

Abstract
In this issue of Cancer Cell, Obeng et al. identify the consequences of expressing the most common mutation in the spliceosomal gene SF3B1 on hematopoiesis. The knockin mouse model described represents a valuable tool to dissect the effects of SF3B1 mutations on transformation, splicing, and less well-characterized functions of SF3B1.
Mutations in genes encoding the spliceosomal proteins SF3B1, SRSF2, U2AF1, and ZRSR2 were discovered in 2011 and are the most common class of mutations in patients with myelodysplastic syndromes (MDS). These mutations occur in a mutually exclusive manner with one another and at highly restricted residues in SF3B1, SRSF2, and U2AF1 that are now known to confer an alteration in mRNA splicing. Despite this mutual exclusivity, each of these mutations is associated with distinct clinical subtypes of MDS and their own spectrum of co-existing mutations and malignancies other than MDS. For example, SF3B1 mutations are present in >80% of patients with refractory anemia with ring sideroblasts (RARS), a form of MDS marked by accumulation of erythroid precursors with characteristic iron-laden mitochondria deposited in a ring around the nucleus. At the same time, SF3B1 mutations are common in chronic lymphocytic leukemia (CLL) and a number of epithelial malignancies.
Research on the functional implications of SF3B1 mutations in cancer thus far has identified that expression of heterozygous SF3B1 mutations impart a characteristic change in splicing distinct from loss of function, namely the use of aberrant 3′ splice sites and cryptic AG dinucleotides 10–30 bp upstream of the canonical AG dinucleotide. Although this finding has been reported in several studies, the effects of SF3B1 mutations on splicing to date have largely relied on data from cell types other than myeloid hematopoietic cells. Moreover, the biological consequences of SF3B1 mutations and their unique functional link to clonal hematopoietic disorders are quite unclear. To this end, Obeng et al. have now generated a conditional knockin mouse model to study the consequences of the most common SF3B1 mutation, SF3B1K700E (Obeng et al., 2016). This joins a wider ongoing effort to generate genetically engineered model systems to study the biological and biochemical consequences of spliceosomal mutations in model systems as diverse as yeast, zebrafish, mouse, and human cells (Figure 1)(Kim et al., 2015; Komeno et al., 2015; Matsunawa et al., 2014; Shirai et al., 2015; Visconte et al., 2012; Wang et al., 2014). Such models are absolutely necessary to study the consequences of expressing mutated splicing factors from their endogenous loci with proper levels of overall expression and mutant allelic frequency. This need is further evidenced by the multiple mechanisms regulating homeostatic expression of spliceosomal genes, even in their wild-type state.
Figure 1. Genetically Engineered Mouse Models Studying the Hematopoietic Effects of Deletion or Mutation of Spliceosomal Genes Commonly Mutated in Hematopoietic Malignancies.
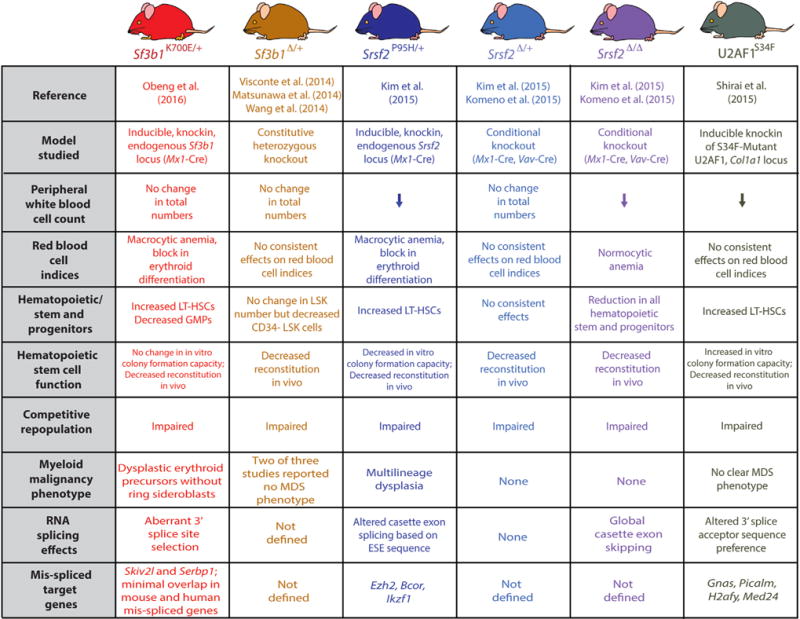
Abbreviations: ESE, exonic splicing enhancer; GMP, granulocyte/macrophage progenitor; LSK, lineage− Sca1+ c-Kit+ cells; LT-HSC, long-term hematopoietic stem cells.
In this paper, Sf3b1K700E/+ mice developed macrocytic anemia associated with a terminal block in erythroid maturation. However, the mice did not exhibit ring sideroblasts (RS), the phenotypic hallmark of RARS. RNA sequencing of myeloid progenitors from Sf3b1K700E/+ mice and controls demonstrated increased usage of aberrant 3′ splice sites, as noted in prior studies of SF3B1 mutations across cancer and also in MDS patient samples. However, there was minimal overlap in mis-spliced genes and events between human MDS and murine samples here, in part due to lack of conservation of intronic DNA sequences between the two species. For example, although ABCB7, a heme transporter mutated in congenital sideroblastic anemia and consistently downregulated in RARS, is mis-spliced in human SF3B1K700E mutant cells due to aberrant 3′ splice site usage, this was not seen in Sf3b1K700E mice because of the distinct intronic sequence at this region of Abcb7. Despite this, it is important to note that ABCB7 mis-splicing is not consistent across the various SF3B1 mutations seen in MDS, and functional evidence for a single or a handful of mis-splicing events that link SF3B1K700E mutations to RARS does not currently exist. Moreover, even Abcb7-deficient mice do not exhibit RS, potentially highlighting an unknown difference between human and murine erythroid precursors with respect to mitochondrial iron metabolism.
Despite differences in splicing events between mouse and human cells expressing K700E mutant SF3B1, Sf3b1K700E/+ mice clearly develop key phenotypic hallmarks of human MDS. This discrepancy suggests a possible effect of SF3B1 mutations on conserved functions of SF3B1 unrelated to splicing. To this end, several studies have identified novel roles for SF3B1 in binding to chromatin or chromatin-modifying proteins. For example, SF3B1 binds to nucleosomes residing on exonic DNA, independently of RNA, and the association of SF3B1 with nucleosomes facilitates the splicing recognition of exons (Kfir et al., 2015). In a similar vein, an interaction between SF3B1 and Polycomb repressive complex 1 (PRC1) components was described over a decade ago (Isono et al., 2005). Understanding how mutations in SF3B1 affect PRC1 function and the compendium of proteins interacting with SF3B1 will be critical to study further. This effort will likely be facilitated by improving the structural and biochemical understanding of the HEAT domains of SF3B1, where the majority of cancer-associated hotspot mutations reside.
As noted earlier, each mutated splicing factor appears to be associated with its own spectrum of co-existing and mutually exclusive mutations. Analyses of the functional basis for these genetic associations are already providing important clues into the mechanistic effects of each splicing factor mutation. Here, Obeng et al. identify that loss of Tet2, which plays a key role in active DNA demethylation through the conversion from 5-methylcytosine to 5-hydroxymethylcytosine, cooperates with Sf3b1K700E to exacerbate the macrocytic anemia and expansion of long-term hematopoietic stem cells further than seen with either mutation alone. While it is still not entirely clear why mutant splicing factors are selected for in human clonal disorders of hematopoiesis (Figure 1), loss of Tet2 in the setting of the Sf3b1K700E was sufficient to rescue the competitive disadvantage caused by Sf3b1K700E alone, possibly suggesting that such combinatorial models may be necessary to elucidate the contribution of spliceosomal mutations to cancer. Moreover, it has been reported that 5-hydroxymethylcytosine is more abundant at constitutive exons than alternatively spliced exons and that splice sites and exons have higher DNA methylation than flanking introns. Thus, efforts to understand how cancer-associated mutations in epigenetic modifiers such as TET2 affect the function of mutations in splicing factors, and vice versa, using models such as this will be critical.
In addition to mechanistic studies, the authors have identified that hematopoietic stem and progenitor cells expressing the Sf3b1K700E mutation have increased sensitivity to pharmacologic spliceosome modulation compared to Sf3b1 wild-type counterparts. This parallels recent work demonstrating preferential effects of this same compound on SRSF2 mutant leukemias (Lee et al., 2016). In this latter study, pharmacologic modulation of spliceosome function resulted in a greater magnitude of splicing inhibition in SRSF2 mutant cells, although E7107 treatment resulted in widespread intron retention and cassette exon skipping regardless of SRSF2 genotype. Considering that E7107 blocks spliceosome assembly by preventing tight binding of U2 small nuclear ribonucleoprotein particle (snRNP) to pre-mRNA and that SF3B1 is required for the ATP-dependent conformational change in U2 snRNP necessary for pre-mRNA binding, it would be interesting to compare the effects of E7107 on splicing in SF3B1 versus SRSF2 mutant contexts.
While this study represents an important step in understanding the physiological consequences of SF3B1 mutations in MDS, the conditional flexibility of this model will also be incredibly useful in hopefully elucidating the roles of mutant SF3B1 in other forms of cancer, such as CLL and epithelial malignancies of the breast, bladder, and pancreas. Moreover, such systems will be very important in understanding the requirement for mutant SF3B1 and/or its downstream transcriptional consequences across various forms of cancer.
Acknowledgments
D.I. is supported by Postdoctoral Fellowships for Research Abroad by the Japan Society for the Promotion of Science, by The YASUDA Medical Foundation, and by the Kanae Foundation for the Promotion of Medical Science. O.A.-W. is supported by grants from the Edward P. Evans Foundation, the Department of Defense Bone Marrow Failure Research Program (BM150092 and W81XWH-12-1-0041), NIH/NHLBI (R01 HL128239), an NIH K08 Clinical Investigator Award (1K08CA160647-01), the Josie Robertson Investigator Program, a Damon Runyon Clinical Investigator Award, an award from the Starr Foundation (I8-A8-075), the Leukemia and Lymphoma Society, and the Pershing Square Sohn Cancer Research Alliance.
References
- Isono K, Mizutani-Koseki Y, Komori T, Schmidt-Zachmann MS, Koseki H. Genes Dev. 2005;19:536–541. doi: 10.1101/gad.1284605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kfir N, Lev-Maor G, Glaich O, Alajem A, Datta A, Sze SK, Meshorer E, Ast G. Cell Rep. 2015;11:618–629. doi: 10.1016/j.celrep.2015.03.048. [DOI] [PubMed] [Google Scholar]
- Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, Li Y, Chung YR, Micol JBB, Murphy ME, et al. Cancer Cell. 2015;27:617–630. doi: 10.1016/j.ccell.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komeno Y, Huang YJ, Qiu J, Lin L, Xu Y, Zhou Y, Chen L, Monterroza DD, Li H, DeKelver RC, et al. Mol Cell Biol. 2015;35:3071–3082. doi: 10.1128/MCB.00202-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC, Dvinge H, Kim E, Cho H, Micol JB, Chung YR, Durham BH, Yoshimi A, Kim YJ, Thomas M, et al. Nat Med. 2016;22:672–678. doi: 10.1038/nm.4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunawa M, Yamamoto R, Sanada M, Sato-Otsubo A, Shiozawa Y, Yoshida K, Otsu M, Shiraishi Y, Miyano S, Isono K, et al. Leukemia. 2014;28:1844–1850. doi: 10.1038/leu.2014.73. [DOI] [PubMed] [Google Scholar]
- Obeng E, Chappell RJ, Seiler M, Chen MC, Campagna DR, Schmidt PJ, Schneider RK, Lord AM, Wang L, Gambe RG, et al. Cancer Cell. 2016;30:404–417. doi: 10.1016/j.ccell.2016.08.006. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirai CL, Ley JN, White BS, Kim S, Tibbitts J, Shao J, Ndonwi M, Wadugu B, Duncavage EJ, Okeyo-Owuor T, et al. Cancer Cell. 2015;27:631–643. doi: 10.1016/j.ccell.2015.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visconte V, Rogers HJ, Singh J, Barnard J, Bupathi M, Traina F, McMahon J, Makishima H, Szpurka H, Jankowska A, et al. Blood. 2012;120:3173–3186. doi: 10.1182/blood-2012-05-430876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Sashida G, Saraya A, Ishiga R, Koide S, Oshima M, Isono K, Koseki H, Iwama A. Blood. 2014;123:3336–3343. doi: 10.1182/blood-2013-12-544544. [DOI] [PubMed] [Google Scholar]