

Abstract
Background
Phorbol 12-myristate 13-acetate (PMA) is often used as an activating phorbol ester of protein kinase C (PKC) to investigate the roles of the kinase in cellular functions. Accumulating lines of evidence indicate that in addition to activating PKC, PMA also produces some regulatory effects in a PKC-independent manner. In this study, we investigated the non-PKC effects of PMA on electrical excitability of rat pancreatic β-cells by using patch-clamp techniques.
Results
In current-clamp recording, PMA (80 nM) reversibly inhibited 15 mM glucose-induced action potential spikes superimposed on a slow membrane depolarization and this inhibition can not be prevented by pre-treatment of the cell with a specific PKC inhibitor, bisindolylmaleimide (BIM, 1 μM). In the presence of a subthreshold concentration (5.5 mM) of glucose, PMA hyperpolarized β-cells in a concentration-dependent manner (0.8–240 nM), even in the presence of BIM. Based on cell-attached single channel recordings, PMA increased ATP-sensitive K+ channel (KATP) activity. Based on inside-out patch-clamp recordings, PMA had little effect on KATP activity if no ATP was in the bath, while PMA restored KATP activity that was suppressed by 10 μM ATP in the bath. In voltage-clamp recording, PMA enhanced tolbutamide-sensitive membrane currents elicited by repetitive ramp pulses from -90 to -50 mV in a concentration-dependent manner, and this potentiation could not be prevented by pre-treatment of cell with BIM. 4α-phorbol 12,13-didecanoate (4α-PDD), a non-PKC-activating phorbol ester, mimicked the effect of PMA on both current-clamp and voltage-clamp recording configurations. With either 5.5 or 16.6 mM glucose in the extracellular solution, PMA (80 nM) increased insulin secretion from rat islets. However, in islets pretreated with BIM (1 μM), PMA did not increase, but rather reduced insulin secretion.
Conclusion
In rat pancreatic β-cells, PMA modulates insulin secretion through a mixed mechanism: increases insulin secretion by activation of PKC, and meanwhile decrease insulin secretion by impairing β-cell excitability in a PKC-independent manner. The enhancement of KATP activity by reducing sensitivity of KATP to ATP seems to underlie the PMA-induced impairment of β-cells electrical excitation in response to glucose stimulation.
Background
Phorbol esters are often used as activators of protein kinase C (PKC) to investigate the role of the kinase in cellular functions [1]. Pancreatic β-cells possess α, β1, δ and ε PKC isotypes [2], and activation of PKC is known to synergize with glucose stimulation in insulin secretion [3-5]. PKC, via protein phosphorylation seems to facilitate exocytosis of insulin granules [6-8]. In glucose-stimulated insulin secretion, ATP-sensitive K+ channels (KATP (SUR1, Kir6.2)) play a crucial role [9,10]. ATP produced by the metabolism of glucose inhibits KATP activity, resulting in development of depolarization responsible for opening of L-type Ca2+ channels, allowing Ca2+ influx. Elevation of intracellular Ca2+ ([Ca2+]i) accelerates exocytosis of insulin granules. The possibility that KATP activity is regulated by PKC has long been studied. In this respect, however, contradictory results have been obtained. For example, in insulin-secreting cell lines, PKC was reported to potentiate KATP activity [11,12], although PKC-dependent inhibition [13] or inhibition followed by activation [14] has been also reported. In primary β-cells from the mouse, activation of PKC by PMA did not cause any change in membrane potential [15,16], although in the same cell type, PMA reduced the glucose-stimulated elevation of [Ca2+]i depending on activation of PKC [16]. On the other hand, recent studies in myocytes suggest that activation of PKC increases the open-time probability of KATP (SUR2, Kir6.1) by reducing the sensitivity of KATP to ATP [17-20]. Therefore, whether regulation of insulin secretion by PMA is mediated through PKC or not is still obscure. Recently, we reported that phorbol ester inhibited Ca2+ influx through PKC-dependent and PKC-independent pathways [21]. In the present study, we further evaluated the non-PKC mechanism of PMA action on β-cell electrical excitation by using patch-clamp techniques. The results indicate that in rat pancreatic β-cells, PMA modulates insulin secretion through a mixed mechanism: increases insulin secretion by activation of PKC, and meanwhile decrease insulin secretion by impairing β-cell excitability in a PKC-independent manner. The enhancement of KATP activity by reducing sensitivity of KATP to ATP seems to underlie the PMA-induced impairment of β-cells electrical excitation in response to glucose stimulation.
Results
PMA inhibits electrical excitation of single rat β-cells
With glucose at 5.5 mM in the extracellular solution, the membrane potential, measured using the nystatin-perforation technique, was stable (-52.9 ± 1.1 mV, mean ± SE, n = 28). Bath-applied 15 mM glucose induced the action potential spikes superimposed on a slow membrane depolarization (Fig. 1 panel A). When PMA (80 nM) was applied to the cell during 15 mM glucose stimulation, both slow depolarization and action potentials was abolished. The effect of PMA was reversible after removal of PMA (Fig. 1 panel A). This inhibitory effect by PMA appeared to be independent of PKC since addition of the PKC blocker, BIM (1 μM) did not prevent PMA-induced inhibition (Fig. 1 panel B). In two experiments, higher concentration (10 μM) of BIM failed to prevent PMA-induced inhibition too (data not shown). In the presence of 5.5 mM glucose, 63% of tested β-cells (5/8) showed spontaneous action potentials superimposed on a slow depolarizing potential, and PMA (80 nM) inhibited these action potentials as well (Fig. 1 panel C).
Figure 1.
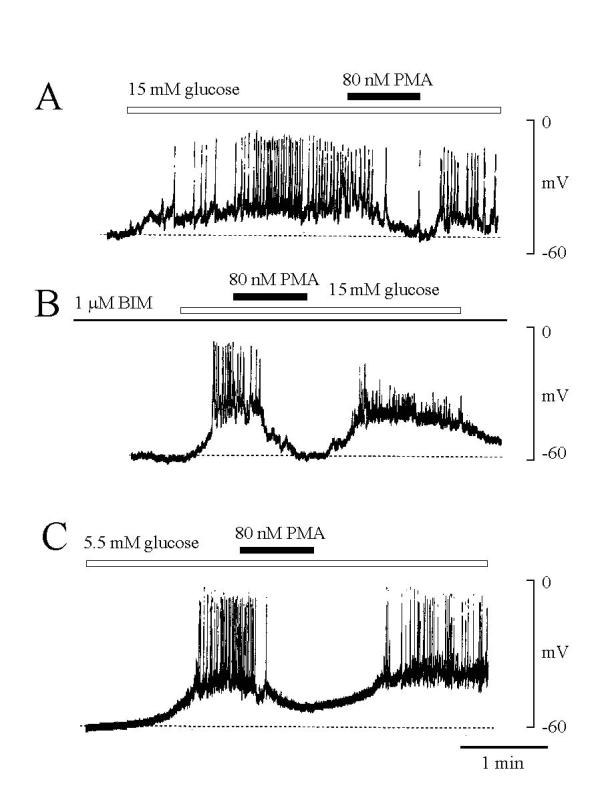
Effect of PMA on electrical excitation of single rat pancreatic β-cells. The membrane potential was measured the nystatin-perforation method. (A) Increases in extracellular glucose concentration from 5.5 to 15 mM induced a slow membrane depolarization superimposed on action potentials. Addition of PMA (80 nM) inhibited action potentials also membrane depolarization. (B) In the cell treated with BIM (1 μM, for one hour), PMA (80 nM) still inhibited extracellular glucose-induced membrane depolarization and action potentials. (C) In the presence of 5.5 mM glucose, some cells (5/8) showed spontaneous electrical excitation, which also inhibited by PMA. The concentration of glucose in the extracellular solution was 5.5 mM. Dotted lines indicate the membrane potential level before stimulation with glucose or PMA. Representative tracings from at least six experiments are shown.
PMA induces hyperpolarization of rat β-cells
In the presence of 5.5 mM glucose, application of PMA (80 nM) alone induced a slow, reversible membrane hyperpolarization (Fig. 2 panel A). The magnitude of the hyperpolarization depended on the concentration of PMA (Fig. 2 panel C). Pretreatment of cells with the PKC inhibitor, BIM (1 μM) for about 60 min did not prevent PMA-induced hyperpolarization (Fig. 2 panel B). Figure 2C summarized that PMA-induced hyperpolarization was not significantly different between BIM-treated and BIM-untreated β-cells (Fig. 2 panel C). These results indicate that PMA-induced hyperpolarization of pancreatic β-cells is not mediated through PKC activation.
Figure 2.
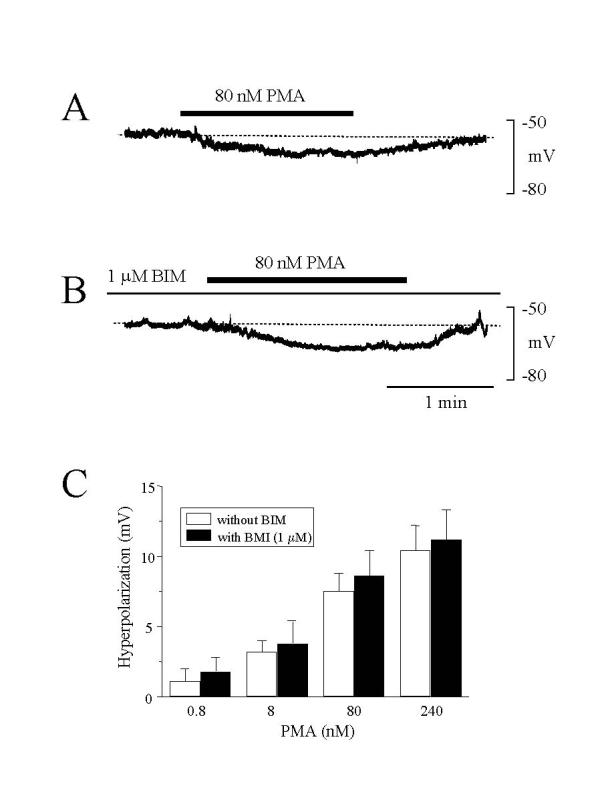
PMA induced membrane hyperpolarization. (A) PMA (80 nM) applied during a steady-state phase of membrane potential in a solution containing 5.5 mM glucose caused a slow hyperpolarization. (B) In cells treated with BIM (1 μM), PMA (80 nM) induced a similar slow hyperpolarization. Representative traces in A and B from at least six experiments are shown. (C) Concentration-response relationships of PMA-induced hyperpolarization in cells treated and untreated with BIM. Results are from 5–8 experiments. The vertical bars represent SEM. The values with PMA at the same concentration are not significantly different in cells treated or untreated with BIM.
PMA increases KATP activity in cell-attached single channel recording
Since KATP plays a crucial role in glucose-stimulated insulin secretion, PMA-induced hyperpolarization could be mediated by changes in KATP activity. To test this hypothesis, we examined the effect of PMA on single channel activities of KATP. In the cell-attached configuration, spontaneous single channel currents were recorded at a pipette potential of 0 mV (Fig. 3). The specific KATP blocker, tolbutamide (0.5 mM) completely inhibited channel activity (Fig. 3), indicating that the recorded single channel currents are passed through KATP channels. Application of 80 nM PMA to the bath solution significantly increased channel activities (Fig. 3). Values of open-time probability for KATP were 0.08 ± 0.01 before application of PMA and 0.11 ± 0.01 during application of PMA (n = 9, Mean ± SEM, P < 0.01). This result indicates that PMA indeed increases KATP activity.
Figure 3.

PMA induces KATP activity recorded in the cell-attached configuration. Single channel recordings in the cell-attached configuration are shown. The pipette potential (Vp) was held at 0 mV. Application of 80 nM PMA to the bath increased channel opening. Tolbutamide (0.5 mM), applied to the bath during PMA, completely inhibited channel activity, suggesting that PMA increased KATP activity.
PMA increases whole-cell membrane current through KATP
In the whole-cell configuration, membrane currents in response to repeatedly applied voltage ramp pulses from -90 to -50 mV were recorded. Tolbutamide (0.5 mM) markedly reduced the size of the current, and after blockade of membrane current, PMA failed to increase the current (Fig. 4 panel A), suggesting that PMA increased membrane currents through KATP. Application of PMA (80 nM) increased the magnitude of membrane current (Fig. 4 panel B) and the magnitude of increased currents depended on the concentrations of PMA (Fig. 4 panel D). In cells treated with BIM (1 μM), PMA still increased membrane current (Fig. 4 panel C). No significant differences were observed between BIM-untreated and treated cells for PMA-induced membrane current increase (Fig. 4 panel D), suggesting that PMA increases KATP-mediated currents in a PKC-independent manner.
Figure 4.
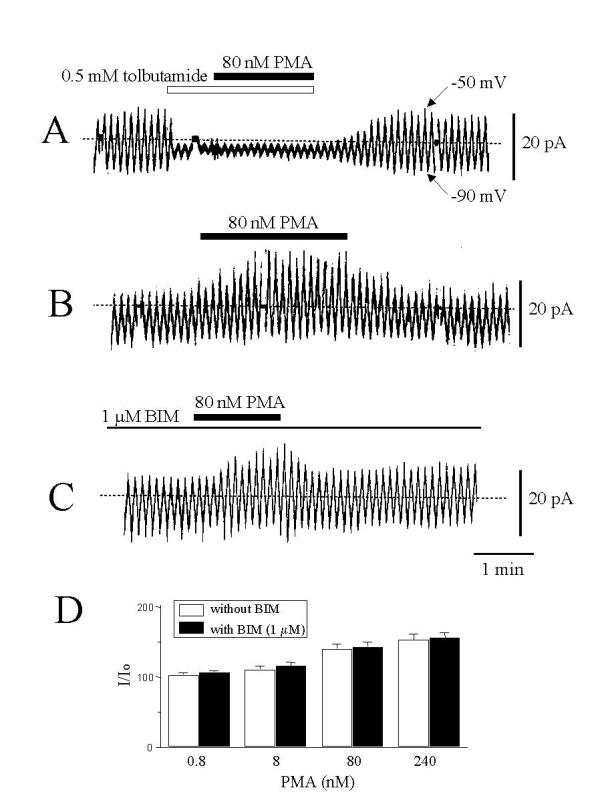
The effect of PMA on membrane current in response to voltage ramp pulses. Whole cell membrane currents in response to voltage ramps from -90 to -50 mV, recorded while clamping the ATP concentration at 1 mM in the pipette solution, are shown. (A) The effect of tolbutamide (0.5 mM), and the effect of PMA (80 nM), applied on top of tolbutamide exposure. (B) The effect of PMA (80 nM) alone. (C) The effect of PMA on cells treated with BIM (1 μM). (D) Concentration-response relationships for the PMA effect on currents in cells treated or untreated with BIM (1 μM). The size of membrane current was expressed relative to that before application of PMA. The values with PMA at the same concentration are not significantly different for cells treated with or without BIM. Results are from 5–7 experiments.
4α-PDD mimics the effect of PMA on electrical properties of rat β-cells
There are two ways to test the hypothesis that the effect of PMA on electrical properties of rat β-cells is independent of PKC activation. One is to use specific PKC blockers, like BIM, to examine whether the effect of PMA can be prevented (Fig. 1,2,4). Another way is to employ PMA analogs, which do not stimulate PKC, to see if they can mimic the PMA effect. 4α-PDD, as a PKC non-stimulating phorbol ester [22], provides a good opportunity to test our hypothesis. As shown in Fig. 5 panel A, 4α-PDD (1 μM) completely abolished action potentials elicited by elevation of glucose concentration from 5.5 to 15 mM, just like the PMA effect in Fig. 1 panel A. In voltage-clamp current recordings (Fig. 5 panel B), 4α-PDD (1 μM) increased membrane current evoked by ramp from -90 to -50 mV, similar to the PMA effect in figure 4 panel B. The similar results have also been obtained by low concentration of 4α-PDD (80 nM, n = 3 for current-clamp data and n = 2 for voltage-clamp data, data not shown). These results indicate that 4α-PDD virtually mimics PMA effects, and these data further support the idea that the increase of KATP activity by PMA does not depend on PKC activation.
Figure 5.

The effect of 4α-PDD (1 μM) on β-cell electrical excitation. (A) Effect of 4α-PDD on glucose (15 mM)-induced depolarization and action potential. (B) effect of 4α-PDD on ramp pulse-induced membrane current. Dotted lines indicate the control level of membrane current. Representative tracings from at least six experiments are shown.
Possible mechanism of PMA increases KATP activity
How does PMA increase KATP activity? Two possible explanations might be addressed. First, PMA reduces the sensitivity of KATP to ATP as reports using myocytes [17-20]. Second, PMA may reduce ATP production. To assess the effects of PMA on KATP activity, we performed two experiments. First, using inside-out recording configuration, spontaneous single channel activities were more prominent when there was no ATP in bath solution (intracellular face), and recorded currents were very sensitive to bath-applied tolbutamide (0.5 mM, Fig. 6 panel A). In the absence of ATP in the bath, application of PMA showed little effect on KATP activity (Fig. 6 panel B). Values of the open-time probability for KATP were 0.19 ± 0.02 (n = 12) without and 0.20 ± 0.02 (n = 5) with 80 nM PMA (P > 0.05). The average single channel conductances were 68 pS without and 66 pS with 80 nM PMA, measured between -90 to 0 mV (Fig. 6 panel D). Application of 10 μM ATP to the bath decreased the open probability of KATP, and under this condition, PMA restored KATP activity (Fig. 6 panel C) even in the presence of 1 μM BIM (Fig. 6 panel E). Values for ATP concentration producing half-maximal inhibition of KATP, taken from the concentration-response relationships of ATP inhibition of KATP activity, were 10.8 ± 1.9 μM (n = 6) without and 24.9 ± 4.9 μM (n = 5) with 80 nM PMA (P <0.05, Fig. 6 panel F). Values of the Hill coefficient were 1.04 ± 0.02 without and 1.08 ± 0.02 with PMA (P > 0.05). These results indicate that PMA increases KATP activity in the presence of intracellular ATP, which implies that PMA may reduce the sensitivity of KATP to ATP. Second, the ATP content of rat islets was measured during PMA treatment. As shown in Table 1,10 min loading of islets with extracellular solution containing 80 nM PMA did not alter ATP content of islets. On the other hand, loading cells with 1 mM NaCN markedly reduced ATP content.
Figure 6.
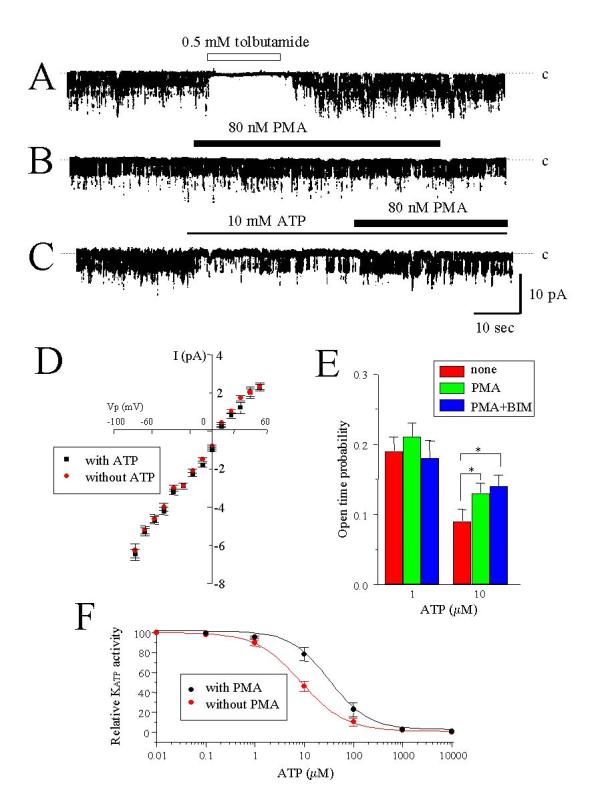
Effect of PMA on the ATP inhibition of KATP activity. (A) Single channel current recordings in the inside-out configuration. The transmembrane potential was -60 mV (inside negative). Tolbutamide was applied to the intracellular side of the membrane at 0.5 mM. Inhibition of the current by tolbutamide indicates that KATP channel currents were recorded. (B) 80 nM PMA was applied inside the membrane. (C) ATP was first applied inside the membrane at 10 μM and then 80 nM PMA was further applied. Note that KATP activity was reduced by ATP, and application of PMA restored activity even in the presence of ATP. Representative tracings at least from five experiments are shown. (D) I-V relationship of KATP in the absence of ATP without (red circles) and with (black circles) 80 nM PMA. Data are from eight (without PMA) and six (with PMA) patches. (E) The values of open-time probability with and without 10 μM ATP inside the membrane are shown. Data with 80 nM PMA or with PMA plus 1 μM BIM are also shown. Each value is the mean of at least five experiments. *P < 0.05, compared with data with 10 μM ATP in the absence of PMA. (F) Concentration-dependence for ATP inhibition of KATP activity with and without 80 nM PMA. A series of experiments with ATP at various concentrations was carried out on the same patch. Data from twelve (without PMA) and six (with 80 nM PMA) patches. Theoretical curves for data with (a black line) or without (a red line) PMA were fitted to the Hill equation.
Table 1.
ATP concentration of rat islets after loading with PMA or NaCN
| Treatment | ATP (pmole/islet) | Pa) |
| none (with ethanolb)) | 0.72 ± 0.07 (n = 13) | |
| PMA (80 nM)c) | 0.75 ± 0.10(n = 13) | >0.05 |
| none (without ethanol) | 0.68 ± 0.08 (n = 8) | |
| NaCN (I mM)d) | 0.31 ± 0.04 (n = 8) | <0.01 |
a)P value, compared with the value of the paired control (none). b)The concentration of ethanol was 0.01%. c)PMA was applied for 10 min. d)NaCN was applied for 10 min. All measurements were performed at 37°C.
Effects of PMA on glucose-induced insulin secretion
The results presented such far indicate that PMA decreases electrical excitation of β-cells and this effect is independent on PKC activation. According to insulin secretion theory, decrease of electrical excitation of β-cells should reduce insulin secretion. To address this question, we measured insulin secretion from rat islets. As shown in Fig. 7, PMA (80 nM) increase both basal (5.5 mM glucose in the extracellular solution) and evoked (16.6 mM glucose in the extracellular solution) insulin secretion rates. However, as we expected, in BIM-treated islets PMA (80 nM) decreased the rates of insulin secretion (Fig. 7). These data indicate that PMA regulates insulin secretion mediated by a mixed mechanism, a PKC-dependent increase of insulin secretion and a PKC-independent decrease of insulin secretion.
Figure 7.
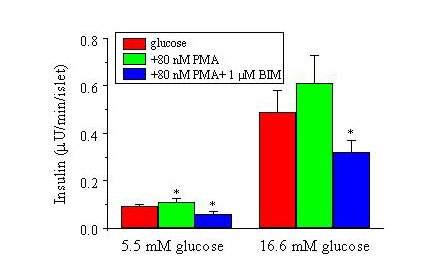
Effect of PMA on insulin secretion in response to glucose stimulation of rat isolated islets. The rates of insulin secretion from rat islets were shown. Islets were separated into two groups, treated and untreated with BIM. The rates of insulin secretion were measured with glucose at 5.5 mM and 16.6 mM in the extracellular solution, respectively. 80 nM PMA or 80 nM PM plus 1 μM BIM was compared with the value obtained in islets without any treatment. Mean values of 5–15 experiments are shown. Vertical bars indicate SE. *P < 0.05. All measurements were performed at 37°C.
Discussion
Phorbol esters are often used as activators of PKC to investigate the role of the kinase in cellular functions [1]. Accumulating lines of evidence indicates that PMA plays a complex role in regulation of cellular function, including PKC-dependent [18-20] and PKC-independent ways [23,24,21]. In the present results, PMA inhibited slow membrane depolarization and action potentials in response to glucose stimulation (Fig. 1), and caused a membrane hyperpolarization in rat β-cells (Fig. 2). These findings are consistent with the observation that 12-O-tetradecanoylphorbol 13-acetate lowers [Ca2+]i at a stimulatory glucose concentration in pancreatic islets from obese-hyperglycaemic mice [16]. Since PMA increased KATP activity (Fig. 3), it is obvious that the hyperpolarization caused by PMA results from activation of this channel type, and PMA inhibition of glucose-induced electrical excitation of rat β-cells is ascribable at least in part to hyperpolarization resulting from an increase in KATP activity. The activity of KATP can be regulated by changes in intracellular concentration of ATP and/or ATP/ADP ratio [9]. In order to examine whether PMA decrease β-cell ATP concentration, we measured ATP content of rat islets, and results indicated that PMA did not change ATP levels in islets (Table 1). It is not clear at present whether ADP concentrations are changed by the phorbol ester. Under a condition where ATP concentration in the β-cell was clamped at a constant level, PMA increased tolbutamide-sensitive currents in response to voltage ramps from -90 to -50 mV (Fig. 4). Furthermore, in inside-out single channel recordings, PMA restored KATP activity, which had been reduced by the preceding application of ATP (Figs. 6 panels C, E). Together, these findings support the idea that PMA activation of KATP does not result from a change in ATP concentration.
In cardiac myocytes, PMA is known to activate KATP through activation of PKC [18-20]. In insulin-secreting cell lines as well, PKC activation by PMA was shown to potentiate KATP activity [11,12]. In the present study, however, the activation of KATP activity by PMA is independent on activation of PKC for two reasons. First, the PKC activation inhibitor, BIM, did not prevent either the PMA-induced hyperpolarization (Figs. 2 panels B & C) or augmentation of whole-cell membrane currents (Fig. 4 panels C & D). Second, the non-PKC-stimulating phorbol ester, 4α-PDD, mimicked the effect of PMA (Fig. 5 panel C).
Pharmacological effects of PMA independent of activation of PKC have been demonstrated in various tissues [23,24]. Since the magnitude of hyperpolarization induced by PMA (Fig. 2 panel C) and the effects of PMA on KATP activity (Fig. 6 panel E) were not significantly different between cells treated or untreated with BIM, activation of PKC seems to show little effect on KATP, at least in this cell type. In this regard, the present findings support the idea derived from previous findings on mouse [15] or obese-hyperglycaemic mouse β-cells [16]. However, PMA-induced PKC-independent hyperpolarization of β-cells was not previously reported [15,16]. The reason for the discrepancy is not clear at the moment. The difference in species (rat in the present study and mouse in previous studies) could be one explanation. In addition, hyperpolarization in response to PMA might be unnoticed under some conditions with intracellular ATP at particular concentrations, because, as shown in Fig. 6 panel F, the effect of PMA on KATP activity, which produces hyperpolarization, varied as a function of intracellular ATP concentration.
There are two possible explanations for the PKC-independent activation of KATP by phorbol esters: 1) phorbol esters may increase KATP activity in an intracellular ATP-independent manner and 2) phorbol esters may reduce sensitivity of KATP to ATP. The first possibility can be discounted because PMA does not affect KATP activity in the absence of ATP (Fig. 6 panel B). On the other hand, PMA restoration of KATP activity which had been reduced by ATP (Fig. 6 panels C, E) supports the conclusion that phorbol esters decrease KATP sensitivity to ATP.
In the present study, application of PMA potentiated insulin secretion from rat islets in response to elevation of extracellular glucose concentration (Fig. 7), supporting the hypothesis that activation of PKC synergizes secretory process activated by the glucose stimulation [3-5]. BIM is known to selectively inhibit α and β PKC isotypes [25], and rat pancreatic β-cells possess at least one PKC isotype [2]. In fact, in this study, the treatment of islets with BIM reversed the PMA effect on insulin secretion (Fig. 7). Therefore, BIM can be used as a PKC inhibitor in rat pancreatic β-cells, as already shown in other tissues including murine macrophages [26], rat adrenal cortex [27], human melanocytes [28] and rabbit vetricular myocytes [20].
Recent reports with a truncated isoform of Kir6.2 have revealed that an ATP-inhibitory site lies on the Kir6.2 subunit [29,30]. In addition, mutations of the C-terminus of Kir6.2 showed that the amino acid sequence of the C-terminus was responsible for regulation of ATP sensitivity to KATP[31]. It is not clear at present whether phorbol ester effects reflection at the C-terminus. In addition, other recent studies have shown that the membrane phospholipid phosohatidylinositol-4,5-bis-phosphate (PIP2) reduces sensitivity of KATP to ATP, and therefore, activates KATP[32-34]. To our knowledge, this is the first report of regulation of KATP activity by phorbol ester in rat β-cells. The present findings provide new insight into mechanisms of PMA regulation of insulin secretion by PMA.
Conclusion
The major and important finding in this study is that PMA impairs electrical excitation of rat pancreatic β-cells through PKC-independent mechanism. PMA increases KATP activity, induces a membrane hyperpolarization, then reduces the electrical excitability of pancreatic β-cells, leading to a decrease of insulin secretion. Reduction of sensitivity of KATP to ATP seems to underlie the PMA-induced impairment of β-cells electrical excitation in response to glucose stimulation. These results provide a novel insight of modulation mechanism of insulin secretion by PMA, increases insulin secretion by activation of PKC, and meanwhile decrease insulin secretion by impairing β-cell excitability in a PKC-independent manner in rat pancreatic β-cells.
Materials and Methods
Islet preparation and β-cell isolation
Isolation of rat islets was performed as previously described [35,36]. In short, male adult rats (Wistar) were anesthetized with diethyl ether, and 10 ml Hank's buffered saline (HBSS) containing collagenase (200 U/ml, Wako Chem., Japan) was injected into the common bile duct. The pancreas swollen with digestion solution was quickly excised and incubated in a plastic culture bottle for 20 min at 37°C. The suspension obtained by shaking the bottle was filtered through 0.5 mm metal mesh and washed with HBSS including 2% bovine serum albumin (BSA). About 100 islets were obtained from one rat with the histopaque (specific gravity 1.077, Sigma, USA) gradient method. After washing with HBSS containing 2% BSA, islets were cultured for 24 hours with 5% C02 in the tissue culture medium. Separation of islets was carried out using dispase (1000 U/ml, Godo Shusei, Japan) as previously described [35]. Separated cells were again cultured for 1–4 days. Only single cells were chosen for experiments. β-cells were identified by detecting the responses of the cells to 15 mM glucose or 0.5 mM tolbutamide (Sigma, USA).
Patch-clamp recordings
Cells were kept in a 35-mm Petri dish, and the dish was placed on the stage of an inverted microscope (IMT-2, Olympus, Tokyo, Japan). Membrane potentials and membrane currents were measured using a patch-clamp amplifier (EPC-7, List Electronic, Darmstadt, Germany). The nystatin-perforation method [37] was used to measure the membrane potential, and the standard method was used to measure whole-cell currents [38]. The resistance of the electrode, filled with the pipette solution, ranged from 2 to 4 MΩ. To measure the whole-cell membrane current, voltage ramp pulses from -90 to -50 mV were repeatedly applied using a ramp pulse generator (SET-2100, Nihon Kohden, Tokyo, Japan). The membrane capacitance ranged from 8 to 14 pF. Series resistances below 12 MΩ were accepted. Single channel current recordings were carried out by the cell-attached and inside-out configurations. All electrophysiological experiments were carried out at room temperature. Data of single channel currents were low-pass filtered at 1 KHz, digitized at 10 KHz and analyzed using a single channel current analysis program (QP-120J, Nihon Kohden). The open time probability (Po) of the single channel was calculated according to the equation Po = 1 - Pc1/N where pc is the total closed time probability and N is the total number of channels. The Po for KATP was shown as function of the concentration of ATP. Theoretical curves for ATP inhibition of KATP activity were fitted to the Hill equation (P/Po)=l/{1+ ([ATP]/ki)h} where P is the open time probability with ATP at each concentration, Po is the open time probability without ATP, [ATP] is the concentration of ATP, ki is the ATP concentration at which the inhibition is half maximal, and h is the Hill coefficient.
Measurement of ATP content
The ATP concentration of islets was measured by the luciferin-luciferase method [39]. Assay kits (FL-ASC) were purchased from Sigma. For ATP assay, 500 isolated rat pancreatic islets were suspended in 1.2 ml standard extracellular solution. After application of PMA at the final concentration of 80 nM, islet suspensions containing 17 islets were transferred to reaction tubes, and the amount of emission was immediately measured using a luminescence reader (BLR-301, Aloka, Japan). Bioluminescence produced by the luciferin-luciferase reaction was amplified and output as the count rate (cpm) after converting pulse signals. Using a calibration curve, values of counting rate were calculated in terms of moles of ATP.
Measurement of insulin secretion
Isolated islets were hand-picked under microscopy, and ten islets were distributed to each 35-mm Petri dish with 3 ml of HBSS containing 5.5 mM glucose, 20 mM HEPES, and 2% BSA. After pre-incubation for 60 min, islets were exposed to 1 μM BIM for 60 min, and then measured insulin secretion rate. The PMA effects with or without BIM on insulin secretion were measured by using immuno-reactive insulin (IRI) and stored at -20°C until the assay. IRI was measured by RIA using anti-human insulin antibody with rat insulin standard (Radio-immunoassay Kit, Insulin "Eiken", Tokyo Japan).
Solutions and Drugs
The standard extracellular solution contained 135 mM NaCl, 5.6 mM KCl, 1.2 mM MgCl2, 1 mM CaCl2, 5 mM glucose and 10 mM HEPES, pH 7.3. For membrane potential recordings, the pipette solution contained 100 mM K-gluconate, 35 mM KCl, 5 mM glucose, 0.5 mM EGTA, 10 mM HEPES and 200 mg/ml nystatin (Sigma, St. Louis, MO, USA), pH 7.2. For whole-cell recordings, the pipette solution contained 100 mM K-gluconate, 35 mM KCl, 1.2 mM MgCl2, 5 mM glucose, 0.5 mM EGTA, 10 mM HEPES, pH 7.2. For cell-attached and inside-out single channel recordings, the pipette solution contained 135 mM KCl, 1.2 mM MgCl2, 5 mM glucose, 0.5 mM EGTA and 10 mM HEPES, pH 7.3. The ionic composition of the solution inside the membrane (bath solution) in inside-out recordings was the same as that of the pipette solution, but the pH of this solution was 7.2. In inside-out recordings, ATP was added to the bath solution at various concentrations. Phorbol 12-myristate 13-acetate (PMA), 4a-phorbol 12,13-didecanoate (4a-PDD), bisindolylmaleimide (BIM) and NaCN were purchased from Sigma. PMA and 4a-PDD were dissolved in ethanol, and the final concentration of ethanol in the experimental solution was less than 0.01%. BIM was dissolved in DMSO, and the final concentration of DMSO in the experimental solution was 0.1%. Cells in the experimental bath were continuously exposed to a stream of the extracellular solution throughout the experiment. In the use of BIM, islets or cells were preincubated in the solution containing BIM (50 μM) for 30–60 min, and BIM was present in the bath solution throughout the experiment
Statistics
Data were expressed as mean ± SE of several experiments, and statistical significance was evaluated by the two-tail paired and unpaired Student's t-tests. A value of P less than 0.05 was accepted as significant.
This study was carried out in accordance with the Guidelines for Animal Experimentation, Hirosaki University, Japan.
List of abbreviations
phorbol 12-myristate 13-acetate (PMA), protein kinase C (PKC), ATP-sensitive K+ channel (KATP), 4a-phorbol 12,13-didecanoate (4a-PDD), bisindolylmaleimide (BIM).
Acknowledgments
Acknowledgments
Authors thank Professor Ronald J. Lukas for his careful reading and discussion. Kevin Ellsworth assists to prepare manuscript. This work was supported in part by the Karouji Memorial Fund for Medical Research in Hirosaki University, Hirosaki, Japan.
Contributor Information
Sechiko Suga, Email: ssuga@yahoo.com.
Jie Wu, Email: jwu2@chw.edu.
Yoshiji Ogawa, Email: yoshi35@cc.hirosaki-u.ac.jp.
Teruko Takeo, Email: ttakeo@cc.hirosaki-u.ac.jp.
Takahiro Kanno, Email: tkanno@cc.hirosaki-u.ac.jp.
Makoto Wakui, Email: mw1224@cc.hirosaki-u.ac.jp.
References
- Nishizuka Y. Protein kinase C and lipid signaling for sustained cellular responses. FASEB J. 1995;9:484–496. [PubMed] [Google Scholar]
- Rivera J, Beaven MA. Regulation of secretion from secretory cells by protein kinase C. in Protein Kinase C (Edited by Parker PJ, Dekker LV) Springer-Verlag, Heidelberg. 1997. pp. 133–166.
- Zawalich WS, Zawalich KC. Regulation of insulin secretion by phospholipase C. Am J Physiol. 1996;271:E409–416. doi: 10.1152/ajpendo.1996.271.3.E409. [DOI] [PubMed] [Google Scholar]
- Deeney JT, Cunningham BA, Chheda S, Bokvist K, Juntti-Berggren L, Lam K, Korchak HM, Corkey BE, Berggren PO. Reversible Ca2+-dependent translocation of protein kinase C and glucose-induced insulin release. J Biol Chem. 1996;271:18154–18160. doi: 10.1074/jbc.271.18.10623. [DOI] [PubMed] [Google Scholar]
- Ravier MA, Gilon P, Henquin JC. Oscillations of insulin secretion can be triggered by imposed oscillations of cytoplasmic Ca2+ or metabolism in normal mouse islets. Diabetes. 1999;48:2374–2382. doi: 10.2337/diabetes.48.12.2374. [DOI] [PubMed] [Google Scholar]
- Wang JL, Corbett JA, Marshall CA, McDaniel ML. Glucose-induced insulin secretion from purified beta-cells: a role for modulation of Ca2+ influx by cAMP-and protein kinase C-dependent signal transduction pathways. J Biol Chem. 1993;268:7785–7791. [PubMed] [Google Scholar]
- Arkhammar P, Berggren LJ, Larsson O, Welsh M, Nanberg E, Sjoholm A, Kohler M, Berggren PO. Protein kinase C modulates the insulin secretory process by maintaining a proper function of β-cell voltage-activated Ca2+ channels. J Biol Chem. 1994;269:2743–2749. [PubMed] [Google Scholar]
- Hughes SJ, Ashcroft SJH. Effects of phorbol ester and clomiphene on protein phosphorylation and insulin secretion in rat pancreatic islets. Biochem J. 1988;249:825–830. doi: 10.1042/bj2490825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft FM, Ashcroft SJH. Mechanism of insulin secretion, in Insulin: Molecular Biology to Pathology (Edited by Ashcroft FM, Ashcroft SJH) Oxford University Press, New York. 1992. pp. 97–150.
- Ashcroft FM, Gribble FM. ATP-sensitive K+ channels and insulin secretion: their role in health and disease. Diabetologia. 1999;42:903–919. doi: 10.1007/s001250051247. [DOI] [PubMed] [Google Scholar]
- Dunne MJ, West-Jordan JA, Abraham RJ, Edwards RHT, Petersen OH. The gating of ATP-sensitive K+ channels in insulin-secreting cells can be modulated by changes in the ratio of ATP4-/ADP3- and by non-hydrolysable analogues of both ATP and ADP. J Membr Biol. 1988;104:165–177. doi: 10.1007/BF01870928. [DOI] [PubMed] [Google Scholar]
- de Weille JR, Schmid-Antomarchi H, Fosset M, Lazdunski M. Regulation of ATP-sensitive K+ channels in insulinoma cells: activation by somatostatin and protein kinase C and the role of cAMP. Proc Natl Acad Sci USA. 1989;86:2971–2975. doi: 10.1073/pnas.86.8.2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollheim CB, Dunne MJ, Peter-Reisch B, Brozzon R, Pozzan T, Petersen OH. Activators of protein kinase C depolarise insulin-secreting cells by closing K+ channels. EMBO J. 1988;7:2443–2449. doi: 10.1002/j.1460-2075.1988.tb03090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunne MJ. Phorbol myristate acetate and ATP-sensitive potassium channels in insulin-secreting cells. Am J Physiol. 1994;267:C501–C506. doi: 10.1152/ajpcell.1994.267.2.C501. [DOI] [PubMed] [Google Scholar]
- Bozem M, Nenquin M, Henquin JC. The ionic, electrical, and secretory effects of protein kinase C activation in mouse pancreatic β-cells: studies with a phorbol ester. Endocrinology. 1987;121:1025–1033. doi: 10.1210/endo-121-3-1025. [DOI] [PubMed] [Google Scholar]
- Arkhammar P, Nilsson T, Welsh M, Welsh N, Berggren PO. Effects of protein kinase C activation on the regulation of the stimulus-secretion coupling in pancreatic β-cells. Biochem J. 1989;264:207–215. doi: 10.1042/bj2640207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Light PE, Sabir AA, Allen BG, Walsh MP, French RJ. Protein kinase C-induced changes in the stoichiometry of ATP binding activate cardiac ATP-sensitive K+ channels. Circ Res. 1996;79:399–406. doi: 10.1161/01.res.79.3.399. [DOI] [PubMed] [Google Scholar]
- Liu Y, Gao WD, O'Rourke B, Marban E. Synergistic modulation of ATP-sensitive K+ currents by protein kinase C and adenosine. Circ Res. 1996;78:443–454. doi: 10.1161/01.res.78.3.443. [DOI] [PubMed] [Google Scholar]
- Hu K, Duan D, Li GR, Nattel S. Protein kinase C activates ATP-sensitive K+ current in human and rabbit ventricular myocytes. Circ Res. 1996;78:492–498. doi: 10.1161/01.res.78.3.492. [DOI] [PubMed] [Google Scholar]
- Hu K, Li GR, Nattel S. Adenosine-induced activation of ATP-sensitive K+ channels in excised membrane patches is mediated by PKC. Am J Physiol. 1999;276:H488–H495. doi: 10.1152/ajpheart.1999.276.2.H488. [DOI] [PubMed] [Google Scholar]
- Nakamura J, Suda T, Ogawa Y, Takeo T, Suga S, Wakui M. Protein Kinase C-dependent and – independent inhibition of Ca2+ influx by phorbol ester in rat pancreatic β-cells. Cell Signal. 2001;13:199–205. doi: 10.1016/S0898-6568(01)00136-X. [DOI] [PubMed] [Google Scholar]
- White JR, Huang CK, Hill JM, Jr, Naccache PH, Becker EL, Sha'afi RI. Effect of phorbol 12-myristate 13-acetate and its analogue 4α-phorbol 12, 13 didecanoate on protein phosphorylation and lysosomal enzyme release in rabbit neutrophils. J Biol Chem. 1994;259:8605–8611. [PubMed] [Google Scholar]
- Su LN, Toscano WA, Jr, Kennedy AR. Suppression of phorbol ester-enhanced radiation-induced malignancy in vitro by protease inhibitors is independent of protein kinase C. Biochem Biophys Res Commun. 1991;176:18–24. doi: 10.1016/0006-291x(91)90883-9. [DOI] [PubMed] [Google Scholar]
- Murphy JJ, Yaxley JC, Norton JD. Evidence for protein kinase C-independent pathways mediating phorbol ester induced plasmacytoid differentiation of B chronic lymphocytic leukemia cells. Biochem Biophys Acta. 1991;1092:110–118. doi: 10.1016/0167-4889(91)90184-Y. [DOI] [PubMed] [Google Scholar]
- Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F, Duhame L, Charon D, Kirilovsky J. The bisindolylmaleimide GF109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem. 1991;266:15771–15781. [PubMed] [Google Scholar]
- Severn A, Wakelam MJ, Liew FY. The role of protein kinase C in the induction of nitric oxide synthesis by murine macrophages. Biochem Biophys Res Commun. 1992;188:997–1002. doi: 10.1016/0006-291x(92)91330-s. [DOI] [PubMed] [Google Scholar]
- Kapas S, Purbrick A, Hinson JP. Role of tyrosine kinase and protein kinase C in steroidogenic actions of angiotensin II, alpha-melanocyte-stimulating hormone and corticotropin in the rat adrenal cortex. Biochem J. 1995;305:433–438. doi: 10.1042/bj3050433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsberg CJ, Warenius HM, Friedmann PS. Ultraviolet radiation-induced melanogenesis in human melanocytes: effects of modulating protein kinase C. J Cell Sci. 1994;107:2591–2597. doi: 10.1242/jcs.107.9.2591. [DOI] [PubMed] [Google Scholar]
- Tucker SJ, Gribble FM, Zhao C, Trapp S, Ashcroft FM. Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature. 1997;387:179–183. doi: 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]
- Proks P, Gribble FM, Adhikar R, Tucker SJ, Ashcroft FM. Involvement of the N-terminus of Kir6.2 in the inhibition of the KATP channel by ATP. J Physiol. 1999;514:19–25. doi: 10.1111/j.1469-7793.1999.019af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker SJ, Gribble FM, Proks P, Trapp S, Ryder TJ, Haug T, Reimann F, Ashcroft FM. Molecular determinants of KATP channel inhibition by ATP. EMBO. 1998;17:3290–3296. doi: 10.1093/emboj/17.12.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CL, Feng S, Hilgemann DW. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbg. Nature. 1998;391:803–806. doi: 10.1038/35882. [DOI] [PubMed] [Google Scholar]
- Shyng SL, Nichols CG. Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science. 1998;282:1138–1141. doi: 10.1126/science.282.5391.1138. [DOI] [PubMed] [Google Scholar]
- Baukrowitz T, Schulte U, Oliver D, Herlitze S, Krauter T, Tucker SJ, Ruppersberg JP, Fakler B. PIP2 and PIP as determinants for ATP inhibition of KATP channels. Science. 1998;282:1141–1144. doi: 10.1126/science.282.5391.1141. [DOI] [PubMed] [Google Scholar]
- Suga S, Kanno T, Nakano K, Takeo T, Dobashi Y, Wakui M. GLP-I (7-36) amide augments Ba2+ current through L-type Ca2+ channel of rat pancreatic β-cell in a cAMP-dependent manner. Diabetes. 1997;46:1755–1760. doi: 10.2337/diab.46.11.1755. [DOI] [PubMed] [Google Scholar]
- Kanno T, Suga S, Nakano K, Kamimura N, Wakui M. Corticotropin-releasing factor modulation of Ca2+ influx in rat pancreatic β-cells. Diabetes. 1999;48:1741–1746. doi: 10.2337/diabetes.48.9.1741. [DOI] [PubMed] [Google Scholar]
- Horn R, Marty A. Muscarinic activation of ion currents measured by a new whole-cell recording method. J Gen Physiol. 1988;92:145–159. doi: 10.1085/jgp.92.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. P flugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Ringer D. Batch analysis of the ATP content of bovine sperm, oocytes, and early embryos using a scintillation counter to measure the chemiluminescence produced by the luciferin-luciferase reaction. Anal Biochem. 1997;246:67–70. doi: 10.1006/abio.1996.9978. [DOI] [PubMed] [Google Scholar]