

Abstract
The aim of this study was to describe the clinical characteristics of ANCA-associated vasculitides (AAV) at presentation, in a wide cohort of Spanish patients, and to analyze the impact of the vasculitis type, ANCA specificity, prognostic factors, and treatments administered at diagnosis, in the outcome.
A total of 450 patients diagnosed between January 1990 and January 2014 in 20 Hospitals from Spain were included. Altogether, 40.9% had granulomatosis with polyangiitis (GPA), 37.1% microscopic polyangiitis (MPA), and 22% eosinophilic granulomatosis with polyangiitis (EGPA). The mean age at diagnosis was 55.6 ± 17.3 years, patients with MPA being significantly older (P < 0.001). Fever, arthralgia, weight loss, respiratory, and ear–nose–throat (ENT) symptoms, were the most common at disease onset. ANCAs tested positive in 86.4% of cases: 36.2% C-ANCA-PR3 and 50.2% P-ANCA-MPO. P-ANCA-MPO was significantly associated with an increased risk for renal disease (OR 2.6, P < 0.001) and alveolar hemorrhage (OR 2, P = 0.010), while C-ANCA-PR3 was significantly associated with an increased risk for ENT (OR 3.4, P < 0.001) and ocular involvement (OR 2.3, P = 0.002). All patients received corticosteroids (CS) and 74.9% cyclophosphamide (CYC). The median follow-up was 82 months (IQR 100.4). Over this period 39.9% of patients suffered bacterial infections and 14.6% opportunistic infections, both being most prevalent in patients with high-cumulated doses of CYC and CS (P < 0.001). Relapses were recorded in 36.4% of cases with a mean rate of 2.5 ± 2.3, and were more frequent in patients with C-ANCA-PR3 (P = 0.012). The initial disease severity was significantly associated with mortality but not with the occurrence of relapses. One hundred twenty-nine (28.7%) patients (74 MPA, 41 GPA, 14 EGPA) died. The mean survival was 58 months (IQR 105) and was significantly lower for patients with MPA (P < 0.001). Factors independently related to death were renal involvement (P = 0.010), cardiac failure (P = 0.029) and age over 65 years old (P < 0.001) at disease onset, and bacterial infections (P < 0.001). An improved outcome with significant decrease in mortality and treatment-related morbidity was observed in patients diagnosed after 2000, and was related to the implementation of less toxic regimens adapted to the disease activity and stage, and a drastic reduction in the cumulated CYC and CS dose.
Keywords: ANCA-associated vasculitides, eosinophilic granulomatosis with polyangiitis, granulomatosis with polyangiitis, infections, microscopic polyangiitis, mortality predictors, outcome, Spanish people
1. Introduction
ANCA-associated vasculitides (AAV) are chronic and relapsing autoimmune diseases, characterized by necrotizing small-vessels inflammation and circulating antineutrophil cytoplasmic autoantibodies (ANCA) in 60% to 90% of patients at disease onset.[1–9]
AAV have been classified according to their clinical phenotype, histological findings, and ANCA specificity.[1–4] Three different clinical forms have been differentiated: granulomatosis with polyangiitis (GPA) formerly known as Wegener granulomatosis (WG), eosinophilic granulomatosis with polyangiitis (EGPA) formerly known as Churg–Strauss syndrome (CSS), and microscopic polyangiitis (MPA). Two fluorescence patterns of ANCAs distinguished by indirect immunofluorescence (IIF) have been associated with AAV: the cytoplasmic-staining pattern (C-ANCA) and the perinuclear-staining pattern (P-ANCA). Most patients with C-ANCA by IIF have ANCAs directed against proteinase-3 (PR3) by antigen-specific enzyme-linked immunosorbent assay (ELISA), while most patients with P-ANCA by IIF have ANCAs directed against myeloperoxidase (MPO). The combination of C-ANCA-PR3 predominates in patients with GPA, whereas the combination of P-ANCA-MPO is predominant in MPA and EGPA.[1]
AAV may occur at any age, with varied and overlapping clinical manifestations at presentation that usually affect more than one organ and make difficult to perform a definitive diagnosis quickly.[1–9] Disease severity ranges from life organ-threatening manifestations to milder forms, and early and appropriate immunosuppressive treatment is crucial to reduce death by major-organ failure and long-term morbidity. The introduction of immunosuppressive therapy with corticosteroids (CS) and cyclophosphamide (CYC) in the 1960s led to a dramatic improvement in AAV prognosis, with over 90% of patients achieving remission compared with a 1-year mortality rate of 80% in untreated patients.[5] However, this therapy was associated with a substantial degree of toxicity.[6–9] In order to reduce treatment-related adverse events, the European Vasculitis Study Group (EUVAS) standardized the definitions and assessment of AAV, and defined 4 stages of the disease according to severity and extent at presentation, tailoring treatment regimens to the different stages[10,11]. Nonetheless, despite these advances, there is still considerable morbidity associated with both the disease and its treatment, that increases over time, as well as an increased mortality compared with the general population.[12,13]
The main clinical and pathological features of AAV have been documented in several studies from different countries, but series derived from Spain are scarce and involve a small number of cases.[14–20] Based on the National Spanish Network of the Internal Medicine Society (SEMI) for systemic vasculitis study, here we describe the clinical and pathological characteristics at the disease onset of the largest cohort of patients derived exclusively from this region of Europe, and we analyze the long-term outcome and the impact on the disease course of the vasculitis type, the ANCA specificity, prognostic factors, and treatments administered at the time of diagnosis.
2. Patients and methods
2.1. Study cohort
The Spanish Registry of Systemic vasculitis “REVAS” (Registro Español de VAsculitis Sistemicas in Spanish nomenclature), was created in 2009 by the Grupo de Enfermedades Autoinmunes Sistémicas (GEAS), from the Spanish Internal Medicine Society (SEMI), with the aim of compiling a large cohort of Spanish patients with these rare diseases. By November 2015 (the censoring date), 20 centers with substantial experience in the management of these diseases participated in the recruitment of more than 1500 patients with different vasculitides that were included in the Registry. Epidemiological (including time and cause of death), clinical, and laboratory data encompassing more than 150 variables were collected from medical records according to a standardized form designed by the REVAS-Study Group, and then entered into a specific database. All participating Centers had obtained Ethics Committee approval.
In the present study, a total of 450 patients with AAV diagnosed between January 1990 and January 2014 and regularly followed-up until the censoring data, were retrospectively analyzed. All patients met the American College of Rheumatology (ACR) classification criteria for WG or CSS[2,3] and the definition of GPA, MPA, and EGPA according to the 2012 revised Chapel Hill consensus criteria.[4]
The following data were collected: date/age at diagnosis; gender; comorbidities; type of AAV; diagnosis delay (time to diagnosis after the occurrence of the first vasculitis-related symptoms); organ systems involvement at disease onset and throughout the disease course; relevant laboratory data (erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), white blood count (WBC), hemoglobin, serum creatinine, estimated glomerular filtration rate (eGFR), urinalysis for urine protein, red blood count and casts, 24-hour urine protein excretion, and ANCA) at diagnosis and during follow-up; histopathology; radiology examinations; duration of illness; relapses; treatment at the time of presentation and relapses; deaths, and the likely causes and comorbidity that may have contributed toward death. ANCA were determined by IIF and ELISA techniques. Disease activity at diagnosis, relapses and last visit, was assessed according to the Birmingham Vasculitis Activity Score version 3 (BVAS v.3).[21] Permanent damage of organs was evaluated according to the Vasculitis Damage Index (VDI). The Five factors score (FFS) developed by the French Vasculitis Study Group[22] was determined for each patient as a prognostic score.
For analysis, severity of organ involvement was defined as the presence of one or more items within each category of the BVAS v.3 form (systemic; cutaneous; mucous membranes and eyes; ear, nose, and throat (ENT); chest; cardiovascular; gastrointestinal tract; kidney and nervous system). Renal disease was defined as the presence of hematuria >10 RBCs/hpf, proteinuria >1 g, hypertension, creatinine >125 μmol or rise in creatinine >30%, or creatinine clearance fall >25%, attributable only to vasculitis, as per the BVAS form. Acute renal failure was defined by the presence of progressively raised serum creatinine or 15% declined clearance rate of serum creatinine on the baseline within days or weeks. eGFR was calculated for each patient for the time of diagnosis and for the last visit of the study period using the modification of diet in renal disease equation. Chronic kidney disease was defined as an eGFR <60 mL/min/1.73 m2. The nervous system involvement was subdivided into central nervous system (stroke, meningitis, cord lesion, and cranial nerve palsy) and peripheral nervous system (sensory peripheral neuropathy and motor mononeuritis multiplex).
According to the European recommendations,[10,11] after 2000 most patients with generalized or severe disease received intravenous pulses of CYC as induction therapy and switched to other less toxic immunosuppressant drugs for maintenance therapy. For this reason, the study was divided in 2 periods (1990–2000 and 2001–2014) in order to analyze the influence of the different therapeutic regimens in the disease outcome.
2.2. Treatment protocols
Therapeutic regimens varied between individual patients and hospitals, depending on the disease severity and developments in treatment protocols over the study period. Induction therapy consisted in CS, usually in conjunction with CYC or methotrexate (MTX), though azathioprine (AZA) or mycophenolate mofetil (MMF) were administered in some cases. Oral prednisone was prescribed at an initial dose of 1 mg/kg/day. CYC was given daily oral (2 mg/kg/day) or intravenously (0.7 g/m2) every month. MTX was given 15 to 25 mg once a week, with folic acid substitution. AZA was given 1 to 2 mg per kg body weight per day, and MMF 2 g daily. Patients with severe disease received 3 to 5 consecutive intravenous pulses of methyl-prednisolone (7–15 mg/kg/day) before standard induction treatment. Some patients with severe alveolar hemorrhage or rapidly progressive glomerulonephritis received additionally plasma exchange (PE). Intravenous immunoglobulin (IVIG) or Rituximab (RTX) were administered in some refractory cases. Oral or intravenous CYC, MTX, AZA, or MMF were given as maintenance treatment in conjunction with CS according to the attending physician's discretion, for at least 18 months after remission had been achieved. There was no strict protocol for trimethoprim–sulfamethoxazole (TM-SX) prophylaxis for Pneumocystis jiroveci.
Remission was defined as the absence of clinical and laboratory evidence of vasculitis activity. Relapses were defined as recurrence of signs or new symptoms after an initial remission had been achieved. Uncontrolled vasculitis was defined as the occurrence of new manifestations or aggravation of manifestations already present despite treatment for the disease.
2.3. Statistical analysis
This was a multicenter observational, longitudinal-retrospective study. Categorical data were summarized as percentages; significant differences or associations were analyzed using the χ2 test or Fisher exact tests. Continuous variables are presented as mean ± standard deviation (SD) or median and interquartile range (IQR), depending on normality demonstrated by Kolmogorov–Smirnov test. Associations of quantitative data were analyzed with Student t test and with the nonparametric Mann–Whitney U test or the Kruskal–Wallis test. For mortality analyses, independent variables that appeared to have statistical significance in the univariate analysis were included in a multivariate logistic regression model using a backward stepwise method. The odds ratios (OR) and their 95% confidence interval (CI) obtained in the adjusted regression analysis were calculated. Survival curves were constructed according to the Kaplan–Meier method and compared using the log-rank test. For all the statistical analyses, P < 0.05 was considered significant. Statistical analysis was performed using the SPSS package (IBM Corp. 2010, Armonk, NY, SPSS Statistics 19.0).
3. Results
3.1. Demographics of all patients at baseline
Four hundred fifty patients (50.4% males) were included in the study: 184 (40.9%) with GPA, 167 (37.1%) with MPA and 99 (22%) with EGPA. Mean age at disease onset was 55.6 ± 17.3 years (range 17–91 years), 35.1% of patients being older than 65 years at disease onset. Altogether, 30.7% of patients had hypertension, 9.8% diabetes, and 18.2% hypercholesterolemia, before AAV diagnosis. The median diagnostic delay was 3 months (IQR 2–5). However, 14.2% of patients were diagnosed 6 months after the occurrence of the first vasculitis-related symptoms, and 6% of patients after more than 12 months, mainly those patients with granulomatous forms of GPA. Demographic data, mean BVAS score at diagnosis, and number of relapses and deaths, for the whole cohort and each type of AAV, are shown in Table 1.
Table 1.
Demographic and epidemiological characteristics at baseline of the total cohort and each type of AAV.

3.2. Clinical characteristics of all patients at diagnosis
Nonspecific symptoms such as fever, fatigue, joint pain, and weight loss were present at diagnosis in over 50% of cases. Renal involvement was detected in 279/450 (62%) patients, hematuria being the most common initial finding (227/279, 81.4%), followed by renal insufficiency (183/279, 40.7%). Severe renal failure (creatinine ≥ 500 μmol/L) was present in 30/279 (6.7%) patients, proteinuria ≥1 g in 99/279 (35.5%), and nephrotic syndrome in 35/279 (12.5%). Renal biopsy was performed in 164/279 (58.8%) cases and showed a crescent glomerulonephritis in 140/164 (85.3%). Lung involvement was present in 245 (54.5%) patients, of whom 84 (34.3%) had hemoptysis, and 90 (36.7%) respiratory failure, 22 (8.9%) of them needing mechanical ventilation, mostly due to alveolar hemorrhage. Chest X-ray and thorax computed tomography showed alveolar infiltrates or interstitial lung disease in 193/245 (78.8%) of patients, and isolated or multiple lung nodules in 106/245 (43.3%). A pulmonary-renal syndrome was observed in 74/450 (16.4%) cases. Neurological involvement was detected in 156/450 (34.7%) patients, of whom 80 (51.3%) had mononeuritis multiplex, 64 (41%) sensory peripheral neuropathy, and 22 (14.1%) cerebrovascular accidents. Cardiac involvement was recorded in 58/450 (12.9%) patients, with pericarditis in 12 (20.7%) of them, cardiomyopathy in 10 (17.2%), coronary artery involvement in 16 (27.6%), and congestive cardiac failure in 33 (56.9%). Two patients developed an acute aortic insufficiency and required surgical intervention. Gastrointestinal involvement manifested by vomiting, abdominal pain, or bloody diarrhea was present in 20/450 (4.4%) patients. ENT involvement manifested by sinusitis, hearing loss, or bloody nasal crusts was found in 203/450 (45.1%) patients. Skin involvement manifested by palpable purpura, maculopapular rash, or erythematous nodules was found in 112/450 (24.9%) patients, and ophthalmic involvement manifested by conjunctivitis, episcleritis, anterior uveitis, retinal vasculitis, or optical nerve vasculitis, in 60/450 (13.3%). Mean BVAS at the time of diagnosis was 17.8 ± 7.7 (range 3–45) in the entire cohort. No gender-related differences in terms of clinical symptoms and BVAS were observed. Clinical characteristics of the entire cohort and each type of AAV are summarized in Tables 1 and 2.
Table 2.
Clinical characteristics and organ involvement at diagnosis in the entire cohort and in each type of AAV.

3.3. Laboratory data of all patients at baseline
ESR and CRP values were increased in more than 75% of patients at the time of diagnosis and serum creatinine was raised in 40.7% of patients, with a mean serum creatinine at presentation of 2.03 ± 2.1 mg/dL (range: 0.3–16.5 mg/dL). Normochromic anemia was present in 68.2% of cases. ANCA tested positive in 86.4% of cases: 36.2% C-ANCA-PR3 and 50.2% P-ANCA-MPO. Laboratory data are summarized in Tables 2–4.
Table 4.
ANCA specificities in patients with AAV.

Table 3.
Laboratory data at baseline.

A total of 521 biopsies (renal, muscle, nerve, skin, nose, ear, throat, liver, intestine, meninges) were done. The diagnosis of vasculitis was supported by histopathology in 368 (81.8%) cases. In the remaining cases, the diagnosis was made on the basis of typical clinical symptoms in conjunction with a positive ANCA test.
3.4. Treatment
Almost all patients received daily oral prednisone (1 mg/kg/day) that was gradually tapered depending on the clinical response and the evolution of laboratory parameters. Intravenous methylprednisolone (3–5 consecutive pulses) was administered to 237 (52.7%) patients with severe disease. For induction therapy 334 (74.2%) patients received CYC (daily oral 168/334, 50.3%), 27 (7.7%) MTX, 19 (4.2%) AZA, and 5 (1.1%) MMF. Additionally, 26 (5.8%) patients received RTX after CYC failure. For maintenance therapy, 143 (31.8%) patients received AZA, 118 (26.2%) CYC (47 IV pulses and 71 oral), 49 (10.9%) MTX, 42 (9.3%) MMF, and 24 (5.3%) RTX. TMT-SX prophylaxis (one 400 mg tablet/day or 800 mg tablet every 2 days) was maintained in 213 (47.3%) patients. Dialysis was required in 60 (13.3%) cases.
3.5. Comparison between MPA, GPA, and EGPA
Patients with MPA were significantly older at diagnosis than patients with GPA and EGPA (P < 0.001). Kidney involvement and pulmonary-renal syndrome were more prevalent in patients with MPA (P < 0.001), neurological and cardiac involvement in patients with EGPA (P < 0.001), and ENT and ocular involvement in patients with GPA (P < 0.001). Of note, 91.9% of patients with EGPA had asthma, with a median period of 60 months (IQR 96) between asthma and vasculitis development. Median BVAS at diagnosis was similar in GPA, MPA, and EGPA. The median time to diagnosis was significantly greater in patients with GPA and EGPA (P = 0.006). Clinical characteristics at disease onset and demographic data are depicted in Tables 1 and 2.
Concerning the laboratory data (Tables 2–4), anemia was more prevalent in patients with MPA (89.2%) than in those with GPA (64.7%) and EGPA (39.4%), P < 0.001; serum creatinine values were higher in patients with MPA than in those with GPA and EGPA (P < 0.001), and the mean eGFR was lower in patients with MPA (P = 0.006). ANCA tested positive in 100% of patients with MPA, 88.6% of patients with GPA, and 60.6% of patients with EGPA. C-ANCA-PR3 autoantibodies were more prevalent in patients with GPA (P < 0.001), and P-ANCA-MPO in patients with MPA and EGPA (P < 0.005). The presence of P-ANCA-MPO was associated with a significant increased risk for renal disease (OR 2.6, 95% CI: 1.7–3.8, P < 0.001) and alveolar hemorrhage (OR 2, 95% CI: 1.8–3.8, P = 0.010), while the presence of C-ANCA-PR3 was related to a significant increased risk for ENT involvement (OR 3.4, 95% CI: 2.3–5.1, P < 0.001) and ocular involvement (OR 2.3, 95% CI: 1.4–4.1, P = 0.002). No significant relationship was found between pulmonary-renal syndrome and ANCA specificity.
In regard to treatment, oral CYC was more frequently administered as induction therapy to patients with GPA (87, 47.3%) than to those with MPA (62, 37%) and EGPA (22, 2.2%), (OR 1.9, 95% CI: 1.3–2.9, P = 0.001), especially to patients with granulomatous manifestations. Similarly, TMT-SX was more frequently given to patients with GPA than to those with MPA and EGPA (99.5% vs 46.1% and 23.1%, respectively, P < 0.001). In contrast, dialysis was mainly required in patients with MPA (41, 24.6%), compared to patients with GPA (18, 9.8%) and EGPA (1, 1%), P < 0.001.
3.6. Outcome
The median follow-up period was 82 months (IQR 104.5). During this period, 175 (39.9%) patients had a total of 219 bacterial infections (106 pneumonias, 73 urinary tract infections, 40 septicemia); 56 (12.4%) suffered a total of 74 opportunistic infections (27 Candida sp., 16 other systemic mycosis, 14 P jirovecii, 12 CMV, 5 atypical mycobacteria), and 24 (6%) developed a solid neoplasm. Furthermore, 99 (22%) patients developed leucopenia and 23 (5.1%) had pancytopenia.
Bacterial infections were more prevalent in patients treated with oral CYC (OR 3.1, 95% CI: 2.1–4.6, P < 0.001), patients who received a cumulative dose of CS >7.5 in the first year of treatment (OR 2.2 95% CI: 1.5–3.3, P < 0.001), and in those who developed leucopenia (OR 4.1 95% CI: 2.6–6.7, P < 0.001). Similarly, opportunistic infections were more prevalent in patients who received oral CYC (OR 2.5, 95% CI: 1.4–4.4, P = 0.002), a cumulative dose of CS >7.5 in the first year of treatment (OR 1.9, 95% CI: 1.1–3.4, P = 0.034), and in those who developed leucopenia (OR 4.6, 95% CI: 2.8–8.4, P < 0.001). Leucopenia was significantly related to oral CYC therapy (OR 6.7, 95% CI: 4.1–11.2, P < 0.001). Septicemia was more prevalent in patients with severe renal failure who needed dialysis (OR 4.0, 95% CI: 1.7–9.3).
A total of 164 (36.4%) patients experienced one or more disease relapses, with a mean rate of 2.5 ± 2.3 (range: 1–15). The number of relapses varied according to the type of AAV (Table 1), being more frequent in patients with GPA (P < 0.005). Regarding the ANCA specificity, patients with C-ANCA-PR3 suffered more relapses (71/147, 48.3%) than those with P-ANCA-MPO (72/204, 35.3%), OR 1.7, 95% CI: 1.1–2.6, P = 0.012. No relationship was observed between the initial disease severity and the occurrence of relapse (BVAS 19 ± 8.4 in the group with relapses and BVAS 17.3 ± 7.5 in the group without, P = 0.083).
One hundred twenty-nine (28.7%) patients died: 74 with MPA, 41 with GPA, and 14 with EGPA. Mortality was significantly higher in patients with MPA than in those with GPA and EGPA (OR 3.3, 95% CI: 2.1–5.1, P < 0.001). The median time to death was 58 months (IQR 105) for the total cohort, and was significantly lower in patients with MPA (P < 0.001), as shown in Table 1 and Fig. 1. With respect to ANCA, mortality was significantly higher in patients with positive ANCA at disease onset (OR 5.3, 95% CI: 2.1–13.4, P < 0.001) than in those with negative ANCA (Fig. 2), as well as in patients with P-ANCA-MPO compared to those with C-ANCA-PR3 (OR 2.0, 95% CI: 1.2–3.04, P < 0.001).
Figure 1.
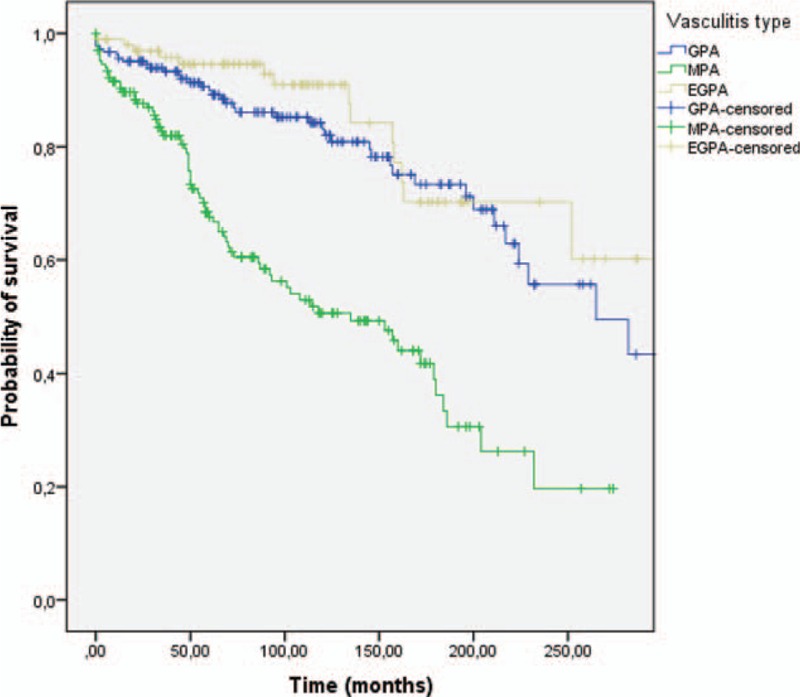
Long-term survival according to the type of AAV. Kaplan–Meier curve showing an association between survival and type of AAV (P < 0.001, log-rank < 0.001). EGPA = eosinophilic granulomatosis with polyangiitis, GPA = granulomatosis with polyangiitis, MPA = microscopic polyangiitis.
Figure 2.

Long-term survival according to the ANCA positivity. Kaplan–Meier curve showing an association between survival and ANCA positivity (P < 0.001). EGPA = eosinophilic granulomatosis with polyangiitis, GPA = granulomatosis with polyangiitis, MPA = microscopic polyangiitis.
Factors associated with death in univariate and multivariate analysis are listed in Table 5. Multivariate Cox regression analysis identified renal involvement (OR 2.5, 95% CI: 1.2–4.9), cardiac failure (OR 3.1, 95% CI: 1.1–8.6), and age higher than 65 years old (OR 3.1, 95% CI: 1.8–5.4) at disease onset as independent factors related to death. Bacterial infections were also independently related to death (OR 3.7, 95% CI: 2.1–6.6). In contrast, ENT involvement and mortality were inversely related (P < 0.001). There were no significant differences related to gender.
Table 5.
Factors related to death according to univariate and multivariate analysis.

3.7. Comparison between patients diagnosed before and after 2000
A total of 135/450 (30%) patients were diagnosed before 2000. No significant differences were noted between patients diagnosed before and after 2000, in terms of age at diagnosis, sex, cardiovascular risk factors, type of vasculitis, diagnosis delay, ANCA positivity and specificity, initial clinical symptoms and organ involvement, except for renal disease (Table 6). Indeed, only kidney involvement was significantly more common in patients diagnosed before 2000 than in those diagnosed later (69.6% vs 58.7%, P = 0.034).
Table 6.
Clinical, laboratory features and treatment, in patients diagnosed before and after 2000.

Patients diagnosed before 2000 received more frequently oral CYC as induction therapy than patients diagnosed after this date (OR 3.3, 95% CI: 2.6–5.2, P < 0.001). In contrast, patients diagnosed after 2000 received more frequently IV pulses of CYC as induction therapy than those diagnosed before 2000 (OR 2.2, 95% CI: 1.3–3.1, P = 0.001), and switched to another less toxic immunosuppressant drug (AZA, MMF) for maintenance therapy. This fact accounted for a reduced cumulated dose of CYC after 2000 (Table 6). In a similar way, most patients diagnosed after 2000 received high-dose of CS (>20 mg/day) for a shorter period of time, followed by a faster tapering, which lead to a cumulative dose of CS < 7.5 g in the first year of treatment in most cases. Indeed, before 2000, the dose of prednisone was usually reduced by 10 mg/day every 4 weeks up to 10 mg/day, and then 2.5 mg/day every month up to 5 mg/day, whereas after 2000 the dose was decreased more rapidly (10 mg/day every 2 weeks up to 20 mg/day, followed by 10 mg/day every 4 weeks up to 10 mg/day, and then 2.5 mg/day every month up to 5 mg/day), with a significant reduction of the total cumulative CS dose in the first year of treatment (from an average of 7.5 g before 2000 to an average of 4.5 g after 2000) (Table 6).
Bacterial infections were significantly more prevalent in patients diagnosed before 2000 (55%) than in those diagnosed later (33.3%) (OR 2.5, 95% CI: 1.6–3.3, P < 0.001), and were significantly related to treatment with oral CYC (P < 0.001).
Finally, mortality was significantly higher in patients diagnosed before 2000 (48%) than in those diagnosed later (20.3%) (OR 3.3, 95% CI: 2.34–5.61, P < 0.001).
4. Discussion
This is the largest series of patients with AAV reported from Spain. The onset, age, gender, and patterns of organ involvement in our patients were similar to those described in the main series in Europe and other countries,[5–9,23–29] though the mean age at the time of diagnosis was significantly higher in patients with MPA in our cohort, in agreement to that recently reported by Mohammad et al[24] and Sada et al.[29]
As already described,[5–9,27–29] the most common symptoms at the disease presentation were nonspecific (fever, fatigue, joint pain, weight loss), followed by respiratory and ENT manifestations. Most patients had multisystem involvement, but kidney and lung were the 2 most vulnerable organs to be involved. Indeed, renal involvement was present in over 60% of patients, and severe renal failure was the major cause for long-term end stage organ damage. Similarly, lung involvement was present in over 50% of patients, of whom 16.2% developed a pulmonary-renal syndrome, pulmonary hemorrhage being the major cause of acute respiratory failure requiring mechanical ventilation.
In line with previous studies,[11,13,23,25–29] we found significant differences in clinical features at disease presentation according to the different AAV categories. In this sense, renal involvement was almost constant in MPA but exceptional in EGPA; ENT involvement was predominant in GPA but rare in MPA; neurological and cardiac involvement was notably more prevalent in EGPA than in GPA and MPA, and pulmonary-renal syndrome was more frequent in MPA than in GPA and exceptional in EGPA. However, these features did not allow differentiating between the particular AAV clinical forms, similarly to the abnormal laboratory findings present at the time of diagnosis, which were nonspecific, though the presence of eosinophilia strongly suggested EGPA.
With respect to ANCA, we confirmed that C-ANCA-PR3 antibodies were mainly associated with GPA, and P-ANCA-MPO antibodies with MPA and EGPA, although this fact was not exclusive. Thus, 14.7% of patients with GPA showed P-ANCA-MPO, and about 10% of patients with MPA and EGPA showed C-ANCA-PR3, in consonance with other studies.[7,25,30] This fact emphasizes the importance of histopathology in AAV diagnosis accuracy, to avoid misdiagnosis.[1–9,27–29] In our series, diagnosis of vasculitis was supported by characteristic histopathologic findings in 81.8% of cases.
The median time to diagnosis was shorter in our series than reported in other studies,[25,27] and similar to that recently described by Mohammad et al.[24] The broad heterogeneity in the clinical features of patients at disease onset and the absence of ANCA, especially in limited forms of GPA and in a significant percentage of patients with EGPA, were probably related to the substantial delay in diagnosis observed in some cases. Misdiagnosis of nasal crusts and hearing loss in patients with GPA, lung infiltrates, and skin lesions in asthmatic patients with EGPA, and differences in the diagnosis work-up (testing for ANCA) in elderly patients with progressive renal failure, may probably explain the diagnostic delay.
The mean BVAS at diagnosis was comparable in our patients to that recorded in other large cohorts,[7,13,23,25,28] and was significantly related to the disease outcome, indicating that the extent of organ involvement is determinant in patient's survival. However, no relationship was found between the initial BVAS and the occurrence of relapses during the follow-up, in line with other studies.[7,9] In this sense, the number of disease relapses varied depending on the type of vasculitis and especially on the ANCA specificity. Relapses were more like to develop in patients with GPA and EGPA than in those with MPA, especially among patients with ENT manifestations, as previously described,[7,9,11,23,27,29] but above all, patients with C-ANCA-PR3 were almost twice as likely to experience a relapse as those with P-ANCA-MPO, consistent with previous reports.[11,24,29–32] These findings suggest that the ANCA specificity classification may be more strongly associated with outcomes such as relapse rate and death than the traditional GPA-MPA separation, as recently pointed out by Mahr et al.[33]
Concerning therapy, we found significant differences between treatment administered to patients diagnosed before and after 2000. In this regard, oral CYC was more commonly used for induction and maintenance therapy in patients diagnosed before 2000 than in those diagnosed later, especially in patients with granulomatous manifestations due to GPA which often are more refractory than those vasculitic. According to the European studies and recommendations,[10–11,34–37] after 2000 most patients with generalized or severe disease received intravenous pulses of CYC for induction therapy and switched to other less toxic immunosuppressant drugs for maintenance therapy, with a drastic reduction of the total cumulative CYC dose per patient. In a similar way, patients diagnosed after 2000 received high-doses of CS (>20 mg/day) for a shorter period, followed by a faster tapering, which lead to an important reduction of the cumulated dose of CS in the first year. Both facts were associated with a significant decrease of bacterial and opportunistic infections, and a lower mortality rate. Of note, we found the strongest relationship between bacterial and opportunistic infections and treatment with oral CYC, as reported by other authors.[6,23,38,39] Our findings confirm that the old CYC-based regimens are associated with a substantial morbidity, and emphasize the importance of tailoring treatment regimens according to the different clinical forms of AAV and disease severity at presentation. Additionally, our findings highlight the need for avoiding high-doses of CS therapy for a long time to avoid infections, as recently proposed.[39,40]
The overall mortality rate in our cohort was 28.7%, quite similar to that observed in the largest series of patients with AAV in other countries.[11,13,23–25,33] A higher mortality rate (33.9%) has recently been reported in Chinese patients, probably due to a higher percentage of patients with MPA.[41] In our study, mortality was clearly related to the disease severity and to secondary bacterial and opportunistic infections associated with treatment. Of particular interest, we found that the 3 clinical variables present at diagnosis with the greatest impact in mortality were renal failure, cardiac involvement, and age older than 65 years at disease onset. In contrast, ENT involvement seemed to be a protective factor, suggesting that vasculitic manifestations are more acute and life threatening than granulomatous manifestations, which are more likely to have a chronic course. Bacterial and opportunistic infections were also independent predictors of death, being both more like to develop in patients who received high doses of oral CYC and CS, especially in those patients who developed leucopenia, as reported by Charlier et al.[39] The most common bacterial infections were from urinary and respiratory tract, followed by generalized septicemia, which was significantly more frequent in patients requiring dialysis. Similar results have been found in former outcome studies.[7,11,13,23–25,35,39–48]
The presence of ANCA at diagnosis conferred an increased risk of death, suggesting that the absence of ANCA may indicate a variation in clinical presentation and prognosis, as already proposed.[11,25] Regarding the ANCA specificities, patients with P-ANCA-MPO had a worse prognosis than patients with C-ANCA-PR3 due to an increased risk for severe renal injury and alveolar hemorrhage, although patients with C-ANCA-PR3 experienced more relapses, our findings being in keeping with prior reports.[13,30]
Finally, we also found significant differences in terms of mortality according to the distinct AAV categories, with a worse prognosis for MPA mainly due to a greater rate of severe renal failure and an older age of patients at disease onset. Previous reports have suggested that MPA has a poorer prognosis due to more severe renal disease.[11,17,24,25,29,41,42] However, the influence of age at diagnosis is controversial[11,13,17,23,29] and discrepancies can only be addressed by larger prospective studies.
In contrast to other series,[7,11,29,41] we did not find a high mortality in patients with lung involvement, neither in the entire cohort nor according to the different clinical forms, although pulmonary-renal syndrome was clearly related to a worse prognosis. Additionally, we did not find significant differences related to gender.
When compared patients diagnosed before and after 2000, we found a significant decrease in renal involvement, whereas the frequency of other organ manifestations did not change significantly as recently reported by Holle et al,[42] and Eriksson et al.[49] Of note, the proportion of patients receiving oral CYC decreased drastically after 2000, and the rate of infections and mortality declined steadily. Both, the reduction in renal involvement, as well as the improved outcome after 2000, may be related to an increased awareness of AAV and their manifestations, and an earlier introduction of therapy together with the implementation of less toxic regimens adapted to the disease activity and stage. Our findings corroborate those from other studies regarding nearly unchanged organ manifestations at AAV diagnosis, but a rise in mean age at disease onset,[24,40,49] and more important, our findings confirm an overall improvement in the prognosis of vasculitis over the last decades, with a clear decline of mortality after 2000, in line with recent studies.[42,48–50]
The main strengths of the present study are the recruitment of a large cohort of patients from the same geographical zone and the very long-term follow-up. However, some limitations are recognized such as the retrospective design, and that patients were treated according to the physician discretion. Despite those limitations, we believe that this study represents a real picture of the Spanish patients with AAV.
In summary, this is the first nationwide survey of Spanish patients with AAV. Patient characteristics and disease activity were similar in our series to those from other world regions. Mortality was significantly associated with disease severity at presentation. In contrast, no relationship was observed between the initial disease severity and the occurrence of disease relapses. Patients with positive ANCA, renal involvement, heart failure, and age older than 65 years at disease onset, had the poorer prognosis. Treatment-related infections were a general and independent predictor of death, and were significantly related to cumulative high doses of CYC and CS. Patients with MPA showed the highest mortality rate while patients with GPA showed the highest rate of relapse. An improved outcome, with reduced morbidity and mortality, was observed in patients diagnosed after 2000 that was primarily related to the implementation of less toxic regimens adapted to disease severity. However, despite these advances a considerable mortality and treatment-related morbidity remained. Our results underscore the need for further immunosuppressant, and especially new therapeutic strategies, which allow reducing CYC and CS cumulative doses, and improve the prognosis of patients with AAV.
Footnotes
Abbreviations: AAV = ANCA-associated vasculitis, ANCA = antineutrophil cytoplasmatic antibody, anti-MPO = antimyeloperoxidase, anti-PR3 = antiproteinase 3, BVAS = Birmingham Vasculitis Activity Score, CI = confidence interval, CRP = C-reactive protein, CS = corticosteroids, CYC = cyclophosphamide, eGFR = estimated glomerular filtration rate, EGPA = eosinophilic granulomatosis with polyangiitis, ELISA = antigen-specific enzyme-linked immunosorbent assay, ENT = ear, nose, and throat, ESR = erythrocyte sedimentation rate, FFS = Five Factors score, GPA = granulomatosis with polyangiitis, IIF = indirect immunofluorescence, IQR = interquartile range, MMF = mycophenolate mofetil, MPA = microscopic polyangiitis, MTX =methotrexate, OR = odds ratio, RTX = Rituximab, WBC = white blood count.
The authors have no conflicts of interest to disclose.
References
- [1].Pagnoux C. Updates in ANCA-associated vasculitis. Eur J Rheumatol 2016;3:122–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Leavitt RY, Fauci AS, Bloch DA, et al. The American College of Rheumatology criteria for the classification of Wegener's Granulomatosis. Arthritis Rheum 1990;33:1101–7. [DOI] [PubMed] [Google Scholar]
- [3].Masi AT, Hunder GG, Lie JT, et al. The American college of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (Allergic granulomatosis and angiitis). Arthritis Rheum 1990;33:1094–100. [DOI] [PubMed] [Google Scholar]
- [4].Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013;65:1–1. [DOI] [PubMed] [Google Scholar]
- [5].Fauci AS, Haynes BF, Katz P, et al. Wegener's granulomatosis: prospective clinical and therapeutic experience with 85 patients for 21 years. Ann Intern Med 1983;98:76–85. [DOI] [PubMed] [Google Scholar]
- [6].Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992;116:488–98. [DOI] [PubMed] [Google Scholar]
- [7].Guillevin L, Durand-Gasselin B, Cevallos R, et al. Microscopic polyangiitis: clinical and laboratory findings in eighty-five patients. Arthritis Rheum 1999;42:421–30. [DOI] [PubMed] [Google Scholar]
- [8].Lanham JG, Elkon KB, Pusey CD, et al. Systemic vasculitis with asthma and eosinophilia: a clinical approach to the Churg Strauss syndrome. Medicine (Baltimore) 1984;63:65–81. [DOI] [PubMed] [Google Scholar]
- [9].Comarmond C, Pagnoux C, Khellaf M, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss). Clinical characteristics and long-term follow-up of the 383 Patients enrolled in the French Vasculitis Study Group Cohort. Arthritis Rheum 2013;65:270–81. [DOI] [PubMed] [Google Scholar]
- [10].Jayne D. Evidence based-treatment of systemic vasculitis. Rheumatology (Oxford) 2000;39:585–95. [DOI] [PubMed] [Google Scholar]
- [11].Mukhtyar C, Flossmann O, Hellmich B, et al. Outcome from studies of antineutrophil cytoplasm antibody associated vasculitis: a systematic review by the European League Against Rheumatism systemic vasculitis task force. Ann Rheum Dis 2008;67:1004–10. [DOI] [PubMed] [Google Scholar]
- [12].Robson J, Doll H, Suppiah R, et al. Damage in the ANCA-associated vasculitides: long-term data from the European Vasculitis Study group (EUVAS) therapeutic trials. Ann Rheum Dis 2015;74:177–84. [DOI] [PubMed] [Google Scholar]
- [13].Flossmann O, Berden A, De Groot K, et al. Long-term patient survival in ANCA-associated vasculitis. Ann Rheum Dis 2011;70:488–94. [DOI] [PubMed] [Google Scholar]
- [14].Solans R, Bosch JA, Perez-Bocanegra C, et al. Churg-Strauss syndrome: outcome and long-term follow-up of 32 patients. Rheumatology (Oxford) 2001;40:763–71. [DOI] [PubMed] [Google Scholar]
- [15].Ríos JJ, Gómez Cerezo J, Súarez I, et al. Churg-Strauss syndrome. Our two-decade experience. Rev Clin Esp 2000;200:597–601. [PubMed] [Google Scholar]
- [16].Solans-Laqué R, Bosch-Gil JA, Canela M, et al. Clinical features and therapeutic management of subglottic stenosis in patients with Wegener's granulomatosis. Lupus 2008;17:832–6. [DOI] [PubMed] [Google Scholar]
- [17].Borao-Cengotita-Bengoa M, Corral-Gudino L, Del Pino-Montes J, et al. Long-term follow-up of microscopic polyangiitis. 17-year experience at a single center. Eur J Intern Med 2010;21:542–7. [DOI] [PubMed] [Google Scholar]
- [18].Merino JL, Galeano C, Espejo B, et al. A retrospective study on outcome of microscopic polyangiitis in chronic replacement therapy. Nephrol Dial Transplant 2011;26:1360–6. [DOI] [PubMed] [Google Scholar]
- [19].Morales-Angulo C, García-Zamora R, Obeso-Agüera S, et al. Ear, nose and throat manifestations of Wegener's granulomatosis (granulomatosis with polyangiitis). Acta Otorrinolaringol Esp 2012;63:206–11. [DOI] [PubMed] [Google Scholar]
- [20].Marco H, Mirapeix E, Arcos E, et al. Long-term outcome of antineutrophil cytoplasmic antibody-associated small vessel vasculitis after renal transplantation. Clin Transplant 2013;27:338–47. [DOI] [PubMed] [Google Scholar]
- [21].Luqmani RA, Bacon PA, Moots RJ, et al. Birmingham Vasculitis Activity Score (BVAS) in systemic necrotizing vasculitis. Q J Med 1994;87:671–8. [PubMed] [Google Scholar]
- [22].Guillevin L, Lhote F, Gayraud M, et al. Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome. A prospective study in 342 patients. Medicine (Baltimore) 1996;75:17–28. [DOI] [PubMed] [Google Scholar]
- [23].Gayraud M, Guillevin L, le Toumelin P, et al. Long-term follow-up of polyarteritis nodosa, microscopic polyangiitis, and Churg-Strauss syndrome: analysis of four prospective trials including 278 patients. Arthritis Rheum 2001;44:666–75. [DOI] [PubMed] [Google Scholar]
- [24].Mohammad J, Jacobsson LTH, Westman KWA, et al. Incidence and survival rates in Wegener's granulomatosis, microscopic polyangiitis, Churg-Strauss syndrome and polyarteritis nodosa. Rheumatology (Oxford) 2009;48:1560–5. [DOI] [PubMed] [Google Scholar]
- [25].Lane SE, Watts RA, Shepstone L, et al. Primary systemic vasculitis: clinical features and mortality. Q J Med 2005;98:97–111. [DOI] [PubMed] [Google Scholar]
- [26].Pavone L, Grasselli C, Chierici E, et al. Outcome and prognostic factors during the course of primary small-vessel vasculitides. J Rheumatol 2006;33:1299–306. [PubMed] [Google Scholar]
- [27].Chen M, Yu F, Zhang Y, et al. Clinical and pathological characteristics of Chinese patients with antineutrophil cytoplasmic autoantibody associated systemic vasculitides: a study of 426 patients from a single centre. Postgrad Med J 2005;81:723–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Shuai ZW, Lv YZ, Zhang MM, et al. Clinical analysis of patients with myeloperoxidase antineutrophil cytoplasma antibody-associated vasculitis. Genet Mol Res 2015;14:5296–303. [DOI] [PubMed] [Google Scholar]
- [29].Sada K, Yamamura M, Harigai M, et al. Classification and characteristics of Japanese patients with antineutrophil cytoplasmic antibody-associated vasculitis, in a nationwide prospective, inception cohort study. Arthritis Res Ther 2014;16:R101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lionaki S, Blyth ER, Hogan SL, et al. Classification of antineutrophil cytoplasmic autoantibody vasculitides: the role of antineutrophil cytoplasmic autoantibody specificity for myeloperoxidase or proteinase 3 in disease recognition and prognosis. Arthritis Rheum 2012;64:3452–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Pagnoux C, Hogan SL, Chin H, et al. Predictors of treatment resistance and relapse in antineutrophil cytoplasmic antibody-associated small-vessel vasculitis: comparison of two independent cohorts. Arthritis Rheum 2008;58:2908–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Walsh M, Flossmann O, Berden A, et al. Risk factors for relapse of antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum 2012;64:542–8. [DOI] [PubMed] [Google Scholar]
- [33].Mahr A, Katsahian S, Varet H, et al. Revisiting the classification of clinical phenotypes of anti-neutrophil cytoplasmic antibody-associated vasculitis: a cluster analysis. Ann Rheum Dis 2013;72:1003–10. [DOI] [PubMed] [Google Scholar]
- [34].Jayne D, Rasmussen N, Andrassy K, et al. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med 2003;349:36–44. [DOI] [PubMed] [Google Scholar]
- [35].De Groot K, Rasmussen N, Bacon PA, et al. Randomized trial of cyclophosphamide versus methotrexate for induction of remission in early systemic antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum 2005;52:2461–9. [DOI] [PubMed] [Google Scholar]
- [36].De Groot K, Harper L, Jayne DR, et al. Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial. Ann Intern Med 2009;150:670–80. [DOI] [PubMed] [Google Scholar]
- [37].Pagnoux C, Mahr A, Hamidou MA, et al. Azathioprine or methotrexate maintenance for ANCA-associated vasculitis. N Engl J Med 2008;359:2790–803. [DOI] [PubMed] [Google Scholar]
- [38].Langford C. Cyclophosphamide as induction therapy for Wegener's granulomatosis and microscopic polyangiitis. Clin Exp Immunol 2011;164(suppl 1):31–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Charlier C, Henegar C, Launay O, et al. Risk factors for major infections in Wegener granulomatosis: analysis of 113 patients. Ann Rheum Dis 2009;68:658–63. [DOI] [PubMed] [Google Scholar]
- [40].McGregor JAG, Hogan SL, Hu Y, et al. Glucocorticoids and relapse and infection rates in anti-neutrophil cytoplasmic antibody disease. Clin J Am Soc Nephrol 2012;7:240–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lai QY, Ma TT, Li ZY, et al. Predictors for mortality in patients with antineutrophil cytoplasmic autoantibody-associated vasculitis: a study of 398 Chinese patients. J Rheumatol 2014;41:1849–55. [DOI] [PubMed] [Google Scholar]
- [42].Holle J, Gross WL, Latza U, et al. Improved outcome in 445 patients with Wegener's granulomatosis in a German Vasculitis Center over four decades. Arthritis Rheum 2011;63:256–66. [DOI] [PubMed] [Google Scholar]
- [43].Bligny D, Mahr A, Toumelin PL, et al. Predicting mortality in systemic Wegener's granulomatosis: a survival analysis based on 93 patients. Arthritis Rheum 2004;51:83–91. [DOI] [PubMed] [Google Scholar]
- [44].Mahr A, Girard T, Agher R, et al. Analysis of factors predictive of survival based on 49 patients with systemic Wegener's Granulomatosis and prospective follow-up. Rheumatology (Oxford) 2001;40:492–8. [DOI] [PubMed] [Google Scholar]
- [45].Koldingsnes W, Nossent H. Predictors of survival and organ damage in Wegener's granulomatosis. Rheumatology (Oxford) 2002;41:572–81. [DOI] [PubMed] [Google Scholar]
- [46].Guillevin L, Pagnoux C, Seror R, et al. The Five-Factor Score Revisited Assessment of Prognoses of Systemic Necrotizing Vasculitides Based on the French Vasculitis Study Group (FVSG) Cohort. Medicine (Baltimore) 2011;90:19–27. [DOI] [PubMed] [Google Scholar]
- [47].Little MA, Nightingale P, Verburgh CA, et al. Early mortality in systemic vasculitis: relative contribution of adverse events and active vasculitis. Ann Rheum Dis 2010;69:1036–43. [DOI] [PubMed] [Google Scholar]
- [48].Abdou NI, Kullman GJ, Hoffman GS, et al. Wegener's Granulomatosis: survey of 701 patients in North America. Changes in outcome in the 1990s. J Rheumatol 2002;29:309–16. [PubMed] [Google Scholar]
- [49].Eriksson P, Jacobsson L, Lindell A, et al. Improved outcome in Wegener's granulomatosis and microscopic polyangiitis? A retrospective analysis of 95 cases in two cohorts. J Intern Med 2009;265:496–506. [DOI] [PubMed] [Google Scholar]
- [50].Stratta P, Marcuccio C, Campo A, et al. Improvement in relative survival of patients with vasculitis: study of 101 cases compared to the general population. Int J Immunopathol Pharmacol 2008;21:631–42. [DOI] [PubMed] [Google Scholar]