

Abstract
CYP121, the cytochrome P450 enzyme in Mycobacterium tuberculosis that catalyzes a single intramolecular C–C cross-linking reaction in the biosynthesis of mycocyclosin, is crucial for the viability of this pathogen. This C–C coupling reaction represents an expansion of the activities carried out by P450 enzymes distinct from oxygen insertion. Although the traditional mechanism for P450 enzymes has been well studied, it is unclear whether CYP121 follows the general P450 mechanism or uses a different catalytic strategy for generating an iron-bound oxidant. To gain mechanistic insight into the CYP121-catalyzed reaction, we tested the peroxide shunt pathway by using rapid kinetic techniques to monitor the enzyme activity with its substrate dicyclotyrosine (cYY) and observed the formation of the cross-linked product mycocyclosin by LC-MS. In stopped-flow experiments, we observed that cYY binding to CYP121 proceeds in a two-step process, and EPR spectroscopy indicates that the binding induces active site reorganization and uniformity. Using rapid freeze-quenching EPR, we observed the formation of a high-spin intermediate upon the addition of peracetic acid to the enzyme–substrate complex. This intermediate exhibits a high-spin (S = 5/2) signal with g values of 2.00, 5.77, and 6.87. Likewise, iodosylbenzene could also produce mycocyclosin, implicating compound I as the initial oxidizing species. Moreover, we also demonstrated that CYP121 performs a standard peroxidase type of reaction by observing substrate-based radicals. On the basis of these results, we propose plausible free radical–based mechanisms for the C–C bond coupling reaction.
Keywords: cytochrome P450, electron transfer, enzyme kinetics, metabolism, tuberculosis, C–C bond coupling, cyclodipeptide, diradical, oxygen activation
Introduction
Mycobacterium tuberculosis causes more deaths annually worldwide than any other known pathogen. As the causative agent of tuberculosis in humans, it is one of the most dangerous and difficult-to-combat bacterial infections. Approximately 10.4 million people suffered from tuberculosis in 2015 with 1.5 million deaths (1). A primary reason for the effectiveness of the pathogen is the recent development of drug- and multidrug-resistant M. tuberculosis strains. Nearly 10% of new infection cases are multidrug-resistant tuberculosis. Resistance to common antibiotics makes treatment very difficult. As the number of strains resistant to frontline drugs grows, pressure is increasing for the identification of potential new targets to combat M. tuberculosis infections and the development of new types of drugs and drug classes (2).
A significant milestone in the molecular biology of M. tuberculosis was the sequencing of the full genome in 1998. The results revealed a large number of genes encoding for cytochrome P450 enzymes (3). A total of 20 different P450-encoding genes were found in M. tuberculosis, far more than any previously sequenced bacterial genome at the time. Two P450 enzymes were quickly identified as potential new drug targets, CYP51 and CYP121. CYP51 was found to possess sterol demethylase activity (4–6). Initial studies found that both CYP51 and CYP121 had low binding constant values to azole- and triazole-based drugs, with CYP121 having a higher affinity (6, 7). A knock-out study of rv2276, the gene encoding for CYP121 in M. tuberculosis, highlighted the physiological importance of CYP121 (8). These findings set the stage for CYP121 as a possible novel target to combat M. tuberculosis infections.
The first breakthrough in the biochemical characterization of CYP121 was the initial determination of the X-ray crystal structure solved at atomic resolution by Munro and co-workers (9). The structure has allowed for the characterization of various small molecules binding to the enzyme active site. The CYP121 structure has complemented the research approach for finding new azole-based inhibitors and characterizing its interactions with current azole-based drugs (10–16). Binding constants for several azole molecules were determined, and these values correspond very closely to minimum inhibition constants against M. tuberculosis, further validating CYP121 as a viable drug target (8).
The next breakthrough in the study of CYP121 was the report identifying the native substrate. The gene encoding CYP121 in M. tuberculosis was found in an operon-like structure with the gene rv2275 (3, 17). Characterization of Rv2275 in Escherichia coli revealed that the products were mainly tyrosine-containing cyclo dipeptides, the majority of which were cyclo-(l-Tyr-l-Tyr) (cYY)3 (18). At the same time, the crystal structure for CYP121 in complex with cYY was also solved by Belin et al. (19). Assays conducted on CYP121 utilizing a ferredoxin and ferredoxin reductase system demonstrated that CYP121 catalyzes multiple turnovers of cYY to form mycocyclosin as the single major product in the presence of NADPH (19). The product exhibits a cross-link between the respective carbons in the ortho position of cYY tyrosine moieties. P450 enzymes are normally known to promote a wide range of catalytic activities of aliphatic and aromatic hydroxylation, dealkylation, desaturation, epoxidation, deamination, dehalogenation, dehydration, and isomerization. The C–C bond formation represents an unusual activity of the P450 enzyme superfamily (20, 21).
However, important chemical and biological questions remain unanswered regarding CYP121. The mechanism for cross-link formation and the identity of the oxidizing species or the physiological relevance of mycocyclosin are still unclear. A quantum mechanics/molecular mechanics study supports the catalytic mechanism via formation of a diradical intermediate species with the cross-link being formed non-enzymatically in solution (22). The current work expands on previous research by investigating the reaction pathway of the CYP121 system. CYP121 is uniquely attractive because of the non-canonical P450 chemistry it catalyzes and the question of whether or not it follows the classical mechanism of P450s. The mechanistic question under investigation includes the “short circuit,” also known as the catalytic “peroxide shunt” pathway, for the formation of a ferric hydroperoxide adduct complex and the subsequent oxo-ferryl species (Fig. 1) (23, 24). Toward this aim, we have carried out rapid kinetics, spectroscopy, and LC-MS analysis to investigate the CYP121 reaction mechanism.
Figure 1.
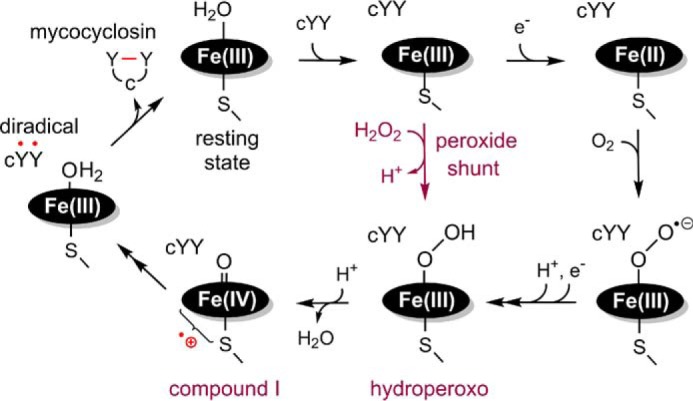
Proposed peroxide shunt pathway of CYP121 reaction within the general P450 mechanism. The shunt pathway directly arrives at the hydroperoxo intermediate without the need for receiving electrons and a proton from an external donor (i.e. NAD(P)H) via a reductase system.
Results
Spectral properties of cYY binding to CYP121
The as-isolated CYP121 exhibits a Soret peak centered at 416 nm. Two absorbance features at 538 and 565 nm in the α/β region and an additional minor band at 648 nm are also present. CYP121 displays type I characteristic spectral changes in the UV-visible heme Soret spectrum upon binding of cYY (Fig. 2A), as is frequently observed in P450 enzymes during the binding of endogenous substrates and xenobiotics (25). After substrate binding, the Soret peak blue-shifts to 395 nm. However, a significant shoulder peak remains even when saturating concentrations of cYY (400 μm, 80:1 ratio of the substrate over enzyme, 20 times the cYY Kd value of 21.3 μm) (19) were used. This phenomenon has previously been observed among the P450 family as well as in CYP121 (19). In the α/β region, additional changes occurred with the loss of the two peaks to generate a new broad feature at 516 nm with shoulders at 541 and 571 nm, whereas the charge transfer peak at 651 nm increased in intensity (19). The spectroscopic changes observed for substrate binding are summarized in Table 1. The spin transition was less pronounced when monitored by low-temperature EPR spectroscopy for samples frozen by liquid ethane. The incomplete spin-state conversion most likely originates from the low temperature used in the EPR studies. A slight decrease in the g-anisotropy of the low-spin species and a new high-spin resonance at g = 8 is observed (Fig. 2B).
Figure 2.
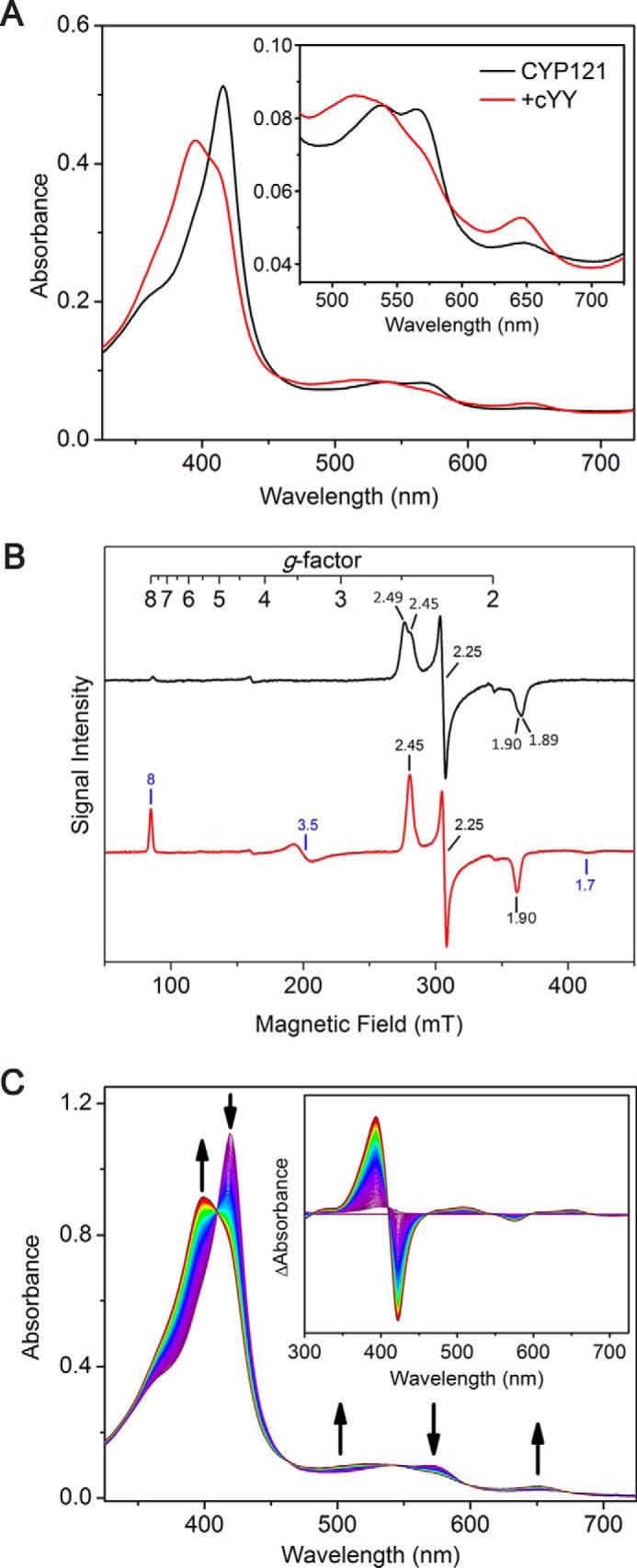
Spectroscopic characterization of substrate cYY binding to CYP121. All substrate binding experiments were monitored using nearly saturating concentrations of cYY, 400 μm. A, UV-visible spectra of CYP121 (10 μm) incubated with cYY; B, EPR characterization of CYP121 (250 μm; black spectrum) binding to cYY (red spectrum); C, stopped-flow UV-visible spectroscopic monitoring of the kinetics of cYY binding to CYP121 shows complete conversion to the ES complex form within the first 250 ms.
Table 1.
Summary of spectra profile changes observed for substrate binding to CYP121 and ES complex reaction with peracetic acid as monitored by stopped-flow UV-visible spectroscopy
| Transition | Positive peak |
Negative peak |
Isosbestic point |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 | 4 | 5 | 6 | |
| nm | nm | nm | ||||||||||
| cYY binding | 393 | 508 | 651 | 423 | 575 | 410 | 460 | 540 | 597 | 674 | ||
| ES + PA | 433 | 573 | 393 | 517 | 651 | 345 | 404 | 473 | 551 | 629 | 672 | |
Stopped-flow kinetics studies of cYY binding
Stopped-flow experiments to determine the microscopic rate constants (kon and koff) for cYY binding to Fe(III)-CYP121 were performed by monitoring the formation of the ES complex at 388 nm using a stopped-flow spectrometer. This wavelength was chosen because in the difference spectra, the largest amplitude change is at 388 nm (Fig. 2C). Fig. 3A shows stopped-flow time traces varying the [cYY] from 20 to 700 μm. Fitting the kinetic traces to single exponential equations resulted in a very poor fit with significant residual amplitudes that show systematic dependence with [cYY]. In contrast, a very good fit was obtained by using a two-exponential equation (Fig. 3A).
Figure 3.

Kinetic characterization of substrate cYY binding to CYP121. A, single-wavelength stopped-flow data collected at 388 nm monitoring formation of enzyme–substrate complex. Reaction conditions were pH 7.5, 21 °C, 5.05 μm heme after mixing. Residuals from fitting stopped-flow data with single- and double-exponential curves are shown. B, plot of reciprocal relaxation times from double-exponential fitting of single-wavelength stopped-flow data collected at pH 7.5 (5 μm heme after mixing). The slower phase shows saturation behavior at high cYY concentrations. Experiments were limited by low solubility of cYY in water. Error bars originate from fitting S.D. values from fitting multiple experimental data sets. C, replots of cYY concentration dependence data showing the sum and product of two observed reciprocal relaxation times from double-exponential fitting of data.
The plots of the observed rates (1/τ1 and 1/τ2), from double-exponential fitting of the [cYY] dependence are shown in Fig. 3B. Whereas 1/τ1 shows linear [cYY] dependence, 1/τ2 shows parabolic concentration dependence, suggesting a stepwise mechanism for cYY binding (Reaction 1) to form the binary ES complex. Fitting both 1/τ1 to a linear equation and 1/τ2 to a hyperbolic equation yielded non-zero y intercepts, suggesting that both steps are reversible. From the replotting of the [cYY] dependence data (Fig. 3C), taking both the sum (1/τ1 + 1/τ2) and the product (1/τ1 × 1/τ2) of the observed reciprocal relaxation times, the microscopic rate constants (k1, k−1, k2, and k−2) can be calculated from the slope and the y intercept from the two graphs (Reaction 1) (26). The second-order rate constant (k1) of 0.065 ± 0.002 μm−1 s−1 is fairly low where the initial binding step is slower than both k−1 and k2 (k1 < k−1 and k2 when [cYY] < 62 μm). The reverse rates (k−1 and k−2) are also on the same order of magnitude and have similar values to k2. These suggest a highly reversible system where ES* and ES are in rapid equilibrium.
| (Eq. 1) |
Reaction 1 shows the proposed multistep mechanism for cYY binding to Fe(III)CYP121, with rate constants obtained from double exponential fitting of stopped-flow data from [cYY] dependence data (Fig. 3).
Transient kinetic studies of the reaction of CYP121 with peracetic acid in the presence and absence of substrate cYY
If CYP121 follows the general cytochrome P450 mechanism, it would be able to generate the enzyme-based key oxidant, compound I, through the well-established peroxide shunt pathway. The peroxide shunt pathway bypasses the need for NAD(P)H and a redox mediator system to supply electrons and protons to the heme-bound O2 (27–29). Among H2O2, cumene peroxide, t-butyl peroxide, meta-chloroperoxybenzoic acid, and peracetic acid (PAA) tried at pH 7.4, only H2O2 and PAA gave significant reactions. Because PAA has the most apparent reaction at lower concentrations during our initial tests, the following work described in this study was mostly focused on PAA. The reaction was initiated by incubating the peroxide oxidant with 5 μm CYP121 premixed with cYY to determine whether the shunt pathway would be a viable route to generate the C–C cross-linked product. A parallel experiment was performed in the absence of substrate as a control to assist the identification of the intermediate species.
The enzyme was first preincubated with the substrate (600 μm) and subsequently mixed with increasing concentrations of PAA, and the reaction was monitored for 30 s by stopped-flow UV-visible spectroscopy (Fig. 4A). Over the course of the reaction, the difference spectra show several transitions (Fig. 4B). During the first 300 ms, the ES complex absorbance at 395 nm decreases concomitantly with an increase at 427 nm. As the reaction continues, the 395-nm Soret peak of the ES complex continues to decrease while the 427 nm peak increases and red-shifts to 433 nm. Finally, when the reaction proceeds for longer than 10 s, the isosbestic points become less clear, suggesting that heme bleaching becomes a contributing factor in the reaction. Monitoring the reaction rate as a function of PAA concentration allows for the determination of the apparent pseudo-first-order rate constant kobs of (7.2 ± 0.1) × 10−4 s−1.
Figure 4.

Transient kinetic analysis of the ES complex (10 μm CYP121 and 400 μm CYY) reacting with peracetic acid (2 mm). A, full spectra of the first 10 s of reaction showing decay of the ES complex. B, difference spectra showing return back to the resting enzyme within 10 s with multiple clear isosbestic points signifying a clear transition back to the resting state during this time period and showing the development of a 427-nm intermediate between 5 and 449 ms. After this time, the intermediate decayed and shifted to 433 nm. C, single-wavelength kinetic traces monitoring the regeneration of the resting enzyme at the expense of the ES complex. D, plots of substrate binding (described in the legend to Fig. 3) showing the formation of the ES complex.
For comparison, when the enzyme is reacted with PAA in the absence of substrate, the first 100 ms generate a new intermediate species (Fig. 5A). The formation of the intermediate species is described by a decrease in the intensity of the Soret peak with an increase in the shoulder peak near 379 nm. Near the α/β region of the spectra, decreases at 543 and 574 nm are observed with the formation of two minor peaks at 616 and 704 nm. The difference spectra clearly show the formation of a new species. Five isosbestic points at 300, 392, 447, 517, and 584 nm are also observed in the difference spectra (Fig. 5A, inset). The clear isosbestic points suggest a direct transition from resting state heme directly to the first intermediate species. From 100 ms to ∼2 s, a second transition occurs, forming a second intermediate species (Fig. 5B). This second intermediate is distinct with isosbestic points at 296, 340, 427, and 580 nm. The second intermediate is highlighted by two noticeable absorption changes at 421 and 440 nm, decreasing and increasing, respectively. The final transition occurs from 2 to 20 s of CYP121 reacting with PAA (Fig. 5C). The difference spectra contain two prominent peaks centered at 412 nm (increasing) and 440 nm (decreasing) (Fig. 5C, inset). A complete profile of the transitions observed is compiled in Table 2. The 433-nm species found in the reaction of ES complex with PAA is not present in the reaction when cYY is absent.
Figure 5.
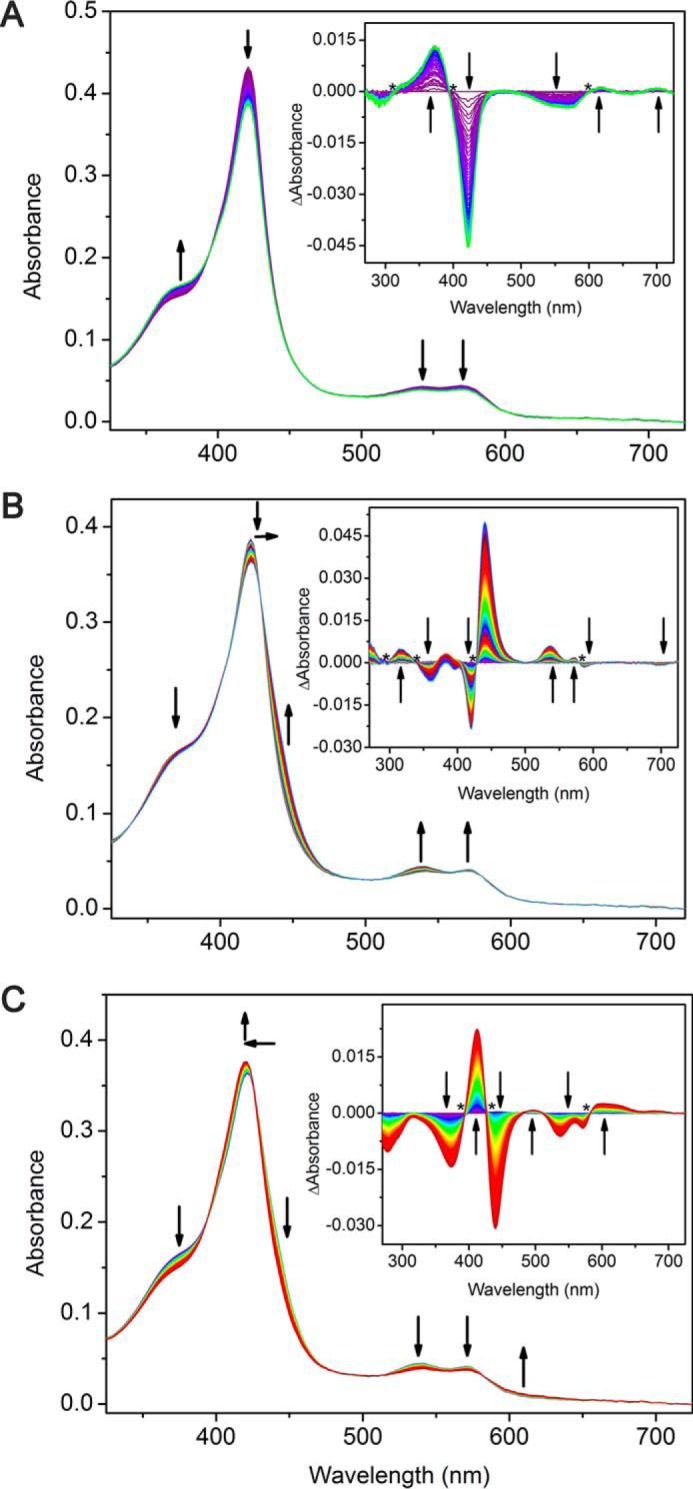
Stopped-flow UV-visible kinetic characterization of CYP121 (5 μm) reacting with peracetic acid (2 mm). A, 0–100 ms; B, 100 ms to 2 s; C, 2–20 s. Insets, difference spectra showing three intermediates observed for each transition.
Table 2.
Summary of intermediate spectrum profiles
Shown are the parameters for the difference spectra obtained from reacting CYP121 and PAA, monitored by stopped-flow UV-visible spectroscopy.
| Transition | Positive peak |
Negative peak |
Isosbestic point |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | |
| nm | nm | nm | |||||||||||
| 100 ms | 379 | 469 | 616 | 704 | 421 | 543 | 574 | 313 | 392 | 601 | |||
| 2 s | 318 | 382 | 440 | 535 | 571 | 359 | 421 | 587 | 296 | 340 | 427 | 580 | |
| 20 s | 412 | 495 | 595 | 624 | 681 | 374 | 440 | 538 | 572 | 393 | 425 | 583 | |
Detection of mycocyclosin from the peroxide shunt pathway
Because the P450 enzyme-mediated C–C bond coupling mechanism has not yet been elucidated, one cannot assume that the shunt pathway in CYP121 will lead to the generation of the reported product. To demonstrate the relevance of PAA as an oxidative source for mechanistic studies, the reaction mixture was characterized by LC-MS. When the substrate was analyzed alone or pairwise in the presence of either PAA or CYP121 only, a single peak elutes with a retention time of ∼8 min (Fig. 6A). This peak possesses an m/z of 325, which is the expected value for the cYY substrate (Fig. 6B). When all three components, CYP121, cYY, and PAA, are combined and allowed to react (see “Experimental procedures”), the reaction mixture contains new peaks, and the peak with a retention time close to 5 min shows an m/z of 323, which is consistent with the cross-linked mycocyclosin product (Fig. 6, C and D). These data demonstrate that CYP121 can utilize the shunt pathway to carry out the C–C cross-linking reaction on cYY and generate mycocyclosin.
Figure 6.

Mass spectrometry analysis of ferric CYP121-mediated cYY cross-linking using peracetic acid as the oxidant. A, total ion count of enzyme assay mixture; B, mass-spectral detection of cYY (325 m/z) at 8.1 min; C, selected ion monitoring of both cYY and mycocyclosin shows a new peak at 4.8 min after incubation of reaction mixture for 10 min; D, molecular mass detection of the cross-linked mycocyclosin (323 m/z) after incubation of the enzyme, cYY, and peracetic acid.
Characterization of the shunt reaction by EPR spectroscopy
To gain more insight into the CYP121 cross-linking reaction, rapid freeze-quench EPR samples were made in which CYP121 was rapidly mixed with PAA in either the presence or absence of cYY before quenching in liquid ethane at various time points. The quenching times chosen were guided by our stopped-flow studies described above. When the reaction of the ES complex with PAA is quenched at 5 ms, EPR data reveal a nearly complete disappearance of the ES complex EPR signal (≥75%). Instead, the EPR spectra from samples trapped at different time points in the millisecond time window show a new high-spin (S = 5/2) ferric heme species with g values of 6.87, 5.77, and 2.00 (Fig. 7). When allowed to react for longer times, the high-spin species decreases in intensity (Table 3). However, the decay of the high-spin species was not accompanied by regeneration of the low-spin ferric signal. Noticeably, a new EPR-silent heme species was formed during this time. The remaining minor low-spin heme signal also continuously decays during this period. During the same time window, the 427-nm species was developed and shifted to 433 nm in the stopped-flow experiments. The final sample quenched at 10 s after mixing gives a spectrum with no sign of the low-spin heme. The high-spin ferric signal intensity is significantly reduced compared with the 5 ms sample. A new significant portion of adventitious iron, presumably from heme degradation, and a free radical species are observed. Minor EPR species at g = 2 are observed in the samples, but they are unlike any characterized tyrosyl radicals. Thus, the radical species are unlikely to be associated with the cYY reaction but are more likely to be intermediates of side reactions toward heme degradation. When the rapid freeze-quench EPR study was performed with 0.4 mm PAA with more time points in the first 180 s, similar results were obtained (i.e. the formation of the g = 6 species maximizes and then later starts to decay). The only difference is that the g = 6 species is lower in intensity compared with the 10 mm PAA, and the heme-bleaching g = 4.3 signal does not occur at the end of the reaction.
Figure 7.
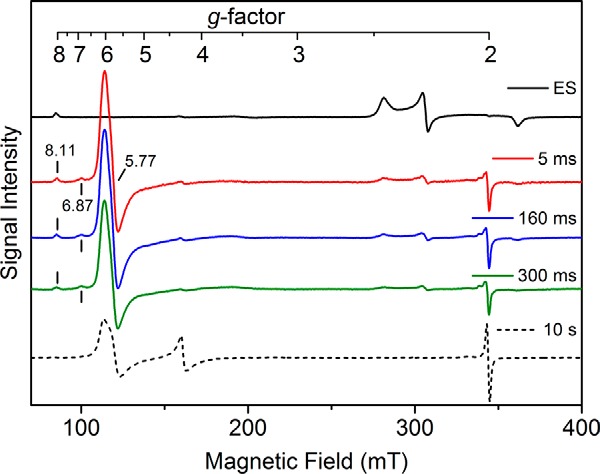
EPR analysis of the enzyme–substrate complex (250 μm) reacting with peracetic acid (10 mm). The black trace shows the ES complex of cYY and CYP121. A new high-spin ferric heme intermediate is formed in 5 ms with concomitant decay of the low-spin ES complex (red trace). The EPR spectra also include the new high-spin intermediate at 160 (blue) and 300 ms (green). The final decayed reaction complex after 10 s of reaction is shown by a dashed line, which does not contain the intermediate. EPR spectra were obtained at 5 K, 9.6-GHz microwave frequency, and 1-milliwatt microwave power. mT, milliteslas.
Table 3.
Summary of the resonance components in the rapid freeze-quench EPR spectra of ES complex reacting with PAA
Values are given as a percentage of the amplitude of the resonance using the 5-ms intermediate sample as a reference.
| g value | ES complex | 5 ms | 160 ms | 300 ms | 10 s |
|---|---|---|---|---|---|
| % | % | % | % | % | |
| 5.77 | 0 | 100 | 98.9 | 80 | 36 |
| 6.87 | 0 | 100 | 77 | 80 | 0 |
| 8.11 | 100 | 100 | 100 | 100 | 0 |
For the reaction of CYP121 alone with PAA, a series of freeze-quench samples were prepared and analyzed by EPR (Fig. 8). After quenching at 100 ms, a significant decrease in the low-spin heme signal was observed with the formation of a minor new high-spin species with g values of 6.67, 5.77, and 2.0. This species is distinctly different from the high-spin heme intermediate observed in the reaction of the ES complex with PAA. After reacting for longer times, the 2-s sample resulted in a further reduction of the low-spin heme and a continued increase of the high-spin species. The high-spin heme was present as a heterogeneous species with three peaks featured on the spectral profile. Allowing the reaction to proceed further and quenching at 20 s causes a greater formation of the high-spin species, with the g = 6.67 feature becoming more prominent and distinguished.
Figure 8.
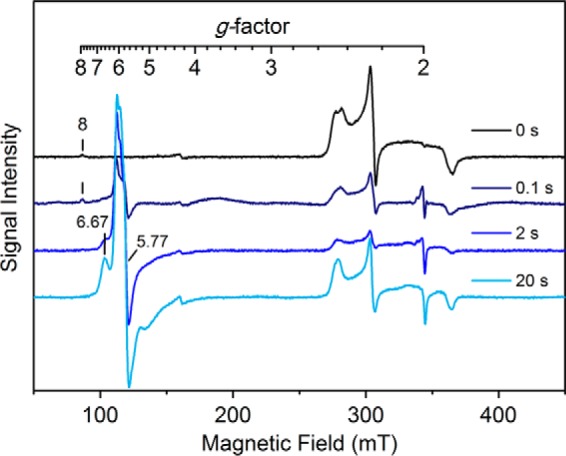
EPR analysis of the resting CYP121 enzyme (250 μm) reacting with peracetic acid (2 mm). The resting CYP121 enzyme is present as a heterogeneous low-spin ferric heme species (black trace). CYP121 was mixed in a 1:1 ratio and quenched in liquid ethane after reaction times of 100 ms (navy trace), 2 s (blue), and 20 s (cyan). The reaction generated at least one high-spin species at g = 6.67, 5.77, and 2 that increased in intensity after longer reaction times. EPR spectra were obtained at 5 K, 9.6-GHz microwave frequency, and 1-milliwatt microwave power. mT, milliteslas.
The shunt reaction using iodosylbenzene
The oxidant of cYY could be a ferric peroxide intermediate or a compound I species. In fact, the fatty acid peroxide-metabolizing P450s, CYP5, CYP8A, and CYP74, are believed to directly utilize a ferric peroxide to initiate substrate oxidation reactions to generate a compound II and a substrate radical (30–33). In C–C bond cleavage reactions catalyzed by 11A1, 17A1, 19A1, and 51A1, the active oxidant has been controversial (34, 35). Our EPR detection of a ferric intermediate prompted us to consider whether the intermediate is an active species or the precursor of a compound I intermediate, as illustrated in Fig. 1. Following the strategy of the CYP17A1 study by Guengerich and co-workers (34) using iodosylbenzene, we performed a comparative study with PAA under the exact same reaction conditions. Fig. 9 shows that the anticipated product was formed using iodosylbenzene as the oxidant, although additional side products were also produced. This result sheds light on the catalytic mechanism and is in favor of the compound I intermediate as the catalytic oxidant, because iodosylbenzene cannot form a peroxy intermediate. However, such evidence does not preclude the possibility of the ferric peroxide intermediate detected in the PAA reaction being an active species.
Figure 9.
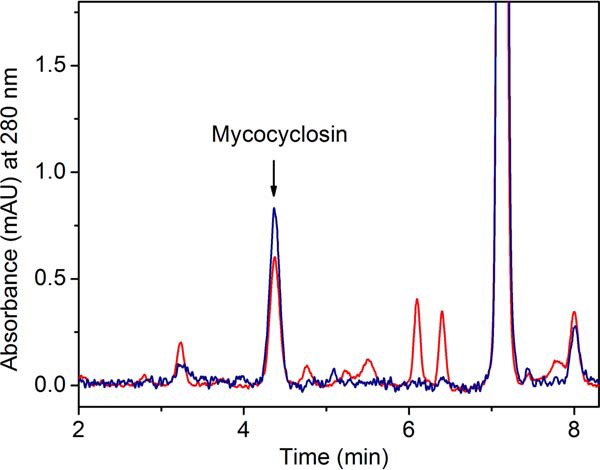
HPLC chromatogram (monitored at 280 nm) showing formation of mycocyclosin from reactions of 5 μm CYP121, 400 μm cYY iodosylbenzene (PhIO, red trace), or 400 μm PAA (blue). The product mycocyclosin elutes at 4.3 min. The PhIO reaction generates more side products than the PAA reaction. For the PhIO reaction, the background at 358 nm was subtracted to correct the baseline. mAU, milliabsorbance units.
The P450 enzyme CYP121 catalyzes a peroxidase reaction
If the cross-linking of cYY is a radical mechanism, then the CYP121-mediated enzymatic action is a peroxidase-like reaction, where two substrate-based radicals would be expected to form by compound I and then compound II in succession. We found that CYP121 is able to perform catalytic turnover of the peroxidase substrate, 2,29-azino-bis(3-ethylbenzothiazoline-6-sulfonate) (ABTS) to generate ABTS cation radical (Fig. 10). When the [PAA] is varied the kinetic parameters, kcat and Km, are 4.1 ± 0.1 s−1 and 1.5 ± 0.1 mm, respectively. Because of the relatively high Km for PAA, the kcat and Km cannot be accurately determined as a function of [ABTS] due to complications of enzyme damage and non-enzymatic reaction between ABTS and PAA at high oxidant concentrations. Nonetheless, the results shown in Fig. 10 show that CYP121 is able to function as a peroxidase and generate substrate-based radicals.
Figure 10.

Steady-state kinetic analysis of the peroxidase activity of CYP121 (100 nm) with ABTS (2 mm) and PAA (0.02–5 mm). A, representative spectra of CYP121 peroxidase activity as monitored by UV-visible spectroscopy for 1.5 min. B, EPR spectroscopic detection of ABTS+·. C, single-wavelength stopped-flow data monitoring formation of ABTS+· at 420 nm. D, Michaelis–Menten fit to the kinetic data as a function of PAA concentration. AU, absorbance units.
Discussion
In addition to CYP121, several other P450 enzymes have been identified as performing C–C coupling reactions. For instance, the biosynthesis of staurosporine requires an intramolecular C–C bond between two indole rings at the C2 positon and is catalyzed by P450 StaP (36). In mammals, several P450s are reported to form C–C bonds in the synthesis of salutaridine, a morphine precursor (37, 38). In plant secondary metabolism, P450 enzymes CYP80G2 and CYP719B1 catalyze intramolecular C–C phenol-coupling reactions (20). The C–C bond formation represents a novel and small, but growing, activity of the P450 enzyme superfamily (21). The work described here represents an initial experimental effort to unveil the C–C bond coupling mechanisms mediated by the P450 enzymes.
Structural reorganization and active site uniformity introduced by substrate binding
The binding of cYY to CYP121 appears to be a complicated multistep process similar to binding studies of other P450 systems (25). There are several possible models to explain these results. In the first model, the high-spin and two different low-spin states of the heme in the active site have different affinities for the substrate. The second model is that the initial binding of cYY is followed by reorientation of the substrate within the active site. The first model is believed to be an unlikely scenario; whereas the heme is present in two conformations, the overall configuration within the active site is highly conserved before and after binding of substrate. The ligand-free and the substrate-bound active site architectures of the enzyme are nearly identical (9, 19). The second model is more likely, due to the bulky nature of the substrate and the conformational freedom at α-carbons of both tyrosine ends in cYY. The as-isolated CYP121 contains two EPR resonance components in the low-spin region: an outer component, species A, and an inner component, species B, with smaller g-anisotropy (Fig. 2B). Species B increases in intensity upon binding of the substrate at the expense of species A. Whereas the UV-visible spectra of CYP121 demonstrate typical type I binding of cYY with a low-spin to high-spin transition, this conversion does not appear to be complete even under saturating cYY concentrations. In EPR experiments, the ratio of the low-spin to high-spin transition of the enzyme–substrate complex appears to be dependent on freezing speed, with faster freezing producing more high-spin heme signal (data not shown), suggesting that a dynamic equilibrium exists between the two spin states.
Mechanistic implications
The ability of CYP121 to react with a hydrogen peroxide analogue (i.e. peracetic acid) was studied to understand the heme-based oxidant behind the unusual intramolecular C–C coupling reaction chemistry carried out by this enzyme. As an established strategy, peracids have been utilized in place of hydrogen peroxide in the catalytic shunt pathway to generate and characterize high-valent ferryl species to probe P450 reaction mechanisms (39, 40). The advantage of using a peracid is that it has an active leaving group, which helps to promote heterolytic cleavage of the O–O bond to generate an oxoferryl porphyrin cation radical (known as compound I) in P450 systems (41). Here, we show that peracetic acid could react with the heme center of CYP121 to produce the C–C bond coupling product mycocyclosin. Based on the shunt pathway illustrated in Fig. 1, either a ferric peroxo, a compound I intermediate, or both should be part of the catalytic cycle. Compound I is a strongly spin-coupled system carrying two oxidizing equivalents on a single heme. The P450 compound I intermediate is unfortunately not a spectroscopically long-lived species and previously eluded EPR and Mössbauer spectroscopic detection for several decades until recent characterization of the thermophilic P450 CYP119 at 4 °C through the catalytic shut reaction using an organic peroxide (42). The understanding of C–C bond coupling of cYY has stagnated for lack of a detectable intermediate in the typical catalytic reaction with O2 and the ferredoxin system (11, 13–15, 19, 43, 44). The slow formation of an active intermediate from the alkylperoxo and its rapid reaction with cYY is the most likely reason for its lack of spectroscopic detection.
The investigation of the peroxide shunt pathway carries several mechanistic implications for understanding the CYP121-mediated C–C bond formation. We established the shunt pathway in the CYP121 reaction, which often suggests the involvement of a high-valent ferryl species in the catalytic cycle. A key mechanistic difference between CYP121 and other P450 enzymes is that CYP121 does not perform a hydroxylation reaction on cYY (19), and thus the quintessential radical-rebound mechanism found in traditional P450 mechanisms does not apply in the C–C bond formation reaction (45).
Our LC-MS analysis confirmed the cYY-to-mycocyclosin conversion. However, this reaction is limited to only a few turnovers, presumably due to complications of enzyme damage from PAA. When monitored by EPR spectroscopy, the reaction of the ES complex with PAA shows that the low-spin heme signal nearly disappears, and a new high-spin intermediate was formed with subsequent slow conversion to an EPR-silent heme species and an organic radical. This high-spin intermediate is different from traditional P450 high-spin systems that are characterized with g-values at 8, 4, and 1.7 (46).
The most plausible candidate for the rapid and nearly full production of the high-spin ferric intermediate from the ES complex reaction with PAA is an alkylperoxo (i.e. a side-on Ac-O-O-Fe(III) intermediate). Typically, ferric hydroperoxo species in heme-based systems are present at the low-spin state, and ferric alkylperoxo is less studied. High-spin, peroxo-bound species have been reported in the study of ferric peroxide complex (47), binuclear cytochrome c oxidase mimics (48, 49), iron complexes with tetra- and pentadentate ligands, and a superoxide reductase mutant with four equatorial histidines and an axial cysteine (50–52). The high-spin state is probably due to the side-on geometry of the peroxo moiety to the iron ion (heptacoordinate) or dianionic (strong π-base) O22− ligand causing weakening of the other axial ligand. It has been known through model complex studies that the spin state of the ferric–alkylperoxo complexes is important for the reactivity. The high-spin state in the nonheme complex presents a barrier for homolytic cleavage of the O–O bond relative to the low-spin complex (53). How the spin state effects the heterolytic cleavage of the O–O bond remains unexplored. The electronic structure of the high-spin ferric intermediate found in CYP121 and corresponding model complexes, once available, deserves further characterizations in subsequent studies.
The peroxide shunt study and our finding of the peroxidase activity of CYP121 provide insights into the mechanism of action for this unusual P450 enzyme. Taken together, we were able to put forth plausible catalytic mechanisms for the C–C bond coupling by the P450 enzyme. Fig. 11 (A and B) illustrates that the compound I intermediate is the likely oxidizing species. It can perform hydrogen atom abstraction on the hydroxyl group of the cYY proximal tyrosine, generating a protonated compound II and a cYY radical. Likewise, two subsequent pathways could take place. The reaction proceeds via an electron tunneling or hopping step to generate a cation radical and phenolate species (Fig. 11A). A second hydrogen atom abstraction at the ortho position by the protonated compound II will generate the second radical on cYY. The two radicals of opposite charge on cYY would be expected to combine rapidly to give rise to a new C–C bond. Finally, deprotonation of the distal tyrosine by the phenolate will re-aromatize the final product. In Fig. 10B, we propose an initial intramolecular proton-coupled electron transfer step to migrate the single radical to the distal tyrosine moiety. The protonated compound II then abstracts a hydrogen atom from the hydroxyl moiety of the proximal tyrosine. The two phenoxyl radicals are delocalized on the aromatic ring system, allowing for the diradical to combine and form the C–C bond. The resulting diketone group can tautomerize to generate mycocyclosin. Because no substrate-based radical intermediate was observed, we therefore assume that the catalytic chemistry is fast and not rate-limiting.
Figure 11.

Proposed mechanistic models for the CYP121-mediated cYY cross-linking reaction using peracetic acid as the oxidant. The top two mechanisms are proposed using compound I as the oxidant (A and B), and the bottom two using the peroxo intermediate as the active oxidant (C and D). In each case, two pathways are proposed for the formation of a second radical and subsequent C–C bond formation.
Our current understanding of the PAA-based CYP121 shunt mechanisms shown in Fig. 11 (A and B) is consistent with all of the observations obtained so far. In the PAA reaction, heterolytic cleavage of the alkylperoxo will be expected to yield a compound I oxidant for the subsequent oxidation reactions. The production of compound I is expected to be rate-limiting based on our stopped-flow and RFQ-EPR data. Additionally, the observation of a species consistent with protonated compound II also supports mechanisms A and B in Fig. 10.
Although compound I is probably the oxidant, our data cannot conclusively eliminate the possibility that the Ac-O-O-Fe(III) intermediate oxidizes cYY directly. If so, an oxoferryl species and a substrate-based radical species that is electronically equivalent to compound I will be generated. After this initial phase, two possible diverging mechanisms for the formation of a second substrate radical and subsequent C–C bond formation are proposed (Fig. 11, C and D). The first is a direct electron tunneling or electron/hole hopping via a protein residue to share the radical character with the remote tyrosine moiety (54–56). Subsequent oxidation of the proximal tyrosine generates a second radical for radical-radical coupling reactions (Fig. 11C). An alternative proposal is proton-coupled electron transfer (57–60). In contrast to the above model, the high-valent iron intermediate will oxidize the hydroxyl group of the closer tyrosine again, yielding a diradical species on the cYY for a rapid C–C bond coupling (Fig. 11D). In this model, an intranuclear tautomerization reaction must take place to produce the final product, mycocyclosin.
Conclusions
In summary, we have studied substrate binding kinetics and characterized the reactivity of CYP121 toward cYY and peracetic acid, an exogenous oxidant that bypasses the need to provide electrons through an exogenous reductase system. Iodosylbenzene was also employed as an oxidant of the reaction to support the catalytic competence of a putative compound I species in CYP121. Furthermore, CYP121 was shown to be able to perform peroxidase chemistry, generating substrate-based radicals. Together, we demonstrate that CYP121 can utilize the well-described shunt pathway of other P450 enzymes to generate the cross-linked product mycocyclosin from the cYY substrate. The formation of the postulated compound I is rate-limiting, whereas all of the subsequent chemical steps are rapid. With these advances, we propose four detailed catalytic mechanisms for future investigations.
Experimental procedures
Expression and purification
The CYP121 gene rv2276 was synthesized using an outsourced gene synthesis company, GenScript. The gene sequence encoding CYP121 was cloned into vector pET28a-TEV and expressed in E. coli strain BL21 (DE3). Conditions used for expression of a CYP121 protein with heme incorporation were incubation at 37 °C under kanamycin selection with constant agitation at 220 rpm until a value of 0.6 A600 is reached. Immediately before induction, the cells were inoculated with 5-aminolevulinic acid and ammonium iron(II) sulfate to a final concentration of 300 and 35 μm, respectively. The cells were then induced with a final concentration of 400 μm IPTG and left to continue shaking overnight at a reduced temperature, 28 °C. Cells were harvested the next morning by centrifugation and stored at −80 °C until further use.
Before purification, cultured cell pellets were resuspended in lysis buffer (50 mm potassium phosphate, pH 8.0, 300 mm NaCl) and passed through an LS-20 cell disruptor (Microfluidics) to lyse cells. The resulting suspension was centrifuged, and the supernatant was collected. Clarified cell extract was applied to a nickel–nitrilotriacetic acid affinity chromatography column, and bound protein was eluted using elution buffer (50 mm potassium phosphate, pH 8.0, 300 mm NaCl, 500 mm imidazole). Fractions were collected during purification and pooled together; the sample was then buffer-exchanged into 50 mm Tris-HCl, pH 7.4, 5% glycerol buffer. The purified protein was flash-frozen in liquid nitrogen and stored in −80 °C until required.
Pre-steady-state kinetics with cYY
Stopped-flow studies were carried out in single kinetic traces and multiwavelength data sets on an Applied Photophysics SX20 stopped-flow system. The assay conditions were 5–25 μm CYP121 and varied concentrations of each oxidant, from 0.1 to 20.0 mm. Assays with cYY used a constant 400 μm concentration of substrate. The stock solution of cYY was made in dimethyl sulfoxide, and a desired amount of the solution was incubated with CYP121 prior to the kinetics experiments (14). The experiments were carried out at room temperature in 50 mm Tris-HCl, pH 7.4, buffer, and dimethyl sulfoxide was less than 2% (v/v).
Rapid freeze-quench EPR spectroscopy
Rapid freeze-quench experiments were carried out using a System 1000 chemical/freeze-quench apparatus made by Update Instruments, Inc. A typical freeze- quench set-up consisted of a liquid ethane bath at −130 °C. Reactions were carried out by mixing 500 μm CYP121 or the enzyme–substrate complex in a 1:1 ratio with 2–10 mm PAA. The reaction was carried out at room temperature in 50 mm Tris-HCl, pH 7.4, buffer. The reaction mixture was rapidly frozen in liquid ethane after reacting for different time intervals. The frozen samples were transferred into EPR tubes, and excess liquid ethane was vacuumed off. Quenched samples were stored in a liquid nitrogen Dewar before analysis.
EPR spectra were recording at 9.6-GHz microwave frequency with a Bruker E560 EPR spectrometer with a dual-mode resonator at 100-kHz modulation frequency. The temperature was maintained at 10 K by a cryogen-free 4 K temperature system. Frozen sample solutions were analyzed in 4-mm quartz EPR tubes.
LC-MS analyses
A typical assay experiment was performed using 20–30 μm CYP121, 40 μm cYY, and 10 eq of oxidant relative to the enzyme. The assay was carried out in 5 mm Tris-HCl buffer, pH 7.4. Oxidant was titrated over 10 additions on ice with 1-min intervals between each addition to avoid pronounced peroxide damage to the enzyme. After 10-min reactions, the final sample was centrifuged for 15 min using an Amicon Ultra centrifugal filter with a 10-kDa cut-off. Chromatographic separations and mass spectrometry analyses were performed on an Acquity UPLC system directly coupled to a triple-quadrupole detector (Waters Corp.). For chromatography, solvent A was an aqueous solution containing H2O plus 0.1% formic acid, whereas solvent B was acetonitrile plus 0.1% FA. Separation was achieved at a flow rate of 0.2 ml/min using a Synergy 4u Fusion-RP 80A column (50 × 2.00 mm, C18, 4-μm particle size) from Phenomenex. For each analysis, 5 μl of sample was injected onto the column. Mass spectra for full scans and selected-ion monitoring were acquired in negative ionization mode in the range 100 ≤ m/z ≤ 1000. Capillary, cone, and extraction potentials were maintained at 3.5, 35, and 3 V, respectively. The source block temperature was 120 °C, and the desolvation temperature was 250 °C. The cone gas flow rate was 50 liters/h. Product detection was performed by selected-ion monitoring in which the quadrupole was used to filter for the substrate (325.1 m/z) and the product (323.1 m/z).
Peroxidase activity assay
Using ABTS as substrate, we tested the ability of CYP121 to generate substrate-based radicals. A representative assay shown in Fig. 10A was conducted using an Agilent 8453 UV-visible spectrometer. The steady-state kinetic analyses were performed with single-wavelength traces monitored at 420 nm for formation of ABTṢ+ using an Applied Photophysics SX20 stopped-flow spectrometer (Leatherhead, UK). The assay conditions for the determination of kinetic parameters were 100 nm CYP121, 2 mm ABTS, and the concentration of PAA was varied between 0.02 and 5 mm. The concentration of ABTS radical formed during the reaction was determined using ϵ420 36,000 m−1 cm−1 (61). The experiments were performed at room temperature in 50 mm Tris-HCl, pH 7.4, buffer.
Author contributions
K. D. and I. D. performed stopped-flow and RFQ-EPR experiments. A. J. F. conducted substrate binding studies. K. D., I. D., and A. J. F. analyzed the results and wrote most of the paper. J. R. T. and L. M. synthesized cYY. K. D., I. D., L. M., and A. L. proposed catalytic models. A. L. conceived the research idea for the project and wrote the paper with K. D. and co-authors. All authors approved of the final version of the manuscript.
Acknowledgments
We thank Dr. Kyle H. Rohde for advice in optimizing CYP121 expression and Dr. Wendell P. Griffith for assistance with LC-MS analyses.
This work was supported in whole or part by National Institutes of Health Grants GM108988 and GM107529 (to A. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- cYY
- dicyclotyrosine
- Ac-O-O-Fe(III)
- acetate-O-O-ferric heme
- P450
- cytochrome P450
- PAA
- peracetic acid
- ABTS
- 2,29-azino-bis(3-ethylbenzothiazoline-6-sulfonate)
- PhIO
- cYY iodosylbenzene.
References
- 1. World Health Organization (2016) Global Tuberculosis Report 2016, World Health Organization, Geneva, Switzerland [Google Scholar]
- 2. Matsumoto M., Hashizume H., Tsubouchi H., Sasaki H., Itotani M., Kuroda H., Tomishige T., Kawasaki M., and Komatsu M. (2007) Screening for novel antituberculosis agents that are effective against multidrug resistant tuberculosis. Curr. Top. Med. Chem. 7, 499–507 [DOI] [PubMed] [Google Scholar]
- 3. Cole S. T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D., Gordon S. V., Eiglmeier K., Gas S., Barry C. E. 3rd, Tekaia F., Badcock K., Basham D., Brown D., Chillingworth T., et al. (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544 [DOI] [PubMed] [Google Scholar]
- 4. Bellamine A., Mangla A. T., Nes W. D., and Waterman M. R. (1999) Characterization and catalytic properties of the sterol 14α-demethylase from Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 96, 8937–8942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Souter A., McLean K. J., Smith W. E., and Munro A. W. (2000) The genome sequence of Mycobacterium tuberculosis reveals cytochromes P450 as novel anti-TB drug targets. J. Chem. Technol. Biotechnol. 75, 933–941 [Google Scholar]
- 6. Guardiola-Diaz H. M., Foster L. A., Mushrush D., and Vaz A. D. (2001) Azole-antifungal binding to a novel cytochrome P450 from Mycobacterium tuberculosis: implications for treatment of tuberculosis. Biochem. Pharmacol. 61, 1463–1470 [DOI] [PubMed] [Google Scholar]
- 7. McLean K. J., Cheesman M. R., Rivers S. L., Richmond A., Leys D., Chapman S. K., Reid G. A., Price N. C., Kelly S. M., Clarkson J., Smith W. E., and Munro A. W. (2002) Expression, purification and spectroscopic characterization of the cytochrome P450 CYP121 from Mycobacterium tuberculosis. J. Inorg. Biochem. 91, 527–541 [DOI] [PubMed] [Google Scholar]
- 8. McLean K. J., Carroll P., Lewis D. G., Dunford A. J., Seward H. E., Neeli R., Cheesman M. R., Marsollier L., Douglas P., Smith W. E., Rosenkrands I., Cole S. T., Leys D., Parish T., and Munro A. W. (2008) Characterization of active site structure in CYP121: a cytochrome P450 essential for viability of Mycobacterium tuberculosis H37Rv. J. Biol. Chem. 283, 33406–33416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Leys D., Mowat C. G., McLean K. J., Richmond A., Chapman S. K., Walkinshaw M. D., and Munro A. W. (2003) Atomic structure of Mycobacterium tuberculosis CYP121 to 1.06 Å reveals novel features of cytochrome P450. J. Biol. Chem. 278, 5141–5147 [DOI] [PubMed] [Google Scholar]
- 10. Sundaramurthi J. C., Kumar S., Silambuchelvi K., and Hanna L. E. (2011) Molecular docking of azole drugs and their analogs on CYP121 of Mycobacterium tuberculosis. Bioinformation 7, 130–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hudson S. A., McLean K. J., Surade S., Yang Y. Q., Leys D., Ciulli A., Munro A. W., and Abell C. (2012) Application of fragment screening and merging to the discovery of inhibitors of the Mycobacterium tuberculosis cytochrome P450 CYP121. Angew. Chem. Int. Ed. Engl. 51, 9311–9316 [DOI] [PubMed] [Google Scholar]
- 12. Duffell K. M., Hudson S. A., McLean K. J., Munro A. W., Abell C., and Matak-Vinković D. (2013) Nanoelectrospray ionization mass spectrometric study of Mycobacterium tuberculosis CYP121-ligand interactions. Anal. Chem. 85, 5707–5714 [DOI] [PubMed] [Google Scholar]
- 13. Hudson S. A., Surade S., Coyne A. G., McLean K. J., Leys D., Munro A. W., and Abell C. (2013) Overcoming the limitations of fragment merging: rescuing a strained merged fragment series targeting Mycobacterium tuberculosis CYP121. ChemMedChem 8, 1451–1456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fonvielle M., Le Du M. H., Lequin O., Lecoq A., Jacquet M., Thai R., Dubois S., Grach G., Gondry M., and Belin P. (2013) Substrate and reaction specificity of Mycobacterium tuberculosis cytochrome P450 CYP121 insights from biochemical studies and crystal structures. J. Biol. Chem. 288, 17347–17359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kavanagh M. E., Coyne A. G., McLean K. J., James G. G., Levy C. W., Marino L. B., de Carvalho L. P., Chan D. S., Hudson S. A., Surade S., Leys D., Munro A. W., and Abell C. (2016) Fragment-based approaches to the development of Mycobacterium tuberculosis CYP121 inhibitors. J. Med. Chem. 59, 3272–3302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Balding P. R., Porro C. S., McLean K. J., Sutcliffe M. J., Maréchal J.-D., Munro A. W., and de Visser S. P. (2008) How do azoles inhibit cytochrome P450 enzymes? A density functional study. J. Phys. Chem. A 112, 12911–12918 [DOI] [PubMed] [Google Scholar]
- 17. Roback P., Beard J., Baumann D., Gille C., Henry K., Krohn S., Wiste H., Voskuil M. I., Rainville C., and Rutherford R. (2007) A predicted operon map for Mycobacterium tuberculosis. Nucleic Acids Res. 35, 5085–5095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gondry M., Sauguet L., Belin P., Thai R., Amouroux R., Tellier C., Tuphile K., Jacquet M., Braud S., Courçon M., Masson C., Dubois S., Lautru S., Lecoq A., Hashimoto S., Genet R., and Pernodet J. L. (2009) Cyclodipeptide synthases are a family of tRNA-dependent peptide bond–forming enzymes. Nat. Chem. Biol. 5, 414–420 [DOI] [PubMed] [Google Scholar]
- 19. Belin P., Le Du M. H., Fielding A., Lequin O., Jacquet M., Charbonnier J. B., Lecoq A., Thai R., Courçon M., Masson C., Dugave C., Genet R., Pernodet J. L., and Gondry M. (2009) Identification and structural basis of the reaction catalyzed by CYP121, an essential cytochrome P450 in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 106, 7426–7431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mizutani M., and Sato F. (2011) Unusual P450 reactions in plant secondary metabolism. Arch. Biochem. Biophys. 507, 194–203 [DOI] [PubMed] [Google Scholar]
- 21. Giessen T. W., and Marahiel M. A. (2015) Rational and combinatorial tailoring of bioactive cyclic dipeptides. Front. Microbiol. 6, 785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dumas V. G., Defelipe L. A., Petruk A. A., Turjanski A. G., and Marti M. A. (2014) QM/MM study of the C—C coupling reaction mechanism of CYP121, an essential cytochrome P450 of Mycobacterium tuberculosis. Proteins 82, 1004–1021 [DOI] [PubMed] [Google Scholar]
- 23. Porter T. D., and Coon M. J. (1991) Cytochrome P-450. Multiplicity of isoforms, substrates, and catalytic and regulatory mechanisms. J. Biol. Chem. 266, 13469–13472 [PubMed] [Google Scholar]
- 24. Sono M., Roach M. P., Coulter E. D., and Dawson J. H. (1996) Heme-containing oxygenases. Chem. Rev. 96, 2841–2888 [DOI] [PubMed] [Google Scholar]
- 25. Isin E. M., and Guengerich F. P. (2008) Substrate binding to cytochromes P450. Anal. Bioanal. Chem. 392, 1019–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Segel I. H. (1993) Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-state Enzyme Systems, Wiley-Interscience, New York [Google Scholar]
- 27. Nordblom G. D., White R. E., and Coon M. J. (1976) Studies on hydroperoxide-dependent substrate hydroxylation by purified liver microsomal cytochrome-P-450. Arch. Biochem. Biophys. 175, 524–533 [DOI] [PubMed] [Google Scholar]
- 28. Gustafsson J. A., Rondahl L., and Bergman J. (1979) Iodosylbenzene derivatives as oxygen donors in cytochrome P-450 catalyzed steroid hydroxylations. Biochemistry 18, 865–870 [DOI] [PubMed] [Google Scholar]
- 29. White R. E., Sligar S. G., and Coon M. J. (1980) Evidence for a homolytic mechanism of peroxide oxygen-oxygen bond cleavage during substrate hydroxylation by cytochrome P-450. J. Biol. Chem. 255, 11108–11111 [PubMed] [Google Scholar]
- 30. Ullrich V. (2003) Thoughts on thiolate tethering: tribute and thanks to a teacher. Arch. Biochem. Biophys. 409, 45–51 [DOI] [PubMed] [Google Scholar]
- 31. Brash A. R. (2009) Mechanistic aspects of CYP74 allene oxide synthases and related cytochrome P450 enzymes. Phytochemistry 70, 1522–1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cho K. B., Lai W., Hamberg M., Raman C. S., and Shaik S. (2011) The reaction mechanism of allene oxide synthase: interplay of theoretical QM/MM calculations and experimental investigations. Arch. Biochem. Biophys. 507, 14–25 [DOI] [PubMed] [Google Scholar]
- 33. Boeglin W. E., and Brash A. R. (2012) Cytochrome P450-type hydroxylation and epoxidation in a tyrosine-liganded hemoprotein, catalase-related allene oxide synthase. J. Biol. Chem. 287, 24139–24147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yoshimoto F. K., Gonzalez E., Auchus R. J., and Guengerich F. P. (2016) Mechanism of 17α,20-lyase and new hydroxylation reactions of human cytochrome P450 17A1: 18O labeling and oxygen surrogate evidence for a role of a perferryl oxygen. J. Biol. Chem. 291, 17143–17164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mak P. J., Gregory M. C., Denisov I. G., Sligar S. G., and Kincaid J. R. (2015) Unveiling the crucial intermediates in androgen production. Proc. Natl. Acad. Sci. U.S.A. 112, 15856–15861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Makino M., Sugimoto H., Shiro Y., Asamizu S., Onaka H., and Nagano S. (2007) Crystal structures and catalytic mechanism of cytochrome P450 StaP that produces the indolocarbazole skeleton. Proc. Natl. Acad. Sci. U.S.A. 104, 11591–11596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Amann T., Roos PH, Huh H, and Zenk M. H. (1995) Purification and characterization of a cytochrome P450 enzyme from pig liver, catalyzing the phenol oxidative coupling of (R)-reticuline to salutaridine, the critical step in morphine biosynthesis. Heterocycles 40, 425–440 [Google Scholar]
- 38. Grobe N., Zhang B., Fisinger U., Kutchan T. M., Zenk M. H., and Guengerich F. P. (2009) Mammalian cytochrome P450 enzymes catalyze the phenol-coupling step in endogenous morphine biosynthesis. J. Biol. Chem. 284, 24425–24431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Spolitak T., Dawson J. H., and Ballou D. P. (2005) Reaction of ferric cytochrome P450cam with peracids: kinetic characterization of intermediates on the reaction pathway. J. Biol. Chem. 280, 20300–20309 [DOI] [PubMed] [Google Scholar]
- 40. Stone K. L., Behan R. K., and Green M. T. (2006) Resonance Raman spectroscopy of chloroperoxidase compound II provides direct evidence for the existence of an iron(IV)-hydroxide. Proc. Natl. Acad. Sci. U.S.A. 103, 12307–12310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bruice T. C. (1983) Leaving group tendencies and the rates of mono-oxygen donation by hydrogen peroxide, organic hydroperoxides, and peroxycarboxylic acids. J. Chem. Soc. Chem. Commun. 10.1039/C39830000014 [DOI] [Google Scholar]
- 42. Rittle J., and Green M. T. (2010) Cytochrome P450 compound I: capture, characterization, and C–H bond activation kinetics. Science 330, 933–937 [DOI] [PubMed] [Google Scholar]
- 43. Dunford A. J., McLean K. J., Sabri M., Seward H. E., Heyes D. J., Scrutton N. S., and Munro A. W. (2007) Rapid P450 heme iron reduction by laser photoexcitation of Mycobacterium tuberculosis CYP121 and CYP51B1: analysis of CO complexation reactions and reversibility of the P450/P420 equilibrium. J. Biol. Chem. 282, 24816–24824 [DOI] [PubMed] [Google Scholar]
- 44. Kavanagh M. E., Gray J. L., Gilbert S. H., Coyne A. G., McLean K. J., Davis H. J., Munro A. W., and Abell C. (2016) Substrate fragmentation for the design of M. tuberculosis CYP121 inhibitors. ChemMedChem 11, 1924–1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Su J., and Groves J. T. (2009) Direct detection of the oxygen rebound intermediates, ferryl Mb and NO2, in the reaction of metmyoglobin with peroxynitrite. J. Am. Chem. Soc. 131, 12979–12988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lipscomb J. D. (1980) Electron paramagnetic resonance detectable states of cytochrome P-450cam. Biochemistry 19, 3590–3599 [DOI] [PubMed] [Google Scholar]
- 47. McCandlish E., Miksztal A. R., Nappa M., Sprenger A. Q., Valentine J. S., Stong J. D., and Spiro T. G. (1980) Reactions of superoxide with iron porphyrins in aprotic solvents: a high spin ferric porphyrin peroxo complex. J. Am. Chem. Soc. 102, 4268–4271 [Google Scholar]
- 48. Ghiladi R. A., Ju T. D., Lee D.-H., Moeenne-Loccoz P., Kaderli S., Neuhold Y.-M., Zuberbuehler A. D., Woods A. S., Cotter R. J., and Karlin K. D. (1999) Formation and characterization of a high-spin heme-copper dioxygen (peroxo) complex. J. Am. Chem. Soc. 121, 9885–9886 [Google Scholar]
- 49. Ghiladi R. A., Hatwell K. R., Karlin K. D., Huang H.-W., Moënne-Loccoz P., Krebs C., Huynh B. H., Marzilli L. A., Cotter R. J., Kaderli S., and Zuberbuehler A. D. (2001) Dioxygen reactivity of mononuclear heme and copper components yielding a high-spin heme-peroxo-Cu complex. J. Am. Chem. Soc. 123, 6183–6184 [DOI] [PubMed] [Google Scholar]
- 50. Li F., Meier K. K., Cranswick M. A., Chakrabarti M., Van Heuvelen K. M., Münck E., and Que L. (2011) Characterization of a high-spin non-heme Fe(III)-OOH intermediate and its quantitative conversion to an Fe(IV)=O complex. J. Am. Chem. Soc. 133, 7256–7259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Roelfes G., Vrajmasu V., Chen K., Ho R. Y., Rohde J.-U., Zondervan C., La Crois R. M., Schudde E. P., Lutz M., Spek A. L., Hage R., Feringa B. L., Münck E., and Que L. Jr. (2003) End-on and side-on peroxo derivatives of non-heme iron complexes with pentadentate ligands: models for putative intermediates in biological iron/dioxygen chemistry. Inorg. Chem. 42, 2639–2653 [DOI] [PubMed] [Google Scholar]
- 52. Mathé C., Mattioli T. A., Horner O., Lombard M., Latour J. M., Fontecave M., and Nivière V. (2002) Identification of iron(III) peroxo species in the active site of the superoxide reductase SOR from Desulfoarculus baarsii. J. Am. Chem. Soc. 124, 4966–4967 [DOI] [PubMed] [Google Scholar]
- 53. Lehnert N., Ho R. Y., Que L. Jr., and Solomon E. I. (2001) Electronic structure of high-spin iron(III)-alkylperoxo complexes and its relation to low-spin analogues: reaction coordinate of O–O bond homolysis. J. Am. Chem. Soc. 123, 12802–12816 [DOI] [PubMed] [Google Scholar]
- 54. Warren J. J., Ener M. E., Vlček A. Jr., Winkler J. R., and Gray H. B. (2012) Electron hopping through proteins. Coord. Chem. Rev. 256, 2478–2487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Geng J., Dornevil K., Davidson V. L., and Liu A. (2013) Tryptophan-mediated charge-resonance stabilization in the bis-Fe(IV) redox state of MauG. Proc. Natl. Acad. Sci. U.S.A. 110, 9639–9644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Geng J., Davis I., and Liu A. (2015) Probing bis-Fe(IV) MauG: experimental evidence for the long-range charge-resonance model. Angew. Chem. Int. Ed. Engl. 54, 3692–3696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cukier R. I., and Nocera D. G. (1998) Proton-coupled electron transfer. Annu. Rev. Phys. Chem. 49, 337–369 [DOI] [PubMed] [Google Scholar]
- 58. Weinberg D. R., Gagliardi C. J., Hull J. F., Murphy C. F., Kent C. A., Westlake B. C., Paul A., Ess D. H., McCafferty D. G., and Meyer T. J. (2012) Proton-coupled electron transfer. Chem. Rev. 112, 4016–4093 [DOI] [PubMed] [Google Scholar]
- 59. Hammes-Schiffer S. (2015) Proton-coupled electron transfer: moving together and charging forward. J. Am. Chem. Soc. 137, 8860–8871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Barry B. A. (2015) Reaction dynamics and proton coupled electron transfer: studies of tyrosine-based charge transfer in natural and biomimetic systems. Biochim. Biophys. Acta 1847, 46–54 [DOI] [PubMed] [Google Scholar]
- 61. Shin K.-S., and Lee Y.-J. (2000) Purification and characterization of a new member of the laccase family from the white-rot basidiomycete Coriolus hirsutus. Arch. Biochem. Biophys. 384, 109–115 [DOI] [PubMed] [Google Scholar]