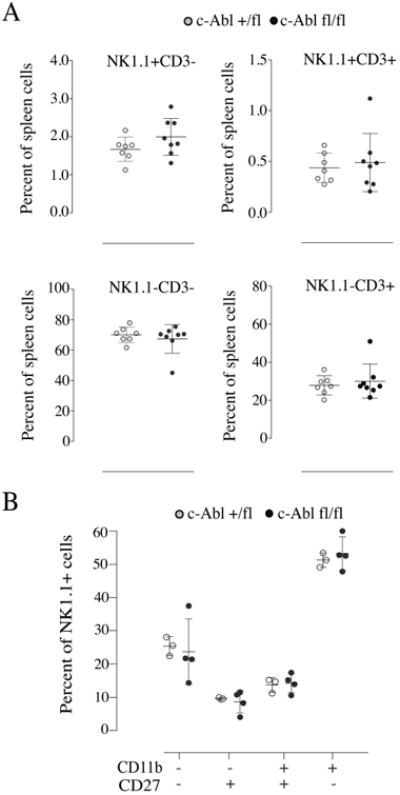
(A) Frequency of splenic lymphocytes in c-Abl deficient mice. Each dot corresponds to one mouse. Data are obtained from three independent experiments. (B) Frequency of different developmental stages of splenic NK cells in c-Abl-deficient mice. Each dot represents data for a single mouse and the graph shows data from two independent experiments. No significant differences were found between c-Abl-deficient and wild type mice.
NK cells acquire maturation markers (CD27/CD11b) during their developmental program, which is crucial for their effector function [25]. Upon analysis of different stages of splenic NK cell maturation, we found similar frequencies of CD27lowCD11blow (immature), CD27highCD11blow (more mature) and CD27lowCD11bhigh (most mature) cells in Cre+/- c-Abl fl/fl mice compared to control mice (Fig. 2B). Thus, c-Abl deletion is dispensable for the generation of normal numbers of NK cells and for their acquisition of conventional maturation markers.
The cell surface expression or activating and inhibitory receptors is unaltered in c-Abl-deficient NK cells
NK cells express various germline-encoded activating and inhibitory receptors, which are crucial for effector function. Activating receptors such as NK1.1, NKp46 and NKG2D are expressed on all splenic NK cells. In contrast, some activating receptors, such as DNAM-1, KLRG-1 and Ly49D are expressed on subsets of NK cells. Individual analysis for these receptors revealed no major differences between Cre+/- c-Abl fl/fl mice and control mice (Fig. S1A, B). In addition to activating receptors, NK cells express a variety of inhibitory receptors whose expression pattern is controlled by NK cell education process [7, 8, 26]. We used a multi-colour flow cytometry panel to investigate the expression pattern of Ly49C, Ly49I, Ly49A, Ly49G2 and NKG2A in Cre+/- c-Abl fl/fl and Cre+/- c-Abl fl/+ NK cells. We focussed on NK cells expressing the self-specific receptors Ly49C and Ly49I as their only receptor, since those subsets are most clearly subject to NK cell education in mice expressing H2Kb, in which where their frequencies are increased compared to mice lacking MHC class I [27]. Our analysis revealed no major differences in the frequency of NK cells expressing any of these five receptors as their only receptor (Fig. S1C).
c-Abl-deficient NK cells display marginally enhanced effector responses after triggering of ITAM-encoded receptors
To test if c-Abl-deficiency would affect the capacity of NK cells to mediate effector functions, we measured the capacity of c-Abl-deficient NK cells to secrete IFN-γ and to degranulate after triggering of the NK1.1 receptor by antibodies. This experiment surprisingly revealed more efficient IFN-γ production and de-granulation in c-Abl-deficient NK cells (Fig. 3A, B). A small difference was noted for CD107 expression also after stimulation of the NKp46 receptor, but the response to NKp46 stimulation in our setup was poor and this difference was not statistically significant. Stimulation with IL-12+IL-18 and with PMA/Ionomycin did not reveal any difference between the genotypes, suggesting that c-Abl deficiency impacted on responses downstream of ITAM signaling specifically. Adding IL-15 as a priming factor increased the response, but did not override the difference between c-Abl-deficient and sufficient NK cells (Fig. 3, B). NK cells from c-Abl-deficient mice also contained slightly more IFN-γ per cell (Fig. 3C), substantiating the increased frequency of responding NK cells.
Fig. 3.
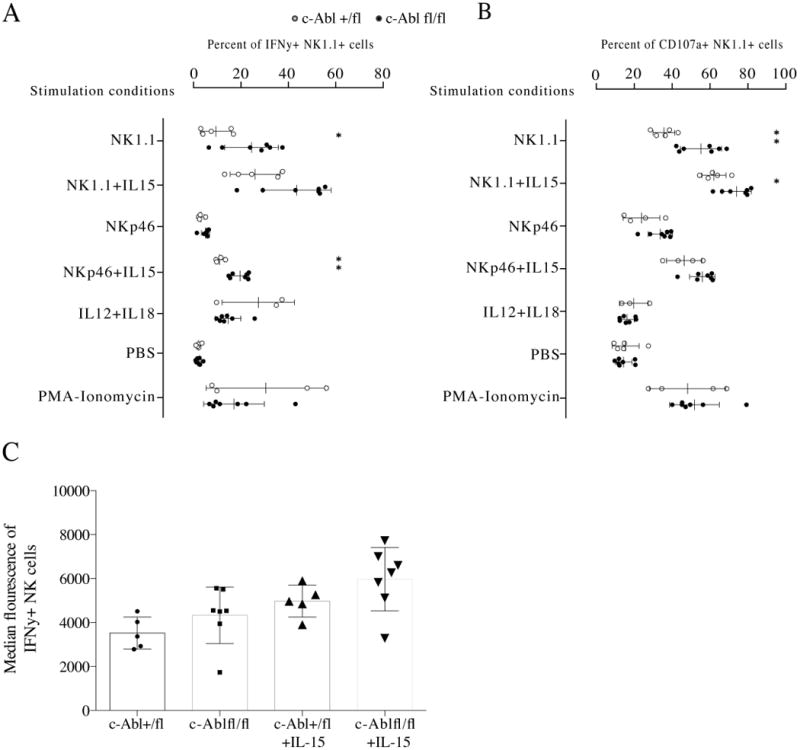
(A) Frequency of IFN-γ+ splenic NK1.1+CD3- cells in c-Abl-deficient mice after stimulation using the agents indicated along the y axis. Each white dot depicts NK cells from a Cre+/- c-Abl +/fl mouse and each black depicts NK cells from a Cre+/- c-Abl fl/fl mouse. Data generated are representative of two independent experiments. (B) Frequency of CD107a+ splenic NK1.1+CD3- cells in c-Abl deficient mice. Data from the same experiments as in A. * p<0,05 and ** p<0,01 using Mann-Whitney test. (C) Median fluorescence intensity of intracellular IFN-γ from the same analysis. The difference between c-Abl-deficient and sufficient NK cells did not reach statistical significance.
We next tested if the enhanced response after NK1.1 triggering was paralleled by an enhanced capacity to flux calcium. We first verified that NK cells from MHC class I-deficient mice showed decreased capacity to release intracellular calcium upon NK1.1 triggering compared to B6 mice (Fig. 4A), confirming previous data showing that lack of NK cell education is manifested at the level of early ITAM signaling [11]. When we compared c-Abl-deficient and sufficient NK cells for calcium flux response after NK1.1 cross-linking, we found that they responded similarly (Fig. 4B), suggesting that c-Abl deficiency may affect the IFN-γ response downstream of calcium flux. Finally, we performed a killing assay against YAC-1 cells. We did not find any major difference in the capacity of c-Abl-deficient NK cells to kill YAC-1 cells, suggesting that the slightly enhanced CD107 degranulation response downstream of ITAM-encoded receptors did not translate to a generally weakened tumour cell killing capacity (Fig. S2).
Fig. 4.
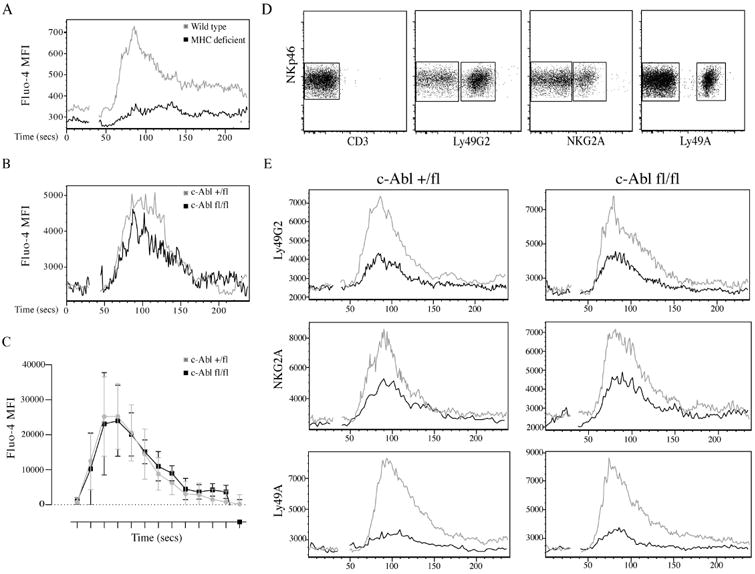
(A) Ca2+ flux in wild type (C57Bl/6) and MHC class I-deficient NK cells after crosslinking of NK1.1. (B) Same analysis as in A using Cre+/- c-Abl+/fl and Cre+/- c-Abl fl/fl NK cells. C) Summary of the area under the curve analysis (performed using the FlowJo software) for four c-Abl+/fl and five c-Ablfl/fl mice from two independent experiments. The X-axis represents time intervals of 240 seconds divided into 13 regions. (D) Gating of three different inhibitory receptors Ly49G2, Ly49A and NKG2A for E. (E) Ca2+ flux in NK cells from c-Abl +/fl and c-Abl fl/fl mice co-stimulation using anti-NK1.1+anti-NKG2A, anti-NK1.1+anti-Ly49G2 and anti-NK1.1+Ly49A antibodies respectively. X-axis represents time interval of 240 seconds. Y- axis represents MFI of Fluo-4 dye. The black line indicates calcium flux for inhibitory receptor positive cells and the grey line indicates calcium flux in inhibitory receptor negative cells. Data represented is from one experiment of three.
Inhibitory receptor signaling in c-Abl-deficient NK cells
Because our data suggested that c-Abl was dispensable for NK cell education, we directly investigated the basic premises for our study, i.e. the involvement of c-Abl in inhibitory signaling in NK cells [21]. To test this, we developed an in vitro assay in which inhibition of early calcium flux after NK1.1 stimulation could be measured after crosslinking of inhibitory receptors. By using flurochrome-conjugated antibodies against the inhibitory receptors, we were able to gate on the inhibitory-receptor positive NK cells and subsequently compare calcium flux on NK cell in the absence and presence of inhibitory signaling (Fig. 4D). Using this setting, we analysed the capacity of three commonly expressed inhibitory receptors, Ly49A, Ly49G2 and NKG2A, to suppress calcium flux induced by the activating receptor NK1.1. A cross-linker was chosen that would cross-link both NK1.1 and the inhibitory receptors in the same setting. Using this assay, we found that all three receptors were able to inhibit ITAM-mediated NK cell activation. In addition, there was no difference in inhibitory capacity of these receptors in c-Abl-deficient NK cells (Fig. 4E), suggesting that c-Abl does not participate in inhibitory signaling in murine NK cells.
Discussion
In the present study, we created mice lacking the c-Abl nonreceptor tyrosine kinase specifically in NK cells to test if c-Abl played an intrinsic role in NK cell education and NK cell function. One starting point for our study was the demonstration that c-Abl was part of the inhibitory signaling cascade in human NK cells, providing a mechanisms for direct disarming of NK cells by means of phosphorylation of the adaptor protein Crk [21]. Because NK cell education is directly controlled by inhibitory receptor signaling, we hypothesized that NK cell education would be perturbed in mice lacking c-Abl specifically in NK cells. In contrast a previous study in the mouse did not find a role for c-Abl in NK cell function [19], and the situation thus seemed unclear.
As expected from its use in many conditional gene deletion models, heterozygous expression of the Cre transgene in NKp46+ cells was sufficient to mediate deletion of the floxed c-Abl gene in NK cells without affecting the genomic structure in T cells. This deletion was accompanied by an NK cell-specific lack of c-Abl transcripts. If c-Abl would have played a role in NK cell education, we thus envisaged reduced function [27, 28] and perturbed inhibitory receptor expression patterns in c-Abl-deficient NK cells [10, 11, 28]. We did not observe any such changes and concluded that c-Abl was dispensable for NK cell education MHC class I molecules, at least as measured by these parameters.
To substantiate our results, we diresctly tested if c-Abl was involved in inhibitory signaling in mouse NK cells, as suggested for human NK cells [21]. To quantify inhibition, the readout for NK cell activation must reflect NK cell activation in a quantitative way and should also be measured as close to the activation event as possible. The release of the Ca2+ from intracellular stores represents an early event in NK cell activation, which rapidly conveys signals that translates into effector functions, such as granule release and translation of cytokine genes [29]. Increase in intracellular Ca2+ can be quantified by flow cytometry by means of the conversion of the dye Fluo-4 from a non-fluorescent form to a fluorescent form [30], allowing quantifying measures of NK cell activation in real time. By adding fluorescently labelled antibodies against both activating and inhibitory antibodies to the cells before crosslinking, NK cell inhibition of Ca2+ flux via Ly49 or NKG2A receptors could potentially be determined. This assay turned out to be surprisingly robust (Ganesan et al., manuscript in preparation), allowing us to conclude that c-Abl deficiency was dispensable for NK cell inhibition via the receptors Ly49A, Ly49G2 and NKG2A. This is the first report to our knowledge in which an assay to directly quantify the inhibitory input on NK cell activation is described.
In the model proposed by Peterson and Long, signaling via inhibitory KIR receptors would lead to inhibition via, on one hand, dephosphorylation of Vav and on the other hand by c-Abl-mediated disruption of a protein complex consisting of Crk, c-Cbl and C3G, which is associated with NK cell activation [21]. We failed to provide evidence for an inhibitory role of c-Abl in NK cell activation, but instead found that c-Abl-deficiency led to slightly enhanced production of IFN-γ and CD107 expression triggered by crosslinking of two ITAM-encoded receptors. Our data thus suggest that c-Abl might downregulate NK cell function also in mouse NK cells, but not in the context of inhibitory receptor signaling, but as part of the activating receptor signaling itself. We did not see a reduction in Ca2+ flux after ITAM-mediated activation of c-Abl-deficient NK cells, suggesting that the dampening effect of c-Abl may lie downstream of calcium signaling. However, because the enhancing effect by c-Abl deficiency on IFN-γ secretion and degranulation is small, it is possible that proximal effects, which would include Ca2+ response, may be affected but are masked by the technical variability of the Ca2+ flux assay. Further work will be required to dissect at which level and via which molecular pathways c-Abl may downregulate NK cell effector responses downstream of ITAM signaling. In any case, we conclude that our data failed to provide any role for the c-Abl nonreceptor tyrosine kinase in inhibitory signaling of mouse NK cells, and consequently it did not play a role in NK cell education driven by these inhibitory receptors. Thus, inhibitory receptor signaling downstream of KIR and Ly49 may be differently regulated, or at least that the role of c-Abl in these two pathway differs between human and mouse NK cells.
Acknowledgments
We thank the staff of animal facility at Karolinska Institutet, Huddinge campus for taking care of the experimental animals. We also thank everyone at HERM for useful discussions in the completion of the project. This work was supported by grants to PH from the Swedish Research Council, the Swedish Cancer Society, Karolinska Institutet and the Stockholm City Council, by grants NRSA F32 AI094905 and K08 AI03036 to DW and by U.S. PHS NIH grant NS089662 to AJK.
References
- 1.Spits H, Artis D, Colonna M, et al. Innate lymphoid cells--a proposal for uniform nomenclature. Nature reviews Immunology. 2013 Feb;13:145–9. doi: 10.1038/nri3365. [DOI] [PubMed] [Google Scholar]
- 2.Cerwenka A, Lanier LL. Natural killer cell memory in infection, inflammation and cancer. Nature reviews Immunology. 2016 Feb;16:112–23. doi: 10.1038/nri.2015.9. [DOI] [PubMed] [Google Scholar]
- 3.Lanier LL. NK cell recognition. Annu Rev Immunol. 2005;23:225–74. doi: 10.1146/annurev.immunol.23.021704.115526. [DOI] [PubMed] [Google Scholar]
- 4.Parham P, Moffett A. Variable NK cell receptors and their MHC class I ligands in immunity, reproduction and human evolution. Nature reviews Immunology. 2013 Feb;13:133–44. doi: 10.1038/nri3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kadri N, Wagner AK, Ganesan S, et al. Dynamic Regulation of NK Cell Responsiveness. Curr Top Microbiol Immunol. 2016;395:95–114. doi: 10.1007/82_2015_485. [DOI] [PubMed] [Google Scholar]
- 6.Ljunggren HG, Karre K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol Today. 1990 Jul;11:237–44. doi: 10.1016/0167-5699(90)90097-s. [DOI] [PubMed] [Google Scholar]
- 7.Hoglund P, Brodin P. Current perspectives of natural killer cell education by MHC class I molecules. Nature reviews Immunology. 2010 Oct;10:724–34. doi: 10.1038/nri2835. [DOI] [PubMed] [Google Scholar]
- 8.Johansson S, Johansson M, Rosmaraki E, et al. Natural killer cell education in mice with single or multiple major histocompatibility complex class I molecules. The Journal of experimental medicine. 2005 Apr 4;201:1145–55. doi: 10.1084/jem.20050167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brodin P, Karre K, Hoglund P. NK cell education: not an on-off switch but a tunable rheostat. Trends Immunol. 2009 Mar 10; doi: 10.1016/j.it.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 10.Gumbleton M, Vivier E, Kerr WG. SHIP1 intrinsically regulates NK cell signaling and education, resulting in tolerance of an MHC class I-mismatched bone marrow graft in mice. J Immunol. 2015 Mar 15;194:2847–54. doi: 10.4049/jimmunol.1402930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Viant C, Fenis A, Chicanne G, Payrastre B, Ugolini S, Vivier E. SHP-1-mediated inhibitory signals promote responsiveness and anti-tumour functions of natural killer cells. Nature communications. 2014 Oct 30;5:5108. doi: 10.1038/ncomms6108. [DOI] [PubMed] [Google Scholar]
- 12.Vivier E, Nunes JA, Vely F. Natural killer cell signaling pathways. Science. 2004 Nov 26;306:1517–9. doi: 10.1126/science.1103478. [DOI] [PubMed] [Google Scholar]
- 13.Stebbins CC, Watzl C, Billadeau DD, Leibson PJ, Burshtyn DN, Long EO. Vav1 dephosphorylation by the tyrosine phosphatase SHP-1 as a mechanism for inhibition of cellular cytotoxicity. Molecular and cellular biology. 2003 Sep;23:6291–9. doi: 10.1128/MCB.23.17.6291-6299.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang JW, Howson JM, Ghansah T, et al. Influence of SHIP on the NK repertoire and allogeneic bone marrow transplantation. Science. 2002 Mar 15;295:2094–7. doi: 10.1126/science.1068438. [DOI] [PubMed] [Google Scholar]
- 15.Tybulewicz VL, Crawford CE, Jackson PK, Bronson RT, Mulligan RC. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 1991 Jun 28;65:1153–63. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- 16.Gu JJ, Zhang N, He YW, Koleske AJ, Pendergast AM. Defective T cell development and function in the absence of Abelson kinases. J Immunol. 2007 Dec 1;179:7334–43. doi: 10.4049/jimmunol.179.11.7334. [DOI] [PubMed] [Google Scholar]
- 17.Zipfel PA, Zhang W, Quiroz M, Pendergast AM. Requirement for Abl kinases in T cell receptor signaling. Curr Biol. 2004 Jul 27;14:1222–31. doi: 10.1016/j.cub.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 18.Huang Y, Comiskey EO, Dupree RS, Li S, Koleske AJ, Burkhardt JK. The c-Abl tyrosine kinase regulates actin remodeling at the immune synapse. Blood. 2008 Jul 01;112:111–9. doi: 10.1182/blood-2007-10-118232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Colucci F, Soudais C, Rosmaraki E, Vanes L, Tybulewicz VL, Di Santo JP. Dissecting NK cell development using a novel alymphoid mouse model: investigating the role of the c-abl proto-oncogene in murine NK cell differentiation. J Immunol. 1999 Mar 1;162:2761–5. [PubMed] [Google Scholar]
- 20.Holcomb M, Rufini A, Barila D, Klemke RL. Deregulation of proteasome function induces Abl-mediated cell death by uncoupling p130CAS and c-CrkII. The Journal of biological chemistry. 2006 Feb 3;281:2430–40. doi: 10.1074/jbc.M508454200. [DOI] [PubMed] [Google Scholar]
- 21.Peterson ME, Long EO. Inhibitory receptor signaling via tyrosine phosphorylation of the adaptor Crk. Immunity. 2008 Oct;29:578–88. doi: 10.1016/j.immuni.2008.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Narni-Mancinelli E, Chaix J, Fenis A, et al. Fate mapping analysis of lymphoid cells expressing the NKp46 cell surface receptor. Proceedings of the National Academy of Sciences of the United States of America. 2011 Nov 08;108:18324–9. doi: 10.1073/pnas.1112064108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moresco EM, Donaldson S, Williamson A, Koleske AJ. Integrin-mediated dendrite branch maintenance requires Abelson (Abl) family kinases. J Neurosci. 2005 Jun 29;25:6105–18. doi: 10.1523/JNEUROSCI.1432-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wetzel DM, McMahon-Pratt D, Koleske AJ. The Abl and Arg kinases mediate distinct modes of phagocytosis and are required for maximal Leishmania infection. Molecular and cellular biology. 2012 Aug;32:3176–86. doi: 10.1128/MCB.00086-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chiossone L, Vitale C, Cottalasso F, et al. Molecular analysis of the methylprednisolone-mediated inhibition of NK-cell function: evidence for different susceptibility of IL-2- versus IL-15-activated NK cells. Blood. 2007 May 1;109:3767–75. doi: 10.1182/blood-2006-07-037846. [DOI] [PubMed] [Google Scholar]
- 26.Brodin P, Lakshmikanth T, Johansson S, Karre K, Hoglund P. The strength of inhibitory input during education quantitatively tunes the functional responsiveness of individual natural killer cells. Blood. 2009 Mar 12;113:2434–41. doi: 10.1182/blood-2008-05-156836. [DOI] [PubMed] [Google Scholar]
- 27.Sternberg-Simon M, Brodin P, Pickman Y, et al. Natural killer cell inhibitory receptor expression in humans and mice: a closer look. Frontiers in immunology. 2013;4:65. doi: 10.3389/fimmu.2013.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brodin P, Lakshmikanth T, Karre K, Hoglund P. Skewing of the NK Cell Repertoire by MHC Class I via Quantitatively Controlled Enrichment and Contraction of Specific Ly49 Subsets. J Immunol. 2012 Mar 1;188:2218–26. doi: 10.4049/jimmunol.1102801. [DOI] [PubMed] [Google Scholar]
- 29.Oh-hora M, Rao A. Calcium signaling in lymphocytes. Current opinion in immunology. 2008 Jun;20:250–8. doi: 10.1016/j.coi.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paredes RM, Etzler JC, Watts LT, Zheng W, Lechleiter JD. Chemical calcium indicators. Methods. 2008 Nov;46:143–51. doi: 10.1016/j.ymeth.2008.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]