

Abstract
The advent of human-induced pluripotent stem cell (hiPSC) technology has provided a unique opportunity to establish cellular models of disease from individual patients, and to study the effects of the underlying genetic aberrations upon multiple different cell types, many of which would not normally be accessible. Combining this with recent advances in genome editing techniques such as the clustered regularly interspaced short palindromic repeat (CRISPR) system has provided an ability to repair putative causative alleles in patient lines, or introduce disease alleles into a healthy “WT” cell line. This has enabled analysis of isogenic cell pairs that differ in a single genetic change, which allows a thorough assessment of the molecular and cellular phenotypes that result from this abnormality. Importantly, this establishes the true causative lesion, which is often impossible to ascertain from human genetic studies alone. These isogenic cell lines can be used not only to understand the cellular consequences of disease mutations, but also to perform high throughput genetic and pharmacological screens to both understand the underlying pathological mechanisms and to develop novel therapeutic agents to prevent or treat such diseases. In the future, optimising and developing such genetic manipulation technologies may facilitate the provision of cellular or molecular gene therapies, to intervene and ultimately cure many debilitating genetic disorders.
Introduction
Current genetic models of disease
Thousands of human diseases are known to have a genetic component, although the penetrance of this effect and the contribution of environmental influences are highly variable. Recent advances in genotyping and DNA sequencing have facilitated the studies of familial inheritance, de novo mutations (Deciphering Developmental Disorders 2015; Wright et al. 2015) and numerous genome-wide association studies (GWAS) (Visscher et al. 2012), which have begun to identify the genetic loci underlying many of these diseases. However, despite such advances in human genetic analysis, unravelling the causative lesions, understanding the underlying molecular and cellular mechanisms and developing ways to prevent or treat such diseases still require experimental models (Nishizaki and Boyle 2016).
The evolutionary conservation of mammalian genomes, especially in protein coding sequence, has enabled the use of many animal models such as mice, rats and non-human primates for studying the effects of genetic lesions upon molecular, cellular, physiological and behavioural phenotypes. This has led to many important insights into disease biology, and their importance in such studies is undeniable. Despite such conservation of function, given the last common ancestor of human and mouse was around 100 million years ago (Mouse Genome Sequencing Consortium et al. 2002), it is unsurprising that there are also differences between these organisms. Around 20% of genes in humans lack an identifiable one-to-one orthologue in mouse (Mouse Genome Sequencing Consortium et al. 2002), and the number of paralogs within an organism is often different, many of which have diverged to provide subtly different functions (Gabaldon and Koonin 2013). Equally, even apparently orthologous genes can play different roles, such as in the case of TDP1, which shows a different subcellular localisation in humans and mice, and mutations in which are linked to the SCAN1 disorder in humans, but lack a clear phenotype in the mouse (Gharib and Robinson-Rechavi 2011). Additionally, there will clearly always be certain differences inherent to a particular species due to their evolutionary adaptation, for instance in cardiac or brain function between human and mouse, making it impossible to study some human-specific phenotypes in animal models.
One of the surprises of the human genome project (Lander et al. 2001) was that only a relatively small proportion of the genome is protein coding (current estimates are around 1.2%) (Pruitt et al. 2009). The remainder of the sequence contains many repetitive sequences and transposon remnants, although a further 3–10% of the human genome displays evidence of evolutionary conservation, implying its functionality (Lunter et al. 2006). There is clearly a role for at least a proportion of this non-coding sequence in regulation of gene expression. In fact, more than 95% of disease-associated single nucleotide polymorphisms (SNPs) lie within the non-coding genome (Maurano et al. 2012). Importantly, such SNPs may be functionally relevant, since they are enriched within enhancer regions (marked by DNAse hypersensitivity) specific for the disease-associated tissue (Maurano et al. 2012), and are often associated with changes in neighbouring gene expression (Degner et al. 2012). It is also beginning to become apparent that such non-coding changes can result in phenotypic effects, and be causative in certain diseases (Soldner et al. 2016). In the context of disease modelling, such sequences are much more poorly conserved between organisms than protein coding sequences, often making it impossible to identify the orthologous region in other species. In this situation, developing a human model of disease becomes even more relevant.
Primary cell cultures from human patients are an invaluable resource to study the molecular and cellular effects of particular mutations, but there are many limitations to this strategy, not least in the inaccessibility of certain tissues, for instance the brain. Even if the tissue is accessible, such cells are often challenging to culture, and cannot be maintained for extended periods of time, making genetic engineering difficult. Equally, many primary cultures consist of heterogeneous cell populations that are not necessarily consistent between samples, often complicating analysis. Although many immortalised cell lines also exist, these necessarily contain genetic aberrations that enable their continued culture, and therefore do not represent a highly physiological model of disease.
The generation of human embryonic stem cell (hESC) lines (Shamblott et al. 1998; Thomson et al. 1998) opened up the exciting possibility of using these pluripotent stem cells to study the function of differentiated derivative cell types. However, due to the technical and ethical difficulties, it is not feasible to produce a large number of such lines or derive them from patients with diseases, limiting their use to studies of normal cellular function, or to introduction of known engineered genetic changes.
Induced pluripotent stem cells (iPSCs) for disease modelling
iPSC technology
The advent of induced pluripotent stem cell (iPSC) technology (Takahashi et al. 2007; Takahashi and Yamanaka 2006) has revolutionised many fields, notably those of disease modelling and cellular therapeutics due to our ability to generate such pluripotent stem cells from essentially any human, including those with disease (Avior et al. 2016). Somatic cells can be reprogrammed to a pluripotent stem cell state similar to that present in very early embryogenesis through transient expression of four transcription factors (Oct4, Sox2, Klf4 and c-Myc) (Takahashi et al. 2007; Takahashi and Yamanaka 2006). Importantly, such cells are diploid and karyotypically normal, can self-renew for many cell divisions and can be differentiated into a broad range of different cell types. These characteristics lend themselves to the study of development and cellular function both in normal and disease states, and also allow large numbers of cells to be produced for high throughput genetic and drug screening as well as cell therapy. This has led to the inception of several large-scale initiatives for deriving iPSCs from thousands of normal and diseased patients (California Institute for Regenerative Medicine (CIRM), Stem Cells for Biological Assays of Novel Drugs and Predictive Toxicology (StemBANCC) (Morrison et al. 2015) and the Human-induced Pluripotent Stem Cell initiative (HiPSCi) (Streeter et al. 2016)). Cell lines have been thoroughly characterised by for example DNA sequencing, SNP genotyping, RNA sequencing and DNA methylation analysis (Soares et al. 2014) and can be accessed through cell banks across the world (such as the European Collection of Cell Cultures (ECACC), the European Bank for induced pluripotent Stem Cells (EBiSC) and the Coriell biorepository). These cell lines have been derived from individuals with a variety of monogenic and polygenic disorders, and provide an invaluable resource for studying genetic contributions to human disease. They can be used to create personalised models of disease, and understand the molecular and cellular phenotypes underlying their pathogenesis. Interestingly, since cells are reprogrammed to a very early stage of development, they can be used to monitor both developmental or differentiation defects as well as the temporal sequence of events in the early stages of disease progression.
Considerations for iPSC disease models
When considering use of iPSCs as a disease model, there are many important considerations; whether the disease is monogenic or polygenic, the penetrance of the mutation, the age of onset, whether differentiation into an appropriate cell type is possible, and if there is an appropriate phenotypic readout at a molecular or cellular level.
Whilst the majority of genetic diseases are due to a small contribution from a large number of genes, such polygenic disorders are inherently more difficult to study than monogenic diseases, since typically both the penetrance and severity of the phenotype due to any single mutation are lower (Wheeler et al. 2016). This is true of any disease model, and our ability to obtain iPSCs from patients with and without a disease makes analysis of polygenic disorders such as autism (DeRosa et al. 2012) or schizophrenia (Brennand et al. 2011) more feasible. However, further genetic manipulations to prove the causal alleles (see below) become more challenging due to the larger number of genes involved, smaller phenotypic effects and the potential for epistasis between different alleles. As with most current models of such diseases, it is often simpler to study the effect of a familial form with higher penetrance and severity, to identify phenotypes that can then be recapitulated in other forms of the disease.
Equally, it is critical with any iPSC disease model to pinpoint a cell type in which the disease manifests, to be able to differentiate effectively into these cells, and to identify a molecular or cellular phenotypic readout of the disease state. Differentiation protocols are now available to efficiently generate a large variety of lineages, and many others are being developed using cocktails of small molecule inhibitors or transcription factor overexpression (Cohen and Melton 2011; Mertens et al. 2016; Murry and Keller 2008). Although such protocols often result in a mixed population, purification of the desired cells by for example fluorescence-activated cell sorting (FACS) using an appropriate marker or reporter gene can be used to enrich for the population of interest (Horikiri et al. 2017; Wu et al. 2016a, b). Perhaps more critical to the success of any cellular disease model is the identification of a molecular or cellular phenotype that correlates with the disease state. In many cases, this can be identified through global gene expression profiling of patient and control samples (at the RNA or protein level), and identification of a profile of gene expression changes that correlate with disease. Alternatively, other cellular phenotypes can be employed such as functional readouts of cell activity (e.g. electrophysiological measurements of neurons, activity of cardiac muscle or response of macrophages to pathogen stimulation), more generic cellular features such as cell shape, subcellular localisation of particular marker genes, endocytic trafficking, or cellular responses to their environment (e.g. secretion or response to signals, sensitivity to drugs or other cellular stresses). In some cases, such as mutations in SCN5A that are linked to cardiac arrhythmia and long QT syndrome, these phenotypes are predictable and directly related to the disease (Davis et al. 2012). However, in other diseases such as autism spectrum disorders (ASD) (DeRosa et al. 2012) or schizophrenia (Brennand et al. 2011) which are classified by complex behavioural phenotypes, how well these correlate to any underlying molecular or cellular changes, and to what extent these are causative in the disease are still largely unexplored.
Another important consideration with the use of iPSCs in disease modelling is that these cells and their differentiated derivatives often resemble those of foetal origin (Hrvatin et al. 2014), and therefore the age of onset of any disease becomes relevant. Indeed, iPSC-derived neurons initially differentiate into an immature state and can require months in culture before they become electrophysiologically active. This complicates analysis of diseases such as neurodegeneration which only show effects late in life. Several strategies exist to circumvent this issue, at least to some extent. Often, rare, early-onset, familial mutations are associated with many normally polygenic late-onset diseases, and these can be useful models to study phenotypes associated with such diseases in general. One example of this is a triplication of a large region including the SNCA locus that leads to an early-onset Parkinson’s disease phenotype (Devine et al. 2011). iPSC-derived dopaminergic neurons derived from these patients show molecular phenotypes characteristic of the disease, suggesting that such pathological events can be detected and monitored (Chung et al. 2013). An alternative strategy is to accelerate ageing or disease progression using stressors such as rotenone, MG-132 or concanamycin A (Cooper et al. 2012; Nguyen et al. 2011), or through expression of Progerin, a truncated form of lamin A that is associated with Hutchinson–Gilford progeria syndrome, a premature ageing disorder (Miller et al. 2013). Whilst Progerin expression has been shown to accelerate cellular markers of ageing such as DNA damage and heterochromatic chromatin modifications (Miller et al. 2013), it is still unclear to what extent such treatments fully recapitulate the effects of old age.
Importantly, it is perhaps unsurprising that even with late-onset diseases, there are pathogenic changes occurring at an early point in disease progression that are detectable in iPS models, and can be reverted by pharmacological intervention (reviewed in (Avior et al. 2016)). Arguably, these early changes are more critical in terms of understanding and treating the disease. Studying such effects would facilitate discovery of biomarkers that identify those patients at risk and allow development of strategies to enable early, targeted intervention to prevent the disease. This is particularly important in situations such as neurodegeneration where, by the time patients present with the disease, they often have irreparable damage such as the loss of neurons, and for whom therapeutic intervention at this late stage may not be possible.
Limitations and developments
Whilst the benefits of iPSC technology are undeniable, there are some limitations in their use for modelling of certain disease states. Such in vitro models have immense power in terms of scalability, and being able to apply techniques such as high throughput genetic or pharmacological screening that would not be possible or be technically difficult in an in vivo setting (Fig. 1). However, they are limited in their ability to recapitulate complex tissue architecture both in terms of the complexity of cell types as well as their spatial organisation, making analysis of many physiological or system-level phenotypes challenging. Highly defined co-culture systems can be beneficial in some situations, for instance where the effects are non-cell autonomous, or rely on cell–cell signalling. This has been successfully applied to modelling of the effects of SOD1 mutation in glial cells on motor neuron survival in cells derived from ALS patients (Di Giorgio et al. 2007, 2008).
Fig. 1.
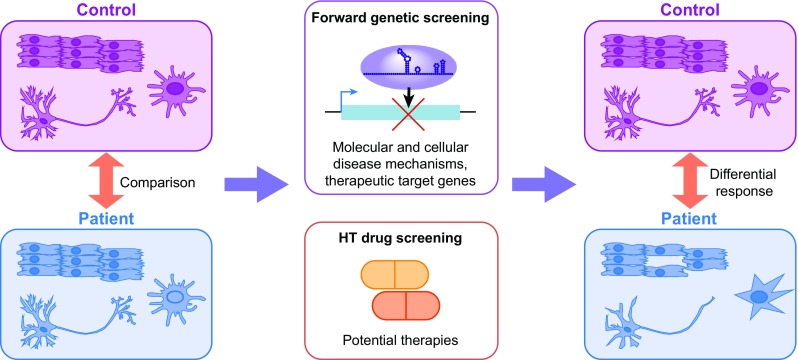
Application of iPS models of disease to high throughput screening. Cells derived from patients with disease and healthy controls can be used to generate disease-relevant cell types, which can be phenotypically compared with each other. Such cells can be generated in sufficient numbers to be able to perform whole genome genetic screens to identify molecular and cellular mechanisms of disease and therapeutic targets, and also for high throughput drug screening to identify compounds that may be able to revert the disease phenotype. Differences between patient-derived and control cells can be used to identify potential therapeutic targets or agents
Exciting developments in terms of three-dimensional culture systems such as intestinal (Dekkers et al. 2013; Schwank et al. 2013) or cerebral (Lancaster et al. 2013) organoids allow analysis of cell–cell interactions in a more complex mixture of cell types with some underlying tissue architecture (Huch and Koo 2015; Lancaster and; Knoblich 2014; Passier et al. 2016). Such systems have been exploited to uncover cellular phenotypes underlying microcephaly (Lancaster et al. 2013) and cystic fibrosis (Dekkers et al. 2013; Schwank et al. 2013), respectively. Although progress is being made in organ reconstruction of structures such as the integumentary system is being made (Takagi et al. 2016), it is unlikely that very complex tissues will be able to be modelled successfully in vitro at least in the near future.
In certain instances, diseases manifest as complex physiological or behavioural phenotypes, which cannot be recapitulated in any in vitro model. An alternative strategy is to use xenograft systems in animal models where the endogenous organ has been genetically ablated (Kobayashi et al. 2010; Lee et al. 2014; Nagashima and Matsunari 2016). These may provide an opportunity to analyse such complex physiological and system-level phenotypes with human patient-derived cells, and potentially provide a source of such organs for transplantation in the future. Whilst such systems may be important for analysis of certain diseases, it is likely that most physiological defects result from inherent underlying molecular or cellular abnormalities. Therefore, cellular phenotypes such as gene expression changes or neuronal electrophysiology will not only help to ascertain the molecular mechanisms underlying complex disease phenotypes such as Alzheimer’s disease but also provide convenient measures for measuring the effects of genetic or pharmacological intervention.
Genome editing in iPSC disease modelling
Importance of genome editing
Many human genetic diseases by their very nature would only be expected to show subtle effects on cellular behaviour, since those individuals show essentially normal differentiation, development and cellular function and only present symptoms of disease after birth, in old age, or upon exposure to environmental triggers. This alongside an inherent variability in both the iPSC derivation process and differentiation into specific cell types makes it necessary to perform comparisons of many independently derived cell lines from multiple healthy and diseased individuals in order to detect such subtle changes. This can be ameliorated by genetic engineering to introduce or repair putative causative alleles to generate isogenic cell line pairs that have identical genetic backgrounds, and differ in only a single genetic change (Fig. 2). This allows detection of subtle phenotypes that would otherwise be masked by variations in cellular phenotype due to the different genetic backgrounds of the donors.
Fig. 2.
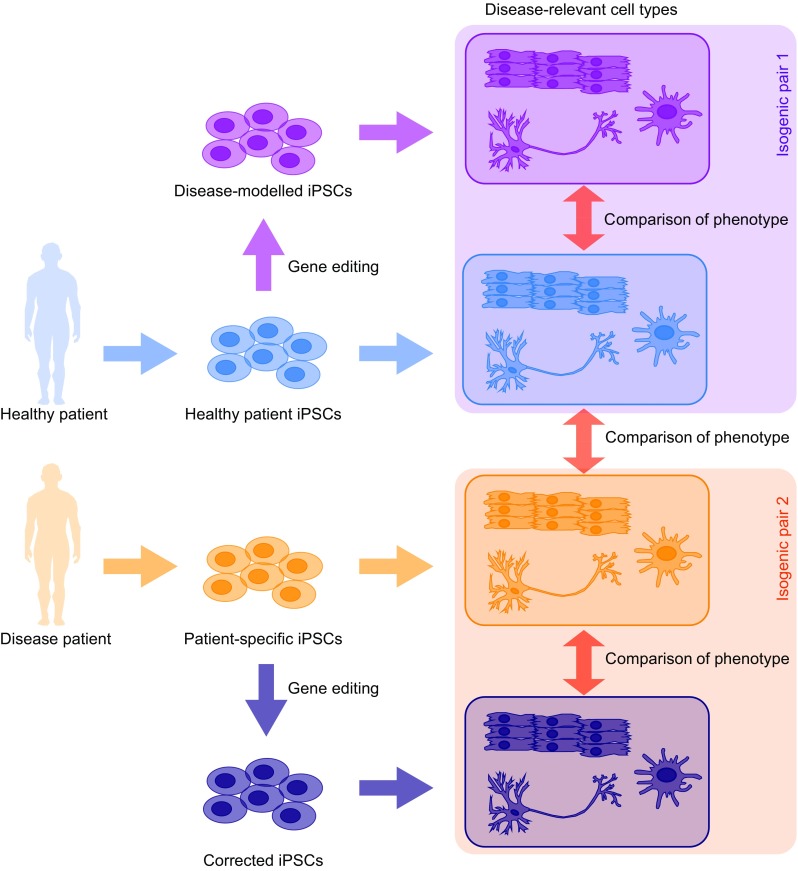
Importance of genome editing in iPS disease modelling. iPSCs can be derived from healthy (blue) and disease (orange) patients, and after differentiation into an appropriate cell type, comparison of molecular or cellular phenotypes can be made. To minimise variability due to genetic background, genome editing can be used to either correct patient-derived cells (dark blue) or to introduce putative causative lesions into cells derived from healthy individuals (purple). This leads to isogenic pairs of cell lines (purple box or orange box) that identify the true impact of the engineered change on the cellular phenotype
Importantly, such experiments also allow the identification of the causative lesion that defines the cellular phenotype and results in the disease (Nishizaki and Boyle 2016). This is not possible from comparisons of iPSC lines derived from patients with the disease and healthy controls, due to the inheritance patterns of linked SNPs. Often several SNPs are in linkage disequilibrium (LD) with each other, and as such are always inherited together (Weiss and Clark 2002) (Fig. 3). Deciphering which of these are causative is therefore a challenge, and although it is possible to infer some information from their position relative to known important genomic features (e.g. protein coding sequence, DNAse hypersensitive sites, etc.), experimentally identifying the important genetic aberration is still not trivial. Genome editing allows the attractive possibility of either repairing the putative causative lesions in patient-derived cells, or introducing them in cells derived from healthy individuals (Fig. 2), to unambiguously identify the mutations involved in the disease phenotype.
Fig. 3.

Linkage disequilibrium (LD) makes identification of causative SNPs challenging. In a typical region of the human genome, many SNPs (orange box) are in strong LD with the tag SNP (red) identified by a GWAS study. Genome editing can be used to identify the causative lesion from within this LD block. LD is measured as R-squared values between pairs of SNPs, and indicated on the heatmap
Genome editing has been used in many examples (Table 1) to create isogenic pairs of cell lines, and this has been successful in both validating the causative lesion and allowing greater sensitivity for phenotypic detection. The importance of such studies is undeniable, and the usage of such isogenic lines in iPS disease modelling will undoubtedly increase in the coming years.
Table 1.
iPSC disease models employing isogenic control lines generated by CRISPR/Cas9. The table lists a number of diseases, the mutation that was reverted or introduced, the differentiated cell type analysed and the molecular or cellular phenotypes observed
| Disease | Editing | Cell type | Phenotype | References |
|---|---|---|---|---|
| Neurological disorders | ||||
| Fragile X syndrome | Removal of triplet repeat (FMR1) | NPC neurons | DNA methylation, gene expression changes | (Boland et al. 2017; Xie et al. 2016) |
| Tetrahydrobiopterin metabolism disorder, Parkinson’s disease | Correction of dopamine synthesis (PHPS, DHPR) | DA neurons | Metabolic (dopamine, BH4), protein expression changes | (Ishikawa et al. 2016) |
| Parkinson’s disease (PD) | Correction of coding point mutations (SNCA) | Cortical neurons | Accumulation of ER-associated degradation substrates | (Chung et al. 2013; Soldner et al. 2011) |
| Correction of coding mutation (LRRK2) | Dopaminergic neurons | Gene expression changes | (Reinhardt et al. 2013) | |
| Alzheimer’s disease (AD) | Point mutations (APP, PSEN1) | Cortical neurons | Protein (Aβ) secretion | (Paquet et al. 2016) |
| Hereditary motor and sensory neuropathy with proximal dominant involvement (HMSN-P) | Correction of point mutations | Spinal motor neurons | Proteasome impairment | (Murakami et al. 2017) |
| Frontotemporal lobar degeneration tauopathy (FTLD-Tau) | Correction of intronic or exonic point mutation (MAPT) | Neurons | Accumulation and release of misfolded tau, cell death, electrical stimulation of calcium transients | (Imamura et al. 2016) |
| Huntington disease (HD) | Correction of expanded CAG repeat (HTT) | Forebrain neurons | Neural rosette formation, mitochondrial respiration, gene expression changes | (Xu et al. 2017) |
| Immunodeficiency, centromeric instability, facial abnormalities (ICF) syndrome | Knockout (DNMT3B) | iPSC | Gene expression, DNA methylation changes | (Horii et al. 2013) |
| Other diseases | ||||
| Chronic granulomatous disease (CGD) | Repair of intronic point mutation, exon 5 replacement (CYBB) | Phagocytes | Oxidative burst function (ROS release) | (Flynn et al. 2015; Sweeney et al. 2017) |
| Duchenne muscular dystrophy (DMD) | Exon skipping, frameshift, exon knock-in, deletion (Dystrophin) | Skeletal muscle | Membrane integrity, protein expression, electrical contraction (Ca overflow) changes | (Li et al. 2015; Young et al. 2016) |
| Barth syndrome | Knockout (TAZ) | Cardiomyocytes | Metabolic and structural changes in muscle, myocardial contraction defects | (Wang et al. 2014a, b) |
| Monocytic and dendritic cell immunodeficiency | Knockout (IRF8) | Dendritic cells | Differentiation, cytokine release, migration, antigen presentation | (Sontag et al. 2017) |
| β-thalassemia | Correction of coding mutation (HBB) | Erythroblast | Gene expression changes, hematopoietic differentiation, reactive oxygen species production | (Niu et al. 2016; Song et al. 2015) |
| Hemophilia A | Reversion of chromosomal inversion | Endothelin cells | Restoration of factor VIII expression, rescue of lethality in mouse xenograft model | (Park et al. 2015) |
| Severe combined immunodeficiency (SCID) | Repair of loss of function mutation (JAK3) | T-cells | Restoration of correct T-cell development | (Chang et al. 2015) |
| Sickle Cell Anaemia | Reversion of point mutation (HBB) | Erythrocytes | HBB expression | (Huang et al. 2015) |
CRISPR genome editing technology
Recent improvements in genome editing have vastly increased our ability to introduce such delicate defined mutations within the genomes of human cells. These are based on designer site-specific nucleases that introduce a double-strand break (DSB) at a desired site in the genome, which is then repaired by the cell, and can be utilised to introduce a variety of different genetic changes at this site. Most recent work focuses on the clustered, regularly interspaced short palindromic repeats (CRISPR) system and the use of the RNA-guided CRISPR-associated 9 (Cas9) endonuclease (Cho et al. 2013; Cong et al. 2013; Jinek et al. 2012, 2013; Mali et al. 2013), predominantly due to the simplicity by which this can be reprogrammed to bind to millions of sites within the genome. This system relies on a short guide RNA molecule to direct its specificity, through base pairing of its first 20 nt with the corresponding DNA sequence in the genome (Jinek et al. 2013). The only limitation to the targeting is a requirement for a protospacer adjacent motif (PAM) sequence adjacent to the target site, which is not present in the guide RNA, but is recognised by the Cas9 protein. In the case of the most widely used Streptococcus pyogenes Cas9 protein, this is NGG, which in a genome with an even base distribution and composition should occur every 8 bp. However, should this be a limitation, orthologues of Cas9 in other species have been discovered with alternative PAM requirements (Hou et al. 2013; Ran et al. 2015; Zetsche et al. 2015) and recently, protein engineering has been used to alter the PAM sequences of several Cas9 nucleases (Kleinstiver et al. 2015a, b) (as discussed elsewhere in this issue).
Introduction of a DSB into the genome allows many different types of genetic manipulation. Usually, this DNA damage is repaired by one of two major pathways, non-homologous end joining (NHEJ) that performs non-templated ligation of the free DNA ends, and homology directed repair (HDR) that utilises homologous DNA to direct a precise repair (Bibikova et al. 2003; Shrivastav et al. 2008). Both of these can be exploited for making site-specific genetic mutations. Repair through NHEJ or related pathways such as microhomology mediated end joining (MMEJ) can result in insertions or deletions (indels) of several nucleotides at the DSB that can be used for instance to introduce a frameshift in protein coding sequence, resulting in a null allele. If two DSBs are made simultaneously, the NHEJ machinery can also ligate the wrong termini together, resulting in deletions, inversions or translocations (Torres et al. 2014; Xiao et al. 2013; Park et al. 2016). In the context of more defined changes to the DNA such as introduction of SNPs, HDR pathways can be utilised by providing an excess of a desired sequence, resulting in this being used in preference to the sister chromatid as the template for repair. Introduction of mutations can be highly efficient, especially for NHEJ-based pathways, which generally predominate in most cells including human iPSCs, and can approach 80–90% in the best examples.
This efficiency, coupled with the simplicity of constructing guide RNAs from synthetic oligonucleotides, which are amenable to being produced in both multi-well plates or in larger pools of tens of thousands, has allowed both arrayed (Hultquist et al. 2016) and pooled screening strategies (Gilbert et al. 2014; Koike-Yusa et al. 2014; Konermann et al. 2015; Shalem et al. 2014; Wang et al. 2014a, b) to be conceived. This provides the exciting prospect of forward genetic screening in human patient-derived cell lines to identify potential therapeutic targets and to better understand molecular and genetic basis of disease (Fig. 1).
Genome editing technologies also have the potential in the future to be utilised as a therapeutic agent in their own right, and to repair the genetic mutations contributing to disease (Cox et al. 2015). Such reagents could not only be applied in an in vivo context, but also to repair causative lesions in patient-derived iPSCs that could subsequently be used to generate specific cell types to use as cellular therapies. Such strategies have shown significant promise in some cases, for example in the treatment of a chemically induced primate model of Parkinson’s disease through injection of autologous iPSC-derived dopaminergic neurons (Emborg et al. 2013; Hallett et al. 2015). Therapies for HIV (Tebas et al. 2014) and cancer (Fesnak et al. 2016) involving genome editing are already in clinical trials, and the next few years will likely herald exciting developments in this area of somatic gene therapy.
Strategies for genome editing in iPS models of disease
There are two main strategies for using genome editing techniques in iPSC models of disease. The first involves repairing a pre-existing, presumed causative allele from an iPSC line derived from a patient with the disease (Fig. 2, isogenic pair 1). This establishes whether this particular genetic change contributes to the disease phenotype, but does not provide any information about whether it is sufficient to cause disease. It also has the substantial benefit that the patient-derived cell line would be expected to express whatever cellular or molecular phenotype that is causing the disease, and therefore reversion of this phenotype in the edited line can be used as a readout. The second strategy involves taking an iPSC line from a healthy patient, and introducing a putatively important lesion (Fig. 2, isogenic pair 2). This is perhaps a more stringent assay, since it establishes whether this single genetic change is sufficient to cause the disease phenotype, since it removes it from the genetic background of the diseased individual. However, if no effect is seen on the molecular or cellular phenotype of interest, it is not possible to infer whether this allele contributes to the disease. Equally, the effect of genetic background can be investigated in this manner by introducing putative causative lesions into a panel of “WT” iPSCs established from healthy donors from diverse genetic ancestries.
These strategies are clearly complementary to each other, and can provide information on potential epistatic interactions with other alleles present in specific genetic backgrounds. It is also worth considering that modification of a “WT” cell line is often simpler technically, since both the genome editing and subsequent downstream differentiation and analysis can be optimised for a particular cell type. Additionally, the “WT” cells can be thoroughly characterised beforehand, and different genetically edited lines involved in the same disease can be directly compared. In the case of patient-derived cells, each cell line will behave somewhat differently, and therefore it is often more difficult to perform such manipulations, especially at higher throughput.
When designing such an experiment, it is also important to consider the inherent variability between patients as well as in the processes of both reprogramming of somatic cells to iPSCs and differentiation into particular cell types. It has been suggested that at least part of this heterogeneity results from an “epigenetic memory” of the DNA methylation signature of the cell type from which the iPSCs were derived (Kim et al. 2010), which may impact upon the phenotypes observed, or the ability to differentiate into particular lineages (Bar-Nur et al. 2011). Reassuringly however, recent studies looking at the origin of heterogeneity within 25 iPSC lines have demonstrated that, at least at the transcriptional level, the majority of variation is due to genetic background as opposed to any epigenetic contribution (Rouhani et al. 2014). In order to account for such inherent variability, it is necessary to analyse multiple (typically at least three) patients, each with independent iPSC derivations, clonally derived lines and differentiation experiment, which rapidly increases the number of samples that need to be analysed (Fig. 2). The number of each that are required depends on multiple factors including the magnitude of the phenotype and the degree of variability within a particular differentiation protocol, but it is unwise to rely at any stage on the results from a single experiment.
Whilst genome editing can be very effectively used to reduce the variability between patients, the process of genome editing itself can introduce artefacts from both off-target mutagenesis (Cradick et al. 2013; Fu et al. 2013; Veres et al. 2014) and clonal variability within a particular iPSC line. One method for controlling the variability introduced by the genome editing process is by re-introducing the disease mutation in the genetically corrected patient line. Similarly, introducing mutations into a consistent “WT” line can reduce the variability between patients and during iPSC derivation. The experimental strategy will depend on the specific question that is being addressed, but these criteria should be taken into account in order to maximise sensitivity for phenotypic changes, and minimise workload.
Genome editing methods
Numerous strategies exist for genome editing using CRISPR, predominantly differing in the method of delivery of the Cas9 and guide RNA components. Inducible Cas9 transgenes (Gonzalez et al. 2014), DNA plasmids (Ding et al. 2013a, b; Kwart et al. 2017; Merkert et al. 2014; Miyaoka et al. 2016, 2014; Yang et al. 2013) or Cas9 ribonucleoprotein (RNP) complexes (Kim et al. 2014; Liang et al. 2015; Lin et al. 2014; Richardson et al. 2016) have all been successful in introducing targeted mutations in hiPS cells (reviewed in more detail in Merkert and Martin 2016; Santos et al. 2016). The choice of system depends on multiple factors including the importance of off-targeting, the type of repair necessary (single nucleotide changes or indel mutations) and prior knowledge of optimal delivery methods into a particular cell line. However, our preference is for delivery of RNPs composed of recombinant, bacterially expressed Cas9 protein and synthetic RNA oligos corresponding to the CRISPR RNA (crRNA) and trans-activating CRISPR RNA (tracrRNA) components of the system. This has many advantages over other systems since the RNP complex is immediately active, when the concentration of any donor HDR template is the highest and is rapidly degraded over a period of around 12 h (Kim et al. 2014), reducing the potential for off-target mutagenesis and re-targeting after successful HDR. The lack of DNA plasmids also eliminates any chances of non-specific integration of DNA vectors into the genome.
Another important consideration is to minimise off-target mutagenesis, the extent of which is still debatable in the field, and likely depends on the exact system used to introduce the CRISPR reagents (Cradick et al. 2013; Fu et al. 2013; Veres et al. 2014). What is clear is that some mismatches between the guide RNA and target DNA can be tolerated, and the degree to which they impact on endonuclease activity depends on their position within the sequence, with those nucleotides closer to the PAM sequence playing a more critical role in target recognition (Hsu et al. 2013). Careful design of crRNA target sites to avoid off-targets of less than 3 mismatches can be readily achieved using a variety of online tools and will certainly minimise any potential problems. Methods for improving specificity have been developed using either pairs of Cas9 enzymes each of which is unable to generate a DSB alone (Guilinger et al. 2014; Ran et al. 2013; Tsai et al. 2014), truncated guide RNAs (Fu et al. 2014) or protein engineering of Cas9 to improve specificity (Kleinstiver et al. 2016; Slaymaker et al. 2016). Some of these strategies including the double nickase approach (Ran et al. 2013) have been successfully used in hiPS cells (Eggenschwiler et al. 2016; Wu et al. 2016b), although these systems will never remove the potential for off-target mutations completely. Therefore at least in the case of disease models, where a small number of lines are produced, it is also possible to sequence the putative off-target sites (or even the whole genome), generate cell lines with independent guides each of which would have a different set of off-targets, or perform another round of genome engineering to repair the introduced genetic change.
Any such manipulations generate a mixed population of cells with different genotypes, and a large proportion of the workload is in clonal growth of cells and genotyping them. Thus, strategies for rapid, scalable and efficient genotyping are paramount to the success of any genome editing experiment. Numerous techniques can be employed, either at the DNA, RNA, protein or functional level. Often selection strategies begin with PCR amplification of regions around the sgRNA target site, and subsequent analysis by restriction enzyme polymorphisms, digital PCR (Mock et al. 2016) and Sanger (Brinkman et al. 2014) or high throughput sequencing (Bell et al. 2014). However, the choice of strategy will largely depend on the efficiency of the process, and the class of mutant introduced.
Gene knockouts
Different classes of allele vary in terms of the ease by which they can be generated, largely due to the bias of repair pathways towards NHEJ, and the resulting indel mutations. Gene knockouts are perhaps the simplest to produce, since such indel mutations can be used to introduce frameshifts into protein coding sequence (Fig. 4b), making the efficiency of mutagenesis very high. It is important to carefully consider the gene structure when designing such a strategy, since alternative promoters, alternative splicing and alternative polyadenylation signals are all critical factors to consider.
Fig. 4.
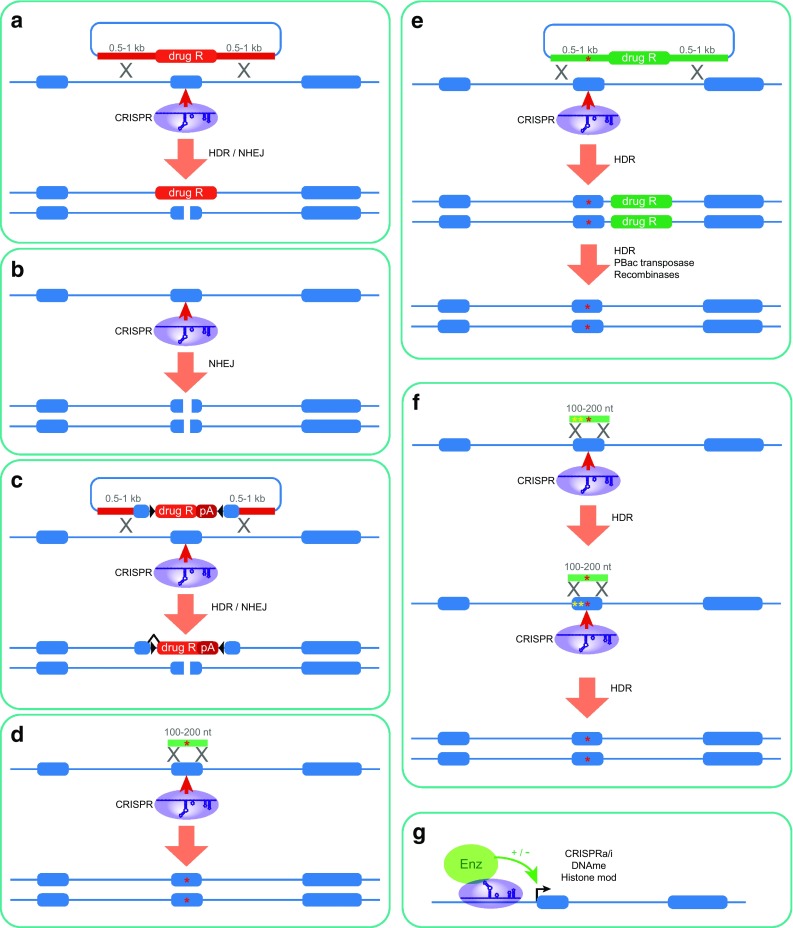
Strategies for genome editing using CRISPR/Cas9. a Gene knockout—CRISPR-enhanced HDR can be employed to replace a critical exon with a selectable drug resistance cassette (drug R), on one allele, relying on NHEJ-dependent indels to disrupt the other allele. b Gene knockout—A CRISPR-induced DSB can be used to efficiently introduce indels on both alleles. c Conditional knockout by inversion (COIN)—CRISPR-enhanced HDR can be used to introduce a Cre-recombinase invertible cassette, flanked by loxP sites (black triangles) into an artificial intron. This contains a splice acceptor site followed by a transcriptional termination signal (pA), so in one orientation it causes premature termination and mutation of the gene. In the opposite orientation, splicing occurs around the cassette, allowing the normal gene product to be produced from this allele. The second allele is disrupted by NHEJ-induced indels as in (a). d SNP introduction—A CRISPR-induced DSB is used to enhance HDR with a 100–200 nt ssDNA oligonucleotide repair template (green) to introduce small defined changes. e Scarless SNP introduction—A selectable marker cassette (drug R, green) is introduced into an intron or non-functional region along with the SNP of interest, and subsequently removed by a further round of HDR, or the piggyBac transposase. f Scarless SNP introduction—A SNP of interest is introduced as in D along with second site mutations necessary to prevent re-cleavage by the Cas9 enzyme. A subsequent second round of editing in a similar manner corrects the secondary mutations to leave only the SNP of interest. g Epigenetic editing—Catalytically dead Cas9 protein is used to recruit a variety of enzymatic activities (Enz, green circle) to specific sites, leading to transcriptional modulation (both positively and negatively), DNA or histone modifications such as DNA methylation, histone acetylation, methylation or phosphorylation, or cytosine deamination
It is important that frameshifts are not made too early in the protein coding sequence, since if the open reading frame (ORF) is too short, reinitiation at a downstream start codon can occur (Zimmer et al. 1994). The efficiency of such reinitiation is dependent on the length of the upstream ORF, and is highly inefficient if this is greater than 30–40 amino acids (Luukkonen et al. 1995). Equally, frameshifts should not be introduced too late in the coding sequence. Normally premature stop codons are recognised by the cell and trigger the process of nonsense mediated decay (NMD), which prevents expression of the entire protein through mRNA degradation (Hug et al. 2016). However, if the premature stop codon is present in the final exon, this process does not occur, and a slightly truncated peptide will be produced, likely with at least partial functionality (Hug et al. 2016). In order to obtain a complete knockout allele, it is therefore optimal to target a constitutive exon at least 30–40 amino acids into the protein coding sequence that is not in the final exon. Even in this situation, a complete knockout cannot be guaranteed, since NMD does not always act efficiently, and aberrant splicing can occur to reconstitute at least part of the protein function. Thus, analysis of protein expression is an important aspect to consider with such mutations.
Many other strategies exist, such as removal of constitutive exons through NHEJ-mediated deletions with pairs of guide RNAs flanking these exons (Liu et al. 2016a, b), or using a homology construct that guides precise exon deletion of one allele (Fig. 4a), coupled with indel mutations on the other. These systems can also be used to make larger deletions to recapitulate for instance copy number variants (CNVs), which could be informative in certain cases. In all cases, it is also important to consider whether the knockout of the gene will be lethal at a cellular level, in which case conditional knockout by for instance flanking constitutive exons with recombinase sites, or conditional by inversion (COIN) or FLIP strategies (Economides et al. 2013; Andersson-Rolf et al. 2017) may be beneficial (Fig. 4c). Inducible CRISPR systems can also be employed to restrict mutations to particular points in time (Bertero et al. 2016), and may be helpful in certain situations.
Single nucleotide polymorphisms
In many cases, genetic changes identified from familial inheritance studies or GWAS are single nucleotide polymorphisms (SNPs). Such mutations often do not result in complete loss of protein function, and frequently cause missense mutations in protein coding sequence, or changes at non-coding regulatory sites that may affect for instance transcription factor binding.
In order to introduce such delicate changes to the genome, it is necessary to employ the HDR pathway of DNA repair, making this process less efficient in general than simple gene knockouts (Fig. 4d). Templates for DNA repair can be supplied as either double-stranded DNA plasmids with approximately 500–1000 nt of homology either side of the introduced mutation, or more typically as chemically synthesised short single-stranded DNA oligonucleotides (ssODN) of 100–200 nt in length. The latter are simple to design and synthesise and have a comparable efficiency of HDR to longer dsDNA fragments due to the higher recombinogenic activity of ssDNA (Chen et al. 2015). Although there is substantial variability in the absolute efficiencies reported in the literature, we find similarly to others that a combination of chemically synthesised crRNA and tracrRNA, recombinant Cas9 protein and approximately 100 nt ssODN highly effective for introduction of SNPs (Kim et al. 2014; Liang et al. 2015; Lin et al. 2014; Niu et al. 2016; Richardson et al. 2016; Song et al. 2015). This obviates the need for any cloning or DNA manipulation, and all components can be purchased from commercial vendors. However, longer homology constructs have also been effectively used to introduce point mutations in other published reports (Wang et al. 2017; Yusa 2013).
In all cases, it is critical to ensure that upon correct HDR, the sgRNA is unable to guide further DSB events on the newly modified allele, since these could result in further undesirable mutagenesis through NHEJ-mediated indels. This ideally requires that the sgRNA is chosen to span the SNP of interest. Since the guide RNA can tolerate up to three mismatches whilst still retaining the ability to direct the Cas9 endonuclease to this site, it is often necessary or advisable to introduce additional base changes outside the SNP of interest to ensure this is the case. For SNPs within protein coding genes, this can be achieved by introducing silent mutations into the DNA whilst maintaining its protein coding capacity. Even in this case, there may still be a role of such synonymous mutations in protein translation efficiency (Quax et al. 2015), exonic transcription factor binding (Stergachis et al. 2013) or other as yet unknown functions. However, for those changes outside of protein coding sequence, it is usually impossible to predict the effects of such secondary mutations. This makes it necessary to either perform completely scarless mutagenesis or alternatively to create two independent alleles with different secondary mutations, which can be used to control for the effects of these mutations.
Scarless mutagenesis is possible in some cases where the SNP happens to fall within the PAM sequence, or within a few nucleotides of the 3′ end of the guide RNA, since even single-point mutations at these sites can prevent re-cleavage. However, this is not the case in many instances, and certain nucleotides are therefore inaccessible to such manipulations at least with Streptococcus pyogenes Cas9. As mentioned above, orthologues from other species (Hou et al. 2013; Ran et al. 2015; Zetsche et al. 2015) or engineered variants (Kleinstiver et al. 2015a, b) are already increasing the range of potential targets, and it is likely that in the future, most SNPs will be amenable to manipulation in this manner. Another strategy is to use a larger dsDNA donor to introduce a selectable marker cassette into a non-functional region neighbouring the SNP of interest (Fig. 4e) (Wang et al. 2017; Yusa 2013). This cassette can subsequently be removed either by site-specific recombinases, or scarlessly by a second round of homologous recombination or the piggyBac transposase (Wang et al. 2017; Yusa 2013). However, in the latter case, there are sequence requirements on where the piggyBac sequences can be integrated that may limit the effectiveness of this strategy (Yusa 2013). A third strategy involves a two-step genome editing strategy whereby in the first step the desired mutation is introduced alongside secondary mutations to prevent recutting, and subsequently, the secondary mutations are removed by a redesigned guide (or alternative Cas9 enzyme) and homology template (Fig. 4f) (Paquet et al. 2016; Kwart et al. 2017). However, these latter strategies involve considerably more complex processes, and at least two stages of clonal selection, increasing the time and cost of producing each mutation.
An additional strategy which has recently been developed is the use of Cas9 to recruit cytosine deaminase enzymes usually involved in somatic hypermutation in immune cells (such as the Apobec and AID enzymes) to edit the sequence of the genome without inducing a DSB (Hess et al. 2016; Komor et al. 2016; Nishida et al. 2016) (Fig. 4g). Whilst this could be informative in terms of screening and for certain specific mutations, its general application in disease modelling is somewhat limited, since only transitions from cytosine to thymine are possible.
Epigenome editing
In addition to genetic manipulations, such as gene knockouts or single nucleotide changes, a growing number of diseases including cancer (Salarinia et al. 2016) and neurodegenerative (Gos 2013; Jakovcevski and Akbarian 2012; Landgrave-Gomez et al. 2015) diseases can be driven by changes in the epigenome of the cell. This can be both in terms of transcriptional levels, or for instance changes in DNA methylation patterns that may have more subtle effects, e.g. to alter how the cell responds to external signals. The impact and importance of such changes can be assessed by using nuclease-deficient forms of Cas9 fused to specific chromatin or DNA modifying factors (Fig. 4g). Two point mutations in the Cas9 protein (D10A, H840A) render it catalytically inactive, whilst retaining its ability to bind to specific DNA sequences. Fusion of domains to the Cas9 protein or binding to modified guide RNA scaffolds then allow recruitment of specific enzymatic activities to desired sites in the DNA (Dominguez et al. 2016; Konermann et al. 2013) (Fig. 4g). This has been used extensively to manipulate gene expression both positively using transcriptional activation domains such as VP16, Rta, p65 or HSF1 and negatively by fusion to KRAB, and genome-wide screens using these reagents have been successfully implemented (Gilbert et al. 2014; Konermann et al. 2015). Such systems have been used to for instance manipulate alpha-synuclein levels both positively and negatively in neurons derived from patients with a triplication of the SNCA locus, allowing manipulation of SNCA levels in this disease model (Heman-Ackah et al. 2016). Equally importantly, a variety of chromatin modifying activities can also be recruited, including the p300 histone acetyltransferase (Hilton et al. 2015) or LSD1 histone demethylase (Kearns et al. 2015) to activate or inactivate enhancers, the Dnmt3 DNA methyltransferases (Amabile et al. 2016; Liu et al. 2016a, b; Vojta et al. 2016) or TET1 demethylase (Choudhury et al. 2016; Liu et al. 2016a, b; Xu et al. 2016) to add or remove DNA methylation marks, or other chromatin modifying enzymes such as G9a or SUV39H1 (Snowden et al. 2002). These reagents will be important in establishing the role and importance of such epigenetic changes in disease progression either by introducing them into healthy cells, or reverting effects observed in disease models.
Conclusions
The recent advances in DNA sequencing technologies have led to an ever increasing number of human genetic studies that have identified numerous candidate loci that are correlated with many diseases. There is therefore a pressing need for simple, robust models of disease that can be applied to understand the functionality of such genetic lesions. It is clear that the intersection of iPSC and genome editing technologies will provide powerful tools to study such diseases in a human cellular system. The use of a human system has many advantages, especially in terms of studying the majority of disease-associated mutations that do not reside within protein coding genes, and for which conservation is not sufficient to allow direct comparisons to be made in other organisms. The ability to be able to derive cells from multiple patient genotypes and the ability to obtain such cell lines from repositories around the world will provide invaluable opportunities to investigate the link between genotype and phenotype. Genome editing will form an essential component of such studies, to revert or introduce desired genetic mutations to understand their function by comparison with a fully isogenic background (Fig. 2). iPSC technologies are also amenable to higher throughput studies of multiple SNPs, identification of the causative lesions, loss- and gain-of-function genetic screens to understand disease mechanisms and identify drug targets and high throughput drug screening, which are likely to be highly informative in the coming years (Fig. 1).
One of the powers of iPSCs is that all of these strategies can also be applied to multiple different cell types, although it remains to be seen to what extent the results obtained in such isolated cell populations recapitulate the effects seen in complex human tissues. As differentiation protocols and three-dimensional cell culture techniques evolve, this can only improve our ability to use such systems to model many aspects of human disease. However, such cellular systems will always be limited in terms of assaying more complex system-level physiological and behavioural phenotypes, although xenograft systems may provide one means of achieving this in an in vivo context. One of the most important aspects of employing iPSC models of human disease is therefore in cellular and molecular phenotyping. Whilst for many diseases, such phenotypes will be known and predictable, for others this will require correlation of the normally complex physiological phenotypes seen in disease to the underlying molecular and cellular defects. Whilst challenging, this will also provide invaluable information about the pathogenic mechanisms leading to such diseases and novel avenues for therapeutic intervention.
The synergy between iPSC and genome editing technologies will no doubt provide many insights into disease mechanisms and therapeutic targets, and enable characterisation and prioritisation of genetic aberrations that cause particular diseases. This information coupled with the applications of iPSC-derived cell types for cellular therapies and CRISPR-based reagents for in vivo therapeutics offer exciting possibilities for personalised genetic medicines in the not-too-distant future.
Acknowledgements
The author would like to acknowledge the Wellcome Trust for funding, and Sarah Cooper for critical reading of the manuscript.
References
- Amabile A, Migliara A, Capasso P, Biffi M, Cittaro D, Naldini L, Lombardo A. Inheritable silencing of endogenous genes by hit-and-run targeted epigenetic editing. Cell. 2016;167(219–232):e214. doi: 10.1016/j.cell.2016.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson-Rolf A, et al. One-step generation of conditional and reversible gene knockouts. Nat Methods. 2017;14:287–289. doi: 10.1038/nmeth.4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avior Y, Sagi I, Benvenisty N. Pluripotent stem cells in disease modelling and drug discovery. Nat Rev Mol Cell Biol. 2016;17:170–182. doi: 10.1038/nrm.2015.27. [DOI] [PubMed] [Google Scholar]
- Bar-Nur O, Russ HA, Efrat S, Benvenisty N. Epigenetic memory and preferential lineage-specific differentiation in induced pluripotent stem cells derived from human pancreatic islet beta cells. Cell Stem Cell. 2011;9:17–23. doi: 10.1016/j.stem.2011.06.007. [DOI] [PubMed] [Google Scholar]
- Bell CC, Magor GW, Gillinder KR, Perkins AC. A high-throughput screening strategy for detecting CRISPR-Cas9 induced mutations using next-generation sequencing. BMC Genomics. 2014;15:1002. doi: 10.1186/1471-2164-15-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertero A, et al. Optimized inducible shRNA and CRISPR/Cas9 platforms for in vitro studies of human development using hPSCs. Development. 2016;143:4405–4418. doi: 10.1242/dev.138081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibikova M, Beumer K, Trautman JK, Carroll D. Enhancing gene targeting with designed zinc finger nucleases. Science. 2003;300:764. doi: 10.1126/science.1079512. [DOI] [PubMed] [Google Scholar]
- Boland MJ, et al. Molecular analyses of neurogenic defects in a human pluripotent stem cell model of Fragile X syndrome. Brain. 2017 doi: 10.1093/brain/aww357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennand KJ, et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 2011;473:221–225. doi: 10.1038/nature09915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman EK, Chen T, Amendola M, van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42:e168. doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CW, et al. Modeling Human Severe Combined Immunodeficiency and Correction by CRISPR/Cas9-Enhanced Gene Targeting. Cell Rep. 2015;12:1668–1677. doi: 10.1016/j.celrep.2015.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Pruett-Miller SM, Davis GD. Gene editing using ssODNs with engineered endonucleases. Methods Mol Biol. 2015;1239:251–265. doi: 10.1007/978-1-4939-1862-1_14. [DOI] [PubMed] [Google Scholar]
- Cho SW, Kim S, Kim JM, Kim JS. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol. 2013;31:230–232. doi: 10.1038/nbt.2507. [DOI] [PubMed] [Google Scholar]
- Choudhury SR, Cui Y, Lubecka K, Stefanska B, Irudayaraj J. CRISPR-dCas9 mediated TET1 targeting for selective DNA demethylation at BRCA1 promoter. Oncotarget. 2016;7:46545–46556. doi: 10.18632/oncotarget.10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CY, et al. Identification and rescue of alpha-synuclein toxicity in Parkinson patient-derived neurons. Science. 2013;342:983–987. doi: 10.1126/science.1245296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen DE, Melton D. Turning straw into gold: directing cell fate for regenerative medicine. Nat Rev Genet. 2011;12:243–252. doi: 10.1038/nrg2938. [DOI] [PubMed] [Google Scholar]
- Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper O, et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci Transl Med. 2012;4:141ra190. doi: 10.1126/scitranslmed.3003985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox DB, Platt RJ, Zhang F. Therapeutic genome editing: prospects and challenges. Nat Med. 2015;21:121–131. doi: 10.1038/nm.3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cradick TJ, Fine EJ, Antico CJ, Bao G. CRISPR/Cas9 systems targeting beta-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 2013;41:9584–9592. doi: 10.1093/nar/gkt714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RP, et al. Cardiomyocytes derived from pluripotent stem cells recapitulate electrophysiological characteristics of an overlap syndrome of cardiac sodium channel disease. Circulation. 2012;125:3079–3091. doi: 10.1161/CIRCULATIONAHA.111.066092. [DOI] [PubMed] [Google Scholar]
- Deciphering Developmental Disorders S Large-scale discovery of novel genetic causes of developmental disorders. Nature. 2015;519:223–228. doi: 10.1038/nature14135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degner JF, et al. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature. 2012;482:390–394. doi: 10.1038/nature10808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekkers JF, et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat Med. 2013;19:939–945. doi: 10.1038/nm.3201. [DOI] [PubMed] [Google Scholar]
- DeRosa BA, et al. Derivation of autism spectrum disorder-specific induced pluripotent stem cells from peripheral blood mononuclear cells. Neurosci Lett. 2012;516:9–14. doi: 10.1016/j.neulet.2012.02.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine MJ, et al. Parkinson’s disease induced pluripotent stem cells with triplication of the alpha-synuclein locus. Nat Commun. 2011;2:440. doi: 10.1038/ncomms1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat Neurosci. 2007;10:608–614. doi: 10.1038/nn1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giorgio FP, Boulting GL, Bobrowicz S, Eggan KC. Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell. 2008;3:637–648. doi: 10.1016/j.stem.2008.09.017. [DOI] [PubMed] [Google Scholar]
- Ding Q, et al. A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell. 2013;12:238–251. doi: 10.1016/j.stem.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Regan SN, Xia Y, Oostrom LA, Cowan CA, Musunuru K. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell. 2013;12:393–394. doi: 10.1016/j.stem.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez AA, Lim WA, Qi LS. Beyond editing: repurposing CRISPR-Cas9 for precision genome regulation and interrogation. Nat Rev Mol Cell Biol. 2016;17:5–15. doi: 10.1038/nrm.2015.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Economides AN, et al. Conditionals by inversion provide a universal method for the generation of conditional alleles. Proc Natl Acad Sci USA. 2013;110:E3179–E3188. doi: 10.1073/pnas.1217812110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggenschwiler R, et al. Improved bi-allelic modification of a transcriptionally silent locus in patient-derived iPSC by Cas9 nickase. Sci Rep. 2016;6:38198. doi: 10.1038/srep38198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emborg ME, et al. Induced pluripotent stem cell-derived neural cells survive and mature in the nonhuman primate brain. Cell Rep. 2013;3:646–650. doi: 10.1016/j.celrep.2013.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fesnak AD, June CH, Levine BL. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer. 2016;16:566–581. doi: 10.1038/nrc.2016.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn R, Grundmann A, Renz P, Hanseler W, James WS, Cowley SA, Moore MD. CRISPR-mediated genotypic and phenotypic correction of a chronic granulomatous disease mutation in human iPS cells. Exp Hematol. 2015;43(838–848):e833. doi: 10.1016/j.exphem.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32:279–284. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabaldon T, Koonin EV. Functional and evolutionary implications of gene orthology. Nat Rev Genet. 2013;14:360–366. doi: 10.1038/nrg3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gharib WH, Robinson-Rechavi M. When orthologs diverge between human and mouse. Brief Bioinform. 2011;12:436–441. doi: 10.1093/bib/bbr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, et al. Genome-Scale CRISPR-mediated control of gene repression and activation. Cell. 2014;159:647–661. doi: 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez F, Zhu Z, Shi ZD, Lelli K, Verma N, Li QV, Huangfu D. An iCRISPR platform for rapid, multiplexable, and inducible genome editing in human pluripotent stem cells. Cell Stem Cell. 2014;15:215–226. doi: 10.1016/j.stem.2014.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gos M. Epigenetic mechanisms of gene expression regulation in neurological diseases. Acta Neurobiol Exp. 2013;73:19–37. doi: 10.55782/ane-2013-1919. [DOI] [PubMed] [Google Scholar]
- Guilinger JP, Thompson DB, Liu DR. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat Biotechnol. 2014;32:577–582. doi: 10.1038/nbt.2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallett PJ, et al. Successful function of autologous iPSC-derived dopamine neurons following transplantation in a non-human primate model of Parkinson’s disease. Cell Stem Cell. 2015;16:269–274. doi: 10.1016/j.stem.2015.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heman-Ackah SM, Bassett AR, Wood MJ. Precision modulation of neurodegenerative disease-related gene expression in human iPSC-derived neurons. Sci Rep. 2016;6:28420. doi: 10.1038/srep28420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess GT, et al. Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells. Nat Methods. 2016;13:1036–1042. doi: 10.1038/nmeth.4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton IB, D’Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, Gersbach CA. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol. 2015;33:510–517. doi: 10.1038/nbt.3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horii T, Tamura D, Morita S, Kimura M, Hatada I. Generation of an ICF syndrome model by efficient genome editing of human induced pluripotent stem cells using the CRISPR system. Int J Mol Sci. 2013;14:19774–19781. doi: 10.3390/ijms141019774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horikiri T, et al. SOX10-nano-lantern reporter human iPS cells; a versatile tool for neural crest research. PLoS ONE. 2017;12:e0170342. doi: 10.1371/journal.pone.0170342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Z, Zhang Y, Propson NE, Howden SE, Chu LF, Sontheimer EJ, Thomson JA. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc Natl Acad Sci USA. 2013;110:15644–15649. doi: 10.1073/pnas.1313587110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrvatin S, et al. Differentiated human stem cells resemble fetal, not adult, beta cells. Proc Natl Acad Sci USA. 2014;111:3038–3043. doi: 10.1073/pnas.1400709111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, et al. Production of gene-corrected adult beta globin protein in human erythrocytes differentiated from patient iPSCs after genome editing of the sickle point mutation. Stem Cells. 2015;33:1470–1479. doi: 10.1002/stem.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huch M, Koo BK. Modeling mouse and human development using organoid cultures. Development. 2015;142:3113–3125. doi: 10.1242/dev.118570. [DOI] [PubMed] [Google Scholar]
- Hug N, Longman D, Caceres JF. Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res. 2016;44:1483–1495. doi: 10.1093/nar/gkw010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultquist JF, et al. A Cas9 Ribonucleoprotein Platform for Functional Genetic Studies of HIV-Host Interactions in Primary Human T Cells. Cell Rep. 2016;17:1438–1452. doi: 10.1016/j.celrep.2016.09.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura K, et al. Calcium dysregulation contributes to neurodegeneration in FTLD patient iPSC-derived neurons. Sci Rep. 2016;6:34904. doi: 10.1038/srep34904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T, et al. Genetic and pharmacological correction of aberrant dopamine synthesis using patient iPSCs with BH4 metabolism disorders. Hum Mol Genet. 2016 doi: 10.1093/hmg/ddw339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakovcevski M, Akbarian S. Epigenetic mechanisms in neurological disease. Nat Med. 2012;18:1194–1204. doi: 10.1038/nm.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. RNA-programmed genome editing in human cells. Elife. 2013;2:e00471. doi: 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearns NA, Pham H, Tabak B, Genga RM, Silverstein NJ, Garber M, Maehr R. Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat Methods. 2015;12:401–403. doi: 10.1038/nmeth.3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, et al. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467:285–290. doi: 10.1038/nature09342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kim D, Cho SW, Kim J, Kim JS. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014;24:1012–1019. doi: 10.1101/gr.171322.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinstiver BP, Prew MS, Tsai SQ, Nguyen NT, Topkar VV, Zheng Z, Joung JK. Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat Biotechnol. 2015;33:1293–1298. doi: 10.1038/nbt.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinstiver BP, et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 2015;523:481–485. doi: 10.1038/nature14592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, Joung JK. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490–495. doi: 10.1038/nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, et al. Generation of rat pancreas in mouse by interspecific blastocyst injection of pluripotent stem cells. Cell. 2010;142:787–799. doi: 10.1016/j.cell.2010.07.039. [DOI] [PubMed] [Google Scholar]
- Koike-Yusa H, Li Y, Tan EP, Velasco-Herrera Mdel C, Yusa K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat Biotechnol. 2014;32:267–273. doi: 10.1038/nbt.2800. [DOI] [PubMed] [Google Scholar]
- Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S, et al. Optical control of mammalian endogenous transcription and epigenetic states. Nature. 2013;500:472–476. doi: 10.1038/nature12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S, et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwart D, Paquet D, Teo S, Tessier-Lavigne M. Precise and efficient scarless genome editing in stem cells using CORRECT. Nat Protoc. 2017;12:329–354. doi: 10.1038/nprot.2016.171. [DOI] [PubMed] [Google Scholar]
- Lancaster MA, Knoblich JA. Organogenesis in a dish: modeling development and disease using organoid technologies. Science. 2014;345:1247125. doi: 10.1126/science.1247125. [DOI] [PubMed] [Google Scholar]
- Lancaster MA, et al. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501:373–379. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Landgrave-Gomez J, Mercado-Gomez O, Guevara-Guzman R. Epigenetic mechanisms in neurological and neurodegenerative diseases. Front Cell Neurosci. 2015;9:58. doi: 10.3389/fncel.2015.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, et al. Engraftment of human iPS cells and allogeneic porcine cells into pigs with inactivated RAG2 and accompanying severe combined immunodeficiency. Proc Natl Acad Sci USA. 2014;111:7260–7265. doi: 10.1073/pnas.1406376111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HL, et al. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Rep. 2015;4:143–154. doi: 10.1016/j.stemcr.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J Biotechnol. 2015;208:44–53. doi: 10.1016/j.jbiotec.2015.04.024. [DOI] [PubMed] [Google Scholar]
- Lin S, Staahl BT, Alla RK, Doudna JA. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife. 2014;3:e04766. doi: 10.7554/eLife.04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XS, et al. Editing DNA methylation in the mammalian genome. Cell. 2016;167(233–247):e217. doi: 10.1016/j.cell.2016.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, et al. Efficient CRISPR/Cas9-Mediated Versatile, Predictable, and Donor-Free Gene Knockout in Human Pluripotent Stem Cells. Stem Cell Rep. 2016;7:496–507. doi: 10.1016/j.stemcr.2016.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunter G, Ponting CP, Hein J. Genome-wide identification of human functional DNA using a neutral indel model. PLoS Comput Biol. 2006;2:e5. doi: 10.1371/journal.pcbi.0020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luukkonen BG, Tan W, Schwartz S. Efficiency of reinitiation of translation on human immunodeficiency virus type 1 mRNAs is determined by the length of the upstream open reading frame and by intercistronic distance. J Virol. 1995;69:4086–4094. doi: 10.1128/jvi.69.7.4086-4094.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano MT, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkert S, Martin U. Targeted genome engineering using designer nucleases: state of the art and practical guidance for application in human pluripotent stem cells. Stem Cell Res. 2016;16:377–386. doi: 10.1016/j.scr.2016.02.027. [DOI] [PubMed] [Google Scholar]
- Merkert S, et al. Efficient designer nuclease-based homologous recombination enables direct PCR screening for footprintless targeted human pluripotent stem cells. Stem Cell Rep. 2014;2:107–118. doi: 10.1016/j.stemcr.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens J, Marchetto MC, Bardy C, Gage FH. Evaluating cell reprogramming, differentiation and conversion technologies in neuroscience. Nat Rev Neurosci. 2016;17:424–437. doi: 10.1038/nrn.2016.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JD, et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell. 2013;13:691–705. doi: 10.1016/j.stem.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyaoka Y, et al. Isolation of single-base genome-edited human iPS cells without antibiotic selection. Nat Methods. 2014;11:291–293. doi: 10.1038/nmeth.2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyaoka Y, et al. Systematic quantification of HDR and NHEJ reveals effects of locus, nuclease, and cell type on genome-editing. Sci Rep. 2016;6:23549. doi: 10.1038/srep23549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mock U, Hauber I, Fehse B. Digital PCR to assess gene-editing frequencies (GEF-dPCR) mediated by designer nucleases. Nat Protoc. 2016;11:598–615. doi: 10.1038/nprot.2016.027. [DOI] [PubMed] [Google Scholar]
- Morrison M, et al. StemBANCC: governing access to material and data in a large stem cell research consortium. Stem Cell Rev. 2015;11:681–687. doi: 10.1007/s12015-015-9599-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouse Genome Sequencing et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- Murakami N, et al. Proteasome impairment in neural cells derived from HMSN-P patient iPSCs. Mol Brain. 2017;10:7. doi: 10.1186/s13041-017-0286-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murry CE, Keller G. Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell. 2008;132:661–680. doi: 10.1016/j.cell.2008.02.008. [DOI] [PubMed] [Google Scholar]
- Nagashima H, Matsunari H. Growing human organs in pigs-A dream or reality? Theriogenology. 2016;86:422–426. doi: 10.1016/j.theriogenology.2016.04.056. [DOI] [PubMed] [Google Scholar]
- Nguyen HN, et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell. 2011;8:267–280. doi: 10.1016/j.stem.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida K, et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science. 2016 doi: 10.1126/science.aaf8729. [DOI] [PubMed] [Google Scholar]
- Nishizaki SS, Boyle AP. Mining the unknown: assigning function to noncoding single nucleotide polymorphisms. Trends Genet. 2016 doi: 10.1016/j.tig.2016.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu X, et al. Combining single strand oligodeoxynucleotides and CRISPR/Cas9 to correct gene mutations in beta-thalassemia-induced pluripotent stem cells. J Biol Chem. 2016;291:16576–16585. doi: 10.1074/jbc.M116.719237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquet D, et al. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature. 2016;533:125–129. doi: 10.1038/nature17664. [DOI] [PubMed] [Google Scholar]
- Park CY, et al. Functional correction of large factor VIII gene chromosomal inversions in hemophilia A patient-derived iPSCs using CRISPR-Cas9. Cell Stem Cell. 2015;17:213–220. doi: 10.1016/j.stem.2015.07.001. [DOI] [PubMed] [Google Scholar]
- Park CY, Sung JJ, Choi SH, Lee DR, Park IH, Kim DW. Modeling and correction of structural variations in patient-derived iPSCs using CRISPR/Cas9. Nat Protoc. 2016;11:2154–2169. doi: 10.1038/nprot.2016.129. [DOI] [PubMed] [Google Scholar]
- Passier R, Orlova V, Mummery C. Complex tissue and disease modeling using hiPSCs. Cell Stem Cell. 2016;18:309–321. doi: 10.1016/j.stem.2016.02.011. [DOI] [PubMed] [Google Scholar]
- Pruitt KD, et al. The consensus coding sequence (CCDS) project: identifying a common protein-coding gene set for the human and mouse genomes. Genome Res. 2009;19:1316–1323. doi: 10.1101/gr.080531.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quax TE, Claassens NJ, Soll D, van der Oost J. Codon bias as a means to fine-tune gene expression. Mol Cell. 2015;59:149–161. doi: 10.1016/j.molcel.2015.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt P, et al. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell. 2013;12:354–367. doi: 10.1016/j.stem.2013.01.008. [DOI] [PubMed] [Google Scholar]
- Richardson CD, Ray GJ, DeWitt MA, Curie GL, Corn JE. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat Biotechnol. 2016;34:339–344. doi: 10.1038/nbt.3481. [DOI] [PubMed] [Google Scholar]
- Rouhani F, Kumasaka N, de Brito MC, Bradley A, Vallier L, Gaffney D. Genetic background drives transcriptional variation in human induced pluripotent stem cells. PLoS Genet. 2014;10:e1004432. doi: 10.1371/journal.pgen.1004432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salarinia R, et al. Epi-Drugs and Epi-miRs: Moving Beyond Current Cancer Therapies. Curr Cancer Drug Targets. 2016;16:773–788. doi: 10.2174/1568009616666151207110143. [DOI] [PubMed] [Google Scholar]
- Santos DP, Kiskinis E, Eggan K, Merkle FT. Comprehensive protocols for CRISPR/Cas9-based gene editing in human pluripotent stem cells. Curr Protoc Stem Cell Biol. 2016 doi: 10.1002/cpsc.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwank G, et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell. 2013;13:653–658. doi: 10.1016/j.stem.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Shalem O, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamblott MJ, et al. Derivation of pluripotent stem cells from cultured human primordial germ cells. Proc Natl Acad Sci USA. 1998;95:13726–13731. doi: 10.1073/pnas.95.23.13726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastav M, De Haro LP, Nickoloff JA. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008;18:134–147. doi: 10.1038/cr.2007.111. [DOI] [PubMed] [Google Scholar]
- Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351:84–88. doi: 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snowden AW, Gregory PD, Case CC, Pabo CO. Gene-specific targeting of H3K9 methylation is sufficient for initiating repression in vivo. Curr Biol. 2002;12:2159–2166. doi: 10.1016/S0960-9822(02)01391-X. [DOI] [PubMed] [Google Scholar]
- Soares FA, Sheldon M, Rao M, Mummery C, Vallier L. International coordination of large-scale human induced pluripotent stem cell initiatives: Wellcome Trust and ISSCR workshops white paper. Stem Cell Rep. 2014;3:931–939. doi: 10.1016/j.stemcr.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldner F, et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell. 2011;146:318–331. doi: 10.1016/j.cell.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldner F, et al. Parkinson-associated risk variant in distal enhancer of alpha-synuclein modulates target gene expression. Nature. 2016;533:95–99. doi: 10.1038/nature17939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song B, et al. Improved hematopoietic differentiation efficiency of gene-corrected beta-thalassemia induced pluripotent stem cells by CRISPR/Cas9 system. Stem Cells Dev. 2015;24:1053–1065. doi: 10.1089/scd.2014.0347. [DOI] [PubMed] [Google Scholar]
- Sontag S, et al. Modelling IRF8 deficient human hematopoiesis and dendritic cell development with engineered iPS cells. Stem Cells. 2017 doi: 10.1002/stem.2565. [DOI] [PubMed] [Google Scholar]
- Stergachis AB, et al. Exonic transcription factor binding directs codon choice and affects protein evolution. Science. 2013;342:1367–1372. doi: 10.1126/science.1243490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streeter I, Harrison PW, Faulconbridge A, Flicek P, Parkinson H, Clarke L, The HipSci C The human-induced pluripotent stem cell initiative-data resources for cellular genetics. Nucleic Acids Res. 2016 doi: 10.1093/nar/gkw928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney CL, et al. Targeted Repair of CYBB in X-CGD iPSCs requires retention of intronic sequences for expression and functional correction. Mol Ther. 2017;25:321–330. doi: 10.1016/j.ymthe.2016.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi R, et al. Bioengineering a 3D integumentary organ system from iPS cells using an in vivo transplantation model. Sci Adv. 2016;2:e1500887. doi: 10.1126/sciadv.1500887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Tebas P, et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med. 2014;370:901–910. doi: 10.1056/NEJMoa1300662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- Torres R, Martin MC, Garcia A, Cigudosa JC, Ramirez JC, Rodriguez-Perales S. Engineering human tumour-associated chromosomal translocations with the RNA-guided CRISPR-Cas9 system. Nat Commun. 2014;5:3964. doi: 10.1038/ncomms4964. [DOI] [PubMed] [Google Scholar]
- Tsai SQ, et al. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol. 2014;32:569–576. doi: 10.1038/nbt.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veres A, et al. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell. 2014;15:27–30. doi: 10.1016/j.stem.2014.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visscher PM, Brown MA, McCarthy MI, Yang J. Five years of GWAS discovery. Am J Hum Genet. 2012;90:7–24. doi: 10.1016/j.ajhg.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vojta A, et al. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016;44:5615–5628. doi: 10.1093/nar/gkw159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat Med. 2014;20:616–623. doi: 10.1038/nm.3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, et al. Efficient, footprint-free human iPSC genome editing by consolidation of Cas9/CRISPR piggyBac technologies. Nat Protoc. 2017;12:88–103. doi: 10.1038/nprot.2016.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss KM, Clark AG. Linkage disequilibrium and the mapping of complex human traits. Trends Genet. 2002;18:19–24. doi: 10.1016/S0168-9525(01)02550-1. [DOI] [PubMed] [Google Scholar]
- Wheeler HE, et al. Survey of the heritability and sparse architecture of gene expression traits across human tissues. PLoS Genet. 2016;12:e1006423. doi: 10.1371/journal.pgen.1006423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright CF, et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. The Lancet. 2015;385:1305–1314. doi: 10.1016/S0140-6736(14)61705-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Hunt SD, Xue H, Liu Y, Darabi R. Generation and characterization of a MYF5 reporter human iPS Cell Line Using CRISPR/Cas9 mediated homologous recombination. Sci Rep. 2016;6:18759. doi: 10.1038/srep18759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Hunt SD, Xue H, Liu Y, Darabi R. Generation and validation of PAX7 reporter lines from human iPS cells using CRISPR/Cas9 technology. Stem Cell Res. 2016;16:220–228. doi: 10.1016/j.scr.2016.01.003. [DOI] [PubMed] [Google Scholar]
- Xiao A, et al. Chromosomal deletions and inversions mediated by TALENs and CRISPR/Cas in zebrafish. Nucleic Acids Res. 2013;41:e141. doi: 10.1093/nar/gkt464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie N, Gong H, Suhl JA, Chopra P, Wang T, Warren ST. Reactivation of FMR1 by CRISPR/Cas9-mediated deletion of the expanded CGG-Repeat of the Fragile X Chromosome. PLoS ONE. 2016;11:e0165499. doi: 10.1371/journal.pone.0165499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, et al. A CRISPR-based approach for targeted DNA demethylation. Cell Discov. 2016;2:16009. doi: 10.1038/celldisc.2016.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, et al. Reversal of Phenotypic Abnormalities by CRISPR/Cas9-Mediated Gene Correction in Huntington Disease Patient-Derived Induced Pluripotent Stem Cells. Stem Cell Rep. 2017 doi: 10.1016/j.stemcr.2017.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Res. 2013;41:9049–9061. doi: 10.1093/nar/gkt555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young CS, et al. A Single CRISPR-Cas9 deletion strategy that targets the majority of DMD patients restores dystrophin function in hiPSC-derived muscle cells. Cell Stem Cell. 2016;18:533–540. doi: 10.1016/j.stem.2016.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusa K. Seamless genome editing in human pluripotent stem cells using custom endonuclease-based gene targeting and the piggyBac transposon. Nat Protoc. 2013;8:2061–2078. doi: 10.1038/nprot.2013.126. [DOI] [PubMed] [Google Scholar]
- Zetsche B, et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163:759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer A, Zimmer AM, Reynolds K. Tissue specific expression of the retinoic acid receptor-beta 2: regulation by short open reading frames in the 5′-noncoding region. J Cell Biol. 1994;127:1111–1119. doi: 10.1083/jcb.127.4.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]